

Summary
Although it is reported that mitochondria-localized nuclear transcription factors (TFs) regulate mitochondrial processes such as apoptosis and mitochondrial transcription/respiration, the functions and mechanisms of mitochondrial dynamics regulated by mitochondria-localized nuclear TFs are yet to be fully characterized. Here, we identify STAT6 as a mitochondrial protein that is localized in the outer membrane of mitochondria (OMM). STAT6 in OMM inhibits mitochondrial fusion by blocking MFN2 dimerization. This implies that STAT6 has a critical role in mitochondrial dynamics. Moreover, mitochondrial accumulation of STAT6 in response to hypoxic conditions reveals that STAT6 is a regulator of mitochondrial processes including fusion/fission mechanisms.
Subject areas: Natural sciences, Biological sciences, Molecular biology, Molecular interaction
Graphical abstract
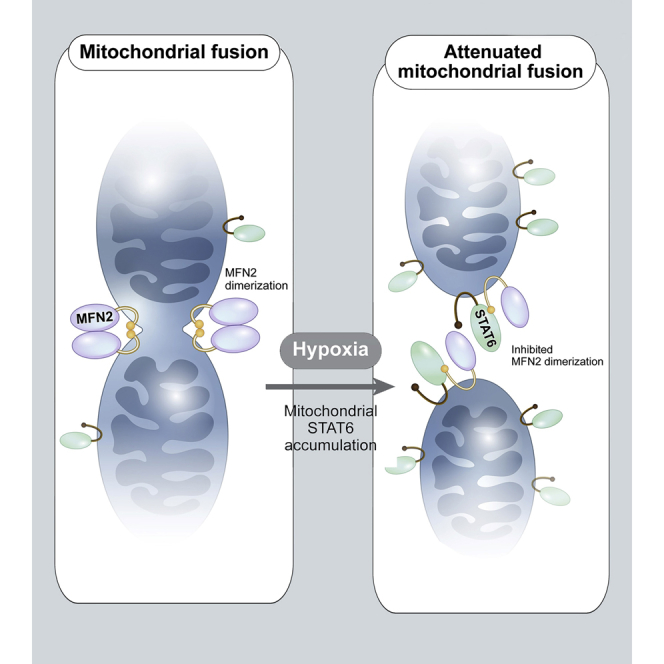
Highlights
-
•
STAT6 has mitochondrial-targeting sequences and anchoring transmembrane segments
-
•
STAT6 in OMM attenuates mitochondrial fusion by blocking MFN2 dimerization
-
•
Hypoxia-induced STAT6 mitochondrial accumulation inhibits tumorigenesis
Natural sciences; Biological sciences; Molecular biology; Molecular interaction;
Introduction
Mitochondria are essential for life as they support metabolic functions and ATP generation via oxidative phosphorylation. At the same time, they also regulate intrinsic apoptotic cell death via mitochondrial outer membrane permeabilization, and subsequent caspase activation driven by effector proapoptotic B cell lymphoma 2 (Bcl-2) family members (prominently BAX or BAK) (Bock and Tait, 2020). Mitochondria are structurally dynamic interconnected organelles that continuously undergo mitochondrial fusion and fission events. Mitochondrial fission is mediated by recruitment of the cytosolic GTPase dynamin 1-like protein (DNM1L/Drp1) on outer mitochondrial membrane (OMM), through binding with mitochondrial fission factor (MFF) (Gandre-Babbe and van der Bliek, 2008), Mid51, and Mid49 (Palmer et al., 2011). Mitochondrial fusion involves three major proteins: optic atrophy 1 (OPA1) mediates inner mitochondrial membrane (IMM) fusion (Ishihara et al., 2006), while mitofusin (MFN) 1 and 2 are responsible for OMM fusion (Filadi et al., 2018). MFN 1 and 2 belong to the large family of mitochondrial transmembrane GTPases and form homomeric/heteromeric complexes to induce mitochondrial outer membrane fusion. The crucial balance of mitochondrial fusion and fission, referred to as mitochondrial dynamics, is closely associated with mitochondrial function as well as establishment of overall mitochondrial architecture. Accordingly, mitochondrial dysfunction and aberrant balance between fusion/fission have been implicated in a number of cellular physiology and pathology, such as neurodegenerative diseases, metabolic disorders, cardiomyopathies (Suárez-Rivero et al., 2016), and cancer (Maycotte et al., 2017).
Although mitochondria contain their own genome, transcription, and translation mechanisms, mitochondrial DNA encodes for only 1% of the total mitochondrial proteome. The rest 99% is encoded by nuclear DNA, translated in ER, and then translocated to the mitochondria (Song et al., 2021). Eventually, the fine tune of mitochondrial DNA and nuclear DNA-encoded mitochondrial gene expressions decides overall mitochondrial biogenesis as well as proper assembly of mitochondrial protein complexes (Lionaki et al., 2016), emphasizing the significance of crosstalk between mitochondria and nucleus. This interorganellar coordination relies on the redistribution of nuclear or mitochondrial proteins between the two organelles. Nuclear proteins with mitochondrial distribution are mainly transcription factors (TFs) that translocate to mitochondria upon a stress stimulus or reside in mitochondria under steady-state conditions. Among TFs localized in mitochondria, signal transducer and activators of transcriptions (STATs), well-known nuclear TF targeting to mitochondria, play key roles in mitochondria (Meier and Larner, 2014). Especially, STAT3, a most studied isoform among them, was reported to localize in mitochondrial matrix and IMM through phosphorylation of serine 727, and regulates the activity of electron transport chain complex influencing cellular metabolism (Wegrzyn et al., 2009), protection against various stresses (Szczepanek et al., 2011), and tumorigenesis (Gough et al., 2009; Zhang et al., 2013). Although other STAT subfamilies, STAT1, 2, 5, and 6, also have been shown to localize to mitochondria (Bourke et al., 2013; Chueh et al., 2010; Goswami et al., 2013; Khan et al., 2013), crucial questions, such as whether STATs possess mitochondrial-targeting sequences (MTS), and how mitochondrial function regulated by individual STAT is linked to human disease, however, remains to be clarified.
We found that mitochondrial fragmentation was triggered in STAT6-GFP-expressing mitochondria, suggesting mitochondrial role of STAT6. This raises a number of important questions regarding STAT6 function and regulation in mitochondria. For example, where is submitochondrial localization of STAT6? What is mitochondrial STAT6 function and derived cellular/physiological events from mitochondrial STAT6?
To address these questions, we dissected the role of STAT6 as a mitochondrial protein. Our findings prove that STAT6 is an OMM protein with MTS and transmembrane segments anchored to OMM. Also, using quantitative proteomic analysis, we found that MFN2 is a crucial binding partner of mitochondrial STAT6 in OMM that mediates inhibition of mitochondrial fusion. Moreover, we demonstrate that mitochondrial accumulation of STAT6 under hypoxic conditions induced Cytochrome c release linked to cell death and tumorigenesis inhibition. Altogether, our findings present insights into the cellular and physiological roles of STAT6 in mitochondria.
Results
STAT6 is localized in mitochondrial outer membrane
Based on the previous finding suggesting the presence of STAT6 within mitochondria (Khan et al., 2013), we first examined it by transiently transfecting STAT6-GFP to U373-MG cell lines (STAT6-GFP). Confocal images demonstrated that STAT6-GFP signals were co-localized with TOM20 (mitochondria marker) at mitochondria (Figure 1A). Biochemical fractionation experiments with homogenates of brain tissues also demonstrated mitochondrial distribution of STAT6 (Figure 1B). Results using cultured primary astrocytes obtained from brain and STAT6-expressing U373-MG (STAT6-U373) showed consistent results, revealing mitochondrial presence of STAT6 (Figure 1C). That was also observed in other cell lines, including several glioma cell lines (Figure S1A), revealing that mitochondrial localization of STAT6 is a ubiquitous phenomenon. Further fractionation assays to determine subcellular localization of STAT6 demonstrated that it was detected in outer membrane of mitochondria (OMM) fraction (Figure 1D). EM images demonstrated immunogold-labeled STAT6 localized along the mitochondrial surface, where TOM20 signals were detected (Figure S1B). Next, to determine whether STAT6 is an integral membrane protein in OMM, mitochondria were isolated from astrocytes and STAT6-U373, and were subjected to high salt (NaCl) or sodium carbonate (Na2CO3) extraction (Figures 1E and 1F). Sodium carbonate extraction is widely accepted as the method to distinguish peripheral from integral membrane protein (Fujiki et al., 1982) and high salt extraction is an alternative method for isolating membranes to identify integral membrane proteins (Okamoto et al., 2001). Specific markers for mitochondrial fractions were served: The signal-anchored outer membrane protein TOM20 with moderate hydrophobicity (Ahting et al., 2005), intermembrane space or loosely associated inner membrane protein CYCS (Cytochrome c), inner membrane protein TIM23, and matrix-located ACO2 (Aconitase 2), respectively. STAT6 was detected in pellet (P) fractions of mitochondria with high salt in both cell types (Figure 1E), unraveling that it is an integral membrane protein similar to TOM20 and TIM23. Although STAT6 was resistant to carbonate treatment at pH range of 9.5–11.5, carbonate extraction at pH 12.5 did result in the release of STAT6 into the supernatant fraction (Figure 1F). This suggests that STAT6 is either partially embedded into the outer membrane or has a moderately hydrophobic transmembrane domain because mitochondrial proteins with moderate hydrophobicity have been shown to be released from the membrane upon sodium carbonate treatment (Kim et al., 2015). These findings were also observed in other cell lines tested (Figures S1C–S1F). To further confirm that STAT6 is an integral protein in OMM, we treated proteinase K (PK) on isolated mitochondria from astrocytes and STAT6-U373, and then performed Western blot using STAT6 antibodies targeted to different sites. While STAT6 was disappeared in PK-treated mitochondria using the STAT6 antibodies against N-terminal, 280–480 residues, and c-terminal (Figure S1G), the truncated STAT6 bands were detected when SH2 domain-targeted STAT6 antibody was applied (Figure 1G), suggesting that SH2 domain of STAT6 is possibly a transmembrane segment of OMM or exposed to mitochondrial intermembrane space. These results were also confirmed in thrombin (T.)-treated mitochondria from astrocytes and STAT6-U373 (Figure S1H), and observed in PK-treated mitochondria from other cell lines (Figure S1I). Furthermore, using membrane protein explorer (MPEx) program, we demonstrated that STAT6 possesses predicted hydrophobic transmembrane segments (293–311, 399–417, 506–524, 537–555, and 688–706 residues) (Figure 1H). In addition, translocon-mediated TM (transmembrane) analysis suggests that 532–552 residues in STAT6 might be transmembrane segments (Figure S1J). Taken together, these data suggest that STAT6 localizes in OMM, not within mitochondria.
