

Abstract
COVID-19 disease caused by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) was declared a global pandemic by the World Health Organization (WHO) in March 2020. Since then, the SARS-CoV-2 virus has impacted millions of lives worldwide. Various preclinical and clinical trials on the treatment of COVID-19 disease have revealed that the drugs that work in combination are more likely to reduce reinfection and multi-organ failure. Considering the combination drug therapy, herein, we performed a systematic computational study starting with the formation of sixty-two combinations of drugs and phytochemicals with 2-deoxy-D-glucose (2-DG). The top nineteen combinations resulting from Drug-Drug Interaction (DDI) analysis were selected for individual and multiple-ligand-simultaneous docking (MLSD) study with a host target Serine Protease (TMPRSS2; PDB ID: 7MEQ) and two viral targets, Main Protease (3CLpro; PDB ID: 6LU7) and Uridylate-Specific Endoribonuclease (NSP15; PDB ID: 6VWW). We found that the resulting drugs and phytochemicals in combination with 2-DG shows better binding than the individual compounds. We performed the re-docking of the top three drug combinations by utilizing the polypharmacology approach to validate the binding patterns of drug combinations with multiple targets for verifying the best drug combinatorial output obtained by blind docking. A strong binding affinity pattern was observed for 2-DG + Ruxolitinib (NIH-recommended drug), 2-DG + Telmisartan (phase 4 clinical trial drug), and 2-DG + Punicalagin (phytochemical) for all the selected targets. Additionally, we conducted multiple-ligand-simultaneous molecular dynamics (MLS-MD) simulations on the selected targets with the 2-DG + Ruxolitinib combination. The MLS-MD analysis of the drug combinations shows that stabilization of the interaction complexes could have significant inhibition potential against SARS CoV-2. This study provides an insight into developing drug combinations utilizing integrated computational approaches to uncover their potential in synergistic drug therapy.
Supplementary Information
The online version contains supplementary material available at 10.1007/s11224-022-02049-0.
Keywords: COVID-19, Computational screening, Multiple ligand simultaneous docking, Synergistic effect, Combination drug therapy, Polypharmacology
Introduction
In March 2020, the World Health Organization (WHO) declared SARS-COV-2 a global pandemic [1]. SARS-CoV-2 access the human body through a droplet/air particle exposure causing severe acute respiratory distress syndrome (ARDS) with pneumonia. As of August 19, 2022, about 591,683,619 cases of COVID-19 registered worldwide, with 6,443,306 deaths (https://covid19.who.int/). With such a large number of infections and deaths, it is critical to develop an effective treatment and affordable therapies to recover from the disease. The majority of SARS-CoV-2 cases are asymptomatic or only minor flu-like symptoms; however, serious cases of viral pneumonia occur between 1.0% of those who are greater than or equal to 20 years old (≥ 20 years) and 18.4% of those who are greater than and equal to 80 years old (≥ 80 years) [2]. Furthermore, patients with pre-existing diseases such as hypertension, cardiovascular, heart failure, obesity, pulmonary diseases, and diabetes heighten the risk of severe SARS-CoV-2 infection [3]. Coronaviruses (CoVs) are a family of beta coronaviruses, a positive-sense, single-stranded RNA virus, which was preliminarily found in bats, and now it can infect humans as well [4]. Many aspects of viral and host proteases have been identified as a result of biological insights into the genomic and structural properties of SARS-CoV-2. The initial phase of SARS-CoV-2 entry in the human body is by linking its spike glycoprotein (S2) to host cellular receptors of angiotensin-converting enzyme 2 (ACE2), serine protease (TMPRSS2), cathepsin L, cathepsin B, and furin coronavirus [5]. When the virus reaches the host cell, it uses the main protease (3CLpro) and uridylate-specific endoribonuclease (NSP15) to begin replication. The viral genome encodes a 3-chymotrypsin-like protease (3CLpro), which is crucial for generating multiple non-structural proteins (NSPs) required for viral replication, including the main protease (Mpro). Mpro plays a vital role in the digestion of polyproteins translated from viral RNA, and SARS-CoV-2 RNA-dependent RNA polymerase (RdRp) is involved in viral genome replication and viral gene transcription [6, 7].
Several repurposed drug molecules have been approved for treating SARS-CoV-2 patients and are available in the NIH-recommended drug guideline 2021 [https://www.covid19treatmentguidelines.nih.gov/whats-new/]. The Drug Controller General of India (DCGI) recently approved 2-DG as an additional medication for emergency usage. Several studies have shown that 2-DG has antiviral properties owing to its dual inhibitor role [https://cdsco.gov.in/opencms/opencms/en/Home/], which are directly linked to 2-DG’s interaction with the virus (avoiding viral entrance into the host cells) as well as glycolysis suppression, which compromises the high energy requirement [8, 9]. It has been observed that host cells undergo metabolic change after virus entry to satisfy the increased nutritional need for virus reproduction interfered by 2-DG. Recently, some studies recommended that 2-DG binds to the protease 3CLpro and NSP15 endoribonuclease to prevent SARS-CoV-2 receptors from attaching to the host cells [10, 11]. Several treatments and therapies are still emerging to treat SARS-CoV-2, such as plasma therapy, repurposed drugs, and combination/synergistic therapy [12]. Distinct repurposed drug compounds have received emergency authorization from the U.S. Food and Drug Administration (USFDA) and other regulatory agencies worldwide [13]. The scarcity of prospective coronavirus therapies increased the demand for identifying potential drug combinations with current medications with well-established safety profiles to reduce reinfection and other complications of multi-organ response. Synergistic therapy can minimize drug doses while reducing the risk of harmful side effects during treatment. Barcitinib/Remedsivir, Lopinavir/Ritonavir, and Hydroxychloroquine/Azithromycin are currently being used as combination drugs for the therapy/treatment of SARS-CoV-2 patients, and clinical trials for the combination of Traditional Chinese Medicine (TCM) and western medicine are going on [14]. An effective combination therapy reported in a recent study on anticoagulation drugs combined with corticosteroids and/or remdesivir resulted in a lower mortality rate [15]. Here, we investigated using integrated framework of computational approaches for developing potential synergetic drug therapeutic combinations for the inhibition of microbial pathogenesis by considering drugs and phytochemicals in combination with 2-deoxy-D-glucose against SARS-CoV-2. In this context, the synergetic effects between the nineteen approved SARS-CoV-2 drugs and selected phytochemicals with 2-DG could act as potential modulators on simultaneous inhibition and disrupting the stability of the selected host (TMPRSS2) and viral targets (3CLpro and NSP15) responsible for viral pathogenies.
