

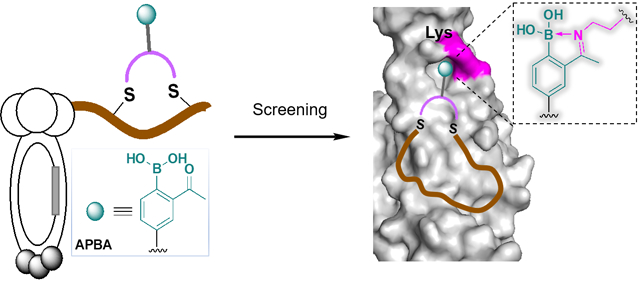
Abstract
Binding via reversible covalent bond formation presents a novel and powerful mechanism to enhance the potency of synthetic inhibitors for therapeutically important proteins. Work on this front has yielded the anticancer drug bortezomib as well as the anti-sickling drug voxelotor. However, rational design of reversible covalent inhibitors remains difficult even when noncovalent inhibitors are available as a scaffold. Herein we report chemically modified phage libraries, both linear and cyclic, that incorporate 2-acetylphenylboronic acid (APBA) as a warhead to bind lysines via reversible iminoboronate formation. To demonstrate their utility, these APBA-presenting phage libraries were screened against sortase A of Staphylococcus aureus, as well as the spike protein of SARS-CoV-2. For both protein targets, peptide ligands were readily identified with single digit micromolar potency and excellent specificity, enabling live cell sortase inhibition and highly sensitive spike protein detection respectively. Furthermore, our structure-activity studies unambiguously demonstrate the benefit of the APBA warhead for protein binding. Overall, this contribution shows for the first time that reversible covalent inhibitors can be developed via phage display for a protein of interest. The phage display platform should be widely applicable to proteins including those involved in protein-protein interactions.
Graphical Abstract
INTRODUCTION
Peptides are reemerging as an appealing modality of therapeutics1–2 due to their ease of synthesis and low vulnerability to structural denaturation in comparison to monoclonal antibodies. On the other hand, peptides have shown advantages over traditional small molecule drugs in inhibiting protein-protein interactions, which play many important roles in human physiology and disease.3 An additional appealing aspect of peptide drug discovery lies in the existence of several high-throughput screening platforms that allow facile construction and screening of peptide libraries with easily over a billion unique sequences.4 A prominent example of such screening platforms is phage display, which presents genetically encoded peptide libraries on the surface of bacteriophage.5 Since its invention, phage display of peptide libraries has revealed a number of peptide probes and inhibitors for various protein targets. However, these peptide hits often show low to modest potency presumably due to their structural simplicity: the ribosomal origin of the phage coat proteins limits phage display to linear or simple disulfide-cyclized peptides with just proteinogenic amino acids. Recently, several strategies have been developed to allow the incorporation of nonnatural structural motifs into phage-displayed peptides.6–11 Characterization of these chemically modified phage libraries has shown exciting promise to deliver potent ligands for challenging protein targets including those involved in protein-protein interactions (PPIs).
We and others have been investigating the use of reversible covalent warheads to improve the potency of synthetic bioactive molecules.12–16 Specifically, we have reported the use of 2-acetylphenylboronic acid (APBA) to covalently bind amine-presenting biomolecules (Figure 1A).17 Under physiologic conditions, APAB reversibly “binds” an amine to give an iminoboronate conjugate.18 The covalent binding mechanism provides –(3–4) kcal/mol of free energy and excellent selectivity for primary amines. While a couple of recent papers showcase the benefit of such a covalent binding mechanism towards protein inhibition,19–20 rational design of protein ligands that incorporate the APBA warhead remains challenging. We envision that screening warhead-bearing peptide libraries can be an efficient way to identify reversible covalent probes and inhibitors for a protein of interest. To test this hypothesis, we carried out a comprehensive evaluation of phage-displayed peptide libraries, both linear and cyclic, that incorporate APBA warheads. Screening of these novel phage libraries yielded potent peptide ligands for sortase A (SrtA)21 and the SARS-CoV-2 spike protein,22 both engage in protein-protein interactions and have frustrated small molecule ligand discovery. Drawing parallel to a recent report of the Bogyo group,23 which nicely demonstrates irreversible covalent inhibitor discovery via phage display, our contribution presents the first example of protein ligand discovery using reversible covalent phage libraries.
Figure 1.
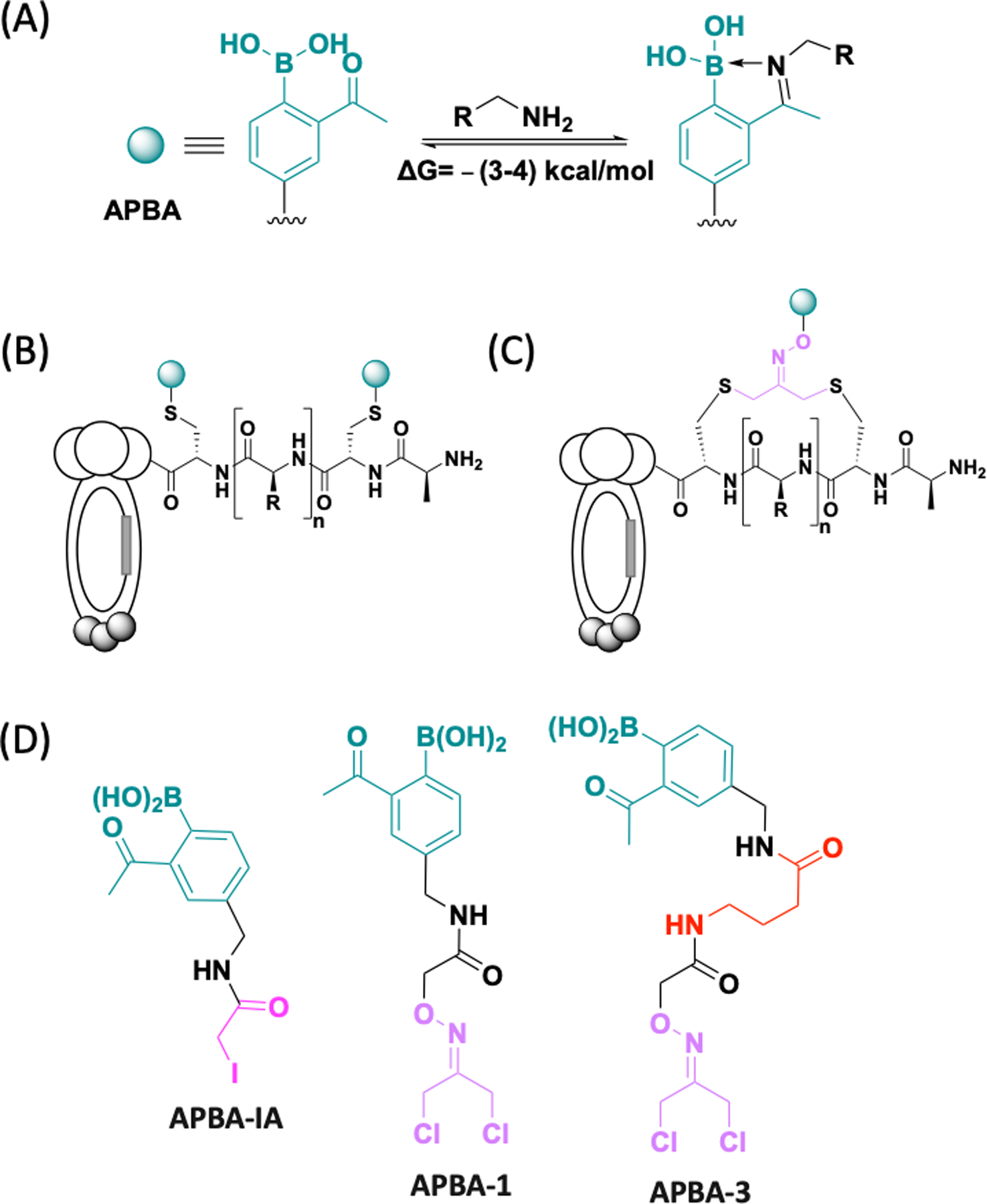
Structural illustration of phage libraries displaying APBA. (A) Illustration of the covalent binding of amines by APBA. (B) APBA-dimer library; (C) Cyclic peptide libraries displaying APBA; (D) Chemical structure of the designed phage modifiers APBA-IA, APBA-1 and APBA-3.
