

Summary
Chronic environmental stress can profoundly impact cell and body function. Although the underlying mechanisms are poorly understood, epigenetics has emerged as a key link between environment and health. The genomic effects of stress are thought to be mediated by the action of glucocorticoid stress hormones, primarily cortisol in humans, which act via the glucocorticoid receptor (GR). To dissect how chronic stress-driven GR activation influences epigenetic and cell states, human fibroblasts underwent prolonged exposure to physiological stress levels of cortisol and/or a selective GR antagonist. Cortisol was found to drive robust changes in cell proliferation, migration, and morphology, which were abrogated by concomitant GR blockade. The GR-driven cell phenotypes were accompanied by widespread, yet genomic context-dependent, changes in DNA methylation and mRNA expression, including gene loci with known roles in cell proliferation and migration. These findings provide insights into how chronic stress-driven functional epigenomic patterns become established to shape key cell phenotypes.
Subject areas: Biological sciences, Endocrinology, Molecular biology, Transcriptomics
Graphical abstract
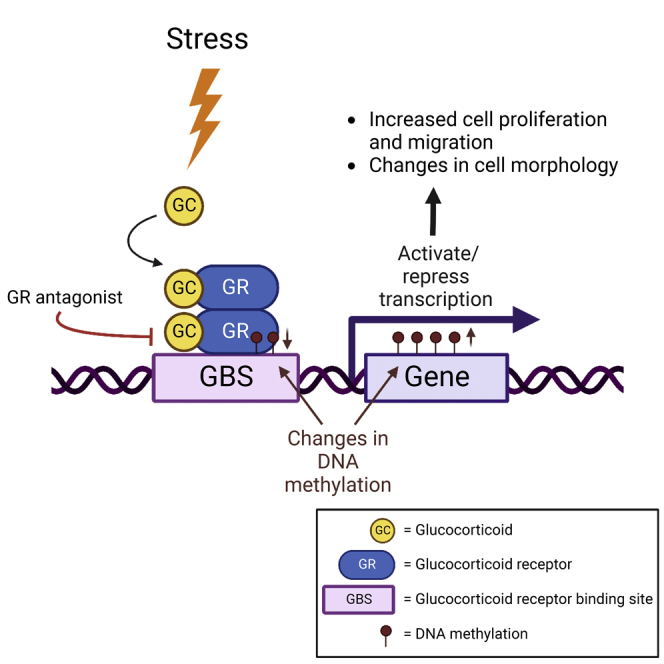
Highlights
-
•
Physiological stress levels of cortisol drive robust changes in key cell phenotypes
-
•
Stress-driven changes in cell phenotypes are abrogated by concomitant GR blockade
-
•
GR activation induces functional and phenotypically relevant epigenomic changes
Biological sciences; Endocrinology; Molecular biology; Transcriptomics
Introduction
Environmental stress is ubiquitous in modern societies and, especially when chronic or excessive, can profoundly impact cell and body function (Hong et al., 2020; Yang et al., 2020; Epel et al., 2004; Joëls et al., 2004). This impact can accumulate throughout human life and contribute to a range of diseases together responsible for 70% of all deaths (World Health Organization, 2017). Although the underlying molecular mechanisms are poorly understood, epigenetics—the chemical changes that regulate genomic function without altering the genetic code—has emerged as a key link between environment and health (Cavalli and Heard, 2019; D'Anna et al., 2020; Lam et al., 2012, National Library of Medicine, 2021; Kim et al., 2020; Nanavaty et al., 2020; Weaver et al., 2004; Zannas, 2019; Zannas et al., 2019). Studying the epigenetic sequelae of prolonged stress exposure can thus yield fundamental insights into determinants of health and disease. Given the complexity of examining stress in humans, such fundamental studies call for cell models with translational value. However, the precise knowledge of how chronic stress acts to establish epigenetic and cell states remains elusive.
Stress can result from a host of heterogeneous stimuli, including physical, mental, and social stressors. Despite their heterogeneity, these stressors share an ability to trigger conserved neuroendocrine responses that culminate in the systemic secretion of glucocorticoid stress hormones, primarily cortisol in humans (Chrousos and Gold, 1992). Systemic glucocorticoids, in turn, can influence genomic function in essentially every human cell by activating the glucocorticoid receptor (GR), a ligand-regulated transcription factor (Zannas and Chrousos, 2017). Importantly, glucocorticoid exposure and GR activation can induce not only acute changes in gene transcription but also lasting epigenetic changes, most notably in DNA methylation (Kress et al., 2006; Seifuddin et al., 2017; Thomassin et al., 2001; Wiechmann et al., 2019; Zannas et al., 2015, 2019; Provencal et al., 2019; Wiench et al., 2011; Zannas, 2021b), which involves the addition of a methyl group onto cytosine, usually in the CpG context. We, therefore, hypothesized that prolonged GR activation can drive functional DNA methylation changes at susceptible genomic sites with potential key roles in cell phenotypes.
To dissect the effects of chronic stress at the cellular and molecular levels, we leveraged a well-established human lung fibroblast cell line (IMR-90) as well as primary human dermal fibroblasts obtained from donors of different ancestry and sex. Fibroblasts are highly suitable for studying the long-term epigenetic sequelae of stress because these cells can be cultured easily for prolonged periods of time (≥2 months) and exhibit well-defined, malleable phenotypes, such as finite proliferative potential, migratory behavior, and distinct morphologies (Deschênes-Simard et al., 2013; Ramilowski et al., 2020b; Zannas et al., 2020). Moreover, fibroblasts are at the forefront of wound healing (Bainbridge, 2013) and are involved in a host of disease states, including fibrotic illness (Wynn and Ramalingam, 2012), cardiovascular disease (Hall et al., 2021), inflammatory states (Chang and Turley, 2015), and cancer (Kalluri, 2016). To model stress in culture, we treated fibroblasts with the endogenous human glucocorticoid cortisol at levels known to be reached in human tissues during in vivo stress (100 nM) (Wust et al., 2005; Bhake et al., 2013; Tarjanyi et al., 2014; Bendel et al., 2008; Hamrahian et al., 2004; Christ-Crain et al., 2007). To mechanistically dissect the role of GR activation, and as the finite proliferative potential of fibroblasts makes the generation of GR knockouts impractical, we further employed as a tool the selective GR modulator relacorilant at a previously established concentration (500 nM) shown to exert robust antagonistic properties (Hunt et al., 2017).
Our findings show that prolonged exposure to physiological stress levels of cortisol drives robust changes in fibroblast phenotypes, including proliferation, migration, and morphology, whereas concomitant GR blockade with relacorilant abrogates these changes. The GR-driven cell phenotypes are accompanied by widespread, yet genomic context-dependent, changes in DNA methylation and mRNA expression, including gene loci with known roles in cell proliferation and migration. Together these findings provide insights into how chronic stress-driven functional epigenomic patterns become established to shape key cell phenotypes.
Results
Toward a more physiological cell model for studying how chronic stress and glucocorticoid receptor activation impact epigenetic and cell states
Glucocorticoids have been shown to extend fibroblast proliferative potential, but studies to date are limited by the use of either large concentrations of the human glucocorticoid cortisol (∼14 μM) or synthetic (and much more potent) glucocorticoids (Cristofalo, 1975; Mawal-Dewan et al., 2003; Cristofalo and Kabakjian, 1975). To establish proof-of-concept for using physiological stress levels of cortisol to drive GR activation and genomic responses in human fibroblasts, we treated IMR-90 cells with either 100 nM cortisol or 500 nM relacorilant, as well as a combination of the two compounds, for 4 h and performed qPCR for three known glucocorticoid-responsive genes, FKBP5, GILZ, and HSD11B1 (Binder, 2009; Zannas et al., 2016; D'Adamio et al., 1997; Hammami and Siiteri, 1991). Cortisol robustly induced the expression of all genes, whereas this induction was abrogated by concomitant relacorilant treatment (Figure S1A). Furthermore, we performed a glucocorticoid receptor element luciferase reporter assay and detected significant induction of luciferase activity with cortisol treatment, which was again abrogated by the addition of relacorilant (Figure S1B). These findings showed that physiological stress levels of cortisol are sufficient to elicit a robust transcriptional response and that relacorilant efficiently antagonizes the GR in this context, supporting the use of this model to dissect how stress and GR activation impact epigenetic and cell states.
Chronic stress-driven glucocorticoid receptor activation programs key cell phenotypes in IMR-90 fibroblasts
To determine the extent to which chronic physiological GR activation can shape cell phenotypes over time, we performed prolonged treatment (up to 81 days) of IMR-90 fibroblasts with 100 nM cortisol and 500 nM relacorilant, while comprehensively tracking key fibroblast phenotypes, including cell proliferation, migration, and morphology (Deschênes-Simard et al., 2013; Ramilowski et al., 2020b; Zannas et al., 2020).
