

Abstract
Beta-blockers are a standard treatment for heart failure and cardiac arrhythmias. There are about 30 commonly used beta-blockers representing a diverse class of drugs with different receptor affinities and pleiotropic properties. We reported that among 14 beta-blockers tested previously, only carvedilol effectively suppressed cardiac ryanodine receptor (RyR2) mediated spontaneous Ca2+ waves during store Ca2+ overload, also known as store-overload induced Ca2+ release (SOICR). Given the critical role of SOICR in arrhythmogenesis, it is of importance to determine whether there are other beta-blockers that suppress SOICR. Here we assessed the effect of other commonly used beta-blockers on RyR2-mediated SOICR in HEK293 cells using single cell Ca2+ imaging. Of the 13 beta-blockers tested, only nebivolol, a beta-1 selective beta-blocker with nitric oxide synthase (NOS) stimulating action, effectively suppressed SOICR. The NOS inhibitor (N-nitro-L-arginine methyl ester) had no effect on nebivolol’s SOICR inhibition, and the NOS activator (histamine or prostaglandin E2) alone did not inhibit SOICR. Hence, nebivolol’s SOICR inhibition was independent of NOS stimulation. Like carvedilol, nebivolol reduced opening of single RyR2 channels and suppressed spontaneous Ca2+ waves in intact hearts and catecholaminergic polymorphic ventricular tachycardia (CPVT) in mice harboring a RyR2 mutation (R4496C). Interestingly, a non-beta-blocking nebivolol enantiomer, (l)-nebivolol, also suppressed SOICR and CPVT without lowering heart rate. These data indicate that nebivolol, like carvedilol, possesses a RyR2-targeted action that suppresses SOICR and SOICR-evoked VTs. Thus, nebivolol represents a promising agent for Ca2+-triggered arrhythmias.
Keywords: beta-blockers, nebivolol, catecholaminergic polymorphic ventricular tachycardia, spontaneous calcium release, sarcoplasmic reticulum, ryanodine receptor
Summary Statement:
Nebivolol, a third generation of β-blocker with NO-dependent vasodilatory property, possesses a ryanodine receptor-targeted action that suppresses spontaneous Ca2+ waves and wave-evoked ventricular tachyarrhythmia. Thus, nebivolol represents a promising agent for Ca2+-triggered arrhythmias.
INTRODUCTION
Ventricular tachyarrhythmia (VT) is a leading cause of sudden cardiac death, especially in patients with heart failure [1]. A variety of anti-VT therapies have been developed over the past decades. Unfortunately, most existing anti-VT drugs provide little or no survival benefits [2,3]. A better understanding of the molecular mechanism underlying VT is needed in order to develop more effective and targeted anti-VT agents.
Abnormal intracellular Ca2+ handling in cardiomyocytes is a hallmark of heart failure [4–8]. It is well established that sarcoplasmic reticulum (SR) Ca2+ overload leads to spontaneous SR Ca2+ release in the form of propagating Ca2+ waves, also known as store-overload induced Ca2+ release (SOICR) [5,9–17]. This SOICR may alter surface membrane potential of cardiac cells sufficiently to evoke delayed afterdepolarizations (DADs), triggered activity, and triggered arrhythmia [17]. These SOICR-evoked DADs and triggered activities frequently occur in failing hearts and are a major cause of VTs in heart failure [18]. These SOICR-evoked DADs are also the cause of catecholaminergic polymorphic ventricular tachycardia (CPVT) that is linked to mutations in the cardiac ryanodine receptor (RyR2) and calsequestrin (CASQ2). These mutations increase the propensity for SOICR [17]. Given the important role of SOICR in arrhythmogenesis, the pharmacological suppression of SOICR is potentially a promising strategy for inhibiting Ca2+-triggered arrhythmias.
A major effort in combating Ca2+-triggered arrhythmias is to identify and develop agents that suppress spontaneous Ca2+ waves or SOICR [19–23]. We previously tested the effect of a subset of beta-blockers on SOICR, and discovered that carvedilol, a non-selective beta-blocker, effectively suppresses SOICR [19]. Carvedilol does so by directly interacting with the RyR2 channel and shortening its mean open time. This SOICR-inhibiting carvedilol also effectively suppresses CPVT in mice [19]. Clinically used carvedilol is a racemic mixture, and interestingly the non-beta-blocking R-carvedilol enantiomer has this RyR2 targeted, SOICR limiting, and CPVT suppressing action in mice [22]. Thus, this anti-SOICR action of carvedilol is independent of beta-blockade, and may account, in part, for its superior anti-arrhythmic and survival benefits.
There are a large number of beta-blockers (~30) that are commonly used in the clinic and just a subset of these have been tested for possible anti-SOICR actions [19]. Thus, it is possible that there are other beta-blockers that are capable of effectively suppressing SOICR. Here, we tested the effect of 13 additional beta-blockers on SOICR. We found that nebivolol, a third generation of highly selective beta-1 adrenergic receptor blocker with nitric oxide dependent vasodilatory property, also effectively suppresses SOICR [24–29]. Like carvedilol [19], nebivolol reduces the mean open time of single RyR2 channels and suppresses CPVT in mice. Interestingly, like R-carvedilol [22], the non-beta-blocking (l)-nebivolol enantiomer [30] suppresses SOICR and CPVT without lowering heart rate. These data reveal a novel action of nebivolol: suppression of RyR2-mediated SOICR. Thus, our work identifies nebivolol and its non-beta-blocking (l)-enantiomer as new anti-arrhythmic agents for Ca2+-triggered arrhythmias.
MATERIALS AND METHODS
Single-cell Cytosolic Ca2+ Imaging in HEK293 Cells
HEK293 cells expressing a CPVT-causing RyR2 mutation R4496C display robust store overload-induced calcium release (SOICR) [15,16]. The cytosolic Ca2+ changes in RyR2-R4496C HEK293 cells were monitored using single-cell Ca2+ imaging. Briefly, the cells were grown on glass coverslips for 20–24 hours after induction by 1 μg/ml tetracycline and then were loaded with fluorescent Ca2+ indicator dye, fura-2 acetoxymethyl ester (fura-2 AM, 5 μM, Invitrogen), in Krebs-Ringer-Hepes (KRH) solution (125 mM NaCl, 5 mM KCl, 1.2 mM NaH2PO4, 6 mM glucose, 1.2 mM MgCl2 and 25 mM Hepes, pH 7.37–7.38) plus 0.02% pluronic F-127 and 0.1 mg/ml BSA for 20 minutes at room temperature (23 °C). The coverslips were mounted in a perfusion chamber (Warner Instruments, Hamden, CT, USA) on an inverted microscope (Nikon Ti). The Ca2+ concentration in KRH solution was stepped from 0.1 mM, 0.5 mM to 1 mM for 5 minutes each. The cells were continuously perfused with KRH solution containing 1 mM Ca2+ and gradually increased dosage of nebivolol (3 μM, 10 μM, 30 μM, and 60 μM) or DMSO as control, each for 8 minutes. Caffeine (15 mM) was applied at the end of each experiment to verify the presence of functioning RyR2 channels. Time-lapse images (0.25 frame/s) were captured and fluorescence intensities were measured from regions of interest centered on individual cells that responded to caffeine. Data were analyzed with NIS 3.20 AR software.
Single-cell Luminal Ca2+ Imaging in HEK293 Cells
The luminal Ca2+ changes in HEK293 cells were measured using single-cell Ca2+ imaging and the Ca2+ sensitive fluorescence resonance energy transfer (FRET) based Ca2+ sensing cameleon protein D1ER [31,32]. HEK293 cells expressing RyR2-R4496C mutation were transfected with the D1ER cDNA 24 hours before induction of RyR2 expression using the Ca2+ phosphate precipitation method. The cells were perfused continuously with KRH buffer containing various concentrations of CaCl2 (0–2 mM) and tetracaine (1 mM) or caffeine (20 mM) at room temperature. Images were captured with Compix Inc. Simple PCI 6 software at 470 nm and 535 nm emission, with excitation at 430 nm, every 2 seconds using an inverted microscope (Nikon TE2000-S) equipped with a S-Fluor 20x/0.75 objective. The amount of FRET was determined from the ratio of the light emission at 535 and 470 nm.
Single Channel Recordings in Lipid Bilayers
Native SR microsomes isolated from rat ventricular muscle were incorporated into bilayers using the method described by Chamberlain et al. [33]. We have previously suggested that cardiac calsequestrin (CASQ2) may or may not be bound to RyR2 after native SR microsomes are fused into planar lipid bilayers under our single channel recording conditions [34,35], leading to different populations of RyRs (bound with or without CASQ2) with different properties. This makes the interpretation of any drug’s effects difficult. Therefore, to ensure that the changes in RyR2 channel activity observed under our experimental conditions are not influenced by CASQ2 dissociation, we applied the CASQ2 stripping process to remove CASQ2 from the RyR2 complex before drug testing as described previously [34]. CASQ2 was stripped from RyR2 using a high luminal Ca2+ (10 mM) wash lasting for at least 15 minutes. Planar lipid bilayers were composed of a 5:4:1 mixture (50 mg/ml in n-decane) of bovine brain phosphatidylethanolamine, phosphatidylserine and phosphatidylcholine. Bilayers were formed across a 100 μm diameter hole in a Teflon partition separating two compartments. The cytosolic recording solution contained 114 mM Tris-Hepes (pH 7.4), 5 mM ATP (total), 1 mM Mg (free), 1 mM EGTA and 10 μM Ca2+ (free). The luminal recording solution contained 200 mM Cs-Hepes (pH 7.4) and 1 mM Ca2+ (free). Channel incorporation always resulted in the cytosolic side of the RyR2 channel facing the cis compartment [34,36]. This compartment is referred to as cytosolic and the other as luminal. Single RyR2 activity was measured 10–13 minutes after the addition of nebivolol (30 μM) to the cytosolic solution. Recordings were made at room temperature (20–22 °C). Analysis was done using pCLAMP9 software (Molecular Devices, Sunnyvale, CA). Currents were sampled at 50 μs/pt and filtered for display at 0.75 kHz (4-pole Bessel). Note that these planar bilayer studies were used to assess how nebivolol affects single RyR2 function. It is impossible to completely and accurately recapitulate the cytosolic and intra-SR cellular milieu in vitro during planar lipid bilayer studies. Consequently, experimental compromises were necessary. Here, the bilayer recording solutions used were analogous to those in cells during systole. Reducing the cytosolic Ca2+ to a diastolic value (~100 nM) would have reduced RyR2 activity to a very low level, one that would make reliable measurement incredibly difficult. Some groups overcome this obstacle by omitting Mg2+ from the cytosolic solution, but this results in very non-physiological RyR2 Ca2+ dependence [37]. This is because cytosolic Mg2+ is not present to compete with Ca2+ for occupancy of the cytosolic RyR2 Ca2+ activation and inactivation sites, unlike the physiological reality in cells. Instead, we elected to use a systolic-like, not a diastolic-like, condition. This assures that single RyR2 open times are long enough to reliably measure a decrease in open time and is consistent with solutions used in our previous single RyR2 studies.
