

Abstract
Objective
Upacicalcet is a new renally excreted and injectable calcimimetic agent. We evaluated the pharmacokinetics, pharmacodynamics, safety, and tolerability of single and multiple intravenous administration of upacicalcet in patients with secondary hyperparathyroidism undergoing hemodialysis.
Methods
This study was a multicenter, randomized, placebo-controlled, double-blinded, dose-escalation study consisting of a single-dose study and a multiple-dose study. The single-dose study consisted of seven dose steps from 0.025 to 0.8 mg. For each step, six patients were randomly assigned 2:1 to receive upacicalcet or a placebo. The multiple-dose study occurred over 3 weeks in three-dose steps from 0.05 to 0.2 mg. For each step, 12 patients were randomly assigned 3:1 to receive upacicalcet or a placebo.
Results
The plasma concentration of upacicalcet increased in a dose-dependent manner and was maintained for the next dialysis. Upacicalcet was approximately 80% removed by a single dialysis and did not increase in the plasma concentration with repeated administration. Serum intact parathyroid hormone and corrected calcium (Ca2+) levels tended to decrease in response to the plasma concentration of upacicalcet. In the single-dose study, upper gastrointestinal symptoms were observed as a non-serious and mild adverse drug reaction in the groups receiving upacicalcet ≥ 0.4 mg. In the multiple-dose study, abdominal discomfort occurred in each patient in the 0.1 mg and 0.2 mg groups.
Conclusions
Upacicalcet for patients with secondary hyperparathyroidism undergoing hemodialysis could be a calcimimetic agent that acts in a dose-dependent manner and persistently until the next dialysis session. No safety or tolerability issues specific to upacicalcet were found.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40262-022-01139-w.
Key Points
| The pharmacokinetics, pharmacodynamics, safety, and tolerability of single and multiple intravenous upacicalcet, a new renally excreted calcimimetic agent, were evaluated in patients with secondary hyperparathyroidism undergoing hemodialysis. |
| Upacicalcet persisted in a dose-dependent manner until the time of the next dialysis, was well removed by dialysis, and constant blood concentrations were maintained when the same doses were administered repeatedly. |
| Pharmacodynamic parameters, such as serum intact parathyroid hormone and calcium levels, also changed according to the plasma upacicalcet concentration. |
| No severe concerns about the safety and tolerability of upacicalcet were observed. |
Introduction
Secondary hyperparathyroidism (SHPT) is a major clinical manifestation of chronic kidney disease-mineral bone disorder caused by abnormally increased parathyroid hormone (PTH) secretion. Advanced SHPT leads to vascular calcification and bone disorders, worsening the patient’s prognosis [1, 2]. The first choice of medical treatment for patients with SHPT undergoing hemodialysis is administration of vitamin D receptor activators that suppress PTH secretion; however, they carry the risk of hypercalcemia and hyperphosphatemia [3].
The launch of cinacalcet hydrochloride (hereafter cinacalcet) has expanded the scope of treatment for SHPT. Cinacalcet is the first-in-class calcimimetic that acts as an allosteric modulator of calcium (Ca2+)-sensing receptors (CaSR) on parathyroid cell membranes. Cinacalcet can lower serum PTH levels as well as serum Ca2+ and phosphorus (P) levels. As a result, cinacalcet has improved the rate of patients achieving therapeutic targets in the chronic kidney disease-mineral bone disorder guidelines [4–8].
In contrast, cinacalcet causes a high incidence of gastrointestinal (GI) symptoms, such as nausea and vomiting. In addition, cinacalcet, an oral formulation, may increase the medication burden and worsen adherence in patients undergoing maintenance dialysis who use many oral drugs. Moreover, cinacalcet should be administered with caution to patients receiving cytochrome P450 3A4 inhibitors or drugs that are substrates of 2D6 as well as to patients with hepatic dysfunction. Thus, there are medical needs for calcimimetic agents with few GI side effects, improved medication adherence, and avoidance of drug–drug interactions [8–12]. In this context, intravenous etelcalcetide hydrochloride and evocalcet oral medications have emerged as drugs that partially overcome the issues with cinacalcet [13, 14].
Upacicalcet sodium hydrate (AJT240/SK-1403, Fig. S1 of the Electronic Supplementary Material [ESM]) is a novel small-molecule calcimimetic developed for an intravenous injection that does not have an inverse effect on patient adherence. Non-clinical studies have shown that upacicalcet is a specific allosteric modulator of human CaSR; it has low substrate properties for hepatic metabolism and is unlikely to cause inhibition or induction of cytochrome P450 enzymes. In addition, the binding rate of upacicalcet to human plasma proteins is approximately 45%. Furthermore, the results of our phase I study indicated that upacicalcet, when administered to healthy adults, showed a dose-dependent calcimimetic activity and underwent renal excretion in an unchanged form. Thus, it is expected that upacicalcet, when used in patients with SHPT undergoing hemodialysis without renal function, could maintain its effect until the next dialysis session. Therefore, we conducted a double-blind placebo-controlled study to evaluate the pharmacokinetics, pharmacodynamics, safety, and tolerability of single and multiple intravenous doses of upacicalcet in patients with SHPT undergoing hemodialysis.
Methods
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee at which the studies were conducted and with the Helsinki Declaration and comparable ethical standards, Good Clinical Practice guidelines, and other regulatory requirements. The study was approved by the institutional review board of each participating center. All subjects provided written informed consent before participating in the study.
Study Design
This study was conducted as a phase I/II, multicenter, randomized, placebo-controlled, double-blinded, dose-escalation study. At the screening test, patients who met all inclusion criteria and did not meet any exclusion criteria were randomly assigned via a centralized randomization system to receive either upacicalcet or a placebo. This study consisted of a single-dose study and a 3-week multiple-dose study. The dose range of upacicalcet in this study was 0.025–0.8 mg, which was considered to be effective, safe, and well tolerated based on the results of the phase I study. The study design is illustrated in Fig. 1.
Fig. 1.

Study design and the target number of patients
For the single-dose study, six patients in each step cohort were enrolled in seven-dose steps and then randomized 2:1 to receive upacicalcet or a placebo. The first-dose step cohort (Step 1) received a 0.025 mg intravenous injection of upacicalcet. After the safety of the Step 1 cohort was confirmed, the second cohort study was initiated. Each subsequent cohort was subjected to the same procedure. Criteria for consideration to each step-up are shown in the ESM.
For the 3-week multiple-dose study, 12 patients in each step cohort were enrolled in three-dose steps and randomized 3:1 to receive upacicalcet or a placebo. The upacicalcet dose for the first multiple-dose step cohort (Step 1) was 0.05 mg. After completing the safety confirmation with the single dose of 0.1 mg, the first multiple-dose step cohort study was initiated. Subsequent multiple-dose step cohort studies were initiated after safety confirmation of both the previous multiple-dose cohort and a higher single-dose cohort than the planned multiple-dose study. Patients who participated in the one step were not allowed to participate in another cohort study.