Cell proliferation was monitored with automated cell counting and documentation of population doubling levels (PDL) at each cell passage. Cortisol promoted the proliferation rate and extended the proliferative potential of IMR-90 cells, whereas these effects were completely abrogated by concomitant relacorilant treatment (Figure 1A). To further cross-validate the effect of cortisol on proliferation, we stained cells with Ki-67, an established cell proliferation marker (Scholzen and Gerdes, 2000), and observed that a significantly greater percentage of cortisol-treated cells are positive for Ki-67 (Figure 1B).
Figure 1.

Chronic stress-driven GR activation programs key cell phenotypes in IMR-90 fibroblasts
(A) Cumulative change in population doubling level (PDL) of IMR-90 cells treated with vehicle (DMSO), cortisol, relacorilant, or cortisol + relacorilant over 81 days. Points represent the average of 3 biological replicates. Error bars represent one SD above and below the mean.
(B) Ki-67 analysis of IMR-90 cells treated with vehicle, cortisol, relacorilant, or cortisol + relacorilant (F3,36 = 7.9, p < 0.001, FC cortisol vs. vehicle = 1.4, Padj < 0.01 for cortisol vs. vehicle comparison). Cells were stained for both DAPI (blue fluorescence) and Ki-67 (green fluorescence), and fluorescence images were then overlaid to determine the percentage of Ki-67-positive nuclei. Post-hoc adjusted p values (Padj) are Tukey-adjusted for multiple pairwise comparisons. Bars represent the average of 8 biological replicates. Error bars represent one SD above and below the mean. Representative vehicle and cortisol Ki-67-stained cells are shown.
(C) Scratch assays of IMR-90 cells pre-treated with vehicle, cortisol, relacorilant, or cortisol + relacorilant for 2 weeks. Cells were imaged for up to 24 h (F3,8 = 7.4, p < 0.05, FC cortisol-vehicle = 2.7, Padj < 0.01 for cortisol vs. vehicle comparison). Scale bar indicates 1000 μm. Bars in the bar graph represent the average of 3 biological replicates. Error bars represent one SD above and below the mean. Representative vehicle, cortisol, and cortisol + relacorilant images are shown. Post-hoc adjusted p values (Padj) are Tukey multiple pairwise-comparison adjusted.
(D) Overlapping density plots of cell surface area of IMR-90 fibroblasts treated with aphidicolin and either vehicle or cortisol.
(E) Overlapping density plots of total pseudopodia length of IMR-90 fibroblasts treated with aphidicolin and either vehicle or cortisol. Image analyses included a total of 980 vehicle-treated cells and 1026 cortisol-treated cells. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
Fibroblasts play central roles in wound healing (Bainbridge, 2013), a process that relies not only on cell proliferation but also migration (Bainbridge, 2013). To determine the extent to which cortisol-driven GR activation influences cell migration, we performed in vitro scratch assays (Liang et al., 2007) on IMR-90 cells pre-treated with cortisol and/or relacorilant for two weeks. Cortisol increased the rate of migration (μm2/h) measured over 24 h and compared to vehicle, whereas concomitant treatment with relacorilant abrogated the effects of cortisol on migration (Figure 1C). To rule out the possibility that the effect of cortisol on wound closure is merely a consequence of increased cell proliferation, cells were pre-treated with aphidicolin, an inhibitor of nuclear DNA replication (Kues et al., 2000), prior to scratch generation. Although aphidicolin pre-treatment inhibited cell proliferation as expected (Figure S2A), cortisol continued to promote cell migration (Figure S2B), thereby confirming that this migratory effect is independent of cell proliferation.
Lastly, we assessed whether cortisol also drives distinct changes in cell morphology. To this end, we applied a machine learning algorithm to cells pre-treated with cortisol or vehicle for two-three weeks. To account for differences in cell cycle between groups and thus minimize the signal-to-noise ratio, all cells were also treated overnight before measurement with the DNA replication inhibitor aphidicolin that synchronizes the cell cycle to the G1/S boundary (Kues et al., 2000). Cortisol robustly increased cell surface area (+18.4% difference; p = 4∗10−23), total pseudopodia length (+24.7% difference; p = 3.9∗10−21), perimeter (+17.6% difference; p = 2.5∗10−22), average pseudopodia length (+11.7% difference; p = 4∗10−7), and number of pseudopodia per cell (+19.605% difference, p = 6.1∗10−8), while decreasing cell circularity (−16.9% difference, p = 0.00814) (Figures 1D, 1E and S3A–S3D).
Taken together, these findings indicate that chronic stress-driven GR activation extensively programs cell proliferation, migration, and morphology in IMR-90 human fibroblasts.
The glucocorticoid receptor-driven cell phenotypes are recapitulated in primary dermal fibroblasts from donors of different ancestry and sex
The findings presented above are intriguing but are limited to IMR-90 fibroblasts, a single cell line derived from a female fetus. To further assess if these findings generalize across different biological contexts, we extended experiments in primary dermal fibroblast strains obtained from four donors of different ancestry and sex groups (White female, age 21; White male, age 21; Black female, age 21; Black male, age 22). We again assessed the effects on cell proliferation by treating fibroblast strains with cortisol continuously for 23 days, while tracking cumulative PDL, and found that cortisol promoted proliferation consistently in all four individuals (Figure 2A). We also performed a scratch assay for two of the donors (White female and Black male) and found that cortisol promoted cell migration for both fibroblast strains, whereas this effect was abrogated with concomitant relacorilant treatment (Figure 2B). Together these experiments extend findings and show that chronic physiological GR activation programs key fibroblast phenotypes across biological contexts such as donor ancestry and sex.
Figure 2.

Chronic stress-driven GR activation and cell phenotypes in primary dermal fibroblasts from donors of different ancestry and sex
(A) Cumulative change in population doubling level (PDL) of dermal fibroblasts (White female, White male, Black female, Black male) treated with vehicle (DMSO) or cortisol over 23 days. Points represent one biological replicate.
(B) Scratch assays of White female and Black male dermal fibroblasts pre-treated with vehicle, cortisol, relacorilant, or cortisol + relacorilant for 2 weeks. Cells were imaged for up to 24 h (White female: F3,8 = 31.6, p < 0.0001, FC cortisol-vehicle = 2.1, padj < 0.0001 for cortisol vs. vehicle comparison; Black male: F3,8 = 13.3, p < 0.001, FC cortisol-vehicle = 1.7, Padj < 0.01 for cortisol vs. vehicle comparison). Scale bar indicates 1000 μm. Post-hoc adjusted p values (Padj) are Tukey-adjusted for multiple pairwise comparisons. Bars in the bar graph represent the average of 3 biological replicates. Error bars represent one SD above and below the mean. Representative White female dermal fibroblasts treated with vehicle, cortisol, and cortisol + relacorilant are shown. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
Chronic stress-driven glucocorticoid receptor activation modulates mRNA levels of genes with functional roles in fibroblast phenotypes
Given the established role of the GR as a transcription factor (Zannas and Chrousos, 2017), we next examined the functional genomic changes underlying the observed GR-driven cell phenotypes. To this end, we performed RNA-sequencing (RNA-seq) on IMR-90 fibroblasts treated with vehicle, cortisol, relacorilant, or cortisol + relacorilant for 51 days, a timepoint of prolonged treatment where we observed extensive cell phenotype establishment. Principal component analysis (PCA) of all sequenced transcripts showed distinct clustering of the biological replicates by treatment group, suggesting that treatment contributed to a distinct transcriptomic profile (Figure 3A). To further characterize these profiles in conjunction with our phenotypic findings, we performed pairwise comparisons between cortisol and each of the other treatment groups (vehicle, relacorilant, and cortisol + relacorilant) and identified differentially expressed genes (DEGs) using DESeq2 (Love et al., 2014) with FDR-adjusted p < 0.01 and fold change (FC) ≥ ∣1.5∣. The overlapping 946 DEGs with the same directionality across these comparisons represent genes specifically driven by GR activation and are thus hereinafter denoted GR-modulated DEGs (Figures 3B and 3C; Table S1). GO-term enrichment analysis of GR-modulated DEGs revealed significant enrichment for extracellular matrix, cell adhesion, migration, and proliferation (FDR-adjusted p ≤ 0.05) (Figure 3D). Of the 946 GR-modulated genes, 401 are downregulated and 545 are upregulated with cortisol treatment. Direction-specific GO enrichment analysis showed that downregulated GR-modulated DEGs are enriched in branching epithelium morphogenesis, cell-cell adhesion, and cell-cell signaling pathways (Figure S4A), whereas upregulated DEGs are enriched in extracellular structure organization, cellular component movement, and cell proliferation pathways (Figure S4B). Notably, the gene undergoing the most robust cortisol-driven change in expression is ZBTB16 (log2 FC = +14.3), a zinc finger transcription factor previously shown to be glucocorticoid-responsive (Wasim et al., 2010) (Figure 3C).