Animal Studies
All animal studies were approved by the Institutional Animal Care and Use Committees at Rush University Medical Center and the University of Calgary, and were performed in accordance with NIH guidelines. Adult (2–3 months) R4496C heterozygous mutant mice were used for all experiments. The RyR2 R4496C heterozygous mutant mice consistently develop VTs after catecholamine and caffeine challenge [19].
Confocal Ca2+ Imaging of Intact Hearts In situ
The hearts from RyR2 R4496C heterozygous mice were dissected and loaded with Rhod-2 AM (0.3 mM) in Kreb-Henseleit’s (KH) solution (120 mM NaCl, 24 mM NaHCO3, 11.1 mM glucose, 5.4 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 0.42 mM NaH2PO4, 10 mM taurine, and 5 mM creatine, oxygenated with 95% O2 and 5% CO2) at room temperature. Nebivolol (3 μM, Sigma) was added in the KH solution. The hearts were loaded in KH solution with nebivolol for 2 hour via retrograde Langendorff perfusion system [38,39]. The hearts were then transferred to another Langendorff apparatus at 37 °C, which was attached to a confocal microscope system. The hearts were continuously perfused with nebivolol (3 μM) throughout the entire experiment. Blebbistatin (10 μM, Sigma) was used in the perfusion solution to minimize the myocardial contraction [38,39]. Sinus nodes of the hearts were cauterized to eliminate the interference of sinus rhythm. To induce Ca2+ waves, Ca2+ concentration of the perfusion solution was progressively increased to 6 mM, and the hearts were then fast paced at 6 Hz for 30 seconds. In situ confocal line scanning imaging of Ca2+ signals arising from epicardial myocytes after fast pacing was captured and Ca2+ waves were assessed.
Electrocardiogram Recording and Induction of Ventricular Tachycardia in Anesthetized Mice
The effects of drugs on ventricular tachycardia induced by epinephrine and caffeine were assessed in R4496C heterozygous mutant mice using ECG recording. Briefly, mice were lightly anesthetized with isoflurane vapor (0.5–1%) and 95% O2. The anesthetized mice were then placed on a heating pad (27 °C), and needle electrodes were inserted subcutaneously into the right upper limb and left lower abdomen (BIOPAC MP System, Goleta, CA). The ECG was continuously monitored until the heart rate of the mice stabilized. Baseline ECG was recorded for about 10 minutes. VTs were induced by intraperitoneal (i.p.) injection of epinephrine (1.2 mg/kg) and caffeine (100 mg/kg). The ECG was continuously recorded for 30 minutes [40,41]. ECG recordings were analyzed using the AcqKnowledge 3.9.1 program (BIOPAC, Goleta, CA). The 30 minutes overall recording period was divided into 10 consecutive 3-min recording periods (i.e. 0–3, 3–6, 6–9, … , 21–24, 24–27, 27–30 min). The VT duration within each 3-min period as well as during the entire 30 minutes recording was calculated in the percentage of time. VT was defined as 3 or more consecutive ectopic beats.
Statistical Analysis
All values shown are mean ± SEM unless indicated otherwise. To test for differences between groups, Student’s t test or one-way ANOVA was used. Statistical analyses were performed using SPSS V.15.0 (SPSS, Chicago, IL). A P value < 0.05 was considered to be statistically significant.
RESULTS
Nebivolol suppresses spontaneous Ca2+ oscillations in HEK293 cells
A large number of beta-blockers (~30) are used clinically. We reported that among a subset of 14 beta-blockers tested, only carvedilol effectively inhibited RyR2-mediated spontaneous Ca2+ oscillations during store Ca2+ overload in HEK293 cells [19]. Given the important role of spontaneous Ca2+ release in arrhythmogenesis, it is important to determine whether there are other beta-blockers that have a carvedilol-like anti-spontaneous Ca2+ release action. To this end, we now tested the action of additional 13 beta-blockers on spontaneous Ca2+ oscillations in HEK293 cells expressing a spontaneous Ca2+ release-promoting CPVT-causing RyR2 mutation (R4496C). These HEK293 cells were perfused with 1.0 mM extracellular Ca2+ to induce spontaneous Ca2+ oscillations as described previously [15,16]. Spontaneous Ca2+ release was then monitored by using a fluorescence Ca2+ indicator, Fura-2 AM, and single cell Ca2+ imaging before and after addition of various beta-blockers. As shown in Fig. 1, of the 13 beta-blockers tested (arotinolol, bevantolol, bucindolol, bufuralol, bupranolol, carteolol, celiprolol, mepindolol, nebivolol, oxprenolol, penbutolol, landiolol, and levobunolol), nebivolol markedly suppressed spontaneous Ca2+ oscillations (>95%) at 30 μM (Fig. 1a). Thus, like carvedilol, nebivolol also effectively suppresses RyR2-mediated spontaneous Ca2+ oscillations in HEK293 cells. Penbutolol also significantly suppressed spontaneous Ca2+ oscillations in HEK293 cells, although the extent of suppression by penbutolol (~40%) was lower than that by nebivolol. We thus focused on the investigation of the action of nebivolol in the current study.
Figure 1. Nebivolol inhibits store-overload induced Ca2+ release (SOICR) in HEK293 cells.

The extent of SOICR inhibition in HEK293 cells expressing RyR2-R4496C mutant treated with (a) various beta-blockers (30μM) or (b) (d)- or (l)-nebivolol enantiomer (30μM), nebivolol (30μM) plus L-NAME (0.1mM), histamine (1μM), PGE2 (28 nM), or DMSO (control). SOICR Inhibition (%) is defined as the percentage of HEK293 cells (194–1086) in which SOICR was completely inhibited. Representative fura-2 ratios in single HEK293 cells treated with nebivolol (c) or DMSO (control) (d). (e) Percentage of cells showing spontaneous Ca2+ oscillations in HEK293 cells treated with various concentrations of nebivolol or DMSO (control). Data shown are means ± SEM (n=3–10; *P < 0.05 vs control).
To assess whether the beta-blocking and NOS stimulating activities of nebivolol contribute to its spontaneous Ca2+ release-inhibiting action, we tested the effect of the two bioactive enantiomers of nebivolol ((d)-nebivolol and (l)-nebivolol) and modulators of the NOS signaling pathway on spontaneous Ca2+ oscillations. As shown in Fig.1, the beta-blocking (d)-nebivolol and the non-beta-blocking (l)-nebivolol enantiomers [30] effectively suppressed spontaneous Ca2+ oscillations (Fig. 1b). Furthermore, neither the NOS inhibitor (N-nitro-L-arginine methyl ester, L-NAME) nor the NOS activators (histamine and prostaglandin E2) had any significant effect on spontaneous Ca2+ oscillations in HEK293 cells (Fig. 1b). The inhibitory effect of nebivolol on spontaneous Ca2+ oscillations was concentration dependent (Fig.1 c–e). Collectively, our data reveal that nebivolol effectively suppresses spontaneous Ca2+ oscillations in HEK293 cells, and that this novel action of nebivolol is independent of its NOS-stimulating and beta-blocking activities.
Nebivolol increases the activation and termination thresholds for SOICR in HEK293 cells
To investigate the mechanism underlying the inhibition of spontaneous Ca2+ release by nebivolol, we assessed the effect of nebivolol on SOICR activation and termination thresholds in HEK293 cells. We monitored the endoplasmic reticulum (ER) luminal Ca2+ dynamics in HEK293 cells before and after the treatment with DMSO (vehicle control) or nebivolol using a fluorescence resonance energy transfer (FRET)-based ER luminal Ca2+ sensing protein D1ER [31,32]. In the absence of drug, increasing extracellular Ca2+ from 0 to 2 mM induced spontaneous ER Ca2+ oscillations in RyR2-R4496C expressing HEK293 cells (Fig 2a; depicted as downward deflections of the FRET signal). SOICR occurred when the ER luminal Ca2+ content increased to a threshold level (FSOICR), and terminated when the ER luminal Ca2+ content fell to another threshold level (Ftermi). After establishing FSOICR and Ftermi in the absence of drug, the same HEK293 cells were perfused with DMSO (Fig. 2a,c–e) or nebivolol (3 μM) (Fig. 2b,f–h), and FSOICR and Ftermi were determined again. Nebivolol significantly increased SOICR activation threshold (before: 85.6 ± 0.4%, after nebivolol treatment: 91.1 ± 0.9%; P<0.01) and termination threshold (before: 47.9 ± 0.3%, after: 50.6 ± 0.5%; P<0.01) (Fig. 2f,g). However, nebivolol had no effect on the fractional Ca2+ release (before: 38.7 ± 0.5%, after: 40.5 ± 0.9%; P>0.05) (Fig. 2h). In vehicle control (DMSO) studies, there was no change in the SOICR thresholds or fractional Ca2+ release (Fig. 2c–e). Further, SOICR did not occur in control HEK293 cells expressing no RyR2, and SOICR was not affected by the IP3R inhibitor, xestospongin C [42], indicating that the SOICR measured was mediated by RyR2. Thus, nebivolol suppresses RyR2-mediated SOICR by raising the SOICR activation and termination thresholds.
