

Abstract
Oncogenes only transform cells under certain cellular contexts, a phenomenon called oncogenic competence. Using a combination of a human pluripotent stem cell-derived cancer model along with zebrafish transgenesis, we demonstrate that the transforming ability of BRAFV600E along with additional mutations depends upon the intrinsic transcriptional program present in the cell of origin. In both systems, melanocytes are less responsive to mutations, whereas both neural crest and melanoblast populations are readily transformed. Profiling reveals that progenitors have higher expression of chromatin modifying enzymes such as ATAD2, a melanoma competence factor that forms a complex with SOX10 and allows for expression of downstream oncogenic and neural crest programs. These data suggest that oncogenic competence is mediated by regulation of developmental chromatin factors, which then allow for proper response to those oncogenes.
Graphical Abstract
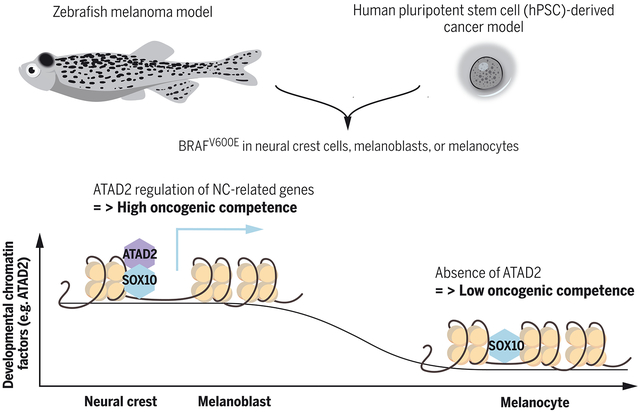
One Sentence Summary:
Developmental factors required for melanoma competence.
DNA mutations are tumorigenic depending on the pre-existing transcriptional programs and only in certain cellular contexts (1), which we refer to as oncogenic competence. In the skin, BRAFV600E activates a neural crest lineage program to initiate tumor formation (2, 3). In particular crestin, a gene that in zebrafish is specifically expressed in neural crest cells, is reactivated in melanoma-initiating cells and maintained in the tumor (2). The activation of neural crest-lineage specific mechanisms (2–6) together with oncogene mutations such as BRAFV600E are fundamental for the acquisition of a malignant state (7). However, it is not known why a neural crest-like state is required and particularly susceptible to oncogenic transformation by BRAF and what factors regulate this state.
Zebrafish models show that neural crest and melanoblasts, but not melanocytes, are oncogenically competent
The melanocytic lineage develops as a hierarchy of cells that starts as undifferentiated neural crest cells, proceeding through a melanoblast stage and then finally differentiating into melanocytes. To understand which cells within this lineage are most sensitive to oncogenic insult, we engineered zebrafish to initiate tumors by using stage-specific promoters to drive BRAFV600E in neural crest cells (sox10 promoter), melanoblasts (mitfa promoter), or melanocytes (tyrp1 promoter). Fish with p53 mutations that expressed BRAFV600E in either the neural crest cells or the melanoblasts developed aggressive tumors (Fig. 1, A to D, fig. S1 and fig. S2, A and B). However, the tyrp1-BRAFV600E p53−/− transgenic animals failed to develop tumors, and instead developed small patches of nevus-like cells (Fig. 1, B, E, H, K and fig. S2C). We analyzed both the neural crest and the melanoblast-derived tumors and found that they stained equally for BRAFV600E and pERK (Fig. 1, F, G, I and J). H&E showed that the neural crest and melanoblast-derived tumors were histologically distinct (fig. S2, A and B). The neural crest derived tumors appeared undifferentiated and they were predominantly positive for the neuronal markers huc/hud and ncam and sparsely for sox10 (Fig. 1, L, N, and P), reflecting the multipotency of the neural crest (8) (fig. S2D). In contrast, the mitfa-driven tumors had an appearance characteristic of typical cutaneous melanoma, and they stained positive for sox10 and mlana (Fig. 1, M, Q and fig. S2F), whereas they were negative for the neuronal markers huc/hud (Fig. 1O). RNA-seq analysis showed that these tumors were transcriptionally distinct (Fig. 1R, fig. S2E and table S1) and confirmed that the neural crest derived tumors expressed neuronal genes while the melanoblast-derived melanomas expressed genes related to the melanocytic lineage (fig. S2F). Gene Set Association Analysis (GSAA) showed that neural crest derived tumors displayed a gene signature linked to poor survival in neuroblastoma (fig. S2G) (9).
Fig. 1. Zebrafish models show that neural crest cells and melanoblasts, but not melanocytes, are cancer competent.
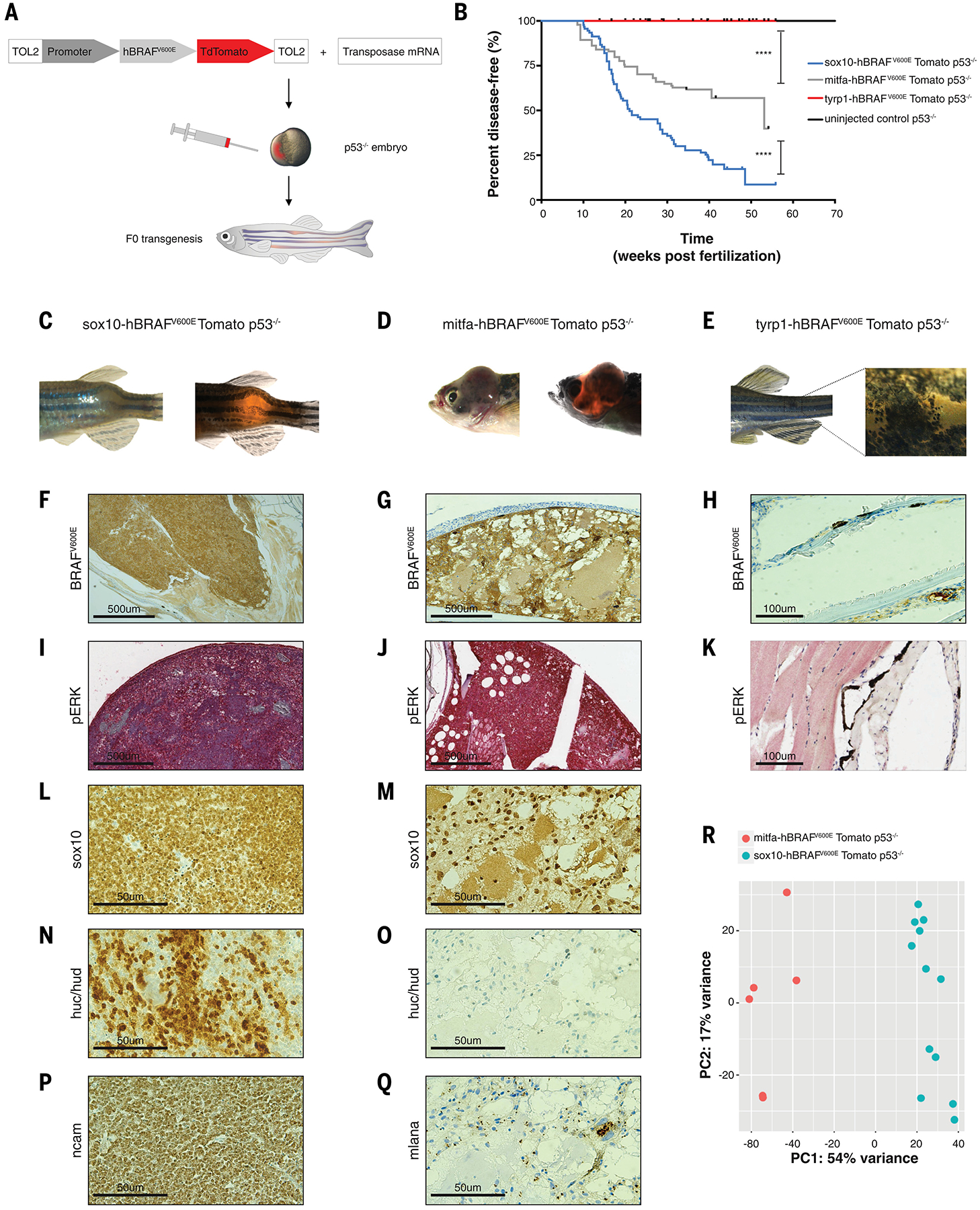
(A) Schematic drawing of zebrafish F0 transgenesis. F0 zebrafish transgenic fish were engineered by injection of p53−/− single-cell embryos with transposase mRNA together with TOL2 flanked plasmids, which encoded a stage-specific promoter (sox10, mitfa, tyrp1) driving BRAFV600E fused to TdTomato.
(B) Kaplan-Meier curves of F0 p53−/− transgenic zebrafish injected with plasmids driving BRAFV600E fused to TdTomato under either the neural crest-specific promoter sox10 (n=92 biological replicates), the melanoblast-specific promoter mitfa (n=94 biological replicates), or the melanocyte-specific promoter tyrp1 (n=49 biological replicates) or uninjected control (n=86 biological replicates). **** = p < 0.0001 for the comparison of the tumor-free survival curves of fish with melanoblast-derived tumors and melanocyte-derived nevus-like structures; **** = p < 0.0001 for the comparison of the tumor-free survival curves of fish with neural crest- and melanoblast-derived tumors; log-rank (Mantel-Cox) test.
