

Abstract
Smoking, inflammation and depression commonly co-occur and may be mechanistically linked. However, key questions remain around the direction of association and the influence of residual confounding. We aimed to characterize the association between lifetime smoking and depression, as well as to assess the role that genetically-predicted C-reactive protein (CRP) level, (an archetypal generalized inflammatory marker) and/or IL-6 activity, as a potential explanation for this association. We performed inverse variance weighted Mendelian randomization (MR) analyses using recently published summary-level GWAS data for lifetime smoking index, CRP levels, and depression. A subset of inflammatory-related genetic variants from the lifetime smoking GWAS were also used to assess the potential inflammatory causal pathways between smoking and depression. The analysis indicated reciprocal relationships of lifetime smoking with depression (ORSmk–Dep = 2.01, 95% CI 1.71–2.37, p < 0.001; ORDep–Smk = 1.09, 95% CI 1.06–1.13, p < 0.001), CRP levels and IL-6 activity (ORSmk–CRP = 1.40, 95% CI 1.21–1.55, p < 0.001; ORCRP–Smk = 1.03, 95% CI 1.02–1.05, p < 0.001, ORIL-6/CRP–Smk = 1.06 (1.03–1.09), p < 0.001). These associations were also supported by the majority of the robust MR methods performed. We did not find evidence for a reciprocal relationship between CRP levels (using > 500 genetic instruments for CRP) and depression (ORCRP–Dep = 1.01, 95% CI 0.99–1.04; ORDep–CRP = 1.03, 95% CI 0.99–1.07). We observed little variation in the IVW estimates between smoking and depression when we limited the genetic variants assessed to those related to measures of generalized inflammation, but we found evidence for an attenuation of the smoking-depression association in multivariable mendelian randomization when adjusting for IL-6 activity, suggesting that the IL-6 pathway may be at least in part responsible for the association of smoking and depression. Our study supports potential bidirectional causal associations between lifetime smoking and depression which may be at least in part explained by the IL-6 signalling pathway. The IL-6 pathway may represent a putative therapeutic target for smoking and to mitigate the effects of smoking on depression.
Subject terms: Epidemiology, Depression, Risk factors, Genetic markers
Introduction
Increasing evidence indicates a role for inflammation, the tissue response to potentially harmful stimuli, in the pathogenesis of mental health disorders, particularly depression1,2. Even though, historically, the central nervous system (CNS) has been considered an immuno-privileged region in the human body, research has shown that microglia in the CNS produce inflammatory cytokines and inflammatory processes outside of the CNS can result inflammatory responses within the CNS3–8. Furthermore, blood levels of peripheral markers of inflammation, such as C-reactive protein (CRP) and interleukin IL62, are elevated in depression in case–control studies and meta-analysis, and diseases associated with inflammation, such as rheumatoid arthritis, diabetes mellitus, coronary heart disease, stroke have been associated with depression9,10.
Modifiable exposures, such as smoking behavior, have been associated with both depression and inflammation. A 2014 study in the US estimated that the smoking prevalence among participants 16% and 40% in patients without a psychiatric and those who had a diagnosis of MDD during the previous year, respectively11. Furthermore, multiple lines of evidence indicate that smoking has pro-inflammatory molecular effects12, and observational studies support a systemic elevation of serum inflammatory markers in smokers12, including increases in CRP levels13–15. However, the evidence regarding direction of association and causality is unclear due to the biases inherent to observational studies16,17.
Mendelian randomization (MR) is an epidemiological approach that uses genetic variants as instruments to untangle the problems of reverse causation (genetic variants are fixed at conception; hence, genetically predicted levels of risk factors must precede any event) and unmeasured confounding (genetic variants show considerably less conventional confounding than phenotypic variables)18. If genetically-predicted values of a risk factor are associated with a specific disease outcome, then it is likely that the association between the risk factor and outcome has a causal basis19–22. In this manner, MR studies act like natural randomized control trials and overcome some of the biases of observational studies23.
Previous studies have shown through MR analyses that smoking and depression may have a bidirectional causal relationship24, although the mechanism through which smoking causes depression is not known. Given phenotypic associations between smoking and inflammation, and inflammation and depression, a plausible pathway from smoking to depression is via smoking’s pro-inflammatory effects. Whilst multiple cytokines are involved in inflammation, the combination of the strength of the evidence for phenotypic associations between CRP and depression, and the fact that GWAS for CRP are much larger than for any other molecular inflammatory biomarker, provide motivation for using CRP as a proxy for inflammation in the investigation of the aetiology of depression. Although previous MR studies using CRP as a proxy for systemic inflammation have shown conflicting evidence regarding the association between higher CRP levels and risk for depression20,25,26, recently, larger and better powered genome wide association studies (GWAS) for depression27 and for CRP28 have become available, and provide an opportunity to use improved genetic instruments, which explain a larger proportion of the variance, for causal inference to resolve prior ambiguities. Furthermore, CRP is a downstream marker of the interleukin (IL)-6 pathway , with IL-6 stimulating the release of CRP from hepatocytes29. The IL-6 pathway has been implicated in the pathogenesis of depression30–32, and smoking may lead to increased activity of the IL-6 signalling pathway33. The IL-6 pathway is therefore a hypothetical mechanism linking smoking behavior with depression.
In this study, the first to examine potential causality and direction of association, we conducted univariable and bidirectional MR analysis testing associations of genetically-predicted smoking behavior with CRP levels (as a measure of generalized inflammation), IL-6 activity, and risk of depression, and vice versa. Second, to examine for a potential mediating role of inflammation between smoking behavior and risk of depression, we conducted univariable MR analyses limiting the smoking exposure genetic variants to those which have been previously associated with inflammatory traits. Finally, we conducted a multivariable Mendelian randomization (MVMR) analysis to examine the associations between genetically-predicted smoking behavior and risk of depression after adjusting for genetically-predicted proxies of generalized inflammation (CRP) and IL-6 activity. We used the latest GWAS data to develop statistically better powered genetic instruments compared to previous studies and used this to investigate if inflammation mediates the potential causal effect of smoking on depression.
Methods
All methods were performed in accordance with the relevant guidelines and regulations.
Data sources
This study used publicly available summary level data obtained from previously published genome-wide association studies (GWAS) for all analyses. The source and description of the GWAS summary statistics used in this report for lifetime smoking, depression, and C-reactive protein (CRP) are presented in Table 1.
Table 1.