Inclusion and Exclusion Criteria and Randomization
Participants who met all the inclusion criteria and did not meet any exclusion criteria at the screening test were enrolled in the study. The inclusion criteria were as follows: age between 20 and 80 years; hemodialysis or hemodiafiltration three times a week for at least 12 weeks prior to the screening test; serum intact PTH (iPTH) level > 240 pg/mL; serum corrected Ca2+ (cCa2+) level ≥ 8.4 mg/dL; and ability to provide written informed consent. The main exclusion criteria were as follows: primary hyperparathyroidism, received cinacalcet or changed the dosage and administration of active vitamin D preparation, Ca preparation, or P adsorbent from 2 weeks prior to the screening test. The other inclusion and exclusion criteria are shown in the ESM. The participants were randomly assigned according to an assignment form prepared by the external organization for case registration (EPS Corporation, Tokyo, Japan).
Interventions
In the single-dose study, allocated subjects were admitted to the hospital the day before or on the day of dosing, subject eligibility was finally confirmed, and upacicalcet or placebo was administered intravenously on day 1 as slowly as possible between 2 and 4 h after the end of dialysis on the day of study drug administration. Patients were hospitalized until day 4. A post-treatment examination was performed on day 8. The planned doses of upacicalcet or placebo were 0.025, 0.05, 0.1, 0.2, 0.4, 0.6, and 0.8 mg.
In the multiple-dose study, allocated subjects were administered with upacicalcet or placebo on the first dialysis day of the week (after the maximum dialysis interval). The study drug was administered intravenously into the venous side of the dialysis circuit immediately before the end of hemodialysis. The dosing interval and duration were three times per week for 3 weeks until day 22. A post-treatment examination was performed on day 29. The planned doses of upacicalcet or placebo were 0.05, 0.1, and 0.2 mg.
Pharmacokinetic Evaluations
To determine the plasma concentrations of upacicalcet and its predominant metabolites (M1, M2, and M3), blood samples were collected at the following timepoints: in the single-dose study, before administration, and 10 and 30 min, and 1, 3, 6, 18, 30, 42, 66, 67, and 70 h after administration; in the multiple-dose study, day 1, 3, 5, 8, 10, 12, 15, 17, 19, and 22 (before dialysis); day 1 (10 and 30 min, and 1 h after administration); day 17 (10 and 30 min and 1 and 20 h after administration); and day 19 (before administration).
Blood samples were collected in a vacuum blood collection tube containing 2 mL of sodium heparin (Insepack II-D, Sekisui Medical Co., Ltd. Tokyo, Japan) at each timepoint. The collected blood was immediately centrifuged, and the separated plasma samples were frozen for storage and transported to Shin Nippon Biomedical Laboratories, Ltd. (Tokyo, Japan). The concentrations of upacicalcet and its metabolites in the plasma were measured by liquid chromatography-tandem mass spectrometry. The pharmacokinetic parameters in the single-dose study were calculated as follows: maximum (peak) plasma drug concentration (Cmax), time to Cmax, area under the plasma concentration–time curve from zero to 66 h, area under the plasma concentration–time curve from time zero to infinity, elimination half-life, mean residence time from zero to time t, mean residence time to infinity, apparent total body clearance of drug from plasma, apparent volume of distribution at steady state, removal rate by hemodialysis, and hemodialysis clearance. The formulas for removal rate and clearance by dialysis are shown in Table 3. In the multiple-dose study, the Cmax and removal rate by hemodialysis were calculated.
Table 3.
The removal rate by dialysis of unchanged upacicalcet and hemodialysis clearance after single and multiple doses of upacicalcet
| A is the removal rate by dialysis of unchanged upacicalcet and hemodialysis clearance after single dose of upacicalcet | ||||
|---|---|---|---|---|
| Step | Group | N | Removal rate by dialysis of unchanged upacicalcet (%) | Hemodialysis clearance (mL/min) |
| Step 1 | 0.025 mg | 4 | 100.00 ± 0.00 | – |
| Step 2 | 0.05 mg | 4 | 94.53 ± 10.95 | – |
| Step 3 | 0.1 mg | 4 | 81.00 ± 14.05 | 61.40 ± 60.25* |
| Step 4 | 0.2 mg | 4 | 78.40 ± 4.47 | 168.25 ± 2.50 |
| Step 5 | 0.4 mg | 4 | 80.40 ± 7.34 | 125.83 ± 39.55** |
| Step 6 | 0.6 mg | 4 | 81.35 ± 3.76 | 183.00 ± 9.90* |
| Step 7 | 0.8 mg | 5 | 83.66 ± 13.37 | 156.00 ± 12.25*** |
| B is the removal rate by dialysis of unchanged upacicalcet after multiple dose of upacicalcet | |||||
|---|---|---|---|---|---|
| Step | Group | N | Removal rate by dialysis of unchanged upacicalcet (%) | ||
| Step 1 | 0.05 mg | 3 | 79.27 ± 3.39 | ||
| Step 2 | 0.1 mg | 6 | 80.68 ± 7.68 | ||
| Step 3 | 0.2 mg | 4 | 86.98 ± 4.97 | ||
The removal rate by dialysis of unchanged upacicalcet in plasma was calculated from the following equation. Removal rate by dialysis (%) = (CBD – CAD)/CBD×100
Hemodialysis clearance was calculated from the following equation. CL (mL/min f0) = (CBI − CBO)/CBI × 200*
AD after dialysis, BD before dialysis, C concentration, BI blood in, BO blood out, C concentration
*Blood flow
Only removal rate by dialysis of unchanged upacicalcet was calculated in multiple-dose study.
Mean ± SD, *n=2, **n=3, ***n=4
Pharmacodynamic Evaluations
As upacicalcet is a calcimimetic developed for the treatment of SHPT, the pharmacodynamic parameters of upacicalcet determined were serum iPTH, serum cCa2+, and P levels. The Payne’s formula was used to calculate cCa2+. As exploratory measurements, we measured whole PTH, fibroblast growth factor 23, 1,25(OH)2 vitamin D, ionized Ca2+, and calcitonin levels in the single-dose study and tartrate-resistant acid phosphatase-5b, bone-specific alkaline phosphatase, and osteocalcin, which are markers of bone metabolism, in the multiple-dose study.
Blood samples were collected in vacuum blood collection tubes (VP series, Terumo Corporation, Tokyo, Japan). Blood samples for the pharmacodynamic parameters were collected at the following timepoints: in the single-dose study, before dialysis, before administration, and 10 and 30 min and 1, 3, 6, 18, 30, 42, 66, and 70 h after administration; in the multiple-dose study, day 1, 3, 5, 8, 10, 12, 15, 17, 19, 22, and 29 (before dialysis); day 1 (30 min and 1 h after administration); day 17 (30 min and 1 and 20 h after administration); and day 19 (before administration) and were measured by SRL Medisearch Inc. (Tokyo, Japan).
