

Summary
Numerous studies have established the involvement of lysosomal and mitochondrial dysfunction in the pathogenesis of neurodegenerative disorders such as Alzheimer’s and Parkinson diseases. Building on our previous studies of the neurodegenerative lysosomal lipidosis Niemann–Pick C1 (NPC1), we have unexpectedly discovered that activation of the mitochondrial chaperone tumor necrosis factor receptor-associated protein 1 (TRAP1) leads to the correction of the lysosomal storage phenotype in patient cells from multiple lysosomal storage disorders including NPC1. Using small compound activators specific for TRAP1, we find that activation of this chaperone leads to a generalized restoration of lysosomal and mitochondrial health. Mechanistically, we show that this process includes inhibition of oxidative phosphorylation and reduction of oxidative stress, which results in activation of AMPK and ultimately stimulates lysosome recycling. Thus, TRAP1 participates in lysosomal-mitochondrial crosstalk to maintain cellular homeostasis and could represent a potential therapeutic target for multiple disorders.
Subject areas: Cell biology, Cellular neuroscience, Neuroscience
Graphical abstract
Highlights
-
•
Small molecules ML405 and 1685 correct the lipid storage in LSDs including NPC1
-
•
These molecules are agonists of the mitochondrial chaperone TRAP1
-
•
TRAP1 agonists ameliorate mitochondrial stress and improve lysosomal function
-
•
TRAP1 agonists reduce lipid storage in a mouse model of Fabry disease
Cell biology; Cellular neuroscience; Neuroscience
Introduction
Lysosomal storage disorders (LSDs) are a group of more than 60 genetic disorders in which the function of cellular endosomes/lysosomes is impaired. They arise owing to defects in lysosomal hydrolases, lysosomal membrane proteins, or other non-lysosomal proteins that participate in lysosomal function (Parenti and Medina, 2021) and collectively constitute the most common genetic defect in man. Neurological impairment is a feature of ∼80% of LSDs, for which there are currently very limited treatment options. A prototypical neurodegenerative LSD is Niemann–Pick type C1 (NPC1) disease, which is characterized by cholesterol and other lipid accumulation in the endosomal/lysosomal (E/L) system (Pentchev et al., 1994). Although the precise function of NPC1 remains ambiguous (Davies et al., 2000; Höglinger et al., 2019; Infante et al., 2008), its loss results in a generalized block in lipid transport from late endosomes to the trans-Golgi network and plasma membrane (PM).
We and others previously showed that overexpression of the small GTPase Rab9 can be beneficial for NPC1 disease, independent of NPC1 protein function (Hao et al., 2009; Kaptzan et al., 2009; Walter et al., 2003). In addition, facilitating the escape of mutant proteins from endoplasmic reticulum (ER)-associated degradation (ERAD) has been proposed as an alternative approach to developing therapeutics for NPC1 and other diseases in which the mutant proteins are partially functional (Carlile et al., 2007; Gelsthorpe et al., 2008). Based on these observations, we developed reporter assays to implement high throughput screens (HTS) to identify Rab9 and NPC1 gene upregulators. Active compounds from the HTS were confirmed in phenotypic screening assays that measured the reduction of lipid storage in NPC1 cells. Lead compounds were subjected to structure activity relation (SAR) studies to improve their efficacy and metabolic properties. Surprisingly, optimized lead compounds, although extremely effective at reducing lysosomal lipid storage, were ultimately found to have no effect on Rab9 and/or NPC1 transcript or protein levels.
Here, in a paradigm shift, we determine that these novel compounds act as agonists of the mitochondrial chaperone tumor necrosis factor receptor-associated protein 1 (TRAP1). TRAP1 is a member of the Hsp90 family of chaperones and appears to play a crucial but poorly understood role in mitochondrial metabolic homeostasis and health (Joshi et al., 2020). Previous reports have focused on its role in promoting the Warburg effect (i.e., aerobic glycolysis) in tumor cells, but there has been no consensus reached, as different and sometimes contradictory data are obtained depending on the tumor type being studied (Masgras et al., 2017; Rasola et al., 2014; Yoshida et al., 2013). Similar to Hsp90, TRAP1 has ATPase activity, although its client proteins, which are required for efficient ATP hydrolysis (McLaughlin et al., 2002; Sung et al., 2016), have not been completely identified (Faienza et al., 2020). In addition to its role in energy metabolism (Yoshida et al., 2013), TRAP1 also serves a protective role against oxidative stress and mitochondrial dysfunction (Costa et al., 2013; Liu et al., 2020; Zhang et al., 2015).
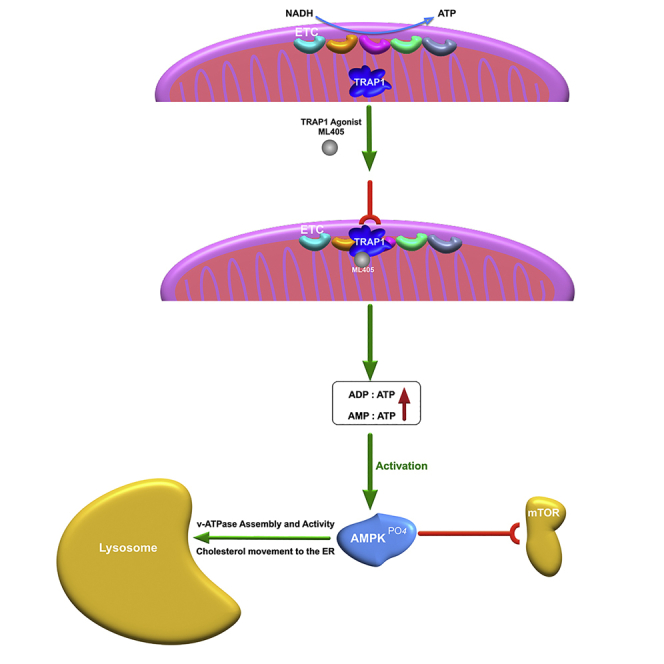
We show that these first-in-class TRAP1 agonists act in a manner that is consistent with increased TRAP1 activity and also modulate processes known to be regulated by TRAP1(Joshi et al., 2020; Liu et al., 2020; Yoshida et al., 2013; Zhang et al., 2015). We confirm that increased TRAP1 expression in NPC1 patient cells corrects their lipid storage phenotype and restores cholesterol transport. In addition, increased TRAP1 expression in patient cells from other LSDs, such as Fabry, Farber, and Wolman diseases, also corrects their lipid storage phenotype, raising an important question: how does the modulation of a mitochondrial chaperone positively impact the E/L storage phenotype of LSDs? These results can potentially be explained by considering the role of the mitochondria-lysosome axis in cellular health (Deus et al., 2020; Raimundo et al., 2016). A growing body of evidence indicates that crosstalk between the two organelles is critical for the maintenance of cellular homeostasis and response to stress, and disruption of this axis is thought to be a major contributor to the pathogenesis of LSDs and neurodegenerative disorders (Audano et al., 2018; Deus et al., 2020; Plotegher and Duchen, 2017; Stepien et al., 2020). Certain proteins, such as adenosine monophosphate-activated protein kinase (AMPK) (Laker et al., 2017), mechanistic target of rapamycin complex 1 (mTORC1) (Morita et al., 2017), and transcription factor EB (TFEB) (Settembre et al., 2011), have been posited as key mediators of this crosstalk. The lysosomal v-ATPase Ragulator complex has been shown to modulate the activity of AMPK and mTORC1 (Zhang et al., 2014), but there have been no reports of mitochondria-associated proteins that perform a similar role. Here we show that the activation of mitochondrial TRAP1 downregulates oxidative phosphorylation, which, in turn, activates AMPK and ultimately stimulates lysosome recycling.
Results
Upregulators of Rab9 and NPC1 expression
Our previous studies identified the small GTPase Rab9 as a suppressor of the NPC1 disease phenotype (Walter et al., 2003). Additionally, we and others observed that transient expression of NPC1 proteins bearing disease-causing mutations also suppresses the disease phenotype in NPC1 cells, presumably because overexpression allows low levels of the mutant NPC1 protein to escape ERAD and localize to late endosomes, where it is partially functional (Gelsthorpe et al., 2008).
Based on these observations, we hypothesized that small chemical compounds that could upregulate the expression of the Rab9 and/or NPC1 genes would be therapeutically relevant for treating NPC1 disease. Thus, we carried out Rab9 and NPC1 reporter gene screens of a library containing ∼320,000 structurally diverse compounds (https://pubchem.ncbi.nlm.nih.gov/bioassay/485297; https://pubchem.ncbi.nlm.nih.gov/bioassay/485313). Selected active molecules from the screen were further optimized for improved potency, physicochemical and metabolic properties through SAR and medicinal chemistry studies (the screening strategy and SAR will be described elsewhere) to yield our two lead compounds, NCGC00348147 (ML405) and NCGC00351685 (1685) (Figure 1A).
Figure 1.

Lead compounds correct the lipid storage phenotype in NPC1 and other LSDs
(A) Structure of lead compounds (see also Figure S1 and Table S1).
(B) Representative images showing correction of cholesterol storage (red; AloD-594) in NPC1 patient cell lines (NPC3, 17, 4) and NPC1 mouse cells (C57-NPC1) treated with 2-μM ML405/85 for 48 h.
(C) Representative western showing CRISPR-generated NPC1-ARPE clones C1, E10, and D12 have varying amounts of NPC1 protein.
(D and E) Representative image showing that E10 has no detectable NPC1 protein and stores large amounts of cholesterol (D, red; AloD-594), which is reduced after treatment with ML405/85 (E).
(F) PM cholesterol efflux is restored after treatment with 5-μM 1685 for 18 h in NPC1 patient (NPC4, NPC3) and mouse (C57-NPC1) cells.
(G) Treatment of NPC1 cells with ML405/85 for 3 h decreases SREBP2 processing. The blot shown is representative of four independent experiments.
(H) PM raft cholesterol levels (probed with the raft probe OlyA) increase after treatment with 1685 in NPC1 patient cell lines in a compound concentration-dependent manner.
(I) Quantitation of lipid storage in NPC1 cells treated with ML405/85 shown in (B) and (D–E).
(J) Treatment of different LSD patient cell lines with 1-μM ML405 reduces lipid storage (as determined by OlyA staining except for Fabry, which was probed with the Fabry lipid-specific probe VTB). At least 150 cells were quantitated for each sample and each experiment was repeated three times. Images for comparison of lipid storage were taken using the same exposure settings and represent the average of at least three independent experiments. Scale bar, 50 μm. Data are represented as mean ± SD ∗∗∗p < 0.0005, ∗∗p < 0.001, ∗p < 0.05.
The pharmacokinetic (PK) profiles of ML405 and 1685 are shown in Figure S1 and their PK parameters are summarized in Table S1. Following single IP injection at 10 mg/kg, systemic exposures measured by Cmax and AUC0-∞ were 6,050 ng/mL and 9,980 ng∗h/mL for ML405, and 4,100 ng/mL and 44,500 ng∗h/mL for 1685, respectively. The higher AUC observed for 1685 may be owing to slower elimination, as the t1/2 of 1685 was ca. 2.5-fold longer (i.e., 3.2 h for 1685 vs 1.3 h for ML405). As the dose increased to 30 mg/kg, in vivo exposures increased with the dose with a longer t1/2 for both compounds.
The liver to plasma AUC ratios were about 7 and 14 for ML405 and 1685, respectively. The brain AUC of ML405 and 1685 was ∼4% of plasma AUC values, which indicated that both compounds had low brain penetration (Table S1).
The lead compounds are active against multiple LSDs, including NPC1
We first evaluated the compounds in NPC1 patient cell lines with different genotypes to assess their ability to correct the NPC1 cellular phenotype irrespective of the causative mutation. We used a fluorescently labeled anthrolysin O (AloD-594) protein to probe for free cholesterol (Endapally et al., 2019), which is abundant in NPC cells but undetectable in wild-type cells. The NPC4 cell line is homozygous for the most common NPC1 patient mutation, I1061T (Millat et al., 1999), whereas the NPC3 (V1165M/X) and NPC17 (I1061T/X) lines each have a missense mutation in one allele and a truncating mutation in the second allele. Treatment of these cell lines with ML405/85 reduced their E/L cholesterol storage (Figure 1B, NPC3, NPC17, NPC4), indicating that the compounds are broadly active against the NPC1 lipid storage defect. In addition, these compounds also ameliorated lipid storage in NPC1 mouse cells (Figure 1B, C57-NPC1), which are homozygous for the D1005G mutation in the region where most human mutations, including the I1061T mutation, are found (Maue et al., 2012). These results suggested that the putative protein target of the compounds is conserved between mice and humans. Of note, the reduction of lipid storage was not equivalent across all cell lines; NPC3 and NPC17 were slightly less responsive to the compounds and exhibited more AloD staining than NPC4 and C57-NPC1 (Figures 1B and 1I), suggesting a minor contribution of the specific NPC1 genotype.
To determine whether the compounds depend on partial NPC1 protein activity for their lipid-lowering effects, we generated stable NPC1-null human retinal pigment epithelial (ARPE) cell lines using CRISPR-Cas9 editing. Clone E10 had no detectable NPC1 protein and stored large amounts of cholesterol (Figures 1C and 1D), so it was selected for use in subsequent studies. ML405/85 reduced the lipid storage in these cells (Figures 1E and 1I), indicating that their effects are independent of the NPC1 protein. To further characterize their activity, we established a cholesterol efflux assay to dynamically determine the amount of E/L cholesterol that can reach the PM, a step that is impaired in NPC1 disease cells (Wojtanik and Liscum, 2003). NPC1 cells were treated with 1685 in the presence of U18666A, an inhibitor that prevents cholesterol egress from the E/L system (Liscum and Faust, 1989). At the end of the treatment, PM cholesterol was first extracted from cells with methyl-β-cyclodextrin to create a baseline from which to measure new transport to the PM. Cells were then grown in lipid-free media for 3 h to release the U18666A block and allow E/L cholesterol to reach the PM. E/L-derived PM cholesterol was extracted as above and quantified. As shown in Figure 1F, 1685 increased cholesterol efflux in multiple NPC1 cell lines, indicating a restoration of intracellular lipid transport (Low et al., 2012). Furthermore, both 1685 and 405 decreased SREBP2 processing similarly to the oxysterol 25-hydroxycholesterol (Figure 1G), which indicates the movement of cholesterol to the ER (Du et al., 2004).
Recent studies have shown that there is only a small amount of PM cholesterol that is “available” (i.e., not complexed with sphingomyelin in lipid rafts) for efflux (Abrams et al., 2020). We developed another assay using a NanoBiT structural complementation reporter system to determine the effect of the compounds on raft-associated PM cholesterol. Cells treated as above were paraformaldehyde-fixed and incubated with an OlyA-SmallBiT probe, which binds to cholesterol-sphingomyelin domains in cell membranes. Luminescence was generated when LargeBiT was added and the fragments formed an active enzyme in the presence of substrate. As shown in Figure 1H, the treatment with 1685 resulted in a concentration-dependent increase in PM-raft cholesterol in three different NPC1 patient cell lines, confirming that the compounds can mobilize different cholesterol pools.
