

Abstract
The pregnane X receptor (PXR; NR1I2) is an important nuclear receptor whose main function is to regulate enzymes within drug metabolism. The main drug metabolizing enzyme regulated by PXR, cytochrome P450 (CYP) 3A4, accounts for the metabolism of nearly 50% of all marketed drugs. Recently, PXR has also been identified as playing a role in energy homeostasis, immune response, and cancer. Due to its interaction with these important roles, alongside its drug-drug interaction function, it is imperative to identify compounds which can modulate PXR. In this study, we screened the Tox21 10,000 compound collection to identify hPXR agonists using a stable hPXR-Luc HepG2 cell line. A pharmacological study in the presence of a PXR antagonist was performed to confirm the activity of the chosen potential hPXR agonists in the same cells. Finally, metabolically competent cell lines - HepaRG and HepaRG-PXR-Knockout (KO) - were used to further confirm the potential PXR activators. We identified a group of structural clusters and singleton compounds which included potentially novel hPXR agonists. Of the 21 selected compounds, 11 potential PXR activators significantly induced CYP3A4 mRNA expression in HepaRG cells. All of these compounds lost their induction when treating HepaRG-PXR-KO cells, confirming their PXR activation. Etomidoline presented as a potentially selective agonist of PXR. In conclusion, the current study has identified 11 compounds as potentially novel or not well-characterized PXR activators. These compounds should further be studied for their potential effects on drug metabolism and drug-drug interactions due to the immense implications of being a PXR agonist.
Keywords: Agonist, Pregnane X receptor, Quantitative high-throughput screening, Tox21
1. Introduction
The pregnane X receptor (PXR) is a nuclear receptor which regulates drug metabolizing enzymes (DMEs) by activation from endogenous, as well as exogenous, ligands [1]. The most notable PXR controlled DME is the extremely abundant liver cytochrome P450 (CYP) 3A4, which accounts for the metabolism of an estimated 50% of clinically used drugs [2, 3]. Due to its promiscuous ligand binding pocket, PXR has an abundant amount of possible ligands which can consequently generate drug-drug interactions (DDIs) through the modulation of DMEs [4]. PXR has a sister nuclear receptor, the constitutive androstane receptor (CAR), that frequently is observed to cross-talk via similar molecular signaling pathways in the management of DMEs [5]. However, CAR and PXR co-modulation may result in differing outcomes, and it is therefore prudent to identify compounds that modulate PXR selectively to identify potential DDI liabilities and therapeutic target interactions. Aside from its classic role, PXR has also been linked with energy homeostasis, cell proliferation, and inflammation [6, 7].
Energy homeostasis serves a fundamental role in the body and is regulated by many different factors, including Vitamin K and PXR [8]. A Vitamin K2 analog, Menaquinone-4, which has been used as a therapeutic agent to treat osteoporosis, was shown to affect the expression levels of genes related to bile acid synthesis and energy homeostasis through PXR activation [8]. In mice, a decrease in blood glucose levels has been observed when using the PXR activator pregnenolone 16α-carbonitrile (PCN), demonstrating that PXR plays some role in modulating energy homeostasis and/or disrupting endogenous metabolism through unintended environmental exposures [6, 9]. Alongside this discovery, a cross-talk between PXR, CAR, and forkhead box protein O1 (FOXO1) was also discovered to modulate gluconeogenesis through direct interaction [9]. These recent studies suggest that PXR regulates energy homeostasis and the knowledge that modulating PXR will have an important effect on the body.
Cancer, an ever-growing public health concern, is known to be caused by genetics, environmental toxins, and certain therapeutics. However, the mechanism by which these factors generate abnormal cell proliferation is constantly being studied. Activation of PXR has produced polarizing opinions as certain studies have shown it to be a tumor suppressor [10–12], while others have identified PXR to potentiate tumor progression [12–16]. Alongside its role in directly suppressing and/or progressing tumor cells, PXR also has an influence on the therapy of cancer through its regulation of DMEs. The metabolism of many cancer drugs occurs through PXR regulated DMEs, meaning activation of PXR could cause a premature clearance or facilitate bioactivation of cancer therapeutics [6, 17]. Therefore, identifying PXR activators may advance therapeutic development and toxicological interpretations.
PXR also plays a role in the inflammation process with respect to inflammatory bowel disease (IBD). Mutation of PXR has exhibited a higher susceptibility to IBD; in fact, activation of this important nuclear receptor results in protection against chemical-induced IBD in mice [18–20]. Alongside its function in IBD, a cross-talk between PXR and nuclear factor-kappa B (NF-κB) has been discovered, implying additional relationships to inflammation [19, 21]. It will be critical to perform future studies to determine how PXR has an impact on inflammation.
Given the diversity and physiological importance of PXR, identification and quantitative estimation of relative potencies of small molecules to activate PXR represents an important landmark in drug discovery and toxicology research. A four-way government collaboration between the National Center for Advancing Translational Sciences (NCATS), the National Toxicology Program (NTP), the Environmental Protection Agency (EPA), and the Food and Drug Administration (FDA) was created to screen 10,000 compounds (10K) for the important interaction they have on pathways in the body, including nuclear receptors [22–26]. In the current study, we performed a Tox21 10K compound library screen using a human PXR cell line and identified a group of PXR agonists.
2. Materials and Methods
2.1. Tox21 chemical library
The Tox21 compound library consisting of ~10,000 (7871 unique) small molecules, is comprised of drugs, pesticides, industrial chemicals, and food additives. This library was identified and compiled by NCATS [27, 28], NTP, and the US EPA. Each small molecule was selected based on properties compatible with high-throughput screens (HTS) (volatility, solubility, molecular weight, logP), possible and definite environmental hazards or exposure concerns, commercial availability, and cost. A diverse group of 88 compounds was also selected to be used as an internal control, and plated in duplicate on each library plate to perform reproducibility analysis as well as to determine positional plate effects [23].
2.2. HepG2-hPXR cell culture
HepG2 cells stably transfected with a CYP3A4-luc promoter construct and hPXR expression plasmid were provided by Dr. Taosheng Chen (Department of Chemical Biology and Therapeutics, St. Jude Children’s Research Hospital) [29]. Cells were thawed in EMEM (ATCC, Rockville, Maryland, USA), supplemented with 10% Hyclone Fetal Bovine Serum (FBS) (GE Healthcare Life Sciences, Logan, Utah, USA) and 100 U/mL of Penicillin and 100 μg/mL of Streptomycin (Invitrogen, Carlsbad, CA, USA) and kept in an incubator at 37 °C and 5% CO2. To culture these HepG2-hPXR cells, 500 μg/mL of Geneticin (GIBCO, Gaithersburg, MD, USA) was added to the thawing media.