Figure 3.

Chronic stress-driven GR activation modulates mRNA levels of genes with functional roles in fibroblast phenotypes
(A) Principal component analysis (PCA) plot of RNA-seq samples of IMR-90 fibroblasts treated with vehicle (DMSO), cortisol, relacorilant, or cortisol + relacorilant, n = 6 biological replicates for each treatment group.
(B) Venn diagram showing overlap of differentially expressed genes (DEGs) across cortisol vs. vehicle, cortisol vs. relacorilant, and cortisol vs. cortisol + relacorilant comparisons. 946 genes represent DEGs that change in the same direction that are denoted by glucocorticoid receptor (GR)-modulated genes and labeled in yellow in the Venn diagram.
(C) Volcano plot showing differences in mRNA levels of all genes in the cortisol vs. vehicle condition. GR-modulated genes are labeled in yellow. Gene names are shown for the top 5 most significant and most fold-changing downregulated and upregulated genes.
(D) Gene ontology (GO) biological process analysis of GR-modulated genes. GO analysis was performed using WebGestalt (WEB-based Gene SeT AnaLysis Toolkit) (Liao et al., 2019).
(E) Normalized count plots of representative GR-modulated genes FKBP5, ZBTB16, and OPCML in vehicle, cortisol, relacorilant, and cortisol + relacorilant samples.
Individual gene count analysis of ZBTB16 but also other representative genes, such as FKBP5 and OPCML, in RNA-seq data, confirmed that the addition of relacorilant abrogates the genomic effects of cortisol (Figure 3E). Moreover, RNA-seq results were cross-validated by performing qPCR for the same RNA samples used in the sequencing experiment as well as in dermal fibroblasts undergoing prolonged treatment with vehicle or cortisol (Figures S4C and S4D, though ZBTB16 gene expression was too low to be detected with qPCR in treatment conditions other than the cortisol group (data not shown).
Chronic stress-driven glucocorticoid receptor activation induces widespread, but genomic context-dependent, methylomic changes
Epigenetic processes can influence gene transcription and phenotypic outcomes (Cavalli and Heard, 2019; D'Anna et al., 2020; Nanavaty et al., 2020; Zannas et al., 2019), and a widely studied epigenetic modification in humans is DNA (CpG) methylation (Yong et al., 2016; Zannas, 2019). To examine whether functional methylomic changes accompany the GR-driven phenotypic and transcriptomic profiles observed above, we measured genome-wide DNA methylation of IMR-90 cells with the Illumina Infinium HumanMethylationEPIC BeadChip at the same time point following prolonged (51-day) exposure to the same four treatment conditions (vehicle, cortisol, relacorilant, or cortisol + relacorilant). Mirroring our phenotypic and transcriptomic analyses, we separately determined the extent to which the methylome-wide patterns of cells treated with cortisol differ from each of the other groups. To increase analytical robustness, significant methylation differences for each comparison and CpG site were defined based on FDR-adjusted p < 0.01 and DNA methylation difference >5%, and the overlapping CpGs with a consistent direction of effect were determined across all comparisons. These analyses identified 21393 CpG sites (12817 hypo- and 8576 hyper-methylated, hereinafter denoted by “GR-modulated CpGs”), which undergo cortisol-driven methylation changes that are abrogated by concomitant relacorilant treatment (Figures 4A and 4B; Table S2). We cross-validated our array data by performing targeted bisulfite sequencing (TBS) in both IMR-90 cells and dermal fibroblasts at the following representative genes: ZBTB16, PTGS1, NRP1, FOXO3, and IL6 (Figures S4C and S4D). GO pathway analysis revealed that the GR-modulated CpGs are enriched in genes that are involved in cellular movement, cell migration, and cell motility (Figure 4C). Because these pathways also overlapped with those found from our RNA-seq analysis, we further investigated the overlapping GR-modulated genes identified from both the RNA-seq and EPIC array analysis (Figure S5A). GO analysis of the 428 overlapping genes showed enrichment for pathways that include cell projection morphogenesis, cellular component movement, and locomotion (Figure S5B).
Figure 4.

Chronic stress-driven GR activation induces widespread, but genomic context-dependent, methylomic changes
(A) Venn diagram showing overlap of differentially methylated CpGs across cortisol vs vehicle, cortisol vs. relacorilant, and cortisol vs. cortisol + relacorilant comparisons. Yellow circle represents GR-modulated CpGs that show a consistent direction in methylation change across all comparisons.
(B) Volcano plot showing differences in CpG methylation levels in the cortisol vs. vehicle condition. GR-modulated CpG sites are labeled in yellow. Dotted vertical lines represent a 5% change in methylation levels. Dotted horizontal lines represent FDR < 0.01.
(C) Gene ontology (GO) biological process pathway analysis of GR-modulated genes identified in the EPIC array analysis. GO analysis was performed using WebGestalt (WEB-based Gene SeT AnaLysis Toolkit) (Liao et al., 2019). The top 10 most enriched pathways are shown.
(D) Enrichment analysis of differentially methylated CpGs localized at previously identified GR binding sites and open sea vs. CpG island sites.
(E) Proportion of CpGs for all EPIC array-covered and GR-modulated sites.
(F) Genic and enhancer annotations of GR-modulated CpG sites. GR-modulated enrichment is shown in black and all EPIC array-covered CpGs are shown in gray.
(G) Metagene plot showing locally estimated scatterplot smoothing of CpG DNA methylation values across the ZBTB16 gene. Locations of CpGs and previously identified GR binding sites are indicated. Yellow points represent DNA methylation in cortisol-treated IMR-90 cells. Black points represent DNA methylation in vehicle-treated IMR-90 cells. Primer locations used for ChIP-qPCR are indicated.
(H) ChIP-qPCR analysis across the ZBTB16 gene. Bars represent the average of 5 biological replicates. Error bars represent one SD above and below the mean. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
Compared to all array-covered CpG sites, the GR-modulated CpGs showed enrichment for localization at previously identified GR ChIP-seq peaks in IMR-90 cells (Starick et al., 2015) (odds ratio (OR) 1.5, 95% CI 1.4 to 1.6) and open sea vs. CpG-island genomic regions (OR 2.1, 95% CI 2.0 to 2.2) (all P-values < 2.2 × 10−16; Figures 4D and 4E), overall supporting local genomic context as an important determinant of GR-driven methylation changes. Furthermore, the GR-modulated CpGs are enriched in enhancers, 1 to 5 kb upstream of genes localization, promoters, 5′UTRs, exons, intron/exon boundaries, and 3′UTRs (Figure 4F).
Lastly, further examining our example GR-regulated gene ZBTB16 showed a characteristic epigenetic landscape of profound gene body hypermethylation (Figure 4G), in line with prior reports of similar epigenetic patterns at upregulated genes (Arechederra et al., 2018; Hernando et al., 2013; Jjingo et al., 2012). This finding prompted us to examine if gene body hypermethylation is a widespread and functional epigenetic sequela of stress. Supporting this hypothesis, GR-driven hypermethylation was noted in the gene bodies of 188 (out of all 946) GR-regulated genes and was further enriched for localization at proliferation and migration genes (p < 0.05). SDK1 is shown as another example gene that undergoes GR-driven gene body hypermethylation (Figure S5C).
Because the GR is recruited to glucocorticoid-responsive genes and can modulate DNA methylation (Zannas et al., 2019; Wiechmann et al., 2019; Provencal et al., 2020; Seifuddin et al., 2017; Weikum et al., 2017), we also asked whether ZBTB16 hypermethylation is associated with enhanced GR recruitment. To address this question, we performed GR ChIP-qPCR across the ZBTB16 gene and found that prolonged exposure to physiological stress levels of cortisol drives GR binding in the ZBTB16 gene body (Figure 4H).
Discussion
Chronic environmental stress can profoundly impact cell and body function (Hong et al., 2020; Yang et al., 2020; Epel et al., 2004; Joëls et al., 2004), but the underlying cellular and molecular mechanisms are poorly understood. Here, we show that prolonged exposure to physiological stress hormone levels drives robust, GR-mediated changes in key fibroblast phenotypes, including cell proliferation, migration, and morphology. Importantly, these findings are present in both IMR-90 cells and in dermal fibroblast strains from donors of different ancestry and sex, indicating that the cellular effects of chronic stress-driven GR activation extend across different biological contexts. Moreover, the GR-driven cell phenotypes are accompanied by concordant functional methylomic and transcriptomic changes.