RESULTS AND DISCUSSION
Library Design.
Recent advances in site-specific protein modification chemistries have made it possible to modify a phage library with nonproteinogenic structural motifs, including designer side chains11, 24 as well as non-disulfide based crosslinks.7, 10 We previously reported an APBA-dimer library (Figure 1B), in which a pair of APBA motifs were grafted onto a linear peptide via cysteine conjugation. Although this library was successfully used in live cell screening to reveal bacterial probes,11, 25 such reversible covalent phage libraries have not been examined towards ligand discovery for specific proteins. To fill this void, we developed several APBA-presenting phage libraries and evaluated them against a protein ligase and a prototypical PPI-engaging protein, both known to frustrate small molecule binding. Specifically, we redesigned the structure of APBA-IA (Figure 1D), the phage modifier to create APBA-dimer libraries, with a shorter and less floppy linker that is entropically favorable for protein ligands. Furthermore, we synthesized two APBA conjugates with dichloroacetone (DCA) via an oxime linkage (APBA-1/3, Figure 1D). The DCA-oxime strategy has been previously used for Cys-Cys crosslinking to give cyclic peptide libraries (Figure 1C).26–27 Cyclic peptides are appealing as potential protein ligands because the cyclic scaffold can provide structural rigidity and project the side chains in desired orientations for target binding. While efficient phage modification has been demonstrated previously for APBA-IA, we probed the phage modification efficiency of APBA-1 and APBA-3 using a DCA-Biotin conjugate as a surrogate crosslinker. To our satisfaction, DCA-Biotin elicited quantitative phage biotinylation under our optimized conditions (Figure S1, S2), while APBA-1 treated phage showed no reaction with DCA-Biotin, suggesting efficient crosslinking by APBA-1 as well. Importantly, the infectivity of the phage library was marginally affected by the chemical modifications (Figure S3).
Discovering reversible covalent inhibitors for sortase A of Staphylococcus aureus.
We chose sortase A (SrtA) as an initial target protein. SrtA is an important enzyme of the bacterium Staphylococcus aureus, responsible for anchoring extracellular proteins onto the bacterial cell wall. SrtA inhibition would eliminate the cell wall-anchored virulence factors, thereby reducing the bacterium’s capability to establish an infection. Furthermore, SrtA inhibition is non-bactericidal and less prone to elicit resistance mechanisms. These factors collectively make SrtA inhibition an appealing strategy for developing novel anti-Staph therapeutics.21, 28 However, it has been challenging to develop small molecule inhibitors for SrtA due to its rather large and shallow active site (Figure 2A), a feature shared by many therapeutically important proteases.29
Figure 2.
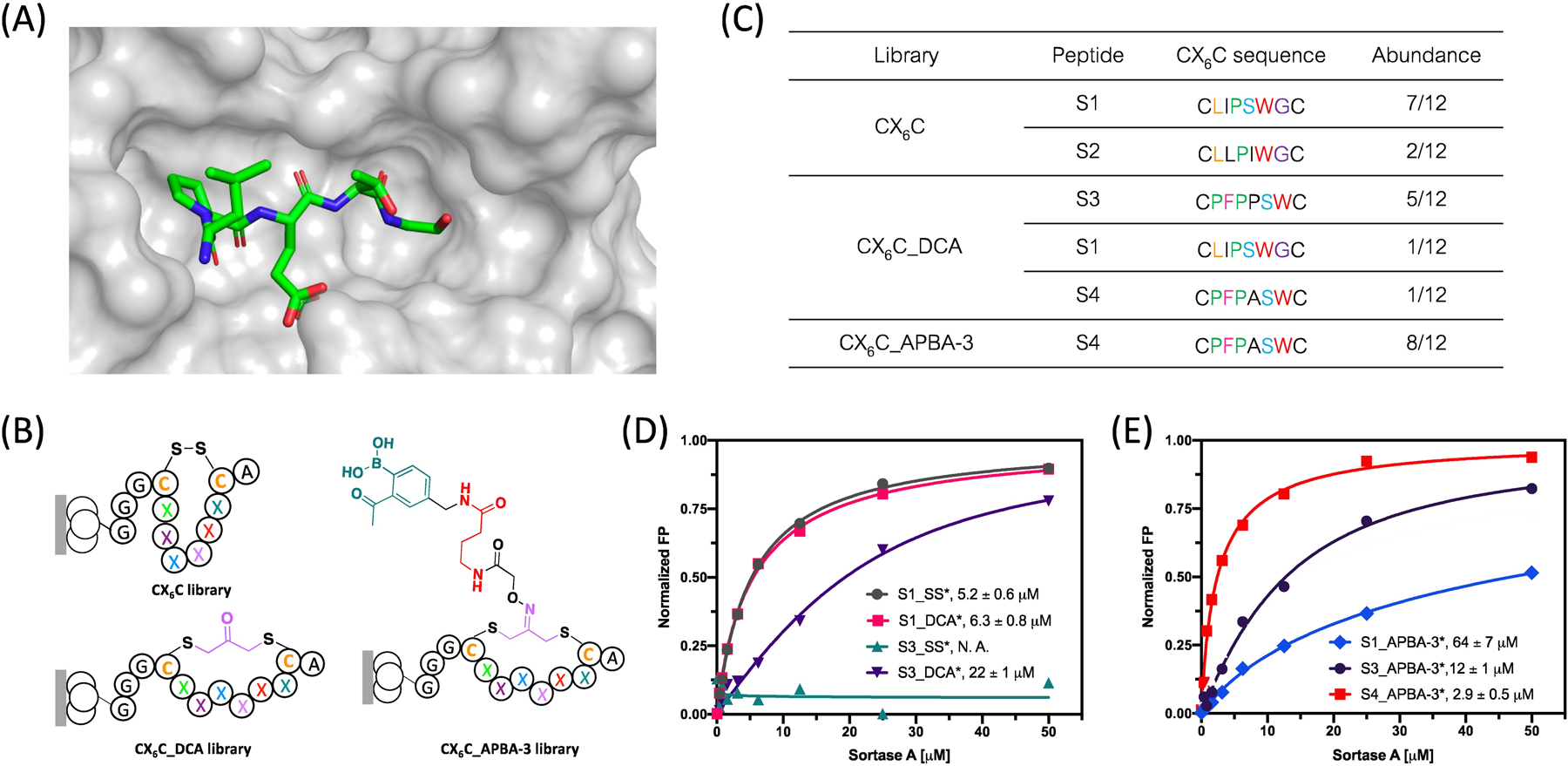
Phage display reveals peptide binders of SrtA. (A) Crystal structure of Sortase A in complex with LPETG motif (pdb 1t2w); (B) Generic structure of the CX6C cyclic peptide libraries with varied crosslinkers. These libraries were screened against SrtA to identify peptide inhibitors; (C) Enriched peptide sequences from various phage libraries. (D-E) SrtA binding curves of various peptide hits generated using a fluorescence polarization assay. The asterisk (*) in the peptide names represents a FAM label incorporated to allow fluorescence polarization measurements. Each data point presents the mean value of three independent measurements. Kd values were extracted via curve fitting and listed in corresponding plots.