2.3. PXR luciferase reporter gene assay
The hPXR-Luc HepG2 cells were plated at 2500 cells/5 μL/well in phenol red-free Dulbecco’s Modified Eagle’s Medium (DMEM) containing 5% charcoal stripped FBS (Invitrogen), 1 mM sodium pyruvate (Invitrogen), and 100 U/mL penicillin and 100 μg/mL streptomycin, into white/solid 1536-well plates (Greiner Bio-One North America, Monroe, NC, USA) using the BioRaptr Flying Reagent Dispenser (FRD) (Beckman Coulter, Brea, CA, USA). After 4–5 h of incubation at 37 °C to allow cell attachment, the positive control (rifampicin (RIF) (Millipore Sigma, St. Louis, MO, USA)) and test compounds were transferred at 23 nL to columns 1–4 and 5–48, respectively, using a Wako Pintool station (Wako Automation, San Diego, CA, USA). The assay plates were incubated for 24 h at 37 °C, after which 1 μL/well of CellTiter-Fluor reagent (Promega, Madison, WI, USA) was added into the assay plates using a BioRaptr FRD and after a 30-minute incubation at 37 °C, the fluorescence intensity was measured using a ViewLux plate reader (Perkin Elmer, Waltham, MA, USA). Data were expressed as relative fluorescence units. Directly after this measurement was taken, 4 μL/well of ONE-Glo™ Luciferase Assay reagent (Promega) was added into the assay plates using an FRD. After a 30-minute incubation at room temperature, the luminescence intensity was measured using the ViewLux plate reader. Data were expressed as relative luminescence units.
2.4. qHTS data analysis
A previous protocol described the method of qHTS data analysis [23]. Briefly, normalization relative to positive control compounds and dimethyl sulfoxide (DMSO)-only wells occurred for each titration point using raw plate reads as follows: % Activity = [(Vcompound−VDMSO)/(Vpos−VDMSO)] × 100, where Vcompound denotes the compound well values, Vpos denotes the median value of the positive control wells, and VDMSO denotes the median values of the DMSO-only wells. Utilizing the DMSO-only compound plates, the data set was corrected by applying an in-house pattern correction algorithm [30]. Using a four-parameter Hill equation, the half maximum effective concentration (EC50) and maximum response (efficacy) values were obtained. Each compound was then assigned to a specific class according to the type of concentration–response curve observed (1.1, 1.2, 1.3, 1.4, 2.1, 2.2, 2.3, 2.4, and 3 for activators; 4 for inactive). These designations were then converted to curve ranks (in integers 1 to 9 for increasing activating abilities; 0 for inactive) according to the criteria previously described [23, 31]. The test compound was then categorized into an activity outcome (active match, inactive match, inconclusive, and mismatch) based on the average curve rank from the triplicate runs and reproducibility calls according to the previously described criterion [23, 31]. The Tox21 10K compound library was put into clusters based on structural similarity (Leadscope® fingerprints; Leadscope, Inc., Columbus, OH, USA) using the self-organizing map (SOM) algorithm [32]. Each hexagon-shaped cluster was evaluated for its amount of active agonists and significance of enrichment as determined by p-values using the Fisher’s exact test.
2.5. HepaRG cell culture
NoSpin™ HepaRG™ cells were acquired from Lonza Group Ltd (Basel, Switzerland) and seeded at 8.0 × 105 cells/well in 12-well collagen-coated plates (Corning Inc., Corning, NY, USA) using Williams’ E Medium (ThermoFisher Scientific, Inc. Grand Island, NY, USA) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin (ThermoFisher Scientific, Inc.) and 1 bottle of HepaRG™ Thawing and Plating Medium Supplement (Lonza Group Ltd) per 100 mL Williams’ E Medium. Cells were treated 72 h later, in fresh media, with vehicle control (DMSO), 6-(4-Chlorophenyl)imidazo[2,1–b][1,3]thiazole-5-carbaldehyde O-(3, 4-dichlorobenzyl)oxime (CITCO: 1 μM) (Millipore Sigma), rifampicin (RIF: 10 μM), or test compounds (1,3-diphenylguanidine (Millipore Sigma): 15 μM; AMI-193 (Tocris Bio-Techne Corporation, Minneapolis, MN, USA): 9 μM; benidipine (Toronto Research, North York, ON, Canada): 20 μM; bensulide (Millipore Sigma): 0.9 μM; bortezomib (Selleck Chemicals, Houston, TX, USA): 16 nM; butachlor (Millipore Sigma): 4 μM; carfilzomib (Sequoia Pharmaceuticals, Gaithersburg, MD, USA): 0.2 μM; clofarabine (Sequoia Pharmaceuticals): 4 μM; dasatinib (Selleck Chemicals) : 0.5 μM; emetine (Sensient Pharmaceutical, St. Louis, MO, USA): 115 nM; etomidoline (Pharmaron, Waltham, MA, USA): 15 μM; FR167356 (Astellas Pharma, Tokyo, Japan): 5 μM; lacidipine (Sequoia Pharmaceuticals): 15 μM; liranaftate (Sequoia Pharmaceuticals): 15 μM; mosapride (Vitas-M Laboratory, Champaign, IL, USA): 15 μM; prodiamine (Chem Service, West Chester, PA, USA): 5 μM; spiperone (Enzo Life Sciences, Farmingdale, NY, USA): 10 μM; thiazopyr (Chem Service): 5 μM; tolindate (Pharmaron): 30 μM; vorinostat (Prestwick Chemical, Illkirch, France): 15 μM; or zosuquidar (Selleck Chemicals): 30 μM) for 24 h before harvesting.
2.6. PXR knockout cell culture
Undifferentiated PXR KO cells were purchased from Millipore Sigma and plated (Day 0) in Williams’ E Medium supplemented with growth supplement (Biopredic International, Saint Grégoire, France), Ala-Gln (Millipore Sigma), and Pen/Strep (Millipore Sigma) in a 24-well TC/Polyd Biocoated plate (Corning, Corning, NY, USA) as directed in the manufacturer’s instructions. Cell culture plates were then moved to an incubator at 37°C/5% CO2 for 48 h. After aspirating culture media (Day 2), 1 mL of fresh medium was added to each well, and the cell plate was returned to the incubator. On Day 4, media were aspirated and fresh medium plus 0.5% DMSO was added to each well. An exchange of media occurred again on day 7 containing 0.5% DMSO. On day 10, old media were aspirated and fresh medium plus 1.5% DMSO was added to each well. Exchanging spent media with fresh medium plus 1.5% DMSO occurred twice a week for two weeks. Cells were then treated (vehicle control (DMSO); CITCO: 1 μM; RIF: 10 μM; benidipine: 20 μM; dasatinib: 0.5 μM; etomidoline: 15 μM; FR167356: 5 μM; lacidipine: 15 μM; liranaftate: 15 μM; mosapride: 15 μM; prodiamine: 5 μM; thiazopyr: 5 μM; tolindate: 30 μM; or zosuquidar: 30 μM) using fresh media (without DMSO supplemented) on day 23. After 24 h of incubation at 37°C/5% CO2, cells were harvested.