(C) Neural crest-derived tumor developed in the sox10-BRAFV600E p53−/− transgenic fish.
(D) Melanoblast-derived tumor developed in the mitfa-BRAFV600E p53−/− transgenic fish.
(E) Nevus-like structure developed in the tyrp1-BRAFV600E p53−/− transgenic fish.
(F−K) Immunohistochemistry for BRAFV600E and phospho ERK in the neural crest- and melanoblast-derived tumors and in the melanocyte-derived nevus-like structure.
(L−Q) Immunohistochemistry staining for sox10, huc/hud, ncam and mlana. Neural crest derived tumors were positive for the neuronal marks huc/hud and ncam (N, P) and weakly positive for sox10 expression (L). MB-derived tumors were melanomas positive for sox10 (M), mlana (Q), and negative for the neuronal marks huc/hud (O).
(R) PCA plot of mitfa-driven tumors (n=6, M, red) and sox10-driven tumors (n=12, S, blue) for whole genome RNA-seq shows a separation at the transcriptional level.
These data suggested that competence to respond to BRAFV600E is biased towards cells of origin exhibiting progenitor gene programs and that those programs allow for formation of distinct tumors. However, because melanocytes have been shown to give rise to melanoma in mouse melanoma models using the Tyrosinase-Cre transgene (10, 11), this raised the question of whether the melanocytes in our model were entirely impervious to melanoma formation. To test this, we used CRISPR to knock out pten in the tyrp1-BRAFV600E p53−/− fish and we observed that 11% of animals developed melanoma (fig. S3). This indicates that both melanoblasts and melanocytes can be competent to give rise to melanoma, but that the melanoblasts do so much more readily.
A hPSC-based cancer model recapitulates the zebrafish models
To mechanistically study oncogenic competence in the precise stages along the differentiation of a melanocyte, we built a human pluripotent stem cell (hPSC)-derived cancer model. We used gene targeting in hPSCs to introduce oncogenic BRAFV600E and to inactivate the tumor suppressors RB1, TP53 (P53), and P16 (referred to hereafter as 3xKO cells) (Fig. 2A). These 3xKO engineered cells were then differentiated into neural crest cells, melanoblasts, and mature melanocytes (12) (Fig. 2A and fig. S4, A and B) and BRAFV600E was induced by doxycycline (dox) (fig. S4C), which caused comparable phosphorylation of ERK (Fig. 2B). Upon subcutaneous injection in NSG mice we found that, similar to the zebrafish, both 3xKO dox neural crest cells and melanoblasts readily formed tumors (Fig. 2, C and D), whereas the 3xKO dox melanocytes largely failed to do so, with only one single animal developing a tumor under this condition (Fig. 2E and fig. S4D). As a control, we also transplanted wildtype (WT) neural crest cells, melanoblasts, and melanocytes and found that these were unable to grow in vivo. Histological analysis of the neural crest and melanoblast-derived tumors (Fig. 2F–S and fig. S4E–L) showed high level of BRAFV600E and pERK expression in both (Fig. 2F–2I). Analogous to the zebrafish tumors, the hPSC-derived 3xKO dox neural crest cells gave rise to tumors that showed a strong preponderance of neuronal markers (Fig. 2, P and R). Previous work has shown that neural crest cells that differentiate into neuronal derivatives downregulate SOX10 expression (13), which is consistent with the fact that we see relatively little SOX10 by immunohistochemistry at this time point. The 3xKO dox melanoblast-derived tumors were instead positive for all the common markers of melanoma whereas they were negative for HuC/HuD and were pathologically categorized as desmoplastic melanomas (Fig. 2, K, M, O, Q, S). GSAA showed that neural crest cells were transcriptionally similar to the neural crest-derived tumors in the fish (fig. S5, A and B) and that melanoblasts were transcriptionally similar to the melanoblast-derived tumors in the fish (fig. S5C), which further corroborated the comparability between the two models. To ensure that our hPSC-derived tumors are relevant to human patients, we performed RNA-seq of the BRAFV600E 3xKO neural crest cells, melanoblasts, and melanocytes and compared their expression profiles to data from TCGA, using a published signature for melanoma subgroups (14). We observed that the hPSC-derived 3xKO dox neural crest cells and 3xKO dox melanoblasts strongly clustered with the human melanoma patient samples (Fig. 2T), whereas the hPSC-derived 3xKO dox melanocytes did not. We found that the 3xKO melanoblasts even without BRAFV600E induction could form tumors in mice indicating that loss of tumor suppressors alone gives these cells enough of a proliferative advantage to grow in this context (fig. S5D).
Fig. 2. A hPSC-based cancer model recapitulates the zebrafish models and demonstrates that human neural crest and melanoblast states are cancer competent, while the differentiated melanocyte state is not.
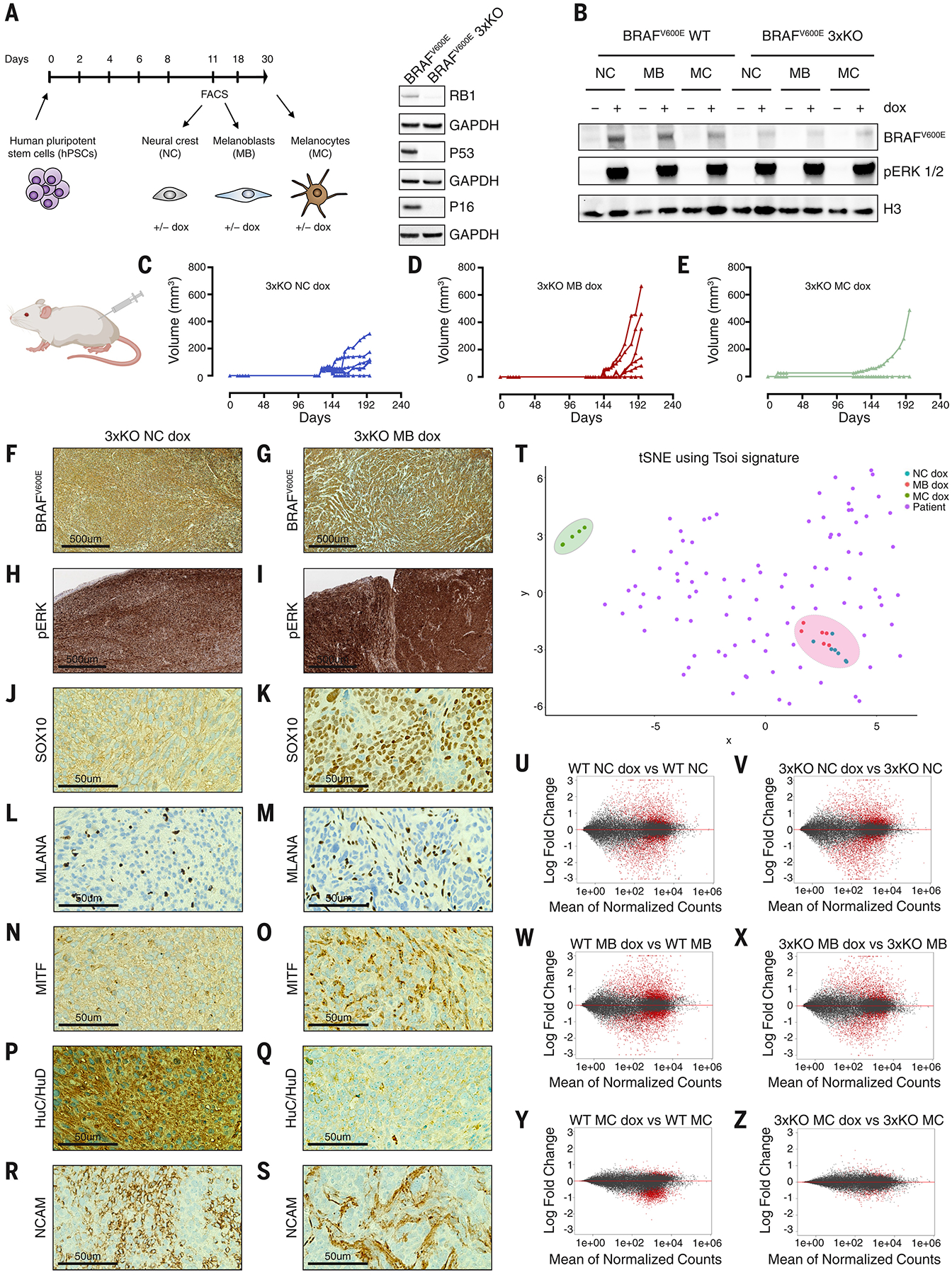
(A) Schematic summary of hPSCs differentiation into neural crest cells, melanoblasts, and melanocytes and Western blot of the dox-inducible BRAFV600E (iBRAFV600E) hPSC line knockout for RB1, P53 and P16 (3xKO) using CRISPR/Cas9 technology.
(B) Western blot of neural crest cells, melanoblasts, and melanocytes differentiated from either the iBRAFV600E WT or the iBRAFV600E 3xKO hPSCs. The cells were exposed to dox (1μg/ml) for 72h.
