

Abstract
Purpose
Janus kinase 1 (JAK1) is implicated in multiple inflammatory pathways that are critical for the pathogenesis of asthma, including the interleukin (IL)-4, IL-5, IL-13, and thymic stromal lymphopoietin cytokine signaling pathways, which have previously been targeted to treat allergic asthma. Here, we describe the development of AZD0449 and AZD4604, two novel and highly selective JAK1 inhibitors with promising properties for inhalation.
Methods
The effects of AZD0449 and AZD4604 in JAK1 signaling pathways were assessed by measuring phosphorylation of signal transducer and activator of transcription (STAT) proteins and chemokine release using immunoassays of whole blood from healthy human volunteers and rats. Pharmacokinetic studies performed on rats evaluated AZD0449 at a lung deposited dose of 52 μg/kg and AZD4604 at 30 µg/kg. The efficacy of AZD0449 and AZD4604 was assessed by evaluating lung inflammation (cell count and cytokine levels) and the late asthmatic response (average enhanced pause [Penh]).
Results
Both compounds inhibited JAK1-dependent cytokine signaling pathways in a dose-dependent manner in human and rat leukocytes. After intratracheal administration in rats, both compounds exhibited low systemic exposures and medium-to-long terminal lung half-lives (AZD0449, 34 hours; AZD4604, 5 hours). Both compounds inhibited STAT3 and STAT5 phosphorylation in lung tissue from ovalbumin (OVA)-challenged rats. AZD0449 and AZD4604 also inhibited eosinophilia in the lung and reduced the late asthmatic response, measured as Penh in the OVA rat model.
Conclusion
AZD0449 and AZD4604 show potential as inhibitors of signaling pathways involved in asthmatic immune responses, with target engagement demonstrated locally in the lung. These findings support the clinical development of AZD0449 and AZD4604 for the treatment of patients with asthma.
Keywords: AZD0449, AZD4604, JAK, STAT
Introduction
Asthma is one of the most common chronic inflammatory diseases worldwide.1,2 Inhaled corticosteroids and β2-adrenergic agonists, the mainstay of treatment, are effective in improving lung function and reducing exacerbations for most individuals with asthma.3 Despite the availability of these therapies, asthma symptoms are not fully controlled in all patients.4–6 Additionally, prolonged use of oral corticosteroid therapy can be associated with side effects, such as adrenal insufficiency, growth delay in children, and osteoporosis and type 2 diabetes in adults.7 Collectively, the data suggest that more effective treatment strategies are required for asthma.
Asthma is a heterogeneous disease with multiple distinct phenotypes, some of which are associated with a lack of response to corticosteroid therapy.8,9 Allergic, or atopic, asthma is the best characterized form of asthma and is commonly driven by type 2 immune responses, typically associated with eosinophil-dominated inflammation and driven by the type 2 cytokines interleukin (IL)-4, IL-5, IL-13, and thymic stromal lymphopoietin (TSLP).10,11 A significant amount of research has focused on targeting type 2 cytokines for a range of investigational and marketed drugs for asthma.12–16 Cytokines such as IL-6 and type I and type II interferons (IFNs) may also be involved in the pathogenesis of some asthma phenotypes.17,18 Cytokines involved in the pathogenesis of asthma exert pleiotropic actions with functional overlap in different immune cell subsets,19 suggesting that targeting one cytokine alone is unlikely to be sufficient for effective asthma treatment.
Cytokines released from stimulated T helper (Th) 1 and Th2 cells bind to their receptors and activate Janus kinases (JAKs). In mammals, there are four JAKs (JAK1, JAK2, JAK3, and tyrosine kinase 2 [TYK2]) and seven signal transducer and activator of transcription (STAT) proteins (STAT1–4, STAT5a, STAT5b, and STAT6). Each type I/II cytokine receptor is associated with two specific JAKs that phosphorylate a specific STAT protein, leading to its dimerization and activation, and, ultimately, to pro-inflammatory downstream transcriptional effects.20 In the context of asthma, IL-4 can signal through two alternative receptor complexes: an IL-4 receptor α (IL-4Rα)/IL-2 common γ-chain receptor complex, which recruits JAK1 and JAK3, or an IL-4Rα/IL-13Rα receptor complex, which recruits JAK1 and JAK2 or TYK2.19,21 IL-13 is also able to signal through the IL-4Rα/IL-13Rα receptor complex.21 The IL-4 and IL-13 pathways both lead to the phosphorylation of STAT6.22 TSLP signals through a receptor complex comprising a TSLP receptor chain and the IL-7Rα chain, which recruits JAK1 and JAK2, leading to phosphorylation of STAT5.23 In dendritic cells, TSLP may also lead to the activation of STAT6.24 IL-6 signals through a receptor complex comprising a gp130 chain and an IL-6Rα chain, with JAK1 as the dominant kinase, and the additional ability to recruit JAK2 and TYK2, leading to phosphorylation of STAT3 and STAT1.25 Aberrant activation of these pathways drives the pathophysiological responses that lead to the characteristic symptoms of asthma including coughing, wheezing, and shortness of breath.26 JAK1 inhibition is therefore an attractive strategy for the treatment of asthma, with the potential to block the activity of multiple cytokines.
Many oral JAK inhibitors have been licensed for the treatment of inflammatory conditions such as rheumatoid arthritis.27 Their use for the treatment of asthma has been limited by potential side effects, including opportunistic infections and cytopenia.28 The use of selective JAK1 inhibitors may avoid the development of cytopenia, given it is associated with the inhibition of JAK2.29 However, systemic JAK1 inhibitor treatment is likely to cause significant immunosuppression and an increased risk of infection as well as herpes zoster reactivation.30 Local delivery of a selective JAK1 inhibitor to the lungs by inhalation may minimize the potential for systemic immunosuppression while providing maximal therapeutic effect in the inflamed lung tissue.
Several inhaled JAK inhibitors have been or are currently being investigated for the treatment of asthma in early Phase 1 clinical trials: GDC-0214/RG6151 (Genentech),31 GDC-4379/RG6244 (Genentech),32,33 TD-8236 (Theravance Biopharma),34,35 AZD0449 (AstraZeneca; NCT03766399),36 AZD4604 (NCT04769869),37 and KN-002 (Kinaset; NCT05006521).38
Among these compounds, GDC-0214 showed a twofold increased selectivity for JAK1 over JAK2 in biochemical assays,31 whereas both TD-8236 and KN-002 are pan-JAK inhibitors. No biochemical selectivity data have been published for GDC-4379 to date; nonetheless, a recent study termed GDC-4379 a JAK1 inhibitor.39 Successful reduction of exhaled nitric oxide levels has been reported for GDC-0214, GDC-4379, and TD-8236 in patients with mild asthma, and for TD-8236 in vitro;31,33–35 however, these compounds are no longer in active development according to the product pipelines of the individual organizations. Suppression of allergic asthma and neutrophil-driven inflammation has also been demonstrated with the JAK inhibitor iJak-381 (Genentech) administered via inhalation in rodent models of asthma, with no effect on systemic JAK1 activity.18 iJak-381 was reported to show a sixfold increased selectivity for JAK1 over JAK2 in biochemical assays, but there are no reports to date of its advancement into clinical trials.
Here, we describe the preclinical testing of two JAK1 inhibitors that are suitable for inhalation, AZD0449 and AZD4604. Both compounds were assessed for their selectivity for JAK1 over JAK2, target engagement, and efficacy in the reduction of inflammation and the late asthmatic response in an established rat model of allergic asthma. This model features allergic inflammation and the late asthmatic response, both of which are features of asthma in humans and endpoints for antigen provocation studies in patients with asthma.40
Materials and Methods
Animal Studies
Male, adult, brown Norway rats (body weight 250–300 g) were obtained from Charles River Laboratories (Sulzfeld, Germany). Animals were housed in temperature-controlled, individually ventilated cages in groups of four, with food and water provided ad libitum. Animals underwent acclimatization for at least 1 week before experimentation.