As the compounds act independently of the NPC1 protein, we speculated that the target of ML405/1685 might be a general regulator of E/L function. To test this hypothesis, we evaluated the effects of ML405 in patient cell lines derived from LSDs both with and without neuronal involvement: Fabry, Gaucher, Niemann-Pick A, Farber, neuronal ceroid lipofuscinosis 2 (CLN2), Pompe, mucopolysaccharidosis type VII (MPS VII), and Wolman diseases. Although the primary storage molecule in these disorders is dissimilar, many LSDs accumulate cholesterol and other lipids as secondary storage molecules (Walkley and Vanier, 2009). Using an mCherry-coupled ostreolysin A (OlyA) protein to probe for cholesterol-rich domains (Shaner et al., 2008), we found that treatment with ML405 reduced the cholesterol storage in these cells, irrespective of the lysosomal enzyme defect (Figure 1J), lending further support to the idea that the protein target of ML405 is a broad modulator of lysosomal function that is therapeutically effective in multiple LSDs.
The mitochondrial chaperone TRAP1 is the protein target
To clarify the mechanism of action of ML405/85, we set out to identify their protein target(s) using the drug affinity responsive target stability (DARTS) approach. This method is based on the rationale that a protein bound to a small molecule compound will exhibit subtle changes in its proteolytic profile, which can then be detected by quantitative proteomic analyses (Lomenick et al., 2011; Pai et al., 2015). After treating cells with ML405, 1685, or vehicle, we performed limited proteolysis of the protein extracts followed by tandem mass spectrometry. As expected, the peptide profiles of the three samples (DMSO-, ML405-, and 1685-treated) were remarkably similar, with 1,016 proteins having the same profile in all three populations (Figure 2A). To ascertain potential targets, we focused on the 69 proteins that had distinct profiles in the compound-treated samples compared with the vehicle-treated sample (Figure 2A); specifically, we identified proteins for which the number of peptides detected in the vehicle-treated sample was altered by more than two-fold in both of the compound-treated samples. Initial analyses yielded 19 proteins that satisfied these criteria (Table 1); interestingly, three candidates (ERP29, HYOU1, and TRAP1) are members of the heat shock family (HSP) of proteins.
Figure 2.

Identification of the protein target of ML405/85
(A) Venn diagram of protein peptide profiles identified by DARTS in DMSO-, ML405-, and 1685-treated cells; 1,016 proteins had essentially the same peptide profile in all three samples, whereas 69 proteins had a different profile from the DMSO control sample.
(B) Representative images for the lack of an effect of candidate target protein expression (green) on the cholesterol storage (red) of NPC3 patient cells.
(C) Representative images showing the effects of transient expression (left panel) or suppression (right panel) of HSP candidates ERP29, HYOU1, and TRAP1 on cholesterol storage in NPC3 patient cells or mouse NPC1 cells. Left panel: cells expressing candidate proteins are red and cholesterol is green. Right panel: cells in which protein is silenced are green, cholesterol is red.
(D) Representative images showing expression of TRAP1 (green) in Fabry disease patient cells decreases the Gb3 storage as determined by VTB staining (red). Likewise, the expression of TRAP1 (red) in Farber and Wolman disease cells decreases the cholesterol storage (green; OlyA) in these cells. Images are representative of at least three independent experiments. Graphs indicate the percentage of transfected cells corrected for lipid storage (n = 50–75) and are represented as mean ± SD Scale bar, 50 μm.
Table 1.
List of potential protein targets for the ML405/1685 compounds
| Protein | kDA | DMSO | 405 | 1685 | |
|---|---|---|---|---|---|
| aEndoplasmic reticulum resident protein 29 | ERP29_HUMAN | 29 | 0 | 10 | 9 |
| Isoform 2 of Tropomyosin alpha-3 | TPM3_HUMAN | 29 | 1 | 11 | 11 |
| Pleckstrin | PLEK_HUMAN | 40 | 0 | 5 | 5 |
| 26S protease regulatory subunit 10B, PSMC6 | PRS10_HUMAN | 44 | 0 | 4 | 5 |
| Importin-7 | IPO7_HUMAN | 120 | 2 | 7 | 7 |
| Synaptic vesicle membrane protein VAT-1 homolog | VAT1_HUMAN | 42 | 3 | 8 | 9 |
| Histone H1.2 | H12_HUMAN | 21 | 1 | 4 | 4 |
| Isoform 3 of Tyrosine-protein phosphatase non-receptor | PTPN6 | 63 | 9 | 18 | 15 |
| Transitional ER ATPase | TERA_HUMAN (VCP) | 89 | 10 | 19 | 19 |
| Isoform 2 of E3 ubiquitin-protein ligase UBR4 | sUBR4_HUMAN | 576 | 1 | 4 | 5 |
| 26S proteasome non-ATPase regulatory subunit 12 | PSD12_HUMAN | 53 | 1 | 3 | 3 |
| Crk-like protein | CRKL_HUMAN | 34 | 3 | 6 | 6 |
| Acetyl-CoA acetyltransferase, mitochondrial | THIL_HUMAN | 45 | 7 | 11 | 14 |
| Peroxiredoxin-1 | PRDX1_HUMAN | 22 | 7 | 13 | 13 |
| Isoform 2 of Proteasome subunit beta type-8 | PSB8_HUMAN | 30 | 3 | 6 | 5 |
| 14-3-3 protein theta | 1433T_HUMAN | 28 | 4 | 7 | 7 |
| aHypoxia up-regulated protein 1, HYOU1 | E9PL22_HUMAN | 105 | 8 | 3 | 3 |
| aHeat shock protein 75 kDa, mitochondrial, TRAP1 | TRAP1_HUMAN | 80 | 7 | 3 | 3 |
| Serpin B6, SERPINB6 | SPB6_HUMAN | 43 | 12 | 5 | 6 |
Following the application of the DARTS technique (see STAR Methods), a list of potential candidates was compiled based on their altered proteomic profile in the presence (405, 1685) or absence (DMSO) of drug compounds.
Heat shock family protein.
To identify the target out of this group of candidates and determine whether ML405/85 act as agonists or antagonists, we transiently expressed each protein in NPC3 patient cells and evaluated their effectiveness in reducing E/L lipid storage. The majority of the candidates, including IPO7, PLEK, PRDX1, PSB8, PSMC6, and TPM3, had little effect on OlyA staining (red) compared with surrounding untransfected (non-green) cells (Figure 2B). Intriguingly, expression (Figure 2C, left panel, red) but not silencing (Figure 2C, right panel, green) of the three chaperones had varying effects on the lipid storage. Both ERP29 and HYOU1 expression reduced the lipid storage slightly, whereas expression of the predominantly mitochondrial TNF receptor-associated protein 1 (TRAP1) consistently and dramatically reduced the lipid storage in human NPC3 cells and also mouse NPC1 cells (Figure 2C, left panel). The inhibition of endogenous TRAP1 expression using shRNA had no effect (Figure 2C, right panel), indicating that ML405/85 act as TRAP1 agonists.
Based on our results that the compounds reduce lipid storage in multiple LSDs (Figure 1J), we hypothesized that if TRAP1 is the correct protein target, then TRAP1 expression in these LSD patient cells should also reduce their lipid storage. Indeed, transient TRAP1 expression in Fabry, Farber, and Wolman disease cells (Figure 2D; Fabry, green; Farber/Wolman, magenta) led to a marked reduction of their Gb3 (Fabry) or cholesterol (Farber, Wolman) storage (Figure 2D; Fabry, magenta; Farber/Wolman, green). These results provided further evidence that TRAP1 is the protein target of ML405/85, albeit raising a significant question: how does a mitochondrial chaperone affect lysosome function?
TRAP1 agonists prevent phosphorylation of c-Src and downregulate the activity of SDH and cytochrome c oxidase
To better understand the mechanism by which TRAP1 activation leads to the restoration of lysosome function, we performed analyses based on reported functions of TRAP1 (Joshi et al., 2020; Liu et al., 2020; Yoshida et al., 2013; Zhang et al., 2015) to determine if ML405/85 are biologically active in the same pathways.
Mitochondrial c-Src is a tyrosine kinase that has been shown to phosphorylate and promote the activity of several components of the mitochondrial respiratory chain, including complex II [succinate dehydrogenase (SDH) A] and complex IV [cytochrome c oxidase (COX)] (Miyazaki et al., 2003; Ogura et al., 2012). TRAP1 inhibits mitochondrial c-Src activity by binding to it and preventing its auto-activation by phosphorylation, and it has been postulated that TRAP1 exerts some of its effects on mitochondrial respiration through its effects on this kinase (Yoshida et al., 2013). Thus, we reasoned that TRAP1 activation by ML405 should lead to reduced hosphor-c-Src levels and a concomitant decrease in SDH and COX activity (Masgras et al., 2017). To confirm this hypothesis, we first expressed a mitochondrial-targeted, Flag-tagged c-Src cDNA (Ogura et al., 2012) in wild-type ARPE cells and then treated the cells with ML405. Flag-immunoprecipitated lysates were separated and the blots probed for total mitochondrial c-Src or the Tyr-416 phosphorylated form of c-Src (Figure 3A). As expected, ML405 treatment led to a detectable decrease in mitochondrial c-Src Tyr-416 phosphorylation (Figures 3A and 3B, Src-pY416), which is consistent with TRAP1 overexpression (Yoshida et al., 2013). Notably, there was no appreciable difference in total c-Src levels (Figure 3A, Input), confirming that ML405 affects c-Src phosphorylation rather than its expression. Similar effects were obtained with endogenous c-Src (not shown).
Figure 3.

ML405/85 mimic TRAP1 actions on mitochondrial respiratory chain enzymes and metabolites
(A) Treatment with ML405 inhibits phosphorylation of transiently expressed mitochondria-targeted c-Src at Tyr-416 (p-Y416) but has no effect on total mitochondrial c-Src (Input) in ARPE cells. The blot shown is representative of four independent experiments.
(B) Densitometry of phospho-c-Src bands in (A) normalized to total c-Src (Input).
(C and D) ML405/85 inhibit complex II (SDH) activity of the mitochondrial respiratory chain in purified ARPE mitochondria (C), which results in the expected increase in cellular succinate levels (D). TTFA: thenoyltrifluoroacetone, complex II inhibitor (positive control).
(E) Treatment with ML405 lowers COX activity in purified ARPE mitochondria.
(F) Treatment with ML405/85 increase cellular and secreted lactate levels in ARPE cells. Data are represented as the mean ± SD ∗∗∗p < 0.0005, ∗∗p < 0.001.
TRAP1 has been shown to inhibit the activity of both SDH and COX (Sciacovelli et al., 2013; Yoshida et al., 2013), so we assessed the compounds’ effects on the activity of those enzymes. Purified mitochondria from treated ARPE cells were used for in vitro enzyme assays. Treatment with ML405/85 resulted in a 40–60% inhibition of SDH activity (Figure 3C), with an attendant increase in cellular succinate levels as expected (Figure 3D) (Sciacovelli et al., 2013). Furthermore, ML405 treatment reduced the COX activity by ∼50% (Figure 3E). These outcomes are predicted to shift cellular metabolism from oxidative phosphorylation to glycolysis, perhaps to allow for the reduction of oxidative stress by TRAP1 (Yoshida et al., 2013). Such a glycolytic shift would result in increased lactate production and secretion, which was confirmed in ML405/85 -treated cells (Figure 3F). Interestingly, 1685 stimulated more than double the amount of lactate production as untreated cells, whereas ML405 only increased production by ∼25%, indicating that the two compounds are not equivalent. Overall, these results suggest that the compounds recapitulate the effects of TRAP1 on components of the mitochondrial respiratory chain.
TRAP1 agonists reduce mitochondrial dysfunction in NPC1 cells
Mitochondrial dysfunction in NPC1 disease has previously been reported (Kennedy et al., 2014; Woś et al., 2016; Yu et al., 2005). Because of the mitochondrial location of TRAP1 and its protective effects on the organelle, we queried if our TRAP1 agonists would improve mitochondrial health in NPC1 cells. To assess mitochondrial health, we used the Mitotimer reporter, which encodes a mitochondrial-targeted green fluorescent protein that shifts irreversibly to red fluorescence under conditions of mitochondrial stress (Laker et al., 2014). Quantifying the number of red and green puncta in a cell produces a red/green fluorescence ratio, which decreases as mitochondrial stress is alleviated. Vehicle-treated NPC1 cells expressing Mitotimer exhibited considerable stress, as shown by the many red puncta (Figure 4A, DMSO), which was lessened in ML405-treated cells, as shown by increased yellow and green fluorescence (Figure 4A, ML405). ML405 treatment resulted in an ∼30–40% reduction in the red/green ratio of these cells (Figure 4B), indicating a reduction of oxidative stress in those mitochondria. Similar results were obtained in live cells with the mitochondrial dye MitoSOX Red, which fluoresces red when oxidized by superoxide. Isogenic wild-type and NPC1 ARPE cells treated with ML405/85 had fewer oxidized mitochondria than vehicle-treated cells, although there was a more dramatic reduction in NPC1 cells, especially with 1685 (Figure 4C). Interestingly, NPC1 ARPE cells do not show appreciably more MitoSOX Red staining compared with their isogenic wt counterparts, as would be expected, given that increased ROS is a feature of NPC1 (Kennedy et al., 2014); this result may be because the reagent only detects superoxide and no other reactive oxygen species or it may be a cell-type-specific issue. However, NPC1 ARPE cells expressing the more generalized oxidation-sensitive Mitotimer marker exhibit clear signs of mitochondrial stress, denoted by red puncta (Figure 4A, DMSO).
Figure 4.

TRAP1 agonists ameliorate mitochondrial and ER stress
(A and B) ML405 decreases the red/green fluorescence ratio in Mitotimer-expressing NPC1 cells, indicating reduced mitochondrial oxidative stress (A, representative image of cell mitochondria quantified in B).
(C) Treatment with ML405/85 reduce superoxide levels (red; MitoSox) in isogenic wt and NPC1-null cells but have no effect in isogenic TRAP1-null cells, indicating that the reduction of mitochondrial superoxide is mediated by TRAP1.
(D) Treatment with ML405 increases ATP levels in both NPC3 and Fabry patient cells.
(E) Treatment with ML405 increases the residual α-galactosidase activity in Fabry patient cells to varying degrees based on the specific α-galactosidase mutation of each cell line.
(F–H) Similarly, treatment with ML405 increases the activity of lysosomal acid lipase in Wolman (F), tripeptidyl peptidase 1 in CLNII (G), and galactocerebrosidase in Krabbe (H) patient cells. Data are represented as the mean ± SD ∗∗∗p < 0.0005, ∗∗p < 0.001.
In isogenic ARPE cells without TRAP1; however, neither compound had an effect (Figure 4C), providing compelling evidence that the compounds mediate their effects through TRAP1. ML405/85 had no effect on the number of mitochondria in treated cells compared with vehicle-treated cells (not shown). Taken together, the effects of the compounds on mitochondria are consistent with the well-documented protective role of TRAP1 against oxidative stress (Costa et al., 2013; Pridgeon et al., 2007; Yoshida et al., 2013).
Concurrent with reduced activity of respiratory chain enzymes, NPC1 mouse brains and neurons are characterized by markedly decreased ATP levels, and the addition of exogenous ATP has been shown to stimulate neurite outgrowth in neurons in these mice (Yu et al., 2005). ATP deficiencies have also been reported for other LSDs such as Fabry disease (Ivanova et al., 2019; Lücke et al., 2004). Based on the above results, we reasoned that ML405 should lead to healthier mitochondria and increased cellular ATP levels. Indeed, ML405-treated NPC1 patient cells had three times more ATP than vehicle-treated cells (Figure 4D, blue bars), whereas treated Fabry cells had almost twice the amount of ATP (Figure 4D, magenta bars). These results are consistent with TRAP overexpression in stressed cells (Zhang et al., 2015) and indicate that TRAP1 agonists can restore the mitochondrial function in multiple LSDs.