Characteristics of the GWAS from which the summary statistics were obtained.
| Variable | GWAS Source | Population Used | Total Participants | Cases | Controls | SNPs Covered | Genome-wide significant SNPs |
|---|---|---|---|---|---|---|---|
| Lifetime Smoking Index | Wootton et al24 | UK Biobank | 462,690* | NA | NA | 7,683,352 | 126 |
| Depression | Howard et al. /PGC27 | UK Biobank & 33 cohorts | 500,199 | 170,756 | 329,443 | 8,483,301 | 50 |
| C-Reactive Protein Levels | Han et al28 | UK Biobank | 418,642 | NA | NA | 8,927,092 | 526 |
NA: Not applicable as continuous variable.
*249 318 never smokers, 164 649 former smokers and 48 723 current smokers.
Lifetime smoking index
Lifetime smoking index was selected as the variable to represent the smoking exposure for all analyses. Wootton et al. generated this lifetime smoking index, encompassing information regarding smoking heaviness, duration, and smoking initiation and cessation. One standard deviation increase in lifetime smoking score is equivalent to an individual smoking 20 cigarettes a day for 15 years and stopping 17 years ago or an individual smoking 60 cigarettes a day for 13 years and stopping 22 years ago24.
Depression
Depression summary statistics were obtained from the Psychiatric Genetic Consortium (PGC), as described by Howard and colleagues27, with 23andMe data excluded as full summary statistics with 23andMe data were not publicly available. The total number of contributing individuals is 500,199 (170,756 cases and 329,443 controls) with 8,483,301 variants analysed. (See Supplementary Table 1 for details of depression definitions in contributing cohorts).
Generalized inflammation (C-reactive Protein)
C-reactive protein (CRP) was selected as a downstream proxy for systemic inflammation6,14,15,34,35. Han et al. (2019) generated the summary statistics used for this study from 418,642 individuals of British ancestry in the UK biobank for whom CRP levels were available and whose levels were lower than 10 mg/L28.
IL-6 activity
Since CRP is a relatively generalized and downstream inflammatory marker, we also included an instrument for the genetically-predicted effects of IL-6 signalling on CRP levels, as a measure of IL-6 activity. This instrument was obtained from Georgakis et al36, and is derived of the effect of SNPs in the IL6R gene region on circulating CRP levels, derived from a large-scale GWAS of 200,402 European participants37.
Selection of genetic instrumental variable
In Mendelian randomization, for a genetic variant to be a valid instrumental variable (IV), it must meet three assumptions: (i) the variant is associated with the exposure, (ii) the variant is not associated with any confounder of the exposure-outcome association, (iii) the variant does not affect the outcome, except via its association with the exposure18. The process for IV selection from the GWAS summary statistics for each of the variables studied was performed following a primarily statistical approach38. However, in some cases, the IV selection was further refined to include biological factors, as described below.
Statistical selection
SNPs were considered significantly associated to the GWAS variable of interest if the GWAS p-value reported on the summary statistics was smaller than 5 × 10−839. Using multiple correlated variants representing the same effect would decrease the efficiency of the analyses and increase the risk of weak instrument bias in the estimates obtained without increasing the power of the study40,41. Consequently, absence of linkage disequilibrium (LD) and independence of the final IVs selected was ascertained using the ld_clump() function from the ieugwasr R package (clumping window kb = 10,000, r2 = 0.001)42.
If an IV selected did not have a match in the outcome GWAS statistics, a proxy IV (in linkage disequilibrium with the original IV; r2 > 0.8), was used instead. Proxy IVs were obtained using the LDlink R package43.
Biological selection
Smoking inflammatory SNPs
Inflammatory traits were defined as those relating to cytokines, acute phase proteins, and immune cells. Out of the lifetime smoking IVs from the Wooton et al. GWAS24 determined significant and in linkage disequilibrium, those that had been associated with inflammatory traits (p < 5 × 10−8) in previously published GWAS were subselected using the Phenoscanner R package44. These inflammatory-related IVs were used to further assess the role of inflammation in the association between smoking and depression.
Generalized inflammation (CRP) cis SNPs
When assessing the role of CRP, four variants—rs1205, rs3093077, rs1130864 and rs1800947—were selected as cis-variants, which are those located in the CRP gene region20. Limiting the analysis to cis-variants allows for more reliable conclusions due to the biological relevance of the variants used38. Clumping was performed using the ld_clump() function from the ieugwasr R package (clumping window kb = 10,000, r2 = 0.001, p = 0.99)42 and the variant rs3093077 was selected as the lead CRP variant. Wald ratio MR analyses38 were performed to assess the effect of this variant on depression and smoking.
IL-6 Activity cis SNPs
As per Georgakis et al36, we used seven variants (rs73026617, rs12083537, rs4556348, rs2228145, rs11264224, rs12059682, rs3469607) which are located within the IL6R coding region. Georgakis and colleagues coded their data to reflect associations of these variants with increased circulating CRP levels. We used the same coding for our analysis.
Univariable reciprocal Mendelian randomization analysis
The Inverse-variance weighted method was selected as the main method to calculate the combined effect of the selected instrumental variables in all univariable MR analyses. The IVW method is similar to a weighted regression of the effect of each specific IV on the outcome on the effect of the same IV on the exposure, restricting the intercept to zero45,46. All of the univariable MR analyses are represented in Fig. 1 and in Table 3. The direction of the relationships was confirmed using Steiger filtering using the steiger_filtering() function from the TwoSampleMR R package47,48. Steiger filtering infers the direction of the effect between two variables for each specific IV by assessing the size of the effect of the IV on each variable and its measurement error47.
Figure 1.
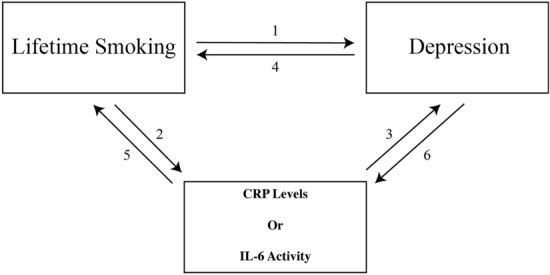
Graphic representation of the different univariable MR analyses performed. The. numbers for the different analyses correspond to those listed in Table 2.
Table 3.