2.7. Real-time reverse transcription polymerase chain reaction (RT-PCR)
Total RNA was extracted from treated HepaRG and PXR-KO cells using an RNeasy Mini Kit (Qiagen, Germantown, MD, USA), homogenized using a QIAshredder (Qiagen), and reverse transcribed with a High-Capacity RNA-to-cDNA™ Kit (ThermoFisher Scientific, Inc.) following the manufacturers’ instructions. CYP3A4 and CYP2B6 mRNA expression were normalized against the housekeeping gene, glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Real-time PCR assays were performed in 384-well plates on a QuantStudio 5 (Applied Biosystems, Foster City, CA, USA) with TaqMan™ Gene Expression Master Mix (ThermoFisher Scientific, Inc.). Probes were purchased from ThermoFisher Scientific, Inc. with the following information: CYP3A4 TaqMan™ – Assay ID: Hs00604506_m1; CYP2B6 TaqMan™ – Assay ID: Hs04183483_g1; Human GAPDH - FAM™/MDB probe, non-primer limited. Induction values were calculated using the equation: Fold = 2−ΔΔCt, where ΔCt represents the differences in cycle threshold numbers between CYP2B6 or CYP3A4 and GAPDH, and ΔΔCt represents the relative change in these differences between control and treatment groups.
2.8. Agonist β-lactamase reporter gene assays
AR-HEK293, ARE-HepG2, ERα-HEK293, ERβ-HEK293, FXR-HEK293, PPARδ-HEK293, and RXRα-HEK293 cells were dispensed at 2000 cells (AR, ARE, ERβ, and RXRα), 3000 cells (PPARδ), or 5000 cells (ERα and FXR) per well in 5 μL (ARE, ERα, ERβ, and FXR), or 6 μL (AR, PPARδ, and RXRα) of assay medium in 1536-well tissue culture treated black-well/clear bottom plates (Greiner Bio-One) using the BioRaptr FRD. After cells were given the allotted time to attach to the plates by being incubated for 5–6 h at 37 °C, 23 nL of compounds or positive controls, both dissolved in DMSO, were transferred to the assay plates via the Wako Pintool station. The assay plates were then incubated for 16 h (AR, ARE, ERβ, FXR, and RXRα), 17 h (PPARδ), or 18 h (ERα) at 37 °C, and 1 μL/well of LiveBLAzer™ FRET-B/G CCF4-AM substrate (ThermoFisher Scientific, Inc.) detection mix was added using a BioRaptr FRD and the plates were incubated at room temperature for 2 h. The fluorescence intensity was then measured by an Envision plate reader (PerkinElmer) at 405 nm excitation as well as 460 and 530 nm emissions. Data were expressed as the ratio of 460/530 nm emission values.
2.9. Agonist luciferase reporter gene assays
AhR-HepG2, AR-MDA-kb2, CAR-HepG2, ER-vMCF7, and PGC/ERR-HEK293 cells were dispensed at 2500 (CAR and PGC/ERR), 3000 (AR), and 4000 (AhR and ER) cells per well in 4 μL (CAR) or 5 μL (AhR, AR, ER, and PGC/ERR) of assay medium in 1536-well tissue culture treated white wall/solid bottom plates (Greiner Bio-One) using a Multidrop Combi dispenser. The assay plates were then incubated at 37 °C for 4–6 h (except ER was incubated for 24 h), followed by the addition of 23 nL of compounds or positive controls (both dissolved in DMSO) via the Wako Pintool station. The CAR assay only, then received 1 μL/well of antagonist (0.75 μM PK11195) using the BioRaptr FRD to decrease the constitutive activity. The assay plates were then incubated at 37 °C and 5% CO2 for 16 h (AR), 18 h (PGC/ERR), 22 h (ER), or 24 h (AhR and CAR). Next, 5 μL/well of ONE-Glo™ Luciferase Assay reagent was added using an FRD followed by a 30 min incubation at room temperature. The luminescence intensity was measured by a ViewLux plate reader. Data were expressed as relative luminescence units.
2.10. Antagonist β-lactamase reporter gene assays
AR-HEK293, ERα-HEK293, ERβ-HEK293, FXR-HEK293, PPARδ-HEK293, PPARγ-HEK293, and PR-HEK293 cells were dispensed at 2,000 cells (AR and ERβ), 3,000 cells (PPARδ, PPARγ, and PR), or 5,000 cells (ERα and FXR) per well in 4 μL (ERβ and PR), 5 μL (FXR, PPARδ, and PPARγ), or 6 μL (AR and ERα) of assay medium in 1536-well tissue culture treated black-well/clear bottom plates (Greiner Bio-One) using the BioRaptr FRD. After cells were allotted time to attach to the plates by being incubated for 5–6 h at 37 °C, 23 nL of compounds dissolved in DMSO or positive controls were transferred to the assay plates via the Pintool station. Assays received 1 μL/well of agonist (10 nM R1881 for AR; 0.5 and 5 nM β-estradiol for ERα and ERβ respectively; 300 μM CDCA for FXR; 0.3 μM L-165,041 for PPARδ; 50 nM rosiglitazone for PPARγ; and 5 nM R5020 for PR) using an FRD. The assay plates were then incubated for 16 h (AR, ERβ, FXR, and PR), 17 h (PPARδ and PPARγ), or 18 h (ERα) at 37 °C, and 1 μL/well of LiveBLAzer™ FRET-B/G CCF4-AM substrate (ThermoFisher Scientific, Inc.) detection mix was added using an FRD and the plates were incubated at room temperature for 2 h. The fluorescence intensity was then measured by an Envision plate reader at 405 nm excitation as well as 460 and 530 nm emissions. Data were expressed as the ratio of 460/530 nm emission values.
2.11. Antagonist luciferase reporter gene assays
AR-MDA-kb2, CAR-HepG2, ER-vMCF7, ERR-HEK293, PGC/ERR-HEK293, RAR-C3H10T1/2, and RORγ-CHO cells were dispensed at 1000 cells (RAR and RORγ), 2500 cells (CAR, ERR, and PGC/ERR), 3000 cells (AR), or 4,000 cells (ER) per well in 4 μL (except ERR and PGC/ERR in 5 μL) of the assay medium in 1,536-well tissue culture treated white wall/solid bottom plates (Greiner Bio-One) using a Multidrop Combi dispenser. The assay plates were then incubated at 37 °C for 4–6 h (AR, CAR, ERR, PGC/ERR, and RORγ), 18 h (RAR), or 24 h (ER). Twenty-three nL of compounds or positive controls, both dissolved in DMSO, were then transferred to the assay plates via a Wako Pintool station. Except for ERR and PGC/ERR, after the compound treatment, the assay plates received 1 μL/well of agonist (0.5 nM R1881 for AR; 50 nM CITCO for CAR; 0.1 nM β-Estradiol for ER; 1 μM retinol for RAR; and 1 μM doxycycline hyclate for RORγ) using an FRD. The assay plates were then incubated for 6 h (RAR), 16 h (AR and RORγ), 18 h (ERR and PGC/ERR), 22 h (ER), or 24 h (CAR) at 37 °C, and 4 μL/well (5 μL/well for ERR and PGC/ERR) of ONE-Glo™ Luciferase Assay reagent was added using an FRD followed by a 30 min incubation at room temperature. The luminescence intensity was measured by a ViewLux plate reader. Data were expressed as relative luminescence units.
2.12. Statistical analysis for RT-PCR
All experimental data are expressed as a mean of triplicate values ± standard deviation unless otherwise noted. Statistical comparisons were made by one-way analysis of variance with post-hoc Dunnett’s analysis. The statistical significance was set at p values of < 0.05 (*), <0.01 (**), or < 0.001 (***).
