

Abstract
The pro-survival effect of VEGF-B has been documented in different in vivo and in vitro models. We have previously shown an enhanced VEGF-B expression in response to candesartan treatment after focal cerebral ischemia. In this study, we aimed to silence VEGF-B expression to assess its contribution to candesartan’s benefit on stroke outcome. Silencing VEGF-B expression was achieved by bilateral intracerebroventricular injections of lentiviral particles containing short hairpin RNA (shRNA) against VEGF-B. Two weeks after lentiviral injections, rats were subjected to either 90 min or 3 h of middle cerebral artery occlusion (MCAO) and randomized to intravenous candesartan (1 mg/kg) or saline at reperfusion. Animals were sacrificed at 24 h or 72 h and brains were collected and analyzed for hemoglobin (Hb) excess and infarct size, respectively. Functional outcome at 24, 48 and 72 h was assessed blindly. Candesartan treatment improved neurobehavioral and motor function, and decreased infarct size and Hb. While silencing VEGF-B expression diminished candesartan’s neuroprotective effect, candesartan-mediated vascular protection was maintained even in the absence of VEGF-B suggesting that this growth factor is not the mediator of candesartan’s vascular protective effects. However, VEGF-B is a mediator of neuroprotection achieved by candesartan and represents a potential drug target to improve stroke outcome. However, further studies are needed to elucidate the underlying molecular mechanisms of VEGF-B in neuroprotection and recovery after ischemic stroke.
Keywords: Angiotensin II Type-1 Receptor Blocker (ARB), Candesartan, Stroke, Vascular Endothelial Growth Factor-B (VEGF-B), Neuroprotection
INTRODUCTION
Stroke is the fifth most common cause of death in the US and a leading cause of adult disability with more than 700,000 stroke cases occurring every year [1]. With the very limited treatment options available, it is of utmost importance to develop new therapies for acute ischemic stroke management.
The mechanisms by which candesartan, an antihypertensive medication of the angiotensin II receptor blocker (ARB) family, provides vascular protection after stroke are not completely known. Recently, we and others demonstrated that candesartan, is neuroprotective and vasculoprotective and improves functional outcome in both permanent and transient models of stroke [2–7]. These beneficial effects of candesartan are mediated through a number of different mechanisms including upregulation of endothelial nitric oxide synthase (eNOS) expression [8], upregulation of growth factor expression [9, 10], and amelioration of oxidative stress [11]. Moreover, we have recently demonstrated the ability of candesartan to increase induction of the brain-derived neurotrophic factor-BDNF and vascular endothelial growth factor-VEGF after ischemic stroke [12, 13], which has been shown to be involved in recovery through induction of neuroplasticity [14, 15]. While stroke clinical trials have been negative [16, 17], it is recognized that the potential benefit of candesartan treatment is complicated by its blood pressure lowering effect. It is, therefore, important to identify the precise mechanisms of neurovascular protection by ARBs in order to develop alternative therapies for acute ischemic stroke.
VEGF-B is a relatively understudied member of the VEGF family and the exact role played by VEGF-B in health and disease remained elusive for a long time. VEGF-B plays an important role on several types of neurons. It is important for the protection of neurons in the retina [18] and motor neuron diseases such as amyotrophic lateral sclerosis [19]. VEGF-A, the more commonly studied isoform, mediates most of its biological activity via VEGF receptor 2 (VEGFR-2), whereas VEGF-B specifically binds VEGFR-1 [20]. These receptors are expressed almost exclusively on endothelial cells, although VEGFR-1 is also found in monocytes, where it mediates migration [21, 22]. VEGFR-1, also known as fms-like tyrosine kinase receptor 1 (Flt-1) participates in tissue revascularization [23], receptor cross-activation [24] and monocyte-mediated arteriogenesis [25]. VEGFR-1–mediated signaling was shown to play a significant role in a variety of pathological conditions including inflammatory diseases [26, 27]. VEGF-B null mice are healthy and fertile, suggesting a redundant role of this molecule [28]. Further studies, however, demonstrated a potent pro-survival effect of VEGF-B. It was found to be neurovascular protective against a wide variety of apoptotic stimuli [18, 29]. VEGF-B administration after stroke decreased infarct size [18] and VEGF-B knockout animals exhibited larger infarcts as compared to their wild type counterparts [30]. Moreover, VEGF-B expression was upregulated in the ischemic border zone after 90-minute focal cerebral ischemia, further supporting its involvement in the endogenous protective/ reparative response to ischemic insult [31]. A recent study also showed that VEGF-B/VEGFR-1 signaling is involved in regulating the function of pericytes after ischemic stroke [32]. Whether VEGF-B is involved in candesartan-mediated protection after ischemic stroke remains completely unknown.
We have previously shown an enhanced VEGF-B production in ischemic brain tissue and brain endothelial cells after candesartan treatment [9, 10]. We hypothesized that silencing VEGF-B would diminish the neurovascular protection by candesartan after ischemic stroke. Our novel results demonstrate a causal relationship between VEGF-B upregulation and improved neurobehavioral outcome after candesartan treatment.
Materials and Methods
VEGF-B shRNA Intracerebroventricular Injections: VEGF-B knockdown
Adult male Wistar rats (Charles River Breeding Company, Wilmington, MA, USA), weighing 180– 200 grams were used. To inject lentiviral particles into the lateral ventricles, rats were anaesthetized with 2–5% isoflurane inhalation and placed in a stereotaxic device. A midline incision was made to expose the skull and a hole was drilled at either side of the brain using the coordinates: anterioposterior (AP) = 0.8mm; mediolateral (ML) = +/− 1.2 mm; dorsoventral (DV) = 3.8 mm with respect to the bregma. Lentiviral particles containing VEGF-B shRNA (SMARTchoice Lentiviral Rat VEGF hCMV-TurboGFP shRNA, 1 × 108 TU/mL, Dharmacon) or non-targeting control vector (NTC) were slowly injected over 5 minutes into each of the lateral ventricles (5 µls/ hemisphere at a rate of 1 µl/ min). The incision was sutured and animals were allowed to recover on a 37°C heated pad.