Figure 2. Nebivolol increases the threshold for SOICR activation and termination in HEK293 cells.
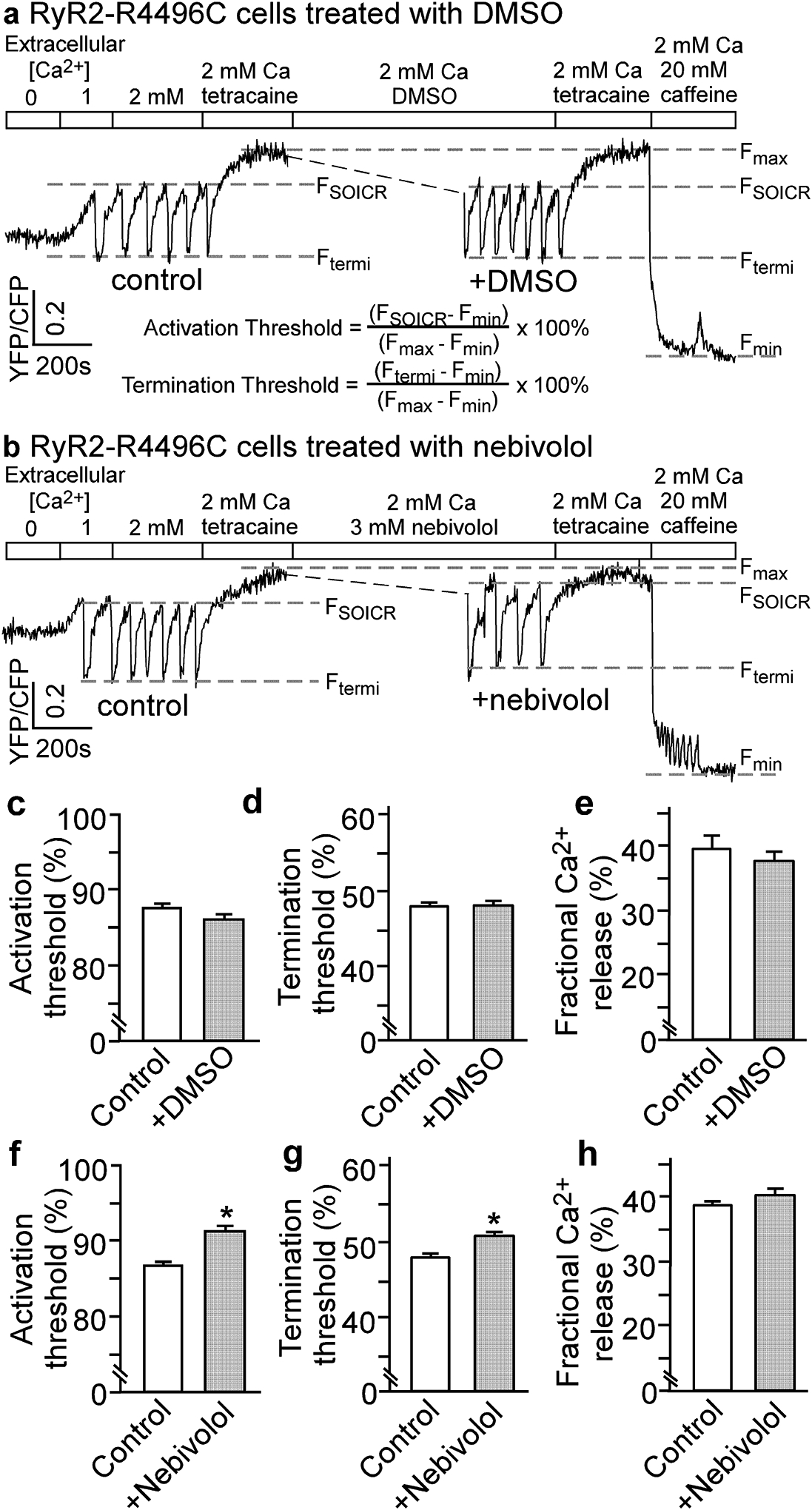
Representative single cell FRET recordings in HEK293 cells expressing RyR2-R4496C mutant before and after the treatment with DMSO (a) or nebivolol (3μM) (b). Threshold for SOICR activation (c, f), threshold for SOICR termination (d, g), and fractional Ca2+ release (e, h) in HEK293 cells before and after the treatment with DMSO (control) or nebivolol (3μM) are shown. The SOICR activation and termination thresholds were determined using the equations shown in (a). Fractional Ca2+ release was calculated by subtracting the termination threshold from the activation threshold. Data shown are means ± SEM (n=3–4; *P < 0.01 vs control).
Nebivolol modifies the function of single RyR2 channels
To further understand the mechanism by which nebivolol suppresses SOICR, we tested the possibility that nebivolol alters the function of the RyR2 channel. We assessed the effect of nebivolol on single RyR2 channels by fusing sarcoplasmic reticulum microsomes into planar lipid bilayers. Figure 3 shows representative single channel recordings before and after the addition of nebivolol to the cytosolic side of the channel (Fig. 3a,b). Nebivolol (30 μM) reduced RyR2 activity, but this action was not immediate. Nebivolol had no significant effect on RyR2 open probability (Po), mean open time (To), or mean closed time (Tc) during the first 3 min following its application (Fig. 3c–e). After 10 min, nebivolol significantly reduced RyR2 Po and To, but not Tc (Fig. 3c–e). This suggests that nebivolol reduces single RyR2 opening by acting at a site(s) not readily/immediately accessible to drug in the aqueous solution.
Figure 3. Nebivolol modifies the gating of single RyR2 channels.
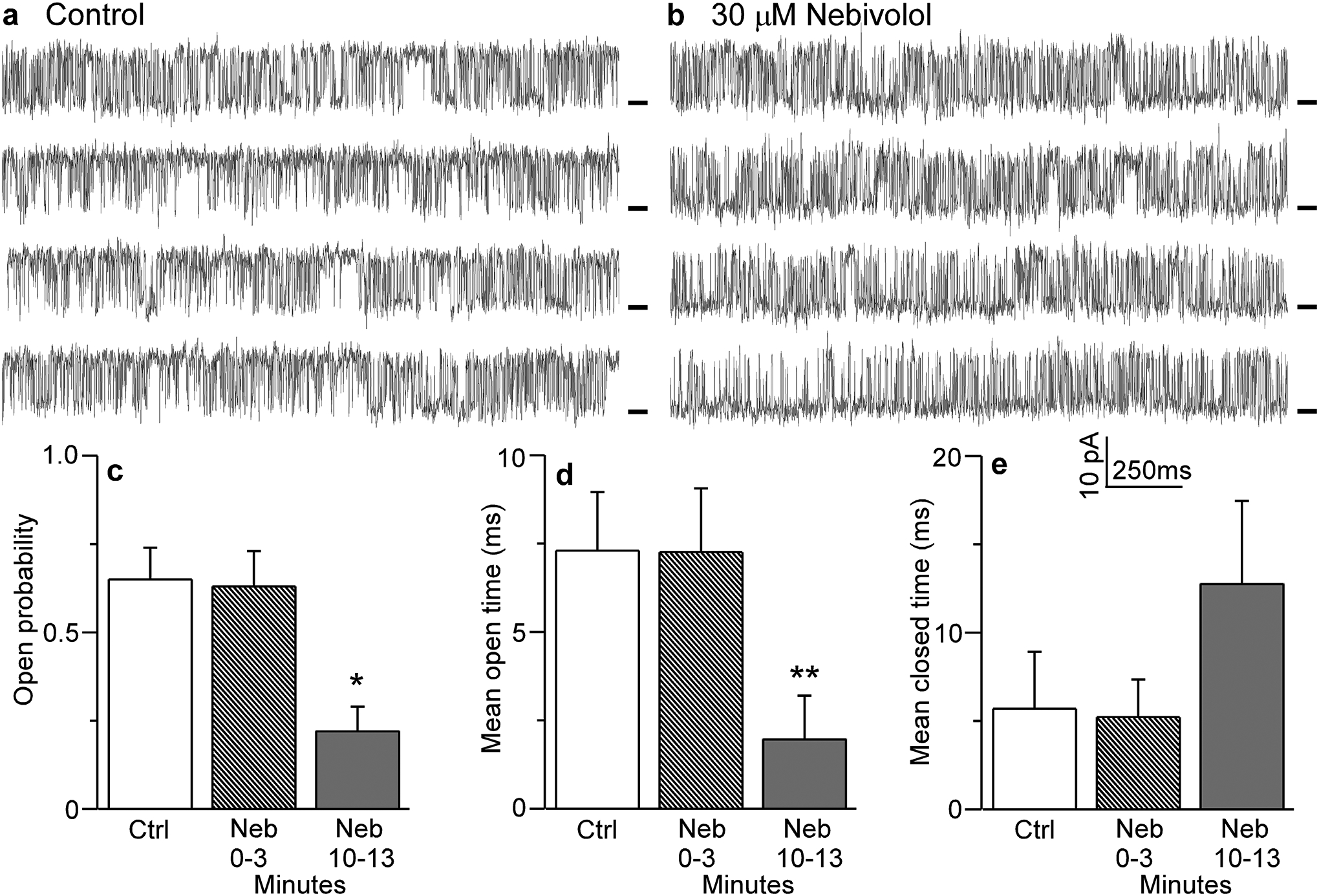
Single RyR2 channels from native rat SR microsomes were fused into planar lipid bilayers and recorded in the absence (control) (a) or 10 minutes after the addition of nebivolol (30μM) (b). Recordings were made at +40 mV. Open events are upward from the marked zero current level (short bars). Opening probability (c), mean open time (d), and mean closed time (e) are shown in control, 0–3 or 3–10 minutes after nebivolol (30μM) addition. Data shown are means ± SEM (n=6–11; *P < 0.01; **P < 0.05 vs control).