3. Results
3.1. qHTS performance and reproducibility
The hPXR-Luc HepG2 cell line was used to perform a primary screen of the Tox21 10 K compound collection to identify hPXR activators. RIF, the positive control, displayed consistent activity throughout the primary screen, containing about four hundred assay plates, with an EC50 of 6.1 ± 3.7 nM. The screening performance statistics indicate an adequate quality of the primary screen with the following values: signal-to-background ratio (S/B), 4.2 ± 0.8; coefficient of variance, 13% ± 5 (DMSO plates only); and Z’ factor, 0.50 ± 0.15.
We further evaluated the reproducibility of the primary screen by assessing the three separate screening runs and Tox21–88 array data. After the primary screening, each compound was sorted into one of four categories based on reproducibility of activity: active match, inactive match, mismatch, or inconclusive [23]. The mismatch rate of the Tox21 10K primary screen was low, with a value of 0.40% indicating a robust assay performance (Table 1).
Table I.
Assay performance of the PXR agonist screen using Tox21-88 duplicates and Tox21 10K compound collection in triplicate runs.
| Active Match | Inactive Match | Inconclusive (%) | Mismatch (%) | EC50 Fold* Change | |
|---|---|---|---|---|---|
|
| |||||
| 10K Screen | 18.55 | 83.37 | 17.67 | 0.40 | 1.45 |
| Tox21-88 | 45.79 | 35.94 | 15.53 | 2.75 | 1.31 |
For each compound with at least one replicate that is not class 4, the standard deviation (SD) of the EC50 values from all replicates was calculated. The SDs were then averaged across these compounds. The EC50 fold was calculated as the inverse log of the average SD.
3.2. Identification of hPXR activators
Following the primary screen, a self-organizing map (SOM) algorithm was used to group the entire Tox21 10K compound collection into 1,041 clusters based on their structural similarities [32]. A heatmap was generated to illustrate the amount of hPXR activators in each structural class (Fig. 1). There were sixty-one structural clusters (maroon in color), encompassing 1,674 samples, which were significantly (p < 0.01) enriched with hPXR activators, including clusters 6.26, 11.12, 18.2, and 42.11 which showed activating hPXR in a concentration dependent manner (Fig. 1).
Fig. 1.

Structural class heat map of hPXR agonist activity. The hPXR-Luc HepG2 stable cell line was screened against the Tox21 10K compound collection and compounds were divided into different hexagons, which represent a class of structurally similar compounds. The enrichment of PXR actives determines the intensity of a specific color in that cluster (negative logarithmic scale of the p-value, −log [p-value]). A maroon color is exhibited when clusters with multiple actives in their class are present, whereas clusters with no activity are a light gray color. The darker gray color represents empty clusters with no available (N/A) compounds in them. The backbone of four representative structural clusters is shown, as well as the dose–response curves for chosen compounds within that structural class. Data are expressed as mean ± SD from three experiments.
Out of the 7,871 unique compounds examined in the primary screen, 246 compounds were selected for cherry-pick confirmation based on efficacy (E > 30% of the positive control) or as a negative control. An 82% confirmation rate was found after these compounds were retested in the same format (data not shown). Based on potency (EC50 < 11 μM), efficacy (E > 80% of the positive control), quality control analysis, cytotoxicity, commercial availability, and cost, 21 compounds were further selected from the cherry-pick confirmation for follow-up studies (Table 2).
Table II.
EC50 and efficacy of potential novel hPXR agonists identified from qHTS primary and follow-up studies
| Chemical Name (CASRN, Supplier) | Structure | PXR |
|
|---|---|---|---|
| Primary EC50 (μM) [Efficacy, (%)] | Follow-up EC50 (μM) [Efficacy, (%)] | ||
|
| |||
| 1,3-Diphenylguanidine (102-06-7, Sigma) |
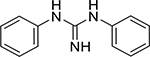
|
8.0 ± 13.7 [461 ± 143] | 2.7 ± 0.3 [393 ± 128] |
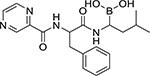
| AMI-193 (510-74-7, Tocris) |
|
5.9 ± 0.6 [363 ± 57.7] | 8.7 ± 1.1 [330 ± 40.1] |
| Bensulide (741-58-2, Sigma) |
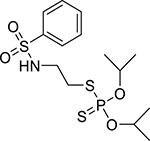
|
5.4 ± 0 [130 ± 20.3] | 3.9 ± 0.2 [114 ± 9.87] |
| Bortezomib (179324-69-7, Selleck) |
|
0.008 ± 0.0005 [97.7 ± 17.9] | 0.02 ± 0.004 [83.8 ± 12.8] |
| Butachlor (23184-66-9, Sigma) |
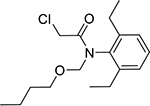
|
4.0 ± 4.0 [119 ± 49.2] | 6.49 ± 2.9 [107 ± 26.0] |
| Carfilzomib (868540-17-4, Sequoia) |
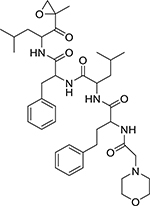
|
0.04 ± 0.024 [110 ± 10.3] | 0.1 ± 0.02 [108 ± 23.9] |
| Clofarabine (123318-82-1, Sequoia) |
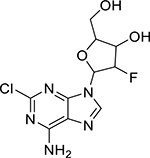
|
0.7 ± 0.2 [112 ± 34.6] | 3.3 ± 0.4 [214 ± 13.9] |
| Dasatinib (302962-49-8, Selleck) |
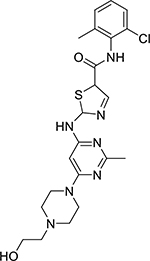
|
0.3 ± 0.5 [84.6 ± 22.2] | 0.6 ± 0.07 [127 ± 12.0] |
| Emetine (483-18-1, Enzo) |
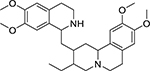
|
0.05 ± 0.04 [211 ± 74.0] | 0.1 ± 0.01 [119 ± 8.89] |
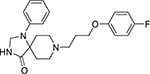
| Etomidoline (21590-92-1, Pharmaron) |
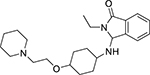
|
2.6 ± 1.3 [175 ± 74.6] | 3.2 ± 0.4 [142 ± 19.4] |
| FR167356 (174185-16-1, Astellas Pharma) |
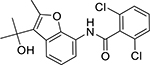
|
3.0 ± 2.2 [178 ± 9.77] | 7.1 ± 1.2 [145 ± 9.86] |
| Lacidipine (103890-78-4, Sequoia) |
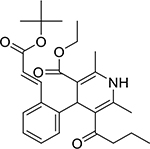
|
5.6 ± 5.5 [155 ± 70.5] | 10.4 ± 4.4 [115 ± 15.0] |
| Liranaftate (88678-31-3, Sequoia) |
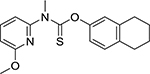
|
5.5 ± 2.6 [148 ± 42.6] | 4.5 ± 0.7 [144 ± 16.0] |
| Mosapride citrate (112885-42-4, Vitas) |
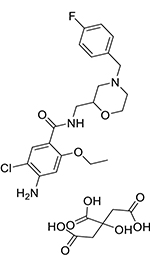
|
8.8 ± 3.3 [222 ± 59.5] | 10.8 ± 1.2 [138 ± 9.00] |
| Prodiamine (29091-21-2, Chemservice) |
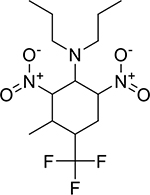
|
4.7 ± 0.8 [168 ± 31.0] | 8.5 ± 0.9 [108 ± 6.61] |
