

Abstract
Maternal stress has been associated with poor birth outcomes, including preterm birth, infant mortality, and low birth weight. Bone development disorders in the embryo as a result of maternal stress are believed to be mediated through oxidative stress damage. Various species of free radicals, such as alkoxyl radicals, can be formed through endogenous redox response or exogenous stimuli in the womb and transmitted to embryos. Yet, whether these free radicals lead to abnormal fetal bone development is unclear. Here, we demonstrate prenatal bone growth retardation and ferroptosis-related signals of chondrocytes were induced by classic alkoxyl radical generators. We also show that alkoxyl radicals lead to significant accumulation of oxidized phospholipids in chondrocytes, through the iron-mediated Fenton reaction in embryos. We further demonstrate a role for the lipid peroxidation end product, 4-HNE, which forms adducts with the pivotal chondrogenesis transcription factor SOX9, leading to its degradation, therefore dampening chondrogenesis. Our data define a critical role for phospholipid peroxidation in alkoxyl radicals-evoked abnormal chondrogenesis, and pinpoint it being a precise target for treating oxidative stress-related bone development disorders.
Keywords: Lipid peroxidation, Ferroptosis, Alkoxyl radicals, Chondrogenic transcription factor, Embryonic bone development, Chondrogenesis
Abbreviations: 4-HNE, 4-hydroxynonenal; SOX9, SRY-related high-mobility group box 9
Graphical abstract

Highlights
-
•
Alkoxyl radicals retard embryonic skeleton development.
-
•
Alkoxyl radicals-induced accumulation of oxidized phospholipids dampen chondrogenesis.
-
•
Alkoxyl radicals trigger phospholipid peroxidation via iron-mediated Fenton reaction.
-
•
Lipid peroxidation end product, 4-HNE, forms adducts with SOX9 to promote its ubiquitin-dependant degradation.
1. Introduction
Prenatal maternal stress is a major concern for fetal and infant health due to its negative consequences in embryonic development [1]. With an estimated birth incidence of 1/10,000 to 1/30,000 [[2], [3], [4]], skeletal dysplasia is a typical developmental disorder and has been implicated in maternal stress [[5], [6], [7], [8], [9]]. By relating with the immature antioxidant system, maternal stress can induce a large amount of free radicals, making the embryos vulnerable to oxidative injury [10,11]. In fact, excessive oxidative injury has been revealed to be an essential pathological factor for maternal stress-provoked bone developmental dysfunction, although the precise mechanisms are enigmatic. Since the discovery of ferroptosis, a type of oxidative cell death, increasing attention has been paid to the pivotal role of phospholipid peroxidation in cell biology and cell pathology [[12], [13], [14]]. The peroxidation of polyunsaturated fatty acids (PUFAs) containing phospholipids (PUFAs-PL) is a free radical-driven chain reaction that can occur in an enzymatic or non-enzymatic manner, comprising of three stages of initiation, propagation, and termination [15,16]. The propagation and amplification of this oxidative reaction largely relies on the accumulation of ferrous iron [17]. The study of ferroptosis opens a new chapter in delineating the pathological mechanisms of oxidative stress-related diseases and greatly contributes to exploiting new targets and therapies [18]. Nevertheless, whether phospholipid peroxidation and ferroptosis are causatively linked to maternal stress-induced bone development defects remain unknown.
During maternal stress, various species of free radicals can be formed through endogenous oxidative stress response and exogenous stimuli, like ionising radiation, ultraviolet radiation, drugs, carcinogenic compounds, and environmental pollutants, among others [[19], [20], [21], [22]]. Of these species, alkoxy radicals play a key role in triggering lipid peroxidation by furnishing PUFA hydroperoxides, which further accelerate the propagation of free radical initiated chain reactions [23]. 2,2′-azobis [2-methylpropionamidine] dihydrochloride (AAPH), a classical generator of alkoxyl radicals, is extensively utilized in oxidative stress-related in vivo and in vitro studies [24,25], including studies on embryonic development [[26], [27], [28]]. In particular, it is worth noting that AAPH has been shown to initiate the oxidation of PUFAs-PL in cell-free conditions [29,30], whereas the presence of iron further enhances the decomposition of lipid hydroperoxides to a wide variety of secondary oxidation products [29,31]. Based on the above, in the present study, we took advantage of AAPH to obtain insights into the precise mechanism of alkoxyl radicals-induced defective bone development in chicken embryos. Our results unravel the essential role of iron-dependant phospholipid peroxidation in fetal bone development. We further show that these defects are facilitated through 4-hydroxynonenal (4-HNE), which forms adducts with the transcription factor Sry-related high-mobility group box 9 (SOX9), which is pivotal to chondrogenesis. Our findings offer a novel insight on a heretofore undefined role of phospholipid peroxidation and ferroptosis in oxidative stress-related fetal skeletal dysplasia, and provide a potential target for therapeutic intervention.
2. Materials and methods
2.1. Reagents
AAPH was purchased from Wako (Osaka, Japan). Erastin and RSL3 were obtained from Calbiochem (Darmstadt, Germany). Fer-1 and DFO were purchased from Selleck Chemicals (Houston, TX, USA). MTT, Z-vad and Nec-1 were obtained from Sigma-Aldrich (Saint Louis, MO, USA). Primary antibodies against SLC7A11, GPX4, SOX9, Ubiquitin (linkage-specific K48), and 4-HNE were purchased from Abcam (Cambridge, UK). Primary antibodies against DMT1 and TFR1 were obtained from Proteintech Group (Rosemont, IL, USA) and GAPDH antibody was obtained from Fude Co., Ltd. (Hangzhou, China). Goat anti-rabbit IgG-HRP and goat anti-mouse IgG-HRP were purchased from Santa Cruz Biotechnology, Inc. (Dallas,TX, USA). MG132, 3-MA, CHX and 4-HNE were purchased from MedchemExpress (New Jersey, USA).
2.2. Cell culture and chondrogenic differentiation
ATDC5 and SW1353 cells were purchased from RIKEN Cell Bank (Ibaraki, Japan) and cultured in Dulbecco's Modified Eagle Medium (Thermo Fisher Scientific, MA, USA) containing 5% fetal bovine serum (Life Technologies, Carlsbad, CA, USA) at 37 °C in a 5% CO2 humidified atmosphere.
The medium was replaced with a chondrogenic-induced medium additionally supplemented with 1 × insulin-transferrin-selenium (ITS, Corning, NY, USA) [32,33]. The chondrogenic culture medium was replaced daily. After differentiation for 14 days, ATDC5 cells were washed with phosphate buffer solution (PBS) and fixed with 4% paraformaldehyde solution for 10 min. Subsequently, stained with 1% alcian blue 8GX (Sigma-Aldrich, Saint Louis, MO, USA). Shanghai, China) dissolved in 0.1 M HCl overnight. Cells were washed with PBS twice and photographed.
2.3. MTT assay
MTT cell viability assay was performed as described previously [34]. Briefly, cells were cultured in 96-well plates and exposed to various concentrations of cytotoxic compounds for the indicated time. Next, add 10 μl of the MTT labeling reagent (5 mg/mL) to each well for 4 h. Then, 150 μL of dimethyl sulfoxide was added to each well and shaken gently for 10 min. The absorbance was measured at 570 nm using a SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA).
2.4. The detection of lactate dehydrogenase release
Lactate dehydrogenase (LDH) release assay is a widely accepted method for the quantitative determination of cell viability, which was performed using a cytotoxicity detection kit (Abcam, Cambridge, UK) following the manufacturer's instructions. Absorbance was quantified at 490 nm using a SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA). Cytotoxicity was then calculated according to the following equation: Cytotoxicity (%) = (experimental value–media control)/(positive control–media control) × 100.
2.5. Establishment of oxidative stress model in chicken embryos
-
(i)
Effect of AAPH on chicken embryos. Fertilized eggs (South China Agricultural University, Guangzhou, China) were incubated in a humidified incubator (Grumbach, Wentzler, Germany) at 37.5 °C and 68% humidity until they reached the desired developmental stage. Then, a range of dosages of AAPH at 0.25, 0.5, 1.0, 2.0, 4.0, 8.0, and 10.0 μmol/egg or avian saline (control, 0.72% sodium chloride) were injected into the albumen near embryos every other day from embryo development day (EDD) 1.5 to EDD 9.5. The embryos were harvested and their mortality and weight recorded at the desired stages.
-
(ii)
Effect of ferroptosis induced oxidative stress on bone development. A range of dosages of RSL3 at 0.1, 1.0, 10.0 nmol/egg were injected into the albumen near embryos every other day from EDD 1.5 to EDD 5.5. The embryos were harvested and assessed for mortality and weight.
-
(iii)
Effects of ferroptosis inhibitors on chicken embryos. Chicken embryos were treated with Fer-1 (50 nmol/egg) for 2 h before administering AAPH (0.50 μmol/egg), and the embryos were harvested on EDD17. At the same time, eggs were pre-treated with DFO (200 nmol/egg) for 2 h before AAPH administration (0.50 μmol/egg) and the embryos harvested on EDD7. The embryo weight and tibia length were determined.
2.6. iTRAQ labeling proteomics
The chicken embryo cartilage tissues were ground with liquid nitrogen. Protein quantification was performed using the BCA assay. Approximately 200 μg of each sample were digested with trypsin and tetraethylammonium bromide overnight at 37 °C. The sample was then mixed and identified by two-dimensional LC-MS/MS analysis. The original MS/MS file data were committed to the ProteinPilot™ Software 5.0.1 for analysis using the Paragon™ algorithm (5.0.1.0, 4874 AB Sciex) against Uniprot chicken database for protein identification. The resulting 5% global fits from FDR corresponding to 95% correct protein identification was used as an initial qualification criterion. For biological replicates or technological replicates, the average fold change induced by treatment relative to the control or AAPH group was defined. Statistically significant variances between AAPH group and control group were determined by the Student's t-test (two-tailed or unpaired), and the minimal value among replicates were regarded as the final p-value. For protein abundance ratios of AAPH to control measured with iTRAQ, P < 0.05 was regarded as the differential protein expression. This research was assisted by the FitGene BioTechnology proteomics platform (http://www.fitgene.com). For bioinformatic analyses, differentially expressed proteins were classified based on annotations from the UniProt knowledge database. Both Gene Ontology (GO) and Kyoto Encyclopedia of Genes and the Genomes (KEGG) pathway enrichment analyses were used to uncover the enriched pathways of significantly altered proteins and to identify their functions.