Experimental Cerebral Ischemia and Candesartan Treatment
Two weeks after ICV injection of lentiviral particles, animals were subjected to middle cerebral artery occlusion (MCAO) using the intraluminal suture model as described previously [9]. Briefly, animals were anesthetized using 2–5 % isoflurane inhalation. Temporary middle cerebral artery occlusion (MCAO) was achieved using 4–0 silicon-coated nylon suture (Doccol, Sharon, MA), advanced into the internal carotid artery to block the origin of the middle cerebral artery. After 90 minutes or 3 h, animals were re-anaesthetized and sutures were removed to allow reperfusion of ischemic brain areas. At reperfusion, animals received intravenous injection of either saline or candesartan (1 mg/ Kg). Candesartan was a kind gift from AstraZeneca Pharmaceuticals (Wilmington, DE). All animals were singly housed before and after surgery, with free access to food and water.
Animals and Treatment Regimen
Male Wistar rats (Charles River Laboratories, Wilmington, MA) were used according to procedures approved by the Institutional Animal Care and Use Committee (IACUC) of the Charlie Norwood VA Medical Center. The study was performed in two separate experiments. A diagram of the experimental design is shown in Figure 1. Experiment I was carried out to evaluate the effects of candesartan treatment on neurological outcome and infarct size on day 3 post-middle cerebral artery occlusion (MCAO). In this 3-day survival study, we used 90-min MCAO to reduce mortality. The groups were: NTC + MCAO saline treatment only group I (NTC-saline only); NTC + MCAO candesartan (1 mg/kg) treatment group II (NTC-cand); VEGF-B shRNA + MCAO saline treatment group III (KD saline); VEGF-B shRNA + MCAO candesartan (1 mg/kg) treatment group IV (KD-cand). Experiment II was carried out to evaluate the effects of candesartan treatment on the vascular damage (hemorrhage and edema) after 3 MCAO followed by 21h reperfusion. The groups were the same as in experiment I.
Figure 1: Schematic diagram of the study:
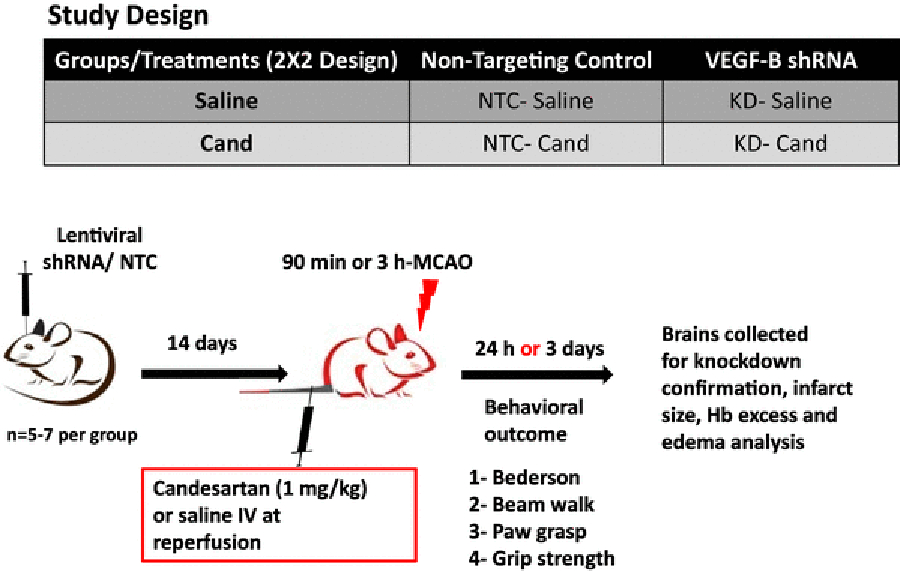
Animals were subjected to 90 min or 3h MCAO after 14 days of bilateral ICV injection and randomized to IV candesartan (1 mg/kg) or saline at reperfusion. NTC: non-targeting control; shRNA: short hairpin RNA; KD: knock-down; ICV: intracerebroventricular; IV: intravenous; Hb: hemoglobin; MCAO: middle cerebral artery occlusion; VEGF: vascular endothelial growth factor.
Neurobehavioral Testing
Neurobehavioral assessment was done 1, 2 and 3 days after the onset of MCAO in a blinded fashion using modified Bederson score, beam walk, paw grasp and grip strength tests.
Modified Bederson score
Animals were assigned a score from 0–3, with lower scores indicating better performance. The animal was given one point for each of the following: forelimb flexion; diminished resistance to lateral push; and contralateral circling [33].
Beam walk
Animals were placed on a beam (60 cm long and 4.5 cm wide) for 60 seconds and assigned a score from 0 to 6 as follows: balances on the beam with a steady posture = 0; grasps side of the beam = 1; hugs the beam with 1 limb falling = 2; hugs the beam with 2 limbs falling =3; falls off the beam within 40 to 60 seconds = 4; falls off the beam within 20 to 40 seconds = 5; falls off the beam in less than 20 seconds = 6 [34].
Paw grasp
Animal were suspended by tail and allowed to grasp a vertical pole. Animals are assigned a score from 1–3 as follows: grasping by both forelimbs= 1; grasping with only one forelimb= 2; failure to grasp= 3 [35].
Grip strength
Grip Strength Meter (Columbus instruments, Columbus, OH) was used to measure baseline and post-stroke neuromuscular function. Three readings were recorded for each animal and averaged. Results are expressed as percentage of baseline grip strength.