Activation of TRAP1 reduces ER stress and allows maturation of mutant LSD enzymes
TRAP1 has been reported to be protective against ER stress by downregulating the unfolded protein response (UPR) and assisting in refolding damaged proteins (Amoroso et al., 2012; Sisinni et al., 2014; Takemoto et al., 2011). To determine if ML405 acts in this pathway, we took advantage of the fact that in many LSDs, a small percentage of residual activity mutant proteins can escape UPR and reach the lysosome, leading to a measurable enzymatic activity (Gelsthorpe et al., 2008). Chemical chaperones or ERAD inhibitors have previously been proposed as methods by which to increase the survival of mutant proteins (Berardi et al., 2014; Yam et al., 2006). We hypothesized that TRAP1 activation by ML405/85 should facilitate the escape of these residual activity mutant enzymes from the ER, leading to increased enzymatic activity and potentially offering an explanation for their therapeutic effects against multiple LSDs. For these studies, we utilized three patient cell lines for Fabry disease (Figure 4E) with different distinct mutations and one each for CLNII, Wolman, and Krabbe disease (Figures 4F–4H) (Aslanidis et al., 1996; Berardi et al., 2014; Fan et al., 1999; Kohan et al., 2013; Yam et al., 2006). As predicted, treatment with ML405 significantly increased the detectable enzyme activity for these four different LSDs (Figures 4E–4H), emphasizing the therapeutic potential of these compounds and their broad applicability toward multiple LSDs.
TRAP1 bridges mitochondria and lysosome health
The above results raise an important and intriguing question: how does activation of a mitochondrial chaperone increase lysosome function and cycling? Clues to answering this question may come from a study showing that acute but not chronic mitochondrial respiratory chain defects trigger lysosome biogenesis via AMPK signaling (Fernández Mosquera et al., 2017). We hypothesized that the brief shutdown of oxidative phosphorylation (OxPhos) by activated TRAP1 would have a similar effect, which could explain its therapeutic activity against multiple LSDs as described above. To test this hypothesis, we first determined whether inhibition of mitochondrial OxPhos could signal the lysosome. Treatment of wild-type cells with antimycin A to inhibit complex III or atpenin A5 to inhibit SDH caused a four- to eight-fold increase in lysosomal lipid storage as determined by OlyA-Cherry staining (Figures 5A and 5B, wt), indicating that respiratory chain defects do affect lysosomal function as has been described (Baixauli et al., 2015). In NPC1 cells, with already a significant accumulation of lysosomal storage, both inhibitors exacerbated the disease phenotype by increasing the amount of storage (Figures 5A and 5B, NPC1).
Figure 5.

Mitochondrial dysfunction affects lysosomal function
(A) Representative images showing that cell treatment with mitochondrial respiratory chain inhibitors antimycin A (complex III) or atpenin A5 (SDH) induces the lysosomal lipid storage in wt cells and exacerbates the storage in NPC1 cells.
(B) Quantitation of the lipid storage seen in wt and NPC1 cells (A). At least 150 cells were quantitated for each sample and each experiment was repeated three times. Images were taken using the same exposure settings.
(C) Schematic showing the importance of mTORC1 as a sensor of glucose availability and a master regulator of mitochondria and lysosome/autophagy function.
(D) Treatment with 1685 or exogenous TRAP1 expression increases the levels of cellular AMPK and phosphorylated AMPK (pAMPK). GAPDH: glyceraldehyde 3-phosphate dehydrogenase.
(E) Effect of ML405/85 on pAMPK levels is rapid, reaching a maximum at about 1 h following treatment. Metform: metformin, known inducer of pAMPK (positive control).
(F) Densitometry of pAMPK bands from (E) normalized to GAPDH signal.
(G) Treatment with 1685 inhibits phosphorylation of the mTOR target p70S6 kinase in a rapid manner. Graph represents levels of normalized phosphorylated p70S6 kinase relative to total p70S6 kinase protein levels. The blots shown are representative of three independent experiments. Data are represented as the mean ± SD ∗∗∗p < 0.0005, ∗∗p < 0.001, ∗p < 0.05.
We next reasoned that inhibition of OxPhos by TRAP1 would shift the cellular AMP:ATP ratio, which is known to activate AMPK (Hardie, 2011, 2014). AMPK activation would, in turn, inhibit mTORC1, which would mobilize lysosomal transport and recycling (Figure 5C) (Laplante and Sabatini, 2009). To characterize this process, we treated cells with 1685 for 3 or 18 h and found a dramatic increase in phospho-AMPK (pAMPK) levels at 3 h of treatment, which was no longer detectable at the later time point, suggesting that the effect of 1685 is not long-lasting (Figure 5D, pAMPK). Transient expression of TRAP1 in these cells also increased pAMPK levels (Figure 5D, Trap), although not to the same extent as 1685, as TRAP1 expression does not equate to TRAP1 activation. The similar effects on AMPK levels and phosphorylation indicate that the effects of 1685 occurred via activation of TRAP1. The above results suggested that AMPK activation is rapid, so we treated cells with ML405/85 for 1 or 3 h and confirmed that induction of pAMPK was rapid, with 1685 showing the highest pAMPK levels at 1 h (Figures 5E and 5F). Interestingly, both TRAP1 agonists induced pAMPK at higher levels than metformin, a ubiquitous diabetes medication that is known to inhibit OxPhos and induce pAMPK (Rena et al., 2017), and at a thousand-fold lower concentration (1 μM for ML405/85 vs. 1 mM for metformin; Figures 5E and 5F). Treatment with 1685 also inhibited the phosphorylation of the mTORC1 target p70S6 kinase (Figure 5G), suggesting that the compound inhibits mTORC1 activity. Similarly to the activation of AMPK by 1685, mTORC1 inhibition is rapid, with about 50% inhibition at 4 h but almost none at 8 h. These results are consistent with recent findings that intervention at the mitochondria results in rapid but not long-lived effects (Long et al., 2022).
TRAP1 agonists show efficacy in mouse LSD models
The results of the cellular studies described above suggested that these molecules could be therapeutically beneficial in LSDs. To evaluate their potential, we first utilized a mouse model of Fabry disease (Ioannou et al., 2001). Mice were treated with either 30 mg/kg ML405 or vehicle by intraperitoneal (IP) injection three times/week for four weeks. Upon the completion of the study, mouse tissues were processed and analyzed for their levels of globotriaosyl ceramide (Gb3), the glycosphingolipid stored in Fabry disease, using an ELISA assay (Zeidner et al., 1999). ML405 reduced Gb3 levels in kidney and heart, the Fabry-relevant tissues, by more than 50% (Figures 6C and 6D) as well as in liver and plasma (Figures 6A and 6B).
Figure 6.

TRAP1 agonists show efficacy in a mouse Fabry model
Age-matched Fabry mice treated with 30 mg/kg ML405 three times/week for four weeks showed a reduced Gb3 storage in the liver (A) plasma (B) kidney (C), and heart (D). Values shown are the average of three replicates, n = 9–10. ∗∗∗p < 0.0005, ∗∗p < 0.001, ∗p < 0.05.
Treatment of a mouse model of NPC1 (Maue et al., 2012) with compound produced only a modest improvement in lifespan (not shown), which could be owing to the poor brain penetration of the compounds as determined in preliminary PK studies. Further modification of the compounds will be required to obtain a true picture of their efficacy for LSDs with neuronal involvement.
Discussion
In recent years, inter-organelle crosstalk has emerged as a critical process for the maintenance of cellular homeostasis. In particular, numerous studies have suggested that the proper function of the mitochondria and lysosomes is interlinked, with deficiencies in mitochondrial function directly impacting the lysosomal function and vice versa (Baixauli et al., 2015; Demers-Lamarche et al., 2016; Fernández Mosquera et al., 2017; Ivanova et al., 2019; Kennedy et al., 2014; Yambire et al., 2019). One way that this bidirectional interaction may be facilitated is by physical contacts between organelles(Wong et al., 2018). The mechanisms of communication between lysosomes and mitochondria are particularly relevant in the context of LSDs and especially those disorders with neuropathology. Whereas the pathogenesis of neurodegeneration in LSDs is not well understood, it has been suggested that mitochondrial dysfunction may be an important contributor (Deus et al., 2020; Saffari et al., 2017; Yu et al., 2005).
It is well established that cholesterol accumulation in NPC1 and other LSDs leads to increased mitochondrial cholesterol, which in turn results in mitochondrial dysfunction ranging from decreased activity of enzymes involved in OxPhos, repressed autophagy, and reduced mitochondrial biogenesis (Ivanova et al., 2019; Kennedy et al., 2014; Lücke et al., 2004; Solsona-Vilarrasa et al., 2019; Yambire et al., 2019; Yu et al., 2005). Similarly, mitochondrial dysfunction disrupts E/L system trafficking and function as well as lysosome biogenesis (Baixauli et al., 2015; Demers-Lamarche et al., 2016; Fernández Mosquera et al., 2017), creating a vicious feedback loop of organelle dysfunction, which disrupts cellular homeostasis. For example, in macrophages, mitochondrial ATP production has been shown to regulate cholesterol efflux, highlighting a direct link between energy metabolism and lipid transport (Karunakaran et al., 2015). In agreement with those results, the present studies show that inhibition of complex II or III of the mitochondrial respiratory chain induces lipid storage even in non-disease cells (Figures 5A and 5B).
Our discovery that agonists of the mitochondrial chaperone TRAP1 ameliorate the E/L lipid storage phenotype in multiple LSDs provides the first example of a mitochondrial protein that can directly affect lysosomal transport and recycling. Not only do the TRAP1 agonists ML405/85 restore the movement of multiple cholesterol pools within the cell and improve lipid storage in a mouse model of Fabry disease, but they also alleviate the mitochondrial dysfunction in NPC1 patient cells, suggesting that TRAP1 may play a hitherto unappreciated role as a modulator of the mitochondria-lysosome axis. Our data indicate that although the agonists are equally effective against TRAP1 in in vitro assays, they do show differences in vivo, possibly owing to their pharmacokinetic properties.
The functions of TRAP1 are only beginning to be elucidated, but multiple studies have shown that it is a key protector against oxidative stress and mitochondrial dysfunction (Costa et al., 2013; Liu et al., 2020; Zhang et al., 2015). Its role as a negative regulator of OxPhos (Yoshida et al., 2013), the main contributor of damaging reactive oxygen species (ROS), may be directly related to this protective function. It is notable that OxPhos is compromised in NPC1 cells (Yu et al., 2005); increased cholesterol at mitochondrial membranes deregulates mitochondrial potential, leading to increased ROS production, oxidative stress, reduced transfer of electrons, and decreased ATP production. The mitochondrial respiratory chain has evolved to produce maximal energy with minimum ROS production by maintaining adequate membrane potential. Reduction of complex II and complex IV activity (Figures 3C–3E) in NPC1 cells by our TRAP1 activators would facilitate a decrease in ROS and oxidative stress (Figures 4A–4C). Restoration of mitochondrial potential will, in turn, lead to a temporal elevation of the AMP/ATP ratio and activation of AMPK (Figures 5D–5F), with a concomitant inhibition of mTORC1 activity (Figure 5G). Activated AMPK promotes lipid homeostasis, lysosome recycling, reduction of ER stress, which ultimately leads to the recovery of cellular ATP levels.
In support of the above hypothesis, ATP levels in both NPC1 and Fabry cells increase after ML405 treatment (Figure 4D), suggesting that energy generation recovers after TRAP1 activation, as shown in cardiomyocytes following injury (Zhang et al., 2015). Furthermore, a distinction must be made between TRAP1 protein levels and its activation, leading to an increase in its ATPase activity. TRAP1 expression studies suffer from the inherent inability to correlate observations to the levels of “active” TRAP1 protein. ML405/85 have no effect on TRAP1 protein levels (not shown) but as shown in these studies, they do recapitulate the cellular effects attributed to active TRAP1, indicating that they act as agonists.
To understand how the activation of a mitochondrial chaperone can alleviate the lysosomal lipid storage in LSDs, we considered proteins that have been shown to act as downstream mediators of the mitochondrial-lysosomal crosstalk. AMPK is a global sensor of cellular energy levels (Herzig and Shaw, 2018), whereas mTORC1 is a nutrient sensor that coordinates cell growth and metabolism (Laplante and Sabatini, 2009); together, they work in a double-negative feedback loop (Holczer et al., 2019) to integrate nutrient and energy signals for the maintenance of cellular homeostasis. AMPK is activated by high AMP:ATP ratios, inhibits mTORC1, and stimulates lysosomal recycling and autophagy, whereas mTORC1 is activated when nutrients are plentiful and acts to oppose active AMPK, inhibit autophagy and lysosome recycling, and promote macromolecule synthesis and cell growth. Interestingly, the NPC1 protein and the lysosomal transmembrane protein SLC3849 act together to inhibit mTORC1 signaling (Castellano et al., 2017), which could explain why, in NPC1 disease, a defective NPC1 protein leads to mTORC1 hyperactivation and reduced autophagy/mitophagy (Davis et al., 2021). In contrast to our results with ML405/85 (Figure 1), direct inhibition of mTORC1 restores lysosomal function and autophagy without correcting the lipid storage in NPC1 cells (Davis et al., 2021), indicating that mTORC1 inhibition is only one of the effects of TRAP1 activation. For example, TRAP1 activation has the added effect of reducing mitochondrial stress, leading to a reduction in autophagic load lysosomal storage. Also, TRAP1 activation reduces ER stress (Sisinni et al., 2014), allowing for more efficient synthesis and processing of lysosomal proteins. These additional effects may be key to facilitating lysosome unloading.
Our data showing that TRAP1 and its agonists activate AMPK sheds light on the partial mechanism by which TRAP1 corrects the lysosomal and mitochondrial dysfunction in LSDs. As discussed above, the activation of AMPK would, in turn, inhibit mTORC1 and stimulate autophagy and lysosomal recycling, as has been described for statins (Bruiners et al., 2020). However, we find that this activation is rapid and short-lived, peaking at ∼1 h and undetectable by ∼18 h (Figures 5D–5F), which is consistent with recent findings that intervention at the mitochondria results in rapid but not long-lived effects (Long et al., 2022). The importance of timing recalls a study in which opposing responses to acute (<12 h) vs. chronic mitochondrial stress led to lysosome biogenesis or complete lysosome repression, respectively (Fernández Mosquera et al., 2017). This difference in timing could explain why the TRAP agonist 1685 induced higher levels of AMPK phosphorylation after 3 h than TRAP expression overnight (Figure 5D), or it may be a difference of expression vs. activation, as discussed above.
The discovery that TRAP1 activation can modulate AMPK and correct the lysosomal and mitochondrial dysfunction in LSDs has implications for other diseases. Most familial forms of Parkinson disease are caused by mutations in genes that regulate either lysosomal or mitochondrial function and furthermore, mutations in one pathway often appear to affect the other (reviewed in Plotegher and Duchen 2017). In addition, the disruption of cellular lipid homeostasis and impaired autophagy/mitophagy has been observed in Parkinson’s as well as Alzheimer disease (Holczer et al., 2019; Roca-Agujetas et al., 2021). The ability of the TRAP1 agonists to increase the residual activity of mutant LSD enzymes (Figure 4E) suggests that they may be of therapeutic value in diseases in which the protein defect is an ER-targeted protein, such as cystic fibrosis or one of the more than 60 LSDs (Carlile et al., 2007; Gelsthorpe et al., 2008). Thus, therapeutics that can target and activate TRAP1 may be of great benefit for many devastating disorders (Davis et al., 2021; Klein and Mazzulli, 2018; Kuk et al., 2021).