2.7. Alcian blue and alizarin red staining
To visualize fetal skeleton, chicken embryos were stained with either alcian blue alone or alcian blue and alizarin red dyes as previously described [35]. Briefly, 7-day-old chicken embryos were fixed in 95% ethanol for 24 h. The whole embryos were stained in 0.1% alcian blue (Sigma-Aldrich, Saint Louis, MO, USA) and washed with PBS. The samples were then treated with 1% KOH solution for 36 h, followed by 50%, 75%, and 100% glycerol ordinally until the embryos sank to the bottom.
Similarly, 17-day-old chicken embryos were fixed in 95% ethanol for 24 h, replacing the liquid with fresh 95% ethanol at 12 h. The embryonic skin and hair were peeled. The whole embryos were stained in 0.1% alcian blue for 10 h, treated with 1% KOH solution until transparency is achieved. Lastly, the embryos were stained with saturated alizarin red dye (Solarbio, Beijing, China) solution in 1% KOH for four days. In order to observe the skeleton more clearly, the fat on the surface of chicken embryos was removed. After the above treatment, the whole embryos were photographed. The tibias were photographed using a MVX10 stereomicroscope (Olympus, Allentown, PA, USA) equipped with an OPTPRO 2007 image acquisition system. The length of the tibia was quantified by IPP 6.0 software.
2.8. Measurement of MDA content, SOD and GPX activities
Malondialdehyde (MDA) content, superoxide dismutase (SOD) and glutathione peroxidase (GPX) activities in the bone tissues on EDD7 or EDD17 were determined using commercially available kits (Nanjing Jiancheng Institute of Biotechnology, Nanjing, China) according to the manufacturer's instructions.
2.9. Determination of GSH and GSSG contents
Levels of reduced glutathione (GSH) and oxidized glutathione (GSSG) were determined by LC-MS using a Dionex Ultimate 3000 HPLC system coupled with a Q-Exactive Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific, MA, USA). The cartilage tissues were removed from the embryonic tibia, washed with pre-cooled PBS, and quickly ground with liquid nitrogen. The powders were mixed with 1 mL of extract solution (80% methanol water plus 2 μl of 50 μg/mL glutamate-d5 (Glu-d5)), filtered using a 0.22 μm filter membrane and stored at −20 °C. For in vitro samples, cells were collected and resuspended in PBS with 1 mL of extract solution, vortexed for 5 min, then sonicated (5 Hz) for 30 s and centrifuged at 4 °C for 25 min at 14,000 rpm.
The LC/MS conditions are as follows: Waters ACQUITY UPLC BEH Amide column (2.1 mm × 100 mm, 17 μm) with acetonitrile. Solvent A was water containing 5 mM ammonium formate and 0.1% formic acid; solvent B was acetonitrile. The flow rate was 300 μL/min, and the column temperature was 40 °C. The GSH and GSSG content in the samples were calculated using a standard curve, and the ratio of GSH/GSSG in each group was then analyzed.
2.10. Hematoxylin and eosin staining (H&E staining)
Chicken embryos were harvested on EDD7 and EDD17, tibias were separated from the embryos and fixed in 4% paraformaldehyde for 24 h, decalcified completely in 4% methanol for 2 weeks, rinsed under running water for 12 h and embedded using paraffin wax. The embedded tibias were sliced at 4 μm thickness and the vertical sections were stained using H&E. The paraffin sections were photographed using a M8 Digital Microscope and Scanner (PreciPoint GmbH, Freising, Germany).
2.11. Western blot analysis
Cells or bone tissues were collected and lysed in lysis buffer containing proteinase inhibitor (Beyotime Institute of Biotechnology, Shanghai, China) for 30 min on ice. Protein concentrations were measured by the BCA assay kit (Beyotime Institute of Biotechnology, Shanghai, China). Proteins were separated by 10% SDS-PAGE and transferred to nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ, USA). After blocking with 5% dried skimmed milk, the membranes were incubated with indicated primary antibodies at 4 °C overnight. The membranes were then incubated with the appropriate HRP-linked secondary antibody for 2 h at room temperature. Enhanced chemiluminescence (ECL) detection kit (Multi Sciences Biotech Co., Ltd, Beijing, China) was used for visualization of the bands. Band density was quantified using Quantity One Analysis Software (Bio-Rad, Hercules, CA, USA).
2.12. Reverse transcription polymerase chain reaction (RT-PCR) assay
Total RNA was extracted from tissue and cells using Trizol kit (Takara, Kyoto, Japan) according to the manufacturer's instructions. Following reverse transcription, cDNA amplification was performed as previously described [36]. For RT-PCR assay, PCR products were separated on 1% agarose gels (Invitrogen, Carlsbad, CA, USA) and visualized by ethidium bromide staining. The band intensity of ethidium bromide was measured by GelDoc XR system (Bio-Rad Corporation, Hercules, CA, USA) and then quantified with the Quantity one analysis software.
Quantitative real-time polymerase chain reaction assay (qRT-PCR) was performed using SYBR Premix ExTaq II (Promega, Madison,USA) on an ABI 7900 sequence detection system (Thermo Fisher Scientific, MA, USA). Relative mRNA expression was calculated using the 2-ΔΔCt method. β-actin was used as an internal control. PCR primers are shown in Supplementary Table 1.
2.13. PI staining and flow cytometry analysis
Cells were seeded in 6-well culture plates. On the day of the experiment, cells were harvested, incubated with propidium iodide (PI, Sigma-Aldrich, Saint Louis, MO, USA) for 20 min, washed three times with PBS and re-suspended in 1 mL PBS. Stained cells were quantified by flow cytometry (BD Biosciences, NJ, USA).
2.14. Measurement of ROS using DCFH-DA
Dichlorofluorescin-diacetate (DCFH-DA) staining is used to measure the total ROS level in cells treated with or without AAPH. After incubation with AAPH, cells were stained with 50 μM DCFH-DA (Sigma-Aldrich, Saint Louis, MO, USA) for 30 min in the dark. Then, cells were resuspended in PBS and assayed by flow cytometry (BD Biosciences, NJ, USA).
2.15. Measurement of lipid peroxidation using C11-BODIPY 581/591
C11-BODIPY 581/591 staining is used to measure the lipid ROS level in cells treated with or without AAPH. Briefly, cells were seeded in 6-well plates and treated with AAPH for the indicated times. Then, cells were collected and stained with 2 μM C11-BODIPY581/591 (Thermo Fisher Scientific, MA, USA) for 30 min at 37 °C in the dark. Subsequently, cells were collected and measured by flow cytometry (BD Biosciences, NJ, USA). The fluorescence intensity of each group was recorded and the data quantified using Flowjo software.
2.16. Measurement of H2O2 using Amplex Red assay
The hydrogen peroxide (H2O2) concentration of extracellular medium was measured by the Amplex Red reagent (Thermo Fisher Scientific, MA, USA). The applied substrate (Amplex Red) reacts with H2O2 in a horseradish peroxidase catalyzed reaction, producing highly fluorescent resorufin. Fluorescence was measured by a fluorescence plate reader (excitation 535 nm, emission 595 nm).
2.17. Determination of thiol levels
Thiol levels were determined by the total sulfhydryl group assay kit (Solarbio, Beijing, China). The principle is that the reduced sulfhydryl groups react with 5,5′-dithiobis-2-nitrobenzoic acid to produce yellow 2-nitro-5-mercaptobenzoic acid [37]. The product then has a characteristic absorption peak at 412 nm and the amount of free thiol is calculated according to the formula given in the total sulfhydryl group assay kit.
2.18. Fe2+ detection by FerroOrange staining
SW1353 cells were seeded in six-well plates or confocal dishes at a density of 1 × 105 cells/mL. Cells were then incubated with the Fe2+ binding fluorescent probe FerroOrange (1 μM, Ex: 543 nm, Em: 580 nm) (Dojindo, Japan) for 30 min at 37 °C under 5% CO2 atmosphere. The culture medium was removed and the cells washed three times with PBS before flow cytometry or observation under a laser scanning confocal microscope (LSM, Zeiss, Germany).
2.19. Measurement of iron levels
Iron levels were measured by an iron assay kit (Abcam, Cambridge, UK) according to the manufacturer's instructions. Cells were collected, homogenized in 10× volume of iron assay buffer on ice and centrifuged (14,000×g, 10 min) at 4 °C. The supernatant was collected and 5 μl iron reducer was added to each sample and incubated at 37 °C for 30 min. Next, add 100 μl iron probe and incubate for 60 min at 37 °C protected from light. Absorbance was analyzed at 593 nm using a microplate reader (Thermo Fisher Scientific, MA, USA).
2.20. Co-immunoprecipitation (CO-IP) assay
SW1353 cells were seeded into a 10 cm dish and processed as indicated. Cells were lysed in RIPA lysis buffer (Beyotime Institute of Biotechnology, Shanghai, China) containing 1 mM PMSF (Beyotime Institute of Biotechnology, Shanghai, China) and protease inhibitor (Abcam, Cambridge, UK) on ice for 30 min. After centrifugation at 12,000×g for 10 min, the supernatant was incubated with indicated primary antibodies overnight. Next, protein A/G beads (Santa Cruz Biotechnology, Dallas,TX, USA) were added to the lysates and the incubation continued on a rotating table for 3-4 h. The beads were washed five times with RIPA lysis buffer, centrifuged at 2500 rpm for 1 min, and loading buffer was added to each sample. Finally, beads were boiled for 5 min at 95 °C.
2.21. Preparation of phospholipids and LC-MS/MS-based phospholipidomics analysis
Preparation and analysis of phospholipids in the chondrocytes were performed as previously described [18]. Dionex UltiMate 3000 DGLC standard system (Thermo Fisher Scientific, MA, USA) was employed to separate phospholipids with an HPLC column (C30 reverse phase column; 2.1 mm × 25 cm, 2.6 μm,Thermo Fisher Scientific, MA, USA). Solvent A was isopropanol: hexane: water (57:43:1); solvent B was isopropanol: hexane: water (57:43:8) containing 10 mM ammonium formate as mobile-phase modifiers. The gradient was as follows: 0-20 min, 10-32% solvent B; 20-30 min, 32-70% solvent B; 30-32 min, 70-100% solvent B; 32-58 min, hold at 100% solvent B; 58-60 min, 100-10% solvent B; 60-75 min, hold at 10% solvent B for equilibration. The flow rate was 200 μL/min, and the column temperature was 40 °C.