Assessment of infarct size, edema and hemorrhage
Infarct size, HT, edema, and hemoglobin content were measured by a blinded investigator. On day 3 post-stroke, animals were deeply anaesthetized with ketamine/ xylazine mixture (85% and 15%, respectively) and transcardially perfused with ice cold PBS. Animals were then decapitated and their brains collected and sliced into seven 2-mm thick coronal sections. Sections were stained with 2% TTC solution (2,3,5-triphenyltetrazolium chloride- Sigma-Aldrich, St. Louis, MO) for 15–20 min and scanned. Infarction areas were measured blindly using Image J software, corrected for edema and expressed as percentage of the contralesional hemisphere using the formula: 100 X [contralateral – (ipsilateral – infarct)]/contralateral. Edema was calculated as a percent (%) area increase in the ischemic hemisphere compared to the contralateral hemisphere.
Bleeding was quantified using two different methods. 1) A colorimetric hemoglobin detection assay kit (QuamtiCrome; BioAssay Systems, Haywood, California, USA) was used at 24 h after ischemic stroke as described previously [36]. Briefly, animals were deeply anaesthetized with ketamine/ xylazine mixture (85% and 15%, respectively) and transcardially perfused with ice cold PBS. Animals were then decapitated and their brains collected. Brain samples were separated into contralateral and ipsilateral hemispheres, and homogenized in a 10 % glycerol-Tris 146 buffered saline solution containing Tween-20. Samples were prepared, read at 562 nm using a standard microplate reader, and hemoglobin concentration was calculated according to the manufacturer’s instructions. 2) Macroscopic Hemorrhagic transformation (HT) was scored in coronal brain slices B to E using a 4-point rubric (0, no hemorrhage; 1, dispersed individual petechiae; 2, confluent petechiae; 3, small diffuse hemorrhage or hematoma; and 4, large diffuse hemorrhage or hematoma), and the total score for each animal was reported as described previously [37].
Western blotting
Brain slices B- E were collected and flash frozen. Brain tissue was then homogenized and protein expression was measured as described previously [9]. Non-specific binding was blocked by incubating the membranes in 5% milk in TBST for 60 minutes prior to overnight incubation with primary antibody against VEGF-B, (1:1000, Abcam, Cambridge, MA), rabbit polyclonal VEGF receptor 1 (VEGFR1) antibody (ab2350; abcam,1: 100, Cambridge, MA), anti-phospho-Flt (Tyr 1213) (07–758; Millipore; 1;750, Billerica, MA), VEGF-A and phosphor-VEGFR2 (1:200, Millipore, Billerica, MA). Beta-actin (Sigma-Aldrich, St. Louis, MO) was used as an endogenous loading control. Protein levels were analyzed densitometrically, using Image J software and were normalized to loading controls.
Statistical Analysis
Two-way ANOVA was conducted for the behavioral data analysis using GraphPad prism software (5.1). Post hoc pair-wise comparisons between groups over time were performed using a Bonferroni adjustment to the overall alpha level. P<0.05 was considered significant. Results are presented as mean ± SEM.
RESULTS
Diminished VEGF-B expression after bilateral ICV injection of VEGF-B shRNA lentiviral particles To knock down VEGF-B protein expression, bilateral intracerebroventricular injection of lentiviral particles encoding short hairpin RNA against VEGF-B (VEGF-B shRNA) was performed. As shown in Figure 2, VEGF-B expression was reduced by 59%, 17 days after injection of VEGF-B shRNA as compared to the injection of the non-targeting control (NTC). In the VEGF-B knockdown group (KD), candesartan treatment was still capable of enhancing VEGF-B expression, but the expression was still 50% less in the KD-candesartan group as compared to the NTC-candesartan group.
Figure 2: Assessment of VEGF-B expression, and its receptor.
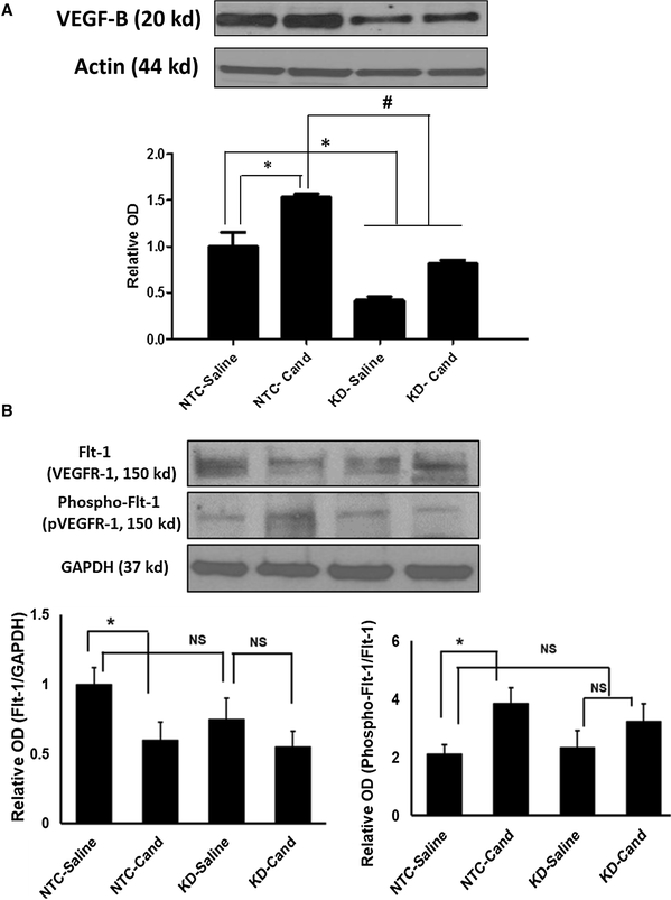
VEGFR1 expression and activation after bilateral intracerebroventricular injection of VEGF-B shRNA lentiviral particles. Quantification of VEGF-B expression (A), and its receptor VEGFR1 expression and activation (B) in the ipsilateral hemispheres after bilateral ICV injection of lentiviral particles containing non targeting control (NTC) or VEGF-B shRNA to knockdown protein expression (KD). Animals were exposed, 72 h before sacrifice, to 90-minute MCAO and received saline or candesartan (1 mg/ Kg) at reperfusion. Ipsilateral hemispheres were homogenized and analyzed for VEGF-B protein by western blot. Data is presented as mean±SEM (n=6/ group, *significantly different from NTC-saline, # significantly different from NTC-cand, P<0.05).