We recombinantly expressed SrtA with an N-terminal AviTag, which allows co-translational biotinylation (Figure S4). The biotinylated SrtA can be readily immobilized on streptavidin-coated magnetic beads, against which phage panning is carried out. Several peptide-based inhibitors of SrtA have been documented in literature.30–31 Using these examples as a reference, we constructed a CX6C library on M13 phage (Figure 2B), which is expected to present a good match in size with the pocket of SrtA. The phage library presents the CX6C peptides as an N-terminal fusion to the phage minor coat protein, pIII. To minimize intramolecular APBA-lysine conjugation, we introduced a K10R mutation to the pIII protein that eliminates the most close-by lysine residue to the CX6C peptides. Our previous study showed that the intramolecular conjugation of APBA with a lysine diminishes when separated by more than nine amino acids.32 The library quality was assessed by subjecting it to four rounds of amplification followed by sequencing. The result showed excellent sequence diversity and absence of any growth-biased sequences (Table S1). To further characterize this library, we screened the library against SrtA without any chemical modifications. Three rounds of panning of the CX6C library revealed a highly enriched sequence S1 (Figure 2C and Table S2). Another clearly enriched peptide S2 presents just a single point mutation (Ser to Ile) from S1. We synthesized peptide S1 with a fluorophore label (S1_SS*: ACLIPSWGCGGGDap(Fam)) and characterized its SrtA binding affinity via a fluorescence polarization assay: titrating SrtA into the peptide solution at increasing concentrations elicited an increase of fluorescence polarization and curve fitting yielded a Kd value of 5.2 μM (Figure 2D). This binding was further validated by a competition assay, in which a non-fluorescently labeled S1_SS was titrated against a fluorescently labeled SrtA ligand (Figure S5). The competition assay yielded an IC50 of 26 μM. The slightly higher IC50 value in comparison to the Kd of the peptide suggests that the fluorescein label may enhance the peptide’s binding to SrtA to some extent. However, this fluorophore effect should not prevent us from comparing the relative potencies of different peptides.
Next, we subjected the CX6C library to reduction and then modification with the APBA-IA, DCA, APBA-1 and APBA-3 respectively. The resulting four libraries were screened against SrtA using similar protocols as described for the CX6C library. We note that the APBA-bearing libraries did show a significantly higher phage retention than the original CX6C library (Figure S6), presumably due to nonspecific iminoboronate formation to some extent. To minimize such nonspecific binding, we adopted a more stringent washing protocol (washing buffer prepared with 0.5% tween-20 instead of 0.1%). Interestingly, in comparison to the unmodified library, screening of the DCA modified library yielded a completely different peptide sequence S3, which dominated the output population after three rounds of panning (Figure 2C and Table S3). The fluorescence polarization-based binding studies revealed a Kd of 22 μM for this DCA-crosslinked peptide (S3_DCA*, Figure 2D). It is important to note that the disulfide cyclized S3 (S3_SS*) showed no binding to SrtA, consistent with the fact that the S3 sequence did not show up in the final output populations of the unmodified CX6C library. The peptide sequence S1 did show up in the output populations of the CX6C_DCA library, albeit at a lower frequency. Interestingly, the peptide S1_DCA* was found to bind SrtA with a slightly higher affinity than S3_DCA* (Figure 2D). These results collectively indicate that our phage panning protocol is effective at enriching true peptide hits for the target protein, although, perhaps not surprisingly, the extent of enrichment does not rigorously correlate with the peptides’ affinity for target binding.
APBA-IA modification of the reduced CX6C library yielded a linear APBA dimer library (CX6C_APBA-dimer), while APBA-1 and APBA-3 modification gave two cyclic APBA-presenting peptide libraries with a slightly different linker length between the APBA warhead and the cyclic peptide scaffold. Interestingly, SrtA screening of the APBA-dimer library as well as the APBA-1 library yielded no clear convergence of the peptide sequences (Table S4 and Table S5). However, panning of the APBA-3 library against SrtA yielded peptide S4 as a clear winner (Table S6). We note that peptide S4 differs from S3 by just a single Pro-to-Ala mutation (Figure 2C). In fact, similar to S3, S4 was enriched from the CX6C_DCA library, albeit to a less extent. Consistently, our binding studies revealed a Kd of 16 μM for S4_DCA*, which is quite similar to that of S3_DCA* (Figure 3A). Excitingly, S4 upon modification with APBA-3 was found to bind SrtA with significantly improved affinity, yielding a Kd value of 2.9 μM (Figure 2E), which is comparable to the best SrtA binders reported in literature.15, 31 For comparison, we synthesized APBA-3 cyclized S1 and S3 and subjected them to SrtA binding studies. Interestingly, compared with S4_APBA-3*(Kd=2.9 μM), S3_APBA-3*(Kd=12 μM) showed a four-fold reduction in SrtA binding affinity despite its high sequence similarity, while S1_APBA-3* (Kd=64 μM) exhibited a more dramatic decrease in its binding affinity, attesting to the uniqueness of the S4 sequence in supporting the APBA warhead for SrtA binding.
Figure 3.