| rac Benidipine hydrochloride (91599-74-5, Toronto Research) |
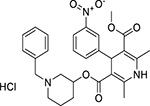
|
5.6 ± 5.4 [141 ± 33.3] | 4.6 ± 2.3 [105 ± 14.7] |
| Spiperone (749-02-0, Enzo) |
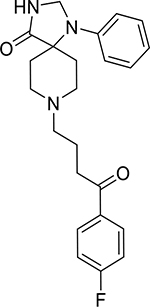
|
3.7 ± 0.4 [341 ± 31.7] | 6.2 ± 3.3 [345 ± 225] |
| Thiazopyr (117718-60-2, Chemservice) |
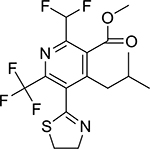
|
1.2 ± 1.0 [108 ± 37.9] | 4.6 ± 0.3 [102 ± 8.66] |
| Tolindate (27877-51-6, Pharmaron) |
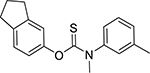
|
8.3 ± 2.3 [201 ± 37.5] | 8.7 ± 1.5 [113 ± 7.13] |
| Vorinostat (SAHA) (149647-78-9, Prestwick Chemical, Inc.) |
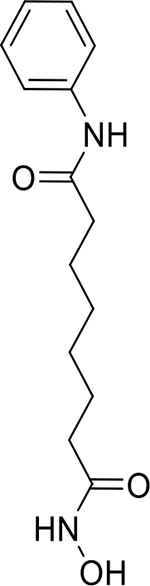
|
7.8 ± 3.8 [182 ± 46.9] | 6.0 ± 1.9 [111 ± 28.5] |
| Zosuquidar trihydrochloride (167465-36-3, Selleck) |
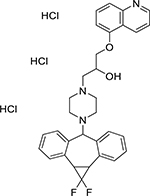
|
3.2 ± 1.7 [188 ± 41.7] | 1.6 ± 0.3 [106 ± 9.09] |
3.3. mRNA induction of CYP3A4 and CYP2B6 in HepaRG cells
Immortalized cell lines, such as HepG2, often do not achieve sufficient differentiation to natively model physiologically-relevant hepatocyte functionality such as nuclear receptor expression/functionality [33]. Moreover, immortalized cell models generally display minimal proficiency for drug metabolism (e.g., CYP3A4, target gene for PXR), yielding responses largely reflective of parent compound effects that do not capture active metabolite contributions. Due to these limitations, it is prudent to use a more physiologically relevant cell model to confirm primary screening data. HepaRG cells, an immortalized hepatocyte progenitor cell model, have been extensively qualified as a surrogate screening model comparable to human primary hepatocyte cultures [34]. Using HepaRG cells, the 21 compounds were treated for 24 h and harvested for mRNA. Among these 21 tested molecules, 11 compounds were found to significantly increase the mRNA expression of CYP3A4 (Fig. 2A) indicating their potential as hPXR activators. Bensulide, butachlor, and spiperone also induced CYP3A4 above a 5-fold increase, but the values were not statistically significant. Etomidoline, lacidipine, liranaftate, and thiazopyr induced CYP3A4 to a greater extent than the positive control, rifampicin. As previously stated, activation of PXR’s sister nuclear receptor CAR also induces CYP3A4 gene expression, often to a lesser extent than CYP2B6. To identify potentially selective PXR activators, the mRNA expression levels of CYP2B6 were also examined (Fig. 2B). All 11 of the identified PXR activators also induced CYP2B6 mRNA content to a significant extent. However, benidipine, dasatanib, and FR167356 induced CYP2B6 less extensively than rifampicin, but in a statistically significant manner implying their potential as selective PXR activators.
Fig. 2.

mRNA induction in HepaRG cells of CYP3A4 and CYP2B6. HepaRG cells were treated with potential PXR agonists selected from the primary screen, CITCO (1 μM), RIF (10 μM), or the vehicle control (0.1% DMSO) as described in Materials and Methods. Real-time PCR was used to analyze the mRNA expression of CYP3A4 (a) and CYP2B6 (b). The dotted line represents 100% activity of RIF for each graph. Each bar represents the mean ± SD in triplicate. *, p < .05; **, p < .01; ***, p < .001.
3.4. Pharmacological inhibition of PXR
To further confirm the compounds for PXR activation, experiments were performed in hPXR HepG2 cells in the presence of SPA70, a known selective PXR antagonist [35]. SPA70 was used to co-treat the cells with each compound in a concentration dependent manner. Fig. 3 demonstrates that this co-treatment generated a right shift on all concentration response curves from the 11 compounds identified as statistically significant CYP3A4 inducers, which suggests that their activity is indeed due to PXR. This further suggests all of these compounds to be true PXR activators, given the deficiencies for CAR functionality with HepG2 cells [5]. Fig. 4 shows the 10 compounds that did not induce CYP3A4 expression to a significant degree after co-treatment with SPA70. Only bensulide and butachlor generated clear shifts in the concentration response curves.
Fig. 3.

Pharmacological concentration response curves of potential PXR agonists. The hPXR-Luc HepG2 stable cells were treated with multiple concentrations of RIF (a), benidipine (b), dasatanib (c), etomidoline (d), FR167356 (e), lacidipine (f), liranaftate, (g), mosapride (h), prodiamine (i), thiazopyr (j), tolindate (k), and zosuquidar (l) and co-treated with DMSO, 0.5 μM SPA70, or 0.75 μM SPA70. The efficacy of each compound was compared to the positive control, RIF (100% = RIF activity). Each curve represents the mean ± SD of three separate experiments.
Fig. 4.

Pharmacological concentration response curves of HepaRG PXR inactives. Concentration response curves were acquired on the 10 compounds (a – j) which did not induce CYP3A4 mRNA in HepaRG cells. Each curve was co-treated with DMSO, 0.5 μM SPA70, and 0.75 μM SPA70 to determine pharmacological activity. All data were compared to the positive control, RIF (100% = RIF activity). Data were expressed as mean ± SD from triplicate experiments.
3.5. HepaRG PXR-KO cell mRNA induction
Due to the amount of crosstalk between PXR and CAR, it is prudent to further identify the extent of CYP3A4 induction attributed to activation of PXR. Therefore, we acquired HepaRG PXR-KO cells and treated them with each of the 11 potential PXR activators confirmed in the HepaRG experiments. The CYP3A4 induction was significantly decreased after treatment for all 11 compounds when using the PXR-KO cells (Fig. 5A). CITCO, a selective CAR activator with much lower potency for PXR activation, was the only compound, out of this set, to lack a significant decrease in CYP3A4 mRNA content in response to chemical exposure at the concentrations examined. Each of the 11 compounds, along with CITCO, significantly decreased the amount of CYP2B6 expression (Fig. 5B).