Material and methods
Drug library generation
A library was generated by collecting different categories of drugs and phytochemicals from an intense literature survey. Fourteen approved SARS-CoV-2 drugs with different categories of antiviral, immunomodulators, kinase inhibitors, and anti-fungal were extracted from NIH-recommended drugs (https://www.covid19treatmentguidelines.nih.gov). Thirty-nine approved SARS-CoV-2 phytochemicals were obtained from the literature [16], and eight drugs from phase 4 of clinical trials (https://clinicaltrials.gov/) along with 2-DG were taken into consideration. We initiated the formation of a drug combination wherein each of the 61 drugs was manually combined with 2-DG. After collecting drugs, we downloaded their 3D chemical structure in.sdf file format from the PubChem database [17] and imported it into Maestro, Schrödinger, 2022 for minimization of the molecule and then saved it into.pdb format.
Drug-drug interaction prediction (DDI)
Drug-drug interaction prediction was carried out based on OpeRationalClassificAtion (ORCA) [18] using the PASS DDI-pred server (http://way2drug.com/PassOnline/) [19]. A dataset of thousands of drug pairs was used to train a model for the DDI-pred tool. This tool uses a set of machine-readable chemical descriptors called PoSMNA (Pairs of Substances Multilevel Neighborhoods of Atoms) to describe drug pairs. The structures (.smi format) of sixty-two compounds along with 2-DG were extracted from PubChem, and combinations of these compounds with 2-DG were created. Further, each drug combination was uploaded to the DDI server to identify the interaction between them.
Selection and preparation of macromolecule
The crystal structure of the SARS-CoV-2 3CLpro (PDB ID: 6LU7), NSP15 (PDB ID: 6VWW), and TMPRSS2 (PDB ID: 7MEQ) was retrieved from RCSB Protein Data Bank (http://www.rcsb.org). Water molecules, heteroatoms, and inbuilt ligand groups already present in crystal structures were removed using the BIOVIA discovery studio [20]. Further, polar hydrogen atoms, charges, and other AutoDock parameters were added to the target protein structures using AutoDock Tools [21] and were saved into.pdbqt format for further analysis.
Molecular docking of individual drug compounds
The blind docking of drugs, phytochemicals along with 2-DG was performed using AutoDock Vina [22]. The files for the ligand and targets were converted into.pdbqt format. Additionally, the configuration file is created with defined grid centers and dimensions, as mentioned in Supplementary Table 3. Furthermore, the lowest binding energy complex was extracted for each compound. Binding interactions between protein–ligand complexes were depicted by using the Discovery studio visualizer.
Multiple ligand simultaneous docking (MLSD) studies
In the presence of ligand, a multi-ligand simultaneous docking method was used to investigate the simultaneous interaction of the macromolecule and the ligands. MLSD computational approach thus can be used to study the interaction of multiple ligands. The current version of MLSD uses AutoDock Vina 1.2.0 (https://github.com/ccsb-scripps/AutoDock-Vina) algorithms, and scoring function were used to evaluate the interaction of various ligands with the target protein. We initiated with MLSD; the ligand and target files were converted into.pdbqt file format. Further, the configuration file was formed with specified grid dimensions and centers, as depicted in Supplementary Table 4. Autodock Vina run was performed using a specific command, and the best complex was chosen for analysis.
Redocking and validation
Redocking studies were executed to validate the compounds showing the best binding energy obtained by blind docking. We analyzed the active site residues of all three targets from available literature [23, 24] and performed individual ligand docking and MLSD with the above protocol [25, 26]. The best poses were selected based on maximum interactions depicted for minimum binding energy conformations, and these interactions between protein–ligand complexes were then visualized using the Discovery studio software.
Multiple ligand simultaneous molecular dynamics simulations
The targets 3CLpro, NSP15, and TMPRSS2 with drugs in combination with Ruxolitinib + 2-DG were simulated using GROMACS 2015 (Groningen Machine for Chemical Simulations) [27], which was used to investigate the compounds interactions, conformational changes, and dynamic binding properties. The generation of a protein topology file and the formation of the molecular topology of the ligands were created using the CGenFF web server (https://cgenff.umaryland.edu). The complex files were created by merging the topologies of both ligand and protein. The complexes in a box with dimensions x-10, y-10, and z-10 were solvated by simple point charge (SPC) water molecules at a distance of 1.0 nm, and they were subsequently neutralized by adding a proper quantity of (Na + /Cl) counter ions. For the systems energy minimization, 50,000 steepest descent algorithm steps were taken with a 10 kJ/mol/nm tolerance. Additionally, systems were calibrated using NVT (canonical) and NPT (isothermal-isobaric) ensembles for 1000 ps at 300 K and 1 bar of pressure. The full-scale equilibrated systems were simulated for 20 ns. Origin software was used to analyze the data [28]. Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), and the radius of gyration (Rg) were used to analyze further the trajectories generated by MLS-MD simulation.
