

Abstract
We recently reported that doxorubicin decreased the expression of calpain-1/2, while inhibition of calpain activity promoted doxorubicin-induced cardiac injury in mice. In this study, we investigated whether and how elevation of calpain-2 could affect doxorubicin-triggered cardiac injury. Transgenic mice with inducible cardiomyocyte-specific expression of calpain-2 were generated. An acute cardiotoxicity was induced in both transgenic mice and their relevant wild-type littermates by injection of a single dose of doxorubicin (20 mg/kg) and cardiac injury was analyzed 5 days after doxorubicin injection. Cardiomyocyte-specific up-regulation of calpain-2 did not induce any adverse cardiac phenotypes under physiological conditions by age 3 months, but significantly reduced myocardial injury and improved myocardial function in doxorubicin-treated mice. Cardiac protection of calpain-2 up-regulation was also observed in a mouse model of chronic doxorubicin cardiotoxicity. Up-regulation of calpain-2 increased the protein levels of mitogen activated protein kinase phosphatase-1 (MKP-1) in cultured mouse cardiomyocytes and heart tissues. Over-expression of MKP-1 prevented, whereas knockdown of MKP-1 augmented doxorubicin-induced apoptosis in cultured cardiomyocytes. Moreover, knockdown of MKP-1 offset calpain-2-elicited protective effects against doxorubicin-induced injury in cultured cardiomyocytes. Mechanistically, up-regulation of calpain-2 reduced the protein levels of phosphatase and tensin homolog and consequently promoted Akt activation, leading to increased MKP-1 protein steady state levels by inhibiting its degradation. Collectively, this study reveals a new role of calpain-2 in promoting MKP-1 expression via phosphatase and tensin homolog/Akt signalling. This study also suggests that calpain-2/MKP-1 signaling may represent new therapeutic targets for doxorubicin-induced cardiac injury.
Keywords: Akt, Calpain-2, Doxorubicin cardiotoxicity, MKP-1, PTEN
Introduction
The anthracycline antibiotic doxorubicin is an effective first-line chemotherapeutic agent for many malignancies (Weiss 1992). However, doxorubicin can cause cardiac injury, leading to cardiomyopathy and heart failure, a fatal condition (Takemura and Fujiwara 2007). Unfortunately, effective therapies and preventive approaches are limited for doxorubicin-induced cardiac injury.
Calpains belong to a family of calcium-dependent cysteine proteases (Goll et al. 2003). Among 15 members in the calpain family, calpain-1 and calpain-2 consist of a distinct large catalytic subunit (CAPN1 and CAPN2) encoded by the genes capn1 and capn2, respectively and a common small regulatory subunit encoded by capns1. Both calpain-1 and calpain-2 are tightly controlled by their natural endogenous inhibitor calpastatin. Calpain activation has been implicated in mediating myocardial injury under various pathological conditions including ischemia/reperfusion (Kang et al. 2010; Zheng et al. 2015), diabetes (Li et al. 2011), and sepsis (Ni et al. 2015), etc. In contrast, a previous study reported that calpain protected the heart against hemodynamic stress in a mouse model of pressure overload-induced myocardial dysfunction (Taneike et al. 2011). We recently showed that inhibition of calpain activity enhanced cardiac apoptosis, and exacerbated myocardial dysfunction and mortality in doxorubicin-injected mice (Wang et al. 2013). Moreover, up-regulation of calpain-2 reduced doxorubicin-induced apoptosis in cultured cardiomyocytes. These findings raised an intriguing possibility that up-regulation of calpain-2 may protect against doxorubicin-induced cardiac injury.
Our recent study revealed that the detrimental effects of calpain inhibition seemed to be associated with disruption of protein kinase B (Akt) signaling in cardiomyocytes during doxorubicin stimulation (Wang et al. 2013). It is well known that Akt signaling is positively regulated by PI(3,4,5)P3 (Sugden 2003), produced by phosphoinositol-3-kinase (PI3K). A reduction in PI(3,4,5)P3 levels results in Akt inactivation. Phosphatase and tensin homolog (PTEN) antagonizes PI3K by converting PI(3,4,5)P3 into PI(4,5)P2, leading to Akt inactivation (Simpson and Parsons 2001). Calpain-2 has been reported to cleave PTEN (Briz et al. 2013). These previous studies suggest that calpain-2 may induce Akt activation by decreasing PTEN in doxorubicin-induced cardiac injury. However, this needs further investigation for clarification.
Activation of Akt signaling has been shown to promote mitogen-activated protein kinase (MAPK) phosphatase-1 (MKP-1) expression (Rastogi et al. 2013). MKP-1 is a protein phosphatase that dephosphorylates both the phosphothreonine and phosphotyrosine residues on activated MAPK including ERK1/2, p38 and JNK1/2 (Liu et al. 2007). Studies have demonstrated that MAPK family members (e.g. p38 and JNK1/2) are activated in cardiomyocytes in response to doxorubicin and contribute to doxorubicin cardiotoxicity (Chang et al. 2011b; Lou et al. 2005; Thandavarayan et al. 2010; Yao et al. 2012; Zhang et al. 2015b). However, it has never been reported whether calpain-2 regulates MKP-1 expression and whether MKP-1 plays a role in doxorubicin-induced cardiac injury.
In this study, we investigated whether and how up-regulation of calpain-2 promoted MKP-1 expression thereby preventing doxorubicin-induced cardiotoxicity.
Results
Characterization of conditional cardiomyocyte-specific CAPN2 expression in Tg-Capn2/tTA double-transgenic mice
Transgenic over-expression of CAPN2 was verified in heart tissues of Tg-Capn2/tTA mice (aged 3 months, Supplementary Fig. 1a1). Casein zymography assay confirmed up-regulation of calpain-2 activity by about one fold in Tg-Capn2/tTA mouse hearts (Supplementary Fig. 1b). In contrast, transgenic CAPN2 protein expression was not detected in lung tissues from Tg-Capn2/tTA mice and their relevant wild-type littermates (Supplementary Fig. 1c). To turn off the transgenic expression of CAPN2, we gave doxycycline to Tg-Capn2/tTA mice. After 14 days of continuous doxycycline administration, the transgenic CAPN2 protein expression was not detected in Tg-Capn2/tTA mouse hearts (Supplementary Fig. 1a2). However, transgenic CAPN2 protein expression was recovered by about 70% after 14 days of doxycycline withdrawal (Supplementary Fig. 1a3). In addition, transgenic over-expression of CAPN2 had no effects on CAPN1 and calpain-1 activity in Tg-Capn2/tTA mouse hearts (Supplementary Fig. 1a and 1b).