Although prolonged exposure to either high non-physiological concentrations of cortisol or synthetic (and much more potent) glucocorticoids was previously found to extend the proliferative potential of fibroblasts (Cristofalo, 1975; Mawal-Dewan et al., 2003; Cristofalo and Kabakjian, 1975), our study is the first to show that physiological stress levels of cortisol are sufficient to promote not only cell proliferation but also cell migration. Because these effects are further abrogated upon co-treatment with the selective GR antagonist relacorilant, we propose that the GR is the primary driver of the observed stress-driven cell phenotypes. Fibroblasts are at the forefront of wound healing (Bainbridge, 2013), a process that relies on their ability to both proliferate and migrate (Bainbridge, 2013). Consequently, our findings suggest that stress-driven GR activation could enhance their wound-healing capability, an adaptation that may be necessary for survival in high-stress situations. Nevertheless, excessive fibroblast activation is also seen in fibrotic diseases (Wynn and Ramalingam, 2012), suggesting that chronic stress could also increase the risk for these disease conditions. Moreover, previous studies have suggested that GR agonism can have detrimental effects on wound healing (Carolina et al., 2018; Jozic et al., 2017; Wang et al., 2013), but these studies involved the use of high-dose or potent synthetic glucocorticoids or were performed in vivo, where the mechanisms and pathways regulating wound healing are more complex. Although these intriguing possibilities could not be addressed by the present study’s focus on cell models, future in vivo studies may elucidate the exact role of GR-driven fibroblast phenotypes in health and disease.
Corroborating our phenotypic findings, we also identified widespread GR-driven transcriptomic and methylomic changes, including gene loci with known roles in cell proliferation and migration. These findings build on previous work showing that glucocorticoids can lead to widespread DNA methylation changes (Zannas et al., 2019; Seifuddin et al., 2017; Wiechmann et al., 2019; Provencal et al., 2020; Zannas, 2021a), while further showing that GR-modulated CpGs are enriched for localization at proliferation and migration genes as identified in our transcriptomic analyses. Interestingly, the GR-modulated CpGs also showed enrichment for localization at open sea regions, suggesting that local genomic context is important for GR-driven methylation changes. This is in line with our previous study showing that prolonged exposure to physiological stress hormone levels drives cumulative epigenomic changes that are also enriched for open sea region localization (Zannas, 2021a). Lastly, our initial unbiased approach, followed by more fine-grained analyses on an example GR-regulated gene (ZBTB16), identified a pattern of GR-driven hypermethylation observed widely in gene bodies. The robust effects of chronic stress on this gene are of particular interest, given that ZBTB16 is a pleiotropic transcription factor that regulates cell proliferation and differentiation and has been previously implicated in diverse disease states including carcinogenesis and metabolic syndrome (Liu et al., 2016; Choi et al., 2014; Šeda et al., 2017). Moreover, our identification of gene body hypermethylation as a widespread signature extends prior reports of similar epigenetic patterns at upregulated genes (Arechederra et al., 2018; Hernando et al., 2013; Jjingo et al., 2012) and suggests a role for this signature in chronic stress-driven epigenetic regulation of gene transcription.
Taken together, our findings lead us to hypothesize a model whereby chronic stress-driven GR activation epigenetically regulates the expression of genes with functional roles in cell proliferation and migration, eventually influencing these key phenotypes in human fibroblasts. We speculate that such stress-driven epigenetic regulation is fine-tuned by both the direction of effects (hypo-vs. hypermethylation) and the specific genomic context where such changes take place (e.g., enhancers and GR binding sites).
Chronic stress has been implicated in increased risk for diverse disease entities, including cardiovascular disease, cancer, and mental illness (Mariotti, 2015). Our study has identified that prolonged exposure to physiological stress hormone levels has a multitude of effects at both the molecular and cellular levels, with potentially important implications for human health. Follow-up studies may extend these findings to other human cell types and examine the implications of chronic stress in regulating cell phenotypes at the systems level to determine their role in health and disease.
Limitations of the study
A limitation of this study is that it is difficult to determine whether epigenetic changes precede or follow alterations in gene expression and the extent to which some of the observed changes represent secondary effects or mere epiphenomena of altered gene expression. Another limitation is the use of EPIC arrays, which are cost-efficient but provide incomplete coverage of the epigenome. Future studies will be needed to provide a more detailed spatiotemporal view of how stress-driven epigenomic patterns become established and shape cell phenotypes.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GR/NR3C1 Antibody (Rabbit polyclonal) | Novus Biologicals | Cat#NBP2-42221 |
| normal rabbit IgG | Santa Cruz Biotechnology | Cat#sc-2027 |
| Ki67 antibody (Mouse monoclonal) | Santa Cruz Biotechnology | Cat#sc-23900 |
| Anti-Mouse IgG Secondary Antibody, Alexa Fluor 488 (Donkey polyclonal) | ThermoFisher Scientific | Cat#A2102 |
| Chemicals, peptides, and recombinant proteins | ||
| Hydrocortisone | Sigma Aldrich | Cat#H4001 |
| DMSO | Sigma Aldrich | Cat#D8418 |
| Relacorilant | Corcept Therapeutics | N/A |
| DMEM | Gibco | Cat#31053-028 |
| Fetal bovine serum (FBS) | VWR Life Science Seradigm | Cat#97068-085 |
| L-glutamine | Thermo Fisher Scientific | Cat#25-030-081 |
| Sodium pyruvate | Gibco | Cat#11360-070 |
| Non-essential amino acids | Gibco | Cat#11140-050 |
| Antibiotic-Antimycotic | Gibco | Cat#15240-062 |
| ezDNase | ThermoFisher Scientific | Cat#11766051 |
| Critical commercial assays | ||
| Dual-Luciferase Reporter Assay System | Promega | Cat#E1910 |
| GenFind V3 DNA extraction and purification kit | Beckman Coulter | Cat#C34880 |
| RNeasy Micro Kit | QIAGEN | Cat#74004 |
| Super-Script IV VILO Master Mix | ThermoFisher Scientific | Cat#11756050 |
| EZ DNA Methylation Kit | Zymo Research | Cat#D5001 |
| Collibri PCR-free DNA Library Prep Kit | ThermoFisher Scientific | Cat#A38609024 |
| Deposited data | ||
| Sequencing and microarray data | Gene Expression Omnibus (GEO) | GEO: GSE210304 |
| Experimental models: Cell lines | ||
| IMR-90 lung fibroblasts | Coriell Institute for Medical Research | Cat#I90-10 |
| Software and algorithms | ||
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/ |
| R 3.6.3 | R Core Team | https://www.r-project.org/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to lead contact Anthony Zannas (anthony_zannas@med.unc.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Cell culture
IMR-90 (human fetal lung fibroblast) cells were obtained from the Coriell Institute for Medical Research and authenticated using Short Tandem Repeat (STR) analysis (Capes-Davis et al., 2013, American Type Culture Collection Standards Development Organization Workgroup et al., 2011). Primary dermal fibroblasts (White female, age 21; White male, age 21; Black female, age 21; Black male, age 22) are derived from an established cohort of previously characterized primary dermal fibroblasts at the National Institute of Environmental Health Sciences (Mackey et al., 2018; Bisogno et al., 2020). Both IMR-90 and dermal fibroblasts were maintained in Dulbecco’s Modified Eagle Medium (DMEM) without phenol red and supplemented with 15% fetal bovine serum (FBS), high glucose, L-glutamine, sodium pyruvate, non-essential amino acids, and Antibiotic-Antimycotic. Media and media supplements were purchased from ThermoFisher Scientific. Cells were maintained at 37°C and 5% CO2 in a humid incubator, seeded at a density of 10,000 cells/cm2, and allowed to proliferate until the cells reached 90% confluency. To get a comprehensive picture of chronic stress-driven cell phenotypes, cell treatments were prolonged to the extent possible (up to 81 days). Given the challenge of performing such prolonged experiments multiple times, however, chronic treatments were shortened (but never below 2 weeks) when validating findings or extending results across paradigms. During treatment, cortisol (ThermoFisher Scientific) and relacorilant (Corcept Therapeutics) were added to cultures for the indicated duration of time at a final concentration of 100 and 500 nM, respectively. Media was changed every 2–3 days. Both compounds were diluted in a very low final amount of DMSO (0.0001%), and the same final DMSO concentration was used as a vehicle control in all experiments.