VEGFB exerts its prosurvival effects predominantly through VEGFR1 (Flt-1). The degree of phosphorylation of Flt-1 is used to quantify their activation. Further, we checked the effect of silencing VEGF-B on VEGFR1 as well as its phosphorylation at 17 days after intracerebroventricular injection of either NTC or VEGF-B shRNA lentiviral particles. Candesartan resulted in a significant decrease in VEGFR1 (Flt-1) expression in the NTC-candesartan compared to NTC-saline group (Figure 2B). Further, candesartan treated animals showed a significant increase in phospho-Flt-1 (activation of VEGFR1) expression in the NTC-candesartan group compared to NTC-saline. Candesartan treatment did not result in any changes in VEGFR1 and its phosphorylation expression in the KD-candesartan group compared to KD-saline (Figure 2B). These data suggest that candesartan’s neuroprotective effect is mediated through VEGF-B and its receptor activation (phosphor-Flt-1) after ischemic stroke.
Silencing VEGF-B expression does not upregulate compensatory VEGF-A expression
We next investigated if VEGF-B silencing elicits a compensatory upregulation of other growth factors such as VEGF-A. We determined the expression of VEGF-A as well as the phosphorylation of its receptor, VEGF receptor-2 at 17 days after intracerebroventricular injection of either NTC or VEGF-B shRNA lentiviral particles (VEGF-B KD). We did not observe any significant change in VEGF-A and phosphorylation of its receptor between the groups (Figure 3). Candesartan treatment showed a trend towards an increased VEGF-A and phosphorylation of VEGR2 in the NTC-candesartan group compared to NTC-saline, as we have previously reported [9].
Figure 3: Silencing VEGF-B expression does not upregulate compensatory VEGF-A and its receptor expression.
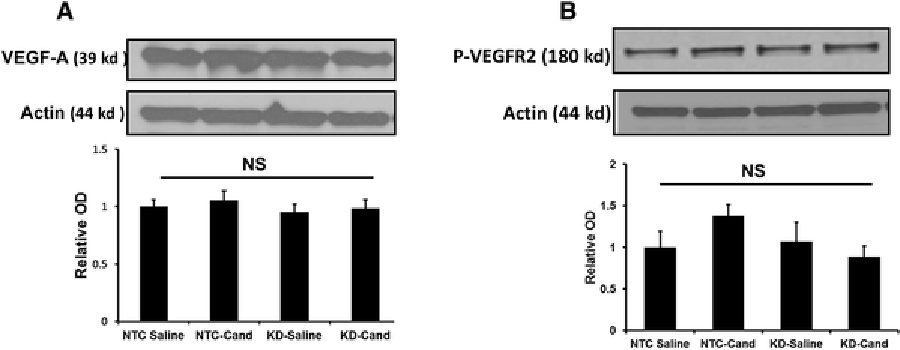
Quantification of VEGF-A expression 17 days after bilateral ICV injection of NTC or VEGF-B shRNA. Animals were exposed, 72h before sacrifice, to 90-minute MCAO. Data is presented as mean±SEM (n=6/ group).
Silencing VEGF-B decreased candesartan-induced improvement of neurobehavioral function after experimental stroke
The role played by VEGF-B in mediating candesartan’s neuroprotective effect was determined by measuring neurobehavioral outcome 1, 2 and 3 days after MCAO. Bederson score shows an improved performance as early as 24 h in the NTC-candesartan animals as compared to their saline-treated counterparts. In the knockdown group, however, candesartan treatment had a tendency to slightly improve neurobehavioral outcome on days 2 and 3 post-stroke. The effect, however, was not significantly different from the KD-saline group (Figure 4A). Beam walk and paw grasp tests followed the same pattern (Figure 4B, C). Candesartan improved animal performance in the NTC group but not in the KD group. Although there was a tendency to improve grip strength by candesartan treatment in the first 48 h in both NTC and KD groups, the improvement was not significant until 72 h and was observed in the NTC-candesartan group only (Figure 4D). It is worth mentioning that saline-treated animals in both NTC and KD groups showed comparable neurobehavioral outcome throughout all the measured time points.
Figure 4: Silencing VEGF-B diminishes candesartan-induced neuroprotective effects after focal cerebral ischemia.