(C-E) In vivo growth curves of 3xKO neural crest cells + dox (n=6 per group) (C); in vivo growth curves of 3xKO melanoblasts + dox (n=6 per group) (D). 3xKO melanocytes + dox were not able to grow in vivo (n=6 per group, 1 outlier) (E), but gave rise to nevus-like structures (fig. S4D). hPSCs-derived cells were injected subcutaneously in immunodeficient NSG mice exposed to a dox-containing diet.
(F-S) Immunohistochemistry of neural crest-derived and melanoblast-derived tumors + dox treatment. Neural crest-derived tumors were undifferentiated and heterogeneous tumors, with strong neuronal features (P, R). Melanoblast-derived tumors were diagnosed as melanomas and they were positive for all the common melanocytic marks (K, M, O).
(T) t-distributed Stochastic Neighbor Embedding (t-SNE) of 3xKO + dox neural crest, melanoblast and melanocyte samples and the TCGA melanoma samples using the Tsoi signature for melanoma subtypes.
(U-Z) MA plots of the RNA-seq of WT neural crest cells, melanoblasts and melanocytes ± dox treatment and 3xKO neural crest cells, melanoblasts and melanocytes ± dox treatment (n=3 per condition). The mean of normalized counts of each gene was plotted against the log fold change following dox-induced BRAFV600E expression within that condition. Adjusted p value cut-off of 0.05 was used for significantly differentially expressed genes (red).
Neural crest and melanoblasts have strong transcriptional responses to BRAF, in contrast to melanocytes
To gain insight into why these cells differed in oncogenic competence, we performed RNA-seq of neural crest cells, melanoblasts, and melanocytes ± BRAFV600E on both the WT and 3xKO background. We observed that dox-induced BRAFV600E expression caused dramatic transcriptional changes in both the neural crest cells and melanoblasts (Fig. 2, U to X, fig. S5, A and E). In contrast, the transcriptional response to BRAF in melanocytes was nearly absent, with few genes being altered (Fig. 2, Y, Z, fig. S5, A, E and table S2). Thus, despite equally robust activation of pERK (Fig. 2B), this indicates that the melanocyte state was refractory to eliciting a transcriptional response following oncogene activation. This raised the question of what was intrinsically different between these cell types. GSAA pathway analysis in WT melanoblasts and WT melanocytes (table S2) showed that multiple pathways related to chromatin modification were significantly enriched in melanoblasts (Fig. 3A) and by examining individual genes, we found the enrichment of specific chromatin modifiers (Fig. 3B). This suggested that melanoblasts express epigenetic-related factors enabling rewiring of their chromatin state in response to BRAFV600E, which renders them competent for melanoma initiation.
Fig. 3. Cancer competence is reflected by a distinct expression of chromatin-related genes and ATAD2 is a key chromatin factor shared between human PSC-derived melanoblasts and patient melanoma cells.
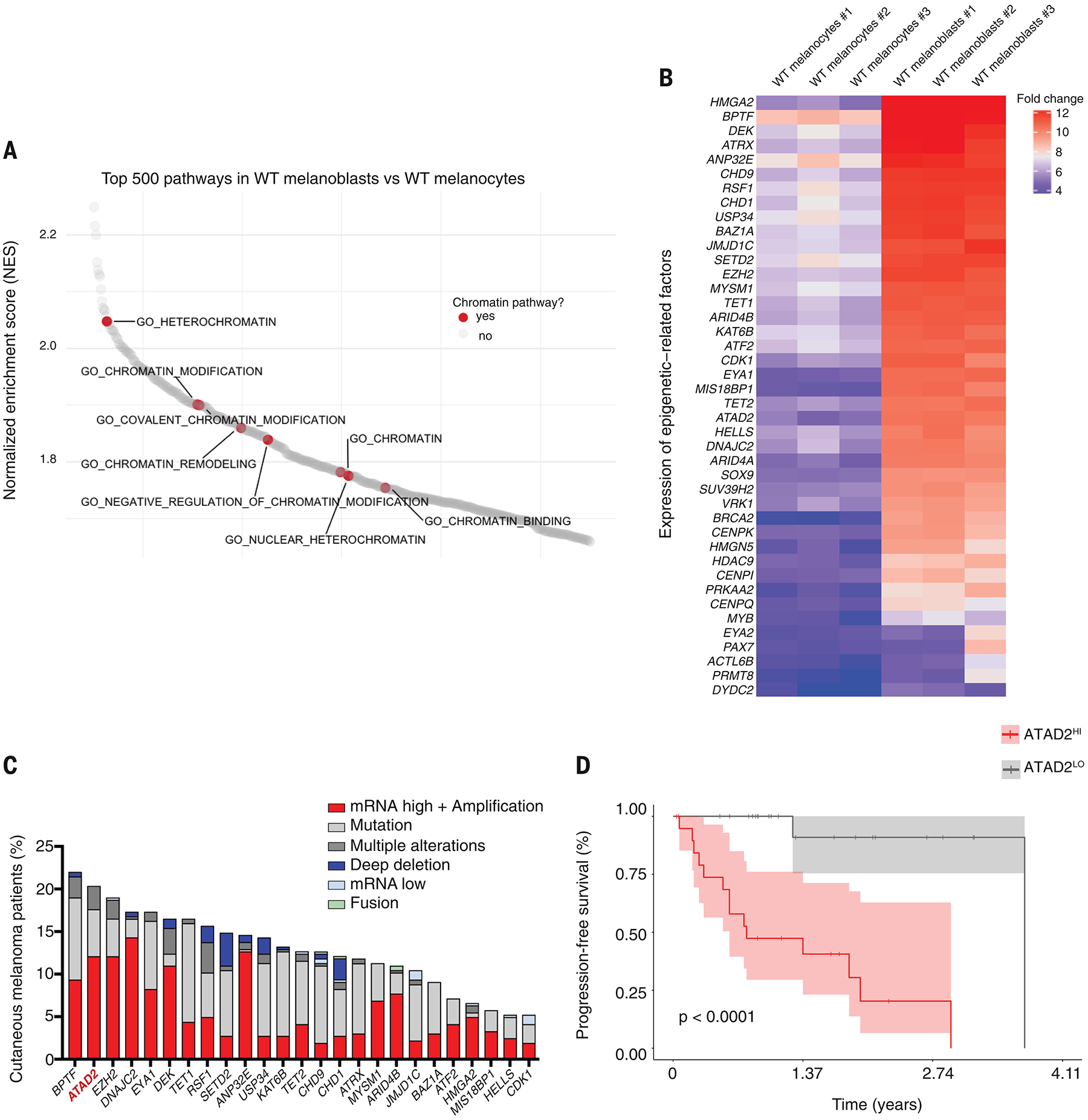
(A) Waterfall plot of the enriched pathways from the GSAA comparing WT melanoblasts to WT melanocytes. Chromatin-related pathways are highlighted in red.
(B) Heatmap depicting the differential expression of chromatin-related genes between WT melanoblasts and WT melanocytes. Normalized log2fold change.
(C) Alteration frequency of the top 25 epigenetic-related factors overexpressed in WT melanoblasts in TCGA SKCM melanoma patient samples.
(D) Kaplan-Meier overall survival curve of TCGA SKCM patients belonging either to the patients group with high levels of ATAD2 expression (ATAD2HI) or with low expression levels (ATAD2LO), log-rank p value reported.
ATAD2 is a chromatin modifier shared between neural crest cells, melanoblasts and melanoma cells
To identify which of these chromatin factors is likely most important in establishing competence, we analyzed the top 25 epigenetic-related factors that are higher in melanoblasts compared to melanocytes (Fig. 3B) and then asked which of these is most commonly amplified or overexpressed in the human melanoma TCGA cohort, which revealed BPTF, ATAD2, and EZH2 at the top of this analysis (Fig. 3C). Although there are known associations between BPTF and EZH2 and melanoma (15, 16), there is no information about ATAD2 in neural crest or melanoma development. ATAD2, an ATPase- and bromodomain-containing protein (17), is known to play roles in chromatin accessibility (18) and it was negatively associated with survival in melanoma, with patients in the highest 20% of expression had significantly worse survival compared to the remaining patients (Fig. 3D and fig. S8A).
ATAD2 acts to reshape chromatin around key neural crest and melanoblast loci
We wished to determine if ATAD2 was required for the establishment of a progenitor signature and subsequent tumorigenesis. We generated a lentivirus that induced ATAD2 expression in the 3xKO melanocytes (Fig. 4A) to a level comparable to what would be found in melanoblasts (fig. S6A). Although melanocytes without ATAD2 are deeply pigmented with melanin, reflecting their differentiated state, we noted that the 3xKO melanocytes expressing ATAD2 decreased their pigmentation (Fig. 4B), possibly because of dedifferentiation (19–21). To test this idea, we performed ATAC-seq on 3xKO dox melanoblasts, 3xKO dox melanocytes, and 3xKO ATAD2 dox melanocytes to assess global changes in chromatin accessibility. Addition of ATAD2 to the 3xKO melanocytes did not lead to a global increase in open chromatin (fig. S6, B and C). Instead, overexpression of ATAD2 in melanocytes led to a significant increase in chromatin accessibility specifically at neural crest-related loci (Fig. 4, C to E). To gain insight into the transcription factors that are binding to these newly opened chromatin regions, we performed HOMER analysis. This revealed that the top motif enriched by ATAD2 was SOX10 itself (Fig. 4, F and H), suggesting that ATAD2 was specifically allowing SOX10 to bind to its target genes. Analogously, we also asked which peaks became closed after expression of ATAD2 and found that these were most highly enriched for the MITF motif (Fig. 4, G and I). Network analysis of the loci most affected by ATAD2 and that carried the SOX10 motif confirmed a strong enrichment for pathways associated with neural precursor proliferation and neural crest migration (Fig. 4J).