Figure 1.

STAT6 is localized in mitochondrial outer membrane (OMM)
(A) Confocal images of mitochondrial morphology in U373-MG transfected with GFP or STAT6-GFP, stained with TOM20 (red, mitochondrial marker). The insets show a magnification of the selected mitochondrial region (white square). 30 images/group were quantified for colocalization. Colocalization (Pearson’s Correlation Coefficient) is shown corresponding to the colocalization of the GFP and TOM20 for each group. Data are presented as means ± SD of 3 independent experiments (∗∗∗p < 0.001), unpaired Student t-test. Scale bar: 10 μm.
(B) Western blot analysis of subcellular fractionations from mouse brain tissue with the indicated antibodies. Cyto: cytosol. MAM: mitochondria-associated membrane. Mito: mitochondria. ACO2 (Aconitase 2): mitochondrial marker. Calnexin: ER and MAM marker. Tubulin: Cytosolic marker.
(C) Sucrose-density gradient centrifugation analysis of mitochondria isolated from primary mouse astrocytes or STAT6-expressing U373-MG. LAMP1: lysosomal marker.
(D) Western blot analysis of mitochondrial subfractionations obtained from mouse brain tissue. OMM: mitochondrial outer membrane. IMM: mitochondrial inner membrane. Catalase: peroxisomal marker.
(E and F) High salt extraction (E) and Sodium carbonate extraction at different pH (F) of mitochondria isolated from mouse astrocytes and STAT6-expressing U373-MG. T: total mitochondrial fraction. P: pellet. S: supernatant. TOM20: signal-anchored outer membrane protein. CYCS (Cytochrome c): soluble protein in intermembrane space or peripherally associated membrane protein in inner membrane. TIM23: integral inner membrane protein. ACO2: soluble matrix protein.
(G) Proteinase K (PK) protection assay of mitochondria isolated from astrocytes and STAT6-expressing U373-MG using STAT6 antibody targeted to SH2 domain. PK: proteinase K. Mito.: mitochondrial fraction. Cyto.: cytoplasmic fraction.
(H) Hydropathy plots of STAT6. The hydrophobicity of STAT6 was analyzed using the membrane protein explorer (MPEx) programme (ver. 3.3).
STAT6 possesses mitochondrial-targeting sequences (MTS) and transmembrane segments
The mitochondrial proteins usually have mitochondrial-targeting sequences (MTS). However, since previous published finding using MitoProt2 algorithm proposed lack of MTS within STAT6 (Khan et al., 2013), we performed experiments to determine it. To identify whether STAT6 has MTSs, we generated domain-deleted STAT6 mutants-expressing stable cell lines and assessed the mitochondrial localization of STAT6 mutants in these stable cell lines using sucrose gradient fractionation and Western blot (Figures 2A and 2B). Interestingly, we found that the 1–260 residues-deleted STAT6 (Δ 1–260) was absent in mitochondrial fraction implying that the MTS of STAT6 exists in 1–260 residues of the TF.
Figure 2.

Identification of mitochondrial targeting sequences (MTS) in STAT6
(A) Schematic of STAT6-WT and domain-deleted mutants with myc-tag. (Δ1–260: 1–260 amino acids-deleted STAT6, ΔDBD: DNA-Binding domain-deleted STAT6, ΔLD: Linker domain-deleted STAT6, ΔSH2: SH2 domain-deleted STAT6, ΔTAD: Transactivation domain-deleted STAT6).
(B) Sucrose-density gradient centrifugation analysis of mitochondria isolated from domain deleted STAT6 mutants-expressing U373-MG stable cell lines. BiP: ER marker.
(C) Sucrose-density gradient centrifugation analysis of mitochondria isolated from 1–50 amino acids-deleted STAT6 (Δ1–50)-expressing U373-MG stable cell line.
(D and E) Confocal images of mitochondria in human U373-MG (D) and mouse B16F10 (E) transfected with GFP or human STAT6 n-terminal fragments with GFP were stained with TOM20. The inset shows a magnification of the selected mitochondrial region (white square). Scale bar, 3μm.
(F and G) At least 18 images/group were quantified for colocalization. Colocalization (Pearson’s Correlation Coefficient) is shown corresponding to the colocalization of the GFP and TOM20 for each group. Data are presented as the means ± SD of 3 independent experiments (∗∗∗p < 0.001). One-way ANOVA with Scheffe’s post-hoc test.
(H) Alignment of the conserved mitochondrial targeting sequences (MTS) identified in STAT6 n-terminal of six mammals. Nucleotide positions that are conserved among the mammals are highlighted with gray rectangle.
(I) Schematic of STAT6-WT and the deletion mutants with myc-tag. (Δ674–847: 674–847 amino acids-deleted STAT6, Δ699–847: 699–847 amino acids-deleted STAT6, and Δ774–847: 774–847 amino acids-deleted STAT6).
(J) PK protection assay of mitochondria isolated from ΔTAD-expressing U373-MG using STAT6 antibody targeted to SH2 domain.
(K) High salt extraction of mitochondria isolated from ΔTAD-expressing U373-MG stable cell lines.
(L) PK protection assay of mitochondria isolated from Δ674-847-expressing U373-MG using STAT6 antibody targeted to SH2 domain.
(M and N) High salt extraction of mitochondria isolated from Δ674–847, Δ699–847, and Δ774-847-expressing U373-MG stable cell lines.
In line with previous reports that MTS of the most mitochondrial proteins exist at N termini, STAT6 mutant without N-terminal 1–50 residues (Δ1–50) did not appear in mitochondrial fraction, indicating that the MTS of STAT6 is in N-terminal region of STAT6 (Figure 2C).
To identify the correct MTS of STAT6, we transfected the different human STAT6 N-terminal fragments with GFP-tag on human U373-MG and mouse B16-F10 (Figures 2D–2G, and S2A). Among them, the human 1–30 residues of STAT6 showed definitive mitochondrial localization in human U373-MG (Figures 2D, F, and S2B), also even in mouse B16–F10 (Figures 2E and 2G). These results also were observed in HEK 293 cells (Figure S2C). Figure 2H shows that residues 1–30 of STAT6 are highly conserved in six species of mammals indicating that MTS of STAT6 is conserved in some species of mammals. Then, how does STAT6 with MTSs get localized in the OMM? Among domain-deleted STAT6 mutants, 624–847 residues-deleted STAT6 (ΔTAD) was found in mitochondrial fraction (Figure 2B), and protected against externally added PK on mitochondria. (Figure 2J). Also, ΔTAD was found in the supernatant fraction upon high salt extraction of mitochondria (Figure 2K), indicating that ΔTAD is localized within mitochondria, not in OMM. We hypothesized that STAT6 localization in OMM needs partial sequences within TAD and the partial sequences might be transmembrane segments. To identify the partial sequences, we generated TAD-partially deleted STAT6 mutants (Δ674–847, Δ699–847, and Δ774–847)-expressing stable cell lines and assessed the mitochondrial localization of these mutants to identify transmembrane segments within TAD of STAT6 (Figures 2L–2N). Interestingly, Δ674–847 was protected against externally added PK on mitochondria (Figure 2L) and still found in supernatant fraction upon high salt extraction of mitochondria (Figure 2M) whereas 699–847 and Δ774–847 were found in the pellet fraction upon high salt extraction of mitochondria (Figure 2N), indicating that 674–698 residues of STAT6 might be transmembrane segments. Taken together, STAT6 is an OMM protein that possesses MTS and transmembrane segments.
MFN2 is a binding protein of STAT6 in OMM
To examine the function of mitochondrial STAT6, we first explored binding partner with mitochondrial STAT6, using proteomic analysis. MS spectrometry analysis following affinity purification identified 89 proteins that putatively bound to STAT6 in mouse brain tissue. GO data demonstrated that 18 candidates were related to mitochondria (Figure 3A and Table S1), where 4 were located in OMM (Figure 3B). And, three among the 4 OMM proteins were involved in mitochondrial fusion and fission (Figure 3B). Among these candidates, MFN2 and MFF, key regulators related to mitochondrial fusion–fission mechanisms in OMM, were identified as STAT6-binding candidates using immunoprecipitation (IP) analysis, revealing that that MFN2 binding with STAT6 was prominent but that with MFF was not evident in 293T and HeLa cells (Figure 3C). To map the domain interaction between MFN2 and STAT6, we constructed individual domain-deleted mutants of STAT6 and MFN2 (Figures 3D and 3F). By IP analysis, we demonstrated that 31–260 residues, DNA-binding (DBD), and linker domain of STAT6 bound to heptad region (HR2) domain of MFN2 (Figures 3E and 3G). Interestingly, IP analysis revealed that ΔTAD failed to interact with MFN2, whereas partially restored ΔTAD (Δ699–847 and Δ774–847) bound to MFN2 (Figure 3E). These results were consistent with mitochondrial re-localization of Δ699–847- and Δ774–847- STAT6 in OMM (Figure 2O).
Figure 3.

MFN2 interacts with STAT6 in OMM
(A and B) Quantitative label-free mass spectrometry identification of STAT6-binding candidates using STAT6 IP product obtained from mouse brain.
(A) Subcellular localization of 89 STAT6-interacting candidates. Those significantly associated (p < 0.05) lists are plotted with the numbers of the binding candidates for each GO term. The STAT6-binding candidates in mitochondrion are marked with red. GO terms with less than 7 binding candidates are not plotted.
(B) The 18 STAT6-binding candidates in mitochondrion were analyzed by DAVID functional annotation corresponding to GO terms, biological function in mitochondria.
(C) Representative Western blot of immunoprecipitation using lysates obtained from HeLa and 293T.
(D) Schematic of STAT6-WT and deletion mutants with myc-tag.
(E) Western blot of immunoprecipitation using lysates from STAT6-WT or deletion mutants-expressing 293T cells.
(F) Schematic of MFN2-WT and deletion mutants with myc-tag. (HR: Heptad region, PR: Proline rich domain, TM: Transmembrane domain).
(G) Western blot of immunoprecipitation using lysates from MFN2-WT or deletion mutants-expressing 293T cells.