Safety Evaluations
For all adverse events (AEs) that occurred after the start of the study treatment, the occurrence and incidence of AEs and adverse drug reactions (ADRs) were summarized for each treatment period and dose. The incidence of AEs and ADRs for each treatment period and dose was summarized using the system organ class and preferred term using Medical Dictionary for Regulatory Activities J version 18.1.
The safety and tolerability profiles of upacicalcet were assessed in terms of occurrence of AEs, vital signs, clinical laboratory measurements, and ophthalmology examination (multiple dose only). Vital signs were monitored at each institution, and 12-lead electrocardiograms were monitored and analyzed by Suzuken Co. Ltd. (Nagoya, Japan).
Statistical Analysis
Patients who received placebo were evaluated collectively as the placebo group without distinguishing the step cohort. Patients who received upacicalcet were evaluated in each dosage group. Patients who experienced an accidental overdose were excluded from pharmacokinetic and pharmacodynamic analyses. The relationship between the measured QT interval as corrected by the Fridericia method (QTcF) and the change in QTcF before dosing and the concentration of upacicalcet and its metabolites (M1, M2, and M3) in plasma were examined for each dose group of upacicalcet by a regression analysis. The relationship between cCa2+ levels and QTcF was also examined in the same manner. SAS version 9.3 (SAS Institute, Cary, NC, USA) was used to perform other statistical analyses and to summarize the data.
Results
Study Population
Single-Dose Study
Forty-six patients were assigned to the placebo or upacicalcet group. Forty-four received the study drug, and two patients assigned to the placebo group did not receive the study drug (Fig. S2A of the ESM). All patients who received the study drugs were included in the analysis.
Patient baseline demographics and clinical characteristics are summarized in Table 1A. The mean age and body mass index of each group were between 57.8 and 67.5 years and between 20.71 and 26.28 kg/m2, respectively. The mean serum iPTH, cCa, and P levels of each group were between 267.3 and 538.7 pg/mL, 9.30 and 10.20 mg/dL, and 2.98 and 4.08 mg/dL, respectively.
Table 1.
Patients baseline demographics and clinical characteristics
| A is patients baseline demographics and clinical characteristics in single dose study | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Step | Placebo | Step 1 | Step 2 | Step 3 | Step 4 | Step 5 | Step 6 | Step 7 | |
| Dose | (Step 1–7) | 0.025 mg | 0.05 mg | 0.1 mg | 0.2 mg | 0.4 mg | 0.6 mg | 0.8 mg | |
| Number of patients (n) | 15 | 4 | 4 | 4 | 4 | 4 | 4 | 5 | |
| Age (years) | 58.9 ± 10.2 | 59.5 ± 10.5 | 60.0 ± 12.9 | 59.0 ± 7.5 | 57.8 ± 8.2 | 67.5 ± 10.1 | 62.0 ± 5.1 | 63.0 ± 9.8 | |
| BMI (kg/m2) | 24.57 ± 4.04 | 21.01 ± 3.13 | 20.71 ± 2.27 | 26.23 ± 4.15 | 26.28 ± 2.85 | 22.27 ± 3.45 | 21.95 ± 4.04 | 26.01 ± 3.61 | |
| Dry weight (kg) | 68.96 ± 14.05 | 58.93 ± 9.91 | 51.23 ± 7.49 | 70.25 ± 11.74 | 76.40 ± 6.21 | 57.83 ± 11.29 | 62.20 ± 14.67 | 65.00 ± 22.71 | |
| Duration of dialysis (years) | 8.82 ± 8.18 | 9.53 ± 7.68 | 13.05 ± 5.18 | 2.44 ± 2.72 | 8.02 ± 2.49 | 3.21 ± 1.75 | 16.26 ± 8.98 | 5.74 ± 5.12 | |
| Dialysate calcium concentration | 2.5 mEq/L | 2 (13.3) | 0 (0.0) | 2 (50.0) | 1 (25.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (20.0) |
| 2.75 mEq/L | 7 (46.7) | 3 (75.0) | 2 (50.0) | 2 (50.0) | 4 (100.0) | 1 (25.0) | 3 (75.0) | 2 (40.0) | |
| 3.0 mEq/L | 6 (40.0) | 1 (25.0) | 0 (0.0) | 1 (25.0) | 0 (0.0) | 3 (75.0) | 1 (25.0) | 2 (40.0) | |
| Serum iPTH (pg/mL) | 510.3 ± 232.2 | 394.8 ± 229.2 | 486.0 ± 127.7 | 301.5 ± 84.5 | 538.7 ± 172.1* | 267.3 ± 166.7 | 404.5 ± 122.7 | 520.8 ± 207.7 | |
| Serum cCa (mg/dL) | 9.71 ± 0.57** | 9.45 ± 0.37 | 9.35 ± 0.35 | 9.30 ± 0.90* | 9.90 ± 0.22 | 9.53 ± 0.50 | 10.20 ± 0.54 | 9.52 ± 0.84 | |
| Serum P (mg/dL) | 3.56 ± 0.94** | 3.45 ± 0.66 | 3.03 ± 0.51 | 3.57 ± 1.00* | 4.08 ± 0.15 | 3.40 ± 0.50 | 2.98 ± 0.29 | 3.76 ± 1.43 | |
| B is patients baseline demographics and clinical characteristics in multiple dose study | |||||
|---|---|---|---|---|---|
| Step | Placebo | Step 1 | Step 2 | Step 3 | |
| Dose | (Step 1–3) | 0.05 mg | 0.1 mg | 0.2 mg | |
| Number of patients (n) | 11 | 9 | 9 | 10 | |
| Age (years) | 65.2 ± 7.9 | 61.9 ± 8.8 | 53.7 ± 14.1 | 56.2 ± 9.8 | |
| BMI (kg/m2) | 23.41 ± 3.73 | 22.63 ± 2.98 | 22.58 ± 2.83 | 23.91 ± 4.13 | |
| Dry weight (kg) | 62.68 ± 13.82 | 60.52 ± 12.18 | 59.70 ± 10.84 | 65.34 ± 12.75 | |
| Duration of dialysis (years) | 12.09 ± 8.19 | 8.61 ± 5.67 | 10.73 ± 13.48 | 6.69 ± 4.29 | |
| Dialysate calcium concentration | 2.5 mEq/L | 1 (9.1) | 4 (44.4) | 1 (11.1) | 2 (20.0) |
| 2.75 mEq/L | 6 (54.5) | 4 (44.4) | 1 (11.1) | 5 (50.0) | |
| 3.0 mEq/L | 4 (36.4) | 1 (11.1) | 7 (77.8) | 3 (30.0) | |
| Serum iPTH (pg/mL) | 368.8 ± 78.9 | 439.0 ± 259.9* | 507.5 ± 226.4* | 393.7 ± 210.4 | |
| Serum cCa (mg/dL) | 9.92 ± 0.70 | 9.43 ± 0.68* | 9.46 ± 0.81* | 9.49 ± 0.63 | |
| Serum P (mg/dL) | 5.64 ± 1.43 | 6.33 ± 0.97* | 5.26 ± 1.14* | 6.00 ± 1.23 | |
A: Mean ± SD, or number of patients (%) *n = 3, **n = 14
B: Mean ± SD, or number of patients (%) *n = 8
Multiple-Dose Study
Thirty-nine patients who were eligible were assigned to the placebo or upacicalcet group (Fig. S2B of the ESM). The number of patients who discontinued the study after administration of the study drug was as follows: one patient in the placebo group, two patients in the group receiving 0.05 mg, three patients in the group receiving 0.1 mg, and six patients in the group receiving upacicalcet 0.2 mg. All patients who received the study drugs were included in the analyses. However, accidental overdose because of an incorrect preparation of the study drug occurred in one patient in the group receiving 0.05 mg and one patient in the group receiving 0.1 mg. These patients discontinued treatment and were excluded from the pharmacokinetic and pharmacodynamic analyses (adopted for the safety evaluation).