Nebivolol suppresses spontaneous Ca2+ waves in ventricular myocytes in intact hearts
To determine whether nebivolol can suppress SOICR in cardiomyocytes, we assessed the effect of nebivolol on spontaneous Ca2+ waves in ventricular myocytes in intact hearts expressing the SOICR-promoting, disease-causing RyR2-R4496C mutation [19]. Spontaneous Ca2+ waves in intact RyR2-R4496C hearts were induced by abrupt cessation of fast pacing (6 Hz) in the presence of elevated extracellular Ca2+ (6 mM). Ca2+ waves were monitored using line-scan confocal Ca2+ imaging. The sinus node was ablated by electro-cautery to eliminate the influence of the intrinsic sinus rhythm on the occurrence of post-cessation spontaneous Ca2+ waves. As shown in Fig. 4, spontaneous Ca2+ waves occurred without nebivolol present (Fig.4a, control), whereas nebivolol (3.0 μM for 2 hours) treatment significantly reduced the frequency of spontaneous Ca2+ waves in cardiomyocytes in intact RyR2-R4496C hearts (1.5 ± 0.1 Hz/100 μm, P<0.01) compared to control treatment (DMSO) (4.0 ± 0.2 Hz/100 μm) (Fig. 4b). Thus, nebivolol also suppresses SOICR in cardiomyocytes in intact hearts.
Figure 4. Nebivolol suppresses Ca2+ waves in intact hearts.
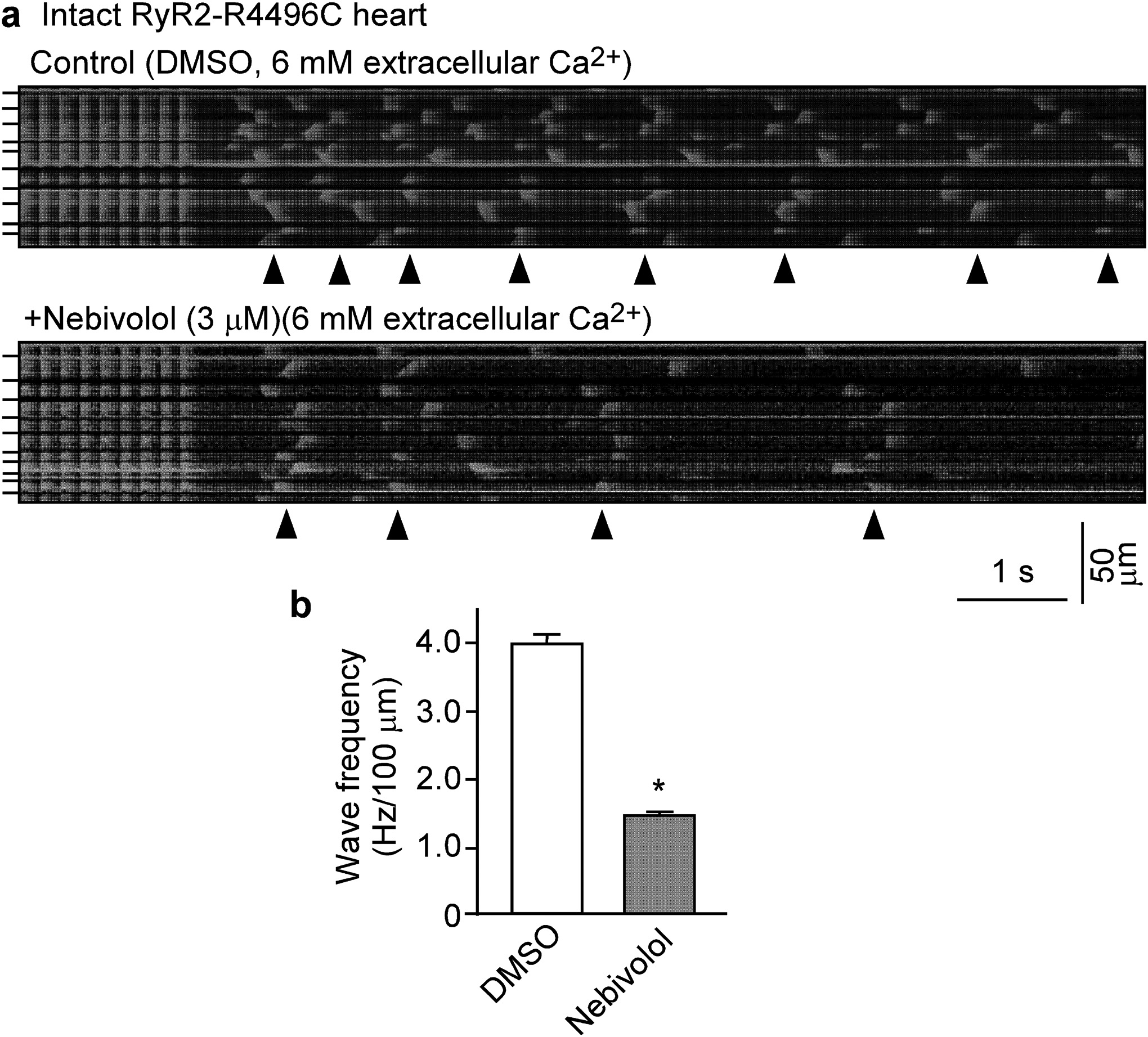
Intact hearts from heterozygous RyR2-R4496C mutant mice were isolated and loaded with Rhod-2 AM, and Langendorff-perfused with KH solution containing 6 mM extracellular Ca2+ to induce SR Ca2+ overload. AV node was ablated by electro-cautery and the hearts were paced at 6Hz, then pacing was stopped. (a) Representative line-scan confocal images of Ca2+ dynamics in hearts 2 hours after perfused with DMSO (control, top panel) or with nebivolol (3μM, bottom panel). (b) Frequency of spontaneous Ca2+ waves. Arrowheads show the occurrence of Ca2+ waves. Short bars to the left indicate cell boundaries within the intact heart. Data shown are means ± SEM from 16–18 scan areas of 3 hearts for each group. (*P < 0.01 vs control).
Nebivolol suppresses CPVT in RyR2-R4496C mutant mice
Enhanced spontaneous Ca2+ waves are closely linked to CPVT [17]. Thus, the anti-SOICR action of nebivolol might be sufficient to suppress Ca2+ wave-evoked CPVT. To test this possibility, we performed stress ECG testing on RyR2-R4496C mutant mice that are prone to SOICR-associated CPVT [19]. As shown in Fig. 5, long lasting (>30 min) ventricular tachyarrhythmias (VTs) (84.0 ± 3.5%), including bidirectional VTs can be pharmacologically induced in RyR2-R4496C mice by injection of epinephrine and caffeine (Fig. 5a,c). This pharmacologically induced VT in mice was significantly reduced in duration after pre-treatment with nebivolol (0.4 or 2.0 mg/kg/day for 5 days) (Fig. 5b,c). At 2.0 mg/kg/day, nebivolol reduced the overall VT duration from 84.0 ± 3.5% to 21.1 ± 4.8% (P<0.01) (Fig. 5d). Nebivolol treatment (2.0 mg/kg/day) also reduced the heart rate (before: 666 ± 11, after treatment: 499 ± 10; P<0.01) (Fig. 5e). Thus, nebivolol effectively suppresses CPVT in RyR2-R4496C mutant mice, but also causes significant bradycardia consistent with this agent’s beta-blocking activity.
Figure 5. Effects of nebivolol on CPVT and heart rate in heterozygous RyR2-R4496C mutant mice.