Limitations of the study
The downstream effects of TRAP1 activation by the agonists described in this study are well characterized and in agreement with other studies investigating TRAP1 activation. However, we cannot totally exclude the possibility that the agonists may have off-target effects that we have not identified. Furthermore, it has been proposed that TRAP1 is also present outside the mitochondria at the ER membrane. This study does not evaluate the effects, if any, of our TRAP1 agonists in this pool of TRAP1 protein.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-NPC1 | Abcam | Cat#ab134113; RRID:AB_2734695 |
| Mouse monoclonal anti-GAPDH | EMD Sigma | Cat#MAB374; RRID:AB_2107445 |
| Mouse monoclonal anti-M2-Flag | EMD Sigma | Cat#1804; RRID:AB_262044 |
| Mouse monoclonal anti-total c-Src (clone GD11) | EMD Sigma | Cat#05-184; RRID:AB_2302631 |
| Mouse monoclonal anti-c-Src phospho-Tyr416 (clone 9A6) | EMD Sigma | Cat#05-677; RRID:AB_309898 |
| Rabbit monoclonal anti-total AMPK α (D5A2) | Cell Signaling Technology | Cat#5831, RRID:AB_10622186 |
| Rabbit monoclonal anti-AMPK α phospho-Thr172 (Clone 40H9) | Cell Signaling Technology | Cat# 2535, RRID:AB_331250 |
| Rabbit monoclonal anti-total-p70S6 kinase (clone 49D7) | Cell Signaling Technology | Cat# 2708, RRID:AB_390722 |
| Rabbit monoclonal anti-p70S6 kinase phospho-Thr389 (108D2) | Cell Signaling Technology | Cat# 9234, RRID:AB_2269803 |
| Anti-DYKDDDDK (M2 Flag) Magnetic Agarose | ThermoFisher | Cat#A36797 |
| Goat anti-rabbit IgG (H+L) secondary antibody, HRP | ThermoFisher | Cat#32460; RRID:AB_1185567 |
| Goat anti-mouse IgG (H+L) secondary antibody, HRP | ThermoFisher | Cat# 32430, RRID:AB_1185566 |
| Rabbit polyclonal anti-β-actin | Abcam | Cat#ab8227; RRID:AB_2305186 |
| Rabbit polyclonal anti-SREBP2 | Abcam | Cat# ab30682; RRID:AB_779079 |
| Bacterial and virus strains | ||
| BL21(DE3) pLysS | ThermoFisher | Cat#C606010 |
| Chemicals, peptides, and recombinant proteins | ||
| 1685 | This paper | |
| ML405 | This paper | |
| Verotoxin B-594 (VTB-594) | This paper | |
| Verotoxin B-StrepTagII (VTB-Strep) | This paper | |
| OlyA-mCherry | This paper | |
| OlyA-AcGFP | This paper | |
| OlyA-Bit | This paper | |
| ALOD-594 | This paper | |
| C11 TopFluor Glucosyl Ceramide | Avanti Polar Lipids | Cat#810267; CAS:1246355-70-3 |
| Amplex Red (1-(3,7-dihydroxy-10H-phenoxazin-10-yl)-ethanone) | Cayman Chemical | Cat#10010469; CAS:119171-73-2 |
| Methyl-ß-cyclodextrin | EMD Sigma | Cat#4555; CAS:128446-36-6 |
| Horseradish peroxidase | EMD Sigma | Cat#P6782; CAS: 9003-99-0 |
| Cholesterol oxidase | EMD Sigma | Cat#C5421; CAS:9028-76-6 |
| Shrimp Alkaline Phosphatase | New England Biolabs | Cat#M0371S |
| 4-methylumbelliferyl oleate | EMD Sigma | Cat#75164; CAS:18323-58-5 |
| 4-Methylumbelliferyl β-D-galactopyranoside-6-sulfate sodium salt | EMD Sigma | Cat#68903; CAS:126938-14-5 |
| Ala-Ala-Phe-7-amido-4-methylcoumarin | EMD Sigma | Cat#A3401; CAS: 62037-41-6 |
| 4-methylumbelliferyl α-D-galactopyranoside | SantaCruz Biotechnology | Cat#sc-280454; CAS: 38597-12-5 |
| Critical commercial assays | ||
| TransIT-X2 dynamic delivery system | Mirus Bio | Cat#MIR-6000 |
| Alexa Fluor 594 C5 maleimide | ThermoFisher | Cat#A10256 |
| HiTrap Talon column | Cytiva | Cat#28953766 |
| MitoSox Red Superoxide Indicator | ThermoFisher | Cat#M36008 |
| Nano-Glo HiBiT Lytic detection system | Promega | Cat#N3030 |
| Cytochrome C oxidase assay kit | EMD Sigma | Cat#CYTOCOX1 |
| Succinate-Glo™ JmjC Demethylase/Hydroxylase Assay | Promega | Cat#V7990 |
| CellTiter-Glo 2.0 Cell Viability Assay | Promega | Cat#G9241 |
| M-PER Mammalian Protein Extraction Reagent | ThermoFisher | Cat#78053 |
| BluePhos microwell substrate kit | Seracare | Cat#5120-0059 |
| Pierce BCA Assay Kit | ThermoFisher | Cat#23227 |
| LiChrolut 500 RP-18 columns | EMD Sigma | Cat#1196870001 |
| Deposited data | ||
| Raw data from screen of NPC1 activators | This paper | PubChem: AID485313 |
| Raw data from screen of Rab9 activators | This paper | PubChem: AID485297 |
| Experimental models: Cell lines | ||
| Human: NPC4 cell line: I1061T/I1061T | Isolated from patient; Denny Porter | N/A |
| Human: NPC17 cell line: I1061T/truncation at codon 962 | Isolated from patient; Denny Porter | N/A |
| Human: NPC3 cell line: V1165M/truncation at codon 1249 | Isolated from patient; Denny Porter | N/A |
| Human: NPC23: R978C/IVS21-22 splice mutation AtoG | Isolated from patient; Denny Porter | N/A |
| Human: NPC25: N701K/truncation at codon 1005 | Isolated from patient; Denny Porter | N/A |
| Human: Fabry 821A: N215S "late" (cardiac variant) | Isolated from patient; Robert Desnick | N/A |
| Human: Gaucher Type II Disease | Coriell | Cat#GM00877; RRID:CVCL_0R27 |
| Human: Niemann-Pick, Type A Disease | Coriell | Cat#GM16195; RRID:CVCL_AX26 |
| Human: Farber Disease | Coriell | Cat#GM18313; RRID:CVCL_8A68 |
| Human: CLN2 Disease | Coriell | Cat#GM16485; RRID:CVCL_DA46 |
| Human: Pompe Disease | Coriell | Cat#GM20124; RRID:CVCL_0H82 |
| Human: MPS Type VII Disease | Coriell | Cat#GM02784; RRID:CVCL_Z830 |
| Human: Wolman Disease | Coriell | Cat#GM06144; RRID:CVCL_X456 |
| Human: ARPE-19 cell line | ATCC | Cat# CRL-2302, RRID:CVCL_0145 |
| Human: ARPE-NPC1 null | This paper | |
| Human: ARPE-TRAP1 null | This paper | |
| Mouse: NPC1 cells: C57BL/6J-Npc1nmf164/J | This paper | |
| Human: Fabry N34S "classic" | Isolated from patient; Robert Desnick | N/A |
| Human: Fabry P250T "classic" | Isolated from patient; Robert Desnick | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: Fabry knockout | Pagant et al., 2021 | N/A |
| Mouse: NPC1 mouse, C57BL/6J-Npc1nmf164/J |
The Jackson Laboratory | Cat#004817; RRID:IMSR_JAX:004817 |
| Mouse: C57BL6 | Charles River | Cat#027; RRID:IMSR_CRL:027 |
| Oligonucleotides | ||
| CRISPR targeting sequence for NPC1: 5′-AAAGAGTTACAATACTACGT-3′ | This paper | https://zlab.bio/guide-design-resources |
| CRISPR targeting sequence for TRAP1: 5′-GCGCCCGCAGCAAAGGCCGC-3′ | This paper | https://zlab.bio/guide-design-resources |
| ERP29 Human shRNA kit | Origene | Cat#: TL313167 |
| HYOU1 Human shRNA kit | Origene | Cat#:TG312275 |
| TRAP1 Human shRNA kit | Origene | Cat#:TG300868 |
| Recombinant DNA | ||
| eSpCas9(1.1)_No_FLAG_ATP1A1_G2_Dual_sgRNA | Agudelo et al., 2017 | Addgene plasmid #86612 |
| pcDNA3-MTS-CA-c-Src-Flag | Ogura et al., 2012 | Addgene plasmid #44654 |
| IPO7 (Myc-DDK-tagged)-Human importin 7 | Origene | Cat#RC215943 |
| PLEK (Myc-DDK-tagged)-Human pleckstrin | Origene | Cat#RC203780 |
| PSMC6 (Myc-DDK-tagged)-Human proteasome (prosome, macropain) 26S subunit, ATPase, 6 | Origene | Cat#RC202809 |
| TPM3 (Myc-DDK-tagged)-Human tropomyosin 3 | Origene | Cat#RC209904 |
| ERP29 (Myc-DDK-tagged)-Human endoplasmic reticulum protein 29 | Origene | Cat#RC210918 |
| PRDX1 (Myc-DDK-tagged)-Human peroxiredoxin 1 | Origene | Cat#RC221235 |
| TRAP1 (Myc-DDK-tagged)-Human TNF receptor-associated protein 1 | Origene | Cat#RC203439 |
| Human PSMB8 cDNA ORF clone | Genscript | Clone ID OHu19302; Acc#NM_004159.5 |
| Human HYOU1 cNDA ORF clone | Genscript | CloneID OHu15674; Acc#NM_001130991.3 |
| pMitoTimer | Laker et al., 2014 | Addgene plasmid #52659 |
| pALOD4 | Endapally et al., 2019 | Addgene plasmid #111026 |
| pRSET-OlyA-mCherry | This paper | |
| pRSET-OlyA-AcGFP | This paper | |
| pRSET-VTB-StrepTagII | This paper | |
| pRSET-OlyA-Bit | This paper | |
| Software and algorithms | ||
| MetaMorph Microscopy Automation and Image Analysis Software | Molecular Devices | RRID:SCR_002368 |
| ImageQuant TL (v.10.1) | Cytiva | RRID:SCR_018374 |
| Mascot, version 1.4.0.288 | Matrix Science | RRID:SCR_014322 |
| X! Tandem, version CYCLONE (2010.12.01.1) | The GPM, thegpm.org | RRID:SCR_015645 |
| Sequest Proteome Discoverer, version 1.4.0.288 | ThermoFisher | RRID:SCR_014477 |
| Scaffold, version 4.11.0 | Proteome Software | RRID:SCR_014345 |
| Phoenix WinNonlin | Certara | RRID:SCR_021370 |
| MassLynx v4.1 | Waters Corporation | RRID:SCR_014271 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yiannis A. Ioannou (yiannis.ioannou@mssm.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact upon completion of a Materials Transfer Agreement.
Experimental model and subject details
Cell lines
The NPC1 gene encodes a 1279 amino acid protein. The NPC1 patient cell lines (NPC3, female 13yrs, V1165M/truncation at codon 1249; NPC4, male 5yrs, I1061T/I1061T; NPC17, male 6yrs, I1061T/truncation at codon 962, NPC23, female 8yrs, R978C/IVS21-22 splice mutation A→G; NPC25, female 7yrs, N701K/truncation at codon 1005) were a generous gift of Denny Porter. Fabry 821A ("late" variant, adult male), Fabry P250T (adult male), and Fabry N34S (adult male) cells were a gift of Robert J. Desnick. All other lysosomal storage disease patient cell lines were from Coriell. The retinal pigment epithelial cell line ARPE-19 was from ATCC. Cells were routinely maintained in DMEM medium supplemented with 10% FBS, 2mM L-glutamine, and 50 μg/mL gentamicin in a humidified incubator at 37°C with 5% CO2.
Generation of cell lines derived from the livers of 3–6 day old C57/Bl6 NPC1 male mice (Maue et al., 2012) was accomplished by trypsin digestion of minced liver tissue and cell dispersal as we have described for another NPC1 mouse model (Chen et al., 2005).
Mice
The Fabry knockout mouse has previously been described (Pagant et al., 2021). Affected mice breed normally; hemizygous adult male mice (age 25–35 days) were used in all studies. Animals were fed ad libitum water and standard chow (Purina, Pico #5053) and housed by cage group on Anderson Bed O Cob 1/4″ inch bedding in filter-topped cages at 68–79°F and 30–70% humidity with 12-hour light/dark cycles. Animal husbandry and all experiments were conducted under the Icahn School of Medicine Institutional Animal Care and Use Committee approved protocols.
Method details
Synthesis of lead compounds
ML405
ML405 is N-(3-(N-(4-bromophenyl)sulfamoyl)-4-methoxyphenyl)-4-methylnicotinamide and was produced as follows (see Figure S1 for reaction scheme):
-
Step 1
A solution of 1-methoxy-4-nitrobenzene A (6.2 g, 41 mmol) in CICH2CH2CI (5 mL) was cooled to 0 °C and treated with dropwise addition of chlorosulfonic acid (4 mL, 6 mmol). The reaction was warmed to RT, refluxed for 2 hr, and then cooled. Water was added carefully to quench excess chlorosulfonic acid. Precipitation of solids was observed that dissolved back on addition and stirring of the mixture with chloroform. The organic layer was separated, dried with MgSO4, filtered, and concentrated to provide 2-methoxy-5-nitrobenzene-1-sulfonyl chloride B (1.78 g, 7.07 mmol, 17.5% yield).
-
Step 2
B (1.7 g, 6.8 mmol) was treated with 4-bromoaniline (1.7 g, 10 mmol) in pyridine (10 mL). The reaction was attached to a reflux condenser and stirred for 16 hr at 90 °C. Most of the pyridine was removed via rotary evaporation; the residue was diluted with EtOAc and then washed with water, and then brine. The organic layer was separated, dried with MgS04, filtered, concentrated, and purified to yield 1.7 g (65%) of C. LC/MS (Agilent system) Retention time ti (short) = 3.50 min, MS (ESI) m/z calculated for C26H22Br2N4NaO10S2 [2M + Na]+ 796.9, found 796.8. 1H NMR (400 MHz, DMS0d6) 8 ppm 10.54 (br. s., 1 H), 8.49 (d, J= 2.7 Hz, 1 H), 8.45 (dd, J = 8.4 Hz, J= 2.8 Hz, 1 H), 7.38 - 7.43 (m, 3 H), 7.09 - 7.02 (m, 2 H), 4.01 (s, 3 H).