Results and discussion
A total of sixty-two selected drugs and phytochemicals were classified into three categories: (a) National Institute of Health (NIH) recommended drugs, (b) phase 4 clinical trial drugs, and (c) phytochemicals. Refer to Supplementary Table 1 for details of selected drugs and phytochemicals. Drug therapeutic activities are frequently altered by drug co-administration, which may result in unavoidable drug-drug interactions (DDIs) and leads to adverse effects [29]. Assessment and identification of drug-drug interactions are vital for the safety of patients and the effectiveness of treatment therapies. Undesired drug reactions may also cause a reduction of pharmacological effects [30]. The DDI-pred a web server of the Way2Drug platform was used for the DDIs predictions. The DDIs were further categorized based on ORCA classification. Classification of DDIs by ORCA is as follows: contraindicated comes under class 1, provisionally contraindicated comes under class 2, conditional comes under class 3, little risk comes under the class 4, and no interaction comes under the class 5. We have made a combination of sixty-two drugs and phytochemicals as variable and 2-DG as constant for prediction of DDI (Table 1). Out of sixty-two combinations, only nineteen of them (Table 2) show no interaction; hence, these combinations were taken into consideration for further study.
Table 1.
OpeRational classification of drug-drug interaction prediction using the PASS server
| S. no. | Drugs | Combinations | DDI |
|---|---|---|---|
| NIH-recommended drugs | |||
| 1 | Antiviral drugs | 2-DG + Remdesvir | Class-2 |
| 2 | 2-DG + Lopinavir | Class-2 | |
| 3 | 2DG + Ritonavir | Class-2 | |
| 4 | 2-DG + Hydroxychloroquine | No-interaction | |
| 5 | 2-DG + Azithromycin | Class-2 | |
| 6 | 2-DG + Ivermectin | No-interaction | |
| 7 | Immunomodulators | 2-DG + Baricitinib | No-interaction |
| 8 | 2-DG + Colchicine | Class-2 | |
| 9 | Kinase inhibitors | 2-DG + Acalabrutinib | Class-2 |
| 10 | 2-DG + Ibrutinib, | Class-2 | |
| 11 | 2-DG + Zanubrutinib | Class-2 | |
| 12 | 2-DG + Ruxolitinib, | No-interaction | |
| 13 | 2-DG + Tofacitinib | No-interaction | |
| 14 | Anti-fungal | 2-DG + Amphotericin B | Class-2 |
| Clinical trial drugs | |||
| 15 | 2-DG + Telmisartan | No-interaction | |
| 16 | 2-DG + Artemisinin | Class-2 | |
| 17 | 2-DG + Cyclosporin A | Class-2 | |
| 18 | 2-DG + Colchicine | Class-2 | |
| 19 | 2-DG + Arbidol | No-interaction | |
| 2-DG + Losartan | Class-2 | ||
| 21 | 2-DG + inhaled budesonide | Class-2 | |
| 22 | 2-DG + Pioglitazone | Class-2 | |
| Phytochemicals | |||
| 23 | SARS-CoV-2 | 2-DG + Berbamine | Class-2 |
| 24 | 2-DG + Cepharanthine | No-interaction | |
| 25 | 2-DG + Emetine | Class-2 | |
| 26 | 2-DG + Brazilin | Class-2 | |
| 27 | 2-DG + Chrysanthemin | No result | |
| 28 | 2-DG + Epigallocatechin-3-gallate | No result | |
| 29 | 2-DG + Herbacetin | Class-2 | |
| 30 | 2-DG + Isorhamnetin | Class-2 | |
| 31 | 2-DG + Kaempferol | Class-2 | |
| 32 | 2-DG + Myricetin | Class-2 | |
| 33 | 2-DG + Naringenin | Class-2 | |
| 34 | 2-DG + Panduratin A | Class-2 | |
| 35 | 2-DG + Pectolinarin | No-interaction | |
| 36 | 2-DG + Quercetagetin | Class-2 | |
| 37 | 2-DG + Quercetin | Class-2 | |
| 38 | 2-DG + Rutin | Class-2 | |
| 39 | 2-DG + Scutellarein | Class-2 | |
| 40 | 2-DG + Theaflavin 3,3'-di-O-gallate | Class-2 | |
| 41 | 2-DG + 6-Gingerol | Class-2 | |
| 42 | 2-DG + Acteoside | Class-2 | |
| 43 | 2-DG + Andrographolide | No-interaction | |
| 44 | 2-DG + Betulin | No-interaction | |
| 45 | 2-DG + Betulinic acid | Class-2 | |
| 46 | 2-DG + Chebulagic acid | No-interaction | |
| 47 | 2-DG + Chlorogenic acid | Class-2 | |
| 48 | 2-DG + Cryptotanshinone | Class-2 | |
| 49 | 2-DG + Curcumin | Class-2 | |
| 50 | 2-DG + Dihydrotanshinone I | Class-2 | |
| 51 | 2-DG + Ellagic acid | Class-2 | |
| 52 | 2-DG + Glycyrrhizin | No-interaction | |
| 53 | 2-DG + Hypericin | Class-2 | |
| 54 | 2-DG + Maclurin | Class-2 | |
| 55 | 2-DG + Maslinic acid | No-interaction | |
| 56 | 2-DG + Nordihydroguaiaretic acid | Class-2 | |
| 57 | 2-DG + Platycodin D | No-interaction | |
| 58 | 2-DG + Punicalagin | No-interaction | |
| 59 | 2-DG + Sennoside B | No-interaction | |
| 60 | 2-DG + Tannic acid | No-interaction | |
| 61 | 2-DG + Tanshinone I | Class 2 | |
| 62 | 2-DG + Ursolic acid | No interaction | |
Table 2.