Under normal conditions, there were no differences in heart weight, body weight, myocardial histology and myocardial function among Tg-Capn2/tTA mice and their relevant wild-type littermates by age 3 months (Fig. 1a, 1c, and 1d, Supplementary Table-1). These results are consistent with a previous report that cardiomyocyte-specific up-regulation of CAPN2 protein and calpain-2 activity is not deleterious to the heart (Galvez et al. 2007) and thus, rule out potential adverse effects of calpain-2 up-regulation and tTA over-expression in Tg-Capn2/tTA mice.
Figure 1. Over-expression of calpain-2 reduces acute doxorubicin cardiotoxicity in Tg-Capn2/tTA mice.

Tg-Capn2/tTA and their relevant wild-type littermates (WT) including wild-type, Tg-tTA, and Tg-Capn2 mice were injected with doxorubicin (20 mg/kg, i.p.) or saline. Five days later, (a) fractional shortening (FS%) was determined by echocardiography. Data are mean ± SD, n = 5-12 mice in each group. *P < 0.05 versus Saline and †P < 0.05 versus Wild-type + Doxorubicin. (b) Troponin I in sera. Data are mean ± SD, n = 5 mice in each group. †P < 0.05 versus Tg-Capn2 + Doxorubicin. (c) Representative cardiac micro-pictures for H&E, Sirius red and Evans blue staining. (d) Quantitation of myocardial collagen deposition determined by Sirius red staining. (e) Quantitation of necrotic cell death determined by Evans blue staining. Data are mean ± SD, n=5 mice in each group. *P < 0.05 versus Tg-Capn2 + Saline and †P < 0.05 versus Tg-Capn2 + Doxorubicin.
Over-expression of calpain-2 attenuates doxorubicin-induced myocardial injury in Tg-Capn2/tTA mice
To determine whether over-expression of calpain-2 provided cardiac protection, we utilized a mouse model of doxorubicin-induced cardiac injury. Tg-Capn2/tTA mice (aged 3 months) and their relevant wild-type littermates including wild-type, Tg-tTA and Tg-Capn2 mice were injected with a single dose of doxorubicin (20 mg/kg, i.p.) or saline. Five days later, doxorubicin induced comparable myocardial dysfunction in different wild-type control mice (wild-type, Tg-tTA and Tg-Capn2) as evidenced by decreased FS% (Fig. 1a, and Supplementary Table-1). However, myocardial function was improved in Tg-Capn2/tTA mice after doxorubicin injection (Fig. 1a, and Supplementary Table-1). No death was observed in both transgenic and wild-type control mice 5 days after doxorubicin injection. Since myocardial function was similar among different wild-type mice (including wild-type, Tg-tTA and Tg-Capn2) under both physiological condition and doxorubicin treatment, we used Tg-Capn2 mice as wild-type control for the following studies.
Doxorubicin injection significantly induced cardiac injury including troponin I and LDH release in sera, patho-histological changes, collagen deposition and cell death in wild-type mouse hearts, all of which were much less in Tg-Capn2/tTA mouse hearts (Fig. 1b-1e, Supplementary Figure-2). These results demonstrate that up-regulation of calpain-2 protects the heart against doxorubicin-induced injury.
To provide direct evidence in support of the protective effects of calpain-2 up-regulation, we isolated and cultured adult cardiomyocytes from adult Tg-Capn2/tTA and Tg-Capn2 mice. Incubation with doxorubicin (1 μmol/L) for 24 hours significantly induced cell death in Tg-Capn2 mouse cardiomyocytes as determined by an increase in the percentage of annexin-V-positive cardiomyocytes (Fig. 2a and 2b). In contrast, doxorubicin induced much less cell death in cardiomyocytes from Tg-Capn2/tTA mice (Fig. 2a and 2b). These results indicate that up-regulation of calpain-2 protects cardiomyocytes against doxorubicin-induced injury.
Figure 2. (a and b) Doxorubicin-induced Cell death is decreased in adult cardiomyocytes from Tg-Capn2/tTA mice.

Adult cardiomyocytes were isolated from Tg-Capn2 and Tg-Capn2/tTA mice, and then incubated with doxorubicin (1 μmol/L) for 24 hours. Cell death was determined by annexin V staining (green color) and nucleus was stained by Hoechst (blue color). (a) Representative pictures for annexin V positive cells. (b) Quantification of cell death (%). Data are mean ± SD, n=10 (repeat 10 times using 10 independent cell cultures). *P < 0.05 versus Tg-Capn2 + saline, †P < 0.05 versus Tg-Capn2 + Doxorubicin. (c, d and e) Chronic doxorubicin-induced cardiotoxicity and the mortality were reduced in Tg-Capn2/tTA mice. Tg-Capn2/tTA mice and their relevant wild-type littermates Tg-Capn2 mice were injected with doxorubicin (DOX, 5 mg/kg/week, i.p.) or saline for 4 consecutive weeks. (c) Four weeks after the last dose of doxorubicin injection, the survival was monitored (*P < 0.05). (d) Fractional shortening (FS%) were analyzed. Data are mean ± SD, n = 6-12. *P < 0.05 vs Saline+ Tg-Capn2 and †P < 0.05 vs Doxorubicin+Tg-Capn2. (e) A representative H&E staining of heart sections from 6 different hearts in each group.
In a mouse model of chronic doxorubicin-induced cardiac injury, transgenic up-regulation of calpain-2 significantly increased the survival rate in Tg-Capn2/tTA mice compared with their wild-type littermates (76.67% vs 57.45%, P < 0.05) (Fig. 2c). Similarly, myocardial function was improved in Tg-Capn2/tTA compared with their wild-type littermates (Fig. 2d). Histological analysis revealed pathological changes including loss of cardiomyocytes and replacement of fibrosis in doxorubicin-injected Tg-Capn2 mouse hearts, which were much less in doxorubicin-injected Tg-Capn2/tTA mouse hearts (Fig. 2e).