Method details
DNA and RNA extraction
DNA was extracted using the GenFind V3 DNA extraction and purification kit (Beckman Coulter). Total RNA was isolated using the RNeasy Micro Kit (QIAGEN) according to the manufacturer’s instructions. DNA and RNA concentrations were measured using the Take3 microplate spectrophotometer (BioTek).
cDNA synthesis and qPCR
200 ng of total RNA was treated with ezDNase (ThermoFisher Scientific) to degrade carry-over genomic DNA following RNA extraction and cDNA was synthesized using the Super-Script IV VILO Master Mix (ThermoFisher Scientific). For qPCR assay, 4 ng of cDNA was added from each sample into individual wells of a Micro-Amp optical 96-well reaction plate (ThermoFisher Scientific). Measurements were then performed using the QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems) under standard conditions. Primers used for qPCR analysis are listed in Table S3.
Machine learning-based analysis of cell morphology
The machine learning pipeline used for morphological analysis was motivated by previous approaches reported by Ramilowski et al. (2020a) and French et al. (2020), herein modified and expanded. IMR-90 fetal lung fibroblast cells were treated for 2–3 weeks prior to imaging with either 100 nM cortisol or vehicle (DMSO) treatment in 6-well plates. After this time, cells were fixed using chilled methanol and stained with a DAPI nuclear staining. Prior to imaging, cells were treated overnight with aphidicolin and then imaged using an EVOS FL microscope under both the Transmitted and DAPI settings, so that 2 images per field were taken to visualize cell body and nucleus. Between 3 and 5 image sets were taken per well and run through a trained program in ilastik (Berg et al., 2019), which identified the cell, cell boundary, and background and binarized the image accordingly into a probability map. The probability maps were then overlaid with DAPI images of the same field to generate composite images that were screened for quality. A total of 12 images from cortisol-treated cells (n = 1026) and 11 images from vehicle-treated cells (n = 980) were run through a CellProfiler pipeline, which first identified the nuclei using the IdentifyPrimaryObjects module and then the cell body using the IdentifySecondaryObjects module. The MeasureObjectSizeShape module was then used to measure area, perimeter, and formfactor (circularity) of the cells and nuclei separately. MeasureObjectSkeleton was also used to quantify cellular projections including the number of pseudopodia projections (Number of Endpoints) and the total length of the cell and its pseudopodia (Total Skeleton Length). These two readouts were also used to calculate the average length per pseudopodia per cell.
Luciferase assay
IMR-90 cells were transfected using Neon system as recommended per manufacturer. Briefly, cells were harvested, washed in PBS and resuspended in resuspension buffer R at a final density of 1.0 × 107 cells/mL. 5 μg of pDNA was added to cell suspension, gently mixed and incubated for 5 min at room temperature, followed by electroporation. Mock transfection was performed without pDNA. Transfected cells were diluted into 10 mL of antibiotic free media and seeded into 96 well tissue culture plate 5,000 cells per well and let rest overnight. Next, cells were treated with vehicle, 100 nM cortisol, 500 nM relacorilant, or cortisol + relacorilant overnight, washed with 100 μL of PBS and lysed with 50 μL of 1 X Passive Lysis Buffer (Promega E1910). The luciferase detection was performed as described previously using a construct that contains 2 glucocorticoid response elements (Hampf and Gossen, 2006).
Ki-67 staining
Lung fibroblasts were seeded into 12-well plates and left undisturbed for 48 h to allow for proliferation. Cells were washed twice with 1 X PBS, fixed and permeabilized in 100% methanol for 10 min at −20°C, washed twice with 1 X PBS, and then blocked with 2% BSA for 2 h at room temperature. Primary antibody Ki-67 (1:100; cat no. sc-23900; Santa Cruz) was added and incubated at room temperature for 90 min. Secondary antibody donkey anti-mouse Alexa 488 (1:500; cat no. A2102 Thermo Fisher) was added and incubated for 48 h at 4°C. DAPI stain was used to counterstain nuclei. Images were taken using Evos Fluorescent Microscope. Ki-67 staining quantification was performed by determining the number of positive nuclei as a proportion of the total number of nuclei in three representative fields of each sample. Images were analyzed using ImageJ.
Scratch assay
Scratch assays were performed according to previous studies (Liang et al., 2007). In short, a 200 μL pipette tip was used to generate a straight line “scratch” on a 90–100% confluent monolayer of cells in a 12-well cell culture plate. Each well was then imaged under a light microscope every 6 h until the scratched region was filled in with cells. The rate of migration was determined by ImageJ analysis.
RNA-sequencing and analysis
RNA-seq libraries were prepared and sequenced on an Illumina HiSeq 4000 with 150 bp paired-end reads at Novogene Co., Ltd. (Sacramento, CA, USA). Reads were aligned to the human reference genome hg38 using STAR at default settings (Dobin et al., 2013). Differentially expressed genes were identified using DESeq2 (Love et al., 2014).
EPIC DNA methylation array measurements
DNA was extracted using the Genfind V3 DNA extraction kit (Beckman Coulter, Brea, CA, USA) according to manufacturer instructions. DNA concentration and purity were determined using the Take3 microplate spectrophotometer (BioTek Instruments, Winooski, VT, USA). Extracted DNA was bisulfite converted using EZ DNA Methylation Kit (Zymo Research, Irvine, CA, USA), and genome-wide DNA methylation was measured with the Illumina Infinium HumanMethylationEPIC BeadChip (EPIC), which assays DNA methylation at >850000 CpG sites (Pidsley et al., 2016). Based on standard quality control procedures, the following probes were removed: (i) previously identified cross-reactive and polymorphic probes (Chen et al., 2013); (ii) CpG probes with less than 3 beads or with a detection p value over 1% in at least 1% of the samples assayed; (iii) CpG probes mapping to the Y chromosome (given that IMR-90 cells originate from a female fetus); and (iv) probes containing SNPs with a minor allele frequency >1% located within 10 bases of the CpG. Following this standardized procedure, a total of 709065 CpG were included in subsequent analyses. Raw signal intensities were normalized with subset quantile normalization available in the minfi package (Aryee et al., 2014). Normalized intensity values were then converted into beta values, which were used in all analyses. To adjust for technical batch effects, EPIC arrays were run after randomizing DNA samples from different treatment conditions across plate, chip, row, and column. The lack of significant batch effects in the data was confirmed with iterative covariate inspection after principal component analysis as implemented in the RaMWAS pipeline (Guintivano et al., 2020; Shabalin et al., 2018).
Targeted bisulfite sequencing (TBS)
DNA was bisulfite-converted using the EZ DNA Methylation Kit (Zymo Research). Methylation-specific PCR was then performed using EpiTaq HS (Takara Bio) to amplify bisulfite-treated template DNA. Libraries were generated using the Collibri PCR-free DNA Library Prep Kit (ThermoFisher Scientific). In brief, adapters with unique dual indexes were ligated to amplicons, pooled, and then sequenced on an Illumina HiSeq4000 with 150 bp paired-end reads at Novogene Co., Ltd. (Sacramento, CA, USA). Reads were aligned to the hg19 genome and methylation calls were performed using Bismark (Krueger and Andrews, 2011). For each gene amplicon, the averages of all CpG methylation levels were calculated and used to compare vehicle and control groups.
ChIP-qPCR
Cells were treated with either vehicle or cortisol for 18 continuous days and then crosslinked with 1% formaldehyde in PBS at 37°C for 10 min. The reaction was quenched with 125 mM glycine at 37°C for 5 min. Cells were washed with PBS and lysed in hypotonic buffer (10 mM HEPES-NaOH, 10 mM KCl, 10 mM MgCl2, 10% glycerol, 0.1% Triton X-100). Chromatin was then sheared to an average size of 150–800 bp in shearing buffer (10 mM Tris pH 8, 1 mM EDTA, 0.5 mM EGTA, 5 mM sodium butyrate, 0.1% SDS). Chromatin was then diluted in IP buffer (2 mM EDTA, 150 mM NaCl, 20 mM Tris pH 8). Anti-GR antibody (NBP2-42221; Novus Biologicals) or IgG (sc-2027; Santa Cruz Biotechnology) were added and the samples were incubated at 4°C overnight with rotation. Protein A and Protein G Dynabeads (ThermoFisher Scientific) were added to each sample and rotated for 2 h at 4°C. Beads were washed once with low salt buffer (20 mM Tris pH 8, 2 mM EDTA, 150 mM NaCl, 0.1% SDS, 1% Triton X-100), high salt buffer (20 mM Tris pH 8, 2 mM EDTA, 500 mM NaCl, 0.1% SDS, 1% Triton X-100), LiCl buffer (10 mM Tris pH 8.0, 250 mM LiCl, 2 mM EDTA, 1% NP-40), and 10:1 TE buffer (10 mM Tris pH 8, 1 mM EDTA). Elution buffer (1% SDS and 100 mM NaHCO3) was added to beads and incubated at 65°C for 30 min. Reverse crosslink buffer (20 mM Tris pH 8, 10 mM EDTA, 5 mM EGTA, 1% SDS, 300 mM NaCl, with RNase A) was added to the samples and incubated overnight at 65°C. The next day Proteinase K was added to the samples and incubated at 55°C for 2 h. DNA was isolated using MinElute columns (QIAGEN). Primers used for ZBTB16 ChIP-qPCR are listed in Table S3.