Fig. 4. ATAD2 expression in melanocytes reshapes the chromatin around neural crest and melanoblast loci and reactivates a developmental signature.
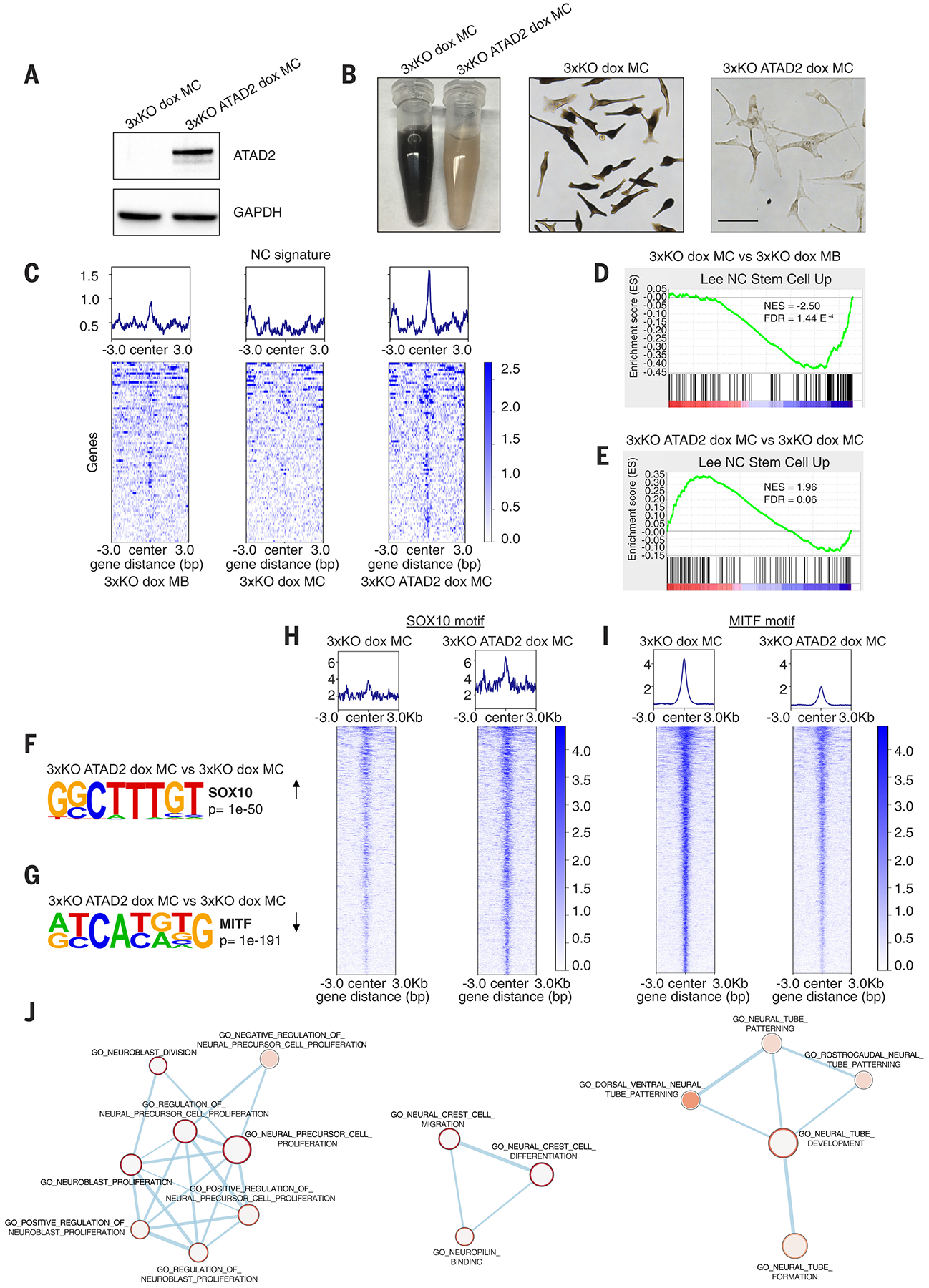
(A) Western blot for lenti-induced ATAD2 expression in 3xKO dox melanocytes.
(B) 3xKO dox melanocytes (right) and 3xKO ATAD2 dox melanocytes (left). Scale bars: 50 μm.
(C) Tornado plots of the Gene Set Enrichment Analysis (GSEA) of the ATAC-seq showing genes belonging to Lee NC Stem Cell Up gene set in 3xKO dox melanoblasts, 3xKO dox melanocytes, and 3xKO ATAD2 dox melanocytes.
(D-E) GSEA of the ATAC-seq of 3xKO dox melanocytes compared to 3xKO dox melanoblasts for Lee NC Stem Cell Up (NES = −2.50, FDR = 1.44 E−4) (D) and of 3xKO ATAD2 dox melanocytes compared to 3xKO dox melanocytes for Lee NC Stem Cell Up (NES = 1.96, FDR = 0.06) (E).
(F) Homer motif discovery shows that the SOX10 motif is one of the most enriched motifs (p value < 1e−50) in 3xKO ATAD2 dox melanocytes compared to 3xKO dox melanocytes.
(G) Homer motif discovery shows that the MITF motif is the most closed motif (p value < 1e−191) in 3xKO ATAD2 dox melanocytes compared to 3xKO dox melanocytes.
(H) Tornado plots depict any dynamic ATAC peak that contains the SOX10 motif, regardless of genomic location or gene annotation.
(I) Tornado plots depict any dynamic ATAC peak that contains the MITF motif, regardless of genomic location or gene annotation.
(J) Network analysis of the genes with increased accessibility for the SOX10 binding motif in 3xKO ATAD2 dox melanocytes compared to 3xKO dox melanocytes.
ATAD2 is necessary for neural crest induction
Because SOX10 is essential for proper neural crest induction, these data suggested that loss of ATAD2 might impair proper neural crest formation. To test this, we utilized an inducible Cas9 system (22) to knockout ATAD2 during the differentiation of hPSCs into neural crest cells (fig. S7). We measured the percentage of neural crest cells and found ~50% reduction in neural crest formation upon loss of ATAD2 (fig. S7D). Overall, these data are consistent with the notion that ATAD2 facilitates access to an early neural crest state.
ATAD2 forms a complex with SOX10 and cMYC allowing for expression of neural crest genes and MAPK-related genes
Previous work has shown that ATAD2 is able to build a protein complex with MYC and in this way regulate a MYC-dependent signature in cancer (23). We hypothesized that ATAD2 might be acting in a similar way with SOX10, by directly binding to it and facilitating transcription of its target genes. In support of this idea, we analyzed genes differentially expressed in the ATAD2HI and ATAD2LO patients from the TCGA cohort (Fig. 5A and fig. S8A). Pathway analysis revealed a strong MYC signature in the ATAD2HI patients (Fig. 5, A, B and fig. S8B), and motif analysis showed enrichment of the SOX motif (Fig. 5C). We confirmed by co-IP that ATAD2 forms a complex with MYC in the 3xKO ATAD2 dox melanocytes and found that it also does so with SOX10 (Fig. 5, D, E and fig. S8C). Based on these findings, we hypothesized that ATAD2 might play a dual role and facilitate the expression of target genes of both MYC and SOX10 transcription factors. To test this, we performed CUT&RUN for ATAD2, SOX10 and MYC in the 3xKO ATAD2 dox melanocytes, comparing it to binding in the control 3xKO dox melanocytes. We created tornado plots to look at significant overlap between these factors and revealed significant co-binding, in which ATAD2 and SOX10 co-bound 43.4% of the ATAD2 target genes (Fig. 5F), ATAD2 and MYC co-bound 6.4%, and 4.0% were co-bound by all 3 factors (Fig. 5G). To further confirm which of these genes are likely transcriptional targets of these complexes, we next performed RNA-seq of the 3xKO ATAD2 dox melanocytes compared to the control 3xKO dox melanocytes (Fig. 5H and table S3). We found significant enrichment for neural crest related genes (i.e. MEIS2, CDH2, CDH11) (24) that were co-bound by ATAD2 and SOX10 or co-bound by ATAD2/SOX10/MYC and upregulated in the RNA-seq dataset (Fig. 5, I, K and L). Furthermore, we also discovered a significant upregulation and binding of regulators of the MAPK pathway including key upstream regulators (i.e. EGFR, FGFR2, Fig. 5, J, L) and key downstream regulators of MAPK including MYC target genes (i.e. RPS6KA6, JUN and E2F, Fig. 5L and fig S6D). Recent work has demonstrated that upregulation of EGFR and FGF, which we find is facilitated by ATAD2, are key mechanisms of MAPK pathway activation in BRAFV600E mutant melanoma, especially in the setting of drug resistance (25, 26). In the 3xKO ATAD2 dox melanocytes, it is possible that this increase in EGFR/FGF explains the increased phosphorylation of ERK1/2 (fig. S8E), and future studies will be required to investigate the potential link between ATAD2 expression, ERK activation and EGFR and FGF in this context. Overall, our data indicate that ATAD2 directly binds to both SOX10 and MYC, facilitates the expression of their target genes, and leads to increased activity of two major tumorigenic mechanisms in melanoma, a neural crest lineage program and MAPK pathway activity.