The baseline patient demographics and clinical characteristics are summarized in Table 1B. The mean age and body mass index of each group were between 53.7 and 65.2 years and between 22.58 and 23.91 kg/m2, respectively. The mean serum iPTH, cCa, and P levels of each group were between 368.8 and 507.5 pg/mL, 9.43 and 9.92 mg/dL, and 5.26 and 6.33 mg/dL, respectively.
Pharmacokinetic Findings
Single-Dose Study
After administration of upacicalcet, the concentration of unchanged upacicalcet in the plasma was maintained up to 66 h after administration and decreased upon dialysis. Plasma concentrations of unchanged upacicalcet tended to increase with increasing dose (Fig. 2A). The pharmacokinetic parameters of the plasma are summarized in Table 2A. The mean Cmax, area under the plasma concentration–time curve from time zero to 66 h, and area under the plasma concentration–time curve from time zero to infinity of unchanged upacicalcet in the plasma increased with increasing dose. The mean elimination half-life was between 65.0 and 122 h. The mean apparent total body clearance of drug from plasma was between 105 and 212 mL/h. The mean apparent volume of distribution at steady state was between 11,700 and 17,700 mL. In addition, the mean dialysis removal rate of unchanged upacicalcet in the plasma was between 78.40 and 100.00%, and the mean hemodialysis clearance was between 61.40 and 183.00 mL/min (Table 3A).
Fig. 2.

Mean plasma concentration-time profiles after single (a) and multiple (b) doses of unchanged upacicalcet. Arrows indicate the initiation of hemodialysis
Table 2.
Plasma concentration-time profiles
| A is plasma concentration-time profiles after single dose of upacicalcet | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Step | Group | N | Cmax (ng/mL) | tmax (h) | AUC0–66h (h·ng/mL) | AUCinf (h·ng/mL) | t1/2 (h) | MRT0–t (h) | MRTinf (h) | CLp (mL/h) | Vdss (mL) |
| Step 1 | 0.025 mg | 4 | 3.20 ± 0.86 | 0.167 ± 0.000 | 85.1 ± 8.21 | 270 ± 161 | 122 ± 80.7 | 29.2 ± 0.250 | 174 ± 113 | 106 ± 43.5 | 14900 ± 741 |
| Step 2 | 0.05 mg | 4 | 6.93 ± 1.62 | 0.167 ± 0.000 | 203 ± 26.6 | 456 ± 63.3 | 77.6 ± 8.87 | 28.8 ± 0.386 | 111 ± 12.2 | 105 ± 13.5 | 11700 ± 1710 |
| Step 3 | 0.1 mg | 4 | 10.5 ± 3.79 | 0.375 ± 0.417 | 261 ± 81.8 | 506 ± 235 | 65.0 ± 30.6 | 26.8 ± 2.83 | 91.5 ± 44.8 | 211 ± 71.5 | 17700 ± 7990 |
| Step 4 | 0.2 mg | 4 | 21.9 ± 2.61 | 0.167 ± 0.000 | 675 ± 99.4 | 1480 ± 257 | 76.6 ± 8.08 | 28.6 ± 0.171 | 109 ± 11.3 | 130 ± 24.7 | 14100 ± 2060 |
| Step 5 | 0.4 mg | 4 | 56.5 ± 8.32 | 0.167 ± 0.000 | 1530 ± 320 | 3150 ± 1080 | 68.1 ± 14.1 | 28.1 ± 0.929 | 97.1 ± 20.7 | 129 ± 37.7 | 11900 ± 1530 |
| Step 6 | 0.6 mg | 4 | 74.2 ± 25.1 | 0.167 ± 0.000 | 2130 ± 542 | 5000 ± 1350 | 83.2 ± 6.88 | 29.1 ± 0.472 | 119 ± 9.42 | 120 ± 35.5 | 14200 ± 3960 |
| Step 7 | 0.8 mg | 5 | 113 ± 42.0 | 0.167 ± 0.000 | 2700 ± 1250 | 6130 ± 3530 | 72.0 ± 33.1 | 26.3 ± 6.23 | 102 ± 47.9 | 212 ± 224 | 13200 ± 4770 |
| B is plasma concentration-time profiles after multiple dose of upacicalcet | |||||
|---|---|---|---|---|---|
| Step | Group | Visit (day) | N | Ctrough (ng/mL) | Cmax (ng/mL) |
| Step 1 | 0.05 mg | 1 | 8 | – | 7.10 ± 1.84 |
| 3 | 8 | 2.21 ± 0.97 | – | ||
| 5 | 8 | 2.42 ± 1.20 | – | ||
| 8 | 8 | 1.97 ± 1.17 | – | ||
| 10 | 8 | 2.66 ± 1.23 | – | ||
| 12 | 8 | 2.46 ± 1.45 | – | ||
| 15 | 7 | 2.04 ± 1.41 | – | ||
| 17 | 7 | 2.67 ± 1.25 | 7.83 ± 1.43 | ||
| 19 | 7 | 2.84 ± 1.28 | – | ||
| 22 | 7 | 1.88 ± 1.17 | – | ||
| Step 2 | 0.1 mg | 1 | 8 | – | 15.6 ± 4.21 |
| 3 | 8 | 5.19 ± 1.02 | – | ||
| 5 | 8 | 6.43 ± 1.10 | – | ||
| 8 | 8 | 5.49 ± 1.24 | – | ||
| 10 | 8 | 6.56 ± 1.24 | – | ||
| 12 | 7 | 6.60 ± 1.29 | – | ||
| 15 | 7 | 5.38 ± 1.27 | – | ||
| 17 | 6 | 6.61 ± 0.82 | 16.7 ± 3.35 | ||
| 19 | 6 | 6.52 ± 1.15 | – | ||
| 22 | 6 | 5.68 ± 1.01 | ± | ||
| Step 3 | 0.2 mg | 1 | 10 | – | 26.3 ± 4.73 |
| 3 | 10 | 8.65 ± 1.98 | – | ||
| 5 | 10 | 10.7 ± 2.78 | – | ||
| 8 | 9 | 8.88 ± 2.83 | – | ||
| 10 | 9 | 10.5 ± 2.52 | – | ||
| 12 | 7 | 11.2 ± 3.69 | – | ||
| 15 | 7 | 9.17 ± 3.12 | – | ||
| 17 | 7 | 11.2 ± 3.35 | 26.4 ± 7.15 | ||
| 19 | 6 | 10.5 ± 3.21 | – | ||
| 22 | 4 | 11.1 ± 2.90 | – | ||
Mean ± SD
M2 was not detected in the groups receiving a dose of 0.025–0.2 mg and was detected immediately after dosing in the group receiving 0.4–0.8 mg. M3 was not detected in the group receiving 0.025 mg and was detected from 18 h after dosing in the groups receiving ≥ 0.05 mg. The plasma concentrations of M2 and M3 were much lower than those of the unchanged drug (Fig. S3A–S3B of the ESM).