Fig. 5.

mRNA induction of CYP3A4 and CYP2B6 in HepaRG-PXR-KO cells. HepaRG and HepaRG-PXR-KO cells were treated with potential PXR agonists, CITCO (1 μM), RIF (10 μM), or the vehicle control (0.1% DMSO) as described in materials and methods. Real-time PCR was used to analyze the mRNA expression of CYP3A4 (a) and CYP2B6 (b). Each bar represents the mean ± SD in triplicate. Statistical analysis was completed between each treatment in HepaRG and HepaRG-PXR-KO cells; *, p < .05; **, p < .01; ***, p < .001.
3.6. Selective activity of compounds
The human body entails a very complex system of pathways working together to maintain homeostasis. Therefore, it is important to note that the outcome for one individual pathway does not necessarily reflect the full scope of the system. To integrate individual nuclear receptors pathways, we generated a primary screening outcome heatmap, from previously performed experiments, including 11 agonist and 12 antagonist primary assays (Fig. 6). All the assays shown here are nuclear receptor assays, except for the ARE/Nrf2 assay performed. ARE/Nrf2 represents an important component of adaptive response to protect against oxidative stress having known interplay with PXR [36]. Three of the 11 compounds were active agonists to both PXR and ARE/Nrf2 pathways, although these three compounds tested in the ARE/Nrf2 assay had much lower potencies than when tested in the PXR assay (data can be found at https://tripod.nih.gov/tox21/assays/), demonstrating the potential of PXR activators to also induce oxidative stress response. Thiazopyr was observed as selective to PXR and CAR activation within this suite of pathways. Finally, a primary goal of this study was to identify selective PXR activators to empower drug capabilities and interactions. Across this suite of 23 individual assay models, etomidoline appeared to be uniquely qualified as an apparent selective activator of PXR at the concentrations examined. This form of selectivity for PXR activation was unique across the thousands of chemical structures examined within our investigation, and has implications as a useful tool for mechanistic and translational drug safety research.
Fig. 6.

Selectivity heatmap of potential PXR agonists. The 11 potential PXR agonists were screened, and their activity outcome in 10 agonist assays and 12 antagonist assays are displayed. For the AR-bla/luc and ER-bla/luc agonist and antagonist assays, the upper left triangle represents the first mentioned assay, while the lower right triangle represents the second assay mentioned.
4. Discussion
Identification and characterization of PXR activators have become an important step in drug development to estimate drug-drug interaction potential and unravel the complexities of drug safety considerations in humans. PXR also plays many important roles in metabolism, energy homeostasis, cell proliferation and repression, and inflammation, which generates the need to fully explore a compound’s ability to activate this nuclear receptor. The current study identified 11 PXR activators, nine of which are potentially novel (etomidoline, FR167356, lacidipine, liranaftate, mosapride, prodiamine, thiazopyr, tolindate, and zosuquidar). The other two compounds, benidipine and dasatinib, found to be PXR agonists in the current study, generated consistent data with previous findings [37, 38]. Among these 11 identified PXR activators, etomidoline appeared to be uniquely selective to PXR activation across a panel of 23 Tox21 assay platforms, which could provide a useful tool for PXR-related drug-drug interaction and drug safety research.
In this study, the Tox21 10K compound collection was screened using a stable HepG2 cell line imbued with CYP3A4-luc and PXR [29, 39]. Sixty-one structural clusters were identified to have a statistically significant amount of PXR activators in Fig. 1. Alongside these clusters, individual actives were also chosen for a follow-up screen. From this cherry pick confirmation, 21 compounds were chosen based on potency (EC50 < 11 μM), efficacy (E > 80%), quality control analysis, cytotoxicity, commercial availability, and cost. Eleven of these 21 compounds significantly induced the mRNA expression of CYP3A4 in HepaRG cells, a physiologically relevant system to human primary hepatocytes [34]. All 11 compounds also had a decreased activity when co-treated with SPA70, the PXR antagonist, as well as in HepaRG PXR-KO cells, further identifying them as PXR activators. The other 10 compounds, not having significantly induced CYP3A4 mRNA, were also co-treated with SPA70, and all but two (bensulide, and butachlor) were found to have a nominal or no EC50 shift, implying an alternative mechanism for assay responses that does not appear to be a direct activation of PXR. Bensulide and butachlor did increase the mRNA expression of CYP3A4 in HepaRG cells, but not in a statistically significant manner, therefore identifying them as very weak potential PXR agonists.
The compounds identified as hPXR activators in this study have a wide variety of therapeutic usage and toxicity. Benidipine and lacidipine are both calcium channel blockers used in the treatment of high blood pressure and other conditions [40, 41]. Dasatinib and zosuquidar both have a therapeutic effect on different types of cancer; the former being a targeted tyrosine-kinase inhibitor used for leukemia and the latter being a p-glycoprotein inhibitor potentially useful in the co-treatment for different cancers [42, 43]. As a vacuolar ATPase inhibitor, FR167356 may play a therapeutic role in bone disease [44]. As a topical therapeutic, liranaftate [45] and tolindate (https://pubchem.ncbi.nlm.nih.gov/compound/34051) are known to be used as an antifungal. A gastroprokinetic agent, mosapride is a 5-hydroxytryptamine (5-HT, serotonin) type 4 (5-HT4) agonist, and contrastingly, is also a 5-HT3 inhibitor [46]. With the therapeutic uses these PXR activators are known for, it is imperative to fully understand their mechanism of action and clearly identify any DDIs that could interfere when taking other medications concurrently. Further studies are therefore needed to reduce the potential for DDIs with these identified PXR agonists.
So far, many pesticides have been found to activate PXR and/or CAR by inducing the expression of CYP3A4 and/or CYP2B6, respectively [47]. It is interesting to note that prodiamine and thiazopyr are both used as pesticides, specifically weed killers. These pesticides have previously exhibited tumorigenic properties and must be used with caution [48]. Due to their identification as PXR activators in this study, further studies are needed to assess these mechanisms and the implications on human health and environmental toxicology. For example, if these pesticides reached sufficiently high internal exposure concentrations to activate PXR and induced liver enzymes, agricultural workers may unknowingly have altered drug clearance capacity and significant human health effects (e.g., lower drug efficacy). In Fig. 5, thiazopyr appears to selectively activate PXR and CAR at the concentrations examined, while prodiamine is an activator and inhibitor of a few different receptors. Therefore, the full mechanism through which these compounds act goes far beyond the scope of nuclear receptors and will need to be further studied to determine the extent of human toxicological potential.
Etomidoline, a known muscle relaxant [49], was identified as a selective PXR agonist from the current study. Due to its promiscuous ligand binding pocket, many compounds bind to PXR as well as other nuclear receptors and it is, therefore, difficult to find a selective PXR agonist. PXR has a sister nuclear receptor known as CAR; many of the compounds identified as modulating PXR generally have an effect on CAR due to the similarity in the ligand binding domain. Therefore, identifying etomidoline as a selective PXR agonist is encouraging when therapeutic usage is a possibility. Further testing needs to be performed to fully ascertain the importance of the modulation of PXR through etomidoline.