Phospholipids were detected on a Q Exactive mass spectrometer (Thermo Fisher Scientific, MA, USA), and then analyzed by Compound Discoverer 2.0 (Thermo Fisher Scientific, MA, USA) with an in-house generated analysis workflow. The species of phospholipid were identified and filtered by retention time.
2.22. Confocal microscopy
Cells were fixed with 4% paraformaldehyde for 10 min at room temperature. After washing with PBS, cells were permeabilized with 0.1% Triton-X for 10 min, blocked with 0.5% BSA for 1 h and incubated with the anti-SOX9 (1:200) and anti-4HNE (1:200) primary antibodies overnight at 4 °C. Cells were washed again and incubated with AlexaFlour 555 and 488 secondary antibodies (1:400) at room temperature for 1 h. DAPI nuclear staining was done at room temperature for 10 min. The samples were examined using a confocal laser scanning microscope (LSM 880 with AiryScan, Carl Zeiss).
2.23. CHX-chase analysis
SOX9 protein degradation was assayed by CHX-chase analysis. SW1353 cells were pre-incubated with or without 4-HNE. Subsequently, 100 μM CHX was added to inhibit protein synthesis. Cells were collected at 0, 1, 2, 4, 6, and 8 h after CHX treatment as previously described.
2.24. Statistical analysis
All data were analyzed and processed using GraphPad Prism 8.3 software. The data were expressed as mean ± SD. The statistical significance of multiple groups was evaluated using one-way analysis of variance (ANOVA) followed by Dunnett's multiple comparison test. The difference between the two groups was tested by the Student's t-test. Non-linear regression was used to estimate the IC50 value. P < 0.05 was considered to be statistically significant.
3. Results
3.1. Alkoxyl radicals exposure retard embryonic skeleton development
We established a chronic oxidative stress model in chicken embryos by injecting AAPH, a generator of alkoxyl radicals, into the albumen on embryo development day (EDD) 1.5. This treatment was given to embryos every other day for a total of five times (Fig. 1A). On EDD17, a dramatic elevation of mortality was observed in AAPH treated embryos (Fig. 1B), which corresponded with a decrease in embryo weight (Fig. 1C) and retardation in the growth of embryonic limbs (Fig. 1D). To determine whether limb deformity has already occurred during endochondral bone formation, cartilage from EDD7 embryos was stained. Consistent with the results at EDD17, embryos at EDD7 also presented shorter limbs in the AAPH treatment group (Fig. 1E).
Fig. 1.

Alkoxyl radicals exposure restrict embryonic limb growth. (A) Schematic diagram establishing a chronic oxidative injury model in chicken embryos by injecting alkoxyl radicals generator AAPH into the albumen of eggs. The first day of incubation is considered as the initiation day of embryonic development and is termed as embryo development day (EDD). (B) The mortality and (C) body weight of chicken embryos were measured on EDD17. (D) Representative images of alcian blue and alizarin red-stained tibia and the quantification of tibia length of EDD17 embryos. Scale bar, 1 cm. (E) Representative images of the alcian blue-stained tibia and the quantification of tibia length of EDD7 embryos. The whole embryos and tibias were photographed. Scale bar, 1 mm. (F) The schematic diagram of the growth plate subregions. PZ indicates proliferative zone. (G) H&E staining of tibial growth plate. (H) H&E staining of the tibial PZ mapping the white double-head arrows on EDD17. Scale bar, 200 μm. Quantification of tibial proliferation zone is shown in the right panel. AAPH, 0.50 μmol/egg. Data are represented as mean ± SD. Comparisons between groups were made using one-way ANOVA (C–E) and Student's t-test (H). *P < 0.05, **P < 0.01, ***P < 0.001 vs the Control group. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Endochondral ossification is a critical step for initial limb development in the fetus. The growth plate, which plays an important role in endochondral bone formation [38], can be divided into stagnant zone, proliferation zone (PZ), hypertrophic zone and calcified zone (Fig. 1F). H&E staining performed on paraffin sections of the tibial growth plates (Fig. 1G) showed that the structure of PZ was destroyed and the length of PZ shortened in the AAPH treatment group (Fig. 1H). Collectively, the above results illustrate that alkoxyl radicals-induced oxidative stress induces abnormal chondrogenic development, thereby impeding embryonic bone growth.
3.2. Ferroptosis is implicated in alkoxyl radicals-induced chondrogenic development defects
To identify the molecular mechanism involved in oxidative stress-induced aberrant chondrogenesis, iTRAQ-labeling proteomics analysis was performed in cartilaginous tissues from control and AAPH-treated chicken embryos (Fig. 2A). In total, 1534 credible proteins were identified (with confidence≥95%), of which 449 proteins were found to be significantly regulated in the AAPH group versus the control group. Of these, sixteen proteins were upregulated and two proteins were downregulated (Fig. 2B). Furthermore, gene set enrichment analysis (GSEA) revealed that the differentially expressed genes were enriched in iron-ion-binding pathways (Fig. 2C). KEGG analysis indicated that the assembled signaling included metabolic pathways, ferroptosis pathways, arachidonic acid metabolism, regulation of actin cytoskeleton, etc. (Fig. 2D and E). The key differentially expressed proteins involved in ferroptosis (Fig. 2F) were further confirmed by western blot analysis of the bone tissues of these embryos. Results showed that embryos exposed to AAPH had elevated expression of DMT1 and TFR1 and dramatically reduced levels of GPX4 and SLC7A11 (Fig. 2G). Additionally, AAPH treatment was also seen to boost the production of MDA, and the depletion of GSH (Fig. 2H and I), and remarkably reduce GPX and SOD activities (Fig. 2J and K) in the bone tissues. Furthermore, ferroptosis inducer RSL3 was administrated to chicken embryos and results showed that RSL3 at doses between 1.0 and 10.0 nmol/egg notably increased embryo mortality (Figure 2L) and decreased embryo weight (Figure 2M). Alcian blue staining demonstrated that RSL3 treatment remarkably hindered the growth of embryonic limbs (Figure 2N). These data offered proof underscoring the essential role of ferroptosis in oxidative stress-induced bone development disorder.
Fig. 2.

Ferroptosis is involved in alkoxyl radicals-induced bone development dysfunction. (A) Schematic diagram of iTRAQ labeling proteomic approach. (B) Volcano plot showing differentially expressed proteins in chicken embryo between the control group and AAPH group. Low expression is depicted in green, while high expression is depicted in red. AAPH, 0.50 μmol/egg. (C) Enrichment score of the iron-ion-binding pathway analyzed by GSEA based on the iTRAQ labeling proteomic datasets. (D) Sankey diagram showing the classifications of proteins based on Kyoto Encyclopedia of Genes and Genomes (KEGG) database. (E) KEGG pathway enrichment analyses. The y-axis represents the main pathways. (F) Cartoon depicting the mechanism of alkoxyl radicals-induced ferroptosis. (G) Immunoblotting analysis was performed to detect the expression of ferroptosis-related proteins. AAPH, 0.50 μmol/egg. (H–K) Effects of AAPH on the contents of MDA (H) and GSH (I), and the activities of GPX (J) and SOD (K) were measured on EDD17. AAPH, 0.50 μmol/egg. (L) The mortality and (M) body weight of chicken embryos were measured after treated with various concentrations of RSL3 on EDD7, respectively. (N) Representative images of alcian blue-stained tibia of RSL3-treated embryos. Scale bar, 1 mm. Histograms represent the quantification of tibia length of EDD7 embryos. Data are represented as mean ± SD. Comparisons between groups were made using one-way ANOVA (L–N) and Student's t-test (H–K).*P < 0.05, **P < 0.01, ***P < 0.001 vs the Control group. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
In line with chicken embryos, AAPH at a dose of 1.00 mM, which showed little impact on cell viability (Fig. 3A), remarkably repressed ITS-induced chondrogenesis in ATDC5 cells (Fig. 3B). A similar trend was also observed in the treatment of the ferroptosis inducer Erastin (Fig. 3C). Both AAPH and Erastin-caused mortality of ATDC5 cells was reversed by ferroptosis inhibitors, but not by the apoptosis inhibitor Z-VAD-FMK, and the necroptosis inhibitor Nec-1 partially protects against cell mortality caused by AAPH (Fig. 3D and E). In SW1353 cells, AAPH and RSL3 caused the release of LDH (Fig. 3F and G), and inhibited cell viability in a dose-dependent manner (Fig. 3H and I), while Fer-1 and DFO attenuated cell death (Fig. 3J). A time-dependent analysis by DCFH-DA, C11-BODIPY and Amplex Red staining, respectively, indicated that the levels of total ROS, lipid ROS and H2O2 were increasingly elevated in chondrogenic SW1353 cells upon AAPH treatment (Figs. S1A–S1C). Moreover, AAPH significantly reduced the content of sulfhydryl groups (Fig. S1D), and diminished the GSH/GSSG ratio (Figs. S1E and S1F). Altogether, these results illustrate that alkoxyl radicals impede the osteogenesis process to prohibit embryonic limb bone growth.
Fig. 3.