Phosphorylation of STAT Proteins and Chemokine Release in Primary Cells from Human or Rat Whole Blood
Whole blood was collected from healthy human volunteers in tubes containing anticoagulant for experiments with human primary cells. Similarly, blood was collected from treatment-naïve brown Norway rats for experiments with rat primary cells. Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using established protocols. Further enrichment for lymphocytes or monocytes was performed for some experiments. Cells were plated in 96-well plates and left to rest. The compound (AZD0449 or AZD4604) was serially diluted in dimethyl sulfoxide (DMSO; vehicle) and added to the cells; only vehicle was added to control wells. After preincubation with compound or vehicle, cells were stimulated with cytokines (recombinant IL-2, IL-4, IL-12, IL-13, IFNα, or TSLP) to induce signaling via JAK1 to various STATs. For the detection of STAT phosphorylation, stimulation was stopped by the addition of formaldehyde-containing fixation buffer and further permeabilized to allow intracellular staining of phosphorylated STATs (STAT1, STAT4, STAT5, and STAT6) relevant for each cytokine using fluorescence-labeled antibodies. Stimulated control samples were stained with the respective isotype control antibodies to define phospho-STAT gates. In some experiments, cell surface markers were stained to discriminate between specific cell types. Cells expressing stained surface markers and the proportion of cells with phosphorylated STAT were quantified by flow cytometry. For the detection of chemokine release, cytokine stimulation was stopped by a short centrifugation to collect cell supernatants. The concentration of the released C-C motif chemokine ligand 17 (CCL17)/thymus- and activation-regulated chemokine was quantified by enzyme-linked immunosorbent assay (ELISA) or MSD U-plex immunoassay (Meso Scale Diagnostics, LLC, Rockville, MD, USA). The compound effect was calculated as the reduction in the percentage of cells containing the specific phospho-STAT or the level of CCL17/thymus- and activation-regulated chemokine in compound-treated cells compared with vehicle-treated, stimulated cells. Further details can be found in the Supplementary Methods.
Pharmacokinetics
Plasma and lung pharmacokinetic profiles up to 24 hours after a single intratracheal (IT) dose were assessed in treatment-naïve rats. AZD0449 was evaluated at a lung deposited dose (LDD) of 52 μg/kg and AZD4604 at 30 µg/kg. Plasma and lung samples were taken at 0.03, 0.5, 2, 6, 8, and 24 hours after dosing from three animals per time point. Terminal concentrations (3 hours and 25 hours after dosing) of each compound in lung and plasma samples from ovalbumin (OVA)-challenged rats were determined after escalating IT doses of AZD0449 (LDD of 100, 300, 1000, or 3000 µg/kg) and AZD4604 (LDD of 100, 300, or 1000 µg/kg).
Physiologically Based Pharmacokinetic Modeling
A whole-body rat-specific physiologically based pharmacokinetic (PBPK) model, which places emphasis on pulmonary drug disposition, was implemented in MATLAB R2013a (MathWorks Inc., Natick, MA, USA).41,42 The lung was divided into 24 airway generations and one extrathoracic region according to the morphometry presented by Lee et al.43 The structural model is illustrated in Supplementary Figure 1, in which airway generation 1 refers to the trachea. Details regarding calculations of regional surface areas, mucociliary clearance, and volumes of epithelial lining fluid (ELF), epithelium, and sub-epithelium are provided by Boger et al.41,42
Each airway generation is divided into three compartments: (1) ELF, (2) epithelium, and (3) sub-epithelium (Supplementary Figure 1). Airway generations belong to either the tracheobronchial region (generations 1–16) or the alveolar region (generations 17–24). Drug particle dissolution (if applicable) in the ELF is modeled by the Nernst–Brunner equation.44,45 In addition, the model assumed the absence of non-specific binding in the ELF compartment. Perfusion rate-limited distribution was assumed to apply for all tissues in the PBPK model. For both AZD0449 and AZD4604, an additional tissue-binding compartment was introduced in each epithelium and sub-epithelium compartment, with model optimized values for kin and kout representing the distribution in and out of a volume-less “deep” compartment. The permeability in the alveolar region was set at 10 times the value for the bronchiolar region. The tracheobronchial region was perfused by the bronchial blood flow and the alveolar region by the entire cardiac output. The local bronchial blood flow to the tracheobronchial generations was calculated according to Boger et al.41,42 Local blood flow to the alveolar generations was assumed to be constant in terms of flow per tissue volume. The deposition pattern following IT dosing is similar to the one reported by Codrons et al,46 with 70% and 30% calculated deposition in the tracheobronchial and alveolar regions, respectively. All ordinary differential equations used in the model were provided by Boger et al.41,42 The final optimized PBPK model parameter values are shown in Supplementary Table 1. Results of PBPK modeling can be found in Supplementary Figure 2.
Assessment of Plasma and Lung Concentrations of AZD0449 and AZD4604
Plasma and lung homogenate concentrations of AZD0449 and AZD4604 were determined by liquid chromatography–tandem mass spectrometry. Blood was spun down at 4°C (1400 × g) and plasma retained at −80°C until compound analysis. Excised post-caval lobes were used for lung compound analysis. Lung pieces were weighed and homogenized in 0.5 mL and 1 mL Ringer’s solution using bead-beating technology (Bertin Technologies, Montigny le Bretonneux, France). For quantification, 50 μL of sample (plasma or lung homogenate) was protein precipitated by the addition of 180 μL of acetonitrile containing 0.2% formic acid and 50 nM internal standard. After vortex mixing and centrifugation at 4000 × g for 20 minutes, supernatants were diluted 1:1 with 0.2% formic acid in water to match the initial mobile phase. The dilution step was accounted for when reporting AZD0449 and AZD4604 concentrations in lung tissue, normalizing for the lung sample weight and Ringer’s solution volume used for homogenization.
OVA-Induced Model of Asthma for Assessment of Efficacy with Respect to Inflammation and the Late Asthmatic Response
For efficacy studies, rats were sensitized on days 0, 14, and 21 by an intraperitoneal (IP) injection of 4 mL/kg (1 mL/rat) of OVA (100 µg/rat) in alum (20 mg/rat aluminum hydroxide and 20 mg/rat magnesium hydroxide) or were sham sensitized (saline in alum). On day 28, rats were challenged with 1% (w/v) aerosolized OVA in isotonic saline or sham challenged with isotonic saline for 30 minutes. On day 28, 1 hour before challenge, rats received IT doses of AZD0449 (100, 300, 1000, or 3000 µg/kg), AZD4604 (100, 300, or 1000 µg/kg), budesonide (3000 µg/kg), or vehicle under isoflurane anesthesia (4% v/v) at 1 mL/kg. The dose ranges used were selected based on a preliminary unpublished dose-ranging study.
Whole-Body Plethysmography—Assessment of Penh
At 30 minutes after OVA challenge, rats were placed unrestrained in perspex whole-body plethysmograph chambers connected to bias flow pumps (DSI, St. Paul, MN, USA), with access to food and water ad libitum. At 55 minutes after OVA challenge, a 5-minute baseline recording was taken; the average enhanced pause (Penh) was subsequently recorded at 10-minute intervals for a total duration of 5 hours and was used for analysis.
Termination and Sampling in the OVA Challenge Study
At 2 hours or 24 hours after OVA challenge, animals were terminally anesthetized using an IP injection of sodium pentobarbitone (200 mg/kg). Terminal whole blood was obtained via cardiac puncture with a heparinized syringe. Following blood collection, rats underwent tracheostomy and were cannulated. Rats underwent manual lavage (2 × 3 mL Roswell Park Memorial Institute [RPMI] medium, 30 seconds; approximately 70% of the volume was recovered), and bronchoalveolar lavage fluid (BALF) was collected in 15 mL centrifuge tubes.
Preparation of BALF Cytospin, Supernatant Collection, and Leukocyte Counts
Slides were prepared using 100 µL of neat BALF and centrifuged (63 × g, 5 minutes, room temperature, low acceleration; Cytospin 2, Shandon, Runcorn, UK). For total leukocyte counts, 800 µL of neat BALF was centrifuged (800 × g, 10 minutes, 4oC; Cytospin 2) and the supernatant was removed, collected, and stored (−80°C). The cell pellet was resuspended (200 µL, RPMI medium) and the total number of leukocytes was quantified using a Sysmex XP-300 automated cell counter (Sysmex Ltd, Milton Keynes, UK).
Lung Collection, Insufflation, Tissue Digest, and Cytospin Preparation
Lung collection and insufflation were performed as described for the assessment of plasma and lung concentrations. The left lobe was finely chopped using a McElwain tissue chopper (Campden Instruments Ltd, Loughborough, UK). In total, 300 mg of finely chopped tissue was weighed and added to 5 mL of RPMI medium plus 10% fetal calf serum (FCS; collagenase 1 mg/mL/DNAse 25 µg/mL), followed by gentle agitation in a water bath (60 minutes, 37°C). Samples were then filtered through a mesh sieve (70 µM Cell Strainer, Corning Inc., Corning, NY, USA) and centrifuged (800 × g, 10 minutes, 4°C). Supernatant was discarded and the cell pellets were washed (RPMI medium plus 10% FCS, 10 mL, ×2) before being resuspended (RPMI medium plus 10% FCS, 1 mL). The total number of leukocytes was measured using a Sysmex XP-300 automated cell counter (Sysmex Ltd). For cytospin preparation, samples were diluted 1:10 (RPMI medium plus 10% FCS) and slides were prepared using 100 µL of diluted sample and centrifugation (63 × g, 5 minutes, room temperature, low acceleration; Cytospin 2, Shandon).