Quantification and statistical analysis
One-way ANOVA analysis followed by Tukey multiple pairwise-comparisons were used to assess group differences in cell proliferation and migration as well as qPCR data between vehicle, cortisol, relacorilant, and cortisol-relacorilant groups. For cell morphology data, outliers were identified and excluded from results using the interquartile range method, and all morphology measures were analyzed using the Mann-Whitney test. Pairwise between-treatment methylation differences at individual CpGs were assessed using Student’s t-test. To assess the localization of CpGs within GR binding sites, analyses used online available GR ChIP-seq peaks previously identified in IMR-90 cells (Starick et al., 2015). Localization for genomic region (open sea, shelf, shore, CpG island) was performed using the EPIC array annotation (manifest file) derived through the minfi package (Aryee et al., 2014). Enrichment analyses were performed using Fisher’s exact test with all EPIC array-covered CpGs as background. All p values were two-tailed, correction for multiple testing for genomic data was done with the false discovery rate method, and results were considered statistically significant based on an adjusted threshold of α = 0.05. All statistical analyses were conducted in R version 3.6.3.
Acknowledgments
This work was in part supported by the National Institute of Mental Health (T32MH093315 to C.S.L.). The following cell line was obtained from the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research: I90-83. Relacorilant was provided by Corcept Therapeutics. We thank Dr. Hazel Hunt (Corcept Therapeutics) for her constructive feedback on this article.
Author contributions
Conceptualization: A.S.Z.; Investigation: C.S.L., O.K., E.M.W., N.D., T.K.A., A.S.Z.; Writing – Original Draft: C.S.L., A.S.Z.; Writing – Review & Editing: C.S.L., E.M.W., N.D, T.K.A, O.K., A.S.Z.; Funding Acquisition and Supervision: A.S.Z.
Declaration of interests
The authors declare no competing interests.
Published: September 16, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104960.
Supplemental information
Data and code availability
The RNA-sequencing, targeted bisulfite sequencing, and EPIC array data reported in this paper are uploaded in Gene Expression Omnibus (GEO) under SuperSeries accession number GEO: GSE210304. This paper does not report original code (if no original code is used in this paper). Requests for any additional information required to reanalyze the data can be directed to the lead contact.
References
- Arechederra M., Daian F., Yim A., Bazai S.K., Richelme S., Dono R., Saurin A.J., Habermann B.H., Maina F. Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer. Nat. Commun. 2018;9:3164. doi: 10.1038/s41467-018-05550-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryee M.J., Jaffe A.E., Corrada-Bravo H., Ladd-Acosta C., Feinberg A.P., Hansen K.D., Irizarry R.A. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Type Culture Collection Standards Development Organization Workgroup. Alston-Roberts C., Barallon R., Bauer S., Butler J., Capes-Davis A., Dirks W., Elmore E., Furtado M., Kerrigan L. 2011. Authentication of Human Cell Lines: Standardization of STR Profiling.http://webstore.ansi.org/RecordDetail.aspx ANSI. ATCC ASN-0002-2011. Copyrighted by ATCC and the American National Standards Institute (ANSI) [Google Scholar]
- Bainbridge P. Wound healing and the role of fibroblasts. J. Wound Care. 2013;22:407–408. doi: 10.12968/jowc.2013.22.8.407. [DOI] [PubMed] [Google Scholar]
- Bendel S., Karlsson S., Pettila V., Loisa P., Varpula M., Ruokonen E. Free cortisol in sepsis and septic shock. Anesth. Analg. 2008;106:1813–1819. doi: 10.1213/ane.0b013e318172fdba. [DOI] [PubMed] [Google Scholar]
- Berg S., Kutra D., Kroeger T., Straehle C.N., Kausler B.X., Haubold C., Schiegg M., Ales J., Beier T., Rudy M., et al. ilastik: interactive machine learning for (bio)image analysis. Nat. Methods. 2019;16:1226–1232. doi: 10.1038/s41592-019-0582-9. [DOI] [PubMed] [Google Scholar]
- Bhake R.C., Leendertz J.A., Linthorst A.C.E., Lightman S.L. Automated 24-hours sampling of subcutaneous tissue free cortisol in humans. J. Med. Eng. Technol. 2013;37:180–184. doi: 10.3109/03091902.2013.773096. [DOI] [PubMed] [Google Scholar]
- Binder E.B. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. 2009;34:S186–S195. doi: 10.1016/j.psyneuen.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Bisogno L.S., Yang J., Bennett B.D., Ward J.M., Mackey L.C., Annab L.A., Bushel P.R., Singhal S., Schurman S.H., Byun J.S., et al. Ancestry-dependent gene expression correlates with reprogramming to pluripotency and multiple dynamic biological processes. Sci. Adv. 2020;6 doi: 10.1126/sciadv.abc3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capes-Davis A., Reid Y.A., Kline M.C., Storts D.R., Strauss E., Dirks W.G., Drexler H.G., MacLeod R.A.F., Sykes G., Kohara A., et al. Match criteria for human cell line authentication: where do we draw the line? Int. J. Cancer. 2013;132:2510–2519. doi: 10.1002/ijc.27931. [DOI] [PubMed] [Google Scholar]
- Carolina E., Kato T., Khanh V.C., Moriguchi K., Yamashita T., Takeuchi K., Hamada H., Ohneda O. Glucocorticoid impaired the wound healing ability of endothelial progenitor cells by reducing the expression of CXCR4 in the PGE2 pathway. Front. Med. 2018;5:276. doi: 10.3389/fmed.2018.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli G., Heard E. Advances in epigenetics link genetics to the environment and disease. Nature. 2019;571:489–499. doi: 10.1038/s41586-019-1411-0. [DOI] [PubMed] [Google Scholar]
- Chang J.E., Turley S.J. Stromal infrastructure of the lymph node and coordination of immunity. Trends Immunol. 2015;36:30–39. doi: 10.1016/j.it.2014.11.003. [DOI] [PubMed] [Google Scholar]
- Chen Y.A., Lemire M., Choufani S., Butcher D.T., Grafodatskaya D., Zanke B.W., Gallinger S., Hudson T.J., Weksberg R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8:203–209. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W.I., Kim M.Y., Jeon B.N., Koh D.I., Yun C.O., Li Y., Lee C.E., Oh J., Kim K., Hur M.W. Role of promyelocytic leukemia zinc finger (PLZF) in cell proliferation and cyclin-dependent kinase inhibitor 1A (p21WAF/CDKN1A) gene repression. J. Biol. Chem. 2014;289:18625–18640. doi: 10.1074/jbc.m113.538751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ-Crain M., Jutla S., Widmer I., Couppis O., Konig C., Pargger H., Puder J., Edwards R., Muller B., Grossman A.B. Measurement of serum free cortisol shows discordant responsivity to stress and dynamic evaluation. J. Clin. Endocrinol. Metab. 2007;92:1729–1735. doi: 10.1210/jc.2006-2361. [DOI] [PubMed] [Google Scholar]
- Chrousos G.P., Gold P.W. The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. JAMA. 1992;267:1244–1252. doi: 10.1001/jama.1992.03480090092034. [DOI] [PubMed] [Google Scholar]
- Cristofalo V.J. The effect of hydrocortisone on DNA synthesis and cell division during aging in vitro. Adv. Exp. Med. Biol. 1975;53:7–22. doi: 10.1007/978-1-4757-0731-1_2. [DOI] [PubMed] [Google Scholar]
- Cristofalo V.J., Kabakjian J. Lysosomal enzymes and aging in vitro: subcellular enzyme distribution and effect of hydrocortisone on cell life-span. Mech. Ageing Dev. 1975;4:19–28. doi: 10.1016/0047-6374(75)90004-4. [DOI] [PubMed] [Google Scholar]
- D'Adamio F., Zollo O., Moraca R., Ayroldi E., Bruscoli S., Bartoli A., Cannarile L., Migliorati G., Riccardi C. A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity. 1997;7:803–812. doi: 10.1016/s1074-7613(00)80398-2. [DOI] [PubMed] [Google Scholar]