Inverse Variance Weighted Estimates for the univariable MR analyses.
| Exposure | Outcome | SNPs | OR | Lower CI | Upper CI | StdErr | p-val |
|---|---|---|---|---|---|---|---|
| Smoking | Depression | 126 | 2.01 | 1.71 | 2.37 | 0.08 | 7.78E−17 |
| Smoking (Inflammatory) | Depression | 15 | 1.89 | 1.23 | 2.91 | 0.22 | 0.004 |
| Smoking (nonInflammatory) | Depression | 114 | 2.07 | 1.73 | 2.47 | 0.09 | 6.97E−16 |
| Smoking | CRP | 126 | 1.4 | 1.27 | 1.55 | 0.05 | 2.89E−11 |
| CRP | Depression | 512 | 1.01 | 0.99 | 1.04 | 0.01 | 0.225 |
| CRP (cis)* | Depression | 1 | 1.00 | 0.92 | 1.09 | 0.04 | 1.000 |
| IL-6 Activity | Depression | 7 | 0.93 | 0.85 | 1.02 | 0.05 | 0.127 |
| Depression | CRP | 50 | 1.03 | 0.99 | 1.07 | 0.02 | 0.110 |
| CRP | Smoking | 521 | 1.03 | 1.02 | 1.05 | 0.01 | 1.71E−10 |
| CRP (cis)* | Smoking | 1 | 0.98 | 0.95 | 1.01 | 0.01 | 0.171 |
| IL-6 Activity | Smoking | 7 | 1.06 | 1.03 | 1.09 | 0.03 | 6.36E−05 |
| Depression | Smoking | 50 | 1.09 | 1.06 | 1.13 | 0.02 | 3.15E−07 |
Abbreviations: Smoking (Inflammatory) indicates smoking IVs previously associated with inflammation. Smoking (non Inflammatory) indicates smoking IVs not previously associated with inflammation. “Low CI” lower limit of 95% confidence interval. “Up CI” upper limit of 95% confidence interval.’
*Wald ratios were used to obtain the estimate for the CRP (cis) variant.
Heterogeneity and sensitivity analysis
For all of the analyses performed, F-statistics were calculated to assess the strength of the instruments49. The heterogeneity between the estimates of the different IVs used was assessed using leave-one-out analyses, and visualizing the data with funnel, scatter, and forest plots48,50,51. The Funnel plots allow for the assessment of unbalanced directional pleiotropy; asymmetric distribution of the variants around the estimate would suggest unbalanced directional pleiotropy and could lead to bias in the results51. The scatterplot includes the SNP effect on the exposure on the x-axis versus the SNP effect on the outcome on the y-axis for every IV. Moreover, linear regression slopes represent the overall estimates of the effect on the exposure on the outcome. These plots highlight the heterogeneity of the Wald ratio estimates51. If all variants are valid IVs, a dose–response relationship should be observed in the scatterplot. If the majority of variants are valid IVs, pleiotropic variants may be detected as outliers in this plot, which would potentially require further investigation into the validity of those IVs based on the IV assumptions. Similarly, forest plots of the Wald ratios of every IV used along the overall IVW estimate allow for the detection of outliers.
The robustness of the overall estimate obtained with the IVW method was assessed using a combination of robust MR methods from different classes and working under a wide range of assumptions. Four additional MR methods were used: weighted median, MR-PRESSO, MR-Egger, and contamination mixture. Weighted median is a median based estimate that provides a valid estimate assuming 50% of the weight is originated from valid instruments. MR-PRESSO52 accounts for pleiotropy by detecting outliers through the comparison of the residual sum of squares of all the IVs with the expected distance under the null hypothesis of no horizontal pleiotropy. The MR-PRESSO results in this study represent the IVW analysis of the remaining IVs after removing the detected outliers.
MR-Egger accounts for pleiotropy by including an intercept term in the IVW model53. MR-Egger is severely biased by violations of the assumption that the variance of the individual IV effect estimates is negligible53. The I2GX is an adaptation of the I2 statistic from meta-analysis, related to the degree of dilution of the causal effect estimate; it ranges between 0 and 1 and values closer to 1 indicate less dilution of the causal effect estimate51. Another useful statistic in MR-Egger analyses is QR, which compares the relative fit of the MR-Egger and IVW models to the estimates of every genetic variant used. When MR-Egger and IVW estimates differ and QR is close to 1, the IVW estimate is preferred, whereas when QR is close to zero, the MR-Egger estimate is preferred54.
Contamination mixture accounts for heterogeneity in causal mechanisms by identifying genetic instruments with similar causal estimates55. Under the assumption that out of all the different values taken by ratio estimates in large samples, the true causal effect is the value taken for the largest number of genetic variants, the contamination mixture method is robust even when more than 50% of the instruments are invalid55.
Although clumping reduces bias due to LD, to address the risk that SNPs located in the same region may be residual in LD with each other and so may bias results by 'double-counting' of effects, we used correlation-corrected IVW as a further sensitivity analysis. Correlations between the variants were obtained from a European sample from the 1000 genomes dataset. LD matrices were obtained using the TwoSampleMR package48, and analysis performed using the MendelianRandomization R package56.
Multivariable Mendelian randomization analysis
Multivariable MR (MVMR) methods allow for the estimation of the proportion of the effect of smoking directly acting on depression and the proportion potentially being explained by either CRP levels (as a measure of generalized inflammation) or IL-6 activity specifically. As for the univariate analysis, we used the 2SampleMR R package for the multivariable analysis. We selected instuments at the same genome-wide threshold of significance for each variable, clumped them, combined them, and clumped again, using the same clumping window as for the univariate analysis. We also conducted a sensitivity analysis using the MendelianRandomization R package56, using correlation correction.
Results
Instrumental variable selection
The results from the IV selection process, including all of the significant SNPs in each GWAS and the final IVs selected for each variable are shown in Supplemental Table 2.
Table 2.
Univariable MR analyses performed in this report. Analyses 1, 2, and 3 are the main analyses in this report and the rest are considered supplemental. Analyses 4, 5, and 6 are the reciprocal MR analysis for 1, 2, and 3, respectively. Analyses 1b, 1c, and 3b include biological factors in the IV selection. Analyses 3b and 5b correspond to those performed using IL-6 activity.
| Analysis | Exposure | Outcome |
|---|---|---|
| 1 | Lifetime Smoking | Depression |
| 1b | Lifetime Smoking – Inflammation | Depression |
| 1c | Lifetime Smoking – non-Inflammation | Depression |
| 2 | Lifetime Smoking | CRP |
| 3 | CRP | Depression |
| 3b | IL-6 Activity | Depression |
| 4 | Depression | Lifetime Smoking |
| 5 | CRP | Smoking |
| 5b | IL-6 Activity | Smoking |
| 6 | Depression | CRP |
IVW MR Analyses testing association of smoking with depression and CRP
The IVW method supported evidence for associations of genetically-predicted lifetime smoking index with risk of depression (ORSmk–Dep = 2.01, 95% CI : 1.71–2.37, p < 0.001), and CRP levels (ORSmk–CRP = 1.40, 95% CI: 1.27 − 1.55, p < 0.001). The IVW method also identified evidence for associations of genetically-predicted depression and lifetime smoking index (ORDep-Smk = 1.09, 95% CI: 1.06–1.13, p < 0.001), and between genetically-predicted CRP levels and lifetime smoking index (ORCRP–Smk = 1.03, 95% CI: 1.02–1.05, p < 0.001) (Supplementary Table 3). However, we did not find strong evidence for causal associations of CRP with depression.