Cytospin Staining and Differential Cell Counts
BALF and lung tissue cytospin slides were placed in an automated stainer (Hematek 2000, Siemens, Erlangen, Germany) and stained with a Modified Wright stain (Siemens). Differential cell counts based on morphological criteria were performed using a light microscope to determine the percentages of macrophages/monocytes, lymphocytes, neutrophils, and eosinophils. Total numbers of each distinct cell type were determined based on total leukocyte numbers.
Whole Protein Extraction and Bradford Assay
Post-caval lobes were powderized in liquid nitrogen to assess protein and nuclear protein levels. For whole protein extraction, 30 mg of tissue was gently homogenized in complete lysis buffer (tris lysis buffer, protease inhibitor solution, phosphatase inhibitors I and II; 10 minutes, 4°C; MSD, Kenilworth, NJ, USA). Samples were centrifuged (15,000 × g, 20 minutes, 4°C). Supernatant (5 µL, 1:50 dilution) was taken for the Bradford assay, which was run according to the manufacturer’s instructions (BioRad, Hercules, CA, USA).
Nuclear Tissue Extraction
Nuclear fractionation of whole lung tissue was performed using a nuclear extraction kit according to the manufacturer’s instructions (Nuclear Extract Kit, Active Motif, Carlsbad, CA, USA).
pSTAT3, pSTAT5a, pSTAT5b, and Protein Kinase B Analysis in Lung Tissue
Protein (10 µg) from the lung tissue extract of samples taken 2 hours after OVA challenge was used in pSTAT3, pSTAT5a, pSTAT5b, and protein kinase B (pAkt) (T308) (Phospho-STAT Panel Kit, Total STAT3 Kit, Total STAT5a, b Kit) MSD assay plates (Meso Scale Diagnostics, LLC), and the assays were run according to the manufacturer’s instructions. Arbitrary electrochemiluminescence signals were read, the blank was subtracted, and values were determined in each plate using an MSD Sector S600 plate reader (Meso Scale Diagnostics).
Nuclear Factor Kappa B p65 Analysis in Lung Tissue
Nuclear tissue extract (10 µg) of samples taken 2 hours after OVA challenge was used in nuclear factor kappa B (NF-κB) p65 TransAm Activation assays (Active Motif) run according to the manufacturer’s instructions. Optical density was determined spectrophotometrically, the blank was subtracted, and values were determined (450 nm; SpectraMax iD5, Molecular Devices, San Jose, CA, USA).
Immunohistochemistry
For immunohistochemistry, formalin-fixed tissues were embedded in paraffin and cut into 4 μm sections. Sections were dewaxed in xylene and rehydrated with decreasing concentrations of alcohol. After rinsing in water, the slides were heat induced for antigen retrieval (24 minutes in a microwave oven, 98–110°C). Samples were then cooled in water for 5 minutes and subjected to endogenous peroxidase block with 1% H2O2 in phosphate-buffered saline (PBS) without Ca2+/Mg2+ for 30 minutes at room temperature, then rinsed with PBS. Endogenous biotin activity was blocked using the Avidin and Biotin Blocking Kit (Vector Laboratories, Burlingame, CA, USA). Slides were then blocked in PBS with 5% goat serum, 1% BSA, and 0.05% Tween, and incubated with the primary antibody (Tyr705, GTX61820, 1:1000; GeneTex, Irvine, CA, USA) overnight at 4°C. After washing with PBS, slides were incubated with the biotinylated secondary antibody (biotinylated anti-rabbit IgG, BA-1000, Vector Laboratories; 1:300) for 1 hour at room temperature. Staining was visualized with the Vectastain ABC Kit (Vector Laboratories) and NovaRed solution (SK-4800, Vector Laboratories). Slides were rinsed with water and counterstained with hematoxylin. An Aperio ScanScope XT (Leica Biosystems, Wetzlar, Germany) was used for imaging. The digital files were analyzed using Visiopharm software (Visiopharm, Hørsholm, Denmark, version 2020.03.0.7330) to identify the large airways, blood vessels, and alveolar epithelium. Furthermore, pSTAT3-positive and -negative cells were quantified with Visiopharm software in the specific region of interest (endothelium, bronchial epithelium, or alveolar epithelium).
Statistics
Statistical analysis of all data from the in vivo studies was performed using log-transformed data and GraphPad Prism (version 8.4.2, GraphPad, San Diego, CA, USA). The treatment window (saline versus the OVA-challenged, vehicle-treated group) was analyzed using an unpaired t-test with Welch’s correction. Analysis of variance (ANOVA) multiple comparisons with Dunnett’s post hoc test were performed to analyze significant effects of treatment with AZD0449 and AZD4604 in the OVA-challenged groups; different levels of significance versus the OVA-challenged, vehicle-treated group were assessed (P < 0.05, P < 0.01, P < 0.001, and P < 0.0001). The efficacy of the model reference compound budesonide (OVA-challenged, vehicle-treated versus budesonide-treated group) was analyzed using an unpaired t-test with Welch’s correction.
Study Approval
Peripheral blood was collected by venous puncture from healthy individuals from Gothenburg and Uppsala, Sweden, after written informed consent had been obtained and in accordance with ethical principles with their origin in the Declaration of Helsinki, consistent with International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use/Good Clinical Practice and applicable regulatory requirements as well as the AstraZeneca policy on Bioethics and Human Biological Samples. The study was approved by the ethical review board/committee in Gothenburg and Uppsala with approval numbers 033–10 and 2009/013, respectively.
Animal handling conformed to standards established by the Council of Europe ETS123 AppA, the Helsinki Convention for the Use and Care of Animals, Swedish legislation, and AstraZeneca global internal standards. Experiments were performed in accordance with the UK Home Office guidelines for animal welfare based on the Animals (Scientific Procedures) Act of 1986 and the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines. All rat experiments were approved by the Gothenburg Ethics Committee for Experimental Animals in Sweden and conformed to Directive 2010/63/EU, ethical license no. 135–2014.
Results
Physicochemical Properties of AZD0449 and AZD4604
Our approach centered on the need to design compounds that have long lung tissue retention and limited systemic exposure. We therefore assessed the physicochemical properties of AZD0449 and AZD4604. The results indicated that each molecule had different mechanisms for lung retention (Table 1). Retention was predicted to be driven by a slow dissolution rate for AZD0449 based on measured solubilities, and through tissue binding for AZD4604 owing to its basic moiety (pKa of 7.9) and lipophilicity (logD of 2.6), which was corroborated by its in vivo steady state volume of distribution.47 The synthesis of AZD0449 and AZD4604 is described in the Supplementary Methods and Supplementary Figures 3–7. AZD0449 was identified through cell-based screening assays, as previously described,48 and AZD4604 was identified in a lead optimization campaign, details of which will be published elsewhere.
Table 1.