Fig. 5. ATAD2 promotes melanoma phenotypes through cMYC and SOX10 in both clinical samples of cutaneous melanoma and in the hPSC-derived cancer model.
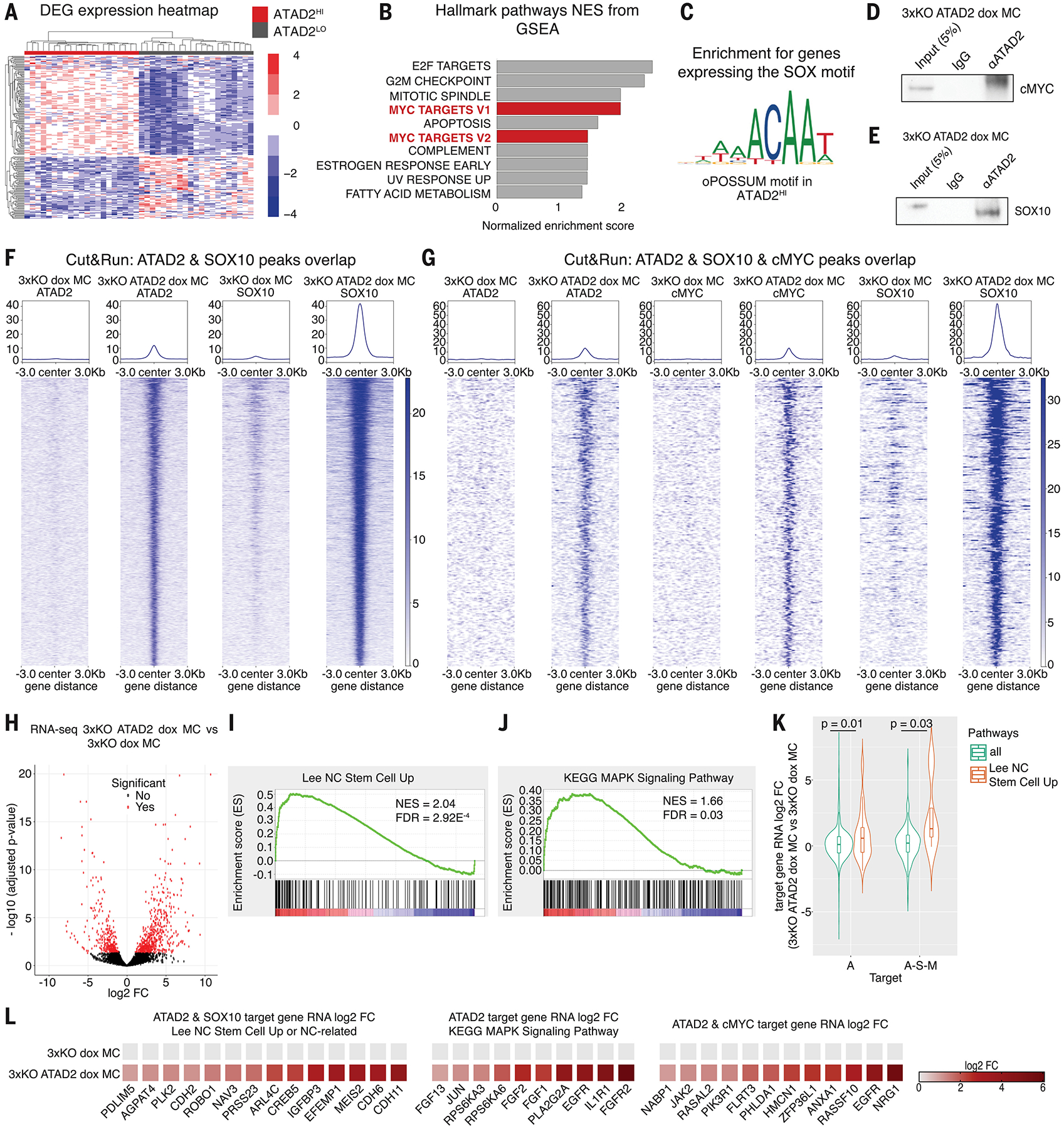
(A) Heatmap plot of differentially expressed genes (DEG) in the ATAD2HI patient group versus the ATAD2LO patient group.
(B) Top 10 hallmark pathways from GSEA enriched in the ATAD2HI patient group compared to the ATAD2LO patient group.
(C) Identification of the SOX binding motif on genes co-expressed in the ATAD2HI patient groups, determined by analysis with the oPOSSUM software tool.
(D-E) Co-IP analysis of protein lysates of 3xKO ATAD2 dox melanocytes using either the ATAD2 or the control IgG antibody and then blotted against cMYC (D) and SOX10 (E).
(F) Tornado plots depict the Cut&Run peaks overlapping between ATAD2 and SOX10 in 3xKO dox melanocytes and 3xKO ATAD2 dox melanocytes.
(G) Tornado plots depict the Cut&Run peaks overlapping between ATAD2, c-MYC and SOX10 in 3xKO dox melanocytes and 3xKO ATAD2 dox melanocytes.
(H) Volcano plot of the RNA-seq depicts the distribution of the transcriptional differences between 3xKO ATAD2 dox melanocytes over 3xKO dox melanocytes.
(I-J) GSEA of the RNA-seq of 3xKO ATAD2 dox melanocytes compared to 3xKO dox melanocytes for Lee NC Stem Cell Up (NES = 2.04, FDR = 2.92 E-4) (I) and for KEGG MAPK Signaling Pathway (NES = 1.66, FDR = 0.03) (J).
(K) Violin plots showing the distribution of Cut&Run target genes that are also significantly upregulated in the RNA-seq dataset. Genes bound by ATAD2 are enriched for RNA upregulation of the Lee NC Stem Cell signature (left, Mann-Whitney U test, p = 0.01), as are genes cobound by ATAD2/SOX10/cMYC (right, Mann-Whitney U test, p = 0.03).
(L) Heatmap depicting expression changes of the ATAD2 and SOX10 co-bound Cut&Run target genes belonging to the Lee NC Stem Cell Up gene set (left). Heatmap depicting expression changes of the ATAD2 Cut&Run target genes belonging to the KEGG MAPK Signaling Pathway (middle). Heatmap depicting expression changes of selected ATAD2 and cMYC co-bound Cut&Run target genes (right).
ATAD2 promotes melanoma phenotypes
EdU incorporation assays showed that 3xKO ATAD2 ± dox melanocytes had a comparable proliferation rate to 3xKO dox melanoblasts and that they were significantly more proliferative than 3xKO dox melanocytes (fig. S8F). Both proliferation rates in 3xKO neural crest cells and melanoblasts increased upon BRAFV600E expression, whereas in the already highly proliferative 3xKO ATAD2 dox melanocytes they did not. Noteworthy, in vitro 3xKO melanocytes were not entering a senescent state upon BRAFV600E expression and their proliferation rate did not change. 3xKO ATAD2 ± dox melanocytes were also more invasive, as measured by the invasion chamber analysis (fig. S9, A and B) and as supported by ATAC-seq gene signatures consistent with an epithelial-to-mesenchymal (EMT) program (fig. S9, C and D). We also assessed the metabolic profile of these hPSC-derived tumor lines and found evidence of significant metabolic rewiring. We used Seahorse assays to measure mitochondrial bioenergetics and glycolysis via oxygen consumption rate (OCR) and the extracellular acidification rate (ECAR). The ratio between OCR and ECAR showed that 3xKO melanoblasts were mostly relying on glycolysis for energy production and that this trend was amplified by dox-induced oncogene expression (fig. S10A) (27). On the contrary, 3xKO melanocytes displayed a profoundly different metabolic profile, with sustained oxidative metabolism. Upon ATAD2 expression, the 3xKO ATAD2 dox melanocytes switched to a more glycolytic state, as evidenced by an increased ECAR/OCR ratio (fig. S10).