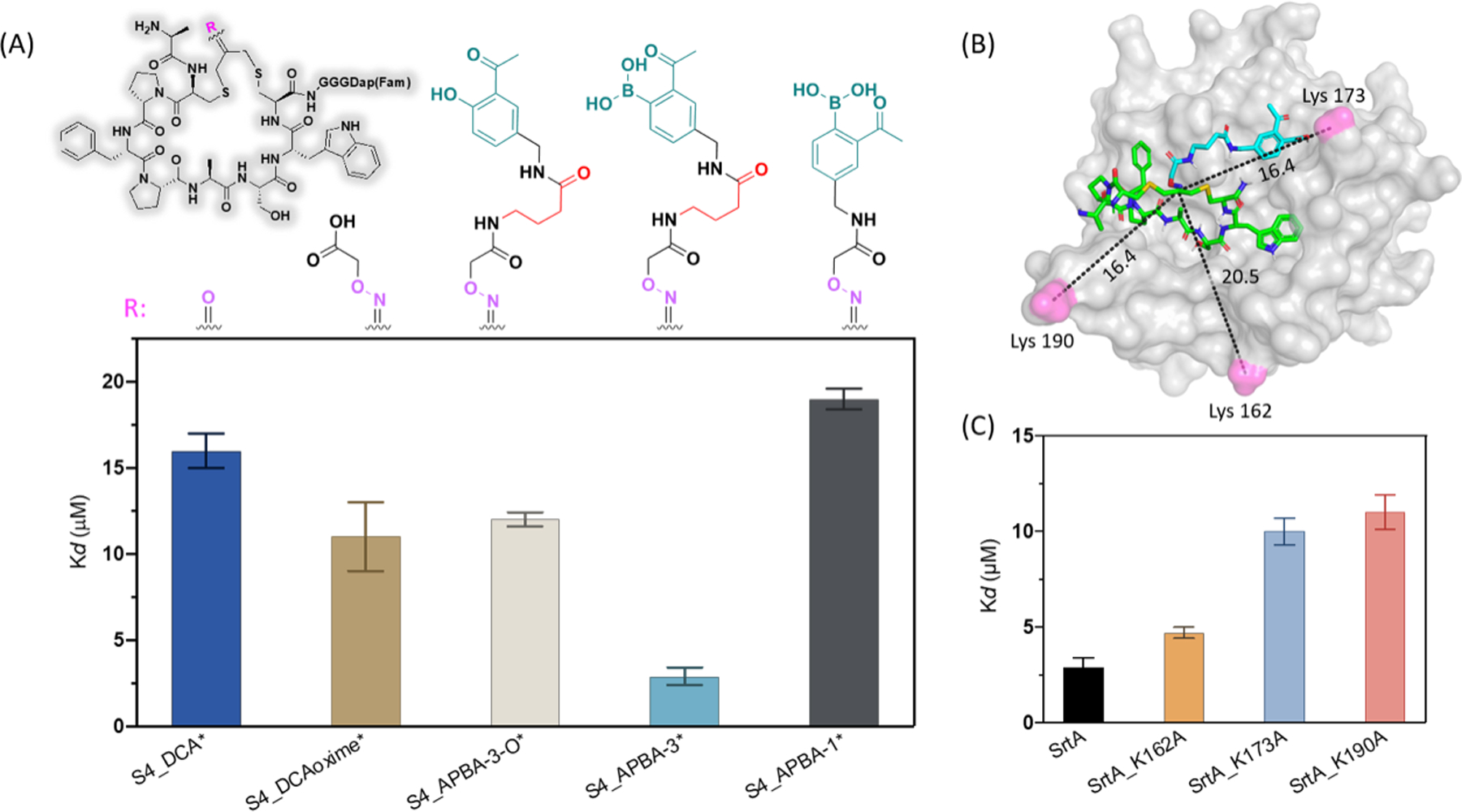
Structure-activity relationship (SAR) studies for S4_APBA-3*. (A) Generic structure of S4_APBA-3* and analogues (Top), and comparison of the SrtA binding affinity of various S4 derivatives generated using a fluorescence polarization assay (Bottom). (B) A docking structure S4_APBA-3 on SrtA. An analogue of APBA warhead was used for docking in which the −B(OH)2 group is replaced with −COOH. (C) Binding affinity of peptide S4_APBA-3* to SrtA and Lys-to-Ala mutants.
To probe the postulated role of APBA as a reversible covalent warhead, we synthesized two negative control peptides carrying just the oxime crosslinker (S4_DCAoxime*) or the oxidized APBA (S4_APBA-3-O*). SrtA binding studies revealed a Kd value of 11 and 12 μM respectively (Figure 3A). The weaker binding of these two control peptides lends strong support to the energetic importance of the reversible covalent binding mechanism of APBA. As an additional control, we synthesized APBA-1 crosslinked S4, which gave a Kd of 19 μM for SrtA binding (Figure 3A). This weakened binding of S4_APBA-1* is presumably due to its shortened linker that prohibits APBA conjugation to a lysine. To gain insight into which lysine residue conjugates with the APBA warhead, we performed molecular docking studies with an analogue of S4_APBA-3, in which −B(OH)2 group is replaced with −COOH (Figure 3B). A majority of the high scoring structures placed the cyclic peptide in the active site of SrtA, which in turn places the warhead moiety close to K173 or K190 for conjugation. To experimentally validate this hypothesis, we individually mutated the lysine residues close to the enzyme active site using site-directed mutagenesis, which gave three single point mutants of SrtA, namely K162A, K173A and K190A. Interestingly, both the K173A and K190A mutation resulted in significant reduction in binding affinity of S4_APBA-3*, while the K162A mutation minimally affected SrtA binding to the peptide (Figure 3C). We further resorted to mass spectrometry analysis to confirm the covalent binding mechanism of our SrtA inhibitors. Unfortunately, the highly dynamic nature of iminoboronate formation made it difficult to trap the APBA-lysine conjugate via reduction (Figure S7). To circumvent this issue, we synthesized an analogue of S4_APBA-3, which installs a salicylaldehyde (SA) as a surrogate warhead of APBA (Figure S8). SA is known to covalently bind lysines to give an imine adduct that is amenable to NaBH4 reduction.33 The peptide S4_SA yielded similar SrtA binding affinity with S4_APBA-3 (Figure S8). When subjected to NaBH4 reduction, the mixture of S4_SA and SrtA yielded a 1:1 covalent adduct as revealed by mass-spectrometry (Figure S9). Interestingly, the covalent adduct was seen with both K173A and K190A mutants (Figure S9), consistent with the docking and SrtA mutant binding studies, which suggest S4_APBA-3 could conjugate with either K73 or K190 (Figure 3C). We further performed trypsin digestion and peptide mapping experiments (Figure S10–S13), which unambiguously revealed the covalent adduct to K190, although the covalent adduct to K173 was not detected. Collectively, these results provide strong support to the covalent binding mechanism of S4_APBA-3 as we intended. We note this APBA-based covalent binder nevertheless exhibit fast equilibrium as seen from our competition studies (Figure S14), which is expected for the highly dynamic iminoboronate formation.
To test whether the SrtA binding peptides inhibit its catalytic activity, we subjected the peptide S4_APBA-3 (no FAM label) to an enzyme inhibition assay, for which a fluorogenic substrate (Dabcyl-LPETG-Edans) was used to report the SrtA activity. As expected, the peptide inhibited the SrtA cleavage of the fluorogenic substrate in a concentration dependent manner. Curve fitting yielded an IC50 of 18 μM (Figure S15). We further tested the SrtA inhibition of S4_APBA-3 against live S. aureus cells. Pentapeptide LPXTG is a recognition motif, which can be recognized and ligated to the cell wall by SrtA. As such, the reactivity of SrtA on live S. aureus cells can be determined with FAM-labeled peptides containing a LPETG motif (Figure S16). Indeed, incubating S. aureus cells with a FAM-labeled peptide substrate resulted in efficient fluorescent labeling of the cells as revealed by flow cytometry (Figure S16). Importantly, the mean fluorescence of cells decreased in a dose-dependent manner with the addition of S4_APBA-3 (Figure S17), indicating S4_APBA-3 effectively inhibited the incorporation of FAM-labeled substrate. Curve fitting yielded an IC50 value of 49 μM for SrtA inhibition on live S. aureus cells.
Developing reversible covalent probes for SARS-CoV-2 spike protein.