MR Analysis using inflammation-related genetic variants for smoking as IVs
Further analysis using the smoking-related genetic variants that are also associated with inflammation as IVs, the evidence for associations of genetically predicted smoking and depression remained for both inflammation-related and unrelated genetic variant sets (Supplementary Table 3).
MR Analysis using cis variants for CRP and IL-6 activity
No association was observed between the lead CRP-cis variant, rs3093077, and depression or smoking. However, there was evidence for an association of a genetically-predicted effect of IL-6 activity on CRP levels with smoking (ORIL-6/CRP–Smk = 1.06 (1.03–1.09), p < 0.001, but not depression (ORIL–6/CRP-Dep = 0.93 (0.85–1.02), p = 0.126 (Table 3 and Supplementary Table 3).
Visual assessment of MR estimates
Visual assessment of the individual IV estimates using scatter, funnel, and forest plots indicated the presence of moderate heterogeneity among these effect estimates (Fig. 2 and Supplemental Figs. 1, 2, 3). However, the symmetrical distribution of individual estimates around the overall estimates suggests that the pleiotropy present is likely balanced.
Figure 2.

Main analyses: Effect of (1) smoking on depression, (2) smoking on CRP, and (3) CRP on depression. (a) Scatterplot showing the relationship between the variant-depression associations (x-axis) and the variant-smoking associations (y-axis) with standard error bars. The slopes of the colored lines correspond to the estimated causal effect obtained with each method used. (b) Funnel plot showing the relationship between the causal effect of the exposure on the outcome estimated using the Wald ratio estimate for each IV (x-axis) against the inverse of the standard error of the such estimate (y-axis). Vertical lines show the causal estimates using all SNPs combined into a single instrument for each of five different methods.
Results for sensitivity analyses
Influential IVs
We did not find evidence for single influential SNPs that may have biased results in leave-one-out tests for all analyses (Supplementary Fig. 2).
Results using robust methods
In the smoking-CRP, smoking-IL-6 activity, smoking-depression, and depression-smoking analyses, all of the global estimates calculated using robust MR methods, excluding the MR-Egger estimate, showed a similar increase in the odds of the outcome and higher levels of the exposure. I2GX values calculated indicate that the MR-Egger was likely to have provided biased estimates for analyses using smoking as an exposure; furthermore, the QR values obtained did not support a better fit of the MR-Egger data to the model when compared to the IVW model (Supplementary Table 5). Although the IVW model did not demonstrate an effect of depression on CRP, MR-PRESSO and contamination mixture analysis did demonstrate this effect (Supplementary Table 3).
Multivariable MR
The estimated effect of smoking on depression remained significant after adjusting for the effect of CRP (OR = 2.00; 95% CI : 1.61–2.48; p = 3.5 * 10–8). When CRP was assessed as a mediator, it did not have a significant effect on depression (OR = 0.98; 95% CI : 0.92–1.04; p = 0.48) (Supplementary Table 4). However, when including the instrument for the effect of IL-6 activity with lifetime smoking in MVMR analysis, estimates for both smoking (IVW OR = 0.94 (95% CI: 0.76–1.17, p = 0.602)) and IL-6 activity (IVW OR = 1.03 (95% CI: 0.89–1.19, p = 0.700)) were attenuated to the null. This indicates that IL-6 activity, rather than generalized inflammation as a whole, could at least in part underly the associations of smoking with depression.
Assessment of bias and reliability of MR-Egger results
As MR-Egger tends to suffer from low statistical power and is particularly susceptible to bias from weak instruments51, we present assessment of reliability of the MR-Egger results. I2GX represents the variants of the SNP-exposure associations. MR-Egger is only recommended when this is large (over 90%). I2GX for the data used in the analyses with smoking as an exposure (Supplementary Table 5), suggested that the results from the MR-Egger analyses with smoking as an exposure may be strongly biased and are not reliable; therefore, in this case the IVW estimate is preferred53,57.
Steiger filtering
According to the Steiger filtering analyses, when lifetime smoking is used as the exposure, 125 out of 126 IVs were more strongly associated to lifetime smoking than to depression, suggesting that the IVs used do represent the causal effect of smoking on depression and do not support reverse causality. Similarly, when using depression as the exposure, 48 out of the 50 IVs assessed were more strongly associated with depression, suggesting that depression may lead to increased lifetime smoking.
Additional analyses with an alternative CRP GWAS58 (not UK Biobank based) are presented in Supplementary Table 6.
Additional analyses taking into account correlation between instruments are presented in Supplementary Tables 7 and 8.
Discussion
Summary of findings
In this study, we present comprehensive Mendelian randomization analyses testing the direction and potential causality of association between smoking, CRP, and depression. Our results suggest potentially causal bi-directional associations of smoking with depression and CRP levels. However, there was no evidence for a potentially causal association between CRP levels and depression, or for the effect of IL-6 activity on depression. We also found no evidence that inflammation-related smoking SNPs were associated with depression, and did not find an attenuation of the effect of smoking on risk of depression when adjusting for CRP in MVMR. However, we did find evidence for an attenuation of the effect of smoking on risk of depression when adjusting for IL-6 activity in MVMR. Together, these results suggest that while generalized inflammation may not be responsible for the associations of smoking and depression, the IL-6 pathway specifically may warrant further investigation as a potential biological mechanism underlying the effect of smoking on depression. The results obtained were consistent through the different sensitivity and robust analyses performed. Overall, the various robust methods performed relying on different assumptions, provide strong evidence to support the potential causal role of lifetime smoking on depression.
Smoking and depression
For the relationship between smoking and depression, our results, using improved genetic instruments compared to prior studies, are consistent with previous evidence supporting a reciprocal association between these variables. Observational evidence has shown that depression can trigger smoking commencement and make its cessation more challenging59–63. Moreover, smoking has been shown to lead to depression, and smoking cessation has been associated with improved depressive outcomes and decreased depression symptomatology63–65. Wootton et al., using IVW methods and using data partially overlapping with that used in this study, obtained an OR of 1.99 (95% CI: 1.71–2.32; p < 0.001) for the causal role of lifetime smoking on depression, and an OR of 1.10 (95% CI : 1.02–1.17; p < 0.001) for the reciprocal relationship between depression and smoking24. The main differences between this analysis and that in Wootton et al. (2019) is that the Howard et al. (2018) depression GWAS that informed our study had a larger sample size and incorporated cases with a broader depression definition, including broad depression (help-seeking for problems with nerves, anxiety, tension, or depression) and probable major depressive disorder. This broader definition allows for an increase in in statistical power resulting from the larger number of cases encompassed may result in a loss of specificity65. Nevertheless, the results obtained are very similar to those from Wootton et al. (2019) (we found an odds ratio of 2.01 for effect of smoking on depression compared to Wootton et al. odds ratio of 1.99), suggesting that a despite the lack of a formal MDD diagnosis in all of the cases used, the broader depression definition used remains relevant for MR analysis27,66. Overall, Steiger filtering results support a bidirectional causal relationship between lifetime smoking and depression, likely through different pathways represented by the two IV sets, rather than simple genetic correlation.