ATAD2 endows melanocytes with oncogenic competence in vivo
To test whether ATAD2 was sufficient for melanoma initiation, we created transgenic zebrafish (Fig. 6A) in which we could overexpress BRAFV600E +/−ATAD2 in the melanocytes using the tyrp1 promoter, a cell type that we showed above (Fig. 1B) is refractory to develop melanoma. In the casper background (28) with rescued melanocytes, none of the tyrp1-BRAFV600E p53−/− animals developed hyperplasia or melanoma (0%, n=0/23). In contrast, we found that 10% (n=2/20) of tyrp1-BRAFV600E tyrp1-ATAD2 p53−/− animals developed melanomas and 15% (n=3/20) developed hyperplastic lesions (Fig. 6, B, C and fig. S11). Conversely, we next wanted to test whether loss of ATAD2 would prevent melanoma initiation. Using the electroporation based TEAZ approach (29) with non-targeting sgRNAs, 65% of the fish developed a patch of GFP+ melanocytes (Fig. 6, D to G), easily discernible from the surrounding normal skin. In contrast, in the animals that were electroporated with sgRNAs against ATAD2, we found that the majority was negative for any GFP (Fig. 6, E, F and H) and the quantification of the GFP+ area revealed a significant decrease in overall tumor size in the ATAD2 knockout compared with the control fish (Fig. 6E). We further tested the role of ATAD2 in the context of PTEN deletion using the MASERATI vector (Fig. S12A) (30), where patches seen at 14 days give rise to fully penetrant melanomas in 78.6% of animals by day 84 (fig. S12, B–H). Similar to the results above, ATAD2 sgRNA led to a significant reduction in melanoma size at day 14 (Fig. S12I). Analogous to the CRISPR knockout of SOX10 (2), over time we observed selection for WT or in-frame ATAD2 clones (Fig. S12, J and K). Taken together, our data supports a model in which high levels of ATAD2 expression, which is found in neural crest cells and melanoblasts, allows for re-expression of a progenitor signature in melanocytes and supports the ability of BRAF to initiate tumors (fig. S13).
Fig. 6. ATAD2 is necessary and sufficient for melanoma initiation.
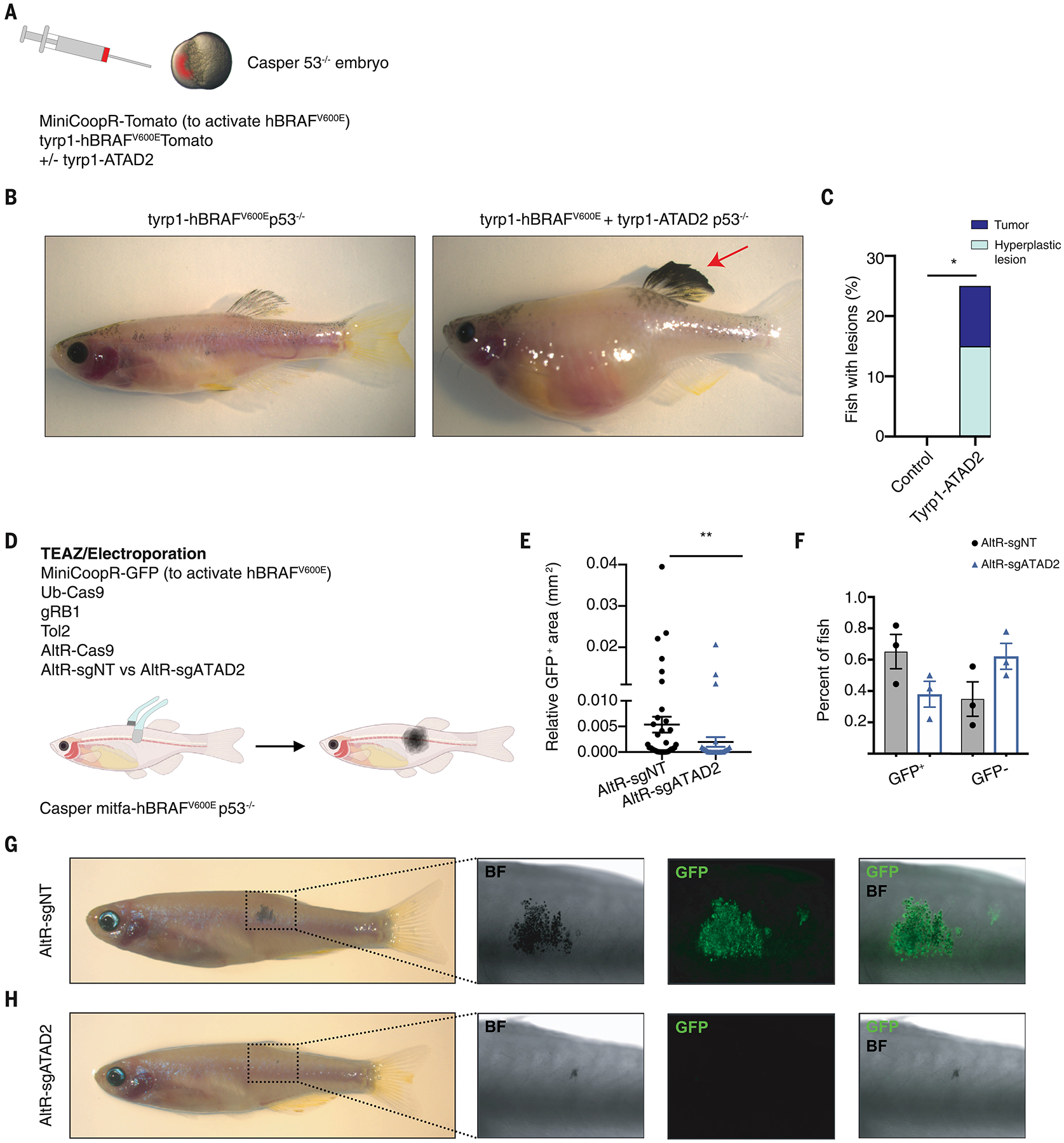
(A) p53−/−;casper single-cell embryos were injected with a plasmid which encoded the melanocyte-specific promoter tyrp1 driving BRAFV600E ± a plasmid that encoded tyrp1 driving ATAD2, along with a MiniCoopR-tdTomato plasmid to rescue melanocytes in the casper background.
(B) Transgenic control fish with tyrp1 driving BRAFV600E (left) and a transgenic fish expressing tyrp1 driving both BRAFV600E and ATAD2, which developed a pigmented tumor (right).
(C) Percentage of fish that developed melanoma with the tyrp1 driving BRAFV600E +/− ATAD2. Control fish expressing tyrp1-BRAFV600E did not develop tumors (n=0/23 fish), whereas fish expressing tyrp1 driving BRAFV600E + ATAD2 developed melanomas (n=2/20 fish) and hyperplastic lesions (n=3/20 fish). Fisher’s exact test, * p < 0.05.
(D) Schematic drawing for the TEAZ/Electroporation experiment. Fish that were p53−/−, casper; mitfa-BRAFV600E were electroporated with MiniCoopR-GFP (mitfa-MITF and mitfa-GFP), Ub-Cas9, gRB1, Tol2, AltR-Cas9, and either AltR-sgNT or a pool of AltR-sgATAD2. The fish were then monitored and quantified for melanoma initiation.
(E) Quantification of the GFP+ area (mm2) in the transgenic fish two weeks after electroporation. Mann-Whitney test with ** p = 0.01.
(F) Percentage of fish that were either GFP+ or GFP− depending on the electroporation of AltR-sgNT or AltR-sgATAD2.
(G) Transgenic fish electroporated with AltR-sgNT, 4 weeks post electroporation. The images depict early lesions characterized by pigmentation and GFP expression.
(H) Transgenic fish electroporated with AltR-sgATAD2, 4 weeks post electroporation.
Discussion
These data show that the ability of a cell to respond to BRAFV600E depends upon the pre-existing transcriptional state of that cell. We find that both the neural crest and the melanoblast stages are able to respond to BRAFV600E, and that ATAD2 is an oncogenic competence factor required for melanoma initiation in melanocytes.
One important difference between our observations and data using genetically engineered mouse models is that we found mature melanocytes (both zebrafish and hPSC-derived) to be relatively resistant to oncogenesis, although not impervious. The most commonly available mouse models of melanoma utilize a tyrosinase-Cre driver to activate BRAFV600E in combination with loss of various tumor suppressors (10, 11). These animals can develop melanoma, although this can be accelerated by inactivation of tumor suppressors such as CDKN2A, TP53, or PTEN (31). Which cells within these mice act as the melanoma cell of origin has not been fully resolved (32–34), but our studies are not precisely comparable to the mouse studies since our zebrafish use tyrp1-driven BRAFV600E. One possible explanation for this discrepancy is that in our system the tyrp1 promoter is actually driving expression in a more fully differentiated melanocyte compared to the tyr promoter used in mouse studies. Another explanation is that these differences may reflect different biological thresholds for tumorigenesis, in that a different number of DNA lesions may be required to transform melanocytes in human versus mice versus zebrafish.
We found that melanocytes can eventually be induced into melanoma formation with additional hits such as PTEN inactivation, albeit still much less efficiently than the melanoblasts. Given that melanocytes are likely more numerically frequent in patient skin compared to melanoblasts- or neural crest-like cells, it is possible that these cells may serve as a cell of origin, but that they would need more genetic lesions compared to melanoblast-like cells.
Our study does not rule out other possible mechanisms that could regulate the response to pro-oncogenic signals such as the timing or the specific order of acquiring mutations. Another aspect of oncogenic competence might be the particular microenvironment in which each of these cells reside. Previous studies have in fact suggested that microenvironmental cues might influence the ability of a cell to transform, (32, 33). It would be important to investigate how oncogenic competence might be modulated in each potential cell of origin depending on the local microenvironment.
Overall, our data suggest a model in which there may not be a discrete cell of origin of melanoma. Instead multiple cells along the spectrum from neural crest- and melanoblast-like cells to melanocytes may be competent to give rise to tumors (fig. S13). This competence appears dependent upon three interrelated factors that cooperate to determine susceptibility: DNA mutations (e.g. BRAFV600E, p53−/−, CDKN2A−/−, PTEN−/−), cell-type specific transcription factors (e.g. SOX10 and MITF), and the inherent levels of developmental chromatin modifiers, which allow for a permissive chromatin landscape (e.g. ATAD2).