Physical and Biochemical Properties of AZD0449 and AZD4604
| Property | AZD0449 | AZD4604 |
|---|---|---|
| Chemical structure | 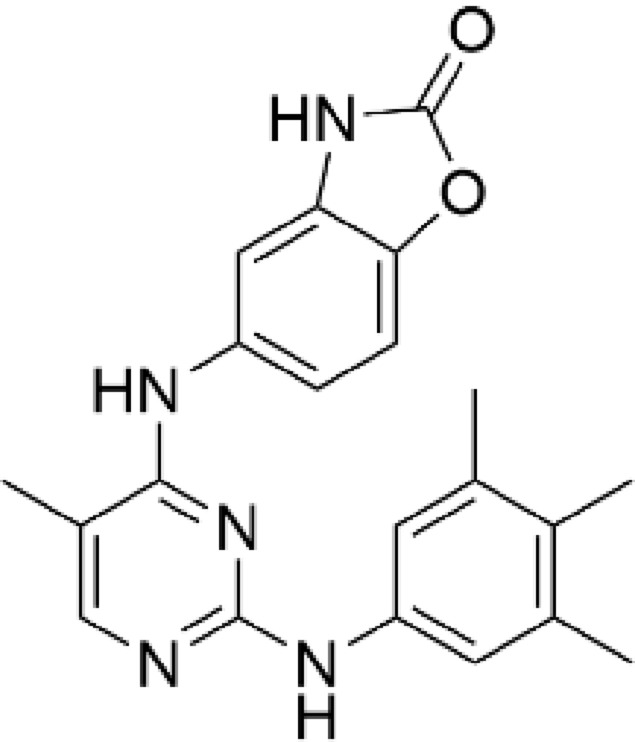 |
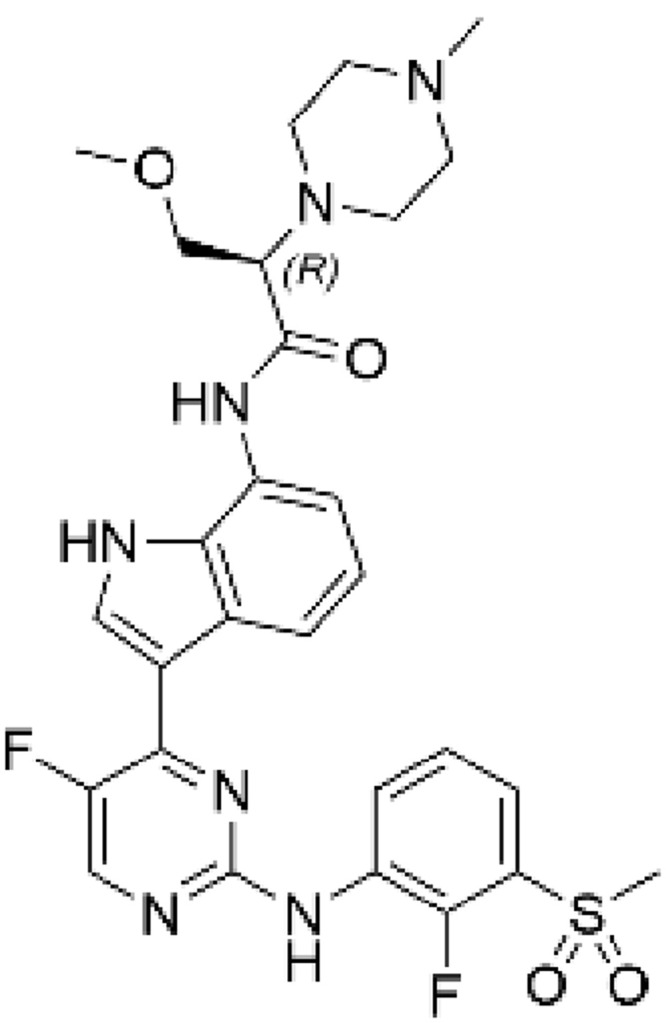 |
| IC50, 1 mM ATP, nMa | ||
| JAK1 | 2.4 | 0.54 |
| JAK2 | 413 | 686 |
| JAK3 | >10,000 | >10,000 |
| TYK2 | 103 | 657 |
| Molecular weight, g/mol | 375.4 | 599.7 |
| LogD, pH 7.4 | 4.2 | 2.6 |
| Basic pKa, measured | 5.9 | 7.9 |
| Thermodynamic solubility, pH 7.4, µM | 0.03 | 6.6 |
| Kinetic solubility, pH 6.8, µM | 1 (hemifumarate salt) | 18 (xinafoate salt) |
| Thermoanalysis, melting endotherm, °C | 355 | 188 |
| Caco-2 permeability, pH 6.5 (Papp A:B), ×10−6 cm/second | 11.2 | 6.7 |
| In vitro rat plasma protein binding, % free | 0.24 | 10.4 |
| In vivo plasma PK after IV dosingb | ||
| Rat plasma clearance, mL/minute/kg | 18 | 33 |
| Vss, L/kg | 1.3 | 9.9 |
| Terminal t½, hours | 3.9 | 4.2 |
Notes: aResults performed in triplicate; bSingle IV bolus dose of AZD0449 (1.0 mg/kg) and AZD4604 (0.5 mg/kg) (n=2).
Abbreviations: ATP, adenosine triphosphate; IC50, half maximal inhibitory concentration; IV, intravenous; JAK, Janus kinase; LogD, distribution coefficient; Papp, permeability coefficient; PK, pharmacokinetics; TYK2, tyrosine kinase 2; Vss, steady state volume of distribution.
Pharmacokinetics of AZD0449 and AZD4604 After Intravenous and Intratracheal Administration
After single intravenous bolus administration of AZD0449 1 mg/kg and AZD4604 0.5 mg/kg in rats, plasma clearance was 18 mL/minute/kg and 33 mL/minute/kg, respectively. The terminal half-life was 3.9 hours for AZD0449 and 4.2 hours for AZD4604, and the steady state volume of distribution was 1.3 L/kg for AZD0449 and 9.9 L/kg for AZD4604 (Table 1). Pharmacokinetics of the inhibitors were also investigated after IT administration in rats. Target lung-deposited doses of AZD0449 and AZD4604 at 52 µg/kg and 30 µg/kg, respectively, were given to naïve rats via single IT administration. The results of this investigation demonstrate that total drug concentrations in lung tissue declined slowly after dosing (Figure 1). Similar results were observed in OVA-challenged rats following increasing IT doses of the compounds, with high total lung concentrations at 3 hours and 25 hours after dosing (Supplementary Figure 8) at biological-effect doses in the herein reported rat efficacy study. In contrast, plasma concentrations were substantially lower. The terminal half-life of AZD0449 was 34 hours in the lung and 29 hours in plasma, and the terminal half-life of AZD4604 was approximately 5 hours in both the lung and plasma. Assuming 100% lung deposition following IT dosing, the proportion of the dose remaining in whole lung tissue 24 hours after administration was 23% for AZD0449 and 0.3% for AZD4604. PBPK modeling indicated that a slow dissolution rate was the main driver for lung retention of AZD0449 and that tissue binding was the main driver for AZD4604, as predicted from their physicochemical properties (Table 1, Supplementary Figures 2 and 8).
Figure 1.

Total lung and plasma concentrations after a single intratracheal dose of (A) AZD0449 52 µg/kg and (B) AZD4604 30 µg/kg in treatment-naïve rats. Data are shown as mean (n=3/time point).
High Selectivity for JAK1 Over Other Kinases in Enzymatic Assays
The in vitro half maximal inhibitory concentrations (IC50s) of AZD0449 and AZD4604 for JAK1 inhibition were 2.4 nM and 0.54 nM, respectively, with 50–1000-fold greater selectivity for JAK1 over JAK2, JAK3, and TYK2 (Table 1). To assess in vitro selectivity further, over 300 kinases were screened using a single concentration of 0.1 µM of AZD0449 or AZD4604 (Supplementary Figure 9A). Kinases that showed a 70% or greater inhibition in the single concentration measurements were prioritized for further assessment. The in vitro IC50 at the physiological adenosine triphosphate concentration was quantified for eight kinases for AZD0449 and 12 kinases for AZD4604. AZD0449 and AZD4604 were 10- and 100-fold more selective for JAK1, respectively, than for any other kinase tested (Supplementary Figure 9B).
Inhibition of Multiple JAK1-Dependent Cytokine Signaling Pathways in Whole Blood-Derived Cells
The ability of AZD0449 and AZD4604 to inhibit JAK1-dependent cytokine signaling in activated human lymphocytes was assessed using immunofluorescent labeling and flow cytometry (Figure 2, Supplementary Figure 10). AZD0449 and AZD4604 inhibited JAK1-dependent signaling, determined by reductions in IL-4-induced STAT6 phosphorylation in human CD4+ T cells, with IC50s of 21.4 nM and 16.9 nM, respectively (Figure 2A). The potency was similar to that of the inhibition of IL-2-induced JAK1 phosphorylation of STAT5 in both human and rat T cells, demonstrating that in vivo rodent target engagement studies would be possible and relevant (Figure 2B). Confirming their selectivity for JAK1, the IC50s for AZD0449 and AZD4604 were 23.47- and 81.7-fold higher, respectively, for the non-JAK1-dependent cytokine signaling pathway than for JAK1-dependent pathways. This was assessed via reductions in IL-12-induced STAT4 phosphorylation in a mixed population of human CD3+ T cells and CD56+ natural killer cells (Figure 2C, Table 2).
Figure 2.

Dose–response curves of AZD0449 and AZD4604 showing mean (A) inhibition of IL-4-induced phosphorylation of STAT6 in human T cells from PBMCs, (B) inhibition of IL-2-induced phosphorylation of STAT5 in human and rat T cells from PBMCs, and (C) inhibition of IFNα- and IL-12-induced phosphorylation of STAT1 and STAT4 in human T cells and NK cells expanded from PBMCs. All assessments were made by intracellular immunofluorescent labeling and flow cytometry. In (B), owing to the discontinuation of the rat pSTAT6 antibody, pSTAT5 was evaluated as a readout. In (C), pSTAT1 inhibition was determined in human cells gated as monocytes and pSTAT4 was determined in human cells gated as T cells or NK cells. Error bars represent SD. IC50s were not adjusted for protein binding.