In conclusion, 11 compounds, including nine potentially novel ones, were identified as PXR agonists through a primary screening using a PXR HepG2 cell line, a pharmacological study co-treating with a PXR inhibitor, and a follow-up study of CYP3A4 induction in HepaRG and HepaRG-PXR-KO cells. It is important to note that etomidoline appears to be PXR selective from this study. Due to the important role that PXR plays in drug metabolism, as well as the newer therapeutic roles in energy homeostasis, cancer, and inflammation, it is imperative to thoroughly study compounds which display PXR agonist abilities. This initial study provides the first step into identifying potential PXR agonists and requires future studies to be done which will provide a better insight into the complete range of biological activity of these compounds.
Acknowledgments
This work was supported in part by the Intramural Research Program of the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health and interagency Agreement IAA #NTR 12003 from the National Institute of Environmental Health Sciences/Division of the National Toxicology Program to the NCATS, NIH. The views expressed in this paper are those of the authors and do not necessarily reflect the statements, opinions, views, conclusions, or policies of the NCATS, the National Institutes of Health, the Environmental Protection Agency, the Division of National Toxicology Program, or the U.S. government. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
References
- [1].Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterstrom RH, Perlmann T, Lehmann JM, An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway, Cell 92(1) (1998) 73–82. [DOI] [PubMed] [Google Scholar]
- [2].Kumar GN, Surapaneni S, Role of drug metabolism in drug discovery and development, Med Res Rev 21(5) (2001) 397–411. [DOI] [PubMed] [Google Scholar]
- [3].Sueyoshi T, Negishi M, Phenobarbital response elements of cytochrome P450 genes and nuclear receptors, Annu Rev Pharmacol Toxicol 41 (2001) 123–43. [DOI] [PubMed] [Google Scholar]
- [4].Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA, The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions, J Clin Invest 102(5) (1998) 1016–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lynch C, Mackowiak B, Huang R, Li L, Heyward S, Sakamuru S, Wang H, Xia M, Identification of Modulators That Activate the Constitutive Androstane Receptor From the Tox21 10K Compound Library, Toxicol Sci 167(1) (2019) 282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mackowiak B, Hodge J, Stern S, Wang H, The Roles of Xenobiotic Receptors: Beyond Chemical Disposition, Drug Metab Dispos 46(9) (2018) 1361–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhou C, Verma S, Blumberg B, The steroid and xenobiotic receptor (SXR), beyond xenobiotic metabolism, Nucl Recept Signal 7 (2009) e001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sultana H, Watanabe K, Rana MM, Takashima R, Ohashi A, Komai M, Shirakawa H, Effects of Vitamin K(2) on the Expression of Genes Involved in Bile Acid Synthesis and Glucose Homeostasis in Mice with Humanized PXR, Nutrients 10(8) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kodama S, Koike C, Negishi M, Yamamoto Y, Nuclear receptors CAR and PXR cross talk with FOXO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes, Mol Cell Biol 24(18) (2004) 7931–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ouyang N, Ke S, Eagleton N, Xie Y, Chen G, Laffins B, Yao H, Zhou B, Tian Y, Pregnane X receptor suppresses proliferation and tumourigenicity of colon cancer cells, Br J Cancer 102(12) (2010) 1753–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Misawa A, Inoue J, Sugino Y, Hosoi H, Sugimoto T, Hosoda F, Ohki M, Imoto I, Inazawa J, Methylation-associated silencing of the nuclear receptor 1I2 gene in advanced-type neuroblastomas, identified by bacterial artificial chromosome array-based methylated CpG island amplification, Cancer Res 65(22) (2005) 10233–42. [DOI] [PubMed] [Google Scholar]
- [12].Yoshinari K, Role of Nuclear Receptors PXR and CAR in Xenobiotic-Induced Hepatocyte Proliferation and Chemical Carcinogenesis, Biol Pharm Bull 42(8) (2019) 1243–1252. [DOI] [PubMed] [Google Scholar]
- [13].Chen Y, Tang Y, Wang MT, Zeng S, Nie D, Human pregnane X receptor and resistance to chemotherapy in prostate cancer, Cancer Res 67(21) (2007) 10361–7. [DOI] [PubMed] [Google Scholar]
- [14].Masuyama H, Hiramatsu Y, Kodama J, Kudo T, Expression and potential roles of pregnane X receptor in endometrial cancer, J Clin Endocrinol Metab 88(9) (2003) 4446–54. [DOI] [PubMed] [Google Scholar]
- [15].Meyer zu Schwabedissen HE, Tirona RG, Yip CS, Ho RH, Kim RB, Interplay between the nuclear receptor pregnane X receptor and the uptake transporter organic anion transporter polypeptide 1A2 selectively enhances estrogen effects in breast cancer, Cancer Res 68(22) (2008) 9338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang H, Venkatesh M, Li H, Goetz R, Mukherjee S, Biswas A, Zhu L, Kaubisch A, Wang L, Pullman J, Whitney K, Kuro-o M, Roig AI, Shay JW, Mohammadi M, Mani S, Pregnane X receptor activation induces FGF19-dependent tumor aggressiveness in humans and mice, J Clin Invest 121(8) (2011) 3220–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhuo W, Hu L, Lv J, Wang H, Zhou H, Fan L, Role of pregnane X receptor in chemotherapeutic treatment, Cancer Chemother Pharmacol 74(2) (2014) 217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cheng J, Shah YM, Gonzalez FJ, Pregnane X receptor as a target for treatment of inflammatory bowel disorders, Trends Pharmacol Sci 33(6) (2012) 323–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ren Y, Yue B, Ren G, Yu Z, Luo X, Sun A, Zhang J, Han M, Wang Z, Dou W, Activation of PXR by alantolactone ameliorates DSS-induced experimental colitis via suppressing NF-kappaB signaling pathway, Sci Rep 9(1) (2019) 16636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rana M, Coshic P, Goswami R, Tyagi RK, Influence of a critical single nucleotide polymorphism on nuclear receptor PXR-promoter function, Cell Biol Int 41(5) (2017) 570–576. [DOI] [PubMed] [Google Scholar]
- [21].Zhou C, Tabb MM, Nelson EL, Grun F, Verma S, Sadatrafiei A, Lin M, Mallick S, Forman BM, Thummel KE, Blumberg B, Mutual repression between steroid and xenobiotic receptor and NF-kappa B signaling pathways links xenobiotic metabolism and inflammation, Journal of Clinical Investigation 116(8) (2006) 2280–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kavlock RJ, Austin CP, Tice RR, Toxicity testing in the 21st century: implications for human health risk assessment, Risk Anal 29(4) (2009) 485–7; discussion 492–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Huang R, A Quantitative High-Throughput Screening Data Analysis Pipeline for Activity Profiling, Methods Mol Biol 1473 (2016) 111–22. [DOI] [PubMed] [Google Scholar]