Encouraged by the phage panning results against SrtA, we set out to test the potential of our APBA libraries for targeting proteins involved in protein-protein interactions. Towards this end, we screened the libraries against the receptor binding domain (RBD) of the SARS-CoV-2 spike protein, whose interaction with human ACE2 (hACE2) receptor is well characterized and an important step in coronavirus etiology.34 Characteristic of difficult-to-inhibit PPIs, the spike RBD binds to hACE2 through a large interface, involving 21 amino acids on RBD and 22 amino acids on hACE2 (Figure 4A). Interestingly, in addition to the hACE2 binding site, the spike protein RBD has been shown to have two additional sites to allow high affinity binding by peptide ligands (Figure 4A).35–36 To target these protein/peptide binding sites of RBD, we resorted to a larger peptide scaffold and created a CX9C phage library, which was then modified with APBA-IA and APBA-3 respectively.
Figure 4.
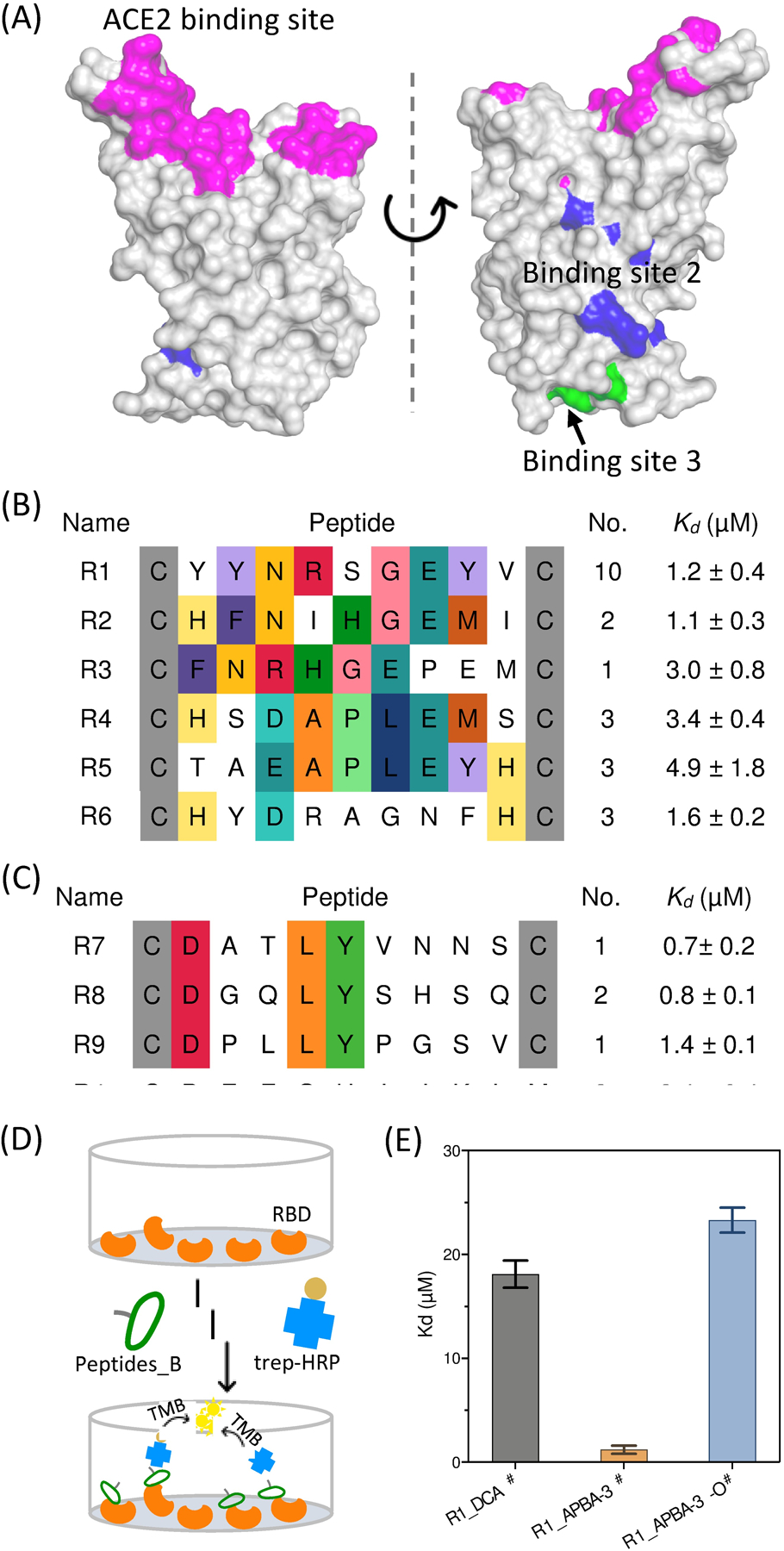
Phage panning yields potent peptide binders of SARS-CoV-2 spike protein. (A) Known peptide binding sites of SARS-CoV-2 RBD (PDB: 6M0J). Highlighted with different colors are the hACE2 binding site (magenta) as well as the two additional peptide binding sites recently reported (site 2 in blue and 3 in green). (B) Sequence alignment of the enriched peptide sequences from CX9C_APBA-3 library. (C) Sequence alignment of the enriched peptide sequences from CX9C_APBA-dimer library. For (B) and (C), conserved amino acid residues are highlighted with color. The number of recurrences seen in our sequencing results as well the corresponding Kd value for RBD binding are listed for each peptide. (D) Schematic representation of the ELISA assay, in which RBD was coated on the plate, blocked with BSA, and then treated with a biotinylated peptide at varied concentrations. After washing, the bound peptide was quantified using streptavidin-HRP. (E) Comparison of R1_APBA-3#, R1_DCA# and R1_APBA-3-O# for RBD binding affinity. The Kd values and corresponding error bars values were generated via fitting titration curves of the ELISA assay. # indicates biotin labeled peptides.
Phage panning against the spike RBD was performed using the same protocol described for the SrtA screening. Briefly, a biotinylated SARS-CoV-2 RBD (L452R, E484Q) was acquired from commercial sources and then immobilized on streptavidin beads for screening. Phage clones were randomly picked from the output population of the fifth round, sequencing of which revealed successful enrichment of certain peptide sequences (Table S7). Specifically, the APBA-dimer library yielded a panel of peptides that share a DXXLY sequence (Figure 4C). The CX9C_APBA-3 library yielded two consensus motifs: three peptide sequences (R1-R3) showed a Y(F)NXXGE motif and two others (R4-R5) shared a D(E)APLE segment at position 4–8 (Figure 4B). To validate the identified hits, we synthesized representative peptides through solid phase peptide synthesis with a biotin installed onto the C-terminal Dap residue. After purification, the precursor peptides were treated with either APBA-IA or APBA-3 to afford the desired peptide hits. The binding affinity of these peptides to RBD were tested using an ELISA assay (Figure 4D). Briefly, the RBD domain is coated into the cells of a 96-well plate. After blocking with BSA, a biotinylated peptide was added into the wells to allow binding to RBD. After 1 h of incubation, the unbound peptide was washed away and the bound peptide was quantified using an HRP-fused streptavidin. Plotting the readout against peptide concentrations afforded the binding curves (Figure S18), curve fitting of which yielded the Kd values for the RBD binding of the peptides.