STAT6 in OMM negatively regulates mitochondrial elongation and mitochondrial fusion, inhibiting MFN2 dimerization
Since STAT6 binds to MFN2 through HR2 region that is essential for MFN2 dimerization/mitochondrial tethering that precedes OMM fusion (Filadi et al., 2018) and mitochondrial fragmentation/abnormal morphology was detected in STAT6-expressing cells (Figures S3A–S3C, and Figure 1A), we hypothesized that STAT6 in OMM inhibits mitochondrial elongation and fusion by blocking MFN2 dimerization. To confirm this hypothesis, we generated FLAG-tagged and myc-tagged MFN2-coexpressing stable cell line, and applied in situ Duolink proximity ligation assay (PLA), which allows us to examine MFN2 dimerization by determining the proximity between FLAG-tagged MFN2 and myc-tagged MFN2. We observed a more than 2-fold increase in the PLA-positive signals of STAT6 siRNA-transfected cells (Figures 4A and 4B). Conversely, we observed the decrease in the PLA signal of STAT6 WT-GFP and SH2 domain-deleted STAT6 (ΔSH2, function of transcription factor is extinct)-GFP-expressing cells compared with Mock-GFP-expressing cells, but restored PLA signal of MTS-deleted STAT6 (ΔMTS, targeting to mitochondria is disabled)-GFP-expressing cells. (Figure 4C) Collectively, STAT6 attenuates MFN2 dimerization.
Figure 4.

STAT6 inhibits MFN2 dimerization
To analyze MFN2 dimerization, MFN2-myc/MFN2-FLAG-coexpressing stable cell line was generated.
(A) Western blot analysis using MFN2-myc/MFN2-FLAG-coexpressing stable cell line transfected with Control siRNA or STAT6 siRNA.
(B) In situ Duolink proximity ligation assay (PLA) in MFN2-myc/MFN2-FLAG-coexpressing stable cell lines transfected with Control siRNA or STAT6 siRNA. MFN2 dimerization was detected using the PLA method. PLA signals (in red) were quantified and displayed. Data are presented as means ± SD of 3 independent experiments. (Control siRNA, n = 284 and STAT6 siRNA, n = 259. ∗∗∗∗p < 0.0001), unpaired Student t-test.
(C) MFN2 dimerization was detected in MFN2-myc/MFN2-FLAG-coexpressing stable cell lines transfected with empty vector, STAT6 WT, ΔSH2, or ΔMTS (mitochondrial targeting sequences-deleted STAT6) with GFP. Data are presented as means ± SD of 3 independent experiments. (GFP, n = 105. STAT6 WT-GFP and ΔSH2-GFP, n = 100. ΔMTS-GFP, n = 101. ∗∗∗∗p < 0.0001) One-way ANOVA with Scheffe’s post-hoc test.
Next, to determine whether mitochondrial morphology and fusion were blocked by STAT6 in OMM, we generated STAT6 KO stable cell lines from 293T and B16-F10 melanoma using CRISPR Cas9 system, and observed mitochondrial elongation in the STAT6 KO stable cell lines. (Figures S3D–S3G). To dissect whether STAT6 in OMM induces mitochondrial fragmentation, we analyzed mitochondrial morphology in STAT6 WT or several STAT6 mutants-expressing cells (Figures 5A–5D). Immunostaining analysis revealed that STAT6 WT and ΔSH2 triggered mitochondrial fragmentation, whereas ΔMTS failed to induce mitochondrial fragmentation. To further test mitochondrial fusion regulated by STAT6 in OMM, we evaluated the effects of mitochondrial STAT6 on extent of mitochondrial fusion using a polyethylene glycol (PEG)-induced cell fusion assay (Figures 5E–5H). STAT6 WT-myc, ΔSH2-myc, and ΔMTS-myc-expressing stable cell lines were generated from STAT6 KO-293T stable cell lines (Figure 5F). These stable cell lines were transfected with mitochondria-targeting green or red fluorescent (mitoGFP or mitoRFP) and were mixed. Cycloheximide was pretreated 30 min prior to cell fusion to inhibit synthesis of the fluorescent protein. The cells were incubated for 3 h post cell fusion, hybrid polykaryons were observed by confocal microscopy, and mitochondrial fusion was determined by mixing of green and red fluorescence signals (yellow) in the polykaryons (Figure 5G). The extent of mitochondrial fusion (colocalization between green and red fluorescence signals) in each polykaryon was further analyzed by Pearson’s correlation coefficient (Figure 5H). A significantly high level of mitochondrial fusion was observed in STAT6 KO-hybrid cells, compared to parental-hybrid cells. In contrast, restoration of STAT6 WT and ΔSH2 expression from STAT6 KO cells resulted in reduced mitochondrial fusion, whereas restoration of ΔMTS expression failed to reduce mitochondrial fusion, implying that STAT6 in OMM attenuates mitochondrial fusion.
Figure 5.

STAT6 in OMM impairs mitochondrial fusion
(A) Schematic of STAT6 WT and deletion mutants with mCherry.
(B–D) Confocal images (C) of mitochondrial morphology in STAT6 WT or deletion mutants-expressing 293T cells stained with TOM20. The insets show a magnification of the selected region (white square). Mitochondrial number per cell (D) and mean area per mitochondrion (B) were quantified from mCherry + cells using ImageJ. (mCherry, n = 21. STAT6 WT-mCherry, n = 23. ΔSH2-mCherry, n = 24. ΔMTS-mCherry, n = 34). Data are presented as means ± SD of 3 independent experiments. (∗∗∗p < 0.001). One-way ANOVA with Scheffe’s post-hoc test.
(E) Illustration of polyethylene glycol (PEG)- mediated cell fusion in 293T transfected with mitochondrial targeted-GFP (mtGFP) or mitochondrial targeted-RFP (mtRFP).
(F) To determine mitochondrial fusion inhibited by STAT6, STAT6-deficient cell line (STAT6 KO) was generated by CRISPR Cas9-mediated knockout from Parental cells (293T). And then STAT6 KO cell line was rescued with empty vector (Mock-myc), domain-deleted STAT6 mutants, and wild-type STAT6 with myc-tag (STAT6 WT-myc). Cell extracts obtained from the stable cell lines mentioned above were analyzed by Western blot using indicated antibodies.
(G) Confocal images of mitochondrial fusion in HEK 293T polykaryons subjected to the PEG-based fusion assay in the stable cell lines mentioned above. These stable cells were subsequently transfected with mtGFP or mtRFP and then co-cultured, treated with PEG for cell fusion. Mitochondrial fusion is indicated by colocalization (yellow mitochondria) of mitoGFP and mitoRFP.
(H) Quantitative analysis of the extent of mitochondrial fusion in individual hybrid cells was performed by the Pearson’s correlation coefficient (PCC). (Parental, n = 39. STAT6 KO, n = 33. STAT6 WT-myc, n = 57. ΔSH2-myc, n = 29. ΔMTS-myc, n = 30). Data are presented as means ± SD of 3 independent experiments. (∗∗∗p < 0.001). One-way ANOVA with Scheffe’s post-hoc test.
Mitochondrial STAT6 accumulation induces cell death, leading to tumorigenesis inhibition
Mitochondrial fragmentation is associated with mitochondrial depolarization and reduced ATP production (Jheng et al., 2012). Various stress, such as hypoxia, induce mitochondrial fragmentation, accompanied by mitochondrial accumulation of DRP1 and BAX (Brooks et al., 2011; Din et al., 2013), that subsequently trigger mitochondrial Cytochrome c release linked to apoptosis (Zhang et al., 2018). To ascertain whether mitochondrial fragmentation by STAT6 in OMM affects ATP production and mitochondrial membrane potential, we measured ATP, oxygen consumption rate (OCR) level, and membrane potential of STAT6 WT, ΔSH2, and ΔMTS-expressing cells relative to Mock-expressing cells. STAT6 WT and ΔSH2 both resulted in a decrease of ATP, OCR level, and mitochondrial membrane potential, whereas ΔMTS recovered ATP, OCR level, and mitochondrial membrane potential (Figures S4A–S4C). Especially during hypoxia, STAT6 accumulation in mitochondria (Figure 6A) and its interaction with MFN2 (Figure 6B) were increased. Moreover, mitochondrial fragmentation and Cytochrome c release into cytoplasm (Figures 6C and 6D) were increased in STAT6-expressing cells compared to Mock-expressing cells. However, cytoplasmic Cytochrome c levels in ΔMTS-expressing cells exposed to hypoxia were alleviated to similar levels found in Mock-expressing cells (Figure 6D). Figure 6E reveals that expression of STAT6 WT and ΔSH2 increased apoptotic cell death compared to Mock-expressing cells, which was decreased in ΔMTS-expressing cells. Also, Figures S4D–S4G indicate the apoptotic cell death induced by hypoxia in parental 293T cells was decreased in STAT6 KO-293T cells (Figure S4D) and mitochondrial fragmentation/apoptotic cell death induced by STAT6 WT was alleviated in MFN2 WT-overexpressing cells (Figures S4E–S4F).
Figure 6.

Mitochondrial STAT6 accumulation under hypoxic condition induces cell death
(A and B) 293T cells were exposed to hypoxia (0.1%) for the indicated times. (A) Mitochondria and cytosol obtained from the 293T were subjected to Western blot. CYCS: mitochondrial marker. HSP90AB1: cytosolic marker.
(B) Western blot of immunoprecipitation using lysates from hypoxia-exposed 293T cells for indicated times.
(C) Representative confocal images of mitochondrial morphology and Cytochrome c release in empty vector- or STAT6-expressing U373-MG stable cell lines exposed to hypoxic condition for indicated times and stained with Cytochrome c and TOM20. Scale bar, 10μm.
(D) Mitochondria and cytosol obtained from STAT6 KO-293T cells transfected with Empty vector-, STAT6 WT, and ΔMTS exposed to hypoxia (0.1% O2, 24 h).
(E) Flow cytometry dotplots of annexin V and propidium iodide (PI) staining in Empty vector-, STAT6 WT, ΔSH2, and ΔMTS-expressing 293T exposed to hypoxic condition. Data are presented as means ± SD of 4 independent experiments. (∗p < 0.05).
(F and G) Clonogenic assay. Representative images of colonies are shown in left, and quantification of the number of colonies is depicted in right. 5 independent experiments (∗∗∗∗p < 0.0001), unpaired Student t-test (F) or One-way ANOVA with Scheffe’s post-hoc test (G).
(H and I) Soft-agar colony formation assay. Representative images of colonies are shown in left, and quantification of the number of colonies is depicted in right. 5 independent experiments (∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001), unpaired Student t-test (H) or One-way ANOVA with Scheffe’s post-hoc test; (I).