Alkoxyl radicals induce ferroptosis in ATDC5 and SW1353 chondrocytes. (A) ATDC5 cells were incubated with various concentrations of AAPH for 24 h. Cell viability was determined by MTT assay. (B) The representative images of ATDC5 cells stained by alcian blue. Scale bar, 50 μm. AAPH, 1.00 mM; Fer-1, 4.00 μM. (C) ATDC5 cells were incubated with various concentrations of Erastin for 24 h. Cell viability was determined by MTT assay. (D–E) ATDC5 cells were treated with AAPH, Erastin (a ferroptosis inducer), Fer-1 and DFO (ferroptosis inhibitors), Z-vad (an apoptosis inhibitor), and Nec-1 (a necroptosis inhibitor), then cell viability were measured by MTT. AAPH, 12.50 mM; Erastin, 32.00 μM; Fer-1, 4.00 μM; DFO, 8.00 μM; Z-vad, 25.00 μM; Nec-1, 25.00 μM. (F–G) SW1353 cells were treated with different concentrations of AAPH and RSL3 for 24 h, and cytotoxicity were evaluated by LDH assay. (H–I) Cell viability of AAPH and RSL3 in SW1353 cells were determined by MTT assay. (J) Cell death was determined by PI staining coupled with flow cytometry in SW1353 cells. AAPH, 20 mM; RSL3, 60 nM; DFO, 200 μM; Fer-1, 5 μM. Data are represented as mean ± SD. Comparisons between groups were made using one-way ANOVA. **P < 0.01, ***P < 0.001 vs the Control group. ###P < 0.001 vs the AAPH-treated group; $$$P < 0.001 vs the Erastin-treated group. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.3. Ferroptosis inhibitors rescue alkoxyl radicals-induced chondrogenic developmental defects
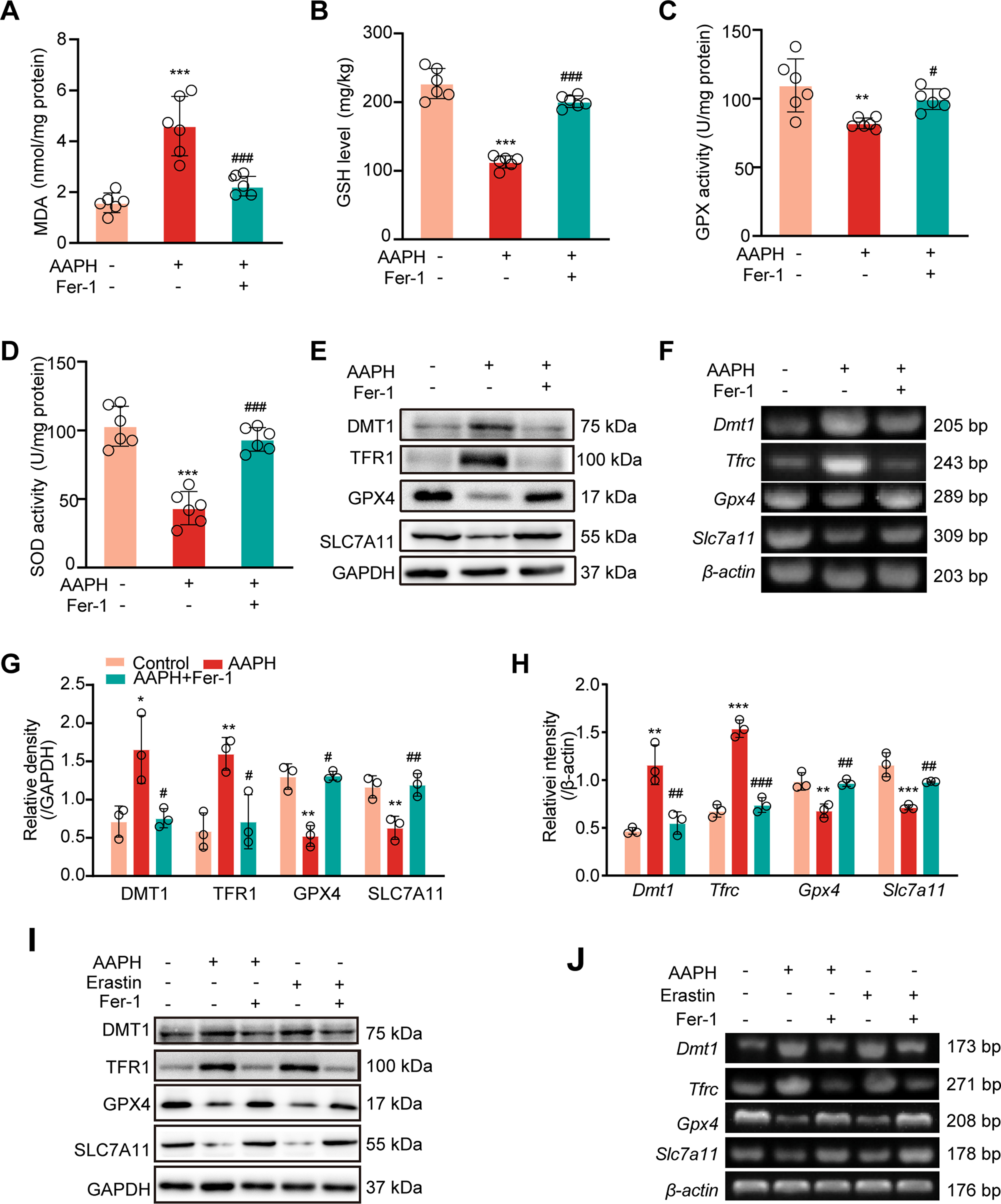
To affirm the role of ferroptosis in abnormal bone development, Fer-1 and DFO, two classic ferroptosis inhibitors, were used in vivo. As expected, Fer-1 significantly rescued AAPH-induced mortality (Fig. 4A) and weight loss of chicken embryos on EDD17 (Fig. 4B). AAPH-impeded growth of the tibia was also attenuated by Fer-1 (Fig. 4C and D). H&E staining showed that Fer-1 treatment notably rescued the shrinkage of tibial PZ in the developing bones (Fig. 4E and F). A similar trend was also observed in chicken embryos treated with the ferroptosis inhibitor DFO. In line with EDD17, EDD7 embryos also showed increased mortality and weight loss after AAPH exposure, which were significantly rescued by DFO (Fig. 4G and H). Moreover, DFO treatment also significantly alleviated the shortening of the tibia caused by AAPH (Fig. 4I and J).
Fig. 4.

Inhibition of ferroptosis rescues alkoxyl radicals-induced bone growth retardation. (A) The mortality and (B) body weight of chicken embryos were measured on EDD17. AAPH, 0.50 μmol/egg; Fer-1, 50 nmol/egg. (C) Representative images of chicken embryos stained with alcian blue and alizarin red on EDD17. The embryos were treated with Fer-1 (50 nmol/egg) and AAPH (0.50 μmol/egg). Scale bar, 1 cm. (D) Quantification of tibia length of EDD17 embryos. (E–F) The tibial paraffin sections were stained by H&E staining. The regions surrounded by white dashed lines were PZ in the growth plate, photographed by a microscope. Scale bar, 200 μm. (G) Mortality and (H) body weight of chicken embryos were measured on EDD7. AAPH, 0.50 μmol/egg; DFO, 200 nmol/egg. (I–J) Representative images and quantification of chicken embryos stained by alcian blue on EDD7. The embryos were treated with DFO (200 nmol/egg) and AAPH (0.50 μmol/egg). Scale bar, 1 mm. Data are represented as mean ± SD. Comparisons between groups were made using one-way ANOVA.**P < 0.01 and ***P < 0.001 vs the Control group. #P < 0.05, ##P < 0.01 and ###P < 0.001 vs the AAPH-treated group. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Next, the effect of ferroptosis inhibition on alkoxyl radicals-initiated redox imbalance was examined in chicken embryos. The studies revealed that Fer-1 and DFO dramatically abrogated AAPH-caused accumulation of MDA (Figs. S2A and S3A), depletion of GSH (Figs. S2B and S3B), attenuation of GPX (Figs. S2C and S3C) and SOD activities (Figs. S2D and S3D). Consistent with the results described above, Fer-1 and DFO restored expressions of DMT1, TFR1, GPX4 and SLC7A11 disturbed by AAPH exposure (Figs. S2E–2H, Figs. S3E and S3F). A similar expression pattern of ferroptosis-related proteins, including DMT1, TFR1, SLC7A11 and GPX4, was also seen in AAPH-exposed ATDC5 cells and SW1353 cells (Figs. S2I and S3G), which were arrested by ferroptosis inhibitors (Figs. S2I and S2J, Figs. S3G and S3H). Overall, these data support a concept linking ferroptosis to alkoxyl radicals-induced bone developmental disorders.
3.4. Exposure to alkoxyl radicals trigger iron overload in chondrocytes
Iron is required for Fenton reaction-propagating phospholipid peroxidation [39]. DMT1 and TFR1 are two major iron transporters that contribute to iron metabolism in cells [[40], [41], [42]]. The expression patterns of DMT1 and TFR1 observed from the ITAQ assay (Fig. 2A) and western blot analysis (Fig. 2G) implied the involvement of iron in alkoxyl radicals-disturbed chondrogenesis. Therefore, we examined changes of intracellular iron by flow cytometry and iron assay kit in chondrogenic cells. High Fe2+ levels were observed in AAPH/RSL3-treated cells, which were abolished by Fer-1 and DFO administration (Fig. 5A-C).To further understand the role of iron in chondrogenesis, siRNA based reduction in cellular DMT1 was achieved (Fig. 5D). It was determined that loss of DMT1 reversed AAPH triggered suppression of osteogenesis-related genes BMP6, COL2A1 and ACAN (Fig. 5E). Taken together, these in vitro findings provide support for a critical role for iron overload in oxidative stress-disturbed embryonic osteogenesis.
Fig. 5.

Alkoxyl radicals trigger iron overload in chondrocytes. (A) Levels of intracellular iron were detected by flow cytometry after incubating with Fe2+ ions fluorescent probe FerroOrange in SW1353 cells subjected to different treatments. AAPH, 15 mM; RSL3, 30 nM; DFO, 200 μM; Fer-1, 5 μM. (B) Levels of intracellular iron were detected by iron assay kit in SW1353 cells subjected to different treatments. (C) Representative confocal microscopy images of FerroOrange staining in SW1353 cells. Scale bar, 20 μm. (D) The verification of DMT1 knockdown with siDMT1 in SW1353 cells. (E) Relative mRNA levels of BMP6, COL2A1 and ACAN in SW1353 cells were determined after transfected with siDMT1. Data are represented as mean ± SD. Comparisons between groups were made using one-way ANOVA. *P < 0.05, **P < 0.01 and ***P < 0.001 vs the Control group. #P < 0.05, ##P < 0.01 and ###P < 0.001 vs the AAPH-treated group.