Elevation of calpain-2 increases MKP-1 protein expression in cardiomyocytes
Studies have demonstrated that MAPK family members (e.g. p38 and JNK1/2) are activated in cardiomyocytes in response to doxorubicin and contribute to doxorubicin cardiotoxicity (Chang et al. 2011b; Lou et al. 2005; Thandavarayan et al. 2010; Yao et al. 2012; Zhang et al. 2015b). It is well known that MKP-1 is a protein phosphatase that dephosphorylates both the phosphothreonine and phosphotyrosine residues on activated MAPK including ERK1/2, p38 and JNK1/2 (Liu et al. 2007). Thus, we determined whether elevation of calpain-2 promoted MKP-1 expression. To address this, we used neonatal mouse cardiomyocytes as about 30% of adult cardiomyocytes died 30 hours after isolation. Neonatal cardiomyocytes were infected with Ad-Capn2 or Ad-gal. Infection of Ad-Capn2 significantly increased calpain-2 activity and induced MKP-1 protein expression in cardiomyocytes (Fig. 3a and 3b). This effect of calpain-2 elevation was abrogated by incubation with calpain inhibitor-III (Fig. 3c), suggesting that induction of MKP-1 protein is due to calpain-2 activity. In contrast, infection with Ad-Capn1 did not increase MKP-1 protein levels in cardiomyocytes (Fig. 3b).
Figure 3. Calpain-2 increases MKP-1 protein levels by inhibiting its proteasomal degradation in cardiomyocytes.
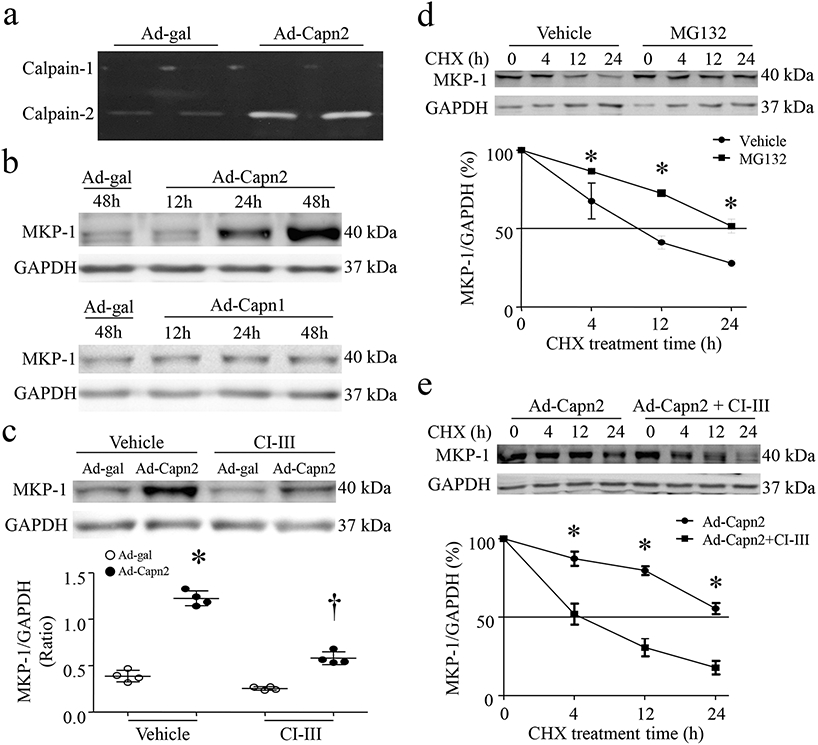
Cultured neonatal cardiomyocytes were infected with Ad-Capn1, Ad-Capn2 or Ad-gal in the presence of calpain inhibitor-III (CI-III, 10μmol/L) or Vehicle for 12-48h. (a) A representative zymography for calpain-1 and calpain-2 activities in duplication from 3 independent cell cultures. (b) Representative western blot for MKP-1 and GAPDH induplication from 4 independent cell cultures. (c) Upper panel; A representative western blot for MKP-1 and GAPDH from 4 independent cell cultures, lower panel; quantitation of MKP-1/GAPDH ratio. Data are mean ± SD, n=4 independent cell cultures. *P < 0.05 versus Ad-gal + Vehicle and †P < 0.05 versus Ad-gal + CI-III. (d) Cardiomyocytes were infected with Ad-MKP1. Forty-eight hours later, the cells were incubated with cycloheximide (CHX, 5 μg/mL) in combination with protease inhibitor MG132 (0.5 μmol/L) or Vehicle for the indicated times. Upper panel; A representative western blot for MKP-1 and GAPDH protein from 3 independent cell cultures, lower panel; quantitation of MKP-1/GAPDH ratio. (e) Forty hours after infection with Ad-Capn2, the cells were incubated with cycloheximide (CHX, 5 μg/mL) in the presence or absence of calpain inhibitor-III (CI-III, 10 μmol/L) for the indicated times. Upper panel; A representative western blots for MKP-1 and GAPDH protein from 3 independent cell cultures, lower panel; quantitation of MKP-1/GAPDH ratio. Data are mean ± SD, n=3. *P < 0.05 versus Vehicle or Ad-Capn2+ CI-III.
Interestingly, elevation of calpain-2 only slightly increased the mRNA levels of MKP-1 in cardiomyocytes without changing the half-life of MPK-1 mRNA (Supplementary Fig. 3a and 3b). Given the significant increase of MKP-1 protein induced by calpain-2 elevation, we hypothesized that calpain-2 might modulate MKP-1 protein degradation. To address this, we infected cultured cardiomyocytes with Ad-Capn2 and determined the half-life of MKP-1 protein. As shown in Fig. 3d, the half-life of calpain-2-induced MKP-1 protein was around 10 hours in cardiomyocytes under normal condition and it was increased to 24 hours in Ad-Capn2-infected cells; however, incubation with calpain inhibitor-III significantly shortened its half-life to about 4 hours (Fig. 3e), suggesting that elevation of calpain-2 increases MKP-1 protein steady state levels in cardiomyocytes. Since it has been shown that MKP-1 protein is targeted for degradation via the ubiquitin-proteasome pathway in cardiomyocytes (Xie et al. 2009), we examined whether inhibition of the proteasome could prevent MKP-1 degradation. Co-combination with MG132, a proteasome inhibitor, significantly increased its half-life to 24 hours (Fig. 3d). Taken together, these results demonstrate that elevation of calpain-2 inhibits proteasomal degradation of MKP-1 protein.