- [24].Shukla SJ, Huang R, Austin CP, Xia M, The future of toxicity testing: a focus on in vitro methods using a quantitative high-throughput screening platform, Drug Discov Today 15(23–24) (2010) 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hsu C-W, Huang R, Attene-Ramos M, Austin C, Simeonov A, Xia M, Advances in high-throughput screening technology for toxicology, International Journal of Risk Assessment and Management X, No. Y (2016) 0–0. [Google Scholar]
- [26].Lynch C, Sakamuru S, Li S, Xia M, High-Throughput Screening and Hazard Testing Prioritization, 2020, pp. 75–86. [Google Scholar]
- [27].Huang R, Southall N, Wang Y, Yasgar A, Shinn P, Jadhav A, Nguyen DT, Austin CP, The NCGC pharmaceutical collection: a comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics, Sci Transl Med 3(80) (2011) 80ps16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Attene-Ramos MS, Miller N, Huang R, Michael S, Itkin M, Kavlock RJ, Austin CP, Shinn P, Simeonov A, Tice RR, Xia M, The Tox21 robotic platform for the assessment of environmental chemicals--from vision to reality, Drug Discov Today 18(15–16) (2013) 716–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lin W, Wu J, Dong H, Bouck D, Zeng FY, Chen T, Cyclin-dependent kinase 2 negatively regulates human pregnane X receptor-mediated CYP3A4 gene expression in HepG2 liver carcinoma cells, J Biol Chem 283(45) (2008) 30650–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang Y, Huang R, Correction of Microplate Data from High-Throughput Screening, Methods Mol Biol 1473 (2016) 123–34. [DOI] [PubMed] [Google Scholar]
- [31].Huang R, Sakamuru S, Martin MT, Reif DM, Judson RS, Houck KA, Casey W, Hsieh JH, Shockley KR, Ceger P, Fostel J, Witt KL, Tong W, Rotroff DM, Zhao T, Shinn P, Simeonov A, Dix DJ, Austin CP, Kavlock RJ, Tice RR, Xia M, Profiling of the Tox21 10K compound library for agonists and antagonists of the estrogen receptor alpha signaling pathway, Sci Rep 4 (2014) 5664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kohonen T, Self-organizing neural projections, Neural Netw 19(6–7) (2006) 723–33. [DOI] [PubMed] [Google Scholar]
- [33].Gerets HH, Tilmant K, Gerin B, Chanteux H, Depelchin BO, Dhalluin S, Atienzar FA, Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins, Cell Biol Toxicol 28(2) (2012) 69–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Jackson JP, Li L, Chamberlain ED, Wang H, Ferguson SS, Contextualizing Hepatocyte Functionality of Cryopreserved HepaRG Cell Cultures, Drug Metab Dispos 44(9) (2016) 1463–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lin W, Wang YM, Chai SC, Lv L, Zheng J, Wu J, Zhang Q, Wang YD, Griffin PR, Chen T, SPA70 is a potent antagonist of human pregnane X receptor, Nat Commun 8(1) (2017) 741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Swales KE, Moore R, Truss NJ, Tucker A, Warner TD, Negishi M, Bishop-Bailey D, Pregnane X receptor regulates drug metabolism and transport in the vasculature and protects from oxidative stress, Cardiovasc Res 93(4) (2012) 674–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Noll EM, Eisen C, Stenzinger A, Espinet E, Muckenhuber A, Klein C, Vogel V, Klaus B, Nadler W, Rosli C, Lutz C, Kulke M, Engelhardt J, Zickgraf FM, Espinosa O, Schlesner M, Jiang X, Kopp-Schneider A, Neuhaus P, Bahra M, Sinn BV, Eils R, Giese NA, Hackert T, Strobel O, Werner J, Buchler MW, Weichert W, Trumpp A, Sprick MR, CYP3A5 mediates basal and acquired therapy resistance in different subtypes of pancreatic ductal adenocarcinoma, Nat Med 22(3) (2016) 278–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Stepankova M, Krasulova K, Doricakova A, Kurka O, Anzenbacher P, Dvorak Z, Optical isomers of dihydropyridine calcium channel blockers display enantiospecific effects on the expression and enzyme activities of human xenobiotics-metabolizing cytochromes P450, Toxicol Lett 262 (2016) 173–186. [DOI] [PubMed] [Google Scholar]
- [39].Dong H, Lin W, Wu J, Chen T, Flavonoids activate pregnane x receptor-mediated CYP3A4 gene expression by inhibiting cyclin-dependent kinases in HepG2 liver carcinoma cells, BMC Biochem 11 (2010) 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kosaka H, Hirayama K, Yoda N, Sasaki K, Kitayama T, Kusaka H, Matsubara M, The L-, N-, and T-type triple calcium channel blocker benidipine acts as an antagonist of mineralocorticoid receptor, a member of nuclear receptor family, Eur J Pharmacol 635(1–3) (2010) 49–55. [DOI] [PubMed] [Google Scholar]
- [41].Lee H, Kim JW, Kim DK, Choi DK, Lee S, Yu JH, Kwon OB, Lee J, Lee DS, Kim JH, Min SH, Calcium Channels as Novel Therapeutic Targets for Ovarian Cancer Stem Cells, Int J Mol Sci 21(7) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dantzig AH, Law KL, Cao J, Starling JJ, Reversal of multidrug resistance by the P-glycoprotein modulator, LY335979, from the bench to the clinic, Curr Med Chem 8(1) (2001) 39–50. [DOI] [PubMed] [Google Scholar]
- [43].Novo G, Di Lisi D, Bronte E, Macaione F, Accurso V, Badalamenti G, Rinaldi G, Siragusa S, Novo S, Russo A, Cardiovascular Toxicity in Cancer Patients Treated with Tyrosine Kinase Inhibitors: A Real-World Single-Center Experience, Oncology (2020) 1–7. [DOI] [PubMed] [Google Scholar]
- [44].Niikura K, Takeshita N, Takano M, A vacuolar ATPase inhibitor, FR167356, prevents bone resorption in ovariectomized rats with high potency and specificity: potential for clinical application, J Bone Miner Res 20(9) (2005) 1579–88. [DOI] [PubMed] [Google Scholar]
- [45].Sulaiman A, Wan X, Fan J, Kasimu H, Dong X, Wang X, Zhang L, Abliz P, Upur H, Analysis on curative effects and safety of 2% liranaftate ointment in treating tinea pedis and tinea corporis & cruris, Pak J Pharm Sci 30(3(Special)) (2017) 1103–1106. [PubMed] [Google Scholar]
- [46].Park YS, Sung KW, Gastroprokinetic agent, mosapride inhibits 5-HT3 receptor currents in NCB-20 cells, Korean J Physiol Pharmacol 23(5) (2019) 419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Abass K, Lamsa V, Reponen P, Kublbeck J, Honkakoski P, Mattila S, Pelkonen O, Hakkola J, Characterization of human cytochrome P450 induction by pesticides, Toxicology 294(1) (2012) 17–26. [DOI] [PubMed] [Google Scholar]
- [48].Hurley PM, Mode of carcinogenic action of pesticides inducing thyroid follicular cell tumors in rodents, Environ Health Perspect 106(8) (1998) 437–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mukai T, Yamaguchi E, Goto J, Takagi K, Smooth muscle relaxing drugs and guinea pig ileum, Jpn J Pharmacol 31(2) (1981) 147–57. [DOI] [PubMed] [Google Scholar]