Chemical structures and their PubChem identifier for selected nineteen drugs/phytochemicals and 2-DG
| S. no. | Name of drug/phytochemicals | PubChem CID | Structures |
|---|---|---|---|
| 1 | 2-d-deoxyglucose (2-DG) | 108223 |
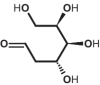
|
| 2 | Baricitinib | 44205240 |
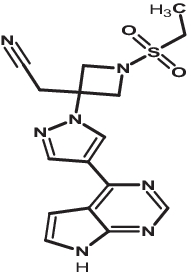
|
| 3 | Hydroxychloroquine | 3652 |
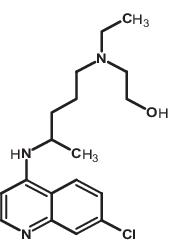
|
| 4 | Ivermectin | 6321424 |
|
| 5 | Ruxolitinib | 25126798 |
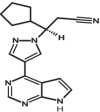
|
| 6 | Tofacitinib | 9926791 |
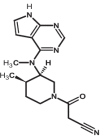
|
| Clinical trial drugs | |||
| 7 | Telmisartan | 65999 |
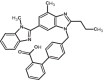
|
| 8 | Arbidol | 131411 |
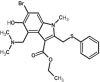
|
| Phytochemicals | |||
| 9 | Glycyrrhizin | 14982 |
|
| 10 | Platycodin D | 162859 |
|
| 11 | Punicalagin | 44584733 |
|
| 12 | Sennoside B | 91440 |
|
| 13 | Andrographolide | 5318517 |
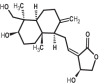
|
| 14 | Betulin | 72326 |
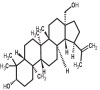
|
| 15 | Cepharanthine | 10206 |
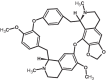
|
| 16 | Chebulagic acid | 442674 |
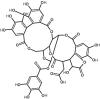
|
| 17 | Maslinic acid | 73659 |
|
| 18 | Pectolinarin | 168849 |
|
| 19 | Tannic acid | 16129778 |
|
| 20 | Ursolic acid | 64945 |
|
An automated individual and MLSD study was performed using Autodock Vina to explore the binding energies, the orientation of ligands in the active pockets, and active residues. The binding energies of individual ligand with specific targets 3CLpro, NSP15, and TMPRSS2 for each category, NIH-recommended drugs (2D-G, Ivermectin, Baricitinib, Ruxolitinib, Hydroxychloroquine Tofacitinib), clinical trial drugs (Telmisartan, Arbidol), and selected phytochemicals are shown in Table 3.
Table 3.
Binding energies (in kcal/mol) obtained for individual drugs/phytochemicals interaction with the viral and host targets
| S. no. | Drug | 3CLpro | NSP15 | TMPRSS2 |
|---|---|---|---|---|
| NIH-recommended drugs | ||||
| 1 | 2-DG | −4.6 | −5 | −4.7 |
| 2 | Ivermectin | −8.1 | −9.3 | −7.8 |
| 3 | Baricitinib | −7.4 | −8 | −6.5 |
| 4 | Ruxolitinib | −6.9 | −8.5 | −6.5 |
| 5 | Hydroxychloroquine | −6 | −7.3 | −6.1 |
| 6 | Tofacitinib | −7.2 | −7.4 | −6.2 |
| Clinical trial drugs | ||||
| 7 | Telmisartan | −8.6 | −10.01 | −8.30 |
| 8 | Arbidol | −5.698 | −7.002 | −5.164 |
| Phytochemicals | ||||
| 9 | Glycyrrhizin | −8.651 | −9.873 | −8.912 |
| 10 | Platycodin_D | −7.275 | −7.256 | −7.621 |
| 11 | Punicalagin | −9.091 | −9.636 | −8.779 |
| 12 | Sennoside_B | −8.164 | −8.735 | −8.075 |
| 13 | Andrographolide | −7.13 | −8.82 | −7.34 |
| 14 | Betulin | −7.57 | −8.35 | −7.29 |
| 15 | Cepharanthine | −8.47 | −9.4 | −8.88 |
| 16 | Chebulagic acid | −8.19 | −9.95 | −9.57 |
| 17 | Maslinic acid | −7.79 | −8.96 | −8.05 |
| 18 | Pectolinarin | −7.96 | −8.96 | −7.76 |
| 19 | Tannic acid | −9.98 | −10.88 | −8.79 |
| 20 | Ursolic acid | −8.36 | −9.08 | −8.22 |
Upon analysis of the docking result of NIH-recommended drugs, Ruxolitinib, a Janus kinase inhibitor, was found to have binding energies with all three selected targets 3CLpro, NSP15, and TMPRSS2, as −6.9 kcal/mol, −8.5 kcal/mol, and −6.5 kcal/mol, respectively (Fig. 1). In addition, the drug from clinical trial drug Telmisartan, which is used to treat high blood pressure, has a binding energy of −8.6 kcal/mol with 3CLpro, −10.01 kcal/mol with NSP15, and −8.30 kcal/mol with TMPRSS2 target. On the other hand, Punicalagin, the primary component of pomegranates that is currently being investigated as a potential antiviral phytochemical, was shown to have binding energies of −9.09 kcal/mol, −9.63 kcal/mol, and −8.77 kcal/mol with targets 3CLpro, NSP15, and TMPRSS2. Furthermore, a multiple ligand simultaneous docking study was performed to understand the synergistic effect of the 2-DG with and drug with phytochemicals in combination provided in Table 4.
Fig. 1.

Binding energy pattern of nineteen drugs and phytochemicals along with 2-DG with three targeted proteins
Table 4.