Over-expression of MKP-1 attenuates apoptosis and silencing of MKP-1 offsets the inhibitory effect of calpain-2 elevation on apoptosis in doxorubicin-stimulated cardiomyocytes
To investigate the role of MKP-1 in doxorubicin-induced apoptosis, we infected neonatal cardiomyocytes with Ad-MKP1 or Ad-gal. Twenty-four hours after infection, cardiomyocytes were incubated with doxorubicin or saline for an additional 24 hours. Infection of Ad-MKP1 increased the protein levels of MKP-1 in cardiomyocytes (Supplementary Fig. 4a). In response to doxorubicin, apoptosis was induced in cardiomyocytes, which was inhibited by over-expression of MKP-1 in cardiomyocytes (Fig. 4a and 4b). Over-expression of MKP-1 also prevented doxorubicin-induced phosphorylation of ERK1/2, p38 and JNK1/2 in cardiomyocytes (Supplementary Fig. 4b-4e). These results demonstrate that over-expression of MKP-1 prevents MAPK activation and apoptosis in doxorubicin-stimulated cardiomyocytes.
Figure 4. Calpain-2 elevation prevents doxorubicin-induced apoptosis through MKP-1 induction.

(A and B) Cultured neonatal cardiomyocytes were infected with Ad-MKP1 or Ad-gal in combination with Ad-Pten. After 24 hours of infection, cardiomyocytes were incubated with doxorubicin (1 μmol/L) or saline for 24 hours. Caspase-3 activity (a) and DNA fragmentation (b) were measured. (c and d) Cultured neonatal cardiomyocytes were transfected with MKP-1 siRNA or a scrambled siRNA as a control (NC siRNA). Cardiomyocytes were then infected with Ad-Capn2 or Ad-gal for 24 hours, followed by incubation with doxorubicin (1 μmol/L) for an additional 24 hours. Caspase-3 activity (c) and DNA fragmentation (d) were measured. Data are mean ± SD, n=5-6 independent cell cultures. *P < 0.05 versus Ad-gal or Ad-gal + NC siRNA, †P < 0.05 versus Doxorubicin + Ad-gal or Doxorubicin +Ad-gal + NC siRNA + Doxorubicin, and ‡P < 0.05 versus Doxorubicin + Ad-Pten or Doxorubicin+Ad-Capn2 + NC siRNA.
To determine whether the anti-apoptotic effect of calpain-2 elevation was mediated through MKP-1 induction, we knocked down MKP-1 expression in neonatal cardiomyocytes using MKP-1 siRNA. A scrambled siRNA served as a control. Transfection of MKP-1 siRNA led to about 50% reduction in MKP-1 protein (Supplementary Fig. 5a). After knockdown of MKP-1, cardiomyocytes were infected with Ad-Capn2 or Ad-gal, and then exposed to doxorubicin for additional 24 hours. Treatment with MKP-1 siRNA enhanced doxorubicin-induced apoptosis in cardiomyocytes. Importantly, knockdown of MKP-1 abrogated the protective effects of calpain-2 elevation on apoptosis in doxorubicin-stimulated cardiomyocytes (Fig. 4c and 4d). Furthermore, infection with Ad-Capn2 inhibited doxorubicin-induced phosphorylation of ERK1/2, p38 and JNK1/2, which was offset by MKP-1 knockdown (Supplementary Fig. 5b-5e). These results suggest that the protective effects of calpain-2 elevation may be mediated through MKP-1 signalling in doxorubicin-stimulated cardiomyocytes.
Elevation of calpain-2 increases MKP-1 protein steady state levels in cardiomyocytes via Akt signaling
Relevant to its potential cardiac protection, calpain-2 has been demonstrated to induce Akt signaling in non-cardiomyocytes (Ho et al. 2012). Since elevated Akt signaling protects against doxorubicin-induced injury (Taniyama and Walsh 2002) and our recent study showed that inhibition of calpain led to inactivation of Akt signalling in doxorubicin-induced cardiomyocytes (Wang et al. 2013), we determined whether elevation of calpain-2 promoted Akt activation in cardiomyocytes. To examine this, we infected neonatal cardiomyocytes with Ad-Capn1, Ad-Capn2 or Ad-gal. Elevation of calpain-2 activity increased the protein levels of phosphorylated Akt at Thr308 and Ser473 in a time-dependent manner (Fig. 5a), indicative of Akt activation, which correlated well with a time-dependent increase in MKP-1 protein levels (Fig. 3b). In contrast, infection with Ad-Capn1 did not change Akt phosphorylation (Supplementary Fig. 6a-6d). These results demonstrate that calpain-2 induces Akt activation.
Figure 5. Calpain-2 elevation promotes MKP-1 protein expression through PTEN/Akt signaling.