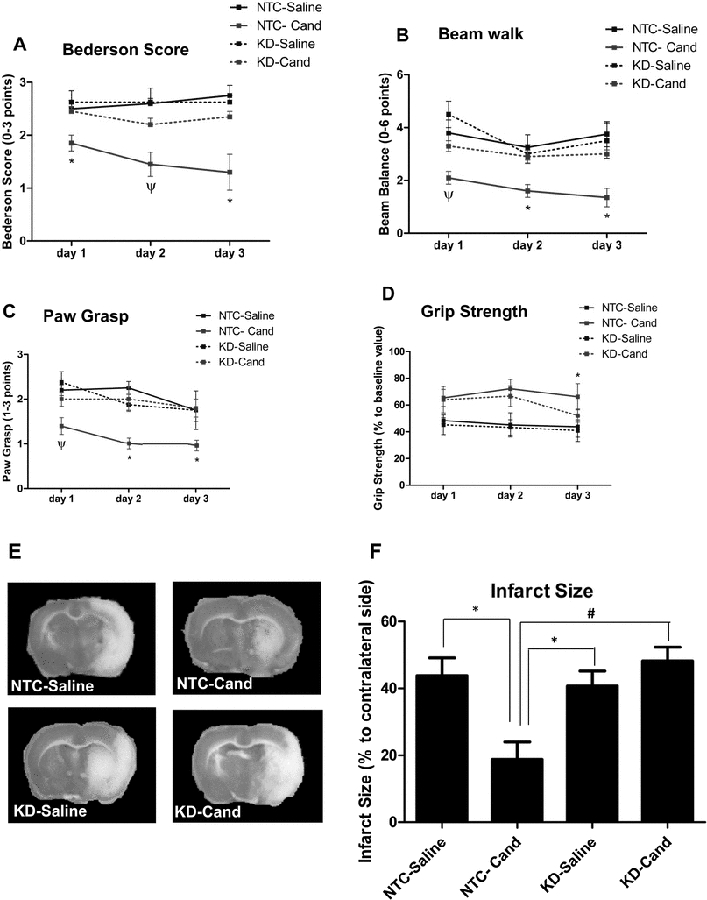
A-D: Assessment of neurobehavioral and motor functions 1, 2 and 3 days after MCAO using modified Bederson score (A), beam balance (B), paw grasp (C) and grip strength tests (D) in saline and candesartan (1 mg/Kg)-treated animals. *significantly different from all three groups, ψ significantly different from saline-treated groups only, P<0.05). E: Representative images of TTC-stained coronal sections 72h after MCAO and treatment with saline or 1 mg/Kg candesartan at reperfusion F: Quantification of infarct size 72 h after MCAO and treatment with saline or candesartan (n=4–5/ group, *significantly different from NTC-saline, # significantly different from NTC-cand, P<0.05). Data is presented as mean±SEM; n=6–8 per group.
Reduction of infarct size by candesartan treatment is mediated by VEGF-B
Infarct size analysis 3 days after MCAO shows a reduction in stroke damage by candesartan treatment in the NTC group only (Figure 4 E, F). Knocking down VEGF-B diminished candesartan’s ability to reduce infarct size. Similar to neurobehavioral tests, no increase in infarct size was observed at 72 h between the saline-treated NTC and KD.
Vascular protective effect of candesartan treatment does not involve VEGF-B
Candesartan treatment showed a trend towards a decreased Hb excess, hemorrhagic score and edema in NTC-candesartan group compared to NTC-Saline after 3 h MCAO followed by 21 h reperfusion. Silencing VEGF-B also decreased hemorrhage with or without candesartan treatment, suggesting that VEGF-B contributes to vascular injury and not protection in our model. This finding also suggests that the vascular protective effect of candesartan is not mediated through VEGF B and its receptor expression/ activation after stroke (Figure 5).
Figure 5: Silencing VEGF-B is vascular protective regardless of candesartan treatment.
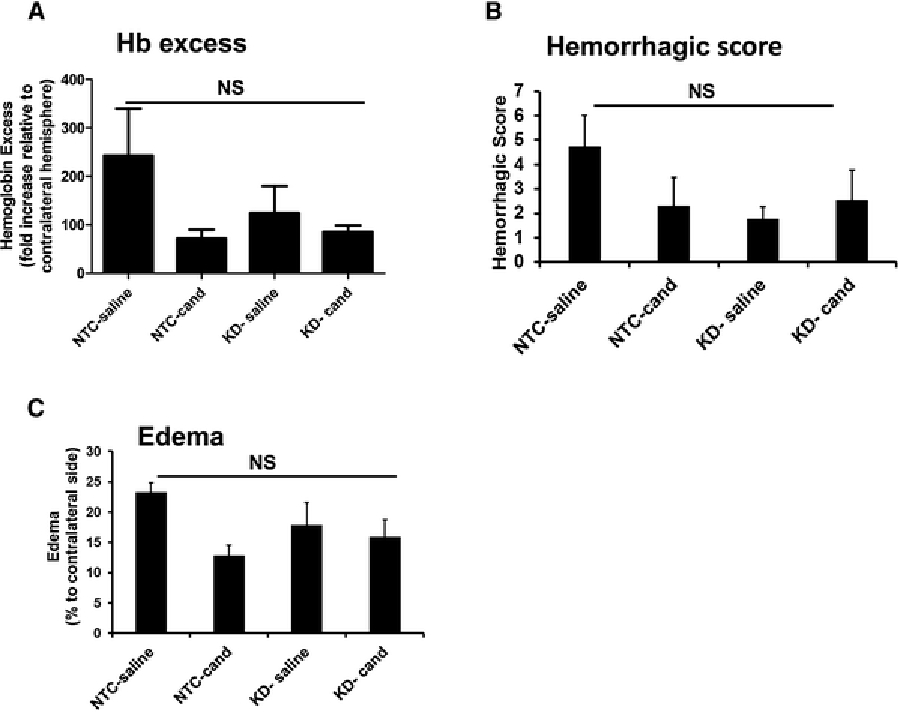
Assessment of Hb excess (A), HT score (B) and edema (C) at 24 h after MCAO. Silencing VEGF-B decreased Hb excess, HT and edema with or without candesartan treatment. There were no statistically significant differences between the groups. Data is presented as mean±SEM (n=5–7 per group).
DISCUSSION
Although a full understanding of the biological functions of VEGF-B is far from complete, it has been recognized as a potent apoptosis inhibitor in different organs and animal models [18]. We have previously shown an enhanced expression of VEGF-B in response to candesartan treatment in the brain tissue and in endothelial cells [9, 10]. Neutralization of VEGF-B in endothelial cell conditioned media had a tendency to decrease its neuroprotective effect, as demonstrated by increased expression of the apoptotic marker, cleaved caspase-3, after 2-h oxygen glucose deprivation (OGD) [10]. The current study builds upon these past studies. Silencing VEGF-B significantly reduced candesartan’s effect on neurobehavioral recovery. Similarly, infarct size was reduced in response to candesartan treatment, an effect that was abrogated in the VEGF-B knockdown animals. The results of this study demonstrate the contribution of VEGF-B to the protective effects of candesartan treatment in experimental ischemic stroke.