Multiple-Dose Study
After 3 weeks of multiple administrations, upacicalcet was present in the plasma mainly in the unchanged form. The plasma upacicalcet trough concentration before dialysis did not increase with multiple administration (Fig. 2B, Table 2B). The dialysis removal rate of unchanged upacicalcet in plasma was between 79.27 and 86.98% (Table 3B).
The trough concentrations of metabolites M1 and M2 were not detected in groups receiving any dose before dialysis. The trough concentrations of metabolite M3 before dialysis were lower than those of the unchanged drug (Fig. S3C of the ESM).
Pharmacodynamic Findings
Single-Dose Study
Serum iPTH levels in the upacicalcet group decreased within 10 min of drug administration, and the effect tended to increase with increasing dose. Pre-dialysis serum cCa2+ levels tended to decrease after 6 or 18 h of treatment. At 66 h of treatment, pre-dialysis serum cCa2+ levels relatively decreased in the group receiving ≥ 0.1 mg. Serum P levels increased over time in all upacicalcet groups, but the degree of increase tended to be smaller in the upacicalcet group than in the placebo group (Fig. 3A–C).
Fig. 3.

Mean percent changes in the serum intact parathyroid hormone (iPTH) level (A), serum corrected calcium level (B), and serum phosphorus level (C) after a single dose of upacicalcet. Mean percent changes in the serum iPTH level (D), serum corrected calcium level (E), and serum phosphorus level (F) after multiple doses of upacicalcet. Arrows indicate the initiation of hemodialysis
The trend of serum whole PTH levels was similar to that of serum iPTH, and the trend of serum ionized Ca2+ levels was similar to that of serum cCa2+ levels. Serum fibroblast growth factor 23 levels tended to increase in the placebo group and the group receiving 0.025 mg, and these increases seemed to be suppressed in the groups receiving ≥ 0.05 mg. There was no difference in 1,25(OH)2 vitamin D levels between the upacicalcet and the placebo groups. Calcitonin levels increased immediately after dosing in all upacicalcet groups compared with the placebo group and tended to decrease over time (Fig. S4A–S4E of the ESM).
Multiple-Dose Study
Serum iPTH and cCa2+ levels decreased in a dose-dependent manner in the upacicalcet groups. The decrease in iPTH and cCa2+ levels in the upacicalcet groups disappeared 1 week after the end of administration (day 29). Serum P levels also tended to decrease in all the upacicalcet groups compared with the placebo group, although the extent of change was not as apparent as for iPTH and cCa2+ levels (Fig. 3D–F). Serum whole PTH, fibroblast growth factor 23, bone-specific alkaline phosphatase, and tartrate-resistant acid phosphatase-5b levels tended to decrease in all the upacicalcet groups compared with the placebo group (Fig. S4F of the ESM).
Safety and Tolerability Analysis
The occurrence of AEs and ADRs, as well as GI disorders and events related to hypocalcemia are summarized in Table 4.
Table 4.
Adverse events and adverse drug reactions in single and multiple dose studies
| Study | Single | Multiple | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Upacicalcet | Upacicalcet | ||||||||||||||
| Group | Placebo | 0.025 mg | 0.05 mg | 0.1 mg | 0.2 mg | 0.4 mg | 0.6 mg | 0.8 mg | Placebo | 0.05 mg | 0.1 mg | 0.2 mg | |||
| N | 15 | 4 | 4 | 4 | 4 | 4 | 4 | 5 | 11 | 9 | 9 | 10 | |||
| Subjects with any AEs | AEs | N | 1 | 2 | 2 | 1 | 1 | 1 | 2 | 2 | 6 | 4 | 5 | 5 | |
| % | 6.7 | 50.0 | 50.0 | 25.0 | 25.0 | 25.0 | 50.0 | 50.0 | 54.5 | 44.4 | 55.6 | 50.0 | |||
| ADRs | N | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 2 | 0 | 2 | 3 | 3 | ||
| % | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 25.0 | 50.0 | 40.0 | 0.0 | 22.2 | 33.3 | 30.0 | |||
| Abdominal discomfort | AEs | N | – | – | – | – | – | – | – | – | 0 | 0 | 1 | 1 | |
| % | – | – | – | – | – | – | – | – | 0.0 | 0.0 | 11.1 | 10.0 | |||
| ADRs | N | – | – | – | – | – | – | – | – | 0 | 0 | 1 | 1 | ||
| % | – | – | – | – | – | – | – | – | 0.0 | 0.0 | 11.1 | 10.0 | |||
| Abdominal pain upper | AEs | N | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | – | – | – | – | |
| % | 0.0 | 0.0 | 0.0 | 0.0 | 25.0 | 0.0 | 0.0 | 0.0 | – | – | – | – | |||
| ADRs | N | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | – | – | – | – | ||
| % | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | – | – | – | – | |||
| Anorectal discomfort | AEs | N | – | – | – | – | – | – | – | – | 1 | 0 | 0 | 0 | |
| Gastrointestinal disorders | % | – | – | – | – | – | – | – | – | 9.1 | 0.0 | 0.0 | 0.0 | ||
| ADRs | N | – | – | – | – | – | – | – | – | 0 | 0 | 0 | 0 | ||
| % | – | – | – | – | – | – | – | – | 0.0 | 0.0 | 0.0 | 0.0 | |||
| Diarrhoea | AEs | N | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | |
| % | 0.0 | 0.0 | 25.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 11.1 | 0.0 | 0.0 | |||
| ADRs | N | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| % | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |||
| Nausea | AEs | N | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | – | – | – | – | |
| % | 0.0 | 25.0 | 0.0 | 0.0 | 0.0 | 0.0 | 25.0 | 0.0 | – | – | – | – | |||
| ADRs | N | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | – | – | – | – | ||
| % | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 25.0 | 0.0 | – | – | – | – | |||
| Vomiting | AEs | N | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | |
| % | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 25.0 | 25.0 | 20.0 | 0.0 | 11.1 | 0.0 | 0.0 | |||
| ADRs | N | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | ||
| % | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 25.0 | 25.0 | 20.0 | 0.0 | 0.0 | 0.0 | 0.0 | |||
| Events related to hypocalcemia | Blood calcium decreased | AEs | N | – | – | – | – | – | – | – | – | 0 | 1* | 0 | 0 |
| % | – | – | – | – | – | – | – | – | 0.0 | 11.1 | 0.0 | 0.0 | |||
| ADRs | N | – | – | – | – | – | – | – | – | 0 | 1* | 0 | 0 | ||
| % | – | – | – | – | – | – | – | – | 0.0 | 11.1 | 0.0 | 0.0 | |||
| Adjusted calcium decreased | AEs | N | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | |
| % | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 20.0 | 0.0 | 11.1 | 0.0 | 10.0 | |||
| ADRs | N | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | ||
| % | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 20.0 | 0.0 | 11.1 | 0.0 | 10.0 | |||
N number of occurrences, % expression rate
*Overdose patient
Single-Dose Study
Among the AEs that occurred in the upacicalcet group, five patients had AEs for which a causal relationship to the study drug could not be ruled out. Of these, vomiting occurred in one patient in the group receiving 0.4 mg, one in the group receiving 0.6 mg, and one in the group receiving 0.8 mg, nausea in one patient in the group receiving 0.6 mg, and adjusted Ca decreased in one patient in the group receiving 0.8 mg. All of these events were non-serious and mild.