3.5. Accumulation of oxidized phospholipids (OxPLs) in alkoxyl radicals-exposed chondrocytes dampens chondrogenesis
Since oxidation of PUFA at the bis-allylic position of phospholipid is a driving factor for peroxidation and ferroptosis, we measured the level of lipid peroxidation by C11-BODIPY staining. The results shown in Fig. 6A demonstrate that AAPH or Erastin treatment leads to accumulation of lipid peroxidation products in cells, which were inhibited by Fer-1 treatment. Redox lipidomics analysis by LC-MS/MS identified five major classes of OxPLs, including phosphatidyl ethanolamine (PE), phosphatidylcholine (PC), phosphatidylserine (PS), phosphatidylglycerol (PG) and phosphatidylinositol (PI) (Fig. 6B). Notably, the AAPH-treated group displayed distinct patterns with more lipid peroxidation compared with the control group (Fig. 6C). A large number of mono-oxygenated and di-oxygenated phospholipid species were increased in the AAPH-treated group (Fig. 6D and E), for example, SAPE-OOH (PE(38:4)+2O). To determine whether SAPE-OOH could directly contribute to the observed effects on bone development, ATDC5 cells were treated with exogenous SAPE-OOH. Consistent with AAPH treatment, incubating cells with SAPE-OOH was found to inhibit Bmp6, Co12a1, and Acan genes, which were associated with bone development (Fig. 6F). These data define the accumulation of OxPLs in alkoxyl radicals-exposed chondrocytes, which suppresses the chondrogenesis process.
Fig. 6.

Oxidized phospholipids accumulates in alkoxyl radicals-treated chondrocytes and suppress bone development. (A) Lipid peroxidation level was detected by flow cytometry after incubating with C11-BODIPY in ATDC5 cells. (B) Normal phase LC/MS chromatogram (grey) and mass spectra of PLs in ATDC5 cells. (C) OxPLs in chondrocytes were extracted and analyzed by principal component analysis, and the 2D score plots display repertoires of control and AAPH-treated embryos. Each point represents a sample, and ellipses represent 95% confidence regions (n = 4). (D–E) Heat map and Volcano plot showing differentially expressed OxPLs between control group and AAPH group. Each dot represents one class of phospholipids and the fold change was set to >2.0. (F) Expressions of chondrogenesis-related genes were measured by qRT-PCR. ATDC5 cells were treated with SAPE-OOH (2 μM). Data are presented as mean ± SD. Comparisons between groups were made using one-way ANOVA (A) and Student's t-test (F). **P < 0.01, ***P < 0.001 vs the Control group. ###P < 0.001 vs the AAPH-treated group. $$P < 0.01 vs the Erastin-treated group.
3.6. 4-HNE modification drives ubiquitin-dependant degradation of SOX9
The transcription factor SOX9 is a key regulator of chondrogenesis [[43], [44], [45]]. When delineating AAPH-induced alteration of chondrogenesis-related pathways, we determined that SOX9 protein levels, rather than mRNA levels, were altered by AAPH treatment (Fig. 7A and B) in chicken embryos and chondrogenic cells (Figs. S4A–S4C). Meanwhile, AAPH substantially perturbed chondrogenesis-related genes, including Bmp6, Co12a1 and Acan, which were rescued by DFO treatment (Fig. 7C). Moreover, Fer-1 and DFO treatment abrogated AAPH-induced down-regulation of SOX9 protein (Figs. S4B and S4C), thus normalizing the expressions of BMP6, COL2A1 and ACAN in SW1353 cells (Fig. S4D).
Fig. 7.

AAPH induces 4-HNE modification of SOX9 and promotes its ubiquitin-proteasomal degradation. (A) Relative mRNA level of Sox9 in chicken embryos. (B) Immunoblotting analysis was performed to detect the level of SOX9 protein after treated with DFO and AAPH in chicken embryos. (C) Expressions of chondrogenesis-related genes were measured by qRT-PCR in chicken embryos treated with DFO and AAPH. (D) Protein expression of 4-HNE was visualized by immunoblotting in SW1353 cells after treatment with AAPH (15 mM). (E) CO-IP assay determined the level of SOX9 conjuncted with 4-HNE in SW1353 cells. (F) Representative confocal images showing the colocalization of SOX9 (green) and 4-HNE (red). Scale bars, 20 μm. (G) CO-IP assay determined the level of SOX9 conjuncted with 4-HNE in siDMT1-treated SW1353 cells. (H) Turnover of SOX9 by CHX chase assay in SW1353 cells with or without 4-HNE. CHX, 100 μM; 4-HNE, 10 μM. (I) Effects of MG132 (10 μM) or 3-MA (20 μM) on the AAPH-induced level of SOX9 protein. The diagram showed quantitative analysis of SOX9 protein levels. (J–K) Ubiquitination of SOX9 was assessed by immunoblotting in SW1353 cells (J) and siDMT1-treated SW1353 cells (K). Data are represented as mean ± SD. Comparisons between groups were made using one-way ANOVA (A–C) and Student's t-test (H). *P < 0.05, **P < 0.01 and ***P < 0.001 vs the Control group. #P < 0.05 and ##P < 0.01 vs the AAPH-treated group. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
4-HNE, an end product of lipid peroxidation, has important electrophilic properties and is able to react with a variety of proteins to form HNE-protein adducts. HNE-protein adducts formation is known to affect the fate of these modified proteins by altering their activities or inducing degradation [46]. In this study, we noticed that 4-HNE-conjugated protein level was considerably increased after AAPH treatment (Fig. 7D). Given this intriguing fact, we proposed whether the decrease of SOX9 at protein level was correlative with 4-HNE modification. To prove this hypothesis, we performed Co-IP assay to determine the interaction between SOX9 and 4-HNE in SW1353 cells, and results indicated that AAPH exposure led to an enhanced level of SOX9 bound to 4-HNE (Fig. 7E). Immunofluorescence staining further confirmed the co-localization of 4-HNE and SOX9 (Fig. 7F). As expected, knockdown of DMT1 by siRNA weakened the conjugation of SOX9 with 4-HNE (Fig. 7G).
Next, a CHX chase experiment was designed to understand 4-HNE modification-induced SOX9 degradation. These experiments revealed that while degradation of SOX9 protein level was dramatically promoted following exogenous 4-HNE treatment (Fig. 7H), the proteasome inhibitor MG132 inhibited AAPH-triggered decline in SOX9 protein levels, while the autophagy inhibitor 3-MA had little effect (Fig. 7I). These data prompted analysis of the ubiquitin-dependant degradation pathways. SW1353 cells were co-transfected with ubiquitin and SOX9 plasmids. AAPH treatment in these cells was seen to promote K48 ubiquitination of SOX9, which was reversed by knockdown of DMT1 (Fig. 7J and K). Altogether, these data suggest that the lipid peroxidation end product 4-HNE commands ubiquitin-proteasomal degradation of SOX9 via protein modification in chondrocytes.
4. Discussion
Oxidative stress has long been deemed as a detrimental factor for embryonic bone developmental disorders [47,48]. Increasing studies have reported that fetal bone is extremely sensitive to environmental influences in pregnancy, during which adverse exposures such as oxidative stress will reduce the weight and growth rate of newborns, thereby increasing the risk of skeletal disorders [7,49,50]. although the precise mechanism has been elusive. Almeida et al. found that the levels of lipid peroxidation and expression of lipoxygenases increased in the murine skeleton with age, which correlated with loss of bone mineral mass [51]. Mutation of GPX4, a phospholipid hydroperoxidase, has also been reported to cause Sedaghatian-type spondylometaphyseal dysplasia [52]. Clinical studies have shown that preterm birth, low birth weight, growth restriction and developmental malformations are associated with lipid peroxidation in the fetus [49,53]. At birth, preterm infants have been reported to have higher levels of lipid peroxidation than full-term infants, as revealed by higher plasma MDA and presence of lipid peroxides [54,55]. These observations suggest that accumulation of lipid peroxides is a risk for preterm birth.
Alkoxyl radicals, which can be endogenously produced in large quantities are known to be important for ferroptosis [18,[56], [57], [58]]. In the present study, by utilizing AAPH, it was revealed that alkoxyl radicals impede bone development by inducing chondrocyte ferroptosis. To our knowledge, this study provides the first evidence demonstrating a negative role for ferroptosis in embryonic bone development.
The propagation of lipid peroxidation ascribes to iron-dependant Fenton reactions and lipoxygenases-catalyzed oxidation [59,60]. Inhibiting iron accumulation inhibits accumulation of lipid peroxides [61,62] and prevents ferroptotic cell death [63]. In the present study, expression analysis of iron transporters (TFR1 and DMT1), FerroOrange staining, iron chelator, and siRNA gene knockdown, were used to demonstrate that iron accumulation is central to alkoxyl radicals mediated disturbance in chondrogenesis. These findings are in line with a previous report showing that iron overload resulting from upregulation of iron transporters, resulted in changes in bone microarchitecture and bone loss [64]. Yamada et al. used a cell-free system to show that while AAPH autoxidation resulted in an excessive accumulation of lipid hydroperoxides, iron and other metal ions further promoted the transformation of lipid hydroperoxides to many secondary oxidative products, thus amplifying lipid peroxidation [29]. These observations are in harmony with the collaborative mode of alkoxyl radicals and iron identified in our study.
We further determined that oxidative stress negatively affects embryonic bone development via 4-HNE dependent conjugation of the transcription factor SOX9, a master regulator in the osteogenesis process. This explosive burst of phospholipid peroxidation products produces a large amount of 4-HNE [65,66], which promotes carbonylation of SOX9, leading to its ubiquitin-dependant degradation. Loss of SOX9 eventually impedes transduction of signaling guiding osteogenesis, thereby impacting normal bone development.
5. Conclusions
In the present study, we characterize an essential role of ferroptosis in dysplasia in embryonic bone development by establishing an oxidative injury model in chicken embryos and chondrocytes. Alkoxyl radicals give rise to the accumulation of lipid peroxidation by a synergic mechanism involving the initiation of auto-oxidative reaction and iron-mediated amplification. The lipid peroxidation end product 4-HNE triggered SOX9 modification, which leads to its ubiquitin-mediated degradation in chondrocytes. Our findings show that ferroptosis is switched on by oxidative stress, which leads to dysfunction in embryonic bone development. These findings provide support for use of anti-ferroptosis agents in treating oxidative stress dependent defects in bone development.
Author contributions
R. R. He, Y.F. Li, and H Kurihara conceived and designed the research. J Niu, X Wan, G.Y. Yu, S Jiang, R.N. Yi, Y.P. Wu, and S·H Ouyang performed the experiments. J Niu, L Liang, X Wan, and W.Y. Sun contributed to the acquisition and analysis of the data. J Niu and W.J. Duan prepared figures, tables and the manuscript. Y.F. Cao, X.F. Zhu, and R.H. Zhang partly advised the research. Y.F. Cao supported the LC-MS platform. R.R. He, Y.F. Li, W.J. Duan, and J.B. R.R. He and Y.F. Li revised and approved the manuscript. All of the authors have approved the final manuscript.