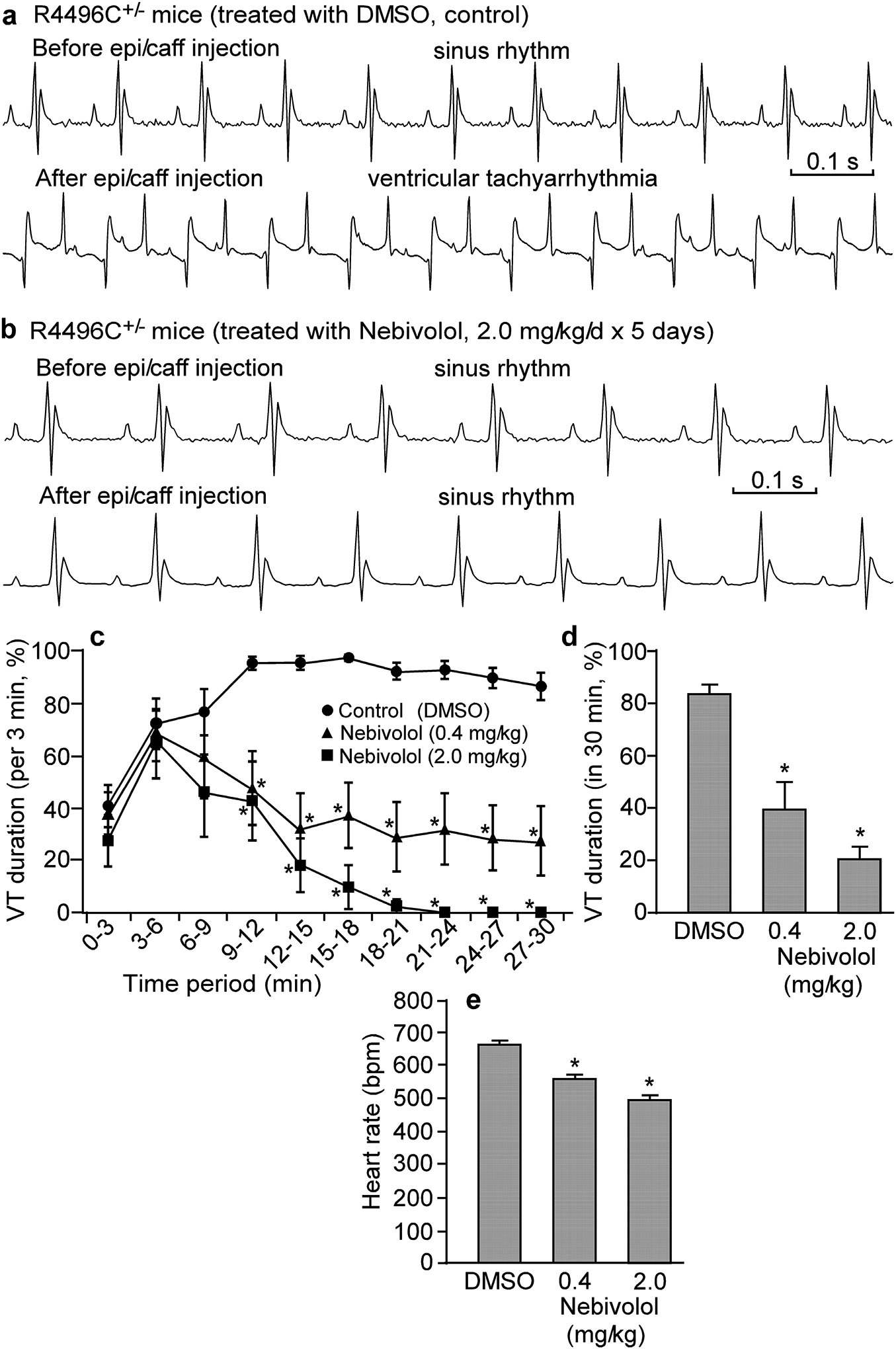
Representative ECG recordings of heterozygous RyR2-R4496C mutant mice treated with DMSO (control) (a) or with nebivolol (2.0 mg/kg/day for 5 days) (b) before (top panel) and 24 minutes after (bottom panel) intraperitoneal injection of epinephrine (1.2 mg/kg) and caffeine (100 mg/kg). VT occurred intermittently. Percentage time in VT (VT duration) in sequential 3-min period post-injection (c) and percentage time in VT (VT duration) over the entire 30-min recording period post-injection (d) were measured in mice treated with nebivolol (0.4 mg/kg/day or 2.0 mg/kg/day) or DMSO (control). (e) Heart rate (pre-injection of epinephrine/caffeine) in mice treated with DMSO (control) or nebivolol (0.4 mg/kg/day or 2.0 mg/kg/day) as determined by ECG recordings. Data shown are means ± SEM (n=8–16; *P < 0.01 vs control).
(l)-nebivolol enantiomer suppresses CPVT without lowering heart rate
To minimize the adverse bradycardia of racemic nebivolol, we assessed the impact of the (l)-nebivolol enantiomer that possesses no beta-blocking activity on CPVT and heart rate. Like the racemic nebivolol, the non-beta-blocking (l)-nebivolol also reduced the VT duration (from 84.0 ± 3.5% to 24.4 ± 6.2%, at 2.0 mg/kg/day; P<0.01) (Fig. 6a,d). Unlike racemic nebivolol, however, the non-beta-blocking (l)-nebivolol at different doses (0.4 or 2.0 mg/kg/day for 5 days) had no significant effect on heart rate (before: 666 ± 11, after treatment: 660 ± 14, at 2.0 mg/kg/day; P>0.05) (Fig. 6e). Thus, the non-beta-blocking (l)-nebivolol enantiomer is able to suppress CPVT without lowering heart rate.
Figure 6. (l)-nebivolol suppresses CPVT in heterozygous RyR2-R4496C mutant mice.
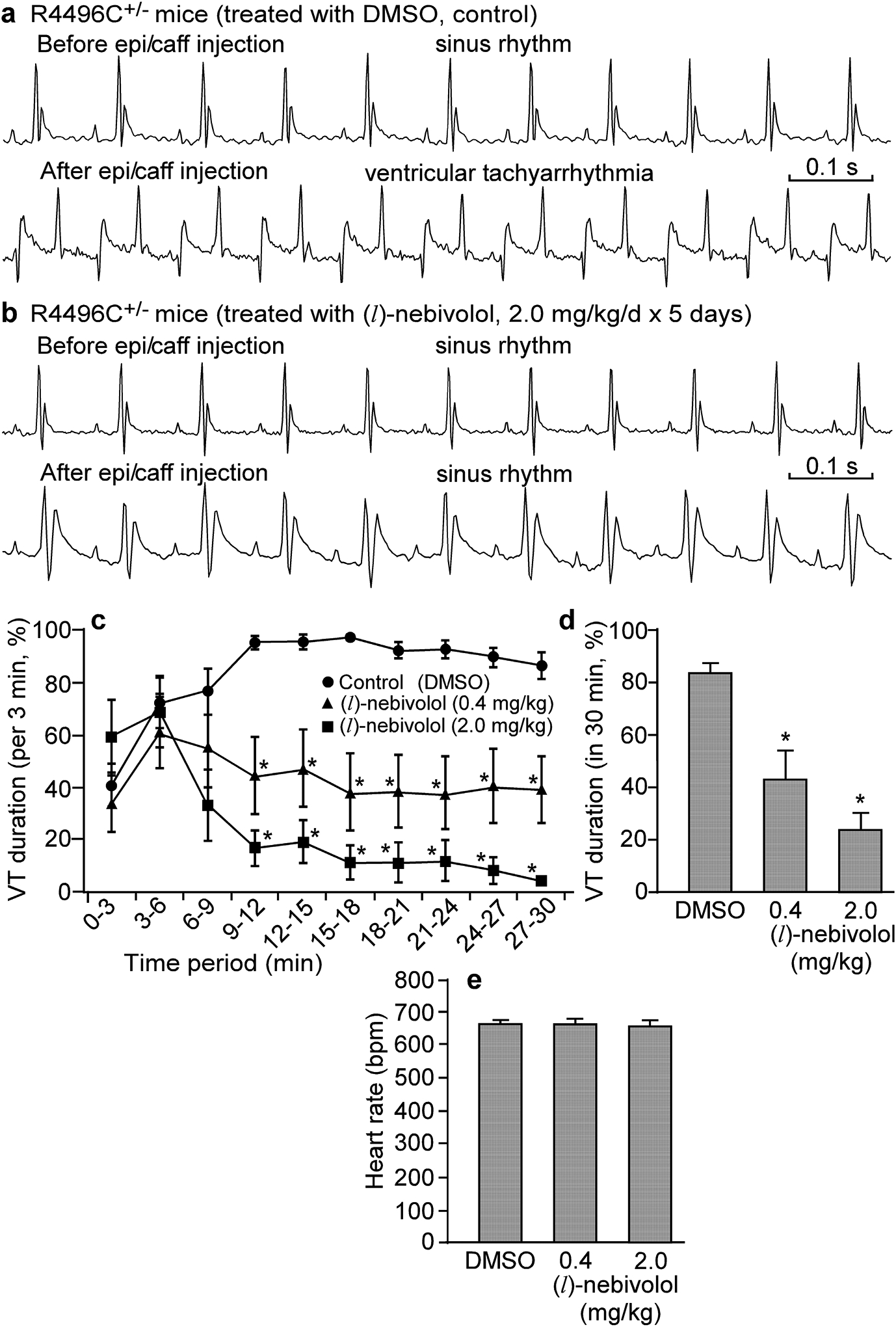
Representative ECG recordings of heterozygous RyR2-R4496C mutant mice treated with DMSO (control) (a) or (l)-nebivolol (2.0 mg/kg/day for 5 days) (b) before and 24 minutes after intraperitoneal injection of epinephrine (1.2 mg/kg) and caffeine (100 mg/kg). VT duration (%) in sequential 3-min period post-injection (c) and over the entire 30-min recording period post-injection (d) were measured in mice treated with (l)-nebivolol (0.4 mg/kg/day or 2.0 mg/kg/day) or DMSO (control). (e) Heart rate (pre-injection of epinephrine/caffeine) in mice treated with DMSO (control) or (l)-nebivolol (0.4 mg/kg/day or 2.0 mg/kg/day) as determined by ECG recordings. Data shown are means ± SEM (n=7–10; *P < 0.01 vs control).
DISCUSSION
Spontaneous Ca2+ waves in cardiac cells as a result of SR Ca2+ overload, also known as store overload induced Ca2+ release (SOICR), are a well-known cause of Ca2+-triggered ventricular tachyarrhythmias (VTs) and sudden death [17]. Thus, inhibiting SOICR is a logical therapeutic approach to limiting Ca2+-triggered VTs. In the present study, we tested the effect of many commonly used beta-blockers on SOICR in HEK293 cells to identify novel SOICR inhibitors. Of the 13 commonly used beta-blockers tested, only nebivolol effectively suppresses SOICR and does so by increasing SOICR activation and termination thresholds in HEK293 cells. Furthermore, we show that nebivolol inhibits SOICR in ventricular myocytes in intact hearts as well as stress-induced VTs in mice harboring a CPVT-causing RyR2 mutation. Our data demonstrate that nebivolol has a previously unappreciated inhibitory action on RyR2-mediated SOICR and SOICR-evoked VT.