Excitingly, all peptides, both cyclic and linear, were found to bind RBD with sub to low micromolar affinity (Figure 4B, C). The APBA-dimer peptides appear to give slightly lower Kd values than the cyclic ones. However, significant binding to the control wells (no RBD, just BSA coating) was observed for the APBA-dimer peptides, indicating a lack of specificity (Figure S19). In contrast, the cyclic peptide hits showed little to no binding to the control wells, suggesting excellent selectivity of these peptides for target protein over the BSA control. To gain mechanistic insights to the peptides’ binding to RBD, we synthesized several control peptides to compare with R1_APBA-3#. First, an APBA-3 crosslinked random CX9C peptide (R0) elicited no binding to RBD under the same experimental conditions (Figure S20), highlighting the importance of the peptide sequences born out of the phage selection. Further, the DCA cyclized R1 peptide (R1_DCA#) gave a Kd of 18 μM, which in comparison to that of R1_APBA-3# (1.2 μM) suggests the critical importance of the APBA warhead. Consistently, the peptide R1_APBA-3-O#, which has the −B(OH)2 group oxidized to −OH, gave a Kd of 23 μM (Figure 4E). The 20-fold erosion of the peptide’s binding to RBD unambiguously demonstrates the energetic significance of the APBA warhead for RBD binding.
To confirm the covalent binding mechanism to RBD, we again resorted to the SA analogue of the peptide hits. Specifically, R1_SA# was synthesized and found to display similar affinity for RBD binding as R1_APBA-3# (Figure S21). The covalent adduct of R1_SA# to RBD was successfully trapped by NaBH4 reduction and the resulting RBD biotinylation was readily detected in a gel-shift assay, where the biotinylated RBD appeared as streptavidin complexes (Figure S22). While the intact RBD failed to be analyzed by mass spectrometry due to heterogenous glycosylation, we were able to obtain meaningful results from peptide mapping experiments, which unambiguously revealed the covalent adduct of R1_SA# to K417 of the RBD protein (Figure S23–S25). To further corroborate this finding, we performed computational docking studies of R1_APBA-3, which revealed a pose where the APBA warhead is placed within the bonding distance with K417 (Figure S26). Although the detailed structure of this peptide-RBD complex needs further exploration, our data above clearly establish the covalent binding mechanism of the R1 peptides and showcase the magnitude of affinity improvement that the APBA warhead can afford.
We further tested whether our peptide hits for RBD could inhibit the RBD binding to its cognate receptor hACE2. Towards this end, we synthesized two cyclic peptide hits, namely R1_APBA-3 and R5_APBA-3, for which the biotin label is not included. These two peptide hits were chosen due to their distinct peptide sequence as well as their superior specificity for RBD binding over the linear APBA-dimers. To measure the peptides’ inhibitory activity, the hACE2 protein was coated into the wells of a plate, and then the biotinylated RBD was added with a peptide ligand at varied concentrations. The amount of bound RBD was quantified using streptavidin-HRP. Unfortunately, neither R1_APBA-3 nor R5_APBA-3 showed inhibitory activity in this assay (Figure S27). This lack of inhibition is nevertheless consistent with the docking structure of R1_APBA-3, where the bulk of the peptide binds outside of the RBD-hACE2 interface (Figure 26).
Even though incapable of inhibiting the RBD - hACE2 interaction, the RBD binding peptides may serve as powerful molecular tools to allow facile detection of the coronavirus spike protein towards COVID diagnosis. To test this hypothesis, we tested the potential of our RBD-binding peptide for spike protein detection in saliva and in serum using flow cytometry as a readout. Specifically, we immobilized the biotinylated peptide R1_APBA-3# on streptavidin beads and blocked the beads with 1% BSA at 4°C for overnight. Then to the peptide-coated beads was added serially diluted Alexa488-labeled spike protein in the presence of 10% saliva (Figure 5A). Excitingly, the fluorophore-labeled spike protein bound to the peptide-coated beads in a dose-dependent manner with a Kd as low as 222 pg/mL (~ 4 pM) (Figure 5B and Figure S28). We attribute this high sensitivity of spike protein capturing to a polyvalent effect: the spike protein is a symmetric trimer, which can engage a 3-to-3 interaction with bead-immobilized peptide probe. Importantly, the streptavidin beads without peptide coating elicited no spike protein binding until much elevated concentrations. Furthermore, we investigated the binding specificity of R1_APBA-3# to spike protein in the presence of human serum. The result showed that 1ng/mL spike protein dissolved in 20% human serum also results in strong signal (Figure 5C). Taken together, the above results indicate that R1_APBA-3 serves as a potent and specific probe for the spike protein RBD. With the benefit polyvalent binding, it may enable novel diagnostic devices that enable detection of the coronavirus spike protein at clinically relevant concentrations.37
Figure 5.
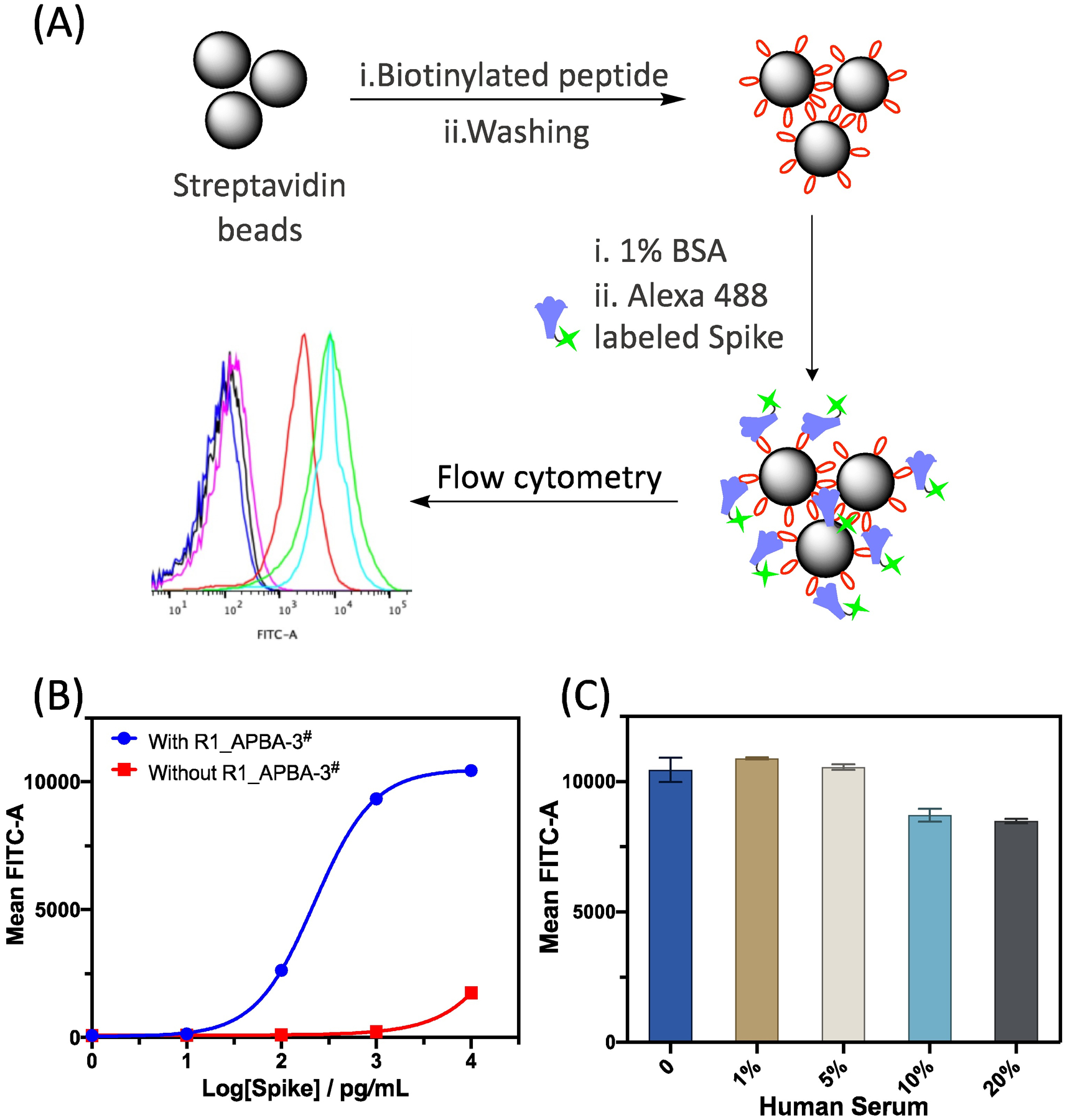
Detecting the coronavirus spike protein using R1_APBA-3# coated beads. (A) Schematic illustration of the use of flow cytometry to detect spike proteins. (B) Dose-dependent binding of the spike protein to R1_APBA-3# immobilized beads. The uncoated beads are used as a negative control. (C) Peptide enabled spike protein (1ng/mL) detection in blood serum. The bar graph presents averaged flow cytometry reading as well as the standard deviations of three independent samples.