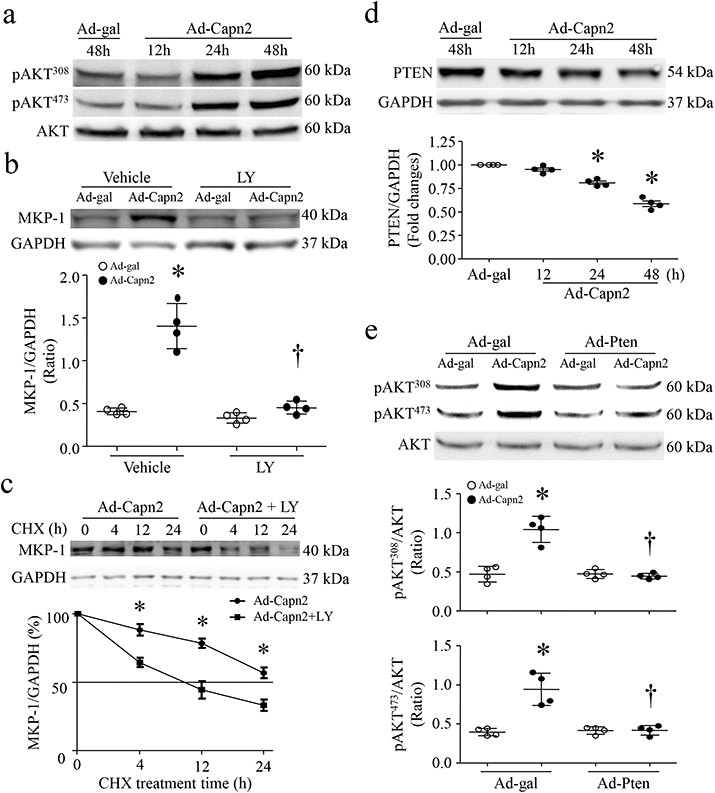
Cultured neonatal cardiomyocytes were infected with Ad-Capn2 or Ad-gal. (a) A representative western blot for Akt and phosphorylated Akt (Ser308 and Thr473) from 3 different cell cultures. (b) Cultured neonatal cardiomyocytes were infected with Ad-Capn2 or Ad-gal in the presence of LY294002 (LY, 2μmol/L) or Vehicle for the indicated times. Upper panel; A representative western blot from 4 different cell cultures for MKP-1 and GAPDH protein, lower panel; quantitation of MKP-1/GAPDH ratio. (c) Twenty-four hours after Ad-Capn2 infection, the cells were incubated with cycloheximide (CHX, 5 μg/mL) for the indicated times. Upper panel; A representative western blot from 4 independent cell cultures for MKP-1 and GAPDH protein, lower panel; quantitation of MKP-1/GAPDH ratio. Data are mean ± SD, n=4 independent cell cultures. *P < 0.05 versus Ad-gal+Vehicle or Ad-Capn2+LY and †P < 0.05 versus Ad-Capn2+Vehicle. (d) Cultured neonatal cardiomyocytes were infected with Ad-Capn2 or Ad-gal. Upper panel; A representative western blot for PTEN and GAPDH from 4 independent cell cultures, lower panel; quantitation of PTEN/GAPDH ratio. (e) Cardiomyocytes were infected with Ad-Pten or Ad-gal, followed by Ad-Capn2. Upper panel; A representative western blot for phosphorylated Akt and total Akt, lower panels; quantitation of phosphorylated Akt/Akt ratio. Data are mean ± SD, n=4 independent cell cultures. *P < 0.05 versus Ad-gal and †P < 0.05 versus Ad-Capn2+Ad-gal.
We then determined whether elevation of calpain-2 protects MKP-1 stability through Akt signaling. To address this, we infected neonatal cardiomyocytes with Ad-Capn2 or Ad-gal in the presence of LY294002 or vehicle. Blockade of Akt signalling with LY294002 prevented calpain-2-induced MKP-1 protein expression in cardiomyocytes (Fig. 5b). Consistently, blocking Akt signalling with LY294002 accelerated MKP-1 protein degradation in Ad-Capn2-infected cardiomyocytes (Fig. 5c). This result supports that elevation of calpain-2 inhibits MKP-1 degradation via Akt signalling.
Over-expression of calpain-2 induces Akt activation by decreasing phosphatase and tensin homolog (PTEN)
Akt signaling is positively regulated by PI(3,4,5)P3 (Sugden 2003), produced by phosphoinositol-3-kinase (PI3K). A reduction in PI(3,4,5)P3 levels results in Akt inactivation. PTEN antagonizes PI3K by converting PI(3,4,5)P3 into PI(4,5)P2, leading to Akt inactivation (Simpson and Parsons 2001). Calpain-2 has been reported to cleave PTEN (Briz et al. 2013). Thus, we hypothesized that calpain-2 promoted Akt activation by reducing PTEN in cardiomyocytes. To examine this hypothesis, we infected neonatal cardiomyocytes with Ad-Capn1, Ad-Capn2 or Ad-gal. Infection with Ad-Capn2 (Fig. 5d) but not Ad-Capn1 decreased the protein levels of PTEN (Supplementary Fig. 6b and 6e). The time-dependent reduction of PTEN protein correlated well with an increase in Akt phosphorylation (Fig. 5a), suggesting that calpain-2 may promote Akt activation by decreasing PTEN. To substantiate this, we infected neonatal cardiomyocytes with Ad-Pten or Ad-gal in combination with Ad-Capn2 or Ad-gal. Forty-eight hours later, infection with Ad-Capn2 induced Akt activation in cardiomyocytes, the effect of which was prevented by over-expression of PTEN with Ad-Pten (Fig. 5e, Supplementary Figure 6f). These results provide direct evidence that calpain-2 promotes Akt activation by decreasing PTEN protein in cardiomyocytes.
Inhibitory effects of calpain-2 elevation or MKP-1 over-expression on apoptosis are prevented by increasing PTEN or blocking Akt signalling in doxorubicin-stimulated cardiomyocytes
To determine whether calpain-2 protected cardiomyocytes against doxorubicin-induced apoptosis through PTEN/Akt/MKP-1 signalling, we infected neonatal cardiomyocytes with Ad-Capn2, Ad-MKP1 or Ad-gal in combination with Ad-Pten or Ad-gal, or in the presence of LY294002 or vehicle, followed by incubation with doxorubicin or saline for 24 hours. Consistently, infection with Ad-Capn2 inhibited doxorubicin-induced apoptosis in cardiomyocytes. However, infection with Ad-Pten or incubation with LY294002 abolished the inhibitory effects of calpain-2 (Fig. 6a-6d) or MKP-1 over-expression (Fig. 4a and 4b) on apoptosis in doxorubicin-stimulated cardiomyocytes. These results support that the protective effects of calpain-2 are mediated through PTEN/Akt/MKP-1 signaling in doxorubicin-stimulated cardiomyocytes.