The association between smoking and depression has important implications; it strengthens the case for primary prevention of smoking and stop-smoking initiatives, and raises the question, for future research, of whether smoking cessation initiatives are effective treatments for depression.
Smoking and CRP
Evidence supporting a causal relationship between smoking and elevated CRP levels has been extensively documented in observational studies: with higher CRP levels in smokers13–15,67–80, and decreased CRP levels after smoking cessation14,15,67,72,74–77,79,80. This study provides new evidence to support a causal relationship between smoking behavior and CRP levels using Mendelian randomization techniques. Furthermore, this study supported a very small, but significant, effect (odds ratio of 1.03) of CRP levels on smoking lifetime index, which may be clinically negligible. Further study would be required to investigate which aspects of lifetime smoking CRP is causally associated with (as lifetime smoking is a composite measure reflecting both initiation and persistence of smoking).
CRP and depression
Our results did not provide strong support for a potentially causal relationship between CRP and depression or vice versa, though several observational studies have demonstrated that patients with depression have significantly higher CRP levels when compared to those without depression26,34,81–84. Of previously published MR studies assessing this relationship20,25,26, our prior study (Khandaker et al. (2020)) found evidence for a potentially causal association between CRP levels and depression using data from the UK Biobank cohort. More recently, using the MR approach Kappelmann and colleagues have reported that inflammatory markers like CRP and IL-6 are associated with specific symptoms of depression, such as suicidality85. Using symptom-level data from the UK Biobank and Dutch NESDA cohorts we have reported observational and MR associations for CRP and IL-6 with somatic/neurovegetative symptoms of depression such as fatigue and sleeping difficulties86. Taken together, current evidence from epidemiological and genetic MR studies is consistent with inflammation being potentially causally related to certain symptoms of depression, namely somatic/neurovegetative symptoms, though MR evidence for a potentially causal role of CRP on the syndrome of depression as outcome is mixed. We note that there are commonly observed phenotypic associations between CRP and heart disease, but mendelian randomisation studies do not indicate those are causal52. On the question of whether depression causes raised CRP, we do note that whilst there was no association between genetically predicted depression and CRP as assessed with the IVW method, the more robust methods of MR-PRESSO and contamination mixture did indicate such an effect; these conflicting results make it hard to draw definitive conclusions on whether depression causes raised CRP.
Overall, these results do not support a role for CRP-indexed inflammation in the development of depression. This null effect contrasts with a prior finding, using the same CRP GWAS and a similar methodology, that serum CRP is causally associated with another multifactorial etiology phenotype—namely age-related macular degeneration—indicating that the CRP genetic instruments we used are capable of revealing positive causal relationships between serum CRP and disease. Randomized control trials have shown that using of anti-inflammatory agents, such as celecoxib or infliximab, can successfully improve depression outcomes in patients with elevated CRP levels and patients with treatment-resistant depression87, strongly suggesting that inflammation has a crucial role in the pathogenesis of at least certain types of depression58,87. If inflammation is only causally relevant in certain subtypes of depression with their own distinct etiology and pathogenesis, our approach would not necessarily detect this. However, our results remain consistent with the possibility that inflammation may cause depression, considered as a unitary entity, via non-CRP mediated processes. Our research strongly suggests that future studies examining the effect of inflammatory cytokines on depression should broaden their scope beyond CRP.
Inflammation as a mediator
The lack of difference in results of the analyses using inflammatory-related and non-inflammatory related smoking IVs, suggest that there is no clear distinct CRP mediated inflammatory causal pathway mediating the causal relationship detected between smoking and depression. Furthermore, MVMR analyses showed that adjusting the effect of smoking on depression for CRP levels did not result in a significant estimate for the effect of CRP levels. However, MVMR analysis did show that adjusting the effect of smoking for IL-6 activity did significantly attenuate the estimate effect of smoking on depression, which is consistent with the hypothesis that smoking’s effects on depression are mediated through IL-6 activity; this hypothesis therefore merits further investigation.
Limitations
Lifetime smoking index and depression are both behavioral variables, with complex etiologies and hard to measure phenotypes. Furthermore, the statistical approach to IV selection use increases the risk of bias from horizontal pleiotropy88. A genetic variant may present horizontal pleiotropy, meaning that it is not only associated with the exposure, but it is additionally associated with one or more risk factors for the outcome, biasing the true association between the exposure and outcome89. However, the robust methods used, including MR-Egger, MR-PRESSO, and MR contamination mixture, allow MR analyses to be conducted in the presence of horizontal pleiotropy. The various sensitivity analyses we performed generally provide supporting evidence for the robustness of the findings.
It is important to note that the smoking and CRP data were only from the UK Biobank, as in a one-sample MR study. One-sample MR IVW analyses using weak instruments, as assessed by the F-statistic, could lead to an overestimation of the causal effect between the exposure and the outcome assessed90,91. Nevertheless, previous studies performed in non-overlapping samples have demonstrated the same direction, with a very similar effect size, of the effect between smoking and depression24. Furthermore, we used a smaller (47 independent IVs), non-overlapping CRP GWAS37, which showed a similar reciprocal associations between smoking and CRP, and similar CRP and depression findings (Supplementary Table 6).
Conclusion
The results from this study add on to the growing body of evidence supporting the bidirectionality of the causal relationship between smoking and depression. Furthermore, this study strengthens the evidence for a causal role of smoking on CRP levels. However, we do not find evidence for a potentially causal role for CRP on depression or for potentially mediating role for CRP on the association between smoking and depression, though we did find evidence for a potentially mediating role for IL-6 activity on this association. Further research is needed to understand potential mechanisms for bidirectional association between smoking and depression and to develop effective interventions.
Supplementary Information
Author contributions
D.G.: Conceptualization, Methodology, Formal Analysis, Writing—Original Draft. B.P.: Resources, Methodology, Formal Analysis, Writing—Review and Editing. V.W.: Conceptualization, Methodology, Writing—Review and Editing. C.D.: Formal Analysis, Writing—Review and Editing. O.S.: Formal analysis. G.K.: Methodology, Writing- Review and Editing. D.E.: Writing—Review and Editing, Supervision. G.M.: Conceptualization, Methodology, Formal Analysis, Writing—Review and Editing, Supervision.