- D'Anna F., Van Dyck L., Xiong J., Zhao H., Berrens R.V., Qian J., Bieniasz-Krzywiec P., Chandra V., Schoonjans L., Matthews J., et al. DNA methylation repels binding of hypoxia-inducible transcription factors to maintain tumor immunotolerance. Genome Biol. 2020;21:182. doi: 10.1186/s13059-020-02087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschênes-Simard X., Gaumont-Leclerc M.F., Bourdeau V., Lessard F., Moiseeva O., Forest V., Igelmann S., Mallette F.A., Saba-El-Leil M.K., Meloche S., et al. Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev. 2013;27:900–915. doi: 10.1101/gad.203984.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epel E.S., Blackburn E.H., Lin J., Dhabhar F.S., Adler N.E., Morrow J.D., Cawthon R.M. Accelerated telomere shortening in response to life stress. Proc. Natl. Acad. Sci. USA. 2004;101:17312–17315. doi: 10.1073/pnas.0407162101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French S.L., Vijey P., Karhohs K.W., Wilkie A.R., Horin L.J., Ray A., Posorske B., Carpenter A.E., Machlus K.R., Italiano J.E., Jr. High-content, label-free analysis of proplatelet production from megakaryocytes. J Thromb Haemost. 2020;18:2701–2711. doi: 10.1111/jth.15012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guintivano J., Shabalin A.A., Chan R.F., Rubinow D.R., Sullivan P.F., Meltzer-Brody S., Aberg K.A., van den Oord E.J.C.G. Test-statistic inflation in methylome-wide association studies. Epigenetics. 2020;15:1163–1166. doi: 10.1080/15592294.2020.1758382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall C., Gehmlich K., Denning C., Pavlovic D. Complex relationship between cardiac fibroblasts and cardiomyocytes in health and disease. J. Am. Heart Assoc. 2021;10:e019338. doi: 10.1161/jaha.120.019338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammami M.M., Siiteri P.K. Regulation of 1lβ-Hydroxysteroid dehydrogenase activity in human skin fibroblasts: enzymatic modulation of glucocorticoid action∗. J. Clin. Endocrinol. Metab. 1991;73:326–334. doi: 10.1210/jcem-73-2-326. [DOI] [PubMed] [Google Scholar]
- Hampf M., Gossen M. A protocol for combined Photinus and Renilla luciferase quantification compatible with protein assays. Anal. Biochem. 2006;356:94–99. doi: 10.1016/j.ab.2006.04.046. [DOI] [PubMed] [Google Scholar]
- Hamrahian A.H., Oseni T.S., Arafah B.M. Measurements of serum free cortisol in critically ill patients. N. Engl. J. Med. 2004;350:1629–1638. doi: 10.1056/nejmoa020266. [DOI] [PubMed] [Google Scholar]
- Hernando H., Shannon-Lowe C., Islam A.B., Al-Shahrour F., Rodríguez-Ubreva J., Rodríguez-Cortez V.C., Javierre B.M., Mangas C., Fernández A.F., Parra M., et al. The B cell transcription program mediates hypomethylation and overexpression of key genes in Epstein-Barr virus-associated proliferative conversion. Genome Biol. 2013;14:R3. doi: 10.1186/gb-2013-14-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J.Y., Lim J., Carvalho F., Cho J.Y., Vaidyanathan B., Yu S., Annicelli C., Ip W.E., Medzhitov R. Long-term programming of CD8 T cell immunity by perinatal exposure to glucocorticoids. Cell. 2020;180:847–861.e15. doi: 10.1016/j.cell.2020.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt H.J., Belanoff J.K., Walters I., Gourdet B., Thomas J., Barton N., Unitt J., Phillips T., Swift D., Eaton E. Identification of the Clinical Candidate (R)-(1-(4-Fluorophenyl)-6-((1-methyl-1H-pyrazol-4-yl)sulfonyl)-4,4a,5,6,7,8-hexah ydro-1H-pyrazolo[3,4-g]isoquinolin-4a-yl)(4-(trifluoromethyl)pyridin-2-yl)methano ne (CORT125134): a Selective Glucocorticoid Receptor (GR) Antagonist. J. Med. Chem. 2017;60:3405–3421. doi: 10.1021/acs.jmedchem.7b00162. [DOI] [PubMed] [Google Scholar]
- Jjingo D., Conley A.B., Yi S.V., Lunyak V.V., Jordan I.K. On the presence and role of human gene-body DNA methylation. Oncotarget. 2012;3:462–474. doi: 10.18632/oncotarget.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joëls M., Karst H., Alfarez D., Heine V.M., Qin Y., Van Riel E., Verkuyl M., Lucassen P.J., Krugers H.J. Effects of chronic stress on structure and cell function in rat hippocampus and hypothalamus. Stress. 2004;7:221–231. doi: 10.1080/10253890500070005. [DOI] [PubMed] [Google Scholar]
- Jozic I., Vukelic S., Stojadinovic O., Liang L., Ramirez H.A., Pastar I., Tomic Canic M. Stress signals, mediated by membranous glucocorticoid receptor, activate PLC/PKC/GSK-3β/β-catenin pathway to Inhibit wound closure. J. Invest. Dermatol. 2017;137:1144–1154. doi: 10.1016/j.jid.2016.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer. 2016;16:582–598. doi: 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- Kim K.P., Choi J., Yoon J., Bruder J.M., Shin B., Kim J., Arauzo-Bravo M.J., Han D., Wu G., Han D.W., et al. Permissive epigenomes endow reprogramming competence to transcriptional regulators. Nat. Chem. Biol. 2020;17:47–56. doi: 10.1038/s41589-020-0618-6. [DOI] [PubMed] [Google Scholar]
- Kress C., Thomassin H., Grange T. Active cytosine demethylation triggered by a nuclear receptor involves DNA strand breaks. Proc. Natl. Acad. Sci. USA. 2006;103:11112–11117. doi: 10.1073/pnas.0601793103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F., Andrews S.R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kues W.A., Anger M., Carnwath J.W., Paul D., Motlik J., Niemann H. Cell cycle synchronization of porcine fetal fibroblasts: effects of serum deprivation and reversible cell cycle Inhibitors1. Biol. Reprod. 2000;62:412–419. doi: 10.1095/biolreprod62.2.412. [DOI] [PubMed] [Google Scholar]
- Lam L.L., Emberly E., Fraser H.B., Neumann S.M., Chen E., Miller G.E., Kobor M.S. Factors underlying variable DNA methylation in a human community cohort. Proc. Natl. Acad. Sci. USA. 2012;109:17253–17260. doi: 10.1073/pnas.1121249109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C.C., Park A.Y., Guan J.L. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- Liao Y., Wang J., Jaehnig E.J., Shi Z., Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47:W199–W205. doi: 10.1093/nar/gkz401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T.M., Lee E.H., Lim B., Shyh-Chang N. Concise review: balancing stem cell self-renewal and differentiation with PLZF. Stem Cell. 2016;34:277–287. doi: 10.1002/stem.2270. [DOI] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackey L.C., Annab L.A., Yang J., Rao B., Kissling G.E., Schurman S.H., Dixon D., Archer T.K. Epigenetic enzymes, age, and ancestry regulate the efficiency of human iPSC reprogramming. Stem Cell. 2018;36:1697–1708. doi: 10.1002/stem.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariotti A. The effects of chronic stress on health: new insights into the molecular mechanisms of brain-body communication. Future Sci OA. 2015;1:FSO23. doi: 10.4155/fso.15.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawal-Dewan M., Frisoni L., Cristofalo V.J., Sell C. Extension of replicative lifespan in WI-38 human fibroblasts by dexamethasone treatment is accompanied by suppression of p21 Waf1/Cip1/Sdi1 levels. Exp. Cell Res. 2003;285:91–98. doi: 10.1016/s0014-4827(03)00013-2. [DOI] [PubMed] [Google Scholar]
- Nanavaty V., Abrash E.W., Hong C., Park S., Fink E.E., Li Z., Sweet T.J., Bhasin J.M., Singuri S., Lee B.H., et al. DNA methylation regulates alternative polyadenylation via CTCF and the cohesin complex. Mol. Cell. 2020;78:752–764.e6. doi: 10.1016/j.molcel.2020.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Library of Medicine . 2021. What is Epigenetics?https://ghr.nlm.nih.gov/primer/howgeneswork/epigenome [Google Scholar]