Abbreviations: IFN, interferon; IL, interleukin; n, number of biological replicates; NK, natural killer; pIC50, phosphorylated half maximal inhibitory concentration; PBMC, peripheral blood mononuclear cell; pSTAT, phosphorylated signal transducer and activator of transcription; SD, standard deviation.
Table 2.
Summary of IC50s of AZD0449 and AZD4604 in Human and Rat Leukocytes
| Cell Type | Cytokine | STAT/Chemokine | AZD0449 IC50 (nM) | AZD4604 IC50 (nM) |
|---|---|---|---|---|
| Human T cells or monocytes | IFNα | STAT1 | 17.0 | 1.2 |
| Human CD3+ T cells and CD56+ NK cells | IL-12 | STAT4 | 399.0 | 98.0 |
| Human CD4+ T cells | IL-4 | STAT6 | 21.4 | 16.9 |
| Human T cells | IL-2 | STAT5 | 17.8 | 21.0 |
| Rat CD4+ T cells | IL-2 | STAT5 | 17.7 | 28.0 |
| Human CD14+ monocytes | IL-13 | STAT6 | 51.7 | 23.1 |
| Human CD14+ monocytes | TSLP | CCL17 | 81.7 | 57.0 |
Abbreviations: IC50, half maximal inhibitory concentration; IFN, interferon; IL, interleukin; NK, natural killer; STAT, signal transducer and activator of transcription; TSLP, thymic stromal lymphopoietin.
In human CD14+ monocytes, AZD0449 and AZD4604 inhibited IL-13-induced phosphorylation of STAT6- and TSLP-induced production of CCL17, confirming the inhibition of multiple JAK1 pathways involved in asthma pathogenesis (Table 2, Supplementary Figure 10). As expected, AZD0449 and AZD4604 both potently inhibited IFNα-induced JAK1-dependent phosphorylation of STAT1 in human T cells (Figure 2C).
Inhibition of JAK/STAT Signaling in Lung Tissue in a Rat Model of Asthma
Target engagement after the IT administration of AZD0449 and AZD4604 was evaluated in the OVA-driven rodent model of allergic asthma.49 First, we determined a time point at which there was a robust signal of OVA-stimulated STAT3, STAT5a, and STAT5b activation in lung tissue. STAT3 and STAT5 levels were significantly increased in whole lung homogenates at 2 hours after OVA challenge (OVA/vehicle) compared with sensitized, non-challenged rats (saline/vehicle) (Supplementary Figure 11). Subsequent target engagement analyses were then conducted at this time point. In this model, IT administration of AZD0449 and AZD4604 dose-dependently inhibited STAT3, STAT5a, and STAT5b phosphorylation in whole lung homogenates of OVA-challenged rats compared with vehicle-treated rats (OVA/vehicle) (Figure 3). pSTAT immunofluorescent staining was used as a marker for further evaluation of the specific localization of AZD0449 and AZD4604 target engagement in lung tissue. The proportion of phosphorylated STAT3-positive cell nuclei in immunohistological sections of the endothelium and bronchial epithelium with AZD4604 was significantly reduced compared with that in vehicle-treated rats (OVA/AZD4604 1 mg/kg versus OVA/vehicle, P < 0.01) (Figure 4). AZD4604 also significantly reduced the proportion of phosphorylated STAT3-positive cell nuclei in the alveolar epithelium compared with vehicle-treated rats (OVA/AZD4604 1 mg/kg versus OVA/vehicle, P < 0.05) (Supplementary Figure 12). Additionally, to determine how inhibition of the JAK pathway may affect other inflammatory pathways known to be associated with the allergic response, NF-κB and pAkt – markers of phosphoinositide 3-kinase [PI3K] activation – were examined. AZD0449 and AZD4604 had no significant effect on NF-κB or pAkt in the OVA rat model (Supplementary Figure 13).
Figure 3.

Inhibition of (A) STAT3 phosphorylation and (B) STAT5 phosphorylation in whole lung homogenate from OVA-challenged rats dosed intratracheally with AZD0449 and AZD4604. Data are shown as bar graphs with mean (n=5–7/group); error bars represent SEM. # denotes statistical significance between saline and OVA-challenged, vehicle-treated rats (P < 0.0001, unpaired t-test with Welch’s correction). Asterisks denote significant differences (*P < 0.05, **P < 0.01, and ***P < 0.001; ANOVA multiple comparisons with Dunnett’s post hoc test for AZD4604 and AZD0449, unpaired t-test with Welch’s correction for model reference budesonide) between OVA-challenged, compound-treated groups and the OVA-challenged, vehicle-treated group.
Abbreviations: ANOVA, analysis of variance; OVA, ovalbumin; pSTAT, phosphorylated signal transducer and activator of transcription; SEM, standard error of the mean.
Figure 4.

Inhibition of STAT3 activation by AZD0449 and AZD4604 (A) in OVA-stimulated lung tissue histology and (B) quantification of pSTAT3 from lung tissue histology in endothelium and bronchial epithelium. In (A), scale bars represent 50 µM. In (B), data are shown as bar graphs with mean (n=5–7/group); error bars represent SEM. # denotes statistical significance between saline and OVA-challenged, vehicle-treated rats (P < 0.0001, unpaired t-test with Welch’s correction). Asterisks denote significant differences (*P < 0.05 and **P < 0.01; ANOVA multiple comparisons with Dunnett’s post hoc test for AZD4604 and AZD0449, unpaired t-test with Welch’s correction for model reference budesonide) between OVA-challenged, compound-treated groups and the OVA-challenged, vehicle-treated group.
Abbreviations: ANOVA, analysis of variance; OVA, ovalbumin; pSTAT, phosphorylated signal transducer and activator of transcription; SEM, standard error of the mean.
AZD0449 and AZD4604 Inhibited Lung Inflammation and Reduced the Late Asthmatic Response
The effect of AZD0449 and AZD4604 on airway and lung inflammation, and Penh during the late asthmatic response, was assessed in the established OVA-driven rodent model of allergic asthma. Both endpoints are features of asthma in humans and are often used to profile new therapeutics.40 The anti-inflammatory potential of AZD0449 and AZD4604 was evaluated in BALF and lung tissue 24 hours after OVA challenge, which is an established time point for assessing eosinophilia in this model (Supplementary Figure 14). Penh was measured 1–6 hours after OVA challenge, representing the late asthmatic response.
Treatment with AZD0449 and AZD4604 numerically reduced eosinophil numbers in BALF in OVA-challenged rats compared with vehicle-treated rats (Figure 5A; OVA/AZD0449 and OVA/AZD4604 versus OVA/vehicle). AZD0449 and AZD4604 treatment also numerically lowered levels of lymphocytes, neutrophils, and macrophages in BALF, as well as the cytokines eotaxin and IL-1β, in OVA/AZD0449 and OVA/AZD4604 versus OVA/vehicle treated rats (Supplementary Figures 15 and 16). Eosinophil numbers in lung tissue were also reduced in OVA-challenged rats treated with AZD0449 and AZD4604 compared with vehicle-treated rats, to levels seen in saline-challenged animals (Figure 5B; OVA/AZD0449 and OVA/AZD4604 versus OVA/vehicle). Finally, IT administration of AZD0449 and AZD4604 indicated a reduction in Penh during the late asthmatic response; however, no significant differences were observed when compared with vehicle-treated rats (Figure 5C).
Figure 5.

Intratracheal dosing of AZD0449 and AZD4604 showing inhibition of lung inflammation in (A) BALF eosinophils and (B) lung eosinophils, and (C) reduction of the late asthmatic response. Data are shown as bar graphs with mean (n=7–10/group); error bars represent SEM. # denotes statistical significance between saline and OVA-challenged, vehicle-treated rats (P < 0.0001, unpaired t-test with Welch’s correction). Asterisks denote significant differences (*P < 0.05, **P < 0.01, and ***P < 0.001; ANOVA multiple comparisons with Dunnett’s post hoc test for AZD4604 and AZD0449, unpaired t-test with Welch’s correction for model reference budesonide) between OVA-challenged, compound-treated groups and the OVA-challenged, vehicle-treated group.
Abbreviations: ANOVA, analysis of variance; BALF, bronchoalveolar lavage fluid; OVA, ovalbumin; Penh (AUC), average enhanced pause area under the curve.