CONCLUSIONS
This contribution describes our development and evaluation of APBA-displaying phage libraries, which we envisioned to be a generally applicable platform for reversible covalent ligand discovery for proteins of interest. APBA represents 2-acetylphenylboronic acid, which has recently emerged as a powerful amine-targeting warhead that can facilitate protein binding through reversible iminoboronate formation. While rational design of reversible covalent inhibitors using this warhead remains challenging, we demonstrate herein that the APBA warhead can be readily installed onto the surface of bacteriophage to give linear and cyclic peptide libraries, screening of which enables facile identification of reversible covalent ligands. The practicality of this approach was demonstrated by developing reversible covalent ligands for the S. aureus sortase A (SrtA) as well as the coronavirus spike protein, both are considered difficult-to-target proteins due to the lack of deep pockets for binding synthetic molecules. Our phage display approach readily yielded single digit micromolar binders for both proteins. Importantly, the peptide hits that we characterized were found to show excellent target specificity, leading to retained activity in complex biological systems as seen in SrtA inhibition on live cells as well as in spike protein detection in saliva or serum. SrtA and the spike RBD represents two distinct classes of proteins engaging protein-protein interactions, with SrtA displaying a large and shallow pocket for protein substrate recognition, while the spike RBD displays no discernable pocket at all. The success for these two initial targets suggests the general applicability of this approach to a wide range of proteins.
It is worth noting that parallel screens of phage libraries with and without a warhead revealed distinct peptide sequences that best support the warhead for target binding. The peptide sequence variations are difficult to predict a priori, showcasing the power of phage selection in evaluating chemically modified peptide libraries. On the other hand, it is important to note that our carefully designed structure-activity studies of select peptide hits revealed the benefit as well as some potential limitation of the APBA warheads towards binding proteins. By comparing the peptide hits to their oxidized analogues (APBA-3 vs APBA-3-O), we show that the covalent binding (iminoboronate formation) can improve the peptides’ affinity by 4 to 20 folds, which is consistent with the free energy gain of 3–4 kcal/mol revealed from small molecule studies. Spike protein RBD binding studies of the APBA-dimer peptides revealed a poor target specificity as the APBA-dimers showed substantial binding to BSA as well. This erosion of target specificity of the APBA dimers may be because the postulated double covalent bond formation gave too strong a force that applied to all proteins.
Finally, we note that our work presents the first examples of phage libraries that carry reversible covalent warheads. A very recent publication23 by Bogyo and coworkers reports phage displayed peptide libraries that carry irreversible covalent warheads that target catalytic cysteines or serines. The phage libraries described herein differ in the reversible nature of the APBA warhead and also in the fact that the covalent binding is no longer limited to just catalytic residues. Finally, we note that the phage library construction and screening should be readily adaptable to include other reversible covalent warheads and to identify inhibitors for essentially any protein of interest. Work along these lines are currently underway in our laboratory.
Supplementary Material
ACKNOWLEDGMENT
We thank Professor Tim van Opijnen and Mr. Stephen Hummel for the help with flow cytometry analysis. We would also like to acknowledge Ms. Yuhan Lyu for her help in preparing the SrtA mutant proteins.
Funding Sources
Financial support of this work is provided by the National Institutes of Health (GM124231), the National Science Foundation (CHE-1904874) and the Ono Pharma Foundation.
Footnotes
SUPPORTING INFORMATION
The Supporting Information is available free of charge on the ACS Publications website: additional phage sequencing results, protein expression data, peptide synthesis and characterization data can be found in the Supporting Information.
REFERENCES
- (1).Muttenthaler M; King GF; Adams DJ; Alewood PF, Trends in peptide drug discovery. Nat. Rev. Drug Discov 2021, 20 (4), 309–325. [DOI] [PubMed] [Google Scholar]
- (2).Henninot A; Collins JC; Nuss JM, The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem 2018, 61 (4), 1382–1414. [DOI] [PubMed] [Google Scholar]
- (3).Lu H; Zhou Q; He J; Jiang Z; Peng C; Tong R; Shi J, Recent advances in the development of protein–protein interactions modulators: mechanisms and clinical trials. Signal Transduct. Target 2020, 5 (1), 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Passioura T; Katoh T; Goto Y; Suga H, Selection-Based Discovery of Druglike Macrocyclic Peptides. Annu. Rev. Biochem 2014, 83, 727–752. [DOI] [PubMed] [Google Scholar]
- (5).Smith GP; Petrenko VA, Phage Display. Chem. Rev 1997, 97 (2), 391–410. [DOI] [PubMed] [Google Scholar]
- (6).Liu CC; Mack AV; Brustad EM; Mills JH; Groff D; Smider VV; Schultz PG, Evolution of Proteins with Genetically Encoded “Chemical Warheads”. J. Am. Chem. Soc 2009, 131 (28), 9616–9617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Heinis C; Rutherford T; Freund S; Winter G, Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol 2009, 5 (7), 502–7. [DOI] [PubMed] [Google Scholar]
- (8).Jafari MR; Deng L; Kitov PI; Ng S; Matochko WL; Tjhung KF; Zeberoff A; Elias A; Klassen JS; Derda R, Discovery of light-responsive ligands through screening of a light-responsive genetically encoded library. ACS Chem. Biol 2014, 9 (2), 443–50. [DOI] [PubMed] [Google Scholar]