Multiple-Dose Study
Serious AEs were as follows: shunt obstruction in one patient in the group receiving 0.1 mg, shunt stenosis in one patient in the placebo group, and lumbar fracture in one patient in the placebo group, all of which were judged to be unrelated to the study drug. The reported ADRs were as follows: supraventricular extrasystoles in one patient in the group receiving 0.1 mg and one in the group receiving 0.2 mg, ventricular extrasystoles in one patient in the group receiving 0.1 mg, abdominal discomfort in one patient in the group receiving 0.1 mg and one in the group receiving 0.2 mg, adjusted Ca decreased in one patient in the group receiving 0.05 mg and one patient in the group receiving 0.2 mg.
One patient in the group receiving 0.05 mg and one in the group receiving 0.1 mg experienced an accidental overdose (1.0 mg). In these patients, blood Ca2+ levels decreased (in the group receiving 0.05 mg) and hypoesthesia occurred (in the group receiving 0.1 mg).
The merged QTcF values obtained from the 12-lead electrocardiogram results in the single-dose and multiple-dose studies are shown in Fig. 4. Although QTcF tended to be prolonged with upacicalcet administration, QTcF values seemed to have a negative relationship with serum cCa2+ levels.
Fig. 4.
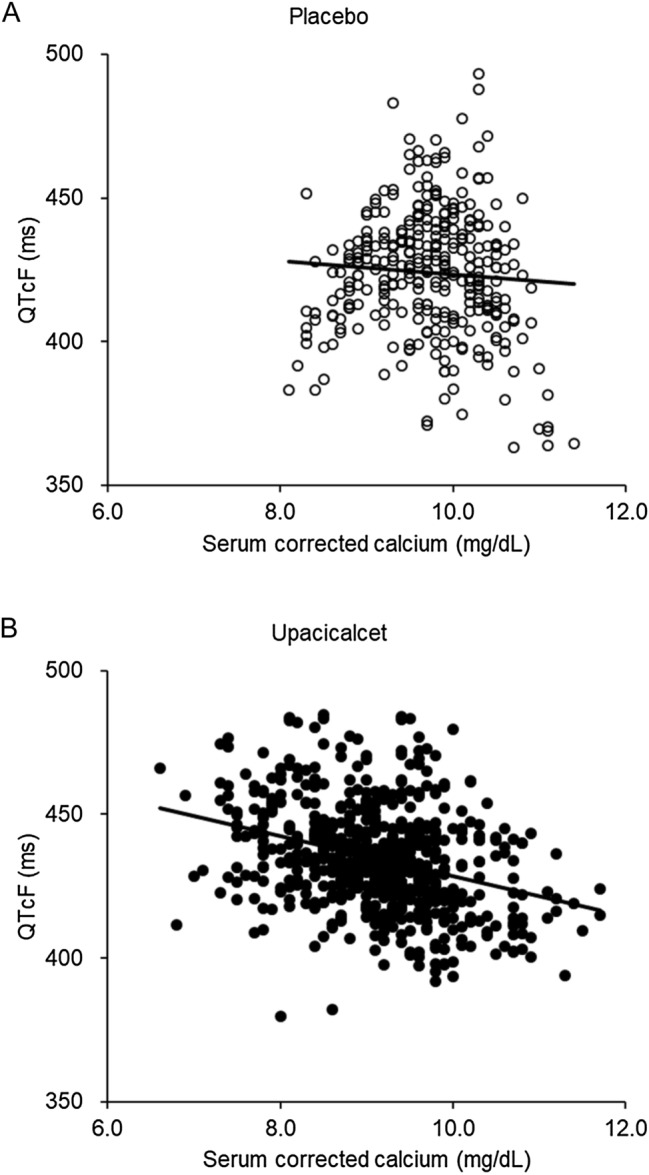
Correlation between the QT interval as corrected by the Fridericia method (QTcF) and serum-corrected calcium in placebo-treated patients (A) and upacicalcet-treated patients (B). The merged QTcF values were obtained from the 12-lead electrocardiogram results in single-dose and multiple-dose studies. 〇 indicates placebo (Pearson’s correlation coefficient = − 0.068), ● indicates upacicalcet (Pearson’s correlation coefficient = − 0.323).
Discussion
The results of this study revealed the pharmacokinetic and pharmacodynamic properties of upacicalcet when administered for the first time to patients with SHPT undergoing hemodialysis. Upacicalcet, when administered after dialysis, remained mainly unchanged in the plasma until the next dialysis session. Plasma upacicalcet concentrations increased in a dose-dependent manner. In addition, upacicalcet was well removed by dialysis and did not accumulate after multiple administration after each dialysis.