Interestingly, we observed significantly more rapid growth in STAT6 KO-293T cells compared to parental cells. So, we investigated whether STAT6 in OMM attenuates regulation of cell growth. The frequency of STAT6 KO-293T colony formation increased dramatically under regular medium culture (Figure 6F). Conversely, recovery of STAT6 WT or ΔSH2 expression from STAT6 KO-293T inhibited cell growth whereas recovery of ΔMTS expression induced cell growth again (Figure 6G). We have confirmed in Figures 6C–6E that STAT6 in OMM triggered mitochondrial fragmentation, Cytochrome c release, and cell death with hypoxic condition, which is a common feature of solid tumor microenvironment (Guha and Avadhani, 2013).
To investigate whether STAT6 in OMM regulates tumorigenesis, the tumorigenic potential was tested in vitro by soft-agar assay. STAT6 KO-293T cells grew faster than parental cells in an anchorage-independent way in soft agar (Figure 6H). However, STAT6 WT and ΔSH2 expression reduced colony formation whereas ΔMTS recovered colony formation again (Figure 6I). We next examined the impact of STAT6 on tumor growth in vivo (Figure 7). STAT6-deficient 293T exhibited stronger cancer progression than parental cells, as evaluated by larger tumor volume over the treatment period (Figures 7A–7C). Interestingly, STAT6 WT or ΔSH2-expressing 293T exhibited inhibition of cancer progression whereas ΔMTS-expressing 293T recovered cancer progression (Figures 7D–7H).
Figure 7.

Mitochondrial STAT6 accumulation leads to tumorigenesis inhibition
In vivo results on xenograft mouse models.
(A–C) Parental or STAT6 KO-293T stable cell line (2 × 106 cells) was injected subcutaneously into the left or right flank of nude mice (n = 18).
(A) Representative images of xenograft tumor.
(B) Tumor growth curve was measured over time. Data are plotted as mean ± SEM with error bars by two-way repeated measures ANOVA. (∗∗∗∗p < 0.0001) (C) Weight of xenograft tumors. Data represent mean ± SD with error bar (n = 18). ∗∗p < 0.01 (D–H) STAT6 KO-293T, STAT6 WT, ΔSH2, and ΔMTS were inoculated subcutaneously into the left (STAT6 KO) or right (STAT6 WT, ΔSH2, or ΔMTS) flank of nude mice.
(D–F) Representative images of xenograft tumors.
(G) Tumor growth curve was measured. Data are plotted as mean ± SEM with error bars by two-way repeated measures ANOVA and Tukey’s post-hoc test. ∗∗∗∗p < 0.0001 (STAT6 WT or ΔSH2 vs STAT6 KO).
(H) Weight of xenograft tumors. Data represent mean ± S.D. with error bars by one-way ANOVA and Tukey’s post-hoc test. (STAT6 KO, n = 35; STAT6 WT, n = 12; ΔSH2, n = 10; ΔMTS, n = 13). ∗∗p < 0.01 (STAT6 WT or ΔSH2 vs STAT6 KO) and ∗∗∗p < 0.001 (STAT6 WT or ΔSH2 vs ΔMTS).
Discussion
Aberrant mitochondrial dynamics causes a vast array of health concerns including metabolic stroke, aging, myopathy, developmental or cognitive disabilities, cancer, and much more. For this reason, understanding the mechanisms of mitochondrial dynamics is of importance especially in providing therapeutic strategies. Despite recent progress in research on the mitochondrial function of STAT subfamily (Meier and Larner, 2014), their submitochondrial localization and underlying mechanisms remain largely unknown.
Our study investigated the role and underlying mechanisms of mitochondrial dynamics regulated by STAT6 as an OMM protein in the pathogenesis of mitochondrial-related diseases.
We also investigated if other STAT family members (STAT1, 2, 3, and 5) localize in OMM of brain tissue, and screened for their subcellular distribution in brain tissue. Figure S5A demonstrates that STAT1, 3, and 5 were detected in mitochondrial fraction, implying mitochondrial localization of these STATs. PK treatment of mitochondria from astrocytes showed that STAT1 likely localizes in OMM (Figure S5B). Furthermore, MPEx program suggests that STAT1 possesses predicted hydrophobic transmembrane segments (300–318, 461–504, and 552–582 residues) (Figure S5C). These results imply mitochondrial role of STAT1 in OMM, as well as STAT6.
Our findings indicate that STAT6 in OMM interacts with MFN2, thereby suppressing mitochondrial fusion (Figure 5). These findings imply that STAT6 is involved in mitochondrial fusion/fission mechanism.
MFN2 is responsible for fusion of OMM, and is composed of N-terminal GTPase domain, heptad-repeat (HR) 1, proline-rich, and C-terminal HR2 exposed to cytosolic face through two transmembrane domains (TM) crossing the OMM (Filadi et al., 2018). To induce OMM fusion, first of all, it is required of membrane tethering that is mediated by the interaction between HR2 of MFN2 on opposite OMM (Koshiba et al., 2004). Next, OMM fusion is triggered by interaction of N-terminal GTPase domains (Hales and Fuller, 1997). Strikingly, we found that STAT6 was bound to HR2 of MFN2 (Figure 3), indicating that STAT6 blocked OMM tethering mediated by HR2 of MFN2, and inhibited mitochondrial fusion as a result (Figure 5). These findings raise an interesting question: What is required to target STAT6 to OMM? Although it was previously reported that STAT6 is localized within mitochondria using immunogold-EM imaging and lacked obvious mitochondrial-targeting sequences (MTS) (Khan et al., 2013), we do not share this opinion. On the basis of our results, we furnish strong arguments against the previous report. First, is STAT6 devoid of MTS? Our data revealed that 1–30 residues at the N-terminal of STAT6 serve as MTS (Figures 2D–2G, and S2) in agreement with a number of reports that ∼90% of mitochondrial proteins have MTS at N-terminal of the proteins (Kang et al., 2018), supporting that STAT6 is as a mitochondrial protein with MTS. Second, as shown in Figures S1B and S1G–S1I, and Figure 1G, we guessed that STAT6 may be localized in OMM through several experiments including immunogold-EM imaging. If STAT6 is imported within mitochondria, the STAT6 might be protected from thrombin or PK. Our data, however, revealed that the large N-terminal (1–480 residues) and the short C-terminal portion of STAT6 in OMM are exposed to cytosolic face whereas SH2 domain of STAT6 may be under OMM. This finding is in line with our result that STAT6 was bound to MFN2 through the large N-terminal region exposed to cytosolic face (Figure 3E). Finally, although the previous report demonstrated that STAT6 N-terminal fragments (1–459 residues) were localized within mitochondria, the result is due to the lack of transmembrane segments (624–698 residues of STAT6) to be anchored in OMM. Thus, we assert STAT6 in OMM interacts with MFN2 and inhibits mitochondrial fusion.
Our MS data (Figure 3B) revealed that MFF and ganglioside-induced differentiation-associated protein 1 (GDAP1) play important roles in mitochondrial fission (Gandre-Babbe and van der Bliek, 2008; Niemann et al., 2005), and also serve as STAT6-binding candidates in OMM. MFF, is a C-tail-anchored protein in OMM, recruits dynamin-related protein 1(Drp1) to the mitochondrial surface (Otera et al., 2010). Recruited DRP1, due to its ability to homo-oligomerize, forms spiral structure around the mitochondrial tubules, and induces mitochondrial fission (Bui and Shaw, 2013). GDAP1, is a single-pass transmembrane protein anchored by its C-tail to OMM (Wagner et al., 2009), also contributes to mitochondrial fragmentation. Therefore, does STAT6 causes excessive mitochondrial fission through interaction with MFF or GDAP1 in OMM? Although our IP data also demonstrated that MFF interacted with STAT6, any STAT6 mutants failed to block or trigger DRP-1 recruitment (data not shown). Underlying mechanisms for how STAT6-MFF interaction affects mitochondrial fission, and whether STAT6 binds to GDAP1 in OMM should be investigated.
Mitochondria are not only the power houses for energy production, they also regulate many aspects of cellular processes (McBride et al., 2006). Mitochondria are regulated by stringent nuclear control (anterograde regulation) that can promote mitochondrial biogenesis. Conversely, mitochondria also generate and transduce signals to the nucleus (retrograde response) to modify cellular function (Xia et al., 2019). These anterograde and retrograde signals are referred to as mitonuclear communication (Quirós et al., 2016), and contribute to maintaining cellular homeostasis under various stress responses including hypoxia (Guha and Avadhani, 2013). Hypoxia, as one of the fundamentally important features of solid tumors (Ruan et al., 2009), triggers mitochondrial ROS production, membrane potential (Δψm) loss, and dysfunction (Chandel et al., 1997; Hamanaka and Chandel, 2009; Solaini et al., 2010; Weinberg et al., 2000; Yang et al., 2006). We observed that hypoxia induced STAT6 accumulation in mitochondria and triggered STAT6-MFN2 interaction (Figures 6A and 6B). Consequently, mitochondrial STAT6 accumulation in hypoxic conditions aggravated mitochondrial fragmentation, cytochrome c release, cell death in vitro (Figures 6C–6E), and inhibited tumorigenesis in vivo (Figure 7), supporting retrograde signaling (Figure S6) and physiological roles derived from mitochondrial STAT6 under hypoxia.
These findings collectively show that mitonuclear communication regulated by STAT6 exerts influence on diverse cellular and physiological events. However, physiological roles/functions of STAT6 OMM protein in different cells and tissues exposed to various conditions other than hypoxia, such as disease models, need to be investigated.
Because abnormal mitochondrial quality control alters mitochondrial metabolism and causes a number of symptoms, we recommend for further additional studies to investigate potential roles of yet to be characterized TFs in influencing mitochondrial quality control systems. This is an important step toward understanding the underlying pathophysiology of mitochondrial diseases.
Limitations of the study
We propose a role for STAT6 in regulation of mitochondrial fusion/fission via inhibition of MFN2 dimerization. Although we show that STAT6 in OMM inhibits mitochondrial fusion through direct interaction with MFN2, we cannot fully exclude the possibilities that STAT6 induces mitochondrial fission through other STAT6-interacting proteins (such as MFF and GDAP1) identified from our proteomic analysis.