Discussion
JAK1-dependent signaling pathways are critically involved in the pathophysiology of asthma, making them an attractive therapeutic target. The results of this study confirmed that AZD0449 and AZD4604 are potent and selective inhibitors of JAK1, with properties allowing high lung concentrations when topically administered but with limited systemic exposure. Data in this study showed that, when using an OVA-induced rat model for allergic responses observed in patients with asthma, both AZD0449 and AZD4604 inhibited JAK1-dependent signaling. This was demonstrated by phosphorylation of STAT3, STAT5a, and STAT5b in lung tissue. In this rat model, a single dose of AZD0449 or AZD4604 was sufficient to affect the measured endpoints. This was consistent with a previously reported compound intervention study on the JAK inhibitor LAS194046 in an OVA rat model.50 Briefly, IT administration of AZD0449 and AZD4604 reduced the number of airway eosinophils and lowered Penh during the late asthmatic response in OVA-challenged rats compared with OVA-challenged vehicle-treated controls. Penh is a variable parameter to be measured. As such, capturing significant differences in the late asthmatic response is often challenging when using Penh as a surrogate measure. Hence, these findings further support the efficacy data from the OVA rat model and indicate the potential efficacy of AZD0449 and AZD4604 for the treatment of eosinophil-high allergic asthma. These findings are also in line with those from previous studies of systemically administered JAK inhibitors showing reduced inflammatory responses in the lungs of allergic mice.48,51
Corticosteroids have a broad anti-inflammatory effect on a wide range of immunological pathways, including NF-κB and PI3K signaling. AZD0449 and AZD4604 did not suppress signaling via NF-κB in the rodent model in this study. It was expected that the compounds may have an impact on PI3K signaling; however, results using the rodent model suggest that there is no effect on this pathway either. In the context of this model, the potent JAK1 specificity of AZD0449 and AZD4604 and functional redundancies in the PI3K pathway may explain the absence of an effect, but the clinical significance of this remains unknown. Additionally, recent models of PI3K/Akt signaling suggest that the pathway may be maintained in T cells independently of JAK1 signaling.52 Selective and local anti-inflammatory actions of inhaled JAK1 inhibitors may provide advantages over pan-JAK inhibitors, which can lead to global suppression of the immune system and side effects, including opportunistic infections and cytopenia, that would not be acceptable to patients with asthma.28,53
This study provides evidence that AZD0449 and AZD4604 inhibit multiple cytokine pathways involved in type 2-high asthma, as demonstrated by inhibition of STAT5 and STAT6 phosphorylation in human immune cells, and by the inhibition of STAT5 phosphorylation in rodent lung tissue. Inhibition of STAT5 signaling may also prevent IL-9- and IL-2-driven allergic asthma.54,55 In addition to the inhibition of type 2-high associated cytokine signaling pathways, the results also demonstrate that AZD0449 and AZD4604 dose-dependently inhibited signaling associated with non-type 2 asthma phenotypes, as shown by the decrease in STAT3 phosphorylation in the allergic rat model. A reduction in STAT3 and STAT1 phosphorylation may possibly, in turn, block Th17- and Th1-driven lung inflammation, respectively. Indeed, there is some evidence from other studies in OVA‑sensitized mice that inhibiting the JAK/STAT pathway may suppress Th1 and Th17 signaling.56,57 Our data do not rule out the possibility that AZD0449 and AZD4604 have an effect on non-type 2 asthma phenotypes, suggesting broad applicability for selective inhaled JAK1 inhibitors, but further studies are required to explore this potential.
Several monoclonal antibodies are available for the treatment of moderate-to-severe asthma, including the IL-4Rα agonist dupilumab, the immunoglobulin E agonist omalizumab, and the IL-5 agonists benralizumab, mepolizumab, and reslizumab.3,58 Given multiple cytokines are involved in asthma pathogenesis, often with functional redundancy, the inhibition of one receptor or cytokine may be insufficient to block downstream signaling cascades. In addition, the heterogeneity of asthma inflammation may result in some patients, especially those with eosinophilic-low phenotypes, being unsuitable for treatment with these biologics. However, a major advantage of these monoclonal antibodies is that they have a steroid-sparing effect, by reducing or eliminating the need for oral corticosteroids. This may limit the risk of steroid-related side effects such as adrenal suppression and delayed growth in children.7,58,59 Development of new small-molecule drugs with broader immunomodulatory effects and suitability for delivery by inhalation may be more effective than blocking the action of a single cytokine while still conferring corticosteroid-sparing advantages, and may potentially expand treatment options for patients with asthma.
Most JAK inhibitors approved to date are systemic drugs for the treatment of rheumatoid arthritis, such as tofacitinib and baricitinib (JAK1/2/3 and JAK1/2 inhibitors, respectively).60 Pan-JAK inhibitors are also in development for the treatment of asthma.34 Cytopenia and an increased risk of infection limit the use of these systemic, first-generation pan-JAK inhibitors; these effects are brought about through inhibition of the essential hemopoietic cytokines erythropoietin, thrombopoietin, and granulocyte colony-stimulating factor, which signal exclusively through JAK2.7,61–63 Compared with patients with rheumatoid arthritis or myelofibrosis, individuals with asthma are often younger, with mild or infrequent symptoms, and are otherwise healthy. The tolerance for medication-related adverse events is therefore very low in this population. Any novel treatment for asthma must have minimal side effects to be considered a viable option for daily preventative therapy. Inhaled administration of asthma therapies can lead to a localized response in the lung, with lower systemic exposure than orally administered medications.64 Crucially, in preclinical models, AZD0449 and AZD4604 have demonstrated both potent and specific JAK1 inhibition in the lung, with a limited systemic response predicted. Therefore, we anticipate that these inhaled JAK1 inhibitors may be less prone to side effects arising from systemic inhibition of cytokine signaling that are commonly seen with oral JAK inhibitors. Additionally, the long lung retention time of AZD0449 may allow once-daily dosing, which is convenient for patients. Further studies are needed to evaluate whether these compounds allow effective treatment of asthma without suppressing non-asthma-related immune responses.
Conclusion
We have developed two highly specific JAK1 inhibitors with low systemic exposure after IT administration. In contrast to conventional JAK inhibitors, AZD0449 and AZD4604 are predicted to be well tolerated and effective, both alone and in conjunction with conventional local and systemic therapies, in the treatment of severe asthma. Our findings were obtained within an acceptable dose range and showed a positive JAK1 target engagement and efficacy in an animal model, which may be relevant to clinically important endpoints. These observations support the decision to progress AZD0449 and AZD4604 to clinical trials.
Acknowledgments
The current affiliation for Suman Mitra: CANTHER: Cancer Heterogeneity, Plasticity, and Resistance to Therapies, CNRS, Inserm, Centre Hospitalier Universitaire de Lille, Lille, France. We thank Laura Drought, PhD, and Barbara Borda d’Agua, PhD, from PharmaGenesis London, London, UK, who provided medical writing support, which was funded by AstraZeneca. We thank John Steele for his previous project leadership. We also thank Petter Svanberg, Anna-Pia Palmgren, and Marie Brännström for their analysis of drug exposure levels. Lastly, we thank Peter Konings for all valuable input and guidance on statistical evaluations of all in vivo data.
Funding Statement
AstraZeneca funded this study and participated in study design, data collection, data analysis, data interpretation, and writing the study report. The Swedish Research Council funded Lars Rönnblom. AstraZeneca develops and markets treatments for asthma. AstraZeneca reviewed the publication, without influencing the opinions of the authors, to ensure medical and scientific accuracy and the protection of intellectual property. The corresponding author had access to all data in the study and had final responsibility for the decision to submit the manuscript for publication.
Data Sharing Statement
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis, and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
Magnus Nilsson, Magdalena Rhedin, Ramon Hendrickx, Susanne Berglund, Antonio Piras, Parmis Blomgran, Anders Cavallin, Mia Collins, Göran Dahl, Therese Ericsson, Ann Aurell Holmberg, Agnes Leffler, Anders J Lundqvist, Thomais Markou, James Pinkerton, Tiiu Wennberg, Dimitrios Zervas, Maria G Belvisi, Mark Birrell, and Annika Borde are employees of AstraZeneca and may own stock or stock options in AstraZeneca. Suman Mitra is a former employee of AstraZeneca and may own stock or stock options in AstraZeneca. Stacey Siu and Vanessa Taylor are employees of Rigel Pharmaceuticals. Magnus Nilsson is an inventor (AZD4604 in WO2018134213, “JAK1 selective inhibitors,” AstraZeneca AB). Lars Rönnblom is a medical advisor for AstraZeneca. Bilel Dekkak, Niklas Hagberg, and Arian D J Laurence have declared that no conflict of interest exists.