Figure 6. Calpain-2 elevation prevents doxorubicin-induced apoptosis via PTEN and Akt signaling.
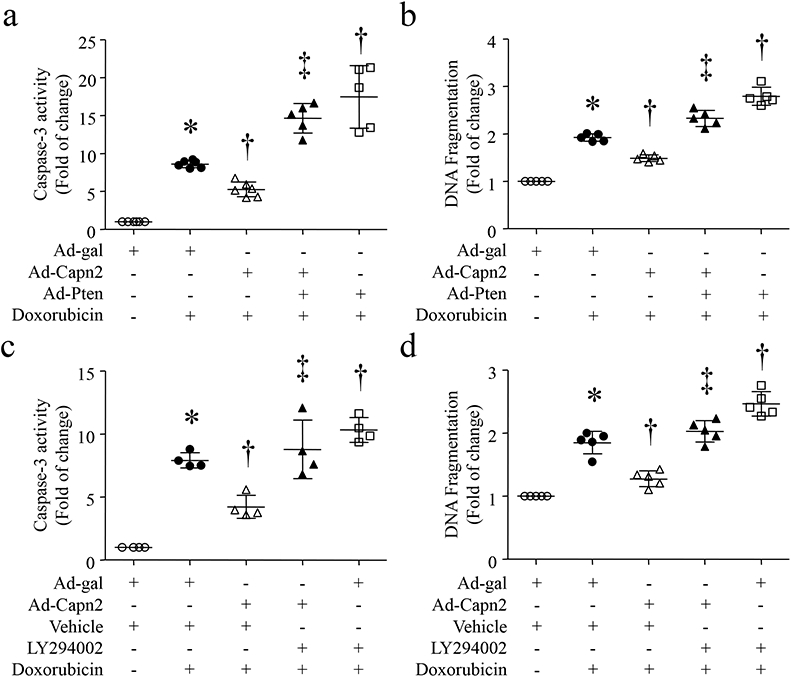
(a and b) Cultured neonatal cardiomyocytes were infected with Ad-Capn2 or Ad-gal in combination with Ad-Pten. After 24 hours of infection, cardiomyocytes were incubated with doxorubicin (1μmol/L) or saline for 24 hours. Caspase-3 activity (a) and DNA fragmentation (b) were measured. (c and d) Cultured neonatal cardiomyocytes were infected with Ad-Capn2 or Ad-gal in combination with LY294002 (2 μmol/L) or Vehicle for 24 hours, and then incubated with doxorubicin (1 μmol/L) for additional 24 hours. Caspase-3 activity (c) and DNA fragmentation (d) were measured. Data are mean ± SD, n=4-6 independent cell cultures. *P < 0.05 versus Ad-gal or Ad-gal + Vehicle, †P < 0.05 versus Ad-gal + Doxorubicin and ‡P < 0.05 versus Ad-Capn2 + Doxorubicin.
Increased calpain-2 reduces the protein levels of PTEN, increases Akt activation and induces MKP-1 protein in transgenic mouse hearts
In line with the results from cultured cardiomyocytes, transgenic up-regulation of calpain-2 reduced the protein levels of PTEN (Fig. 7a), increased phosphorylation of Akt (Fig. 7b) and induced MKP-1 protein expression (fig. 7c) in mouse hearts under normal condition. Upon doxorubicin injection, the protein levels of PTEN were elevated, and phosphorylation of Akt and MKP-1 protein expression were decreased in wild-type mouse hearts. However, these effects of doxorubicin were prevented in calpain-2 transgenic mouse hearts (Fig. 7a-7c). Thus, these results confirm the role of calpain-2 in regulation of PTEN/Akt/MKP-1 signaling in in vivo hearts.
Figure 7. Over-expression of calpain-2 reduces PTEN protein levels and increases Akt activation and MKP-1 protein expression in Tg-Capn2/tTA mouse hearts.

Tg-Capn2/tTA (TG) and Tg-Capn2 mice (WT) received doxorubicin or saline. Five days later, heart tissues were analyzed for PTEN, Akt and MKP-1 protein expression. (a) Upper panel; representative western blot for PTEN and GAPDH from 2 out of 5 different hearts from WT and TG at 5 days, lower panel: quantitation of MKP-1/GAPDH ratio. (b) Upper panel; representative western blot for Akt and phosphorylated Akt (Ser308 and Thr473) from 2 out of 6 different hearts form WT and TG, lower panel; quantitation of phosphorylated Akt/Akt ratio. (c) Upper panel; representative western blot for MKP-1 and GAPDH from 2 out of 5 different hearts from WT and TG, lower panel: quantitation of MKP-1/GAPDH ratio. Data are mean ± SD, n=5-6 different hearts. *P < 0.05 versus WT + saline and †P < 0.05 versus WT + Doxorubicin.
Discussion
The major finding of this study is that up-regulation of calpain-2 reduced doxorubicin-induced cardiac injury and that the protective effect of calpain-2 up-regulation was mediated through MKP-1 up-regulation. To the best of our knowledge, this is the first study that demonstrates that up-regulation of calpain-2 increases MKP-1 protein steady state levels by inhibiting its degradation, leading to cardiac protection in doxorubicin cardiotoxicity.
Calpain-2 has been suggested to induce death and survival signaling (Abeyrathna et al. 2016; Chang et al. 2011a; Ho et al. 2012; Wang et al. 2016; Yuasa et al. 2016). Our recent study reported that over-expression of CAPN2 decreased doxorubicin-induced apoptosis in cardiomyocytes (Wang et al. 2013). In this study, we extend those data in mouse models of both acute and chronic doxorubicin-induced cardiac injury. Similar cardio-protection of calpain-2 up-regulation was also observed in heat stress-induced injury (Liu et al. 2018) but not in ischemia/reperfusion injury and septic mice (data not shown), suggesting that the cardioprotective role of calpain-2 may be limited to distinct conditions. The protective effect of calpain-2 in doxorubicin-induced cardiac injury may be clinically important because dexrazoxane, the only drug available to mitigate doxorubicin cardiotoxicity in clinical settings, was reported to induce calpain-2 expression in doxorubicin-injected rat hearts (Zhang et al. 2015a). In contrast to calpain-2, activation of calpain-1 is deleterious to the heart and sufficiently induces cardiomyopathic features in transgenic mice (Galvez et al. 2007). In response to doxorubicin, a recent study demonstrated that calpain-1 was activated and promoted apoptosis in cardiac muscles (Min et al. 2015). Thus, calpain-2 may counteract the detrimental role that calpain-1 plays in certain conditions.