Nebivolol is a third generation of beta-blocker that is highly selective for beta-1 adrenergic receptors. Nebivolol also has a nitric oxide potentiating vasodilatory action [24–29]. The beta-1 specificity and vasodilatory action of nebivolol are thought to contribute to its antihypertensive property. Recently, nebivolol has also been shown to have significant benefits for patients with myocardial ischemia and heart failure [43–47]. Consistent with this view, nebivolol has been shown to reduce the incidence of ischemia/reperfusion-induced VT and ventricular fibrillation in various animal models in vivo [48]. Furthermore, nebivolol protects against ouabain- and aconitine-induced cardiotoxicity in rats and guinea pigs [48], which is known to involve SR Ca2+ overload. This suggests that nebivolol may also suppress Ca2+-mediated cardiotoxicity. However, the mechanisms underlying nebivolol’s benefits on ischemia, heart failure, arrhythmias and cardiotoxicity are unclear.
We have previously shown that carvedilol, one of the most effective beta-blockers in reducing the incidence of VTs and sudden cardiac death in patients with heart failure, effectively suppresses RyR2-mediated SOICR and SOICR-evoked CPVT [19]. Our novel finding that nebivolol, like carvedilol, suppresses SOICR suggests that nebivolol may also have a carvedilol-like RyR2-targeted anti-arrhythmic action. Indeed, we showed that nebivolol, like carvedilol, reduces RyR2 opening and suppresses SOICR-evoked CPVT in mice. These actions are distinct from nebivolol’s beta-blocking and nitric oxide synthase enhancing activities. This supports the notion that nebivolol possesses RyR2-targeted anti-arrhythmic activity. Given the link between enhanced SOICR and CPVT, and nebivolol’s novel anti-SOICR property and relatively long half-life, it will be of interest to assess the benefits of nebivolol for patients with CPVT. Further, clinical assessments may also be warranted to determine the efficacy of nebivolol in reducing the incidence of VTs and sudden death especially in patients with heart failure.
Clinically used nebivolol contains two bioactive enantiomers, (d)-nebivolol and (l)-nebivolol [30]. The (d)-nebivolol enantiomer is responsible for the selective beta-1 blocking activity of racemic nebivolol, whereas the (l)-nebivolol lacks beta blocking activity, but has a nitric oxide potentiating vasodilatory action [24–29]. We show that racemic nebivolol suppresses stress-induced VTs with a concomitant reduction in heart rate due to its beta-blocking activity. Interestingly, the non-beta-blocking (l)-nebivolol enantiomer alone also effectively suppressed stress-induced VTs, but did not lower heart rate. Similarly, clinically used carvedilol is also a racemic mixture of two enantiomers: the beta-blocking S-carvedilol and the non-beta-blocking R-carvedilol [49,50]. Like the (l)-nebivolol enantiomer, the non-beta-blocking R-carvedilol suppresses SOICR and SOICR-evoked VTs without lowering heart rate [22]. Further, we found that non-beta-blocking carvedilol derivatives (VK-II-86, CS-I-34, and CS-I-59) also suppress stress-induced VTs without reducing heart rate [19]. Thus, there is a growing family of RyR2-targeted, SOICR-inhibiting, non-beta-blocking anti-VT drugs without potentially adverse bradycardia that is generally associated with beta blockers, including racemic nebivolol or carvedilol. These RyR2-targeted anti-SOICR agents represent a novel class of promising antiarrhythmic drugs.
We have recently shown that RyR2 contains a store Ca2+ sensing gate that is responsible for the regulation of RyR2 by store luminal Ca2+ [40]. Ablating this store sensing gate abolishes SOICR and completely protects against Ca2+-triggered VTs [40]. Thus, RyR2 is a critical determinant of SOICR and SOICR-associated VTs as well as a key therapeutic target for Ca2+-triggered arrhythmias. We have shown previously that carvedilol exerts a direct effect on RyR2 gating, reducing mean open time and this in turn suppresses SOICR and CPVT [19]. Here, we found that nebivolol also suppresses SOICR by reducing the open duration of single RyR2 channels. Thus, targeting RyR2 represents an effective therapeutic approach to combatting SOICR-associated VTs.
Nebivolol is highly lipophilic, which may in part explain its unusual time course of action on single RyR2 channels we observed (Fig. 3). The drug may partition into the membrane to reach its site of action. Wang et al. [51] reported that the plasma level of nebivolol in C57B6 mice following 3 weeks of treatment with nebivolol (3 mg/kg/day) delivered through their drinking water was ~20 ng/ml. Interestingly, the level of nebivolol found in the brain tissue (~120 ng/ml) was much higher than that found in the plasma [51]. This suggests that nebivolol can accumulate in tissues, which is consistent with its highly lipophilic nature and a large volume of distribution [52]. It is then possible that nebivolol may also accumulate in the heart. These properties of nebivolol will likely have a substantial influence on the concentration dependence and time course of action of nebivolol on the RyR2 channel.
To identify beta-blockers that are capable of suppressing spontaneous Ca2+ oscillations, we employed the spontaneous Ca2+ release promoting, disease-causing RyR2 mutant (R4496C) HEK293 cells as a drug screening platform. We used relatively high concentrations of beta-blockers (up to 30–60 μM) for screening purpose to ensure that we did not miss beta-blockers that have a relatively weak inhibitory action on spontaneous Ca2+ release. Using this strategy, we found that of the 13 beta-blocker tested, only nebivolol nearly completely abolished spontaneous Ca2+ oscillations in HEK293 cells at 30 μM. It is important to note that in our drug screening experiments, we measured the fraction of RyR2-R4496C mutant HEK293 cells in which their robust spontaneous Ca2+ oscillations were completely blocked by a given beta-blocker. A higher concentration of drug would be needed to completely block spontaneous Ca2+ oscillations than that needed to just suppress spontaneous Ca2+ oscillations. Indeed, we found that nebivolol at 3 μM significantly increased the activation and termination thresholds for spontaneous Ca2+ release in these RyR2-R4496C HEK293 cells. Therefore, the concentrations of nebivolol and other beta-blockers used for drug screening purpose are relatively high and may not be clinically relevant.
Among the 27 commonly used beta-blockers tested, only carvedilol and nebivolol effectively suppress SOICR. This functional similarity suggests the possibility of common structural features in carvedilol and nebivolol. Indeed, a preliminary examination of their ball-and-spoke models indicates that carvedilol and nebivolol conformations exist that enable the close superimposition of their key polar functionalities (ether or hydroxyl groups, as well as the secondary amino groups) (Fig. 7). This also allows one of the hydrophobic aromatic rings of the carbazole moiety of carvedilol to overlap with one of the aromatic rings of nebivolol (Fig. 7). Thus, nebivolol and carvedilol share common structural features, which may confer their similar anti-SOICR action.
Figure 7. Superimposition of carvedilol and nebivolol.
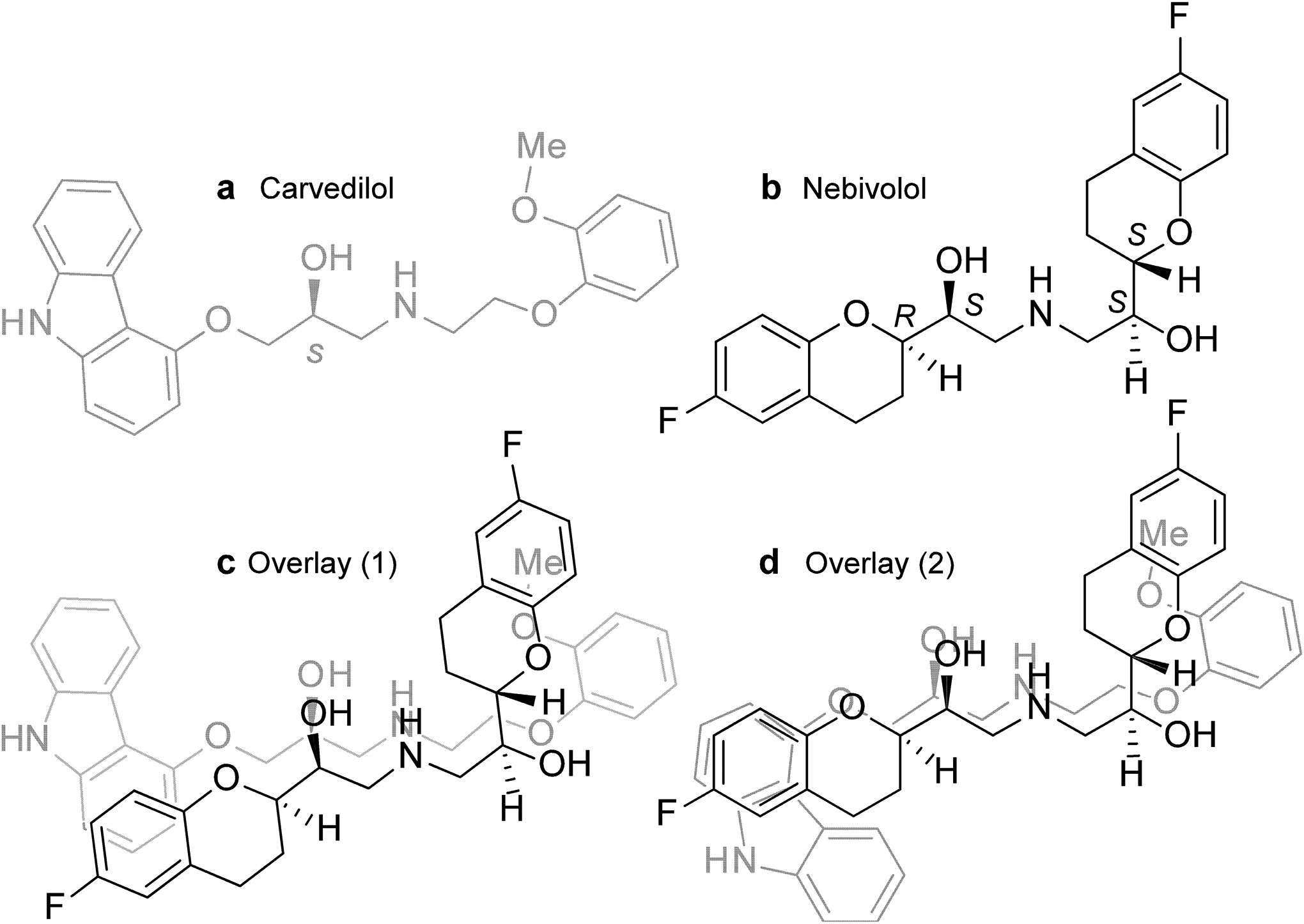
The structures of one of the carvedilol enantiomers, S-carvedilol (a) and one of the nebivolol enantiomers, (l)-nebivolol (b) are shown. (c, d) Overlay of (l)-nebivolol with (S)-carvedilol in two conformations.