Mitochondrial proteins, similar to nuclear and cytoplasmic proteins, can also undergo diverse post-translational modifications (PTMs), leading to altered mitochondrial function (Stram and Payne, 2016). In this context, it is required to identify the PTMs of mitochondrial STAT6 and establish the underlying mechanism of the STAT6 PTMs in mitochondrial function.
Although our study findings reveal that mitochondrial STAT6 is involved in tumorigenesis inhibition, its physiological roles in various disease models remain to be determined.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| STAT6 rabbit monoclonal (C-terminal -targeted) | Abcam | Cat#ab32520; RRID:AB_778113 |
| LAMP1 rabbit polyclonal | Abcam | Cat#ab24170; RRID:AB_775978 |
| BiP rabbit polyclonal | Abcam | Cat#ab21685; RRID:AB_2119834 |
| Catalase rabbit polyclonal | Abcam | Cat#ab52477; RRID:AB_868694 |
| HSP90AB1 mouse monoclonal | Abcam | Cat#ab53497; RRID:AB_881097 |
| Phospho-PKC alpha (S657) rabbit monoclonal | Abcam | Cat#ab180848; RRID:AB_2783796 |
| Myc rabbit monoclonal | Cell signaling | Cat#2278; RRID:AB_490778 |
| Myc mouse monoclonal | Cell signaling | Cat#2276; RRID:AB_331783 |
| FLAG rabbit monoclonal | Cell signaling | Cat#14793; RRID:AB_2572291 |
| FLAG mouse monoclonal | Cell signaling | Cat#8146; RRID:AB_10950495 |
| STAT1 rabbit polyclonal | Cell signaling | Cat#9172; RRID:AB_2198300 |
| STAT2 rabbit polyclonal | Cell signaling | Cat#4594; RRID:AB_2271323 |
| STAT3 rabbit monoclonal | Cell signaling | Cat#12640; RRID:AB_2629499 |
| STAT5 rabbit monoclonal | Cell signaling | Cat#94205; RRID:AB_2737403 |
| STAT6 rabbit monoclonal (SH2 -targeted) | Cell signaling | Cat#5397; RRID:AB_11220421 |
| MFN2 rabbit monoclonal | Cell signaling | Cat#11925; RRID:AB_2750893 |
| COXⅣ rabbit monoclonal | Cell signaling | Cat#4850; RRID:AB_2085424 |
| Phospho-JNK rabbit monoclonal | Cell signaling | Cat#4668; RRID:AB_823588 |
| JNK rabbit monoclonal | Cell signaling | Cat#9258; RRID:AB_2141027 |
| Phospho-ERK rabbit monoclonal | Cell signaling | Cat#4370; RRID:AB_2315112 |
| ERK rabbit monoclonal | Cell signaling | Cat#4695; RRID:AB_390779 |
| Phospho-p38 rabbit monoclonal | Cell signaling | Cat#4511; RRID:AB_2139682 |
| P38 rabbit polyclonal | Cell signaling | Cat#9212; RRID:AB_330713 |
| Phospho-CREB rabbit monoclonal | Cell signaling | Cat#9198: RRID:AB_2561044 |
| Phospho-NFkB rabbit monoclonal | Cell signaling | Cat#3033; RRID:AB_331284 |
| PKC alpha rabbit polyclonal | Cell signaling | Cat#2056; RRID:AB_2284227 |
| STAT6 rabbit polyclonal (280-480 a.a.-targeted) | SantaCruz | Cat#sc-1698; RRID:AB_2197235 |
| TOM20 rabbit polyclonal | SantaCruz | Cat#sc-11415; RRID:AB_2207533 |
| TOM40 rabbit polyclonal | SantaCruz | Cat#sc-11414; RRID:AB_793274 |
| Aconitase 2 rabbit polyclonal | SantaCruz | Cat#sc-130677; RRID:AB_2221514 |
| GAPDH goat polyclonal | SantaCruz | Cat#sc-48167; RRID:AB_1563046 |
| Alpha-tubulin mouse monoclonal | SantaCruz | Cat#sc-5286; RRID:AB_628411 |
| Actin mouse monoclonal | SantaCruz | Cat#sc-47778; RRID:AB_626632 |
| Cytochrome c mouse monoclonal | SantaCruz | Cat#sc-13156; RRID:AB_627385 |
| TIM23 mouse monoclonal | SantaCruz | Cat#sc-514463; RRID:AB_2923126 |
| Calnexin mouse monoclonal | SantaCruz | Cat#sc-46669; RRID:AB_626784 |
| Alexa Fluor Plus 488, anti-Mouse | Invitrogen | Cat#A32766; RRID:AB_2762823 |
| Alexa Fluor Plus 488, anti-Rabbit | Invitrogen | Cat#A32790; RRID:AB_2762833 |
| Alexa Fluor 546, anti-Rabbit | Invitrogen | Cat#A10040; RRID:AB_2534016 |
| Bacterial and virus strains | ||
| E.Coli. (DH5alpha) | RBC | Cat#RH617 |
| E.Coli. (Stbl3) | invitrogen | Cat#C737303 |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | WELGENE | Cat#LM 001-05 |
| Trypsin-EDTA | WELGENE | Cat#LS 015-10 |
| Antibiotic-antimycotic solution | Gibco | Cat#15240-062 |
| PBS | Gibco | Cat#21600-010 |
| FBS, premium | GenDEPOT | Cat#F0600-050 |
| Protease inhibitor cocktail | GenDEPOT | Cat#P3100-005 |
| G418 | Sigma-Aldrich | Cat#A1720 |
| Puromycin | Sigma-Aldrich | Cat#P8833 |
| Polyethylene glycol (PEG) | Sigma-Aldrich | Cat#P7181 |
| Cycloheximide solution | Sigma-Aldrich | Cat#C4859 |
| Paraformaldehyde | Sigma-Aldrich | Cat#P6148 |
| Saponin | Sigma-Aldrich | Cat#47036 |
| PMSF | Sigma-Aldrich | Cat#10837091001 |
| TCA | Sigma-Aldrich | Cat#T8657 |
| Percoll | Sigma-Aldrich | Cat#P1644 |
| Duolink-PLA | Sigma-Aldrich | Cat#DUO92007 |
| Crystal violet solution | Sigma-Aldrich | Cat#V5265 |
| Nitro blue tetrazolium | Sigma-Aldrich | Cat#N5514 |
| Agarose, low-gelling temperature | Sigma-Aldrich | Cat#A9045 |
| Lipofectamine3000 | Invitrogen | Cat#L3000015 |
| TMRM | Invitrogen | Cat#T668 |
| Protein G Dynabeads | Invitrogen | Cat#10003D |
| mitochondria-GFP, BacMam 2.0 | Invitrogen | Cat#C10508 |
| mitochondria-RFP, BacMam 2.0 | Invitrogen | Cat#C10505 |
| DSP | Thermo Scientific | Cat#A35393 |
| Proteinase K | BIONEER | Cat#KB-0111 |
| Mounting medium (with DAPI) | Vector Laboratories | Cat#H-1200 |
| 6nm colloidal gold particle, anti-Rabbit | Jackson ImmunoResearch | Cat#711-195-152 |
| Critical commercial assays | ||
| ATP assay | Abcam | Cat#ab83355 |
| OCR assay | Cayman | Cat#600800 |
| Deposited data | ||
| Raw and analyzed data | This paper | Mendeley Data: https://doi.org/10.17632/44fxxng896.6 |
| Experimental models: Cell lines | ||
| 293T | ATCC | Cat#CRL-3216; RRID:CVCL_0063 |
| HeLa | Korean Cell Line Bank | Cat#10002; RRID:CVCL_0030 |
| B16-F10 | Korean Cell Line Bank | Cat#80008; RRID:CVCL_0159 |
| Hep3B | Korean Cell Line Bank | Cat#88064; RRID:CVCL_0326 |
| HEK293 | Korean Cell Line Bank | Cat#21573; RRID:CVCL_0045 |
| T98-MG | Korean Cell Line Bank | Cat#21690; RRID:CVCL_0556 |
| U373-MG | Korean Cell Line Bank | Cat#30017; RRID:CVCL_2219 |
| U87-MG | Korean Cell Line Bank | Cat#30014; RRID:CVCL_0022 |
| STAT6 WT –U373-MG | This paper | N/A |
| Δ 1-260 –U373-MG | This paper | N/A |
| Δ DBD –U373-MG | This paper | N/A |
| Δ LD –U373-MG | This paper | N/A |
| Δ SH2 –U373-MG | This paper | N/A |
| Δ TAD –U373-MG | This paper | N/A |
| Δ 1-50 –U373-MG | This paper | N/A |
| Δ 674-847 –U373-MG | This paper | N/A |
| Δ 699-847 –U373-MG | This paper | N/A |
| Δ 774-847 –U373-MG | This paper | N/A |
| MFN2-Myc/MFN2-Flag –293T | This paper | N/A |
| STAT6 KO –B16-F10 | This paper | N/A |
| STAT6 KO –293T | This paper | N/A |
| STAT6 WT-Myc –293T (generated from STAT6 KO –293T) | This paper | N/A |
| Δ SH2-Myc –293T (generated from STAT6 KO –293T) | This paper | N/A |
| Δ MTS-Myc –293T (generated from STAT6 KO –293T) | This paper | N/A |
| Experimental models: Organisms/strains | ||
| C57BL/6J | OrientBio | N/A |
| BALB/c nude (CAnN.Cg-Foxn1nu/ CrljOri) | OrientBio | N/A |
| Oligonucleotides | ||
| See Table S2 | See Table S2 | |
| Recombinant DNA | ||
| pCMV6-Entry (Myc-DDK-tagged) | ORIGENE | Cat#PS100001 |
| STAT6 WT (Myc-DDK-tagged) | (Park et al., 2019) | N/A |
| Δ 1-260 (Myc-DDK-tagged) | (Park et al., 2019) | N/A |
| Δ DBD (Myc-DDK-tagged) | (Park et al., 2019) | N/A |
| Δ LD (Myc-DDK-tagged) | (Park et al., 2019) | N/A |
| Δ SH2 (Myc-DDK-tagged) | (Park et al., 2019) | N/A |
| Δ TAD (Myc-DDK-tagged) | (Park et al., 2019) | N/A |
| Δ 1-50 (Myc-DDK-tagged) | This paper | N/A |
| Δ 674-847 –U373-MG | This paper | N/A |
| Δ 699-847 (Myc-DDK-tagged) | This paper | N/A |
| Δ 774-847 (Myc-DDK-tagged) | This paper | N/A |
| Δ 31-260 (Myc-DDK-tagged) | This paper | N/A |
| pLenti-C-mGFP | ORIGENE | Cat#PS100071 |
| pLenti-STAT6 WT-mGFP | ORIGENE | Cat#RC210065L2V |
| pLenti-Δ SH2-mGFP | This paper | N/A |
| pLenti-Δ MTS-mGFP | This paper | N/A |
| pCMV6-AC-GFP | ORIGENE | Cat#PS100010 |
| 31-50 (STAT6 N-terminal fragment)-GFP | This paper | N/A |
| 1-30 (STAT6 N-terminal fragment)-GFP | This paper | N/A |
| 1-20 (STAT6 N-terminal fragment)-GFP | This paper | N/A |
| 16-25 (STAT6 N-terminal fragment)-GFP | This paper | N/A |
| 16-30 (STAT6 N-terminal fragment)-GFP | This paper | N/A |
| 21-30 (STAT6 N-terminal fragment)-GFP | This paper | N/A |
| 11-30 (STAT6 N-terminal fragment)-GFP | This paper | N/A |
| pCMV6-AN-GFP | ORIGENE | Cat#PS100019 |
| GFP-1-30 | This paper | N/A |
| MFN2 WT (Myc-DDK-tagged) | ORIGENE | Cat#RC226143 |
| Δ HR1 (Myc-DDK-tagged) | This paper | N/A |