It is important to mention although calpain-2 requires mili-molar concentration of Ca2+ for its activation, which may never be reached within cells, certain modifications of calpain-2 protein either lower Ca2+ requirement for its activation or directly induce its activation without increasing Ca2+. For example, phosphorylation of calpain-2 by ERK1/2 induces its activation in the absence of Ca2+ (Glading et al. 2004). Indeed, our data shows that ERK1/2 is activated in doxorubicin-stimulated cardiomyocytes. This may provide important basis for examining effects of calpain-2 over-expression in cardiac patho-physiology.
The present study reveals a novel role for calpain-2 in promoting MKP-1 protein expression and further demonstrates that the inhibitory effect of calpain-2 on apoptosis is mediated through MKP-1 induction in doxorubicin-induced cardiomyocytes. It is well known that MKP-1 de-phosphorylates and inactivates MAPK members (e.g. ERK1/2, JNK1/2 and p38) (Liu et al. 2007). Doxorubicin induces activation of ERK1/2, p38 and JNK1/2 in cardiomyocytes and inhibition of them reduces doxorubicin-induced cardiac injury (Chang et al. 2011b; Lou et al. 2005; Thandavarayan et al. 2010; Yao et al. 2012; Zhang et al. 2015b). Similarly, the present study shows that up-regulation of calpain-2 or MKP-1 decreases phosphorylation of ERK1/2, p38 and JNK1/2. These data provide additional evidence in support of the notion that elevation of calpain-2 protects against doxorubicin-induced cardiac injury through MKP-1 signaling. MKP-1 has also been implicated in cardioprotective effects of insulin and dexamethasone. These previous reports together with our present finding suggest a cardioprotective role of MKP-1 under stress. In contrast, a previous study showed that age-associated increase in MKP-1 might result in loss of infarct size reduction with ischemic post-conditioning. We currently do not known whether MKP-1 induction provides similar cardioprotection in old mouse hearts upon doxorubicin injection. Future studies are needed to determine the implications of calpain-2-mediated MKP-1 expression in cardiac patho-physiology using the transgenic Tg-Capn2/tTA mice.
It is worthwhile to mention that MKP-1 has been reported to mediate chemoresistance in human breast cancer. Thus, any approaches to enhance MKP-1 expression to reduce doxorubicin cardiotoxicity may potentially impede its cancer-killing efficacy.
The mechanisms underlying calpain-2 induction of MKP-1 expression have never been reported. MKP-1 expression is tightly regulated transcriptionally, post-transcriptionally and post-translationally (Kuwano and Gorospe 2008). In this study, elevation of calpain-2 slightly increased the mRNA levels of MKP-1, which may not explain a dramatic elevation of MKP-1 protein levels, suggesting the involvement of translational and/or post-translational mechanisms. In support of this, the present study provides evidence demonstrating that elevation of calpain-2 increases MKP-1 protein steady state levels by inhibiting its degradation. Previous studies have demonstrated that MKP-1 protein is degraded via the ubiquitin proteasome pathway (Kuwano and Gorospe 2008; Xie et al. 2009). Indeed, the present study further shows that inhibition of proteasome prevents MKP-1 protein degradation, suggesting that calpain-2 increases MKP-1 protein levels possibly by inhibiting the ubiquitin proteasome pathway.
In pursuit of the signaling mechanisms by which calpain-2 inhibits MKP-1 protein degradation, our data demonstrate that calpain-2 induces a reduction of PTEN protein and subsequently promotes Akt activation, supporting that PTEN is a target of calpain-2 (Briz et al. 2013). This is also supported by our recent study which showed that inhibition of calpain blocked Akt activation in cardiomyocytes (Wang et al. 2013). Activation of Akt signaling has been suggested to promote MKP-1 expression (Rastogi et al. 2013). Similarly, the present study shows that elevation of calpain-2 induces Akt activation by increasing its phosphorylation (Ser473 and Thr308). Moreover, blocking Akt signaling abrogates the effects of calpain-2 elevation in preventing MKP-1 protein degradation. Thus, we argue that calpain-2 promotes MKP-1 protein stability by activating Akt signaling. In fact, Akt activation was reported to inhibit artrogin-1 expression (Sandri et al. 2004; Yoshida et al. 2010), a muscle-specific ubiquitin-ligase, which has been reported to promote MKP-1 ubiquitination and degradation (Xie et al. 2009).
In summary, this study provides direct evidence demonstrating that up-regulation of calpain-2 protects the heart against doxorubicin-induced injury. The cardio-protective effect of calpain-2 up-regulation may be mediated, at least partially by increasing MKP-1 via decreased PTEN and consequent Akt activation. Thus, calpain-2/MKP-1 signaling may represent new therapeutic targets for doxorubicin-induced cardiac injury.
Materials and Methods
Animals
This investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication, 8th Edition, 2011). All experimental procedures were approved by the Animal Use Subcommittee at the Western University, Canada. Breeding pairs of C57BL/6 mice were purchased from the Jackson Laboratory. Transgenic mice with cardiomyocyte-specific over-expression of tetracycline transactivator (Tg-tTA) were generated as described previously (Sanbe et al. 2003). All animals were housed in a temperature and humidity controlled facility at a 12-hour light and dark cycles with water and food ad libitum.
A novel line of transgenic mice with human CAPN2 expression driven by tTA inducible mouse α-myosin heavy chain (MHC) promoter (Tg-Capn2) was generated as described (Sanbe et al. 2003). The Tg-Capn2 mice were crossed with Tg-tTA mice to produce wild-type, Tg-tTA, Tg-Capn2 and Tg-Capn2/tTA mice, which were genotyped by polymerase chain reaction (PCR) with both tTA and human capn2 primers.