Availability of data and materials
The datasets used to support the findings of this study are available from the corresponding author upon request.
Declaration of competing interest
The authors declare no conflicts of interest.
Acknowledgments
This study was partly supported by National Key Research and Development Program of China (grant number 2018YFC2002500), Natural Science Foundation of China (grant number 82004012, 82125038, 81873209, 81973718 and 81903821), Natural Science Foundation of Guangdong (2019A1515010909, 2021A1515011297, 2021B1515120023), the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (grant number 2017BT01Y036), and the Innovation Team Project of Guangdong Provincial Department of Education (2020KCXTD003) and GDUPS (2019). The authors (Rong-Rong He and Yi-Fang Li) also gratefully acknowledge the support of K.C. Wong Education Foundation.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102437.
Contributor Information
Jian-Bo He, Email: hejianbo9@163.com.
Wen-Jun Duan, Email: duanwj@jnu.edu.cn.
Yi-Fang Li, Email: liyifang706@jnu.edu.cn.
Rong-Rong He, Email: rongronghe@jnu.edu.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
AAPH boosts oxidative stress in SW1353 cells. (A) ROS generation and lipid peroxidation levels were detected by flow cytometry after incubation with DCFH-DA or C11-BODIPY in AAPH-treated SW1353 cells. AAPH, 15 mM. (B) Statistical plots showing ROS level and lipid peroxidation level in AAPH-treated SW1353 cells at different time. AAPH, 15 mM. (C) The concentration of H2O2 in the extracellular medium was calculated by the Amplex Red assay. (D) Thiol generation was measured by the Total Sulfhydryl Group Content Assay Kit after AAPH treatment (15 mM). (E) The ratio of GSH/GSSG was measured after treatment with AAPH in SW1353 cells (15 mM). (F) The extracted ion current of GSH and GSSG. Glu-d5 has been used as an internal standard. Data are represented as mean ± SD. Comparisons between groups were made using one-way ANOVA (B and C) and Student's t-test (D and E). ***P < 0.001 vs Control group (DCFH-DA staining). ##P < 0.01 and ###P < 0.001 vs the Control group (C11-BODIPY staining). @P < 0.05, @@P < 0.01 vs the Control group (Amplex Red assay). $P < 0.05, $$P < 0.01 vs the AAPH-treated group.
{kind=link}
AAPH-induced oxidative stress is alleviated by ferroptosis inhibitor Fer-1. (A-D) Effects of co-treatment with AAPH and Fer-1 on the contents of MDA and GSH, and the activities of GPX and SOD were measured on EDD17. Fer-1 (50 nmol/egg) and AAPH (0.50 μmol/egg). (E-H) The effects of Fer-1 on AAPH-induced alterations in ferroptosis-related markers determined by western blot assay and RT-PCR on EDD17. (I) Protein levels of DMT1, TFR1, GPX4, and SLC7A11 were determined by western blotting in ATDC5 cells. (J) Relative mRNA levels determination of Dmt1, Tfrc, Gpx4 and Slc7a11 by RT-PCR in ATDC5 cells. Data are represented as mean ± SD. Comparisons between groups were made using one-way ANOVA. *P < 0.05, **P < 0.01 and ***P < 0.001 vs the Control group. #P < 0.05, ##P < 0.01 and ###P < 0.001 vs the AAPH-treated group.
{kind=link}
Inhibition of ferroptosis rescues AAPH-induced oxidative stress. (A-D) MDA, GSH levels and GPX and SOD activities in EDD7 chicken embryos. (E) Expressions of ferroptosis-related protein (DMT1, TFR1, GPX4, SLC7A11) in EDD7 chicken embryos were determined by western blotting analysis. (F) Levels of ferroptosis-related gene (Dmt1, Tfrc, Gpx4, Slc7a11) in EDD7 chicken embryos were detected by qRT-PCR. (G) Expressions of ferroptosis markers (DMT1, TFR1, GPX4 and SLC7A11) were determined by western blotting analysis in SW1353 cells. (H) Relative mRNA levels of DMT1, TFRC, GPX4 and SLC7A11 in SW1353 cells as determined by qRT-PCR. Data are represented as mean ± SD. Comparisons between groups were made using one-way ANOVA.*P < 0.05, **P < 0.01 and ***P < 0.001 vs the Control group. #P < 0.05, ##P < 0.01 and ###P < 0.001 vs the AAPH-treated group.
{kind=link}
Ferroptosis is engaged in AAPH-induced differentiation disruption of chondrogenic cells in vitro. (A) The mRNA level of SOX9 was detected by qRT-PCR in SW1353 cells. (B–C) Level of SOX9 was determined by western blotting analysis in SW1353 cells. AAPH, 15 mM; RSL3, 30 nM; DFO, 200 μM; Fer-1, 5 μM. (D) The mRNA levels of BMP6, COL2A1 and ACAN were measured by qRT-PCR in SW1353 cells. Data are represented as mean ± SD. Comparisons between groups were made using one-way ANOVA.*P < 0.05, **P < 0.01 and ***P < 0.001 vs the Control group. #P < 0.05, ##P < 0.01 and ###P < 0.001 vs the AAPH-treated group.
{kind=link}
Data availability
Data will be made available on request.
References
- 1.Fleming T.P., Watkins A.J., Velazquez M.A., Mathers J.C., Prentice A.M., Stephenson J., Barker M., Saffery R., Yajnik C.S., Eckert J.J., et al. Origins of lifetime health around the time of conception: causes and consequences. Lancet (London, England) 2018;391(10132):1842–1852. doi: 10.1016/S0140-6736(18)30312-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoover-Fong J., Scott C.I., Jones M.C. Health supervision for people with achondroplasia. Pediatrics. 2020;145(6) doi: 10.1542/peds.2020-1010. [DOI] [PubMed] [Google Scholar]
- 3.Savarirayan R., Tofts L., Irving M., Wilcox W., Bacino C.A., Hoover-Fong J., Ullot Font R., Harmatz P., Rutsch F., Bober M.B., et al. Once-daily, subcutaneous vosoritide therapy in children with achondroplasia: a randomised, double-blind, phase 3, placebo-controlled, multicentre trial. Lancet (London, England) 2020;396(10252):684–692. doi: 10.1016/S0140-6736(20)31541-5. [DOI] [PubMed] [Google Scholar]
- 4.Zhou F., Mei J., Han X., Li H., Yang S., Wang M., Chu L., Qiao H., Tang T. Kinsenoside attenuates osteoarthritis by repolarizing macrophages through inactivating NF-κB/MAPK signaling and protecting chondrocytes. Acta Pharm. Sin. B. 2019;9(5):973–985. doi: 10.1016/j.apsb.2019.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kimura T., Hino K., Kono T., Takano A., Nitta N., Ushio N., Hino S., Takase R., Kudo M., Daigo Y., et al. Maternal undernutrition during early pregnancy inhibits postnatal growth of the tibia in the female offspring of rats by alteration of chondrogenesis. Gen. Comp. Endocrinol. 2018;260:58–66. doi: 10.1016/j.ygcen.2017.12.008. [DOI] [PubMed] [Google Scholar]
- 6.Wang A., Zsengellér Z.K., Hecht J.L., Buccafusca R., Burke S.D., Rajakumar A., Weingart E., Yu P.B., Salahuddin S., Karumanchi S.A. Excess placental secreted frizzled-related protein 1 in maternal smokers impairs fetal growth. J. Clin. Invest. 2015;125(11):4021–4025. doi: 10.1172/JCI80457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snow M.E., Keiver K. Prenatal ethanol exposure disrupts the histological stages of fetal bone development. Bone. 2007;41(2):181–187. doi: 10.1016/j.bone.2007.04.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu H.L., Shi X.T., Xu X.F., Zhou G.X., Xiong Y.W., Yi S.J., Liu W.B., Dai L.M., Cao X.L., Xu D.X., et al. Melatonin protects against environmental stress-induced fetal growth restriction via suppressing ROS-mediated GCN2/ATF4/BNIP3-dependent mitophagy in placental trophoblasts. Redox Biol. 2021;40 doi: 10.1016/j.redox.2021.101854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hedegaard M., Henriksen T.B., Sabroe S., Secher N.J. Psychological distress in pregnancy and preterm delivery. Br. Med. J. 1993;307(6898):234–239. doi: 10.1136/bmj.307.6898.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kimura T., Hino K., Kono T., Takano A., Nitta N., Ushio N., Hino S., Takase R., Kudo M., Daigo Y., et al. Maternal undernutrition during early pregnancy inhibits postnatal growth of the tibia in the female offspring of rats by alteration of chondrogenesis. Gen. Comp. Endocrinol. 2018;260:58–66. doi: 10.1016/j.ygcen.2017.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Tipple T.E., Ambalavanan N. Oxygen toxicity in the neonate: thinking beyond the balance. Clin. Perinatol. 2019;46(3):435–447. doi: 10.1016/j.clp.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang X., Stockwell B.R., Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021;22(4):266–282. doi: 10.1038/s41580-020-00324-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan H.F., Zou T., Tuo Q.Z., Xu S., Li H., Belaidi A.A., Lei P. Ferroptosis: mechanisms and links with diseases. Signal Transduct. Targeted Ther. 2021;6(1):49. doi: 10.1038/s41392-020-00428-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Espinós C., Galindo M.I., García-Gimeno M.A., Ibáñez-Cabellos J.S., Martínez-Rubio D., Millán J.M., Rodrigo R., Sanz P., Seco-Cervera M., Sevilla T., et al. Oxidative stress, a crossroad between rare diseases and neurodegeneration. Antioxidants. 2020;9(4) doi: 10.3390/antiox9040313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liang D., Minikes A.M., Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol. Cell. 2022;82(12):2215–2227. doi: 10.1016/j.molcel.2022.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bayır H., Anthonymuthu T.S., Tyurina Y.Y., Patel S.J., Amoscato A.A., Lamade A.M., Yang Q., Vladimirov G.K., Philpott C.C., Kagan V.E. Achieving life through death: redox biology of lipid peroxidation in ferroptosis. Cell chemical biology. 2020;27(4):387–408. doi: 10.1016/j.chembiol.2020.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng Z., Li Y. What is responsible for the initiating chemistry of iron-mediated lipid peroxidation: an update. Chem. Rev. 2007;107(3):748–766. doi: 10.1021/cr040077w. [DOI] [PubMed] [Google Scholar]