-
Step 3
C (0.90 g, 2.30 mmol) was dissolved in EtOH (24 mL), treated with tin (II) chloride dihydrate (2.1 g, 9.3 mmol). The reaction was refluxed for 1 hr, cooled, and treated with 1 N NaOH till pH∼6. A white suspension, presumably consisting of tin salts, was observed. EtOAc was added and the mixture stirred overnight vigorously. The aqueous layer was still a white suspension. The mixture was filtered through celite, the organic layer was separated, dried with MgSO4, filtered, and concentrated. Purification by flash silica gel chromatography with an isocratic 35% EtOAc/hexanes solvent system separated a closely eluting non polar compound to provide pure aniline D (0.56 g, 1.57 mmol, 67% yield). LC/MS (Agilent system) Retention time t1 (short) = 2.78 min, MS (ESI) m/z calculated for C13H14BrN2O3S [M + H]+ 357.0, found 356.9. 1HNMR (400MHz, DMSO-d6) 9.96 (s, 4H), 7.44 - 7.33 (m, 8H), 7.09 - 6.97 (m, 12H), 6.87 (d, J=8.8 Hz, 4H), 6.72 (dd, J= 8.7, 2.8 Hz, 4H), 5.02 (s, 10H), 4.03 (q, J= 7.1 Hz, 1H), 3.71 (s, 12H), 1.99 (s, 1H), 1.17 (t, J= 7.1 Hz, 1H). HNMR (400 MHz, DMS0-d6) 6 9.96 (s, 1H), 7.42 - 7.33 (m, 2H), 7.07 - 6.98 (m, 3H), 6.87 (d, J= 8.8 Hz, 1H), 6.72 (dd, J= 8.7,2.8 Hz, 1H), 5.02 (s, 2H), 3.71 (s, 311).
-
Step 4
5-Amino-N-(4-bromophenyl)-2-methoxybenzenesulfonamide D (70 mg, 0.20 mmol) was dissolved in DMF and treated with 4-methylnicotinic acid (81 mg, 0.59 mmol), 2,4,6-tripropyl-l,3,5,2,4,6-trioxatriphosphinane 2,4,6-trioxide (0.37 g, 0.59 mmol) (T3PR), and triethylamine (0.11 mL, 0.78 mmol). The reaction stirred at 60 °C for 48 hr. After cooling the mixture, which had turned into a gel, was diluted with water and extracted with EtOAc. The organic layer was separated, concentrated, and purified by C18 reverse phase column chromatography to yield N-(3-(N-(4-bromophenylsulfamoyl)-4-methoxyphenyl)-4-methylnicotinamide (ML 405): 32 mg (0.07 mmol, 34%): LC/MS (Agilent system) Retention time t1 (long) = 4.13 min, MS (ESI) m/z calculated for C20H18BrN3O4S [M + H]+ 476.0 found 476.0 ; 1H NMR (400 MHz, DMSO-d6) δ 10.58 (s, 1H), 10.17 (s, 1H), 8.72 (s, 1H), 8.58 (d, 7 = 5.1 Hz, 1H), 8.25 (d, J = 2.6 Hz, 1H), 7.88 (dd, J = 9.0, 2.7 Hz, 1H), 7.45 (d, J = 5.2 Hz, 1H), 7.43 — 7.37 (m, 2H), 7.19 (d, J = 9.0 Hz, 1H), 7.10 — 7.03 (m, 2H), 3.86 (s, 3H), 2.44 (s, 311); HRMS (ESI) m/z calculated for C20H19BrN3O4S [M + H]+ 476.0274 found 476.0284.
1685
NCGC00351685 (1685) is N-(3-(N-(4-bromophenyl)sulfamoyl)-4-methoxyphenyl)-2-phenyl-1H-imidazole-4-carboxamide and was produced as follows (see Figure S1 for reaction scheme):
A mixture of 5-amino-N-(4-bromophenyl)-2-methoxybenzenesulfonamide D (0.5 g, 1.400 mmol), 2-phenyl-1H-imidazole-4-carboxylic acid (0.790 g, 4.20 mmol), HOBt (0.729 g, 4.76 mmol) was treated with DMF/CH2Cl2 1:1 (Volume: 2.80 mL). To the solution was added DIEA (2.445 mL, 14.00 mmol) followed by EDC (0.805 g, 4.20 mmol). The resulting solution was microwaved at 80°C for 20 minutes then stirred at 50°C overnight. After cooling to room temperature, the reaction solution was diluted with water and extracted with EtOAc. The organic layer was separated, concentrated and purified by C18 reversed phase column chromatography to yield N-(3-(N-(4-bromophenyl)sulfamoyl)-4-methoxyphenyl)-2-phenyl-1H-imidazole-4-carboxamide (1685): 100mg (0.19 mmol, 14%) LC/MS (Agilent system) Retention time t1 (long) = 4.86 min, MS (ESI) m/z calculated for C23H19BrN4O4S [M + H]+ 527.4 found 527.0 1H NMR (400 MHz, DMSO-d6) δ 10.22 (s, 1H), 10.11 (s, 1H), 8.33 (d, J = 2.7 Hz, 1H), 8.10 – 8.03 (m, 2H), 8.03 – 7.96 (m, 2H), 7.55 – 7.47 (m, 2H), 7.47 – 7.40 (m, 1H), 7.40 – 7.34 (m, 2H), 7.19 – 7.13 (m, 1H), 7.07 – 7.01 (m, 2H), 3.84 (s, 3H).
Production and purification of lipid binding toxins
The ostreolysin A (OlyA) (Skočaj et al., 2014) fusions were custom synthesized by Gene Art (Life Technologies) into the prokaryotic expression vector pRSET-A, with mCherry or AcGFP (Shaner et al., 2008) added to the 3′ end of the vector, followed by a 6-His tag for purification. For the OlyA-NanoBiT construct, OlyA-BiT (Promega) was custom synthesized in pRSET with the 6-His and small BiT tag (VSGWRLFKKIS) at the 3′ end of the OlyA gene. The anthrolysin O (AloD) fusion construct, pALOD4, was a gift from Arun Radhakrishnan (Addgene plasmid # 111026 ; http://n2t.net/addgene:111026 ; RRID:Addgene_111026) (Endapally et al., 2019).
Fusions were transformed into E. coli strain BL21 pLysS for protein expression as we have described (Liapis et al., 2012). Briefly, bacteria were pelleted, resuspended in binding buffer (300mM NaCl, 50mM NaH2PO4, pH 8, 1mM PMSF), and lysed using a French press. Cell debris was removed by centrifugation twice at 33,000 xg for 15 min. Proteins were purified on a 5mL HiTrap Talon column (Cytiva) followed by Mono Q ion exchange chromatography using an ÄKTA pure system (Cytiva). AloD was subsequently coupled to Alexa Fluor 594 using the Alexa Fluor 594 C5 maleimide reagent (ThermoFisher) according to the manufacturer’s recommendations.
Verotoxin B (VTB) was produced as we have previously described (Zeidner et al., 1999), purified as above, and coupled to Alexa Fluor 594 as above (VTB-594). The verotoxin-StrepTagII fusion (VTB-Strep) was custom synthesized by Gene Art to contain the StrepTag II sequence (WSHPQFEK) (Schmidt and Skerra, 2007) at the 3′ end of VTB in the pRSET-A vector and then expressed and purified as described above.
Microscopy
For all studies, cells were grown on glass coverslips in 6-well dishes to 75% confluence and treated with compound or DMSO (vehicle) for 17–20 hrs. For multi-day experiments, cells were given fresh media containing compound or DMSO each day. Cell monolayers were rinsed with PBS and fixed with phosphate buffered formalin for 10 min at room temperature (RT).
Cell transfections were accomplished using TransIT-X2 reagent (Mirus Bio) according to the manufacturer’s recommendations unless otherwise indicated.
Cholesterol/sphingolipid (OlyA) staining
Cells were permeabilized with 6mM CHAPSO in PBS for 10 minutes at RT, washed with PBS, and blocked with 1% BSA for 30 min at RT with gentle rocking. Cells were incubated with OlyA-mCherry in PBS for 1hr at RT with rocking, then washed with PBS and counterstained with Hoechst 33342 for 5 min. Coverslips were mounted onto glass slides using Fluoromount G (Southern Biotech) and fluorescence visualized on a Nikon Eclipse microscope fitted with a charge-coupled-device camera driven by Metamorph software. For storage comparisons, images were acquired using the same exposure time for samples being compared.
Free cholesterol (AloD) staining
Cells were permeabilized with 0.025% Triton X-100 in PBS for 5 min at RT, blocked, and then incubated with AloD-594 before processing for microscopy as above.
Globotriaosyl ceramide (Gb3) staining
Cells were permeabilized with 50 μg/mL digitonin in PBS, blocked, and then incubated with VTB-594 before processing for microscopy as above.
Detection of superoxide
Live cells were stained with MitoSox Red reagent (ThermoFisher) according to the manufacturer’s recommendation and Hoechst 33342 simultaneously and then mounted with PBS and imaged as above.
Generation of stable NPC- and TRAP-null ARPE cell lines
The CRISPR targeting sequences for NPC1 (5′-AAAGAGTTACAATACTACGT-3′) or TRAP1 (5′-GCGCCCGCAGCAAAGGCCGC-3′) were cloned into the vector eSpCas9(1.1)_No_FLAG_ATP1A1_G2_Dual_sgRNA (a gift from Yannick Doyon: Addgene plasmid #86612; http://n2t.net/addgene:86612; RRID:Addgene_86612) (Agudelo et al., 2017). Plasmids were transfected into ARPE-19 cells, and stable NPC1-null and TRAP1-null ARPE cells were generated by selection using 0.2μM ouabain for 2 weeks or until individual colonies could be distinguished. Individual clones were assessed by western blot analyses to confirm knockout of NPC1 or TRAP1 before expansion.
Western blot analyses
NPC1, AMPK, and p70S6 kinase
ARPE cells were collected by scraping into ice-cold PBS and centrifugation at 450xg for 5 min. The cells were lysed in buffer (100mM NaPi, pH 7.5, 150mM NaCl, 2mM EDTA and 1% Igepal, with protease inhibitors) for 15 min at 4°C and centrifuged at 20,000 xg to remove insoluble debris. For detection of AMPK and p70S6 kinase, phosphatase inhibitors were added to the lysis buffer. Protein concentration was determined using a BCA assay kit (ThermoFisher). Approximately 20μg protein was resuspended in NuPage LDS loading dye (ThermoFisher) containing 50mM DTT, heated at 70°C for 10 min, and subjected to electrophoresis and transfer using a Novex 4–12% BisTris gel (Invitrogen) in MOPS buffer as we have described (Chen et al., 2005). Blots were probed using primary antibodies against: NPC1 (ab134113; Abcam), total AMPK (5831; Cell Signaling Technology), AMPK phospho-Thr172 (2535; Cell Signaling Technology), p70S6 kinase (2708; Cell Signaling Technology), or p70S6 kinase phospho-Thr389 (9234; Cell Signaling Technology) and GAPDH (MAB374; EMD Sigma) and secondary anti-rabbit and anti-mouse antibodies conjugated with horseradish peroxidase (ThermoFisher). Proteins were visualized by chemiluminescence using SuperSignal West Dura substrate (ThermoFisher) according to the manufacturer’s recommendation as we have described (Chen et al., 2010) on an ImageQuant 800 system (Cytiva).
Flag-tagged c-Src
ARPE cells were grown in 10 cm dishes to 75% confluence and then transfected with 12μg pcDNA3-MTS-CA-c-Src-Flag (a gift from Yoshimi Homma; Addgene plasmid #44654; http://n2t.net/addgene:44654; RRID:Addgene_44654) (Ogura et al., 2012) using 18μL Fugene reagent (Promega). At 24 hr post-transfection, cells were treated with DMSO or 1μM ML405 for 24 hrs. Cells were collected and lysed as above, with the addition of phosphatase inhibitors to the lysis buffer. Flag-tagged c-Src in the lysate was immunoprecipitated using M2-Flag/agarose beads (ThermoFisher). After washing, beads were processed as above for electrophoresis and transfer. c-Src was detected using primary antibodies against M2 Flag (1804; EMD Sigma), total c-Src (05-184; EMD Sigma), or c-Src phospho-Tyr416 (05-677; EMD Sigma) and secondary anti-mouse antibodies conjugated with horseradish peroxidase. Proteins were visualized as above.
SREBP2
Cells were treated for 3 hrs with 5μM ML405, 1685, or 20μM 25-hydroxycholesterol and then collected as above. Cells were lysed in RIPA buffer for 1hr at 4°C and then centrifuged at 20,000 xg. Protein concentration was determined using the BCA method and 50μg was used for Western blot analysis as above using antibodies against SREBP2 (ab30682; Abcam) and β-actin (8227; Abcam).
Cholesterol efflux assay
PM cholesterol efflux was determined using an Amplex Red-based assay (Amundson and Zhou, 1999; Low et al., 2012). Cells were plated at 20,000 cells/well in a 96-well plate and treated with compounds for 16–20 hrs in full media containing 5μM U18666A to prevent cholesterol transport. Cells were then washed with serum-free media (SFM) and incubated with 1mM methyl-ß-cyclodextrin and 5μM U18666A in SFM for 15 min at 37°C to remove available plasma membrane cholesterol from cells and inhibit new transport. Cells were washed with SFM and then incubated in SFM for 3 hrs at 37°C in a 5% CO2 incubator. At the end of that time, media was removed from wells and cells were incubated with fresh SFM containing 1mM methyl-ß-cyclodextrin for 30 min at 37°C in a 5% CO2 incubator. To quantify the amount of cholesterol, media was transferred to a black 96-well plate and mixed with reaction solution (final concentrations: 150mM Amplex, 1U/mL horseradish peroxidase, 0.125U cholesterol oxidase in buffer: 50mM potassium phosphate, pH 7.4, 25mM NaCl, 2.5mM cholic acid, 0.05% Triton X-100) for 30 min at 37°C. Newly effluxed cholesterol in the media was determined by reading plates in a Bio-Tek Synergy H1 microplate reading using excitation 554nm/emission 593nm. The amount of cholesterol in the media was calculated by comparing values to a standard curve generated using cholesterol reference standards.
Determination of PM raft cholesterol
The Nano-Glo HiBiT Lytic detection system (Promega) was used to measure the amount of cholesterol in PM rafts (and thus unavailable for efflux as determined above). Cells were plated at 10,000 cells/well in white plates and treated for 20 hr. Cells were fixed in 4% paraformaldehyde for 20 min at RT, washed, blocked with 1% BSA in PBS for 30 min, and then incubated with OlyA-BiT for 1 hr at RT. For detection, cells were washed and incubated with LargeBit according to the manufacturer’s recommendation. Luminescence was read in a Modulus microplate multimode reader (Turner Biosystems).
Drug affinity responsive target stability (DARTS)
ARPE cells were treated for 20hr with 5μM ML405 or 1685 and then subjected to DARTS essentially as described (Lomenick et al., 2011). Briefly, cells were pelleted, washed with cold PBS, and lysed for 10min on ice in freshly made M-PER buffer (M-PER reagent, protease inhibitor cocktail, 50mM sodium fluoride, 10mM ß-glycerophosphate, 10mM sodium pyrophosphate, 0.1mM sodium orthovanadate). The lysate was centrifuged for 10min at 18,000xg at 4°C and then 10X TNC buffer (500mM Tris-HCl, pH 8.0, 500mM NaCl, 100mM CaCl2) was added to produce a final concentration of 1X TNC buffer in the lysate. The lysate was divided into aliquots of 99μL and mixed with 1μL vehicle or compound stock solutions and rocked for 15min at room temperature. Limited proteolysis was achieved by adding 4μL of 1.25mg/mL pronase in 1X TNC buffer (1:100 solution) to the lysate for exactly 30min at room temperature. Digestion was stopped by addition of protease inhibitor solution. Samples were incubated with 0.1U benzonase for 30 min at 37°C to digest nucleic acids. Proteins were precipitated by mixing with 100% trichloroacetic acid (TCA) to a final concentration of 20% TCA and incubating for 1 hr on ice. Samples were centrifuged at 20,000xg for 15 min at 4°C, and the pellet was washed twice with ice-cold acetone before being air-dried and processed for mass spectrometry.