Funding
This work was supported by a Wellcome Trust award to GMK (201486/Z/16/Z), and a National Institute for Health Research award to BIP (DRF-2018-11-ST2-018), and by the NIHR Cambridge Biomedical Research Centre. VW is supported by the Bowring Research Fellowship (St. Catharine’s College, Cambridge) and a Wellcome Trust Collaborative Grant (214322\Z\18\Z).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: D. Galan and B. I. Perry
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-022-19214-4.
References
- 1.Hashmi AM, Butt Z, Umair M. Is depression an inflammatory condition? A review of available evidence. J. Pak. Med. Assoc. 2013;63:899–906. [PubMed] [Google Scholar]
- 2.Miller AH, Raison CL. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016;16:22–34. doi: 10.1038/nri.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banks WA. The blood-brain barrier in psychoneuroimmunology. Neurol. Clin. 2006;24:413–419. doi: 10.1016/j.ncl.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 4.Dunn AJ, Wang J, Ando T. Effects of cytokines on cerebral neurotransmission. Comparison with the effects of stress. Adv. Exp. Med. Biol. 1999;461:117–127. doi: 10.1007/978-0-585-37970-8_8. [DOI] [PubMed] [Google Scholar]
- 5.Konsman JP, Vigues S, Mackerlova L, Bristow A, Blomqvist A. Rat brain vascular distribution of interleukin-1 type-1 receptor immunoreactivity: Relationship to patterns of inducible cyclooxygenase expression by peripheral inflammatory stimuli. J. Comp. Neurol. 2004;472:113–129. doi: 10.1002/cne.20052. [DOI] [PubMed] [Google Scholar]
- 6.Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol. Psychiat. 2009;65:732–741. doi: 10.1016/j.biopsych.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pace TW, Hu F, Miller AH. Cytokine-effects on glucocorticoid receptor function: Relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav. Immun. 2007;21:9–19. doi: 10.1016/j.bbi.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vitkovic L, et al. Cytokine signals propagate through the brain. Mol. Psychiatry. 2000;5:604–615. doi: 10.1038/sj.mp.4000813. [DOI] [PubMed] [Google Scholar]
- 9.Gallo JJ, et al. Depression, cardiovascular disease, diabetes, and two-year mortality among older, primary-care patients. Am. J. Geriatr. Psychiatry. 2005;13:748–755. doi: 10.1176/appi.ajgp.13.9.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson JL, DeZee K, Berbano E. Can treating depression improve disease outcomes? Ann. Intern. Med. 2004;140:1054–1056. doi: 10.7326/0003-4819-140-12-200406150-00017. [DOI] [PubMed] [Google Scholar]
- 11.Smith PH, Mazure CM, McKee SA. Smoking and mental illness in the U.S. population. Tob Control. 2014;23:e147–153. doi: 10.1136/tobaccocontrol-2013-051466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonçalves RB, et al. Impact of smoking on inflammation: Overview of molecular mechanisms. Inflamm. Res. 2011;60:409–424. doi: 10.1007/s00011-011-0308-7. [DOI] [PubMed] [Google Scholar]
- 13.Arnson Y, Shoenfeld Y, Amital H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J. Autoimmun. 2010;34:J258–265. doi: 10.1016/j.jaut.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 14.Bermudez EA, Rifai N, Buring JE, Manson JE, Ridker PM. Relation between markers of systemic vascular inflammation and smoking in women. Am. J. Cardiol. 2002;89:1117–1119. doi: 10.1016/s0002-9149(02)02284-1. [DOI] [PubMed] [Google Scholar]
- 15.Wannamethee SG, et al. Associations between cigarette smoking, pipe/cigar smoking, and smoking cessation, and haemostatic and inflammatory markers for cardiovascular disease. Eur. Heart J. 2005;26:1765–1773. doi: 10.1093/eurheartj/ehi183. [DOI] [PubMed] [Google Scholar]
- 16.Greenland S, Morgenstern H. Confounding in health research. Annu. Rev. Public Health. 2001;22:189–212. doi: 10.1146/annurev.publhealth.22.1.189. [DOI] [PubMed] [Google Scholar]
- 17.Spieth PM, et al. Randomized controlled trials—a matter of design. Neuropsychiatr. Dis. Treat. 2016;12:1341–1349. doi: 10.2147/ndt.S101938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith GD, et al. Clustered environments and randomized genes: A fundamental distinction between conventional and genetic epidemiology. PLoS Med. 2007;4:e352. doi: 10.1371/journal.pmed.0040352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: A guide, glossary, and checklist for clinicians. BMJ. 2018 doi: 10.1136/bmj.k601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khandaker GM, et al. Shared mechanisms between coronary heart disease and depression: Findings from a large UK general population-based cohort. Mol. Psychiatry. 2020;25:1477–1486. doi: 10.1038/s41380-019-0395-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lawlor DA, Harbord RM, Sterne JAC, Timpson N, Davey Smith G. Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Stat. Med. 2008;27:1133–1163. doi: 10.1002/sim.3034. [DOI] [PubMed] [Google Scholar]
- 22.Smith GD, Ebrahim S. 'Mendelian randomization': Can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 2003;32:1–22. doi: 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
- 23.Thanassoulis G, O'Donnell CJ. Mendelian randomization: Nature's randomized trial in the post-genome era. JAMA. 2009;301:2386–2388. doi: 10.1001/jama.2009.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wootton RE, et al. Evidence for causal effects of lifetime smoking on risk for depression and schizophrenia: A Mendelian randomisation study. Psychol. Med. 2019 doi: 10.1017/s0033291719002678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dardani C, et al. Disentangling causal relationships between inflammatory markers and depression: A bidirectional Mendelian randomization analysis. Biorxiv. 2019;5:712133. doi: 10.1101/712133. [DOI] [Google Scholar]
- 26.Wium-Andersen MK, Ørsted DD, Nordestgaard BG. Elevated C-reactive protein, depression, somatic diseases, and all-cause mortality: A mendelian randomization study. Biol. Psychiat. 2014;76:249–257. doi: 10.1016/j.biopsych.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 27.Howard DM, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neuro. 2019;22(3):343–352. doi: 10.1038/s41593-018-0326-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han X, et al. Using Mendelian randomization to evaluate the causal relationship between serum C-reactive protein levels and age-related macular degeneration. Eur. J. Epidemiol. 2020;35:139–146. doi: 10.1007/s10654-019-00598-z. [DOI] [PubMed] [Google Scholar]
- 29.Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015;16:448–457. doi: 10.1038/ni.3153. [DOI] [PubMed] [Google Scholar]
- 30.Kappelmann N, et al. Dissecting the association between inflammation, metabolic dysregulation, and specific depressive symptoms: A Genetic Correlation and 2-Sample Mendelian randomization study. JAMA Psychiat. 2021;78:161–170. doi: 10.1001/jamapsychiatry.2020.3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khandaker, G. M., Pearson, R. M., Zammit, S., Lewis, G. & Jones, P. B. Association of serum interleukin 6 and C-reactive protein in childhood with depression and psychosis in young adult life: A population-based longitudinal study (2014). [DOI] [PMC free article] [PubMed]