Binding energies (in kcal/mol) obtained for the interaction of viral and host targets with selected combination drugs
| S. no. | Drug combinations | 3CLpro | NSP15 | TMPRSS2 |
|---|---|---|---|---|
| NIH-recommended drugs | ||||
| 1 | Barcitinib + 2-DG | −7.96 | −9.60 | −8.23 |
| 2 | Hydroxychloroquine + 2-DG | −7.11 | −8.88 | −7.06 |
| 3 | Ivermectin + 2-DG | −8.60 | −9.70 | −9.14 |
| 4 | Ruxolitinib + 2-DG | −7.52 | −10.11 | −8.05 |
| 5 | Tofacitinib + 2-DG | −7.04 | −8.71 | −7.69 |
| Clinical trial drugs | ||||
| 6 | Telmisartan + 2-DG | −9.88 | −10.94 | −9.14 |
| 7 | Arbidol + 2-DG | −7.136 | −8.202 | −7.438 |
| Phytochemicals | ||||
| 8 | Glycyrrhizin + 2-DG | −9.710 | −10.419 | −10.005 |
| 9 | Platycodin_D + 2-DG | −7.738 | −8.421 | −8.298 |
| 10 | Punicalagin + 2-DG | −10.402 | −11.045 | −9.649 |
| 11 | Sennoside_B + 2-DG | −9.037 | −10.008 | −8.873 |
| 12 | Andrographolide + 2-DG | −7.68 | −10.32 | −8.16 |
| 13 | Betulin + 2-DG | −9.02 | −9.89 | −9.87 |
| 14 | Cepharanthine + 2-DG | −9.93 | −10.99 | −10.12 |
| 15 | Chebulagic acid + 2-DG | −10.05 | −10.91 | −10.05 |
| 16 | Maslinic acid + 2-DG | −9.37 | −8.99 | −9.01 |
| 17 | Pectolinarin + 2-DG | −8.95 | −9.55 | −8.65 |
| 18 | Tannic acid + 2-DG | −9.28 | −11.1 | −10.27 |
| 19 | Ursolic acid + 2-DG | −9.50 | −10.5 | −9.14 |
According to the docking study, Ruxolitinib with 2-DG interacts with the viral target 3CLpro, which has a binding energy of −7.52 kcal/mol. 2-DG interacts with PHE140 and GLY170, while Ruxolitinib interacts with residues CYS145, ARG188, MET165, ALA191, GLN192, GLN192, and PRO168 (Supplementary Fig. 1A). The same combination interacts with NSP15, found to have a binding energy of −10.11 kcal/mol, the drug 2-DG interacts with residues GLU42, THR31, LYS47, ASP46, and Ruxolitinib interacts with ALA55, TRP59, PHE56, VAL152, ALA7, and VAL10 interactive amino acid residues (Supplementary Fig. 1B). Ruxolitinib + 2-DG has a binding energy of −8.05 kcal/mol when interacting with TMPRSS2. Ruxolitinib interacts with residues ASP482, TRP169, PRO369, and MET478. 2-DG interacts with residues GLY245, ASN247, TRP453, and SER448 (Supplementary Fig. 1C).
While interacting with viral target 3CLpro, the drug Telmisartan, in combination with 2-DG, was discovered to have a binding energy of −9.88 kcal/mol. Telmisartan interacts with residues THR26, MET49, ASN142, MET165, and PRO168, while 2-DG interacts with residues LEU287 and THR199 (Supplementary Fig. 2A). The binding energy of the combination drugs was found to be −10.94 kcal/mol while interacting with NSP15. The 2-DG interacts with the viral target residues LYS277, SER274, LYS71, THR196, ASP324, and ASP237, while Telmisartan interacts with the active residues ASN30, ASN29, and ILE28 (Supplementary Fig. 2B). TMPRSS2 has a binding energy of −9.14 kcal/mol when interacting with Telmisartan + 2-DG. Telmisartan interacts with LEU163, ALA262, ASN247, LEU248, TRP380, and TRP453, while 2-DG interacts with active residues ASP277, HIS279, and GLN317 (Supplementary Fig. 2C).
Punicalagin, in conjunction with 2-DG, has a binding energy of −10.40 kcal/mol when docked simultaneously with 3CLpro. Punicalagin interacts with active amino acid residues MET276, LEU272, TYR237, LYS236, THR199, and ASP289, while 2-DG interacts only with ARG188 (Supplementary Fig. 3A). When the combined drug interacts with NSP15, the binding energy is found to be −11.04 kcal/mol. While the drug 2-DG interacts with ASP184, ARG139, and ASN140 residues in combination, the phytochemical Punicalagin interacts with LEU252, LYS277, TYR89, SER162, VAL67, GLN160, and ALA161 (Supplementary Fig. 3B). Punicalagin + 2-DG also interacts with the host target TMPRSS2, which has a binding energy of −9.64 kcal/mol. Punicalagin interacts with ASN192, ASN358, PHE194, and 2-DG interacts with TRP306, GLN327, GLN276, and THR309 residues (Supplementary Fig. 3C). Overall, the analysis of individual and multiple ligands blind docking of all the selected compounds considered in the study revealed that the combined drugs have more preferred binding energy pattern as compared to individual drugs and phytochemicals, while interacting with selected viral and host targets could have a better synergistic effect for hindering the viral growth. To validate the combinations with the best binding energy acquired by blind docking, namely 2-DG + Ruxolitinib, 2-DG + Telmisartan, and 2-DG + Punicalagin, a redocking experiment was conducted by specifying the active sites of the selected receptors as mentioned in Supplementary Table 2.