Materials and methods summary
Zebrafish transgenesis
Zebrafish lines used in these studies includes the wild-type (AB), casper (mitfa−/−;mpv17−/−), sox10-BRAFV600E p53−/−, mitfa-BRAFV600E p53−/− and the tyrp1-BRAFV600E p53−/− line. For TEAZ-based transgenics, the plasmids were electroporated directly into the skin of adult animals, as previously described (29).
hPSCs engineering and differentiation
To derive neural crest cells and melanoblasts, hPSCs were plated at day −1 on matrigel-coated dishes in E8 medium and 10μM ROCKi. From day0 to day2 cells were cultured in E6 medium with 1ng/ml BMP4,10μM SB and 600nM CHIR. From day2 to day4, cells were cultured in E6 medium with 10μM SB + 1.5μM CHIR. From day 4 to day 6, cells were cultured in E6 medium with 1.5μM CHIR. From day 6 to day 11, cells were cultured in E6 medium with 1.5μM CHIR, 5ng/ml BMP4 and 100nM EDN3. Cells were FACS-sorted for cKIT and P75 expression, and double positive cells were further differentiated into melanocytes using melanocytes medium.
Detailed materials and methods are available in the supplementary materials.
Supplementary Material
Acknowledgments:
We thank J. Ablain for the MASERATI vector. Fig. 2, Fig.6, and fig. S12 contain images created with BioRender.
This work was supported by awards from the Melanoma Research Alliance (R.M.W. and L.S.), NIH Research Program Grant R01CA229215, NIH Director’s New Innovator Award DP2CA186572, NIH Mentored Clinical Scientist Research Career Development Award K08AR055368, The Pershing Square Sohn Foundation, The Alan and Sandra Gerry Metastasis Research Initiative at the Memorial Sloan Kettering Cancer Center, The Harry J. Lloyd Foundation, Consano, and the Starr Cancer Consortium (to R.M.W.); NYSTEM award DOH01-STEM5-2016-00300 and the Starr Stem cell initiative (to L.S.); Kirschstein-NRSA predoctoral fellowship F31CA196305, Joanna M. Nicolay Melanoma Foundation Research Scholar Award and the Robert B. Catell Fellowship (to S.J.C.), Kirschstein-NRSA predoctoral fellowship F30CA220954, Melanoma Research Foundation, Medical Scientist Training Program T32GM007739 (to N.R.C.); NRSA (F32) 5F32MH116590-02 (to R.M.W); Frank Lappin Horsfall, Jr. Fellowship (to Y.F); Kirschstein-NRSA predoctoral fellowship F30CA236442, Molecular and Cell Biology Teaching Grant T32GM008539, Medical Scientist Training Program T32GM007739 (to J.M.W); Individual Predoctoral to Postdoctoral Fellow Transition Award (F99/K00) 5K00CA223016-04 (to E.M.); the Swiss National Science Foundation Postdoc.mobility fellowship P2ZHP3_171967 and P400PB_180672 (to A.B.) and P30 CA008748 (NCI Core Facility Grant).
Footnotes
Competing interests:
L.S. is co-founder and consultant of BlueRock Therapeutics and is listed as inventor on patent application by MSKCC related to melanocyte differentiation from human pluripotent stem cells (WO2011149762A2). R.M.W. is a consultant to N-of-One, a subsidiary of Qiagen. All other authors declare no competing interests.
Data and materials availability:
Datasets deposited at GEO (RNA-seq, ATAC-seq, Cut&Run): GSE172069.
All zebrafish lines are available upon request from the authors or via the ZIRC zebrafish stock center (https://zebrafish.org/home/guide.php). WA-09 lines (NC, MB, MC, 3xKO NC, 3xKO MB, 3xKO MC) will be made available upon request from the Studer lab at Memorial Sloan Kettering Cancer Center under a Material Transfer Agreement with the institute.
References:
- 1.Haigis KM, Cichowski K, Elledge SJ, Tissue-specificity in cancer: The rule, not the exception. Science 363, 1150–1151 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Kaufman CK, Mosimann C, Fan ZP, Yang S, Thomas AJ, Ablain J, Tan JL, Fogley RD, van Rooijen E, Hagedorn EJ, Ciarlo C, White RM, Matos DA, Puller AC, Santoriello C, Liao EC, Young RA, Zon LI, A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science 351, aad2197 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.White RM, Cech J, Ratanasirintrawoot S, Lin CY, Rahl PB, Burke CJ, Langdon E, Tomlinson ML, Mosher J, Kaufman C, Chen F, Long HK, Kramer M, Datta S, Neuberg D, Granter S, Young RA, Morrison S, Wheeler GN, Zon LI, DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature 471, 518–522 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Varum S, Baggiolini A, Zurkirchen L, Atak ZK, Cantu C, Marzorati E, Bossart R, Wouters J, Hausel J, Tuncer E, Zingg D, Veen D, John N, Balz M, Levesque MP, Basler K, Aerts S, Zamboni N, Dummer R, Sommer L, Yin Yang 1 Orchestrates a Metabolic Program Required for Both Neural Crest Development and Melanoma Formation. Cell stem cell 24, 637–653.e639 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Heppt MV, Wang JX, Hristova DM, Wei Z, Li L, Evans B, Beqiri M, Zaman S, Zhang J, Irmler M, Berking C, Besch R, Beckers J, Rauscher FJ 3rd, Sturm RA, Fisher DE, Herlyn M, Fukunaga-Kalabis M, MSX1-Induced Neural Crest-Like Reprogramming Promotes Melanoma Progression. The Journal of investigative dermatology 138, 141–149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shakhova O, Zingg D, Schaefer SM, Hari L, Civenni G, Blunschi J, Claudinot S, Okoniewski M, Beermann F, Mihic-Probst D, Moch H, Wegner M, Dummer R, Barrandon Y, Cinelli P, Sommer L, Sox10 promotes the formation and maintenance of giant congenital naevi and melanoma. Nature cell biology 14, 882–890 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Kunz M, Oncogenes in melanoma: an update. European journal of cell biology 93, 1–10 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Bronner-Fraser M, Fraser SE, Cell lineage analysis reveals multipotency of some avian neural crest cells. Nature 335, 161–164 (1988). [DOI] [PubMed] [Google Scholar]
- 9.Asgharzadeh S, Pique-Regi R, Sposto R, Wang H, Yang Y, Shimada H, Matthay K, Buckley J, Ortega A, Seeger RC, Prognostic significance of gene expression profiles of metastatic neuroblastomas lacking MYCN gene amplification. Journal of the National Cancer Institute 98, 1193–1203 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr., You MJ, DePinho RA, McMahon M, Bosenberg M, Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nature genetics 41, 544–552 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dhomen N, Reis-Filho JS, da Rocha Dias S, Hayward R, Savage K, Delmas V, Larue L, Pritchard C, Marais R, Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer cell 15, 294–303 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Mica Y, Lee G, Chambers SM, Tomishima MJ, Studer L, Modeling neural crest induction, melanocyte specification, and disease-related pigmentation defects in hESCs and patient-specific iPSCs. Cell Rep 3, 1140–1152 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim J, Lo L, Dormand E, Anderson DJ, SOX10 maintains multipotency and inhibits neuronal differentiation of neural crest stem cells. Neuron 38, 17–31 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X, Braas D, Grasso CS, Palaskas N, Ribas A, Graeber TG, Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer cell 33, 890–904.e895 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dar AA, Nosrati M, Bezrookove V, de Semir D, Majid S, Thummala S, Sun V, Tong S, Leong SP, Minor D, Billings PR, Soroceanu L, Debs R, Miller JR 3rd, Sagebiel RW, Kashani-Sabet M, The role of BPTF in melanoma progression and in response to BRAF-targeted therapy. Journal of the National Cancer Institute 107, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zingg D, Debbache J, Schaefer SM, Tuncer E, Frommel SC, Cheng P, Arenas-Ramirez N, Haeusel J, Zhang Y, Bonalli M, McCabe MT, Creasy CL, Levesque MP, Boyman O, Santoro R, Shakhova O, Dummer R, Sommer L, The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nature communications 6, 6051 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Boussouar F, Jamshidikia M, Morozumi Y, Rousseaux S, Khochbin S, Malignant genome reprogramming by ATAD2. Biochimica et biophysica acta 1829, 1010–1014 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Morozumi Y, Boussouar F, Tan M, Chaikuad A, Jamshidikia M, Colak G, He H, Nie L, Petosa C, de Dieuleveult M, Curtet S, Vitte AL, Rabatel C, Debernardi A, Cosset FL, Verhoeyen E, Emadali A, Schweifer N, Gianni D, Gut M, Guardiola P, Rousseaux S, Gerard M, Knapp S, Zhao Y, Khochbin S, Atad2 is a generalist facilitator of chromatin dynamics in embryonic stem cells. Journal of molecular cell biology 8, 349–362 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoek KS, Eichhoff OM, Schlegel NC, Dobbeling U, Kobert N, Schaerer L, Hemmi S, Dummer R, In vivo switching of human melanoma cells between proliferative and invasive states. Cancer research 68, 650–656 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A, Frederick DT, Barzily-Rokni M, Straussman R, Haq R, Fisher DE, Mesirov JP, Hahn WC, Flaherty KT, Wargo JA, Tamayo P, Garraway LA, A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer discovery 4, 816–827 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C, Kong X, Possik PA, Cornelissen-Steijger PD, Geukes Foppen MH, Kemper K, Goding CR, McDermott U, Blank C, Haanen J, Graeber TG, Ribas A, Lo RS, Peeper DS, Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nature communications 5, 5712 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gonzalez F, Zhu Z, Shi ZD, Lelli K, Verma N, Li QV, Huangfu D, An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell stem cell 15, 215–226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ciro M, Prosperini E, Quarto M, Grazini U, Walfridsson