Mass spectrometry
The pellets generated from DARTS were resuspended in 8M urea/100mM Tris-HCl, pH 8.5 and then reduced with 5mM tris(2-carboxyethyl)phosphine, alkylated with 10mM iodoacetamide, and digested with trypsin. The tryptic peptides were purified using PepClean C18 spin columns (ThermoFisher) and then the eluates were dried under vacuum.
The LC-MS/MS analysis was performed on a Waters NanoAcquity UPLC system (Waters) interfaced to a Thermo LTQ-Orbitrap mass spectrometer (ThermoFisher). Reversed-phase LC separation was performed on a Waters BEH300 C18 column (75 μm × 250 mm, 1.7 μm particle size). Samples were trapped and washed on a Waters Symmetry C18 trap column (180 μm × 100 mm, 5 μm particle size) prior to separation on the nanocolumn. Gradient elution was performed with 0.1% formic acid in water as solvent A and in ACN as solvent B, with solvent B raised from 7 to 30% in 300 min, then 30 to 80% in the next 40 min, followed by a 10-min hold at 80%, and a return to 5% for 20 min at a flow rate of 0.25 ul/min. The mass spectrometer was operated in positive mode with spray voltage at 1.4 kV, capillary voltage at 40 V and ion transfer tube temperature at 140°C. A normalized collision energy of 35% and activation time of 30 ms were applied in MS/MS acquisitions. The top eight most intense ions were selected for fragmentation in the LTQ analyzer. The following dynamic exclusion settings were applied to precursor ions chosen for MS/MS analysis: repeat count – 1; repeat duration – 30 s and exclusion duration – 120 s.
Compound candidate expression and silencing studies
NPC3 cells were transfected with Flag-tagged expression plasmids: IPO7 (Origene RC215943), PLEK (Origene RC203780), PRDX1 (Origene RC221235), PSMB8 (Genscript), PSMC6 (Origene RC202809), TPM3 (Origene RC209904), ERP29 (Origene RC210918), HYOU1 (GenScript), TRAP1 (Origene RC203439). Cells were processed at 48 hrs post-transfection as described above for cholesterol storage except that following the blocking step, cells were incubated with M2 Flag antibody for 1 hr at RT and then washed 3 × 5 times with PBS before incubation with anti-mouse secondary antibodies conjugated to Alexa Fluor 488 and OlyA-mCherry simultaneously for 1 hr at RT in the dark. Alternatively, mouse secondary antibodies conjugated to Alexa Fluor 594 and OlyA-acGFP were used. After washing with PBS, cells were counterstained with Hoechst 33342 and coverslips mounted as described above.
Gene silencing experiments were accomplished using shRNAs cloned into the pGFP-V-RS vector (sequences determined by Origene). Vectors were validated to knock down genes of interest by Western blot prior to their use in expression experiments. Cellular expression of the shRNA was confirmed by GFP fluorescence and lipid storage was determined with OlyA-mCherry as described above.
Mitotimer studies
Cells were transfected with pMitoTimer (a gift from Zhen Yan; Addgene plasmid #52659; http://n2t.net/addgene: 52659; RRID:Addgene_52659) (Laker et al., 2014). Compound was added 5 hrs post transfection and cells were evaluated at 24 hrs post-transfection after fixing with 4% paraformaldehyde. MitoTimer images were acquired at 100x magnification using the green (excitation/emission 488/518 nm) and red (excitation/ emission 543/572 nm) channels with identical acquisition parameters for all samples to ensure no signal saturation and similar intensity of the green and red channels in control samples. Each channel was saved as an individual file.
Respiratory chain enzyme assays
Mitochondria were purified from ARPE cells grown in 15cm dishes as follows: cells were collected by scraping into ice-cold PBS and centrifuging at 450xg for 5 min. The pellet was resuspended in mitochondrial isolation buffer (200mM sucrose, 10mM Tris/MOPS pH 7.4, 1mM EGTA/Tris) using ten strokes of a Potter–Elvejehn homogenizer at 700 rev/min, followed by centrifugation at 600 xg for 10 min at 4°C. The supernatant containing mitochondria was removed and centrifuged at 10,000 xg for 5 min at 4°C. The pellet containing mitochondria was resuspended in isolation buffer and protein concentration was determined using the Bradford method.
Succinate dehydrogenase (complex II) activity was determined as described (Trounce et al., 1996). Briefly, mitochondria were incubated for 10 min at 30°C in buffer (50 mM potassium phosphate, pH 7.4, 20 mM succinate). Antimycin A (2 μg/mL), rotenone (2 μg/mL), KCN (2 mM), and 50μM 2,6-dichlorophenolindophenol (DCPIP) were added and the reduction of DCPIP was measured at 600 minus 520 nm for 1 min to record the blank rate. The reaction was initiated by adding 50μM decylubiquinone (DB) and the change in absorbance was measured for 3 min.
Cytochrome C oxidase (complex IV) activity was determined by monitoring the decrease in ferrocytochrome c absorbance at 550nm using the cytochrome C oxidase assay kit (Sigma) according to the manufacturer’s recommendations.
Metabolite quantitation
ARPE cells were plated and treated as for microscopy (above). Cells were trypsinized, washed in full medium, and centrifuged at 1,000xg for 5 min. The pellet was resuspended, washed in PBS, and collected by centrifugation.
Succinate
The pellet was resuspended in 500μL boiling water and placed on a heat block at 95°C for 5 min, after which it was placed on ice for 10 min. Following centrifugation at 10,000xg for 10 min, the supernatant was collected and incubated with 2 units of shrimp alkaline phosphatase (New England Biolabs) at 37°C for 30 min to remove ATP, followed by incubation at 65°C for 10 min to inactivate the phosphatase. Succinate was measured using the Succinate-Glo assay kit (Promega) according to the manufacturer’s recommendations. A succinate standard curve from 1–15μM was run concurrently.
Lactate
Cells were subjected to perchlorate extraction as described (Zhu et al., 2010). Briefly, the pellet was resuspended in 250μL ice-cold 0.25M perchloric acid solution, incubated on ice for 5 min, and then neutralized with 6μL 5M potassium carbonate, pH 6.5. The solution was centrifuged at 10,000 xg for 5 min and 10μL of the supernatant was added to wells of a black 96-well plate in triplicate and mixed with 90μL assay buffer (50mM Tris, pH 7.5, 0.5 units/mL lactate oxidase, 0.5 units/mL horseradish peroxidase, 50μM Amplex Red) with shaking for 5 min at RT. Lactate was determined by reading plates in a Bio-Tek Synergy H1 microplate reader using excitation 554nm/emission 593nm. For media lactate, media was collected and centrifuged at 800xg for 5 min to remove cell debris, and then 10μL was used for quantitation as above.
ATP
ARPE cells were plated at 20,000 cells/well in white plates and treated with 2μM ML405 or DMSO for 20 hrs. Cellular ATP levels were determined using CellTiter-Glo (Promega) according to the manufacturer’s recommendation.
Lysosomal enzyme assays
Cells were plated at 90,000 cells/well in 12 well plates in triplicate and treated with 2μM compound for 48 hr. At the end of that time, cells were collected and lysed in buffer (150mM NaCl, 50mM Na2HPO4, pH 6.9, 1mM EDTA, 1% Igepal containing protease inhibitor cocktail) for 30 min on ice and then centrifuged at 20,000xg for 10min at 4°C. Three-fourths of the lysate was assayed in 96-well plates for 1 hr at 37°C essentially as described (Ioannou et al., 2001; Ribbens et al., 2013; Sheriff et al., 1995; Sohar et al., 2000). Briefly, for Wolman disease, lysosomal acid lipase activity was determined by mixing lysate with 1mM 4-methylumbelliferyl oleate in citrate-phosphate buffer, pH 4.4 and the reaction was stopped with 0.1M Tris-HCl, pH 7.6. For Krabbe disease, β-galactocerebrosidase activity was determined by mixing lysate with 0.4mM 6-hexadecanoylamino-4-methylumbelliferyl-β-D-galactopyranoside and 1 mg/mL sodium taurocholate in citrate-phosphate buffer, pH 4.6. The reaction was stopped with 0.4M glycine, pH 10.8. For Batten disease, tripeptidyl peptidase II activity was determined by mixing lysate with 250μM Ala-Ala-Phe-7-amido-4-methylcoumarin in citrate-phosphate buffer, pH 4.4 and the reaction was stopped with 0.3% trifluoroacetic acid. For Fabry disease, a-galactosidase A activity was determined by mixing lysate with an equal volume of 4 mM 4-methylumbelliferyl α-D-galactopyranoside in PBS/0.2 M acetic acid, pH 4.2 (1:1), and the reaction was stopped with 1M glycine/1M NaOH, pH 10. The fluorescence signal for each assay was read in a Tecan Infinite F200 Pro using the 360/460 nm excitation/emission filter pair. One-tenth of each lysate was used for protein determination using a BCA assay kit (Pierce) and enzyme activity for each sample was normalized to protein concentration.
In vivo mouse studies
ML405 was formulated in saline/solutol/dimethylacetamide (70/25/5) as described (Strickley, 2004). 30mg/kg ML405 or vehicle was injected through intraperitoneal (IP) route of administration 3 times/week for 4 weeks in age-matched adult male Fabry mice (n = 10 for vehicle group, n = 9 for treated group), after which mice were sacrificed. Organs and plasma were collected and snap frozen in liquid nitrogen for further analyses.
Gb3 ELISA
Neutral glycolipids were extracted and purified from mouse organs and plasma essentially as we described (Zeidner et al., 1999), except that C11 TopFluor Glucosyl Ceramide (Avanti Lipids) was added to each sample as an extraction control. Briefly, a part of each organ was weighed and homogenized in chloroform: methanol (2:1) at 10mL/100mg wet weight using a Tissue Tearor homogenizer (Biospec Products) at setting 17, after which 0.5mL was removed and 1μL TopFluor was added. Plasma (25μL) was extracted with 20 volumes of chloroform: methanol (2:1) and processed similarly. Extracts were incubated at 37°C for 15–30 minutes with rocking and then centrifuged at 20,000 xg for 5 min at 4°C. One-fifth volume water was added to the supernatant and the samples were incubated for at least 30 min at 4°C. Samples were then centrifuged at 20,000 xg for 5 min at 4°C and the bottom layer was removed and dried under nitrogen using a Techne sample concentrator and heat block. The dry residue was resuspended in chloroform and applied to a LiChrolut 500 RP-18 column (EMD Sigma) which had been preequilibrated with chloroform. The bound glycolipids were washed with chloroform and eluted with acetone-methanol (9:1). The eluted neutral glycolipids were dried under nitrogen and resuspended in 0.5 mL methanol. TopFluor values were obtained by reading 50μL of each sample in a black plate in a Tecan Infinite F200 Pro using the 485/535nm excitation/emission pair.
For determination of Gb3 levels, 50μL of each sample was spotted 3 times in a 96-well microplate. Pure Gb3 was used to create a standard curve. The plate was placed on a 37°C heating block until the samples were dry (∼2hrs). The plate was washed with Tris-buffered saline (TBS) and then incubated with blocking buffer (1% casein in TBS) for 1 h at 37°C. The plate was washed once in TBS and then incubated with VTB-Strep in TBS at 37°C for 90 min. Plates were washed with TBS 3 × 5min and then incubated with streptavidin-conjugated alkaline phosphatase in TBS for 45 min at 37°C. Plates were washed with TBS 3 × 5min and developed with KPL BluePhos substrate (Seracare) according to the manufacturer’s instructions. Gb3 values were normalized using TopFluor values for each sample.
Pharmacokinetic studies of ML405 and 1685 in mice
Adult male C57Bl6 mice (n = 3/sampling time point) were obtained from Charles River Laboratories (Wilmington, MA). All experimental procedures were approved by the Animal Care and Use Committee (ACUC) of the NIH Division of Veterinary Resources (DVR). A single dose of 10 and 30 mg/kg was administered IP. Dosing solutions were freshly prepared on the day of administration in 50% PEG200/50% water. Mouse blood samples were collected in K2EDTA tubes at 0.167, 0.5, 1, 1.5, 2, 4, 7 and 24 hr after drug administration, and plasma was harvested after centrifugation at 3000 rpm for 10 min. Liver and brain tissues were collected, flash frozen in liquid nitrogen and transferred to 48-well plates. All plasma and tissue samples were stored at −80°C until analysis.
The pharmacokinetic parameters were calculated using the non-compartmental approach (Model 200) of the pharmacokinetic software Phoenix WinNonlin, version 6.2 (Certara, St. Louis, MO). The area under the plasma concentration versus time curve (AUC) was calculated using the linear trapezoidal method. The slope of the apparent terminal phase was estimated by log linear regression using at least 3 data points and the terminal rate constant (λ) was derived from the slope. AUC0-∞ was estimated as the sum of the AUC0-t (where t is the time of the last measurable concentration) and Ct/λ. The apparent terminal half-life (t½) was calculated as 0.693/λ.
Ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) methods were developed to determine ML405 and 1685 concentrations in mouse plasma and tissue samples. Mass spectrometric analysis was performed on a Waters Xevo TQ-S triple quadrupole instrument using electrospray ionization in positive mode with the selected reaction monitoring. The separation of test compounds from endogenous components was performed on an Acquity BEH C18 column (50 × 2.1 mm, 1.7 μm) using a Waters Acquity UPLC system with 0.6 mL/min flow rate and gradient elution. The mobile phases were 0.1%formic acid in water and 0.1% formic acid in acetonitrile. The calibration standards and quality control samples were prepared in the blank mouse plasma and tissue homogenate. Aliquots of 10 μL samples were mixed with 200 μL internal standard in acetonitrile to precipitate proteins in a 96-well plate. 0.5 μL supernatant was injected for the UPLC-MS/MS analysis. Data were analyzed using MassLynx v4.1 (Waters Corp., Milford, MA).
Quantification and statistical analysis
Microscopic data analyses
All experiments were performed 3 or more times. For quantitation of lipid storage in cell lines, fluorescence intensity was determined using the integrated intensity function of Metamorph software as we have described (Chen et al., 2010); between 100-150 cells were quantitated for each sample. For determination of lipid storage correction in transfected cells, the lipid intensity in 50–75 individual transfection-positive cells was compared to the average lipid intensity of the same number of transfection-negative cells in the same population; cells were considered "corrected" if they had at least 25% less lipid staining than untransfected cells. For Mitotimer ratiometric analysis, the green and red signal intensity was quantified using the integrated intensity function of Metamorph. The ratio of red to green was calculated for each individual mitochondrion and the signals for each condition were averaged.
Western blot analyses
All western blots were performed 3 or more times. Quantitation of western blots was achieved with ImageQuantTL software (v10.1, Cytiva) using rolling ball (radius 2) background subtraction.