In this study, we demonstrate the role played by VEGF-B in mediating candesartan’s neuroprotective effect in an acute ischemic stroke animal model. The findings of this study include an improved functional recovery after candesartan treatment in the animals expressing VEGF-B. Silencing VEGF-B significantly reduced candesartan’s effect on neurobehavioral recovery. Similarly, infarct size was reduced in response to candesartan treatment, an effect that was abrogated in the VEGF-B knockdown animals. The results of this study demonstrate the contribution of VEGF-B to the protective effects of candesartan treatment in experimental ischemic stroke.
Previous studies have shown the neuroprotective effect of VEGF-B in experimental stroke. Recombinant human VEGF-B protein treatment reduced infarct size by 32% in wild-type animals [18]. VEGF-B knockout animals demonstrated 50% larger infarcts in a permanent occlusion model [30]. Nonetheless, we have not observed worsening of the functional outcome or infarct size in the saline-treated KD animals as compared to their NTC counterparts. A possible explanation of such a discrepancy is the use of VEGF-B knockout model versus silencing using lentiviral particle injection, which did not abolish VEGF-B expression completely. Another plausible explanation is experimental limitation. Due to the tight scale of outcome measures, further worsening by knocking down VEGF-B could not be observed. Our results, however, suggest a beneficial but expendable role of VEGF-B in recovery after MCAO.
We have previously demonstrated the involvement of other growth factors in mediating candesartan’s actions after an ischemic insult [38]. Brain-derived neurotrophic factor (BDNF) [39] and vascular endothelial growth factor-A (VEGF-A) [9, 10] were upregulated in response to candesartan treatment. Neutralization of either growth factor significantly reduced the candesartan-mediated proangiogenic effect in vitro [10, 39]. Since angiogenesis and neurorestorative processes are coupled in the ischemic brain [40], we expect the involvement of BDNF and VEGF-A in candesartan-mediated neurobehavioral recovery after experimental stroke. We, therefore, do not exclude the involvement of other growth factors besides VEGF-B in mediating neurobehavioral recovery after candesartan treatment. Current evidence suggests that candesartan works by enhancing endogenous reparative mechanisms in the brain and the benefits of treatment are possibly mediated through an orchestrated action of several growth factors.
We have previously reported that angiotensin receptor blockade reduces vascular damage in stroke models, despite increasing matrix metalloproteinase (MMP-9) activity and VEGF-A expression [9, 36, 41]. We further examined whether VEGF-B is involved in candesartan-induced vascular protection. Consistent with previous studies, candesartan treatment decreased vascular damage in NTC-candesartan group compared to NTC-Saline after 3 h MCAO followed by 21 h reperfusion. Interestingly, silencing VEGF-B decreased vascular damage independent of candesartan treatment, suggesting that VEGF-B mediates vascular damage, not protection, under these experimental conditions.
In conclusion, our results suggest an important role played by VEGF-B in mediating some of the neuroprotective effects of candesartan after stroke. Identifying growth factors that mediate recovery after ischemic stroke present possible targets for stroke therapeutics.
Acknowledgements
This work was supported by the National Institute of Health [RO1-NS063965 (SCF), RO1-NS083559 (AE), R01NS097800–01 (TI)], Veterans Affairs Merit Review [BX-000891 (SCF), BX- BX000347 (AE)], and the American Heart Association (12PRE12030197 (SS)). Adviye Ergul is a Research Career Scientist at the Charlie Norwood Veterans Affairs Medical Center in Augusta, Georgia. The authors gratefully acknowledge Abdelrahman Fouda for the technical assistance with animal tissue collection.
Abbreviations:
- ARBs
Angiotensin II Type-1 Receptor Blockers
- KD
VEGF-B Knockdow
- MCAO
Middle Cerebral Artery Occlusion
- NTC
Non-targeting Control
- shRNA
Short Hairpin RNA
- VEGF-B
Vascular Endothelial Growth Factor-B
Footnotes
Disclosures/ Conflict of interest
The contents do not represent the views of the Department of Veterans Affairs or the United States government.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER 3rd, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB (2014) Heart disease and stroke statistics−-2014 update: a report from the american heart association. Circulation 129:e28–e292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Engelhorn T, Goerike S, Doerfler A, Okorn C, Forsting M, Heusch G, Schulz R (2004) The angiotensin II type 1-receptor blocker candesartan increases cerebral blood flow, reduces infarct size, and improves neurologic outcome after transient cerebral ischemia in rats. J Cereb Blood Flow Metab 24:467–474 [DOI] [PubMed] [Google Scholar]
- 3.Brdon J, Kaiser S, Hagemann F, Zhao Y, Culman J, Gohlke P (2007) Comparison between early and delayed systemic treatment with candesartan of rats after ischaemic stroke. Journal of hypertension 25:187–196 [DOI] [PubMed] [Google Scholar]
- 4.Kozak A, Ergul A, El-Remessy AB, Johnson MH, Machado LS, Elewa HF, Abdelsaid M, Wiley DC, Fagan SC (2009) Candesartan augments ischemia-induced proangiogenic state and results in sustained improvement after stroke. Stroke 40:1870–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elewa HF, Kozak A, Johnson MH, Ergul A, Fagan SC (2007) Blood pressure lowering after experimental cerebral ischemia provides neurovascular protection. J Hypertens 25:855–859 [DOI] [PubMed] [Google Scholar]