- 32.Khandaker GM, Zammit S, Burgess S, Lewis G, Jones PB. Association between a functional interleukin 6 receptor genetic variant and risk of depression and psychosis in a population-based birth cohort. Brain Behav. Immun. 2017 doi: 10.1016/j.bbi.2017.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sunyer J, et al. Interaction between smoking and the interleukin-6 gene affects systemic levels of inflammatory biomarkers. Nicotine Tob Res. 2009;11:1347–1353. doi: 10.1093/ntr/ntp144. [DOI] [PubMed] [Google Scholar]
- 34.Gimeno D, et al. Associations of C-reactive protein and interleukin-6 with cognitive symptoms of depression: 12-year follow-up of the Whitehall II study. Psychol. Med. 2009;39:413–423. doi: 10.1017/S0033291708003723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tibuakuu M, et al. The association between cigarette smoking and inflammation: The Genetic Epidemiology Network of Arteriopathy (GENOA) study. PLoS ONE. 2017;12:e0184914. doi: 10.1371/journal.pone.0184914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Georgakis MK, et al. Interleukin-6 Signaling effects on ischemic stroke and other cardiovascular outcomes: A mendelian randomization study. Circulation. Genom Precision Med. 2020;13:e002872. doi: 10.1161/CIRCGEN.119.002872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ligthart S, et al. Genome analyses of >200,000 individuals identify 58 Loci for Chronic Inflammation and highlight pathways that link inflammation and complex disorders. Am. J. Hum. Genet. 2018;103:691–706. doi: 10.1016/j.ajhg.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burgess S, et al. Guidelines for performing Mendelian randomization investigations [version 2; peer review: 2 approved] Wellcome Open Res. 2020;4:1–5. doi: 10.12688/wellcomeopenres.15555.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fadista J, Manning AK, Florez JC, Groop L. The (in)famous GWAS P-value threshold revisited and updated for low-frequency variants. Eur. J. Hum. Genet. 2016;24:1202–1205. doi: 10.1038/ejhg.2015.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burgess S, Dudbridge F, Thompson SG. Combining information on multiple instrumental variables in Mendelian randomization: Comparison of allele score and summarized data methods. Stat. Med. 2016;35:1880–1906. doi: 10.1002/sim.6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swerdlow DI, et al. Selecting instruments for Mendelian randomization in the wake of genome-wide association studies. Int. J. Epidemiol. 2016;45:1600–1616. doi: 10.1093/ije/dyw088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hemani, G. ieugwasr: R interface to the IEU GWAS database API. R package (2020).
- 43.Myers TA, Chanock SJ, Machiela MJ. LDlinkR: An R package for rapidly calculating linkage disequilibrium statistics in diverse populations. Front. Genetics. 2020;11:5–9. doi: 10.3389/fgene.2020.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamat MA, et al. PhenoScanner V2: An expanded tool for searching human genotype-phenotype associations. Bioinformatics. 2019;35:4851–4853. doi: 10.1093/bioinformatics/btz469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 2013;37:658–665. doi: 10.1002/gepi.21758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gage SH, et al. Assessing causality in associations between cannabis use and schizophrenia risk: A two-sample Mendelian randomization study. Psychol. Med. 2017;47:971–980. doi: 10.1017/S0033291716003172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hemani G, Tilling K, Davey Smith G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 2017;13:e1007081. doi: 10.1371/journal.pgen.1007081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hemani G, et al. The MR-Base platform supports systematic causal inference across the human phenome. Life. 2018;7:e34408. doi: 10.7554/eLife.34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burgess S, Thompson SG. Avoiding bias from weak instruments in mendelian randomization studies. Int. J. Epidemiol. 2011;40(3):755–764. doi: 10.1093/ije/dyr036. [DOI] [PubMed] [Google Scholar]
- 50.Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiology. 2017;28:30–42. doi: 10.1097/EDE.0000000000000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng J, et al. Recent developments in mendelian randomization studies. Current Epidemiol. Rep. 2017;4:330–345. doi: 10.1007/s40471-017-0128-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Verbanck M, Chen C-Y, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from mendelian randomization between complex traits and diseases. Nat. Genet. 2018;50(5):693–698. doi: 10.1038/s41588-018-0099-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur. J. Epidemiol. 2017;32:377–389. doi: 10.1007/s10654-017-0255-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan N, Thompson J. A framework for the investigation of pleiotropy in two-sample summary data mendelian randomization. Stat. Med. 2017;36(11):1783–1802. doi: 10.1002/sim.7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Slob EAW, Burgess S. A comparison of robust mendelian randomization methods using summary data. Genet. Epidemiol. 2020;44(4):313–329. doi: 10.1002/gepi.22295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yavorska OO, Stephen Burgess S. MendelianRandomization: An R package for performing Mendelian randomization analyses using summarized data. Int. J. Epidemiol. 2017;46(6):1734–1739. doi: 10.1093/ije/dyx034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bowden J, et al. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: The role of the I2 statistic. Int. J. Epidemiol. 2016;45:1961–1974. doi: 10.1093/ije/dyw220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kohler O, Krogh J, Mors O, Benros ME. Inflammation in depression and the potential for anti-inflammatory treatment. Curr. Neuropharmacol. 2016;14:732–742. doi: 10.2174/1570159x14666151208113700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Akambase JA, et al. Depression outcomes in smokers and nonsmokers: Comparison of collaborative care management versus usual care. J. Prim. Care Commun. Health. 2019;10:2150132719861265. doi: 10.1177/2150132719861265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Breslau N, Kilbey MM, Andreski P. Nicotine dependence and major depression. New evidence from a prospective investigation. Arch. Gen. Psychiatry. 1993;50:31–35. doi: 10.1001/archpsyc.1993.01820130033006. [DOI] [PubMed] [Google Scholar]
- 61.Hitsman B, et al. Past major depression and smoking cessation outcome: A systematic review and meta-analysis update. Addiction. 2013;108:294–306. doi: 10.1111/add.12009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kendler KS, et al. Smoking and major depression. A causal analysis. Arch. Gen. Psychiatry. 1993;50:36–43. doi: 10.1001/archpsyc.1993.01820130038007. [DOI] [PubMed] [Google Scholar]