Mass spectrometric data analysis
MS/MS data were analyzed using Mascot (Matrix Science; version 1.4.0.288), Sequest (ThermoFisher; version 1.4.0.288) and X! Tandem (The GPM, thegpm.org; version CYCLONE (2010.12.01.1)). Peptide parent mass tolerance was set to 10 ppm, and the fragment ion mass tolerance was 0.6 Da. The enzyme was set to ‘‘trypsin’’ with two maximum missed cleavage sites and the search was against uniprot_Homo_sapiens_20140220 database. Carbamidomethylation of cysteine was specified as a fixed modification. Pyroglutamic acid conversion of N-terminal glutamic acid or glutamine, deamidation of asparagine and glutamine, oxidation of histidine, methionine and tryptophan were specified as variable modifications. The output files were integrated into Scaffold (version Scaffold 4.11.0, Proteome Software), which was used to validate MS/MS based peptide and protein identifications.
Statistical analyses
Differences between 2 groups were analyzed using Student’s unpaired two-tailed t tests using GraphPad Prism 5.0 software. Data are presented as mean ±S.D. and statistical significance was set at p < 0.05. Details for individual analyses are provided in the figure legends.
Acknowledgments
This work was supported by grants from the National Institute of Health (NS058335 to Y.A.I.) and the National Center for Advancing Translational Sciences (MH089375 and MH089537 to Y.A.I.).
Author contributions
F.W.C., J.P.D., R.C., S.P., R.W., J.J.M., and Y.A.I. designed experiments; F.W.C.: cholesterol and enzyme assays, DARTS, mouse studies; western blot analyses; J.P.D.: cloning and expression of lipid binding toxins, generation of knockout cell lines, western blot analyses; J.C.: microscopy; G.D. and R.W.: mass spectrometry analyses; R.C., S.P., J.D., R.M., P.D., A.W., X.X., E.H., N.S., M.F., and J.J.M.: compound library screening, chemical syntheses, PK, and ADME studies; F.W.C. and Y.A.I. wrote the manuscript with input from other authors. Y.A.I. supervised and directed the overall concept and studies.
Declaration of interests
F.W.C. and Y.A.I. at the Icahn School of Medicine at Mount Sinai and S.P., R.C., J.J.M., N.S., M.F., J.D., and P.D. at NCATS (NIH) are inventors on a patent (WO16/130,774) for the TRAP1 activating compounds described in these studies, which has been licensed to Amathus Therapeutics for clinical development. Y.A.I. and the Icahn School of Medicine at Mount Sinai are founders of Amathus Therapeutics.
Published: September 16, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104941.
Contributor Information
Juan J. Marugan, Email: maruganj@mail.nih.gov.
Yiannis A. Ioannou, Email: yiannis.ioannou@mssm.edu.
Supplemental information
Data and code availability
-
•
The results of the reporter screens that led to development of the lead compounds have been deposited and are publicly available (https://pubchem.ncbi.nlm.nih.gov/bioassay/485297 and https://pubchem.ncbi.nlm.nih.gov/bioassay/485313). Western blot, microscopy, and enzymatic data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the reported data is available from the lead contact upon request.
References
- Abrams M.E., Johnson K.A., Perelman S.S., Zhang L.-S., Endapally S., Mar K.B., Thompson B.M., McDonald J.G., Schoggins J.W., Radhakrishnan A., et al. Oxysterols provide innate immunity to bacterial infection by mobilizing cell surface accessible cholesterol. Nat. Microbiol. 2020;5:929–942. doi: 10.1038/s41564-020-0701-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agudelo D., Duringer A., Bozoyan L., Huard C.C., Carter S., Loehr J., Synodinou D., Drouin M., Salsman J., Dellaire G., et al. Marker-free coselection for CRISPR-driven genome editing in human cells. Nat. Methods. 2017;14:615–620. doi: 10.1038/nmeth.4265. [DOI] [PubMed] [Google Scholar]
- Amoroso M.R., Matassa D.S., Laudiero G., Egorova A.V., Polishchuk R.S., Maddalena F., Piscazzi A., Paladino S., Sarnataro D., Garbi C., et al. TRAP1 and the proteasome regulatory particle TBP7/Rpt3 interact in the endoplasmic reticulum and control cellular ubiquitination of specific mitochondrial proteins. Cell Death Differ. 2012;19:592–604. doi: 10.1038/cdd.2011.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amundson D.M., Zhou M. Fluorometric method for the enzymatic determination of cholesterol. J. Biochem. Biophys. Methods. 1999;38:43–52. doi: 10.1016/s0165-022x(98)00036-0. [DOI] [PubMed] [Google Scholar]
- Aslanidis C., Ries S., Fehringer P., Büchler C., Klima H., Schmitz G. Genetic and biochemical evidence that CESD and Wolman disease are distinguished by residual lysosomal acid lipase activity. Genomics. 1996;33:85–93. doi: 10.1006/geno.1996.0162. [DOI] [PubMed] [Google Scholar]
- Audano M., Schneider A., Mitro N. Mitochondria, lysosomes, and dysfunction: their meaning in neurodegeneration. J. Neurochem. 2018;147:291–309. doi: 10.1111/jnc.14471. [DOI] [PubMed] [Google Scholar]
- Baixauli F., Acín-Pérez R., Villarroya-Beltrí C., Mazzeo C., Nuñez-Andrade N., Gabandé-Rodriguez E., Ledesma M.D., Blázquez A., Martin M.A., Falcón-Pérez J.M., et al. Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. 2015;22:485–498. doi: 10.1016/j.cmet.2015.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardi A.S., Pannuzzo G., Graziano A., Costantino-Ceccarini E., Piomboni P., Luddi A. Pharmacological chaperones increase residual β-galactocerebrosidase activity in fibroblasts from Krabbe patients. Mol. Genet. Metab. 2014;112:294–301. doi: 10.1016/j.ymgme.2014.05.009. [DOI] [PubMed] [Google Scholar]
- Bruiners N., Dutta N.K., Guerrini V., Salamon H., Yamaguchi K.D., Karakousis P.C., Gennaro M.L. The anti-tubercular activity of simvastatin is mediated by cholesterol-driven autophagy via the AMPK-mTORC1-TFEB axis. J. Lipid Res. 2020;61:1617–1628. doi: 10.1194/jlr.ra120000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlile G.W., Robert R., Zhang D., Teske K.A., Luo Y., Hanrahan J.W., Thomas D.Y. Correctors of protein trafficking defects identified by a novel high-throughput screening assay. Chembiochem. 2007;8:1012–1020. doi: 10.1002/cbic.200700027. [DOI] [PubMed] [Google Scholar]
- Castellano B.M., Thelen A.M., Moldavski O., Feltes M., van der Welle R.E.N., Mydock-McGrane L., Jiang X., van Eijkeren R.J., Davis O.B., Louie S.M., et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science. 2017;355:1306–1311. doi: 10.1126/science.aag1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F.W., Gordon R.E., Ioannou Y.A. NPC1 late endosomes contain elevated levels of non-esterified (“free”) fatty acids and an abnormally glycosylated form of the NPC2 protein. Biochem. J. 2005;390:549–561. doi: 10.1042/bj20050236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F.W., Li C., Ioannou Y.A. Cyclodextrin induces calcium-dependent lysosomal exocytosis. PLoS One. 2010;5:e15054. doi: 10.1371/journal.pone.0015054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A.C., Loh S.H.Y., Martins L.M. Drosophila Trap1 protects against mitochondrial dysfunction in a PINK1/parkin model of Parkinson's disease. Cell Death Dis. 2013;4:e467. doi: 10.1038/cddis.2012.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J.P., Chen F.W., Ioannou Y.A. Transmembrane molecular pump activity of Niemann-Pick C1 protein. Science. 2000;290:2295–2298. doi: 10.1126/science.290.5500.2295. [DOI] [PubMed] [Google Scholar]
- Davis O.B., Shin H.R., Lim C.-Y., Wu E.Y., Kukurugya M., Maher C.F., Perera R.M., Ordonez M.P., Zoncu R. NPC1-mTORC1 signaling couples cholesterol sensing to organelle homeostasis and is a targetable pathway in Niemann-pick type C. Dev. Cell. 2021;56:260–276.e7. doi: 10.1016/j.devcel.2020.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demers-Lamarche J., Guillebaud G., Tlili M., Todkar K., Bélanger N., Grondin M., Nguyen A.P., Michel J., Germain M. Loss of mitochondrial function impairs lysosomes. J. Biol. Chem. 2016;291:10263–10276. doi: 10.1074/jbc.m115.695825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deus C.M., Yambire K.F., Oliveira P.J., Raimundo N. Mitochondria-lysosome crosstalk: from physiology to neurodegeneration. Trends Mol. Med. 2020;26:71–88. doi: 10.1016/j.molmed.2019.10.009. [DOI] [PubMed] [Google Scholar]
- Du X., Pham Y.H., Brown A.J. Effects of 25-hydroxycholesterol on cholesterol esterification and sterol regulatory element-binding protein processing are dissociable: implications for cholesterol movement to the regulatory pool in the endoplasmic reticulum. J. Biol. Chem. 2004;279:47010–47016. doi: 10.1074/jbc.m408690200. [DOI] [PubMed] [Google Scholar]
- Endapally S., Infante R.E., Radhakrishnan A. Monitoring and modulating intracellular cholesterol trafficking using ALOD4, a cholesterol-binding protein. Methods Mol. Biol. 2019;1949:153–163. doi: 10.1007/978-1-4939-9136-5_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faienza F., Rizza S., Giglio P., Filomeni G. TRAP1: a metabolic hub linking aging pathophysiology to mitochondrial S-Nitrosylation. Front. Physiol. 2020;11:340. doi: 10.3389/fphys.2020.00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J.Q., Ishii S., Asano N., Suzuki Y. Accelerated transport and maturation of lysosomal alpha-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat. Med. 1999;5:112–115. doi: 10.1038/4801. [DOI] [PubMed] [Google Scholar]
- Fernández-Mosquera L., Diogo C.V., Yambire K.F., Santos G.L., Luna Sánchez M., Bénit P., Rustin P., Lopez L.C., Milosevic I., Raimundo N. Acute and chronic mitochondrial respiratory chain deficiency differentially regulate lysosomal biogenesis. Sci. Rep. 2017;7:45076. doi: 10.1038/srep45076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelsthorpe M.E., Baumann N., Millard E., Gale S.E., Langmade S.J., Schaffer J.E., Ory D.S. Niemann-Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding. J. Biol. Chem. 2008;283:8229–8236. doi: 10.1074/jbc.m708735200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y., Zhang M., Xu J., Bu B., Wei J. Construction of lentiviral vector carrying Rab9 gene and its expression in mouse brain. Front. Med. China. 2009;3:141–147. doi: 10.1007/s11684-009-0041-6. [DOI] [Google Scholar]
- Hardie D.G. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–1908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie D.G. AMPK--sensing energy while talking to other signaling pathways. Cell Metab. 2014;20:939–952. doi: 10.1016/j.cmet.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S., Shaw R.J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018;19:121–135. doi: 10.1038/nrm.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holczer M., Hajdú B., Lőrincz T., Szarka A., Bánhegyi G., Kapuy O. A double negative feedback loop between mTORC1 and AMPK kinases guarantees precise autophagy induction upon cellular stress. Int. J. Mol. Sci. 2019;20:E5543. doi: 10.3390/ijms20225543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höglinger D., Burgoyne T., Sanchez-Heras E., Hartwig P., Colaco A., Newton J., Futter C.E., Spiegel S., Platt F.M., Eden E.R. NPC1 regulates ER contacts with endocytic organelles to mediate cholesterol egress. Nat. Commun. 2019;10:4276. doi: 10.1038/s41467-019-12152-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante R.E., Abi-Mosleh L., Radhakrishnan A., Dale J.D., Brown M.S., Goldstein J.L. Purified NPC1 protein. I. Binding of cholesterol and oxysterols to a 1278-amino acid membrane protein. J. Biol. Chem. 2008;283:1052–1063. doi: 10.1074/jbc.m707943200. [DOI] [PubMed] [Google Scholar]
- Ioannou Y.A., Zeidner K.M., Gordon R.E., Desnick R.J. Fabry disease: preclinical studies demonstrate the effectiveness of alpha-galactosidase A replacement in enzyme-deficient mice. Am. J. Hum. Genet. 2001;68:14–25. doi: 10.1086/316953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova M.M., Changsila E., Iaonou C., Goker-Alpan O. Impaired autophagic and mitochondrial functions are partially restored by ERT in Gaucher and Fabry diseases. PLoS One. 2019;14:e0210617. doi: 10.1371/journal.pone.0210617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A., Dai L., Liu Y., Lee J., Ghahhari N.M., Segala G., Beebe K., Jenkins L.M., Lyons G.C., Bernasconi L., et al. The mitochondrial HSP90 paralog TRAP1 forms an OXPHOS-regulated tetramer and is involved in mitochondrial metabolic homeostasis. BMC Biol. 2020;18:10–23. doi: 10.1186/s12915-020-0740-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaptzan T., West S.A., Holicky E.L., Wheatley C.L., Marks D.L., Wang T., Peake K.B., Vance J., Walkley S.U., Pagano R.E. Development of a Rab9 transgenic mouse and its ability to increase the lifespan of a murine model of Niemann-Pick type C disease. Am. J. Pathol. 2009;174:14–20. doi: 10.2353/ajpath.2009.080660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunakaran D., Thrush A.B., Nguyen M.-A., Richards L., Geoffrion M., Singaravelu R., Ramphos E., Shangari P., Ouimet M., Pezacki J.P., et al. Macrophage mitochondrial energy status regulates cholesterol efflux and is enhanced by anti-miR33 in atherosclerosis. Circ. Res. 2015;117:266–278. doi: 10.1161/circresaha.117.305624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy B.E., Madreiter C.T., Vishnu N., Malli R., Graier W.F., Karten B. Adaptations of energy metabolism associated with increased levels of mitochondrial cholesterol in Niemann-Pick type C1-deficient cells. J. Biol. Chem. 2014;289:16278–16289. doi: 10.1074/jbc.m114.559914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein A.D., Mazzulli J.R. Is Parkinson's disease a lysosomal disorder? Brain. 2018;141:2255–2262. doi: 10.1093/brain/awy147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohan R., Carabelos M.N., Xin W., Sims K., Guelbert N., Cismondi I.A., Pons P., Alonso G.I., Troncoso M., Witting S., et al. Neuronal ceroid lipofuscinosis type CLN2: a new rationale for the construction of phenotypic subgroups based on a survey of 25 cases in South America. Gene. 2013;516:114–121. doi: 10.1016/j.gene.2012.12.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuk M.U., Lee Y.H., Kim J.W., Hwang S.Y., Park J.T., Park S.C. Potential treatment of lysosomal storage disease through modulation of the mitochondrial-lysosomal Axis. Cells. 2021;10:420. doi: 10.3390/cells10020420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laker R.C., Drake J.C., Wilson R.J., Lira V.A., Lewellen B.M., Ryall K.A., Fisher C.C., Zhang M., Saucerman J.J., Goodyear L.J., et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017;8:548. doi: 10.1038/s41467-017-00520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laker R.C., Xu P., Ryall K.A., Sujkowski A., Kenwood B.M., Chain K.H., Zhang M., Royal M.A., Hoehn K.L., Driscoll M., et al. A novel MitoTimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J. Biol. Chem. 2014;289:12005–12015. doi: 10.1074/jbc.m113.530527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M., Sabatini D.M. mTOR signaling at a glance. J. Cell Sci. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liapis A., Chen F.W., Davies J.P., Wang R., Ioannou Y.A. MLN64 transport to the late endosome is regulated by binding to 14-3-3 via a non-canonical binding site. PLoS One. 2012;7:e34424. doi: 10.1371/journal.pone.0034424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liscum L., Faust J.R. The intracellular transport of low density lipoprotein-derived cholesterol is inhibited in Chinese hamster ovary cells cultured with 3-beta-[2-(diethylamino)ethoxy]androst-5-en-17-one. J. Biol. Chem. 1989;264:11796–11806. doi: 10.1016/S0021-9258(18)80136-3. [DOI] [PubMed] [Google Scholar]