- 6.Fagan SC, Kozak A, Hill WD, Pollock DM, Xu L, Johnson MH, Ergul A, Hess DC (2006) Hypertension after experimental cerebral ischemia: candesartan provides neurovascular protection. J Hypertens 24:535–539 [DOI] [PubMed] [Google Scholar]
- 7.Guan W, Kozak A, El-Remessy AB, Johnson MH, Pillai BA, Fagan SC (2011) Acute treatment with candesartan reduces early injury after permanent middle cerebral artery occlusion. Transl Stroke Res 2:179–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamakawa H, Jezova M, Ando H, Saavedra JM (2003) Normalization of endothelial and inducible nitric oxide synthase expression in brain microvessels of spontaneously hypertensive rats by angiotensin II AT1 receptor inhibition. J Cereb Blood Flow Metab 23:371–380 [DOI] [PubMed] [Google Scholar]
- 9.Guan W, Somanath PR, Kozak A, Goc A, El-Remessy AB, Ergul A, Johnson MH, Alhusban A, Soliman S, Fagan SC (2011) Vascular protection by angiotensin receptor antagonism involves differential VEGF expression in both hemispheres after experimental stroke. PLoS One 6:e24551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soliman S, El-Remessy A, Ishrat T, Pillai A, Somanath P, Ergul A, Fagan S (2014) Candesartan Induces A Prolonged Proangiogenic Effect and Augments Endothelium-Mediated Neuroprotection after Oxygen and Glucose Deprivation: Role of VEGF-A And B. Journal of Pharmacology and Experimental Therapeutics [DOI] [PMC free article] [PubMed]
- 11.Hamai M, Iwai M, Ide A, Tomochika H, Tomono Y, Mogi M, Horiuchi M (2006) Comparison of inhibitory action of candesartan and enalapril on brain ischemia through inhibition of oxidative stress. Neuropharmacology 51:822–828 [DOI] [PubMed] [Google Scholar]
- 12.Ishrat T, Pillai B, Soliman S, Fouda AY, Kozak A, Johnson MH, Ergul A, Fagan SC (2015) Low-dose candesartan enhances molecular mediators of neuroplasticity and subsequent functional recovery after ischemic stroke in rats. Mol Neurobiol 51:1542–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fouda AY, Alhusban A, Ishrat T, Pillai B, Eldahshan W, Waller JL, Ergul A, Fagan SC (2017) Brain-Derived Neurotrophic Factor Knockdown Blocks the Angiogenic and Protective Effects of Angiotensin Modulation After Experimental Stroke. Mol Neurobiol 54:661–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greenberg ME, Xu B, Lu B, Hempstead BL (2009) New insights in the biology of BDNF synthesis and release: implications in CNS function. J Neurosci 29:12764–12767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun Y, Jin K, Xie L, Childs J, Mao XO, Logvinova A, Greenberg DA (2003) VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Invest 111:1843–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He J, Zhang Y, Xu T, Zhao Q, Wang D, Chen CS, Tong W, Liu C, Ju Z, Peng Y, Peng H, Li Q, Geng D, Zhang J, Li D, Zhang F, Guo L, Sun Y, Wang X, Cui Y, Li Y, Ma D, Yang G, Gao Y, Yuan X, Bazzano LA, Chen J (2014) Effects of immediate blood pressure reduction on death and major disability in patients with acute ischemic stroke: the CATIS randomized clinical trial. JAMA : the journal of the American Medical Association 311:479–489 [DOI] [PubMed] [Google Scholar]
- 17.Sandset EC, Bath PM, Boysen G, Jatuzis D, Korv J, Luders S, Murray GD, Richter PS, Roine RO, Terent A, Thijs V, Berge E, Group SS (2011) The angiotensin-receptor blocker candesartan for treatment of acute stroke (SCAST): a randomised, placebo-controlled, double-blind trial. Lancet 377:741–750 [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Zhang F, Nagai N, Tang Z, Zhang S, Scotney P, Lennartsson J, Zhu C, Qu Y, Fang C, Hua J, Matsuo O, Fong GH, Ding H, Cao Y, Becker KG, Nash A, Heldin CH, Li X (2008) VEGF-B inhibits apoptosis via VEGFR-1-mediated suppression of the expression of BH3-only protein genes in mice and rats. J Clin Invest 118:913–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poesen K, Lambrechts D, Van Damme P, Dhondt J, Bender F, Frank N, Bogaert E, Claes B, Heylen L, Verheyen A, Raes K, Tjwa M, Eriksson U, Shibuya M, Nuydens R, Van Den Bosch L, Meert T, D’Hooge R, Sendtner M, Robberecht W, Carmeliet P (2008) Novel role for vascular endothelial growth factor (VEGF) receptor-1 and its ligand VEGF-B in motor neuron degeneration. J Neurosci 28:10451–10459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamazaki Y, Morita T (2006) Molecular and functional diversity of vascular endothelial growth factors. Mol Divers 10:515–527 [DOI] [PubMed] [Google Scholar]
- 21.Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marme D (1996) Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood 87:3336–3343 [PubMed] [Google Scholar]
- 22.Clauss M, Weich H, Breier G, Knies U, Rockl W, Waltenberger J, Risau W (1996) The vascular endothelial growth factor receptor Flt-1 mediates biological activities. Implications for a functional role of placenta growth factor in monocyte activation and chemotaxis. J Biol Chem 271:17629–17634 [DOI] [PubMed] [Google Scholar]
- 23.Luttun A, Tjwa M, Moons L, Wu Y, Angelillo-Scherrer A, Liao F, Nagy JA, Hooper A, Priller J, De Klerck B, Compernolle V, Daci E, Bohlen P, Dewerchin M, Herbert JM, Fava R, Matthys P, Carmeliet G, Collen D, Dvorak HF, Hicklin DJ, Carmeliet P (2002) Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat Med 8:831–840 [DOI] [PubMed] [Google Scholar]