The comprehensive analysis of docking results highlighted that binding energy obtained after re-docking was almost similar to the binding energies obtained by blind docking of all three targets with the individual drug/phytochemical. For instance, the binding energy of phytochemical, Punicalagin for the target 3CLpro was −9.091 kcal/mol with blind docking, and −9.056 kcal/mol was obtained after re-docking performed post the selection of active residues. Similarly, the resulting binding energies of the remaining targets were shown in Table 5. The binding energy value of drug ruxolitinib for 3CLpro −9.124 kcal/mol improved from −7.52 kcal/mol obtained on blind docking. The binding energy values of the remaining 2-DG combination have been provided in Table 5. Further, it has been observed that re-docking ameliorated the result in the case of MLSD, wherein the drugs and phytochemicals are combined with 2-DG.
Table 5.
Binding energies (in kcal/mol) obtained for the interaction of viral and host targets with selected combination drugs after redocking study
| Drug | 3CLpro | NSP15 | TMPRSS2 | |
|---|---|---|---|---|
| Individual drug | 2-DG | −4.42 | −4.96 | −5.00 |
| Ruxolitinib | −9.124 | −8.852 | −6.480 | |
| Telmisartan | −8.997 | −10.399 | −8.837 | |
| Punicalagin | −9.056 | −10.125 | −8.79 | |
| Drugs in combination | Ruxolitinib + 2-DG | −9.124 | −9.310 | −8.803 |
| Telmisartan + 2-DG | −9.363 | −11.336 | −9.771 | |
| Punicalagin + 2-DG | −10.283 | −11.461 | −10.826 |
The results also inferred that the non-covalent interactions obtained from blind docking of Ruxolitinib with 3CLpro are not distinguishable from the ones obtained from re-docking as shown in Figs. 2, 3 and 4. At the same time, some new residues involved in the interaction of 2-DG with the target are also revealed by defining the active site.
Fig. 2.

Three dimensional interaction of top score drug Ruxolitinib + 2-DG with A 3CLpro, B NSP15, C TMPRSS2
Fig. 3.

Three-dimensional interaction of top score drug Telmisartan + 2-DG with A 3CLpro, B NSP15, C TMPRSS2
Fig. 4.

Three-dimensional interaction of top score phytochemical Punicalagin + 2-DG with A 3CLpro, B NSP15, C TMPRSS2
When blind docking was considered, Ruxolitinib interacts with residues CYS145, ARG188, MET165, ALA191, GLN192, and PRO168, and 2-DG interacts with residues PHE140 and GLY170, while after re-docking, the drug interacts with CYS145, GLN192, PRO168, and MET49 while 2-DG interacts with LYS137, ASN238, LEU287, and ASP289. The non-covalent interactions for remaining targets and drug combinations, along with the bond distance obtained after docking of each drug and 2-DG combinations with all the three targets under consideration, are mentioned in Supplementary Tables 5, 6 and 7 referred from earlier reports [31, 32].
Conclusively, it has been observed that the binding pattern attained in the case of MLSD shows some similarity with the blind docking results. According to this study, combination drugs have lower binding energies than individual drugs and phytochemicals. When they interact with specific potential viral and host targets, they may have a more effective synergistic effect on suppressing viral proliferation.
We performed multi-ligand simultaneous molecular dynamics simulation for 20 ns to better understand the interactions between the chosen targets 3CLpro, NSP15, and TMPRSS2 with drugs in combination with Ruxolitinib + 2-DG. The average RMSD values were as follows: 0.19, 0.46, and 0.20 nm for 3CLpro-Ruxolitinib + 2-DG, NSP15-Ruxolitinib + 2-DG, and TMPRSS2- Ruxolitinib + 2-DG, respectively (Fig. 5A). The result suggests that the binding of the drug in combination Ruxolitinib + 2-DG with viral target 3CLpro and TMPRSS2 was profoundly more stable when compared to NSP15.
Fig. 5.

Graph showing the MD simulation analysis. A RMSD plot, B RMSF plot, and C radius of gyration of the protein backbone over the course of the simulation. 3CLpro, 3CLpro–2-DG, 3CLpro–Ruxolitinib, and 3CLpro–Ruxolitinib + 2-DG are represented in orange, purple, blue, and green colors, respectively
RMSF values indicate the backbone of the atoms in the residues versus the simulated trajectory. The graphs aid us in determining the structural flexibility of the system. For complexes, average RMSF was measured and analyzed. The plot of RMSF (Fig. 5B) primarily displays global changes, allowing us to determine each residue’s flexibility during simulation. The RMSF fluctuation value of 3CLpro-Ruxolitinib + 2-DG lies in the range of 0.03 to 0. 64 nm and the fluctuation range of the NSP15-Ruxolitinib + 2-DG and TMPRSS2-Ruxolitinib + 2-DG drugs in combination in complex with protein; the RMSF values were found to be in the range of 0.08–0.65 nm and 0.04–0.41 nm as represented in Fig. 5B. The rate of amino acid fluctuation has been noticed for complexes, and it was found that 3CLpro-Ruxolitinib + 2-DG and NSP15-Ruxolitinib + 2-DG have comparatively higher amino acid fluctuation than with the complex TMPRSS2- Ruxolitinib + 2-DG.
Moreover, to understand the compactness of the structure, we analyzed their respective radius of gyration or Rg values, as shown in the graph in Fig. 5C. The average Rg values of protein 3CLpro along with 3CLpro-Ruxolitinib + 2-DG, NSP15-Ruxolitinib + 2-DG, and TMPRSS2- Ruxolitinib + 2-DG were 2.23, 2.41, and 2.12 nm respectively. Minor differences in the average values observed in all simulated complexes are discovered, which can be attributed to conformational alterations in the protein-drug interaction.
Overall, observed trend from MLS-MD simulation analysis suggests that drugs in combination Ruxolitinib + 2-DG are found to be relatively stable while interacting with the viral and host target.