- 18.Sun W.Y., Tyurin V.A., Mikulska-Ruminska K., Shrivastava I.H., Anthonymuthu T.S., Zhai Y.J., Pan M.H., Gong H.B., Lu D.H., Sun J., et al. Phospholipase iPLA(2)β averts ferroptosis by eliminating a redox lipid death signal. Nat. Chem. Biol. 2021;17(4):465–476. doi: 10.1038/s41589-020-00734-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al-Gubory K.H. Environmental pollutants and lifestyle factors induce oxidative stress and poor prenatal development. Reprod. Biomed. Online. 2014;29(1):17–31. doi: 10.1016/j.rbmo.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 20.Sreetharan S., Stoa L., Cybulski M.E., Jones D.E., Lee A.H., Kulesza A.V., Tharmalingam S., Boreham D.R., Tai T.C., Wilson J.Y. Cardiovascular and growth outcomes of C57Bl/6J mice offspring exposed to maternal stress and ionizing radiation during pregnancy. Int. J. Radiat. Biol. 2019;95(8):1085–1093. doi: 10.1080/09553002.2019.1589025. [DOI] [PubMed] [Google Scholar]
- 21.Shivananjappa M.M., Muralidhara Differential oxidative stress induction and lethality of rat embryos after maternal exposure to t-butyl hydroperoxide during postimplantation period. Drug Chem. Toxicol. 2013;36(2):209–216. doi: 10.3109/01480545.2012.710622. [DOI] [PubMed] [Google Scholar]
- 22.Xiong Y.W., Zhu H.L., Nan Y., Cao X.L., Shi X.T., Yi S.J., Feng Y.J., Zhang C., Gao L., Chen Y.H., et al. Maternal cadmium exposure during late pregnancy causes fetal growth restriction via inhibiting placental progesterone synthesis. Ecotoxicol. Environ. Saf. 2020;187 doi: 10.1016/j.ecoenv.2019.109879. [DOI] [PubMed] [Google Scholar]
- 23.Miotto G., Rossetto M., Di Paolo M.L., Orian L., Venerando R., Roveri A., Vučković A.M., Bosello Travain V., Zaccarin M., Zennaro L., et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020;28 doi: 10.1016/j.redox.2019.101328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barygina V., Becatti M., Lotti T., Moretti S., Taddei N., Fiorillo C. ROS-challenged keratinocytes as a new model for oxidative stress-mediated skin diseases. J. Cell. Biochem. 2019;120(1):28–36. doi: 10.1002/jcb.27485. [DOI] [PubMed] [Google Scholar]
- 25.Li Y.F., Ouyang S.H., Tu L.F., Wang X., Yuan W.L., Wang G.E., Wu Y.P., Duan W.J., Yu H.M., Fang Z.Z., et al. Caffeine protects skin from oxidative stress-induced senescence through the activation of autophagy. Theranostics. 2018;8(20):5713–5730. doi: 10.7150/thno.28778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He R.R., Li Y., Li X.D., Yi R.N., Wang X.Y., Tsoi B., Lee K.K., Abe K., Yang X., Kurihara H. A new oxidative stress model, 2,2-azobis(2-amidinopropane) dihydrochloride induces cardiovascular damages in chicken embryo. PLoS One. 2013;8(3) doi: 10.1371/journal.pone.0057732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y., Wang X.Y., Zhang Z.L., Cheng X., Li X.D., Chuai M., Lee K.K., Kurihara H., Yang X. Excess ROS induced by AAPH causes myocardial hypertrophy in the developing chick embryo. Int. J. Cardiol. 2014;176(1):62–73. doi: 10.1016/j.ijcard.2014.06.044. [DOI] [PubMed] [Google Scholar]
- 28.Wu T., Yu G.Y., Xiao J., Yan C., Kurihara H., Li Y.F., So K.F., He R.R. Fostering efficacy and toxicity evaluation of traditional Chinese medicine and natural products: chick embryo as a high throughput model bridging in vitro and in vivo studies. Pharmacol. Res. 2018;133:21–34. doi: 10.1016/j.phrs.2018.04.011. [DOI] [PubMed] [Google Scholar]
- 29.Matsuoka Y., Takahashi M., Sugiura Y., Izumi Y., Nishiyama K., Nishida M., Suematsu M., Bamba T., Yamada K.I. Structural library and visualization of endogenously oxidized phosphatidylcholines using mass spectrometry-based techniques. Nat. Commun. 2021;12(1):6339. doi: 10.1038/s41467-021-26633-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xin S., Mueller C., Pfeiffer S., Kraft V.A.N., Merl-Pham J., Bao X., Feederle R., Jin X., Hauck S.M., Schmitt-Kopplin P., et al. MS4A15 drives ferroptosis resistance through calcium-restricted lipid remodeling. Cell Death Differ. 2022;29(3):670–686. doi: 10.1038/s41418-021-00883-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuoka Y., Izumi Y., Takahashi M., Bamba T., Yamada K.I. Method for structural determination of lipid-derived radicals. Anal. Chem. 2020;92(10):6993–7002. doi: 10.1021/acs.analchem.0c00053. [DOI] [PubMed] [Google Scholar]
- 32.Kurakazu I., Akasaki Y., Hayashida M., Tsushima H., Goto N., Sueishi T., Toya M., Kuwahara M., Okazaki K., Duffy T., et al. FOXO1 transcription factor regulates chondrogenic differentiation through transforming growth factor β1 signaling. J. Biol. Chem. 2019;294(46):17555–17569. doi: 10.1074/jbc.RA119.009409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dong X., Xu X., Yang C., Luo Y., Wu Y., Wang J. USP7 regulates the proliferation and differentiation of ATDC5 cells through the Sox9-PTHrP-PTH1R axis. Bone. 2021;143 doi: 10.1016/j.bone.2020.115714. [DOI] [PubMed] [Google Scholar]
- 34.Vu T.O., Tran P.T., Seo W., Lee J.H., Min B.S., Kim J.A. Triterpenoids from Celastrus orbiculatus Thunb. inhibit RANKL-induced osteoclast formation and bone resorption via c-Fos signaling. J. Nat. Med. 2021;75(1):56–65. doi: 10.1007/s11418-020-01444-3. [DOI] [PubMed] [Google Scholar]
- 35.Gabner S., Böck P., Fink D., Glösmann M., Handschuh S. The visible skeleton 2.0: phenotyping of cartilage and bone in fixed vertebrate embryos and foetuses based on X-ray microCT. Development. 2020;147(11) doi: 10.1242/dev.187633. [DOI] [PubMed] [Google Scholar]
- 36.Fang D., Kong L.Y., Cai J., Li S., Liu X.D., Han J.S., Xing G.G. Interleukin-6-mediated functional upregulation of TRPV1 receptors in dorsal root ganglion neurons through the activation of JAK/PI3K signaling pathway: roles in the development of bone cancer pain in a rat model. Pain. 2015;156(6):1124–1144. doi: 10.1097/j.pain.0000000000000158. [DOI] [PubMed] [Google Scholar]
- 37.Radbakhsh S., Ganjali S., Moallem S.A., Guest P.C., Sahebkar A. Antioxidant effects of trehalose in an experimental model of type 2 diabetes. Adv. Exp. Med. Biol. 2021;1328:473–480. doi: 10.1007/978-3-030-73234-9_32. [DOI] [PubMed] [Google Scholar]
- 38.Hoshino M., Kaneko K., Miyamoto Y., Yoshimura K., Suzuki D., Akaike T., Sawa T., Ida T., Fujii S., Ihara H., et al. 8-Nitro-cGMP promotes bone growth through expansion of growth plate cartilage. Free Radic. Biol. Med. 2017;110:63–71. doi: 10.1016/j.freeradbiomed.2017.05.022. [DOI] [PubMed] [Google Scholar]
- 39.He Y.J., Liu X.Y., Xing L., Wan X., Chang X., Jiang H.L. Fenton reaction-independent ferroptosis therapy via glutathione and iron redox couple sequentially triggered lipid peroxide generator. Biomaterials. 2020;241 doi: 10.1016/j.biomaterials.2020.119911. [DOI] [PubMed] [Google Scholar]
- 40.Gao G., Li J., Zhang Y., Chang Y.Z. Cellular iron metabolism and regulation. Adv. Exp. Med. Biol. 2019;1173:21–32. doi: 10.1007/978-981-13-9589-5_2. [DOI] [PubMed] [Google Scholar]
- 41.Feng H., Schorpp K., Jin J., Yozwiak C.E., Hoffstrom B.G., Decker A.M., Rajbhandari P., Stokes M.E., Bender H.G., Csuka J.M., et al. Transferrin receptor is a specific ferroptosis marker. Cell Rep. 2020;30(10):3411–3423. doi: 10.1016/j.celrep.2020.02.049. e3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang S., He X., Wu Q., Jiang L., Chen L., Yu Y., Zhang P., Huang X., Wang J., Ju Z., et al. Transferrin receptor 1-mediated iron uptake plays an essential role in hematopoiesis. Haematologica. 2020;105(8):2071–2082. doi: 10.3324/haematol.2019.224899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Gastel N., Stegen S., Eelen G., Schoors S., Carlier A., Daniëls V.W., Baryawno N., Przybylski D., Depypere M., Stiers P.J., et al. Lipid availability determines fate of skeletal progenitor cells via SOX9. Nature. 2020;579(7797):111–117. doi: 10.1038/s41586-020-2050-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lefebvre V., Angelozzi M., Haseeb A. SOX9 in cartilage development and disease. Curr. Opin. Cell Biol. 2019;61:39–47. doi: 10.1016/j.ceb.2019.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song H., Park K.H. Regulation and function of SOX9 during cartilage development and regeneration. Semin. Cancer Biol. 2020;67(Pt 1):12–23. doi: 10.1016/j.semcancer.2020.04.008. [DOI] [PubMed] [Google Scholar]