- 24.Autiero M, Waltenberger J, Communi D, Kranz A, Moons L, Lambrechts D, Kroll J, Plaisance S, De Mol M, Bono F, Kliche S, Fellbrich G, Ballmer-Hofer K, Maglione D, Mayr-Beyrle U, Dewerchin M, Dombrowski S, Stanimirovic D, Van Hummelen P, Dehio C, Hicklin DJ, Persico G, Herbert JM, Communi D, Shibuya M, Collen D, Conway EM, Carmeliet P (2003) Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat Med 9:936–943 [DOI] [PubMed] [Google Scholar]
- 25.Pipp F, Heil M, Issbrucker K, Ziegelhoeffer T, Martin S, van den Heuvel J, Weich H, Fernandez B, Golomb G, Carmeliet P, Schaper W, Clauss M (2003) VEGFR-1-selective VEGF homologue PlGF is arteriogenic: evidence for a monocyte-mediated mechanism. Circ Res 92:378–385 [DOI] [PubMed] [Google Scholar]
- 26.Autiero M, Luttun A, Tjwa M, Carmeliet P (2003) Placental growth factor and its receptor, vascular endothelial growth factor receptor-1: novel targets for stimulation of ischemic tissue revascularization and inhibition of angiogenic and inflammatory disorders. J Thromb Haemost 1:1356–1370 [DOI] [PubMed] [Google Scholar]
- 27.Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M, Wu Y, Bono F, Devy L, Beck H, Scholz D, Acker T, DiPalma T, Dewerchin M, Noel A, Stalmans I, Barra A, Blacher S, VandenDriessche T, Ponten A, Eriksson U, Plate KH, Foidart JM, Schaper W, Charnock-Jones DS, Hicklin DJ, Herbert JM, Collen D, Persico MG (2001) Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med 7:575–583 [DOI] [PubMed] [Google Scholar]
- 28.Li X, Lee C, Tang Z, Zhang F, Arjunan P, Li Y, Hou X, Kumar A, Dong L (2009) VEGF-B: a survival, or an angiogenic factor? Cell adhesion & migration 3:322–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang F, Tang Z, Hou X, Lennartsson J, Li Y, Koch AW, Scotney P, Lee C, Arjunan P, Dong L, Kumar A, Rissanen TT, Wang B, Nagai N, Fons P, Fariss R, Zhang Y, Wawrousek E, Tansey G, Raber J, Fong GH, Ding H, Greenberg DA, Becker KG, Herbert JM, Nash A, Yla-Herttuala S, Cao Y, Watts RJ, Li X (2009) VEGF-B is dispensable for blood vessel growth but critical for their survival, and VEGF-B targeting inhibits pathological angiogenesis. Proceedings of the National Academy of Sciences of the United States of America 106:6152–6157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun Y, Jin K, Childs JT, Xie L, Mao XO, Greenberg DA (2004) Increased severity of cerebral ischemic injury in vascular endothelial growth factor-B-deficient mice. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 24:1146–1152 [DOI] [PubMed] [Google Scholar]
- 31.Xie L, Mao X, Jin K, Greenberg DA (2013) Vascular endothelial growth factor-B expression in postischemic rat brain. Vascular cell 5:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jean LeBlanc N, Guruswamy R, ElAli A (2017) Vascular Endothelial Growth Factor Isoform-B Stimulates Neurovascular Repair After Ischemic Stroke by Promoting the Function of Pericytes via Vascular Endothelial Growth Factor Receptor-1. Mol Neurobiol [DOI] [PubMed]
- 33.Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H (1986) Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke; a journal of cerebral circulation 17:472–476 [DOI] [PubMed] [Google Scholar]
- 34.Watanabe T, Okuda Y, Nonoguchi N, Zhao MZ, Kajimoto Y, Furutama D, Yukawa H, Shibata MA, Otsuki Y, Kuroiwa T, Miyatake S (2004) Postischemic intraventricular administration of FGF-2 expressing adenoviral vectors improves neurologic outcome and reduces infarct volume after transient focal cerebral ischemia in rats. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 24:1205–1213 [DOI] [PubMed] [Google Scholar]
- 35.Machado LS, Sazonova IY, Kozak A, Wiley DC, El-Remessy AB, Ergul A, Hess DC, Waller JL, Fagan SC (2009) Minocycline and tissue-type plasminogen activator for stroke: assessment of interaction potential. Stroke; a journal of cerebral circulation 40:3028–3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishrat T, Pillai B, Ergul A, Hafez S, Fagan SC (2013) Candesartan reduces the hemorrhage associated with delayed tissue plasminogen activator treatment in rat embolic stroke. Neurochem Res 38:2668–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fagan SC, Lapchak PA, Liebeskind DS, Ishrat T, Ergul A (2013) Recommendations for preclinical research in hemorrhagic transformation. Transl Stroke Res 4:322–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fouda AY, Alhusban A, Ishrat T, Pillai B, Eldahshan W, Waller JL, Ergul A, Fagan SC (2016) Brain-Derived Neurotrophic Factor Knockdown Blocks the Angiogenic and Protective Effects of Angiotensin Modulation After Experimental Stroke. Mol Neurobiol [DOI] [PMC free article] [PubMed]
- 39.Alhusban A, Kozak A, Ergul A, Fagan SC (2013) AT1 Receptor Antagonism Is Proangiogenic in the Brain: BDNF a Novel Mediator. The Journal of pharmacology and experimental therapeutics 344:348–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiong Y, Mahmood A, Chopp M (2010) Angiogenesis, neurogenesis and brain recovery of function following injury. Curr Opin Investig Drugs 11:298–308 [PMC free article] [PubMed] [Google Scholar]
- 41.Ishrat T, Kozak A, Alhusban A, Pillai B, Johnson MH, El-Remessy AB, Ergul A, Fagan SC (2014) Role of matrix metalloproteinase activity in the neurovascular protective effects of Angiotensin antagonism. Stroke Res Treat 2014:560491. [DOI] [PMC free article] [PubMed] [Google Scholar]