| Δ PR (Myc-DDK-tagged) | This paper | N/A |
| Δ HR2 (Myc-DDK-tagged) | This paper | N/A |
| pLenti-C-Myc-DDK-IRES-Puro | ORIGENE | Cat#PS100069 |
| pLenti-MFN2-Myc (-IRES-Puro) | This paper | N/A |
| pLenti-MFN2-Flag (-IRES-Puro) | This paper | N/A |
| pLenti-STAT6 WT (C-Myc-DDK-IRES-Puro) | This paper | N/A |
| pLenti-Δ SH2 (C-Myc-DDK-IRES-Puro) | This paper | N/A |
| pLenti-Δ MTS (C-Myc-DDK-IRES-Puro) | This paper | N/A |
| pLenti-C-mCherry | This paper | N/A |
| pLenti-STAT6 WT-mCherry | This paper | N/A |
| pLenti-Δ SH2- mCherry | This paper | N/A |
| pLenti-Δ MTS- mCherry | This paper | N/A |
| Mouse STAT6 CRISPR/Cas9 KO Plasmid | Santa Cruz | Cat#sc-423180 |
| Human STAT6 CRISPR/Cas9 KO Plasmid | Santa Cruz | Cat#sc-418159 |
| Software and algorithms | ||
| Image J | National Institutes of Health, USA | https://imagej.nih.gov/ij/ |
| DAVID | The Laboratory of Immunopathogenesis and Bioinformatics | https://david.ncifcrf.gov/home.jsp |
| Prism 9.0 | GraphPad | N/A |
| SPSS 25.0 | IBM | N/A |
| membrane protein explorer (MPEx) | The Stephen White laboratory at UC Irvine | https://blanco.biomol.uci.edu/mpex/ |
Resource availability
Lead contact
Further information and requests for resources and reagents may be directed to, and will be fulfilled by the lead contact, Ilo Jou (ilojou@aumc.ac.kr).
Materials availability
Plasmids and stable cell lines generated in this study are available from the authors in accordance with the Material Transfer Agreement.
Experimental model and subject details
Cell culture
Primary mouse astrocytes were cultured from the cerebral cortices of 1-day-old C57BL/6J mice (KOATECH, Korea). Cell suspensions were prepared by mechanical digestion of the cortices in DMEM (Sigma, St. Louis, MO, USA) containing 10% FBS (GenDepot, Barker, TX, USA) and 0.1% antibiotic-antimycotic solution (Gibco/Life Technologies, Grand Island, NY, USA). The cells were dispensed to 75-cm2 T-flasks and cultured for 2 weeks. For astrocyte cultures, the flasks were shaken at 250 rpm for 18 h at 37 °C to separate the microglia and oligodendrocytes from astrocytes. The cells were washed with PBS, trypsinized, and cultured in DMEM containing 10% FBS. U373-malignant glioma (U373-MG), B16-F10, and 293T were grown in DMEM containing 10% FBS.
Plasmids and stable cell lines
The human wild type (WT) STAT6, 1–260 residues-deleted (Δ 1–260), DNA binding domain-deleted (ΔDBD), linker domain-deleted (ΔLD), SH2 domain-deleted (ΔSH2), and transactivation domain-deleted STAT6 (ΔTAD) were a kind gift from Soo Jung Park (Ajou University, Korea). The 1–50 residues-deleted (Δ1–50), 31–260 residues-deleted (Δ31–260), 674–847 residues-deleted (Δ674–847), 699–847 residues-deleted (Δ699–847), and 774–847 residues-deleted (Δ774–847) STAT6 were generated from WT STAT6 plasmid. These plasmids were transfected to 293T to confirm MFN2-STAT6 interaction and transfected to human U373-MG to generate stable cell lines. The stable cell lines were maintained in 1 mg/mL of G418 and used for analyzing mitochondrial localization.
The human WT STAT6, ΔSH2, ΔMTS (mitochondria targeting sequences-deleted STAT6), and STAT6 n-terminal fragments (31–50, 1–30, 1–20, 16–25, 16–30, 21–30, and 11–30 residues) were cloned into pCMV6-AC-GFP vector (OriGene, Rockville, USA) and 1–30 fragment was cloned into pCMV6-AN-GFP vector (OriGene). These plasmids were transfected to human U373-MG, 293T, HEK 293, and mouse B16-F10 melanoma.
The human WT MFN2 plasmid (Cat# RC226143) was purchased from OriGene. The Heptad region 1-deleted (ΔHR1), Proline rich-deleted (ΔPR), and Heptad region 2-deleted (ΔHR2) MFN2 plasmids were generated from human WT MFN2 plasmid. These plasmids were transfected to 293T to confirm MFN2-STAT6 interaction.
The MFN2-Myc and MFN2-Flag (MFN2-DDK) were generated from human WT MFN2 plasmid (Cat# RC226143), cloned into pLenti-IRES-Puro vector (OriGene) and co-transfected to 293T to generate stable cell line. The cell lines were maintained in 2 μg/mL of puromycin. This stable cell line was used for analyzing MFN2 dimerization.
The human WT STAT6, ΔSH2, and ΔMTS were cloned to mCherry plasmid. These plasmids were transfected to 293T to analyze mitochondrial mass and morphology.
The human and mouse STAT6 CRISPR/Cas9 plasmids (Santa cruz) were used to generate STAT6 KO-293T and STAT6 KO-B16F10 stable cell lines.
The human WT STAT6, ΔSH2, and ΔMTS were cloned to pLenti-C-Myc-DDK-IRES-Puro vector (OriGene). These plasmids were transfected to STAT6 KO-293T to generated stable cell lines. The cell lines were maintained in 2 μg/mL of puromycin and were used for analyzing PEG-mediated mitochondrial fusion and tumorigenesis.
The oligonucleotides used in generation of plasmids mentioned above, were described in Table S2.
Tumor models
All animal experiments were approved by the Ajou University Medical Center - Institutional Animal Care and Use Committee (Approval number: 2019–0048). Five-week-old, female BALB/c nude mice (CAnN.Cg-Foxn1nu/CrljOri) were purchased from OrientBio. For establishment of solid tumor models, the mice (9-week-old) were subcutaneously inoculated with 2 × 106 cells of 293T-stable cell lines mentioned above. After 4 weeks, the mice were sacrificed and analyzed tumor size. Tumor growth was monitored by digital caliper twice a week. Tumor volume was calculated using a standard solid tumor formula. Volume = (length × width2 × 0.52)
Method details
Hypoxia
The cells were seed on dishes and incubated overnight at 37°C in a humidified atmosphere of 95% air and 5% CO2. For hypoxia treatment, cells were incubated for indicated times in an oxygen control hypoxia chamber (Coy Laboratory Products, Grass Lake, MI, USA) at 37°C in a humidified 5% CO2 environment, with the balance provided by N2.
Flow cytometry
The cells were trypsinized, stained with TMRM (200 nM) and annexin V-PI for indicated time at 37 °C, and analyzed by flow cytometry using a FACS DIVA flow cytometer (BD Biosciences, San Jose, CA, USA) at the Three-Dimensional Immune System Imaging Core Facility of Ajou University.
Mitochondrial fusion by PEG
The stable cell lines were transfected with mitochondria-GFP, BacMam 2.0 (mitoGFP) or mitochondria-RFP, BacMam 2.0 (mitoRFP) (ThermoFisher), mixed, and co-cultured overnight on glass coverslips. Cycloheximide (20 μg/mL, Sigma) was added in DMEM without serum 30 min before cell fusion. PEG-mediated cell fusion was performed by treatment with pre-warmed PEG 1500 (Sigma) solution for 90s and washed three times with DMEM containing 10% serum for 10 min per wash, followed by incubation in DMEM containing 10% serum and cycloheximide for 3h. The cells on glass coverslips were fixed and analyzed by confocal microscopy.
Immunocytochemistry and confocal microscopy
The cells were cultured on coverslips were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) at 25 °C for 10 min and permeabilized by incubating in PBS containing 0.1% Saponin for 10 min before blocking for 1 h with PBS containing 1% BSA and 0.1% Saponin. All primary antibodies were incubated overnight for staining at 4 °C in same blocking solution. Immunoreactive proteins were visualized by incubation with Alexa Fluor 488- or Alexa Fluor 546-conjugated secondary antibodies (Invirtogen). Cells were mounted with DAPI-containing mounting solution and observed under a confocal microscope (TCS, DMi8; Leica, Germany). The colocalization (Pearson’s Correlation Coefficient) was analyzed by Las X software (Leica). The number and mean area of mitochondria were calculated by Image J software 1.42.
Immuno-gold EM
The cells were collected in an Eppendorf tube, fixed using 4% paraformaldehyde for 1h, permeabilized, and incubated with myc or TOM20 antibody. The immunoreactivity was visualized using Protein A- 6nm gold particles. The cells were collected as a pellet, embedded, and performed according to the manufacturer’s instructions.
Transmission electron microscopy
The cells were fixed, washed with cacodylate buffer, dehydrated in a series of graded ethanol, and then embedded in Epon mixture. Ultrathin sections were cut using a Reichert Jung Ultracut S (Leica), mounted on copper grids, stained with uranyl acetate and lead citrate, and analyzed under Zeiss Sigma 500 electron microscope.