Conclusions
The rise in SARS-CoV-2 cases around the globe is of serious concern. The current study demonstrates the utilization of computational framework for developing synergistic therapeutics for effective disease treatment. Predicted trends on drug-drug interactions and analysis of the binding of approved drugs with the potential SARS-CoV-2 targets, namely 3CLpro, NSP15, and human target TMPRSS2, are helpful in selecting the optimal combinations. The combination of 2-DG with drugs and phytochemicals showed no interaction, and was considered for the molecular docking studies. Further, these screened drugs were used for comparative single and multiple ligands docking analysis. Interestingly, results revealed that combinations of drugs and phytochemicals with 2-DG show better binding energy than the individual compound. We re-docked the top three drug combinations, used a polypharmacology approach, and performed MLS molecular dynamics simulation to analyze the binding patterns of drug combinations with multiple targets to reverify the best drug combinatorial output obtained by blind docking. Based on the overall comparison, the top 3 drug combinations, namely, NIH-recommended drug, Ruxolitinib, clinical trial drug, Telmisartan, and phytochemical, Punicalagin all along with 2-DG were showing better binding energies than the selected individual compound. Additionally, MLS-MD study coincided with specific targets with the NIH-recommended drugs combination 2-DG + Ruxolitinib, indicating that it may be sufficient to regulate SARS CoV-2 synergistically. Thus, this study suggests that these combination strategies might provide an excellent synergistic therapy that could improve and enhance the efficacy rate of drug repurposing for the treatment of the current and future coronavirus outbreaks. The outcome of the study suggests polypharmacology of drug combinations for multiple targets of both SARS-CoV-2 and host. Our approaches and the obtained results make it easier to design and synthesize new drug combinations against COVID-19.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
All the authors are thankful for the support from the Council of Scientific and Industrial Research-Indian Institute of Toxicology Research (CSIR-IITR), Lucknow, for providing computer resources. The manuscript communication number is IITR/SEC/2021-2022/36.
Author contribution
AG: conceptualized the study, carried out drug-drug interaction work, individual drug docking, data analysis for tabulation, and drafting of the figures, and wrote the manuscript. SSC: conceptualized the study, carried out the multiple-ligand-simultaneous study, and wrote the manuscript. ASG: performed data analysis and editing of the manuscript. RP: conceptualized the study and edited the final manuscript. All authors reviewed the manuscript.
Funding
The financial support for this work is provided by the CSIR-Mission Mode Programme on Immuno Modulatory Function of Nutritionals and Nutraceuticals for Health and Wellness (HCP-35 WP-09).
Availability of data and material
Available in the article and included in the supplementary information.
Code availability
Not applicable.
Declarations
Ethics approval
This article does not contain any studies with human participants or animals performed by any authors.
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Anshika Gupta and Shweta Singh Chauhan contributed equally.
References
- 1.WHO (2020) Coronavirus disease (COVID-19) situation dashboard. World Health Organization website. https://who.sprinklr.com/. Accessed April 17
- 2.Ward D, Higgins M, Phelan JE, Hibberd ML, Campino S, Clark TG. An integrated in silico immuno-genetic analytical platform provides insights into COVID-19 serological and vaccine targets. Genome Med. 2021;13:1–12. doi: 10.1186/s13073-020-00822-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang X, Yu Y, Xu J, Shu H, Liu H, Wu Y, Zhang L, Yu Z, Fang M, Yu T. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. 2020;8:475–481. doi: 10.1016/S2213-2600(20)30079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zambrana C, Xenos A, Böttcher R, Malod-Dognin N, Przulj N (2021) Between viral targets and differentially expressed genes in COVID-19: the sweet spot for therapeutic intervention [DOI] [PMC free article] [PubMed]
- 5.Deng Q, Ur Rasool R, Russell RM, Natesan R, Asangani IA. Targeting androgen regulation of TMPRSS2 and ACE2 as a therapeutic strategy to combat COVID-19. IScience. 2021;24:102254. doi: 10.1016/j.isci.2021.102254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trougakos IP, Stamatelopoulos K, Terpos E, Tsitsilonis OE, Aivalioti E, Paraskevis D, Kastritis E, Pavlakis GN, Dimopoulos MA. Insights to SARS-CoV-2 life cycle, pathophysiology, and rationalized treatments that target COVID-19 clinical complications. J Biomed Sci. 2021;28:1–18. doi: 10.1186/s12929-020-00703-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mody V, Ho J, Wills S, Mawri A, Lawson L, Ebert MC, Fortin GM, Rayalam S, Taval S. Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Commun Biol. 2021;4:1–10. doi: 10.1038/s42003-020-01577-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verma A, Adhikary A, Woloschak G, Dwarakanath BS, Papineni RV. A combinatorial approach of a polypharmacological adjuvant 2-deoxy-D-glucose with low dose radiation therapy to quell the cytokine storm in COVID-19 management. Int J Radiat Biol. 2020;96:1323–1328. doi: 10.1080/09553002.2020.1818865. [DOI] [PubMed] [Google Scholar]