References
- 1.Global Asthma Network. The global asthma report. Available from: http://www.globalasthmareport.org/. Accessed July 16, 2021.
- 2.Holgate ST, Wenzel S, Postma DS, Weiss ST, Renz H, Sly PD. Asthma. Nat Rev Dis Primers. 2015;1(1):15025. doi: 10.1038/nrdp.2015.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Global Initiative for Asthma. Global strategy for asthma management and prevention. Available from: https://ginasthma.org/reports/. Accessed July 16, 2021.
- 4.Chen H, Gould MK, Blanc PD, et al. Asthma control, severity, and quality of life: quantifying the effect of uncontrolled disease. J Allergy Clin Immunol. 2007;120(2):396–402. doi: 10.1016/j.jaci.2007.04.040 [DOI] [PubMed] [Google Scholar]
- 5.Chen S, Golam S, Myers J, Bly C, Smolen H, Xu X. Systematic literature review of the clinical, humanistic, and economic burden associated with asthma uncontrolled by GINA Steps 4 or 5 treatment. Curr Med Res Opin. 2018;34(12):2075–2088. doi: 10.1080/03007995.2018.1505352 [DOI] [PubMed] [Google Scholar]
- 6.Peters SP, Ferguson G, Deniz Y, Reisner C. Uncontrolled asthma: a review of the prevalence, disease burden and options for treatment. Respir Med. 2006;100(7):1139–1151. doi: 10.1016/j.rmed.2006.03.031 [DOI] [PubMed] [Google Scholar]
- 7.Bourdin A, Adcock I, Berger P, et al. How can we minimise the use of regular oral corticosteroids in asthma? Eur Respir Rev. 2020;29(155):190085. doi: 10.1183/16000617.0085-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moore WC, Meyers DA, Wenzel SE, et al. Identification of asthma phenotypes using cluster analysis in the severe asthma research program. Am J Respir Crit Care Med. 2010;181(4):315–323. doi: 10.1164/rccm.200906-0896OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lefaudeux D, De Meulder B, Loza MJ, et al. U-BIOPRED clinical adult asthma clusters linked to a subset of sputum omics. J Allergy Clin Immunol. 2017;139(6):1797–1807. doi: 10.1016/j.jaci.2016.08.048 [DOI] [PubMed] [Google Scholar]
- 10.Sze E, Bhalla A, Nair P. Mechanisms and therapeutic strategies for non-T2 asthma. Allergy. 2020;75(2):311–325. doi: 10.1111/all.13985 [DOI] [PubMed] [Google Scholar]
- 11.Georas SN, Donohue P, Connolly M, Wechsler ME. JAK inhibitors for asthma. J Allergy Clin Immunol. 2021;148(4):953–963. doi: 10.1016/j.jaci.2021.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corren J, Lemanske RF, Hanania NA, et al. Lebrikizumab treatment in adults with asthma. N Engl J Med. 2011;365(12):1088–1098. doi: 10.1056/NEJMoa1106469 [DOI] [PubMed] [Google Scholar]
- 13.Corren J, Parnes JR, Wang L, et al. Tezepelumab in adults with uncontrolled asthma. N Engl J Med. 2017;377(10):936–946. doi: 10.1056/NEJMoa1704064 [DOI] [PubMed] [Google Scholar]
- 14.FitzGerald JM, Bleecker ER, Menzies-Gow A, et al. Predictors of enhanced response with benralizumab for patients with severe asthma: pooled analysis of the SIROCCO and CALIMA studies. Lancet Respir Med. 2018;6(1):51–64. doi: 10.1016/S2213-2600(17)30344-2 [DOI] [PubMed] [Google Scholar]
- 15.Pavord ID, Korn S, Howarth P, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet. 2012;380(9842):651–659. doi: 10.1016/S0140-6736(12)60988-X [DOI] [PubMed] [Google Scholar]
- 16.Wenzel S, Castro M, Corren J, et al. Dupilumab efficacy and safety in adults with uncontrolled persistent asthma despite use of medium-to-high-dose inhaled corticosteroids plus a long-acting β2 agonist: a randomised double-blind placebo-controlled pivotal phase 2b dose-ranging trial. Lancet. 2016;388(10039):31–44. doi: 10.1016/S0140-6736(16)30307-5 [DOI] [PubMed] [Google Scholar]
- 17.Hinks TSC, Levine SJ, Brusselle GG. Treatment options in type-2 low asthma. Eur Respir J. 2021;57(1):2000528. doi: 10.1183/13993003.00528-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dengler HS, Wu X, Peng I, et al. Lung-restricted inhibition of Janus Kinase 1 is effective in rodent models of asthma. Sci Transl Med. 2018;10(468):eaao2151. doi: 10.1126/scitranslmed.aao2151 [DOI] [PubMed] [Google Scholar]
- 19.LaPorte SL, Juo ZS, Vaclavikova J, et al. Molecular and structural basis of cytokine receptor pleiotropy in the interleukin-4/13 system. Cell. 2008;132(2):259–272. doi: 10.1016/j.cell.2007.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seif F, Khoshmirsafa M, Aazami H, Mohsenzadegan M, Sedighi G, Bahar M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun Signal. 2017;15(1):23. doi: 10.1186/s12964-017-0177-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. THE IL-4 RECEPTOR: signaling mechanisms and biologic functions. Annu Rev Immunol. 1999;17(1):701–738. doi: 10.1146/annurev.immunol.17.1.701 [DOI] [PubMed] [Google Scholar]
- 22.Zak M, Dengler HS, Rajapaksa NS. Inhaled Janus Kinase (JAK) inhibitors for the treatment of asthma. Bioorg Med Chem Lett. 2019;29(20):126658. doi: 10.1016/j.bmcl.2019.126658 [DOI] [PubMed] [Google Scholar]
- 23.Zhong J, Sharma J, Raju R, et al. TSLP signaling pathway map: a platform for analysis of TSLP-mediated signaling. Database (Oxford). 2014;2014:bau007. doi: 10.1093/database/bau007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arima K, Watanabe N, Hanabuchi S, Chang M, Sun SC, Liu YJ. Distinct signal codes generate dendritic cell functional plasticity. Sci Signal. 2010;3(105):ra4. doi: 10.1126/scisignal.2000567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morris R, Kershaw NJ, Babon JJ. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018;27(12):1984–2009. doi: 10.1002/pro.3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Athari SS. Targeting cell signaling in allergic asthma. Signal Transduct Target Ther. 2019;4:45. doi: 10.1038/s41392-019-0079-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. 2015;66(1):311–328. doi: 10.1146/annurev-med-051113-024537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harigai M. Growing evidence of the safety of JAK inhibitors in patients with rheumatoid arthritis. Rheumatology. 2019;58(Suppl 1):i34–i42. doi: 10.1093/rheumatology/key287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Virtanen AT, Haikarainen T, Raivola J, Silvennoinen O. Selective JAKinibs: prospects in inflammatory and autoimmune diseases. BioDrugs. 2019;33(1):15–32. doi: 10.1007/s40259-019-00333-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clarke B, Yates M, Adas M, Bechman K, Galloway J. The safety of JAK-1 inhibitors. Rheumatology. 2021;60(Suppl 2):ii24–ii30. doi: 10.1093/rheumatology/keaa895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Braithwaite IE, Cai F, Tom JA, et al. Inhaled JAK inhibitor GDC-0214 reduces exhaled nitric oxide in patients with mild asthma: a randomized, controlled, proof-of-activity trial. J Allergy Clin Immunol. 2021;148(3):783–789. doi: 10.1016/j.jaci.2021.02.042 [DOI] [PubMed] [Google Scholar]
- 32.Australian New Zealand Clinical Trials Registry. Phase I trial to evaluate the safety and tolerability of GDC-4379 in healthy volunteers and patients with mild asthma (ACTRN12619000227190). Available from: http://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=376919. Accessed July 16, 2021.
- 33.Chen H, Kunder R, Zou Y, Gugelmann H, et al. Inhaled JAK inhibitor GDC-4379 reduces FeNO in patients with mild asthma. Eur Respir J. 2021;58:65. [Google Scholar]
- 34.Kaufman E, Kleinschek M, Moran E, et al. TD-8236, a lung-selective inhaled pan-JAK inhibitor, inhibits gene expression related to severe asthma and exhaled nitric oxide (FeNO), in 3-D airway epithelium liquid interface (ALI) cultures derived from asthmatic donors [abstract]. J Allergy Clin Immunol. 2020;145(2):AB116. doi: 10.1016/j.jaci.2019.12.570 [DOI] [Google Scholar]
- 35.Theravance Biopharma. Theravance Biopharma reports positive results from phase 1 clinical trial of TD-8236, an investigational, lung-selective, inhaled pan-Janus kinase (JAK) inhibitor for inflammatory lung diseases. Available from: https://investor.theravance.com/news-releases/news-release-details/theravance-biopharma-reports-positive-results-phase-1-clinical-0. Accessed July 16, 2021.