- Pidsley R., Zotenko E., Peters T.J., Lawrence M.G., Risbridger G.P., Molloy P., Van Djik S., Muhlhausler B., Stirzaker C., Clark S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17:208. doi: 10.1186/s13059-016-1066-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provencal N., Arloth J., Cattaneo A., Anacker C., Cattane N., Wiechmann T., Roh S., Kodel M., Klengel T., Czamara D., et al. Glucocorticoid exposure during hippocampal neurogenesis primes future stress response by inducing changes in DNA methylation. Proc. Natl. Acad. Sci. USA. 2019;107:73–74. doi: 10.1016/j.psyneuen.2019.07.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provencal N., Arloth J., Cattaneo A., Anacker C., Cattane N., Wiechmann T., Roh S., Kodel M., Klengel T., Czamara D., et al. PREDO team Glucocorticoid exposure during hippocampal neurogenesis primes future stress response by inducing changes in DNA methylation. Proc. Natl. Acad. Sci. USA. 2020;117:23280–23285. doi: 10.1073/pnas.1820842116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramilowski J.A., Yip C.W., Agrawal S., Chang J.C., Ciani Y., Kulakovskiy I.V., Mendez M., Ooi J.L.C., Ouyang J.F., Parkinson N., et al. Functional annotation of human long noncoding RNAs via molecular phenotyping. Genome Res. 2020;30:1060–1072. doi: 10.1101/gr.254219.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramilowski J.A., Yip C.W., Agrawal S., Chang J.C., Ciani Y., Kulakovskiy I.V., Mendez M., Ooi J.L.C., Ouyang J.F., Parkinson N., et al. Functional annotation of human long noncoding RNAs via molecular phenotyping. Genome Res. 2020;30:1060–1072. doi: 10.1101/gr.254219.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholzen T., Gerdes J. The Ki-67 protein: from the known and the unknown. J. Cell. Physiol. 2000;182:311–322. doi: 10.1002/(sici)1097-4652(200003)182:3<311::aid-jcp1>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Šeda O., Šedová L., Včelák J., Vaňková M., Liška F., Bendlová B. ZBTB16 and metabolic syndrome: a network perspective. Physiol. Res. 2017;66:357–365. doi: 10.33549/physiolres.933730. [DOI] [PubMed] [Google Scholar]
- Seifuddin F., Wand G., Cox O., Pirooznia M., Moody L., Yang X., Tai J., Boersma G., Tamashiro K., Zandi P., Lee R. Genome-wide Methyl-Seq analysis of blood-brain targets of glucocorticoid exposure. Epigenetics. 2017;12:637–652. doi: 10.1080/15592294.2017.1334025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalin A.A., Hattab M.W., Clark S.L., Chan R.F., Kumar G., Aberg K.A., van den Oord E.J.C.G. RaMWAS: fast methylome-wide association study pipeline for enrichment platforms. Bioinformatics. 2018;34:2283–2285. doi: 10.1093/bioinformatics/bty069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starick S.R., Ibn-Salem J., Jurk M., Hernandez C., Love M.I., Chung H.R., Vingron M., Thomas-Chollier M., Meijsing S.H. ChIP-exo signal associated with DNA-binding motifs provides insight into the genomic binding of the glucocorticoid receptor and cooperating transcription factors. Genome Res. 2015;25:825–835. doi: 10.1101/gr.185157.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarjanyi Z., Montsko G., Kenyeres P., Marton Z., Hagendorn R., Gulyas E., Nemes O., Bajnok L., L Kovács G., Mezosi E. Free and total cortisol levels are useful prognostic markers in critically ill patients: a prospective observational study. Eur. J. Endocrinol. 2014;171:751–759. doi: 10.1530/eje-14-0576. [DOI] [PubMed] [Google Scholar]
- Thomassin H., Flavin M., Espinas M.L., Grange T. Glucocorticoid-induced DNA demethylation and gene memory during development. EMBO J. 2001;20:1974–1983. doi: 10.1093/emboj/20.8.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A.S., Armstrong E.J., Armstrong A.W. Corticosteroids and wound healing: clinical considerations in the perioperative period. Am. J. Surg. 2013;206:410–417. doi: 10.1016/j.amjsurg.2012.11.018. [DOI] [PubMed] [Google Scholar]
- Wasim M., Carlet M., Mansha M., Greil R., Ploner C., Trockenbacher A., Rainer J., Kofler R. PLZF/ZBTB16, a glucocorticoid response gene in acute lymphoblastic leukemia, interferes with glucocorticoid-induced apoptosis. J. Steroid Biochem. Mol. Biol. 2010;120:218–227. doi: 10.1016/j.jsbmb.2010.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver I.C.G., Cervoni N., Champagne F.A., D'alessio A.C., Sharma S., Seckl J.R., Dymov S., Szyf M., Meaney M.J. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Weikum E.R., Knuesel M.T., Ortlund E.A., Yamamoto K.R. Glucocorticoid receptor control of transcription: precision and plasticity via allostery. Nat. Rev. Mol. Cell Biol. 2017;18:159–174. doi: 10.1038/nrm.2016.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiechmann T., Röh S., Sauer S., Czamara D., Arloth J., Ködel M., Beintner M., Knop L., Menke A., Binder E.B., Provençal N. Identification of dynamic glucocorticoid-induced methylation changes at the FKBP5 locus. Clin. Epigenetics. 2019;11:83. doi: 10.1186/s13148-019-0682-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiench M., John S., Baek S., Johnson T.A., Sung M.H., Escobar T., Simmons C.A., Pearce K.H., Biddie S.C., Sabo P.J., et al. DNA methylation status predicts cell type-specific enhancer activity. EMBO J. 2011;30:3028–3039. doi: 10.1038/emboj.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization . World Health Organization; 2017. Noncommunicable Diseases Progress Monitor. [Google Scholar]
- Wust S., Federenko I.S., Van Rossum E.F., Koper J.W., Hellhammer D.H. Habituation of cortisol responses to repeated psychosocial stress-further characterization and impact of genetic factors. Psychoneuroendocrinology. 2005;30:199–211. doi: 10.1016/j.psyneuen.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Wynn T.A., Ramalingam T.R. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat. Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.H., Istomine R., Alvarez F., Al-Aubodah T.A., Shi X.Q., Takano T., Thornton A.M., Shevach E.M., Zhang J., Piccirillo C.A. Salt sensing by serum/glucocorticoid-regulated kinase 1 promotes Th17-like inflammatory adaptation of Foxp3(+) regulatory T cells. Cell Rep. 2020;30:1515–1529.e4. doi: 10.1016/j.celrep.2020.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong W.S., Hsu F.M., Chen P.Y. Profiling genome-wide DNA methylation. Epigenet. Chromatin. 2016;9:26. doi: 10.1186/s13072-016-0075-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannas A.S. Decoding the life story of our epigenome. Epigenomics. 2019;11:1233–1236. doi: 10.2217/epi-2019-0155. [DOI] [PubMed] [Google Scholar]
- Zannas A.S. Naturalistic stress hormone levels drive cumulative epigenomic changes along the cellular lifespan. Int. J. Mol. Sci. 2021;22:8778. doi: 10.3390/ijms22168778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannas A.S. Naturalistic stress hormone levels drive cumulative epigenomic changes along the cellular lifespan. Int. J. Mol. Sci. 2021;22:8778. doi: 10.3390/ijms22168778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannas A.S., Arloth J., Carrillo-Roa T., Iurato S., Roh S., Ressler K.J., Nemeroff C.B., Smith A.K., Bradley B., Heim C., et al. Lifetime stress accelerates epigenetic aging in an urban, African American cohort: relevance of glucocorticoid signaling. Genome Biol. 2015;16:266. doi: 10.1186/s13059-015-0828-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannas A.S., Chrousos G.P. Epigenetic programming by stress and glucocorticoids along the human lifespan. Mol. Psychiatry. 2017;22:640–646. doi: 10.1038/mp.2017.35. [DOI] [PubMed] [Google Scholar]
- Zannas A.S., Wiechmann T., Gassen N.C., Binder E.B. Gene-stress-epigenetic regulation of FKBP5: clinical and translational implications. Neuropsychopharmacology. 2016;41:261–274. doi: 10.1038/npp.2015.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannas A.S., Jia M., Hafner K., Baumert J., Wiechmann T., Pape J.C., Arloth J., Kodel M., Martinelli S., Roitman M., et al. Epigenetic upregulation of FKBP5 by aging and stress contributes to NF-κB–driven inflammation and cardiovascular risk. Proc. Natl. Acad. Sci. USA. 2019;116:11370–11379. doi: 10.1073/pnas.1816847116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannas A.S., Kosyk O., Leung C.S. Prolonged glucocorticoid exposure does not accelerate telomere shortening in cultured human fibroblasts. Genes (Basel) 2020;11:1425. doi: 10.3390/genes11121425. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-sequencing, targeted bisulfite sequencing, and EPIC array data reported in this paper are uploaded in Gene Expression Omnibus (GEO) under SuperSeries accession number GEO: GSE210304. This paper does not report original code (if no original code is used in this paper). Requests for any additional information required to reanalyze the data can be directed to the lead contact.