- 63.Stepankova L, et al. Depression and smoking cessation: Evidence from a smoking cessation clinic with 1-Year Follow-Up. Ann. Behav. Med. 2017;51:454–463. doi: 10.1007/s12160-016-9869-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rodríguez-Cano R, et al. Smoking cessation and depressive symptoms at 1-, 3-, 6-, and 12-months follow-up. J. Affect Disord. 2016;191:94–99. doi: 10.1016/j.jad.2015.11.042. [DOI] [PubMed] [Google Scholar]
- 65.Taylor G, et al. Change in mental health after smoking cessation: Systematic review and meta-analysis. BMJ British Med. J. 2014;348:g1151. doi: 10.1136/bmj.g1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wray NR, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018;50:668–681. doi: 10.1038/s41588-018-0090-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bakhru A, Erlinger TP. Smoking cessation and cardiovascular disease risk factors: Results from the Third National Health and Nutrition Examination Survey. PLoS Med. 2005;2:e160. doi: 10.1371/journal.pmed.0020160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bazzano LA, He J, Muntner P, Vupputuri S, Whelton PK. Relationship between cigarette smoking and novel risk factors for cardiovascular disease in the United States. Ann. Intern. Med. 2003;138:891–897. doi: 10.7326/0003-4819-138-11-200306030-00010. [DOI] [PubMed] [Google Scholar]
- 69.Collaboration TERF. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: An individual participant meta-analysis. The Lancet. 2010;375:132–140. doi: 10.1016/S0140-6736(09)61717-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Conen D, et al. Smoking, smoking cessation, [corrected] and risk for symptomatic peripheral artery disease in women: A cohort study. Ann. Intern. Med. 2011;154:719–726. doi: 10.7326/0003-4819-154-11-201106070-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Eugen-Olsen J, Ladelund S, Sørensen LT. Plasma suPAR is lowered by smoking cessation: A randomized controlled study. Eur. J. Clin. Invest. 2016;46:305–311. doi: 10.1111/eci.12593. [DOI] [PubMed] [Google Scholar]
- 72.Hastie CE, Haw S, Pell JP. Impact of smoking cessation and lifetime exposure on C-reactive protein. Nicotine Tob Res. 2008;10:637–642. doi: 10.1080/14622200801978722. [DOI] [PubMed] [Google Scholar]
- 73.Kawada T. Relationships between the smoking status and plasma fibrinogen, white blood cell count and serum C-reactive protein in Japanese workers. Diabetes Metab. Syndr. 2015;9:180–182. doi: 10.1016/j.dsx.2015.02.010. [DOI] [PubMed] [Google Scholar]
- 74.Lowe GD, Yarnell JW, Rumley A, Bainton D, Sweetnam PM. C-reactive protein, fibrin D-dimer, and incident ischemic heart disease in the Speedwell study: Are inflammation and fibrin turnover linked in pathogenesis? Arterioscler Thromb. Vasc. Biol. 2001;21:603–610. doi: 10.1161/01.atv.21.4.603. [DOI] [PubMed] [Google Scholar]
- 75.Ohsawa M, et al. CRP levels are elevated in smokers but unrelated to the number of cigarettes and are decreased by long-term smoking cessation in male smokers. Prev. Med. 2005;41:651–656. doi: 10.1016/j.ypmed.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 76.Shiels MS, et al. Cigarette smoking and variations in systemic immune and inflammation markers. JNCI J. National Cancer Inst. 2014 doi: 10.1093/jnci/dju294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tonstad S, Cowan JL. C-reactive protein as a predictor of disease in smokers and former smokers: A review. Int. J. Clin. Pract. 2009;63:1634–1641. doi: 10.1111/j.1742-1241.2009.02179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lao XQ, et al. Smoking, smoking cessation and inflammatory markers in older Chinese men: The Guangzhou Biobank Cohort Study. Atherosclerosis. 2009;203:304–310. doi: 10.1016/j.atherosclerosis.2008.06.028. [DOI] [PubMed] [Google Scholar]
- 79.McEvoy John W, et al. Relationship of cigarette smoking with inflammation and subclinical vascular disease. Arteriosclerosis, Thrombosis Vascular Biol. 2015;35:1002–1010. doi: 10.1161/ATVBAHA.114.304960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gallus S, et al. Effect of tobacco smoking cessation on C-reactive protein levels in a cohort of low-dose computed tomography screening participants. Sci. Rep. 2018;8:12908. doi: 10.1038/s41598-018-29867-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dowlati Y, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67:446–457. doi: 10.1016/j.biopsych.2009.09.033. [DOI] [PubMed] [Google Scholar]
- 82.Haapakoski R, Mathieu J, Ebmeier KP, Alenius H, Kivimäki M. Cumulative meta-analysis of interleukins 6 and 1β, tumour necrosis factor α and C-reactive protein in patients with major depressive disorder. Brain Behav. Immun. 2015;49:206–215. doi: 10.1016/j.bbi.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Howren MB, Lamkin DM, Suls J. Associations of depression with C-reactive protein, IL-1, and IL-6: A meta-analysis. Psychosom Med. 2009;71:171–186. doi: 10.1097/PSY.0b013e3181907c1b. [DOI] [PubMed] [Google Scholar]
- 84.Valkanova V, Ebmeier KP, Allan CL. CRP, IL-6 and depression: A systematic review and meta-analysis of longitudinal studies. J. Affect. Disord. 2013;150:736–744. doi: 10.1016/j.jad.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 85.Kappelmann N, et al. Dissecting the association between inflammation, metabolic dysregulation, and specific depressive symptoms. JAMA Psychiat. 2020 doi: 10.1001/jamapsychiatry.2020.3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Milaneschi, Y., K. N., Lamers, F., Moser, S., Jones, P. B., Burgess, S., Pennix, B. W. J. H., Khandaker, G.M. (2021) Association of inflammation with depression and anxiety: Evidence for symptom-specificity and potential causality from UK Biobank and NESDA Cohorts. (Under Review). [DOI] [PMC free article] [PubMed]
- 87.Raison CL, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: The role of baseline inflammatory biomarkers. JAMA Psychiat. 2013;70:31–41. doi: 10.1001/2013.jamapsychiatry.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Koellinger PD, De Vlaming R. Mendelian randomization: The challenge of unobserved environmental confounds. Int. J. Epidemiol. 2019;48:665–671. doi: 10.1093/ije/dyz138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Burgess S, Thompson SG. Multivariable mendelian randomization: The use of pleiotropic genetic variants to estimate causal effects. Am. J. Epidemiol. 2015;181:251–260. doi: 10.1093/aje/kwu283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Haycock PC, et al. Best (but oft-forgotten) practices: The design, analysis, and interpretation of Mendelian randomization studies. Am. J. Clin. Nutr. 2016;103:965–978. doi: 10.3945/ajcn.115.118216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Taylor AE, et al. Mendelian randomization in health research: Using appropriate genetic variants and avoiding biased estimates. Econ. Hum. Biol. 2014;13:99–106. doi: 10.1016/j.ehb.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.