Protein extraction and mitochondrial isolation from cells
For whole cell extracts preparation, the cells were lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5% NP-40, 1 mM DTT, 1 mM PMSF, protease inhibitor cocktail, phosphatase inhibitor cocktail, 5 mM NaF, 1 mM sodium vanadate, 1 mM EDTA). For mitochondrial isolation, the cells were harvested and homogenized in mitochondrial isolation buffer (225 mM mannitol, 75 mM sucrose, 0.1 mM EGTA, 30 mM Tris-HCl pH 7.4) using a Dounce tissue grinder (Wheaton, Millville, NJ, USA). Nuclei and debris were removed by centrifuging the homogenate twice at 600 ×g for 10 min at 4 °C, after which the collected supernatant was centrifuged at 8,000 ×g for 10 min at 4 °C. The resulting pellet was washed twice with mitochondrial isolation buffer and centrifuged at 15,000 ×g for 15 min at 4 °C. The pellet was collected as the crude mitochondrial fraction.
Proteinase K (PK) protection assay
The isolated mitochondrial fraction was treated with Proteinase K on ice for 10 min. The reaction was terminated by adding 10 mM phenylmethylsulfonyl fluoride (PMSF) on ice for 10 min and centrifuged at 15,000 ×g for 15 min at 4 °C. The pellet was lysed in RIPA buffer and subjected to western blotting using indicated antibodies.
Sodium carbonate and high salt extraction
The isolated mitochondrial pellets were resuspended in 0.1–0.2M Na2CO3 (pH 9.5–12.5) or 0–2 M NaCl in 50 mM Tris, pH 7.4 respectively followed by 10 min incubation on ice prior to centrifugation at 100,000 ×g. All protein samples were TCA precipitated and resuspended into loading dye for electrophoresis and western blotting.
Subcellular fractionation from brain tissue
Brain tissues were homogenized in mitochondrial isolation buffer using a Dounce tissue grinder. Nuclei and debris were removed by centrifuging the homogenate twice at 600 ×g for 10 min at 4 °C, after which the collected supernatant was centrifuged at 8000 ×g for 10 min at 4 °C. The resulting pellet was collected as the crude mitochondrial fraction. The supernatant was centrifuged at 20,000 ×g for 1 h, then again at 100,000 ×g for 1 h, after which the resulting pellet was resuspended as the ER fraction. The supernatant was kept as the cytosolic fraction. For pure mitochondria and MAM fractions, the crude mitochondrial pellet was resuspended in 2 mL mitochondrial resuspension buffer (MRB; 250 mM mannitol, 5 mM HEPES pH 7.4, 0.5 mM EGTA), layered over 30% Percoll medium in a centrifuge tube, and centrifuged at 95,000×g for 30 min at 4 °C. The lower layer (pure mitochondria) and intermediate layer (MAM) between the light membrane and pure mitochondria fractions were then collected. The pure mitochondria fraction was diluted in MRB buffer and further centrifuged at 10,000 ×g, after which the pellet was resuspended in 300 μL of MRB buffer. The MAM fraction was diluted 10× in MRB buffer and centrifuged at 100,000 ×g for 1 h at 4 °C, after which the pellet was resuspended in a small volume of MRB buffer.
Submitochondrial fractionation from brain tissue
Mitochondria obtained from brain tissue were resuspended in 450 μL of hypotonic buffer (5 mm Tris-HCl and 1 mm EDTA, pH 7.4) and incubated on ice for 15 min to generate mitoplasts. The solution was centrifuged at 20,000 ×g for 10 min at 4°C to pellet mitoplasts. Then the supernatant was centrifuged for 2 h at 100,000 ×g at 4 °C to separate the OMM-enriched fraction. The mitoplasts were resuspended in 450 μL of hypotonic buffer and sonicated for 2 min (30 s off and 30 s on at 150 watts, Branson Sonifer) on ice. The solution was then spun at 100,000 ×g for 2 h. The resultant pellet contains the IMM-enriched fraction, whereas the supernatant contains matrix-enriched fraction.
Sucrose density-gradient centrifugation of crude mitochondrial fraction
The crude mitochondrial pellet was washed with SEM buffer (250 mM sucrose, 1 mM EDTA, 10 mM Mops, pH 7.2) and centrifuged at 15,000 ×g for 15 min at 4 °C. The resulting pellet was resuspended in SEM buffer and subsequently loaded onto a three-step sucrose gradient [1.5 mL of 60%, 4 mL of 32%, 1.5 mL of 23%,1.5 mL of 15% (w/v) sucrose in EM buffer (10 mM MOPs, pH 7.2, 1 mM EDTA)]. After centrifugation at 134,000 ×g in a SW41 rotor (Beckman Instruments, Fullerton, CA) for 1 h at 4 °C. From the top of each gradient, 0.65-mL fractions were collected, yielding a total of 14 fractions. The each collected fractions were diluted by 2 volumes SEM buffer and pelleted at 15,000 ×g for 15 min at 4 °C. The fractions were separated by SDS-PAGE and Western blotting.
Mass spectrometry
The mouse brain tissues were homogenized with PBS and added chemical crosslinking agent DSP to a final concentration of 1 mM for 30 min and subsequent quenching with 50 mM Tris-HCl, pH 7.5 for 15 min on ice. The cells were added with SDS to a final concentration of 1%, boiled at 95°C for 5 min, and centrifuged at 15,000 ×g for 15 min at 4 °C. The brain lysates were mixed with indicated antibodies and incubated at 4 °C on a rocker overnight. Immune complexes were collected with magnetic protein G Dynabeads. Immunoprecipitated proteins were digested with trypsin and quantified on a nanoLC-MS/MS platform. The raw MS files were analyzed and searched against mouse protein database based on the species of the samples using Maxquant (1.6.2.6). The parameters were set as follows: the enzyme specificity was set to trypsin; the maximum missed cleavages were set to 2; the precursor ion mass tolerance was set to 10 ppm, and MS/MS tolerance was 0.5 Da. Gene Ontology (GO) and Functional Annotation were analyzed by DAVID. The information of detailed proteins, identified, and quantified peptides was listed in Table S2.
Immunoprecipitation (IP)
Lysates were mixed with indicated antibodies and incubated at 4 °C on a rocker overnight. Immune complexes were collected with magnetic protein G Dynabeads. After washing five times with lysis buffer, proteins were eluted from beads with 2X Laemmli buffer at 95°C for 10 min, followed by resolution by SDS-PAGE.
Western blotting
Cells were lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5% NP-40, 1 mM DTT, 1 mM PMSF, protease inhibitor cocktail, phosphatase inhibitor cocktail, 5 mM NaF, 1 mM sodium vanadate, and 1 mM EDTA). Protein concentration in lysates was determined using the Bio-Rad protein assay (Bio-Rad) according to the manufacturer’s instructions. Proteins (10–20 μg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 10% polyacrylamide gels and transferred to a nitrocellulose membrane. Membranes were probed using the indicated primary and secondary antibodies and developed using an enhanced chemiluminescence detection kit (WESTSAVE Gold; AbFrontier, Seoul, Korea).
Proximity ligation assay (PLA staining)
Fixed cells were stained with the indicated rabbit and mouse antibodies. Duolink-PLA (Olink Bioscience, Sweden) procedure was performed according to the manufacturer’s instructions, each discrete red spot represents a protein-protein complex. The number of spot per cell was calculated by Las X software.
Quantification of ATP levels using glucose and galactose conditioned media
The cells were incubated with DMEM containing either glucose (25mM glucose; 1 mM pyruvate; 2 mM glutamine, and 10% FBS) or galactose (10mM galactose, 1 mM pyruvate; 6 mM glutamine 5 mM HEPES, and 10% FBS) overnight. ATP was measured using ATP assay kit (ab83355, Abcam, Cambridge, MA) following the manufacturer’s instructions. The plate was read at 535nm/587nm (Ex/Em) using Cytation 5 Cell Imaging Multi-Mode Reader (Biotek).
Oxygen consumption assay
Oxygen consumption assay was performed using Oxygen Consumption Rate Assay Kit (Cayman, 600800) following manufacturer’s protocol.
Colony formation assay
For colony formation assay, the cells were seed on 35 mm dishes at a density of 1 × 103/dish and incubated at 37°C with 5% CO2 before counting. The cells on dishes were then fixed and stained with 0.1% crystal violet in 20% methanol for 20 min. The number of colony was calculated by Image J software.
Soft agar colony formation assay
For soft agar colony formation, 1% melted agarose was mixed equally with 2X DMEM containing 20% FBS, 2% antibiotics, and was coated into 35mm dishes, which was then incubated at room temperature for 30 min to allow the bottom layer to solidify. The cells (1 × 104 cells) were added to mixture of 0.7% melted agarose and 2X DMEM containing 20% FBS, and then placed on the bottom layer. The cells were feed 2 times per week with DMEM containing 10% FBS, and were incubated at 37°C with 5% CO2 for 3 weeks before counting. The cells on dishes were incubated with Nitro Blue Tetrazolium (NBT) dye overnight at 37°C.
The stained cells were analyzed using montage-stitching option by Cytation 5 Cell Imaging Multi-Mode Reader (Biotek)
Quantification and statistical analysis
Statistical analyses were conducted using the SPSS 25.0 software and GraphPad Prism 9.0 software (GraphPad Software, Inc., La Jolla, CA, USA). Statistical significance was determined using the Student’s t-test, one-way ANOVA with Scheffe’s post-hoc test, or two-way repeated measures ANOVA with Tukey’s post-hoc test. Results are presented as the means, standard deviation (S.D.) or SE of the mean (SEM) of at least three independent experiments. Values are statistically significant at ∗p < 0.05, ∗p > 0.01, ∗∗∗p > 0.001, and ∗∗∗∗p > 0.0001.
Acknowledgments
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2018R1D1A1B07041750), and Korea Initiative for fostering University of Research and Innovation Program of the NRF funded by the MSIT (No. NRF2021M3H1A104892211). This research was also supported by Korea Basic Science Institute (National research Facilities and Equipment Center) grant funded by the Ministry of Education (grant No. 2019R1A6C1010003).
Author contributions
H.K. and I.J. contributed to the conception and experimental design of the study. H.K. performed the biological experiments, analyzed the data, and prepared the figures and table. H.K. and I.J. wrote and edited the manuscript. S.J.P. advised on the experiment about cell death.
Declaration of interests
The authors declare no competing interests.
Published: September 16, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104923.