- 36.ClinicalTrials.gov. A study in healthy volunteers and patients with mild asthma to investigate the safety, anti-inflammatory effect of inhaled AZD0449 (NCT03766399). Available from: https://clinicaltrials.gov/ct2/show/NCT03766399. Accessed May 11, 2022.
- 37.ClinicalTrials.gov. A clinical trial in healthy volunteers and patients with mild asthma to investigate a new medicine (AZD4604) for the treatment of asthma (NCT04769869). Available from: https://clinicaltrials.gov/ct2/show/NCT04769869. Accessed May 11, 2022.
- 38.ClinicalTrials.gov. Single and multiple ascending dose study of KN-002 (KN-002) (NCT05006521). Available from: https://clinicaltrials.gov/ct2/show/NCT05006521. Accessed May 11, 2022.
- 39.Stumpf A, Burkhard J, Xu D, et al. Efficient, protecting group free kilogram-scale synthesis of the JAK1 inhibitor GDC-4379. Org Process Res Dev. 2021;25(11):2537–2550. doi: 10.1021/acs.oprd.1c00302 [DOI] [Google Scholar]
- 40.Raemdonck K, de Alba J, Birrell MA, et al. A role for sensory nerves in the late asthmatic response. Thorax. 2012;67(1):19–25. doi: 10.1136/thoraxjnl-2011-200365 [DOI] [PubMed] [Google Scholar]
- 41.Boger E, Evans N, Chappell M, et al. Systems pharmacology approach for prediction of pulmonary and systemic pharmacokinetics and receptor occupancy of inhaled drugs. CPT Pharmacomet Syst Pharmacol. 2016;5(4):201–210. doi: 10.1002/psp4.12074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boger E, Fridén M. Physiologically based pharmacokinetic/pharmacodynamic modeling accurately predicts the better bronchodilatory effect of inhaled versus oral salbutamol dosage forms. J Aerosol Med Pulm Drug Deliv. 2019;32(1):1–12. doi: 10.1089/jamp.2017.1436 [DOI] [PubMed] [Google Scholar]
- 43.Lee D, Srirama PK, Wallis C, Wexler AS. Postnatal growth of tracheobronchial airways of Sprague-Dawley rats. J Anat. 2011;218(6):717–725. doi: 10.1111/j.1469-7580.2011.01372.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Noyes AA, Whitney WR. The rate of solution of solid substances in their own solutions. J Am Chem Soc. 1897;19(12):930–934. doi: 10.1021/ja02086a003 [DOI] [Google Scholar]
- 45.Nernst W. Theorie der reaktionsgeschwindigkeit in heterogenen systemen. Zeitschrift für Physikalische Chemie. 1904;47U(1):52–55. doi: 10.1515/zpch-1904-4704 [DOI] [Google Scholar]
- 46.Codrons V, Vanderbist F, Ucakar B, Préat V, Vanbever R. Impact of formulation and methods of pulmonary delivery on absorption of parathyroid hormone (1–34) from rat lungs. J Pharm Sci. 2004;93(5):1241–1252. doi: 10.1002/jps.20053 [DOI] [PubMed] [Google Scholar]
- 47.Cooper AE, Ferguson D, Grime K. Optimisation of DMPK by the inhaled route: challenges and approaches. Curr Drug Metab. 2012;13(4):457–473. doi: 10.2174/138920012800166571 [DOI] [PubMed] [Google Scholar]
- 48.Ashino S, Takeda K, Li H, et al. Janus Kinase 1/3 signaling pathways are key initiators of TH2 differentiation and lung allergic responses. J Allergy Clin Immunol. 2014;133(4):1162–1164. doi: 10.1016/j.jaci.2013.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hele DJ, Birrell MA, Webber SE, Foster ML, Belvisi MG. Mediator involvement in antigen-induced bronchospasm and microvascular leakage in the airways of ovalbumin sensitized Brown Norway rats. Br J Pharmacol. 2001;132(2):481–488. doi: 10.1038/sj.bjp.0703847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Calbet M, Ramis I, Calama E, et al. Novel inhaled pan-JAK inhibitor, LAS194046, reduces allergen-induced airway inflammation, late asthmatic response, and pSTAT activation in Brown Norway Rats. J Pharmacol Exp Ther. 2019;370(2):137–147. doi: 10.1124/jpet.119.256263 [DOI] [PubMed] [Google Scholar]
- 51.Kudlacz E, Conklyn M, Andresen C, Whitney-Pickett C, Changelian P. The JAK-3 inhibitor CP-690550 is a potent anti-inflammatory agent in a murine model of pulmonary eosinophilia. Eur J Pharmacol. 2008;582(1–3):154–161. doi: 10.1016/j.ejphar.2007.12.024 [DOI] [PubMed] [Google Scholar]
- 52.Ross SH, Cantrell DA. Signaling and function of interleukin-2 in T lymphocytes. Annu Rev Immunol. 2018;36(1):411–433. doi: 10.1146/annurev-immunol-042617-053352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schnepf D, Crotta S, Thamamongood T, et al. Selective Janus Kinase inhibition preserves interferon-λ-mediated antiviral responses. Sci Immunol. 2021;6(59):59. doi: 10.1126/sciimmunol.abd5318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hondowicz BD, An D, Schenkel JM, et al. Interleukin-2-dependent allergen-specific tissue-resident memory cells drive asthma. Immunity. 2016;44(1):155–166. doi: 10.1016/j.immuni.2015.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koch S, Sopel N, Finotto S. Th9 and other IL-9-producing cells in allergic asthma. Semin Immunopathol. 2017;39(1):55–68. doi: 10.1007/s00281-016-0601-1 [DOI] [PubMed] [Google Scholar]
- 56.Chen X, Yue R, Li X, Ye W, Gu W, Guo X. Surfactant protein A modulates the activities of the JAK/STAT pathway in suppressing Th1 and Th17 polarization in murine OVA-induced allergic asthma. Lab Invest. 2021;101(9):1176–1185. doi: 10.1038/s41374-021-00618-1 [DOI] [PubMed] [Google Scholar]
- 57.Huang Q, Han L, Lv R, Ling L. Magnolol exerts anti-asthmatic effects by regulating Janus kinase-signal transduction and activation of transcription and Notch signaling pathways and modulating Th1/Th2/Th17 cytokines in ovalbumin-sensitized asthmatic mice. Korean J Physiol Pharmacol. 2019;23(4):251–261. doi: 10.4196/kjpp.2019.23.4.251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kroes JA, Zielhuis SW, van Roon EN, Ten Brinke A. Prediction of response to biological treatment with monoclonal antibodies in severe asthma. Biochem Pharmacol. 2020;179:113978. doi: 10.1016/j.bcp.2020.113978 [DOI] [PubMed] [Google Scholar]
- 59.Gurnell M, Heaney LG, Price D, Menzies-Gow A. Long-term corticosteroid use, adrenal insufficiency and the need for steroid-sparing treatment in adult severe asthma. J Intern Med. 2021;290(2):240–256. doi: 10.1111/joim.13273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gadina M, Johnson C, Schwartz D, et al. Translational and clinical advances in JAK-STAT biology: the present and future of jakinibs. J Leukoc Biol. 2018;104(3):499–514. doi: 10.1002/JLB.5RI0218-084R [DOI] [PubMed] [Google Scholar]
- 61.Witthuhn BA, Quelle FW, Silvennoinen O, et al. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell. 1993;74(2):227–236. doi: 10.1016/0092-8674(93)90414-L [DOI] [PubMed] [Google Scholar]
- 62.Drachman JG, Millett KM, Kaushansky K. Thrombopoietin signal transduction requires functional JAK2, not TYK2. J Biol Chem. 1999;274(19):13480–13484. doi: 10.1074/jbc.274.19.13480 [DOI] [PubMed] [Google Scholar]
- 63.Watanabe S, Itoh T, Arai K. Roles of JAK kinases in human GM-CSF receptor signal transduction. J Allergy Clin Immunol. 1996;98(6 Pt 2):S183–191. doi: 10.1016/S0091-6749(96)70065-9 [DOI] [PubMed] [Google Scholar]
- 64.Rau JL. The inhalation of drugs: advantages and problems. Respir Care. 2005;50(3):367–382. [PubMed] [Google Scholar]