- Liu L., Zhang L., Zhao J., Guo X., Luo Y., Hu W., Zhao T. Tumor necrosis factor receptor-associated protein 1 protects against mitochondrial injury by preventing high glucose-induced mPTP opening in diabetes. Oxid. Med. Cell. Longev. 2020;2020:6431517. doi: 10.1155/2020/6431517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomenick B., Jung G., Wohlschlegel J.A., Huang J. Target identification using drug affinity responsive target stability (DARTS) Curr. Protoc. Chem. Biol. 2011;3:163–180. doi: 10.1002/9780470559277.ch110180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M., Sanchez-Martinez A., Longo M., Suomi F., Stenlund H., Johansson A.I., Ehsan H., Salo V.T., Montava-Garriga L., Naddafi S., et al. DGAT1 activity synchronises with mitophagy to protect cells from metabolic rewiring by iron depletion. Embo. J. 2022;109390 doi: 10.15252/embj.2021109390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low H., Hoang A., Sviridov D. Cholesterol efflux assay. J. Vis. Exp. 2012:e3810. doi: 10.3791/3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lücke T., Höppner W., Schmidt E., Illsinger S., Das A.M. Fabry disease: reduced activities of respiratory chain enzymes with decreased levels of energy-rich phosphates in fibroblasts. Mol. Genet. Metab. 2004;82:93–97. doi: 10.1016/j.ymgme.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Masgras I., Sanchez-Martin C., Colombo G., Rasola A. The chaperone TRAP1 as a modulator of the mitochondrial adaptations in cancer cells. Front. Oncol. 2017;7:58. doi: 10.3389/fonc.2017.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maue R.A., Burgess R.W., Wang B., Wooley C.M., Seburn K.L., Vanier M.T., Rogers M.A., Chang C.C., Chang T.-Y., Harris B.T., et al. A novel mouse model of Niemann-Pick type C disease carrying a D1005G-Npc1 mutation comparable to commonly observed human mutations. Hum. Mol. Genet. 2012;21:730–750. doi: 10.1093/hmg/ddr505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin S.H., Smith H.W., Jackson S.E. Stimulation of the weak ATPase activity of human hsp90 by a client protein. J. Mol. Biol. 2002;315:787–798. doi: 10.1006/jmbi.2001.5245. [DOI] [PubMed] [Google Scholar]
- Millat G., Marçais C., Rafi M.A., Yamamoto T., Morris J.A., Pentchev P.G., Ohno K., Wenger D.A., Vanier M.T. Niemann-Pick C1 disease: the I1061T substitution is a frequent mutant allele in patients of Western European descent and correlates with a classic juvenile phenotype. Am. J. Hum. Genet. 1999;65:1321–1329. doi: 10.1086/302626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T., Neff L., Tanaka S., Horne W.C., Baron R. Regulation of cytochrome c oxidase activity by c-Src in osteoclasts. J. Cell Biol. 2003;160:709–718. doi: 10.1083/jcb.200209098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M., Prudent J., Basu K., Goyon V., Katsumura S., Hulea L., Pearl D., Siddiqui N., Strack S., McGuirk S., et al. mTOR controls mitochondrial dynamics and cell survival via MTFP1. Mol. Cell. 2017;67:922–935.e5. doi: 10.1016/j.molcel.2017.08.013. [DOI] [PubMed] [Google Scholar]
- Ogura M., Yamaki J., Homma M.K., Homma Y. Mitochondrial c-Src regulates cell survival through phosphorylation of respiratory chain components. Biochem. J. 2012;447:281–289. doi: 10.1042/bj20120509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagant S., Huston M.W., Moreira L., Gan L., St Martin S., Sproul S., Holmes M.C., Meyer K., Wechsler T., Desnick R.J., et al. ZFN-mediated in vivo gene editing in hepatocytes leads to supraphysiologic α-Gal A activity and effective substrate reduction in Fabry mice. Mol. Ther. 2021;29:3230–3242. doi: 10.1016/j.ymthe.2021.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai M.Y., Lomenick B., Hwang H., Schiestl R., McBride W., Loo J.A., Huang J. Drug affinity responsive target stability (DARTS) for small-molecule target identification. Methods Mol. Biol. 2015;1263:287–298. doi: 10.1007/978-1-4939-2269-7_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parenti G., Medina D.L., Ballabio A. The rapidly evolving view of lysosomal storage diseases. EMBO Mol. Med. 2021;13:e12836. doi: 10.15252/emmm.202012836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentchev P.G., Brady R.O., Blanchette-Mackie E.J., Vanier M.T., Carstea E.D., Parker C.C., Goldin E., Roff C.F. The Niemann-Pick C lesion and its relationship to the intracellular distribution and utilization of LDL cholesterol. Biochim. Biophys. Acta. 1994;1225:235–243. doi: 10.1016/0925-4439(94)90001-9. [DOI] [PubMed] [Google Scholar]
- Plotegher N., Duchen M.R. Crosstalk between lysosomes and mitochondria in Parkinson's disease. Front. Cell Dev. Biol. 2017;5:110. doi: 10.3389/fcell.2017.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pridgeon J.W., Olzmann J.A., Chin L.-S., Li L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007;5:e172. doi: 10.1371/journal.pbio.0050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimundo N., Fernández-Mosquera L., Yambire K.F., Diogo C.V. Mechanisms of communication between mitochondria and lysosomes. Int. J. Biochem. Cell Biol. 2016;79:345–349. doi: 10.1016/j.biocel.2016.08.020. [DOI] [PubMed] [Google Scholar]
- Rasola A., Neckers L., Picard D. Mitochondrial oxidative phosphorylation TRAP(1)ped in tumor cells. Trends Cell Biol. 2014;24:455–463. doi: 10.1016/j.tcb.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rena G., Hardie D.G., Pearson E.R. The mechanisms of action of metformin. Diabetologia. 2017;60:1577–1585. doi: 10.1007/s00125-017-4342-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribbens J., Whiteley G., Furuya H., Southall N., Hu X., Marugan J., Ferrer M., Maegawa G.H.B. A high-throughput screening assay using Krabbe disease patient cells. Anal. Biochem. 2013;434:15–25. doi: 10.1016/j.ab.2012.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca-Agujetas V., Barbero-Camps E., de Dios C., Podlesniy P., Abadin X., Morales A., Marí M., Trullàs R., Colell A. Cholesterol alters mitophagy by impairing optineurin recruitment and lysosomal clearance in Alzheimer's disease. Mol. Neurodegener. 2021;16:15–26. doi: 10.1186/s13024-021-00435-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffari A., Kölker S., Hoffmann G.F., Ebrahimi-Fakhari D. Linking mitochondrial dysfunction to neurodegeneration in lysosomal storage diseases. J. Inherit. Metab. Dis. 2017;40:631–640. doi: 10.1007/s10545-017-0048-0. [DOI] [PubMed] [Google Scholar]
- Schmidt T.G.M., Skerra A. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2007;2:1528–1535. doi: 10.1038/nprot.2007.209. [DOI] [PubMed] [Google Scholar]
- Sciacovelli M., Guzzo G., Morello V., Frezza C., Zheng L., Nannini N., Calabrese F., Laudiero G., Esposito F., Landriscina M., et al. The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab. 2013;17:988–999. doi: 10.1016/j.cmet.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C., Di Malta C., Polito V.A., Garcia Arencibia M., Vetrini F., Erdin S., Erdin S.U., Huynh T., Medina D., Colella P., et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner N.C., Lin M.Z., McKeown M.R., Steinbach P.A., Hazelwood K.L., Davidson M.W., Tsien R.Y. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat. Methods. 2008;5:545–551. doi: 10.1038/nmeth.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheriff S., Du H., Grabowski G.A. Characterization of lysosomal acid lipase by site-directed mutagenesis and heterologous expression. J. Biol. Chem. 1995;270:27766–27772. doi: 10.1074/jbc.270.46.27766. [DOI] [PubMed] [Google Scholar]
- Sisinni L., Maddalena F., Lettini G., Condelli V., Matassa D.S., Esposito F., Landriscina M. TRAP1 role in endoplasmic reticulum stress protection favors resistance to anthracyclins in breast carcinoma cells. Int. J. Oncol. 2014;44:573–582. doi: 10.3892/ijo.2013.2199. [DOI] [PubMed] [Google Scholar]
- Skočaj M., Resnik N., Grundner M., Ota K., Rojko N., Hodnik V., Anderluh G., Sobota A., Maček P., Veranič P., et al. Tracking cholesterol/sphingomyelin-rich membrane domains with the ostreolysin A-mCherry protein. PLoS One. 2014;9:e92783. doi: 10.1371/journal.pone.0092783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohar I., Lin L., Lobel P. Enzyme-based diagnosis of classical late infantile neuronal ceroid lipofuscinosis: comparison of tripeptidyl peptidase I and pepstatin-insensitive protease assays. Clin. Chem. 2000;46:1005–1008. doi: 10.1093/clinchem/46.7.1005. [DOI] [PubMed] [Google Scholar]
- Solsona-Vilarrasa E., Fucho R., Torres S., Nuñez S., Nuño-Lámbarri N., Enrich C., García-Ruiz C., Fernández-Checa J.C. Cholesterol enrichment in liver mitochondria impairs oxidative phosphorylation and disrupts the assembly of respiratory supercomplexes. Redox Biol. 2019;24:101214. doi: 10.1016/j.redox.2019.101214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepien K.M., Roncaroli F., Turton N., Hendriksz C.J., Roberts M., Heaton R.A., Hargreaves I. Mechanisms of mitochondrial dysfunction in lysosomal storage disorders: a review. J. Clin. Med. 2020;9:2596. doi: 10.3390/jcm9082596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickley R.G. Solubilizing excipients in oral and injectable formulations. Pharm. Res. (N. Y.) 2004;21:201–230. doi: 10.1023/b:pham.0000016235.32639.23. [DOI] [PubMed] [Google Scholar]
- Sung N., Lee J., Kim J.-H., Chang C., Joachimiak A., Lee S., Tsai F.T.F. Mitochondrial Hsp90 is a ligand-activated molecular chaperone coupling ATP binding to dimer closure through a coiled-coil intermediate. Proc. Natl. Acad. Sci. USA. 2016;113:2952–2957. doi: 10.1073/pnas.1516167113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto K., Miyata S., Takamura H., Katayama T., Tohyama M. Mitochondrial TRAP1 regulates the unfolded protein response in the endoplasmic reticulum. Neurochem. Int. 2011;58:880–887. doi: 10.1016/j.neuint.2011.02.015. [DOI] [PubMed] [Google Scholar]
- Trounce I.A., Kim Y.L., Jun A.S., Wallace D.C. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- Walkley S.U., Vanier M.T. Secondary lipid accumulation in lysosomal disease. Biochim. Biophys. Acta. 2009;1793:726–736. doi: 10.1016/j.bbamcr.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter M., Davies J.P., Ioannou Y.A. Telomerase immortalization upregulates Rab9 expression and restores LDL cholesterol egress from Niemann-Pick C1 late endosomes. J. Lipid Res. 2003;44:243–253. doi: 10.1194/jlr.m200230-jlr200. [DOI] [PubMed] [Google Scholar]
- Wojtanik K.M., Liscum L. The transport of low-density lipoprotein-derived cholesterol to the plasma membrane is defective in NPC1 cells. J. Biol. Chem. 2003;278:14850–14856. doi: 10.1074/jbc.m300488200. [DOI] [PubMed] [Google Scholar]
- Wong Y.C., Ysselstein D., Krainc D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature. 2018;554:382–386. doi: 10.1038/nature25486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woś M., Szczepanowska J., Pikuła S., Tylki-Szymańska A., Zabłocki K., Bandorowicz-Pikuła J. Mitochondrial dysfunction in fibroblasts derived from patients with Niemann-Pick type C disease. Arch. Biochem. Biophys. 2016;593:50–59. doi: 10.1016/j.abb.2016.02.012. [DOI] [PubMed] [Google Scholar]
- Yam G.H.-F., Bosshard N., Zuber C., Steinmann B., Roth J. Pharmacological chaperone corrects lysosomal storage in Fabry disease caused by trafficking-incompetent variants. Am. J. Physiol. Cell Physiol. 2006;290:C1076–C1082. doi: 10.1152/ajpcell.00426.2005. [DOI] [PubMed] [Google Scholar]
- Yambire K.F., Fernández Mosquera L., Steinfeld R., Mühle C., Ikonen E., Milosevic I., Raimundo N. Mitochondrial biogenesis is transcriptionally repressed in lysosomal lipid storage diseases. Elife. 2019;8:e39598. doi: 10.7554/elife.39598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S., Tsutsumi S., Muhlebach G., Sourbier C., Lee M.-J., Lee S., Vartholomaiou E., Tatokoro M., Beebe K., Miyajima N., et al. Molecular chaperone TRAP1 regulates a metabolic switch between mitochondrial respiration and aerobic glycolysis. Proc. Natl. Acad. Sci. USA. 2013;110:E1604–E1612. doi: 10.1073/pnas.1220659110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W., Gong J.-S., Ko M., Garver W.S., Yanagisawa K., Michikawa M. Altered cholesterol metabolism in Niemann-Pick type C1 mouse brains affects mitochondrial function. J. Biol. Chem. 2005;280:11731–11739. doi: 10.1074/jbc.m412898200. [DOI] [PubMed] [Google Scholar]
- Zeidner K.M., Desnick R.J., Ioannou Y.A. Quantitative determination of globotriaosylceramide by immunodetection of glycolipid-bound recombinant verotoxin B subunit. Anal. Biochem. 1999;267:104–113. doi: 10.1006/abio.1998.2966. [DOI] [PubMed] [Google Scholar]
- Zhang C.-S., Jiang B., Li M., Zhu M., Peng Y., Zhang Y.-L., Wu Y.-Q., Li T.Y., Liang Y., Lu Z., et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 2014;20:526–540. doi: 10.1016/j.cmet.2014.06.014. [DOI] [PubMed] [Google Scholar]
- Zhang P., Lu Y., Yu D., Zhang D., Hu W. TRAP1 provides protection against myocardial ischemia-reperfusion injury by ameliorating mitochondrial dysfunction. Cell. Physiol. Biochem. 2015;36:2072–2082. doi: 10.1159/000430174. [DOI] [PubMed] [Google Scholar]
- Zhu A., Romero R., Petty H.R. A sensitive fluorimetric assay for pyruvate. Anal. Biochem. 2010;396:146–151. doi: 10.1016/j.ab.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The results of the reporter screens that led to development of the lead compounds have been deposited and are publicly available (https://pubchem.ncbi.nlm.nih.gov/bioassay/485297 and https://pubchem.ncbi.nlm.nih.gov/bioassay/485313). Western blot, microscopy, and enzymatic data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the reported data is available from the lead contact upon request.