- 9.Ardestani A, Azizi Z. Targeting glucose metabolism for treatment of COVID-19. Signal Transduct Target Ther. 2021;6:1–2. doi: 10.1038/s41392-021-00532-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balkrishna A, Thakur P, Singh S, Dev S, Jain V, Varshney A, Sharma RK (2020) Glucose antimetabolite 2-Deoxy-D-Glucose and its derivative as promising candidates for tackling COVID-19: Insights derived from in silico docking and molecular simulations. Authorea [Preprint server]
- 11.Gualdoni GA, Mayer KA, Kapsch A-M, Kreuzberg K, Puck A, Kienzl P, Oberndorfer F, Frühwirth K, Winkler S, Blaas D. Rhinovirus induces an anabolic reprogramming in host cell metabolism essential for viral replication. Proc Natl Acad Sci. 2018;115:E7158–E7165. doi: 10.1073/pnas.1800525115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abd El-Mageed H, Abdelrheem DA, Ahmed SA, Rahman AA, Elsayed KN, Ahmed SA, El-Bassuony AA, Mohamed HS (2021) Combination and tricombination therapy to destabilize the structural integrity of COVID-19 by some bioactive compounds with antiviral drugs: insights from molecular docking study. Struct Chem 1–16 [DOI] [PMC free article] [PubMed]
- 13.Chakraborty C, Sharma AR, Bhattacharya M, Agoramoorthy G, Lee S-S (2021) The drug repurposing for COVID-19 clinical trials provide very effective therapeutic combinations: lessons learned from major clinical studies. Front Pharmacol 12 [DOI] [PMC free article] [PubMed]
- 14.WHO (2004) SARS: clinical trials on treatment using a combination of traditional Chinese medicine and Western medicine: report of the WHO International Expert Meeting to review and analyse clinical reports on combination treatment for SARS, 8–10 Oct 2003, Beijing, People's Republic of China
- 15.Coppock D, Baram M, Chang AM, Henwood P, Kubey A, Summer R, Zurlo J, Li M, Hess B. COVID-19 treatment combinations and associations with mortality in a large multi-site healthcare system. PLoS ONE. 2021;16:e0252591. doi: 10.1371/journal.pone.0252591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Españo E, Kim J, Lee K, Kim J-K. Phytochemicals for the treatment of COVID-19. J Microbiol. 2021;59:959–977. doi: 10.1007/s12275-021-1467-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, Han L, He J, He S, Shoemaker BA. PubChem substance and compound databases. Nucleic Acids Res. 2016;44:D1202–D1213. doi: 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hansten PD. Horn JR and Hazlet TK (2001) ORCA: OpeRational ClassificAtion of drug interactions. J Am Pharm Assoc. 1996;41:161–165. doi: 10.1016/s1086-5802(16)31244-x. [DOI] [PubMed] [Google Scholar]
- 19.Dmitriev A, Filimonov D, Rudik A, Pogodin P, Karasev D, Lagunin A, Poroikov V. Drug-drug interaction prediction using PASS. SAR QSAR Environ Res. 2019;30:655–664. doi: 10.1080/1062936X.2019.1653966. [DOI] [PubMed] [Google Scholar]
- 20.Discovery Studio (2016) Dassault Systèmes BIOVIA. Discovery studio modelling environment, release 4
- 21.Goodsell DS, Morris GM, Olson AJ. Automated docking of flexible ligands: applications of AutoDock. J Mol Recognit. 1996;9:1–5. doi: 10.1002/(SICI)1099-1352(199601)9:1<1::AID-JMR241>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 22.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jamir E, Sarma H, Priyadarsinee L, Nagamani S, Kiewhuo K, Gaur AS, Rawal RK, Murugan NA, Subramanian V, Sastry GN. Applying polypharmacology approach for drug repurposing for SARS-CoV2. J Chem Sci. 2022;134:1–24. doi: 10.1007/s12039-022-02046-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manjunathan R, Periyaswami V, Mitra K, Rosita AS, Pandya M, Selvaraj J, Ravi L, Devarajan N, Doble M. Molecular docking analysis reveals the functional inhibitory effect of Genistein and Quercetin on TMPRSS2: SARS-COV-2 cell entry facilitator spike protein. BMC Bioinform. 2022;23:1–15. doi: 10.1186/s12859-022-04724-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Badavath VN, Sinha BN, Jayaprakash V. Design, in-silico docking and predictive ADME properties of novel pyrazoline derivatives with selective human MAO inhibitory activity. Int J Pharm Pharm Sci. 2015;7:277–282. [Google Scholar]
- 26.Mujwar S, Deshmukh R, Harwansh RK, Gupta JK, Gour A. Drug repurposing approach for developing novel therapy against mupirocin-resistant Staphylococcus aureus. Assay Drug Dev Technol. 2019;17:298–309. doi: 10.1089/adt.2019.944. [DOI] [PubMed] [Google Scholar]
- 27.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 28.Edwards PM. Origin 7.0: scientific graphing and data analysis software. J Chem Inf Comput Sci. 2002;42:1270–1271. doi: 10.1021/ci0255432. [DOI] [Google Scholar]
- 29.Ferdousi R, Safdari R, Omidi Y. Computational prediction of drug-drug interactions based on drugs functional similarities. J Biomed Inform. 2017;70:54–64. doi: 10.1016/j.jbi.2017.04.021. [DOI] [PubMed] [Google Scholar]
- 30.Tilson H, Hines LE, McEvoy G, Weinstein DM, Hansten PD, Matuszewski K, Le Comte M, Higby-Baker S, Hanlon JT, Pezzullo L. Recommendations for selecting drug–drug interactions for clinical decision support. Am J Health Syst Pharm. 2016;73:576–585. doi: 10.2146/ajhp150565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dagur P, Rakshit G, Sheikh M, Biswas A, Jha P, Al-Khafaji K, Ghosh M (2022) Target prediction, computational identification, and network-based pharmacology of most potential phytoconstituent in medicinal leaves of Justicia adhatoda against SARS-CoV-2. J Biomol Struct Dyn 1–17 [DOI] [PubMed]
- 32.Rakshit G, Dagur P, Satpathy S, Patra A, Jain A, Ghosh M (2021) Flavonoids as potential therapeutics against novel coronavirus disease-2019 (nCOVID-19). J Biomol Struct Dyn 1–13 [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Available in the article and included in the supplementary information.
Not applicable.