Nebivolol is mainly used for the treatment of hypertension [24–29]. However, an increased body of evidence indicates that nebivolol exerts general benefits on endothelial dysfunction and cardiovascular protection as well [53]. These benefits are often attributed to nebivolol’s nitric oxide synthase stimulating action and anti-oxidant properties [24–29]. Intracellular Ca2+ dysregulation is a common feature of endothelial and cardiovascular dysfunction, including arterial hypertension, atherosclerosis, heart failure, ischemia/reperfusion injury, cardiac arrhythmias and Ca2+-associated cardiotoxicity. Our study suggests that the RyR2-targeted action of nebivolol may also contribute to nebivolol’s pleiotropic benefits.
In summary, the present study reveals previously unappreciated actions of nebivolol, namely reduction of single RyR2 channel opening, inhibition of RyR2-mediated spontaneous Ca2+ waves (or SOICR), and suppression of SOICR-evoked CPVT. These findings suggest that in addition to nebivolol’s well-known anti-hypertensive action, nebivolol also possesses anti-arrhythmic activity, especially on Ca2+ triggered arrhythmias. With its unique nitric oxide releasing action and anti-SOICR activity, nebivolol represents a promising agent for the treatment of a variety of endothelial and cardiovascular dysfunctions.
ACKNOWLEDGEMENTS
This work was supported by research grants from National Institutes of Health (HL75210 to TGB, MF, and SRWC, and HL090905 to LSS), Canadian Institutes of Health Research (MOP-123506 to SRWC), Canada Foundation for Innovation (23881 to SRWC), Heart and Stroke Foundation/Libin Professorship in Cardiovascular Research (to SRWC), and the Alberta Innovates-Health Solutions (to SRWC).
THE ABBREVIATIONS USED
- VT
Ventricular tachyarrhythmia
- CPVT
catecholaminergic polymorphic ventricular tachycardia
- VF
ventricular fibrillation
- ECG
electrocardiogram
- CICR
Ca2+-induced Ca2+ release
- SOICR
store overload induced Ca2+ release
- DAD
delayed afterdepolarization
- SR
sarcoplasmic reticulum
- RyR2
cardiac ryanodine receptor
Footnotes
CONFLICT OF INTEREST The authors declare that they have no conflicts of interest with the contents of this article.
REFERENCES
- 1.Koplan BA and Stevenson WG (2009) Ventricular tachycardia and sudden cardiac death. Mayo Clin. Proc 84, 289–297. doi: 10.1016/S0025-6196(11)61149-X [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller L (2003) Limitations of current medical therapies for the treatment of heart failure. Rev Cardiovasc Med 4 Suppl 2, S21–9 [PubMed] [Google Scholar]
- 3.Kamath GS and Mittal S (2008) The role of antiarrhythmic drug therapy for the prevention of sudden cardiac death. Prog. Cardiovasc. Dis 50, 439–448. doi: 10.1016/j.pcad.2007.12.001 [DOI] [PubMed] [Google Scholar]
- 4.Vermeulen J, McGuire M, Opthof T, Coronel R, de Bakker J, Klopping C and Janse M (1994) Triggered activity and automaticity in ventricular trabeculae of failing human and rabbit hearts. Cardiovasc. Res 28, 1547–1554 [DOI] [PubMed] [Google Scholar]
- 5.Bers DM (2002) Calcium and cardiac rhythms: Physiological and pathophysiological. Circ. Res 90, 14–7. [PubMed] [Google Scholar]
- 6.Baartscheer A, Schumacher C, Belterman C, Coronel R and Fiolet J (2003) SR calcium handling and calcium after-transients in a rabbit model of heart failure. Cardiovasc. Res 58, 99–108 [DOI] [PubMed] [Google Scholar]
- 7.Shannon TR, Pogwizd SM and Bers DM (2003) Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ. Res 93, 592–594 [DOI] [PubMed] [Google Scholar]
- 8.Bers DM (2014) Cardiac sarcoplasmic reticulum calcium leak: Basis and roles in cardiac dysfunction. Annu. Rev. Physiol 76, 107–127. doi: 10.1146/annurev-physiol-020911-153308; 10.1146/annurev-physiol-020911–153308 [DOI] [PubMed] [Google Scholar]
- 9.Kass RS and Tsien RW (1982) Fluctuations in membrane current driven by intracellular calcium in cardiac purkinje fibers. Biophys. J 38, 259–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orchard C, Eisner D and Allen D (1983) Oscillations of intracellular Ca2+ in mammalian cardiac muscle. Nature. 304, 735–738 [DOI] [PubMed] [Google Scholar]
- 11.Stern M, Kort A, Bhatnagar G and Lakatta E (1983) Scattered-light intensity fluctuations in diastolic rat cardiac muscle caused by spontaneous Ca2+-dependent cellular mechanical oscillations. J. Gen. Physiol 82, 119–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wier W, Kort A, Stern M, Lakatta E and Marban E (1983) Cellular calcium fluctuations in mammalian heart: Direct evidence from noise analysis of aequorin signals in purkinje fibers. Proc. Natl. Acad. Sci. U. S. A 80, 7367–7371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marban E, Robinson SW and Wier WG (1986) Mechanisms of arrhythmogenic delayed and early afterdepolarizations in ferret ventricular muscle. J. Clin. Invest 78, 1185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lakatta EG (1992) Functional implications of spontaneous sarcoplasmic reticulum Ca2+ release in the heart. Cardiovasc. Res 26, 193–214. [DOI] [PubMed] [Google Scholar]
- 15.Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H and Chen SRW (2004) RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc.Natl.Acad.Sci.U.S.A 101, 13062–13067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang D, Wang R, Xiao B, Kong H, Hunt DJ, Choi P, Zhang L and Chen SRW (2005) Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ. Res 97, 1173–1181 [DOI] [PubMed] [Google Scholar]
- 17.Priori SG and Chen SR (2011) Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res 108, 871–883. doi: 10.1161/CIRCRESAHA.110.226845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pogwizd SM and Bers DM (2004) Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc. Med 14, 61–66 [DOI] [PubMed] [Google Scholar]
- 19.Zhou Q, Xiao J, Jiang D, Wang R, Vembaiyan K, Wang A, Smith CD, Xie C, Chen W, Zhang J, Tian X, Jones PP, Zhong X, Guo A, Chen H, Zhang L, Zhu W, Yang D, Li X, Chen J, Gillis AM, Duff HJ, Cheng H, Feldman AM, Song LS, Fill M, Back TG and Chen SR (2011) Carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2+ release. Nat. Med 17, 1003–1009. doi: 10.1038/nm.2406; 10.1038/nm.2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith CD, Wang A, Vembaiyan K, Zhang J, Xie C, Zhou Q, Wu G, Chen SR and Back TG (2013) Novel carvedilol analogues that suppress store-overload-induced Ca2+ release. J. Med. Chem 56, 8626–8655. doi: 10.1021/jm401090a; 10.1021/jm401090a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shinohara T, Kim D, Joung B, Maruyama M, Vembaiyan K, Back TG, Wayne Chen SR, Chen PS and Lin SF (2014) Carvedilol analog modulates both basal and stimulated sinoatrial node automaticity. Heart Vessels. 29, 396–403. doi: 10.1007/s00380-013-0378-2 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Zhou Q, Smith CD, Chen H, Tan Z, Chen B, Nani A, Wu G, Song LS, Fill M, Back TG and Chen SR (2015) Non-beta-blocking R-carvedilol enantiomer suppresses Ca2+ waves and stress-induced ventricular tachyarrhythmia without lowering heart rate or blood pressure. Biochem. J 470, 233–242. doi: 10.1042/BJ20150548 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malig T, Xiao Z, Chen SR and Back TG (2016) Suppression of store overload-induced calcium release by hydroxylated metabolites of carvedilol. Bioorg. Med. Chem. Lett 26, 149–153. doi: 10.1016/j.bmcl.2015.11.008 [doi] [DOI] [PubMed] [Google Scholar]
- 24.Bowman AJ, Chen CP and Ford GA (1994) Nitric oxide mediated venodilator effects of nebivolol. Br. J. Clin. Pharmacol 38, 199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Boer RA, Voors AA and van Veldhuisen DJ (2007) Nebivolol: Third-generation beta-blockade. Expert Opin. Pharmacother 8, 1539–1550. doi: 10.1517/14656566.8.10.1539 [doi] [DOI] [PubMed] [Google Scholar]
- 26.Cheng JW (2009) Nebivolol: A third-generation beta-blocker for hypertension. Clin. Ther 31, 447–462. doi: 10.1016/j.clinthera.2009.03.007 [doi] [DOI] [PubMed] [Google Scholar]