Pharmacodynamic evaluation in the single-dose study showed that upacicalcet rapidly lowers serum iPTH levels and subsequently lowers serum Ca. The time of lowest serum Ca2+ under upacicalcet administration was the next pre-dialysis. It was an important implication of this study for the management of hypocalcemic status in patients undergoing dialysis. In contrast, a decrease in serum P was not apparent. In the single-dose study, the baseline values are post-dialysis values. The interdialytic changes in serum P suggest that the decrease in serum P due to upacicalcet was obscured by the increase in serum P due to diet. In the multiple-dose study, changes in pre-dialysis serum iPTH, Ca, and P levels responded to plasma upacicalcet concentrations and returned to pre-administration levels when upacicalcet was eliminated from the body. These results suggest upacicalcet produces a dose-dependent decreasing effect on iPTH in patients with SHPT undergoing hemodialysis and that the effect disappears within a week after withdrawal.
Meanwhile, it is known that etelcalcetide, an intravenous calcimimetic similar to upacicalcet, forms a covalent serum albumin peptide conjugate and remains in the body after dialysis [11]. Therefore, it is estimated that it takes 32 days for etelcalcetide to reach steady state in patients undergoing dialysis three times a week for 6 h [12]. It has also been reported that the serum Ca-lowering effect of etelcalcetide peaks > 7 weeks after administration [15, 16]. Although upacicalcet is in the same class of injectable calcimimetics as etelcalcetide, it can have a different therapeutic response profile from that of etelcalcetide owing to differences in pharmacokinetic properties. This assumption should be confirmed by further clinical studies in the future.
Concerning the safety assessment of this study, we considered that upacicalcet showed no serious safety or tolerability issues. However, we observed an increase in upper GI disturbances, especially with upacicalcet doses ≥ 0.4 mg, in the single-dose study. It has been pointed out that CaSR stimulation of the GI tract is involved in the pathogenic mechanism of calcimimetic-induced upper GI disorders [17]. Nevertheless, it has been reported that etelcalcetide resulted in no difference in the incidence of upper GI symptoms in direct comparison with cinacalcet, an oral calcimimetic [16]. Hence, even injectable calcimimetics may stimulate CaSR in the GI tract if their plasma concentration is increased. The results of our study support this notion. In contrast, evocalcet is thought to have reduced upper GI adverse events by increasing bioavailability and reducing GI exposure compared with cinacalcet [18, 19]. These reports are consistent with the idea that calcimimetics inhibit gastric peristalsis by mainly stimulating CaSR in gastric parietal cells or gastrin-secreting cells. Our issue would be to control the plasma concentrations of upacicalcet so that it stimulates the parathyroid CaSR specifically but not the CaSR of the GI tract. We should determine the appropriate dosing regimen of upacicalcet to reduce upper GI symptoms as much as possible and demonstrate its efficacy in further studies.
Regarding the correlation between serum cCa2+ levels and QT time prolongation, we assumed that upacicalcet had a serum Ca-lowering effect that inhibited bone resorption when administered to patients undergoing dialysis, similar to other calcimimetics. Calcimimetics do not directly prolong QT but do cause QT prolongation with Ca2+ lowering. The threshold for management of the hypocalcemic state in the treatment of chronic kidney disease-mineral bone disorder is inconclusive [20, 21]. However, it is widely known that a low serum Ca2+ state causes intra-dialytic hypotension, decreased myocardial stroke volume, and QT prolongation [22–24]. Moreover, low serum Ca2+ levels have been suggested to be associated with sudden cardiac arrest [25]. Consequently, patients should be monitored to avoid hypocalcemia when administering calcimimetics.
There are some limitations to this study, the major one being that the maximum duration of administration was only 3 weeks, and no information was available on the effect of dose change. The condition of patients with SHPT undergoing dialysis is heterogeneous, and for this reason, calcimimetics are generally used with dosage adjustments according to the PTH and Ca2+ levels of each patient. The efficacy and safety of long-term dose-adjusted administration of upacicalcet in a large number of patients with SHPT undergoing hemodialysis should be evaluated in future studies.
Conclusions
We present the results of the first study on upacicalcet injection in patients with SHPT undergoing hemodialysis. Upacicalcet persisted in a dose-dependent manner until the time of the next dialysis, was well removed by dialysis, and constant blood concentrations were maintained when the same doses were administered repeatedly. Pharmacodynamic parameters, such as serum iPTH and Ca2+ levels, also changed according to the plasma upacicalcet concentration. In addition, there were no severe concerns about the safety and tolerability of upacicalcet in this study. The efficacy and safety of upacicalcet, as a new calcimimetic agent, should be assessed extensively in further clinical studies.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgments
We thank Editage (www.editage.com) for English language editing.
Declarations
Funding
Sanwa Kagaku Kenkyusho Co., Ltd. (SKK) sponsored this study. SKK was involved in the collection, management, analysis, and interpretation of the study data.
Conflicts of interest/competing interests
Fumihlko Koiwa, Keitaro Yokoyama, and Masafumi Fukagawa received honoraria from SKK. Kenji Asano and Daisuke Honda are employees of SKK. Tadao Akizawa received consulting fees from SKK.
Ethics approval
The study was approved by the institutional review board of each participating center.
Consent to participate
All subjects provided written informed consent before participating in the study.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Authors’ contributions
JJK, KY, MF, and DH substantially contributed to the analysis and interpretation of the data. FK, KA, and TA substantially contributed to the study design and the acquisition and interpretation of the data. All authors critically revised the report, commented on drafts of the manuscript, and approved the final report.