- 46.Hawkins C.L., Davies M.J. Detection, identification, and quantification of oxidative protein modifications. J. Biol. Chem. 2019;294(51):19683–19708. doi: 10.1074/jbc.REV119.006217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu X., Xi C., Thomas B., Pace B.S. Loss of NRF2 function exacerbates the pathophysiology of sickle cell disease in a transgenic mouse model. Blood. 2018;131(5):558–562. doi: 10.1182/blood-2017-10-810531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yao Y., Liu Q., Adrianto I., Wu X., Glassbrook J., Khalasawi N., Yin C., Yi Q., Dong Z., Geissmann F., et al. Histone deacetylase 3 controls lung alveolar macrophage development and homeostasis. Nat. Commun. 2020;11(1):3822. doi: 10.1038/s41467-020-17630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prater M.R., Laudermilch C.L., Liang C., Holladay S.D. Placental oxidative stress alters expression of murine osteogenic genes and impairs fetal skeletal formation. Placenta. 2008;29(9):802–808. doi: 10.1016/j.placenta.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ponsonby A.L., Symeonides C., Saffery R., Mueller J.F., O'Hely M., Sly P.D., Wardrop N., Pezic A., Mansell T., Collier F., et al. Prenatal phthalate exposure, oxidative stress-related genetic vulnerability and early life neurodevelopment: a birth cohort study. Neurotoxicology. 2020;80:20–28. doi: 10.1016/j.neuro.2020.05.006. [DOI] [PubMed] [Google Scholar]
- 51.Jilka R.L., Almeida M., Ambrogini E., Han L., Roberson P.K., Weinstein R.S., Manolagas S.C. Decreased oxidative stress and greater bone anabolism in the aged, when compared to the young, murine skeleton with parathyroid hormone administration. Aging Cell. 2010;9(5):851–867. doi: 10.1111/j.1474-9726.2010.00616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith A.C., Mears A.J., Bunker R., Ahmed A., MacKenzie M., Schwartzentruber J.A., Beaulieu C.L., Ferretti E., Majewski J., Bulman D.E., et al. Mutations in the enzyme glutathione peroxidase 4 cause Sedaghatian-type spondylometaphyseal dysplasia. J. Med. Genet. 2014;51(7):470–474. doi: 10.1136/jmedgenet-2013-102218. [DOI] [PubMed] [Google Scholar]
- 53.Abd El-Aziz G.S., El-Fark M.M., Saleh H.A. The prenatal toxic effect of methylmercury on the development of the appendicular skeleton of rat fetuses and the protective role of vitamin E. Anat. Rec. 2012;295(6):939–949. doi: 10.1002/ar.22485. Hoboken, NJ : 2007. [DOI] [PubMed] [Google Scholar]
- 54.Katti K., Ayasolla K.R., Iurcotta T., Potak D., Codipilly C., Weinberger B. Lipid peroxidation products as predictors of oxidant-mediated disease in preterm infants. J. Matern. Fetal Neonatal Med. 2021:1–6. doi: 10.1080/14767058.2020.1869934. [DOI] [PubMed] [Google Scholar]
- 55.Torres-Cuevas I., Parra-Llorca A., Sánchez-Illana A., Nuñez-Ramiro A., Kuligowski J., Cháfer-Pericás C., Cernada M., Escobar J., Vento M. Oxygen and oxidative stress in the perinatal period. Redox Biol. 2017;12:674–681. doi: 10.1016/j.redox.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen X., Yu C., Kang R., Kroemer G., Tang D. Cellular degradation systems in ferroptosis. Cell Death Differ. 2021;28(4):1135–1148. doi: 10.1038/s41418-020-00728-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Anthonymuthu T.S., Tyurina Y.Y., Sun W.Y., Mikulska-Ruminska K., Shrivastava I.H., Tyurin V.A., Cinemre F.B., Dar H.H., VanDemark A.P., Holman T.R., et al. Resolving the paradox of ferroptotic cell death: ferrostatin-1 binds to 15LOX/PEBP1 complex, suppresses generation of peroxidized ETE-PE, and protects against ferroptosis. Redox Biol. 2021;38 doi: 10.1016/j.redox.2020.101744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Y., Song Z., Liu Y., Ma X., Wang W., Ke Y., Xu Y., Yu D., Liu H. Identification of ferroptosis as a novel mechanism for antitumor activity of natural product derivative a2 in gastric cancer. Acta Pharm. Sin. B. 2021;11(6):1513–1525. doi: 10.1016/j.apsb.2021.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu J., Kang R., Tang D. Signaling pathways and defense mechanisms of ferroptosis. FEBS J. 2021;6:1–13. doi: 10.1111/febs.16059. [DOI] [PubMed] [Google Scholar]
- 60.Kong N., Chen X., Feng J., Duan T., Liu S., Sun X., Chen P., Pan T., Yan L., Jin T., et al. Baicalin induces ferroptosis in bladder cancer cells by downregulating FTH1. Acta Pharm. Sin. B. 2021;11(12):4045–4054. doi: 10.1016/j.apsb.2021.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wan J., Ren H., Wang J. Iron toxicity, lipid peroxidation and ferroptosis after intracerebral haemorrhage. Stroke and vascular neurology. 2019;4(2):93–95. doi: 10.1136/svn-2018-000205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamada N., Karasawa T., Kimura H., Watanabe S., Komada T., Kamata R., Sampilvanjil A., Ito J., Nakagawa K., Kuwata H., et al. Ferroptosis driven by radical oxidation of n-6 polyunsaturated fatty acids mediates acetaminophen-induced acute liver failure. Cell Death Dis. 2020;11(2):144. doi: 10.1038/s41419-020-2334-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tang D., Chen X., Kang R., Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107–125. doi: 10.1038/s41422-020-00441-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsay J., Yang Z., Ross F.P., Cunningham-Rundles S., Lin H., Coleman R., Mayer-Kuckuk P., Doty S.B., Grady R.W., Giardina P.J., et al. Bone loss caused by iron overload in a murine model: importance of oxidative stress. Blood. 2010;116(14):2582–2589. doi: 10.1182/blood-2009-12-260083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park M.W., Cha H.W., Kim J., Kim J.H., Yang H., Yoon S., Boonpraman N., Yi S.S., Yoo I.D., Moon J.S. NOX4 promotes ferroptosis of astrocytes by oxidative stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in Alzheimer's diseases. Redox Biol. 2021;41 doi: 10.1016/j.redox.2021.101947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhong H., Yin H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: focusing on mitochondria. Redox Biol. 2015;4:193–199. doi: 10.1016/j.redox.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
AAPH boosts oxidative stress in SW1353 cells. (A) ROS generation and lipid peroxidation levels were detected by flow cytometry after incubation with DCFH-DA or C11-BODIPY in AAPH-treated SW1353 cells. AAPH, 15 mM. (B) Statistical plots showing ROS level and lipid peroxidation level in AAPH-treated SW1353 cells at different time. AAPH, 15 mM. (C) The concentration of H2O2 in the extracellular medium was calculated by the Amplex Red assay. (D) Thiol generation was measured by the Total Sulfhydryl Group Content Assay Kit after AAPH treatment (15 mM). (E) The ratio of GSH/GSSG was measured after treatment with AAPH in SW1353 cells (15 mM). (F) The extracted ion current of GSH and GSSG. Glu-d5 has been used as an internal standard. Data are represented as mean ± SD. Comparisons between groups were made using one-way ANOVA (B and C) and Student's t-test (D and E). ***P < 0.001 vs Control group (DCFH-DA staining). ##P < 0.01 and ###P < 0.001 vs the Control group (C11-BODIPY staining). @P < 0.05, @@P < 0.01 vs the Control group (Amplex Red assay). $P < 0.05, $$P < 0.01 vs the AAPH-treated group.
AAPH-induced oxidative stress is alleviated by ferroptosis inhibitor Fer-1. (A-D) Effects of co-treatment with AAPH and Fer-1 on the contents of MDA and GSH, and the activities of GPX and SOD were measured on EDD17. Fer-1 (50 nmol/egg) and AAPH (0.50 μmol/egg). (E-H) The effects of Fer-1 on AAPH-induced alterations in ferroptosis-related markers determined by western blot assay and RT-PCR on EDD17. (I) Protein levels of DMT1, TFR1, GPX4, and SLC7A11 were determined by western blotting in ATDC5 cells. (J) Relative mRNA levels determination of Dmt1, Tfrc, Gpx4 and Slc7a11 by RT-PCR in ATDC5 cells. Data are represented as mean ± SD. Comparisons between groups were made using one-way ANOVA. *P < 0.05, **P < 0.01 and ***P < 0.001 vs the Control group. #P < 0.05, ##P < 0.01 and ###P < 0.001 vs the AAPH-treated group.
Inhibition of ferroptosis rescues AAPH-induced oxidative stress. (A-D) MDA, GSH levels and GPX and SOD activities in EDD7 chicken embryos. (E) Expressions of ferroptosis-related protein (DMT1, TFR1, GPX4, SLC7A11) in EDD7 chicken embryos were determined by western blotting analysis. (F) Levels of ferroptosis-related gene (Dmt1, Tfrc, Gpx4, Slc7a11) in EDD7 chicken embryos were detected by qRT-PCR. (G) Expressions of ferroptosis markers (DMT1, TFR1, GPX4 and SLC7A11) were determined by western blotting analysis in SW1353 cells. (H) Relative mRNA levels of DMT1, TFRC, GPX4 and SLC7A11 in SW1353 cells as determined by qRT-PCR. Data are represented as mean ± SD. Comparisons between groups were made using one-way ANOVA.*P < 0.05, **P < 0.01 and ***P < 0.001 vs the Control group. #P < 0.05, ##P < 0.01 and ###P < 0.001 vs the AAPH-treated group.
Ferroptosis is engaged in AAPH-induced differentiation disruption of chondrogenic cells in vitro. (A) The mRNA level of SOX9 was detected by qRT-PCR in SW1353 cells. (B–C) Level of SOX9 was determined by western blotting analysis in SW1353 cells. AAPH, 15 mM; RSL3, 30 nM; DFO, 200 μM; Fer-1, 5 μM. (D) The mRNA levels of BMP6, COL2A1 and ACAN were measured by qRT-PCR in SW1353 cells. Data are represented as mean ± SD. Comparisons between groups were made using one-way ANOVA.*P < 0.05, **P < 0.01 and ***P < 0.001 vs the Control group. #P < 0.05, ##P < 0.01 and ###P < 0.001 vs the AAPH-treated group.
Data Availability Statement
The datasets used to support the findings of this study are available from the corresponding author upon request.
Data will be made available on request.