- 27.Munzel T and Gori T (2009) Nebivolol: The somewhat-different beta-adrenergic receptor blocker. J. Am. Coll. Cardiol 54, 1491–1499. doi: 10.1016/j.jacc.2009.05.066 [doi] [DOI] [PubMed] [Google Scholar]
- 28.Toblli JE, DiGennaro F, Giani JF and Dominici FP (2012) Nebivolol: Impact on cardiac and endothelial function and clinical utility. Vasc. Health. Risk Manag 8, 151–160. doi: 10.2147/VHRM.S20669 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howlett JG (2014) Nebivolol: Vasodilator properties and evidence for relevance in treatment of cardiovascular disease. Can. J. Cardiol 30, S29–37. doi: 10.1016/j.cjca.2014.03.003 [doi] [DOI] [PubMed] [Google Scholar]
- 30.Ignarro LJ (2008) Different pharmacological properties of two enantiomers in a unique beta-blocker, nebivolol. Cardiovasc. Ther 26, 115–134. doi: 10.1111/j.1527-3466.2008.00044.x [doi] [DOI] [PubMed] [Google Scholar]
- 31.Palmer AE, Jin C, Reed JC and Tsien RY (2004) Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc. Natl. Acad. Sci. U. S. A 101, 17404–17409. doi: 10.1073/pnas.0408030101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones PP, Jiang D, Bolstad J, Hunt DJ, Zhang L, Demaurex N and Chen SR (2008) Endoplasmic reticulum Ca2+ measurements reveal that the cardiac ryanodine receptor mutations linked to cardiac arrhythmia and sudden death alter the threshold for store-overload-induced Ca2+ release. Biochem. J 412, 171–178. doi: 10.1042/BJ20071287 [DOI] [PubMed] [Google Scholar]
- 33.Chamberlain BK, Volpe P and Fleischer S (1984) Calcium-induced calcium release from purified cardiac sarcoplasmic reticulum vesicles. general characteristics. J. Biol. Chem 259, 7540–7546 [PubMed] [Google Scholar]
- 34.Qin J, Valle G, Nani A, Nori A, Rizzi N, Priori SG, Volpe P and Fill M (2008) Luminal Ca2+ regulation of single cardiac ryanodine receptors: Insights provided by calsequestrin and its mutants. J. Gen. Physiol 131, 325–334. doi: 10.1085/jgp.200709907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gaburjakova M, Bal NC, Gaburjakova J and Periasamy M (2013) Functional interaction between calsequestrin and ryanodine receptor in the heart. Cell Mol. Life Sci 70, 2935–2945. doi: 10.1007/s00018-012-1199-7 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zima AV, Qin J, Fill M and Blatter LA (2008) Tricyclic antidepressant amitriptyline alters sarcoplasmic reticulum calcium handling in ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol 295, H2008–16. doi: 10.1152/ajpheart.00523.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qin J, Valle G, Nani A, Chen H, Ramos-Franco J, Nori A, Volpe P and Fill M (2009) Ryanodine receptor luminal Ca2+ regulation: Swapping calsequestrin and channel isoforms. Biophys. J 97, 1961–1970. doi: 10.1016/j.bpj.2009.07.030 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen B, Guo A, Gao Z, Wei S, Xie YP, Chen SR, Anderson ME and Song LS (2012) In situ confocal imaging in intact heart reveals stress-induced Ca2+ release variability in a murine catecholaminergic polymorphic ventricular tachycardia model of type 2 ryanodine receptor(R4496C+/−) mutation. Circ. Arrhythm Electrophysiol 5, 841–849. doi: 10.1161/CIRCEP.111.969733; 10.1161/CIRCEP.111.969733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bai Y, Jones PP, Guo J, Zhong X, Clark RB, Zhou Q, Wang R, Vallmitjana A, Benitez R, Hove-Madsen L, Semeniuk L, Guo A, Song LS, Duff HJ and Chen SR (2013) Phospholamban knockout breaks arrhythmogenic Ca2+ waves and suppresses catecholaminergic polymorphic ventricular tachycardia in mice. Circ. Res 113, 517–526. doi: 10.1161/CIRCRESAHA.113.301678; 10.1161/CIRCRESAHA.113.301678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen W, Wang R, Chen B, Zhong X, Kong H, Bai Y, Zhou Q, Xie C, Zhang J, Guo A, Tian X, Jones PP, O’Mara ML, Liu Y, Mi T, Zhang L, Bolstad J, Semeniuk L, Cheng H, Zhang J, Chen J, Tieleman DP, Gillis AM, Duff HJ, Fill M, Song LS and Chen SR (2014) The ryanodine receptor store-sensing gate controls Ca2+ waves and Ca2+-triggered arrhythmias. Nat. Med 20, 184–192. doi: 10.1038/nm.3440; 10.1038/nm.3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang J, Chen B, Zhong X, Mi T, Guo A, Zhou Q, Tan Z, Wu G, Chen AW, Fill M, Song LS and Chen SR (2014) The cardiac ryanodine receptor luminal Ca2+ sensor governs Ca2+ waves, ventricular tachyarrhythmias and cardiac hypertrophy in calsequestrin-null mice. Biochem. J 461, 99–106. doi: 10.1042/BJ20140126; 10.1042/BJ20140126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang Y, Tian X, Wang R, Fill M and Chen SR (2012) Abnormal termination of Ca2+ release is a common defect of RyR2 mutations associated with cardiomyopathies. Circ. Res 110, 968–977. doi: 10.1161/CIRCRESAHA.111.256560; 10.1161/CIRCRESAHA.111.256560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nodari S, Metra M and Dei Cas L (2003) Beta-blocker treatment of patients with diastolic heart failure and arterial hypertension. A prospective, randomized, comparison of the long-term effects of atenolol vs. nebivolol. Eur. J. Heart Fail 5, 621–627. doi:S1388984203000540 [pii] [DOI] [PubMed] [Google Scholar]
- 44.Flather MD, Shibata MC, Coats AJ, Van Veldhuisen DJ, Parkhomenko A, Borbola J, Cohen-Solal A, Dumitrascu D, Ferrari R, Lechat P, Soler-Soler J, Tavazzi L, Spinarova L, Toman J, Bohm M, Anker SD, Thompson SG, Poole-Wilson PA and SENIORS Investigators. (2005) Randomized trial to determine the effect of nebivolol on mortality and cardiovascular hospital admission in elderly patients with heart failure (SENIORS). Eur. Heart J 26, 215–225. doi:ehi115 [pii] [DOI] [PubMed] [Google Scholar]
- 45.Dery AS, Hamilton LA and Starr JA (2011) Nebivolol for the treatment of heart failure. Am. J. Health. Syst. Pharm 68, 879–886. doi: 10.2146/ajhp100309 [doi] [DOI] [PubMed] [Google Scholar]
- 46.Ambrosio G, Flather MD, Bohm M, Cohen-Solal A, Murrone A, Mascagni F, Spinucci G, Conti MG, van Veldhuisen DJ, Tavazzi L and Coats AJ (2011) Beta-blockade with nebivolol for prevention of acute ischaemic events in elderly patients with heart failure. Heart. 97, 209–214. doi: 10.1136/hrt.2010.207365 [doi] [DOI] [PubMed] [Google Scholar]
- 47.Karabacak M, Dogan A, Tayyar S, Ozaydin M and Erdogan D (2015) Carvedilol and nebivolol improve left ventricular systolic functions in patients with non-ischemic heart failure. Anatol J. Cardiol 15, 271–276. doi: 10.5152/akd.2014.5337 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu HR, Remeysen P and De Clerck F (1994) Antiarrhythmic effects of nebivolol in experimental models in vivo. J. Cardiovasc. Pharmacol 24, 986–993 [DOI] [PubMed] [Google Scholar]
- 49.Frishman WH (1998) Carvedilol. N. Engl. J. Med 339, 1759–1765 [DOI] [PubMed] [Google Scholar]
- 50.Stoschitzky K, Koshucharova G, Lercher P, Maier R, Sakotnik A, Klein W, Liebmann PM and Lindner W (2001) Stereoselective effects of (R)- and (S)-carvedilol in humans. Chirality. 13, 342–346. doi: 10.1002/chir.1042 [pii] [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Wright HM, Vempati P, Li H, Wangsa J, Dzhuan A, Habbu K, Knable LA, Ho L and Pasinetti GM (2013) Investigation of nebivolol as a novel therapeutic agent for the treatment of alzheimer’s disease. J. Alzheimers Dis 33, 1147–1156. doi: 10.3233/JAD-2012-120904 [doi] [DOI] [PubMed] [Google Scholar]
- 52.Mangrella M, Rossi F, Fici F and Rossi F (1998) Pharmacology of nebivolol. Pharmacol. Res 38, 419–431. doi:S1043–6618(98)90387–5 [pii] [DOI] [PubMed] [Google Scholar]
- 53.Sorrentino SA, Doerries C, Manes C, Speer T, Dessy C, Lobysheva I, Mohmand W, Akbar R, Bahlmann F, Besler C, Schaefer A, Hilfiker-Kleiner D, Luscher TF, Balligand JL, Drexler H and Landmesser U (2011) Nebivolol exerts beneficial effects on endothelial function, early endothelial progenitor cells, myocardial neovascularization, and left ventricular dysfunction early after myocardial infarction beyond conventional beta1-blockade. J. Am. Coll. Cardiol 57, 601–611. doi: 10.1016/j.jacc.2010.09.037 [doi] [DOI] [PubMed] [Google Scholar]