References
- 1.Cunningham J, Locatelli F, Rodriguez M. Secondary hyperparathyroidism: pathogenesis, disease progression, and therapeutic options. Clin J Am Soc Nephrol. 2011;6(4):913–921. doi: 10.2215/CJN.06040710. [DOI] [PubMed] [Google Scholar]
- 2.Fraser WD. Hyperparathyroidism. Lancet. 2009;374(9684):145–158. doi: 10.1016/S0140-6736(09)60507-9. [DOI] [PubMed] [Google Scholar]
- 3.Goodman WG, Goodman WG. Recent developments in the management of secondary hyperparathyroidism. Kidney Int. 2001;59(3):1187–1201. doi: 10.1046/j.1523-1755.2001.0590031187.x. [DOI] [PubMed] [Google Scholar]
- 4.Fukagawa M, Yokoyama K, Koiwa F, Taniguchi M, Shoji T, Kazama JJ, CKD-MBD Guideline Working Group; Japanese Society for Dialysis Therapy et al. Clinical practice guideline for the management of chronic kidney disease-mineral and bone disorder. Ther Apher Dial. 2013;17(3):247–288. doi: 10.1111/1744-9987.12058. [DOI] [PubMed] [Google Scholar]
- 5.Quarles LD, Sherrard DJ, Adler S, Rosansky SJ, McCary LC, Liu W, et al. The calcimimetic AMG 073 as a potential treatment for secondary hyperparathyroidism of end-stage renal disease. J Am Soc Nephrol. 2003;14(3):575–583. doi: 10.1097/01.asn.0000050224.03126.ad. [DOI] [PubMed] [Google Scholar]
- 6.Block GA, Martin KJ, de Francisco AL, Turner SA, Avram MM, Suranyi MG, et al. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N Engl J Med. 2004;350(15):1516–1525. doi: 10.1056/NEJMoa031633. [DOI] [PubMed] [Google Scholar]
- 7.Lindberg JS, Culleton B, Wong G, Borah MF, Clark RV, Shapiro WB, et al. Cinacalcet HCl, an oral calcimimetic agent for the treatment of secondary hyperparathyroidism in hemodialysis and peritoneal dialysis: a randomized, double-blind, multicenter study. J Am Soc Nephrol. 2005;16(3):800–807. doi: 10.1681/ASN.2004060512. [DOI] [PubMed] [Google Scholar]
- 8.Fukuma S, Kurita N, Fukagawa M, Akizawa T, Fukuhara S. Impact of cinacalcet introduction on MBD management: the MBD-5D study in Japan. Kidney Int Suppl. 2013;3(5):436–441. doi: 10.1038/kisup.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukagawa M, Yumita S, Akizawa T, Uchida E, Tsukamoto Y, Iwasaki M, KRN1493 Study Group et al. Cinacalcet (KRN1493) effectively decreases the serum intact PTH level with favorable control of the serum phosphorus and calcium levels in Japanese dialysis patients. Nephrol Dial Transplant. 2008;23(1):328–335. doi: 10.1093/ndt/gfm534. [DOI] [PubMed] [Google Scholar]
- 10.Gincherman Y, Moloney K, McKee C, Coyne DW. Assessment of adherence to cinacalcet by prescription refil1 rates in hemodialysis patients. Hemodial Int. 2010;14(1):68–72. doi: 10.1111/j.1542-4758.2009.00397.x. [DOI] [PubMed] [Google Scholar]
- 11.Edson KZ, Wu BM, Iyer A, Goodman W, Skiles GL, Subramanian R. Determination of etelcalcetide biotransformation and hemodialysis kinetics to guide the timing of its dosing. Kidney Int Rep. 2016;1(1):24–33. doi: 10.1016/j.ekir.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen P, Melhem M, Xiao J, Kuchimanchi M, Perez Ruixo JJ. Population pharmacokinetics analysis of AMG 416, an allosteric activator of the calcium-sensing receptor, in subjects with secondary hyperparathyroidism receiving hemodialysis. J Clin Pharmacol. 2015;55(6):620–628. doi: 10.1002/jcph.460. [DOI] [PubMed] [Google Scholar]
- 13.Bellasi A, Cozzolino M, Malberti F, Cancarini G, Esposito C, Guastoni CM, et al. New scenarios in secondary hyperparathyroidism: etelcalcetide. Position paper of working group on CKD-MBD of the Italian Society of Nephrology. J Nephrol. 2020;33(2):211–221. doi: 10.1007/s40620-019-00677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akizawa T, Ikejiri K, Kondo Y, Endo Y, Fukagawa M. Evocalcet: a new oral calcimimetic for dialysis patients with secondary hyperparathyroidism. Ther Apher Dial. 2020;24(3):248–257. doi: 10.1111/1744-9987.13434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patel J, Bridgeman MB. Etelcalcetide (Parsabiv) for secondary hyperparathyroidism in adults with chronic kidney disease on hemodialysis. Pharm Ther. 2018;43(7):396–399. [PMC free article] [PubMed] [Google Scholar]
- 16.Block GA, Bushinsky DA, Cheng S, Cunningham J, Dehmel B, Drueke TB, et al. Effect of etelcalcetide vs cinacalcet on serum parathyroid hormone in patients receiving hemodialysis with secondary hyperparathyroidism: a randomized clinical trial. JAMA. 2017;317(2):156–164. doi: 10.1001/jama.2016.19468. [DOI] [PubMed] [Google Scholar]
- 17.Ray JM, Squires PE, Curtis SB, Meloche MR, Buchan AM. Expression of the calcium-sensing receptor on human antral gastrin cells in culture. J Clin Investig. 1997;99(10):2328–2333. doi: 10.1172/JCI119413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawata T, Tokunaga S, Murai M, Masuda N, Haruyama W, Shoukei Y, et al. A novel calcimimetic agent, evocalcet (MT-4580/KHK7580), suppresses the parathyroid cell function with little effect on the gastrointestinal tract or CYP isozymes in vivo and in vitro. PLoS ONE. 2018;13(4):e0195316. doi: 10.1371/journal.pone.0195316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyazaki H, Ikeda Y, Sakurai O, Miyake T, Tsubota R, Okabe J, et al. Discovery of evocalcet, a next-generation calcium-sensing receptor agonist for the treatment of hyperparathyroidism. Bioorg Med Chem Lett. 2018;28(11):2055–2060. doi: 10.1016/j.bmcl.2018.04.055. [DOI] [PubMed] [Google Scholar]
- 20.Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Update Work Group. KDIGO 2017 clinical practice guideline update for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). Kidney Int Suppl. 2017;7(1):1–59. 10.1016/j.kisu.2017.04.001. [DOI] [PMC free article] [PubMed]
- 21.Isakova T, Nickolas TL, Denburg M, Yarlagadda S, Weiner DE, Gutiérrez OM, et al. KDOQI US commentary on the 2017 KDIGO clinical practice guideline update for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD) Am J Kidney Dis. 2017;70(6):737–751. doi: 10.1053/j.ajkd.2017.07.019. [DOI] [PubMed] [Google Scholar]
- 22.Maynard JC, Cruz C, Kleerekoper M, Levin NW. Blood pressure response to changes in serum ionized calcium during hemodialysis. Ann Intern Med. 1986;104(3):358–361. doi: 10.7326/0003-4819-104-3-358. [DOI] [PubMed] [Google Scholar]
- 23.van der Sande FM, Cheriex EC, van Kuijk WH, Leunissen KM. Effect of dialysate calcium concentrations on intradialytic blood pressure course in cardiac-compromised patients. Am J Kidney Dis. 1998;32(1):125–131. doi: 10.1053/ajkd.1998.v32.pm9669433. [DOI] [PubMed] [Google Scholar]
- 24.Newman DB, Fidahussein SS, Kashiwagi DT, Kennel KA, Kashani KB, Wang Z, et al. Reversible cardiac dysfunction associated with hypocalcemia: a systematic review and meta-analysis of individual patient data. Heart Fail Rev. 2014;19(2):199–205. doi: 10.1007/s10741-013-9371-1. [DOI] [PubMed] [Google Scholar]
- 25.Yarmohammadi H, Uy-Evanado A, Reinier K, Rusinaru C, Chugh H, Jui J, et al. Serum calcium and risk of sudden cardiac arrest in the general population. Mayo Clin Proc. 2017;92(10):1479–1485. doi: 10.1016/j.mayocp.2017.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.