J, McBlane F, Nucifero P, Pacchiana G, Capra M, Christensen J, Helin K, ATAD2 is a novel cofactor for MYC, overexpressed and amplified in aggressive tumors. Cancer research 69, 8491–8498 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Williams RM, Candido-Ferreira I, Repapi E, Gavriouchkina D, Senanayake U, Ling ITC, Telenius J, Taylor S, Hughes J, Sauka-Spengler T, Reconstruction of the Global Neural Crest Gene Regulatory Network In Vivo. Developmental cell 51, 255–276 e257 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun C, Wang L, Huang S, Heynen GJ, Prahallad A, Robert C, Haanen J, Blank C, Wesseling J, Willems SM, Zecchin D, Hobor S, Bajpe PK, Lieftink C, Mateus C, Vagner S, Grernrum W, Hofland I, Schlicker A, Wessels LF, Beijersbergen RL, Bardelli A, Di Nicolantonio F, Eggermont AM, Bernards R, Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 508, 118–122 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Wang VE, Xue JY, Frederick DT, Cao Y, Lin E, Wilson C, Urisman A, Carbone DP, Flaherty KT, Bernards R, Lito P, Settleman J, McCormick F, Adaptive Resistance to Dual BRAF/MEK Inhibition in BRAF-Driven Tumors through Autocrine FGFR Pathway Activation. Clinical cancer research : an official journal of the American Association for Cancer Research 25, 7202–7217 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hall A, Meyle KD, Lange MK, Klima M, Sanderhoff M, Dahl C, Abildgaard C, Thorup K, Moghimi SM, Jensen PB, Bartek J, Guldberg P, Christensen C, Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the (V600E)BRAF oncogene. Oncotarget 4, 584–599 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.White RM, Sessa A, Burke C, Bowman T, LeBlanc J, Ceol C, Bourque C, Dovey M, Goessling W, Burns CE, Zon LI, Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell stem cell 2, 183–189 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Callahan SJ, Tepan S, Zhang YM, Lindsay H, Burger A, Campbell NR, Kim IS, Hollmann TJ, Studer L, Mosimann C, White RM, Cancer modeling by Transgene Electroporation in Adult Zebrafish (TEAZ). Disease models & mechanisms 11, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ablain J, Durand EM, Yang S, Zhou Y, Zon LI, A CRISPR/Cas9 vector system for tissue-specific gene disruption in zebrafish. Developmental cell 32, 756–764 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perez-Guijarro E, Day CP, Merlino G, Zaidi MR, Genetically engineered mouse models of melanoma. Cancer 123, 2089–2103 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kohler C, Nittner D, Rambow F, Radaelli E, Stanchi F, Vandamme N, Baggiolini A, Sommer L, Berx G, van den Oord JJ, Gerhardt H, Blanpain C, Marine JC, Mouse Cutaneous Melanoma Induced by Mutant BRaf Arises from Expansion and Dedifferentiation of Mature Pigmented Melanocytes. Cell stem cell 21, 679–693.e676 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Moon H, Donahue LR, Choi E, Scumpia PO, Lowry WE, Grenier JK, Zhu J, White AC, Melanocyte Stem Cell Activation and Translocation Initiate Cutaneous Melanoma in Response to UV Exposure. Cell stem cell 21, 665–678.e666 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun Q, Lee W, Mohri Y, Takeo M, Lim CH, Xu X, Myung P, Atit RP, Taketo MM, Moubarak RS, Schober M, Osman I, Gay DL, Saur D, Nishimura EK, Ito M, A novel mouse model demonstrates that oncogenic melanocyte stem cells engender melanoma resembling human disease. Nature communications 10, 5023 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berghmans S, Murphey RD, Wienholds E, Neuberg D, Kutok JL, Fletcher CD, Morris JP, Liu TX, Schulte-Merker S, Kanki JP, Plasterk R, Zon LI, Look AT, tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proceedings of the National Academy of Sciences of the United States of America 102, 407–412 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ceol CJ, Houvras Y, Jane-Valbuena J, Bilodeau S, Orlando DA, Battisti V, Fritsch L, Lin WM, Hollmann TJ, Ferre F, Bourque C, Burke CJ, Turner L, Uong A, Johnson LA, Beroukhim R, Mermel CH, Loda M, Ait-Si-Ali S, Garraway LA, Young RA, Zon LI, The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 471, 513–517 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.D’Agati G, Beltre R, Sessa A, Burger A, Zhou Y, Mosimann C, White RM, A defect in the mitochondrial protein Mpv17 underlies the transparent casper zebrafish. Developmental biology 430, 11–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dutton JR, Antonellis A, Carney TJ, Rodrigues FS, Pavan WJ, Ward A, Kelsh RN, An evolutionarily conserved intronic region controls the spatiotemporal expression of the transcription factor Sox10. BMC Dev Biol 8, 105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patton EE, Widlund HR, Kutok JL, Kopani KR, Amatruda JF, Murphey RD, Berghmans S, Mayhall EA, Traver D, Fletcher CD, Aster JC, Granter SR, Look AT, Lee C, Fisher DE, Zon LI, BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Current biology : CB 15, 249–254 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Zou J, Beermann F, Wang J, Kawakami K, Wei X, The Fugu tyrp1 promoter directs specific GFP expression in zebrafish: tools to study the RPE and the neural crest-derived melanophores. Pigment Cell Res 19, 615–627 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perez AR, Pritykin Y, Vidigal JA, Chhangawala S, Zamparo L, Leslie CS, Ventura A, GuideScan software for improved single and paired CRISPR guide RNA design. Nature biotechnology 35, 347–349 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Labun K, Montague TG, Krause M, Torres Cleuren YN, Tjeldnes H, Valen E, CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic acids research 47, W171–w174 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L, Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nature biotechnology 27, 275–280 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee G, Chambers SM, Tomishima MJ, Studer L, Derivation of neural crest cells from human pluripotent stem cells. Nature protocols 5, 688–701 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Zhu Z, Gonzalez F, Huangfu D, The iCRISPR platform for rapid genome editing in human pluripotent stem cells. Methods in enzymology 546, 215–250 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haeussler M, Schonig K, Eckert H, Eschstruth A, Mianne J, Renaud JB, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, Joly JS, Concordet JP, Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome biology 17, 148 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eisenberg E, Levanon EY, Human housekeeping genes, revisited. Trends in genetics : TIG 29, 569–574 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Sanjana NE, Shalem O, Zhang F, Improved vectors and genome-wide libraries for CRISPR screening. Nature methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bolger AM, Lohse M, Usadel B, Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics (Oxford, England) 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England) 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DeLuca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire MD, Williams C, Reich M, Winckler W, Getz G, RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics (Oxford, England) 28, 1530–1532 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xiong Q, Ancona N, Hauser ER, Mukherjee S, Furey TS, Integrating genetic and gene expression evidence into genome-wide association analysis of gene sets. Genome research 22, 386–397 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiong Q, Mukherjee S, Furey TS, GSAASeqSP: a toolset for gene set association analysis of RNA-Seq data. Scientific reports 4, 6347 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hu Y, Flockhart I, Vinayagam A, Bergwitz C, Berger B, Perrimon N, Mohr SE, An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC bioinformatics 12, 357 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ, Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Skene PJ, Henikoff JG, Henikoff S, Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nature protocols 13, 1006–1019 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM, The Cancer Genome Atlas Pan-Cancer analysis project. Nature genetics 45, 1113–1120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, Omberg L, Wolf DM, Shriver CD, Thorsson V, Hu H, An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 173, 400–416.e411 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I, Ceccarelli M, Bontempi G, Noushmehr H, TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic acids research 44, e71 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Silva TC, Colaprico A, Olsen C, D’Angelo F, Bontempi G, Ceccarelli M, Noushmehr H, TCGA Workflow: Analyze cancer genomics and epigenomics data using Bioconductor packages. F1000Research 5, 1542 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Risso D, Schwartz K, Sherlock G, Dudoit S, GC-content normalization for RNA-Seq data. BMC bioinformatics 12, 480 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets deposited at GEO (RNA-seq, ATAC-seq, Cut&Run): GSE172069.
All zebrafish lines are available upon request from the authors or via the ZIRC zebrafish stock center (https://zebrafish.org/home/guide.php). WA-09 lines (NC, MB, MC, 3xKO NC, 3xKO MB, 3xKO MC) will be made available upon request from the Studer lab at Memorial Sloan Kettering Cancer Center under a Material Transfer Agreement with the institute.