Supplemental information
Data and code availability
-
•
The raw data have been deposited at Mendeley Data and are publicly available as of the date of publication. DOI is listed in key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ahting U., Waizenegger T., Neupert W., Rapaport D. Signal-anchored proteins follow a unique insertion pathway into the outer membrane of mitochondria. J. Biol. Chem. 2005;280:48–53. doi: 10.1074/jbc.M410905200. [DOI] [PubMed] [Google Scholar]
- Bock F.J., Tait S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020;21:85–100. doi: 10.1038/s41580-019-0173-8. [DOI] [PubMed] [Google Scholar]
- Bourke L.T., Knight R.A., Latchman D.S., Stephanou A., McCormick J. Signal transducer and activator of transcription-1 localizes to the mitochondria and modulates mitophagy. JAK-STAT. 2013;2:e25666. doi: 10.4161/jkst.25666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks C., Cho S.G., Wang C.Y., Yang T., Dong Z. Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Am. J. Physiol. Cell Physiol. 2011;300:C447–C455. doi: 10.1152/ajpcell.00402.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui H.T., Shaw J.M. Dynamin assembly strategies and adaptor proteins in mitochondrial fission. Curr. Biol. 2013;23:R891–R899. doi: 10.1016/j.cub.2013.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel N.S., Budinger G.R., Choe S.H., Schumacker P.T. Cellular respiration during hypoxia. Role of cytochrome oxidase as the oxygen sensor in hepatocytes. J. Biol. Chem. 1997;272:18808–18816. doi: 10.1074/jbc.272.30.18808. [DOI] [PubMed] [Google Scholar]
- Chueh F.Y., Leong K.F., Yu C.L. Mitochondrial translocation of signal transducer and activator of transcription 5 (STAT5) in leukemic T cells and cytokine-stimulated cells. Biochem. Biophys. Res. Commun. 2010;402:778–783. doi: 10.1016/j.bbrc.2010.10.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Din S., Mason M., Völkers M., Johnson B., Cottage C.T., Wang Z., Joyo A.Y., Quijada P., Erhardt P., Magnuson N.S., et al. Pim-1 preserves mitochondrial morphology by inhibiting dynamin-related protein 1 translocation. Proc. Natl. Acad. Sci. USA. 2013;110:5969–5974. doi: 10.1073/pnas.1213294110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filadi R., Pendin D., Pizzo P. Mitofusin 2: from functions to disease. Cell Death Dis. 2018;9:330. doi: 10.1038/s41419-017-0023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki Y., Hubbard A.L., Fowler S., Lazarow P.B. Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J. Cell Biol. 1982;93:97–102. doi: 10.1083/jcb.93.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandre-Babbe S., van der Bliek A.M. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell. 2008;19:2402–2412. doi: 10.1091/mbc.e07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami R., Majumdar T., Dhar J., Chattopadhyay S., Bandyopadhyay S.K., Verbovetskaya V., Sen G.C., Barik S. Viral degradasome hijacks mitochondria to suppress innate immunity. Cell Res. 2013;23:1025–1042. doi: 10.1038/cr.2013.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough D.J., Corlett A., Schlessinger K., Wegrzyn J., Larner A.C., Levy D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–1716. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha M., Avadhani N.G. Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion. 2013;13:577–591. doi: 10.1016/j.mito.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales K.G., Fuller M.T. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell. 1997;90:121–129. doi: 10.1016/s0092-8674(00)80319-0. [DOI] [PubMed] [Google Scholar]
- Hamanaka R.B., Chandel N.S. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr. Opin. Cell Biol. 2009;21:894–899. doi: 10.1016/j.ceb.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N., Fujita Y., Oka T., Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966–2977. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jheng H.F., Tsai P.J., Guo S.M., Kuo L.H., Chang C.S., Su I.J., Chang C.R., Tsai Y.S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell Biol. 2012;32:309–319. doi: 10.1128/mcb.05603-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y.C., Son M., Kang S., Im S., Piao Y., Lim K.S., Song M.Y., Park K.S., Kim Y.H., Pak Y.K. Cell-penetrating artificial mitochondria-targeting peptide-conjugated metallothionein 1A alleviates mitochondrial damage in Parkinson's disease models. Exp. Mol. Med. 2018;50:1–13. doi: 10.1038/s12276-018-0124-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan R., Lee J.E., Yang Y.M., Liang F.X., Sehgal P.B. Live-cell imaging of the association of STAT6-GFP with mitochondria. PLoS One. 2013;8:e55426. doi: 10.1371/journal.pone.0055426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., Botelho S.C., Park K., Kim H. Use of carbonate extraction in analyzing moderately hydrophobic transmembrane proteins in the mitochondrial inner membrane. Protein Sci. 2015;24:2063–2069. doi: 10.1002/pro.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba T., Detmer S.A., Kaiser J.T., Chen H., McCaffery J.M., Chan D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–862. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- Lionaki E., Gkikas I., Tavernarakis N. Differential protein distribution between the nucleus and mitochondria: implications in aging. Front. Genet. 2016;7:162. doi: 10.3389/fgene.2016.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maycotte P., Marín-Hernández A., Goyri-Aguirre M., Anaya-Ruiz M., Reyes-Leyva J., Cortés-Hernández P. Mitochondrial dynamics and cancer. Tumour Biol. 2017;39 doi: 10.1177/1010428317698391. 1010428317698391. [DOI] [PubMed] [Google Scholar]
- McBride H.M., Neuspiel M., Wasiak S. Mitochondria: more than just a powerhouse. Curr. Biol. 2006;16:R551–R560. doi: 10.1016/j.cub.2006.06.054. [DOI] [PubMed] [Google Scholar]
- Meier J.A., Larner A.C. Toward a new STATe: the role of STATs in mitochondrial function. Semin. Immunol. 2014;26:20–28. doi: 10.1016/j.smim.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann A., Ruegg M., La Padula V., Schenone A., Suter U. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot–Marie–Tooth disease. J. Cell Biol. 2005;170:1067–1078. doi: 10.1083/jcb.200507087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto T., Schwab R.B., Scherer P.E., Lisanti M.P. Analysis of the association of proteins with membranes. Curr. Protoc. Cell Biol. 2001 doi: 10.1002/0471143030.cb0101s05. [DOI] [PubMed] [Google Scholar]
- Otera H., Wang C., Cleland M.M., Setoguchi K., Yokota S., Youle R.J., Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C.S., Osellame L.D., Laine D., Koutsopoulos O.S., Frazier A.E., Ryan M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011;12:565–573. doi: 10.1038/embor.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.J., Kim H., Kim S.H., Joe E.H., Jou I. Epigenetic downregulation of STAT6 increases HIF-1α expression via mTOR/S6K/S6, leading to enhanced hypoxic viability of glioma cells. Acta. Neuropathol. Commun. 2019;7:149. doi: 10.1186/s40478-019-0798-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirós P.M., Mottis A., Auwerx J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016;17:213–226. doi: 10.1038/nrm.2016.23. [DOI] [PubMed] [Google Scholar]
- Ruan K., Song G., Ouyang G. Role of hypoxia in the hallmarks of human cancer. J. Cell. Biochem. 2009;107:1053–1062. doi: 10.1002/jcb.22214. [DOI] [PubMed] [Google Scholar]
- Solaini G., Baracca A., Lenaz G., Sgarbi G. Hypoxia and mitochondrial oxidative metabolism. Biochim. Biophys. Acta. 2010;1797:1171–1177. doi: 10.1016/j.bbabio.2010.02.011. [DOI] [PubMed] [Google Scholar]
- Song J., Herrmann J.M., Becker T. Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell Biol. 2021;22:54–70. doi: 10.1038/s41580-020-00300-2. [DOI] [PubMed] [Google Scholar]
- Stram A.R., Payne R.M. Post-translational modifications in mitochondria: protein signaling in the powerhouse. Cell. Mol. Life Sci. 2016;73:4063–4073. doi: 10.1007/s00018-016-2280-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez-Rivero J.M., Villanueva-Paz M., de la Cruz-Ojeda P., de la Mata M., Cotán D., Oropesa-Ávila M., de Lavera I., Álvarez-Córdoba M., Luzón-Hidalgo R., Sánchez-Alcázar J.A. Mitochondrial dynamics in mitochondrial diseases. Diseases. 2016;5:E1. doi: 10.3390/diseases5010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepanek K., Chen Q., Derecka M., Salloum F.N., Zhang Q., Szelag M., Cichy J., Kukreja R.C., Dulak J., Lesnefsky E.J., Larner A.C. Mitochondrial-targeted Signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J. Biol. Chem. 2011;286:29610–29620. doi: 10.1074/jbc.M111.226209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner K.M., Rüegg M., Niemann A., Suter U. Targeting and function of the mitochondrial fission factor GDAP1 are dependent on its tail-anchor. PLoS One. 2009;4:e5160. doi: 10.1371/journal.pone.0005160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegrzyn J., Potla R., Chwae Y.J., Sepuri N.B.V., Zhang Q., Koeck T., Derecka M., Szczepanek K., Szelag M., Gornicka A., et al. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–797. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg J.M., Venkatachalam M.A., Roeser N.F., Nissim I. Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc. Natl. Acad. Sci. USA. 2000;97:2826–2831. doi: 10.1073/pnas.97.6.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia M., Zhang Y., Jin K., Lu Z., Zeng Z., Xiong W. Communication between mitochondria and other organelles: a brand-new perspective on mitochondria in cancer. Cell Biosci. 2019;9:27. doi: 10.1186/s13578-019-0289-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B., Fan L., Fang L., He Q. Hypoxia-mediated fenretinide (4-HPR) resistance in childhood acute lymphoblastic leukemia cells. Cancer Chemother. Pharmacol. 2006;58:540–546. doi: 10.1007/s00280-006-0197-6. [DOI] [PubMed] [Google Scholar]
- Zhang D., Liu Y., Tang Y., Wang X., Li Z., Li R., Ti Z., Gao W., Bai J., Lv Y. Increased mitochondrial fission is critical for hypoxia-induced pancreatic beta cell death. PLoS One. 2018;13 doi: 10.1371/journal.pone.0197266. e0197266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Raje V., Yakovlev V.A., Yacoub A., Szczepanek K., Meier J., Derecka M., Chen Q., Hu Y., Sisler J., et al. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J. Biol. Chem. 2013;288:31280–31288. doi: 10.1074/jbc.M113.505057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The raw data have been deposited at Mendeley Data and are publicly available as of the date of publication. DOI is listed in key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.