- (9).Tharp JM; Hampton JT; Reed CA; Ehnbom A; Chen P-HC; Morse JS; Kurra Y; Pérez LM; Xu S; Liu WR, An amber obligate active site-directed ligand evolution technique for phage display. Nat. comm 2020, 11 (1), 1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Owens AE; Iannuzzelli JA; Gu Y; Fasan R, MOrPH-PhD: An Integrated Phage Display Platform for the Discovery of Functional Genetically Encoded Peptide Macrocycles. ACS Cent. Sci 2020, 6 (3), 368–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).McCarthy KA; Kelly MA; Li K; Cambray S; Hosseini AS; van Opijnen T; Gao J, Phage Display of Dynamic Covalent Binding Motifs Enables Facile Development of Targeted Antibiotics. J. Am. Chem. Soc 2018, 140 (19), 6137–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Bandyopadhyay A; Gao J, Targeting biomolecules with reversible covalent chemistry. Curr. Opin. Chem. Biol 2016, 34, 110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Serafimova IM; Pufall MA; Krishnan S; Duda K; Cohen MS; Maglathlin RL; McFarland JM; Miller RM; Frodin M; Taunton J, Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol 2012, 8 (5), 471–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bold R, “Development of the proteasome inhibitor Velcade (TM) (Bortezomib)” by Julian Adams, Ph.D., and Michael Kauffman, MD, Ph.D. Cancer Invest. 2004, 22 (2), 328–329. [DOI] [PubMed] [Google Scholar]
- (15).Reja RM; Wang W; Lyu Y; Haeffner F; Gao J, Lysine-Targeting Reversible Covalent Inhibitors with Long Residence Time. J. Am. Chem. Soc 2022, 144 (3), 1152–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Guo W-H; Qi X; Yu X; Liu Y; Chung C-I; Bai F; Lin X; Lu D; Wang L; Chen J; Su LH; Nomie KJ; Li F; Wang MC; Shu X; Onuchic JN; Woyach JA; Wang ML; Wang J, Enhancing intracellular accumulation and target engagement of PROTACs with reversible covalent chemistry. Nat. Comm 2020, 11 (1), 4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bandyopadhyay A; McCarthy KA; Kelly MA; Gao J, Targeting bacteria via iminoboronate chemistry of amine-presenting lipids. Nat. Comm 2015, 6, 6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Li K; Kelly MA; Gao J, Biocompatible conjugation of Tris base to 2-acetyl and 2-formyl phenylboronic acid. Org. Bio. Chem 2019, 17 (24), 5908–5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Akcay G; Belmonte MA; Aquila B; Chuaqui C; Hird AW; Lamb ML; Rawlins PB; Su N; Tentarelli S; Grimster NP; Su Q, Inhibition of Mcl-1 through covalent modification of a noncatalytic lysine side chain. Nat. Chem. Biol 2016, 12 (11), 931–936. [DOI] [PubMed] [Google Scholar]
- (20).Quach D; Tang G; Anantharajan J; Baburajendran N; Poulsen A; Wee J; Retna P; Li R; Liu B; Tee D; Kwek P; Joy J; Yang W-Q; Zhang C-J; Foo K; Keller T; Yao SQ, Strategic Design of Catalytic Lysine-Targeting Reversible Covalent BCR-ABL Inhibitors. Angew. Chem. Int. Ed 2021, 60, 17131–17137. [DOI] [PubMed] [Google Scholar]
- (21).Cascioferro S; Raffa D; Maggio B; Raimondi MV; Schillaci D; Daidone G, Sortase A Inhibitors: Recent Advances and Future Perspectives. J. Med. Chem 2015, 58 (23), 9108–23. [DOI] [PubMed] [Google Scholar]
- (22).Lan J; Ge J; Yu J; Shan S; Zhou H; Fan S; Zhang Q; Shi X; Wang Q; Zhang L; Wang X, Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581 (7807), 215–220. [DOI] [PubMed] [Google Scholar]
- (23).Chen S; Lovell S; Lee S; Fellner M; Mace PD; Bogyo M, Identification of highly selective covalent inhibitors by phage display. Nat. Biotechnol 2021, 39 (4), 490–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ng S; Jafari MR; Matochko WL; Derda R,. ACS Chem. Biol 2012, 7 (9), 1482–7. [DOI] [PubMed] [Google Scholar]
- (25).Kelly M; Cambray S; McCarthy KA; Wang W; Geisinger E; Ortiz-Marquez J; van Opijnen T; Gao J, Peptide Probes of Colistin Resistance Discovered via Chemically Enhanced Phage Display. ACS Infect. Dis 2020, 6 (9), 2410–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Assem N; Ferreira DJ; Wolan DW; Dawson PE, Acetone-Linked Peptides: A Convergent Approach for Peptide Macrocyclization and Labeling. Angew. Chem. Int. Ed 2015, 54 (30), 8665–8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ng S; Derda R, Phage-displayed macrocyclic glycopeptide libraries. Org. Bio. Chem 2016, 14 (24), 5539–5545.. [DOI] [PubMed] [Google Scholar]
- (28).Zhang J; Liu H; Zhu K; Gong S; Dramsi S; Wang Y-T; Li J; Chen F; Zhang R; Zhou L; Lan L; Jiang H; Schneewind O; Luo C; Yang C-G, Antiinfective therapy with a small molecule inhibitor of Staphylococcus aureus sortase. Proc. Natl. Acad. Sci. U.S.A 2014, 111 (37), 13517–13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Drag M; Salvesen GS, Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov 2010, 9 (9), 690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wang J; Li H; Pan J; Dong J; Zhou X; Niu X; Deng X, Oligopeptide Targeting Sortase A as Potential Anti-infective Therapy for Staphylococcus aureus. Front. Microbio 2018, 9 (245). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Rentero Rebollo I; McCallin S; Bertoldo D; Entenza JM; Moreillon P; Heinis C, Development of Potent S aureus Sortase A Inhibitors Based on Peptide Macrocycles. ACS Med. Chem. Lett 2016, 7 (6), 606–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bandyopadhyay A; Gao J, Iminoboronate-Based Peptide Cyclization That Responds to pH, Oxidation, and Small Molecule Modulators. J. Am. Chem. Soc 2016, 138 (7), 2098–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Yang T; Cuesta A; Wan X; Craven GB; Hirakawa B; Khamphavong P; May JR; Kath JC; Lapek JD; Niessen S; Burlingame AL; Carelli JD; Taunton J, Reversible lysine-targeted probes reveal residence time-based kinase selectivity. Nat. Chem. Biol 2022, 10.1038/s41589-022-01019-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Taylor PC; Adams AC; Hufford MM; de la Torre I; Winthrop K; Gottlieb RL, Neutralizing monoclonal antibodies for treatment of COVID-19. Nat. Rev. Immunol 2021, 21 (6), 382–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Pomplun S; Jbara M; Quartararo AJ; Zhang G; Brown JS; Lee Y-C; Ye X; Hanna S; Pentelute BL, De Novo Discovery of High-Affinity Peptide Binders for the SARS-CoV-2 Spike Protein. ACS Cent. Sci 2021, 7 (1), 156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Norman A; Franck C; Christie M; Hawkins PME; Patel K; Ashhurst AS; Aggarwal A; Low JKK; Siddiquee R; Ashley CL; Steain M; Triccas JA; Turville S; Mackay JP; Passioura T; Payne RJ, Discovery of Cyclic Peptide Ligands to the SARS-CoV-2 Spike Protein Using mRNA Display. ACS Cent. Sci 2021, 7 (6), 1001–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Bar-On YM; Flamholz A; Phillips R; Milo R, SARS-CoV-2 (COVID-19) by the numbers. Elife 2020, 9, e57309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.