Experimental protocol
Acute cardiotoxicity was induced by injection of a single dose of doxorubicin (20 mg/kg, i.p., Sigma) in male mice (aged 10-12 weeks) (Wang et al. 2013). Mice injected with sterile saline were used as controls. After five days of injection, animals were subjected to echocardiography or heart tissue collection for further analyses.
Chronic doxorubicin-induced cardiac injury was induced by multiple-injections of doxorubicin (5 mg/kg/week for 4 consecutive weeks, total 20 mg/kg, i.p., Sigma) in male mice (aged 2 months). Control mice were injected with sterile saline. After 4 weeks of the last injection, myocardial function was assessed, and heart tissues were collected for histological analyses.
Echocardiography
Animals were lightly anaesthetized with inhaled isoflurane (1%) and imaged on a warm handling platform using a 40 MHz linear array transducer attached to a preclinical ultrasound system (Vevo 2100, FUJIFILM Visual Sonics, Canada) with nominal in-plane spatial resolution of 40 μm (axial) × 80 μm (lateral) as we described recently (Li et al. 2016; Ma et al. 2012; Ni et al. 2015; Wang et al. 2013). Left ventricular (LV) end-systolic inner diameter (LVIDs), LV end-diastolic inner diameter (LVIDd), and fractional shortening (FS) were analyzed. The pulsed wave Doppler measurements of maximal early (E) and late (A) transmitral velocities in diastole were performed in the apical view with the cursor at mitral valve inflow.
Cell membrane permeability to evans blue dye (EBD)
EBD was dissolved in saline and injected into mice (100 mg/kg body weight, i.p.). Twenty-four hours later, the heart was harvested and embedded in optimal cutting temperature (OCT) compound (Sakura), snap frozen in liquid nitrogen and cut into 5-μm cryosections. EBD uptakes (red) were visualized under fluorescent microscope.
Histological analyses
Heart tissues were routinely fixed, embedded, processed and sectioned. Tissue sections were processed either for H&E staining or for assessments of total collagen content as described in our recent report (Ma et al. 2012).
Lactate dehydrogenase (LDH) and troponin I release
The levels of LDH and troponin I in sera were measured using commercially available kits from Sigma-Aldrich (Canada) and MyoBioSource.com (USA) following the manufacturer’s instructions, respectively.
Mouse cardiomyocyte cultures
Neonatal mice (born in 48 h) were euthanized by decapitation. Neonatal cardiomyocytes were prepared and cultured according to methods we described previously (Peng et al. 2003).
Adult mice were anaesthetized with a mixture of ketamine (100 mg/kg, i.p.) and xylazine (5 mg/kg, i.p.). Hearts were isolated and perfused after ensuring that mice did not respond to needle-punch to the skin. Mouse ventricle cardiomyocytes were isolated according to methods we described previously (Ni et al. 2015; Wang et al. 2013).
Adenoviral infection of cardiomyocytes
Cultured neonatal cardiomyocytes were infected with adenoviral vectors containing capn1, capn2, pten and mkp-1 gene (Ad-Capn2, Ad-Capn1, Ad-Pten, Ad-MKP-1, SignaGen, USA) or β-gal (Ad-gal, Vector Biolabs, USA) as a control at a multiplicity of infection of 100 plaque forming units/cell (Peng et al. 2003).
Caspase-3 activity
Caspase-3 activity in cell lysates was measured with a caspase-3 fluorescence assay kit (Biomol Research Laboratories, USA).
Determination of cellular DNA fragmentation
Cultured neonatal cardiomyocytes were pre-labeled with BrdU (1 μg/ml). DNA fragmentation was determined using a Cellular DNA Fragmentation ELISA kit (Roche Applied Science, Germany) according to the manufacturer’s instructions.
Real-time reverse transcriptase-PCR
Total RNA was extracted from cultured neonatal cardiomyocytes and heart tissues using the Trizol Reagent (Sigma-Aldrich, USA) following the manufacturer's instruction (Peng et al. 2003). Real-time reverse transcriptase-PCR was performed for analyzing mRNA levels of MKP-1, 18S rRNA and GAPDH (Li et al. 2010).
Western blot analysis
The protein levels of CAPN1, CAPN2, PTEN, p38, phosphorylated p38, ERK1/2, phosphorylated ERK1/2, JNK1/2, phosphorylated JNK1/2, Akt, phosphorylated Akt (Thr308 and Ser473), MKP-1, ubiquitin and GAPDH were determined by western blot analysis using their specific antibodies (Peng et al. 2003) (Cell Signaling Technology, USA, 1:1000 or Santa Cruz Biotechnology, USA, 1:500).
Treatment of siRNA
Cholesterol and OMe-conjugated siRNAs for MKP-1 were purchased from Guangzhou Ribobio Co., Ltd. (China). A scrambled siRNA conjugated with cholesterol and OMe served as a control. Cultured neonatal cardiomyocytes were incubated with siRNAs (50 nmol/L) in normal culture medium. Our recent study showed that cholesterol-conjugated siRNAs enter cardiomyocytes directly with an efficiency of more than 90% (Li et al. 2016).
Calpain activity
Calpain-1 and calpain-2 activities in cell lysates were measured by casein zymography as described previously (Li et al. 2011).
Determination of MKP-1 mRNA stability
At different time-points after addition of actinomycin D (2 μg/ml, Sigma), the mRNA levels of MKP-1 and GAPDH were measured by RT-qPCR, normalized to 18S rRNA. The time required for mRNA to reach one-half of its initial abundance (or half-life) was determined by plotting the results of RT-qPCR on a logarithmic scale.
Assessment of MKP-1 protein degradation
After addition of cycloheximide (5 μg/ml, Sigma), a protein translation inhibitor to block de novo protein synthesis in combination with MG132 (0.5 μmol/L, Sigma), calpain inhibitor III (10 μmol/L, Sigma), LY294002 (2 μmol/L, Sigma) or vehicle, western blot analysis was performed to quantify the protein levels of MKP-1 and GAPDH at different time points.
Statistical analysis
Data were given as Mean ± SD. Student’s t test was employed for comparisons within two groups. ANOVA followed by Newman-Keuls test was performed for multi-group comparisons. Survival curves were created by the method of Kaplan and Meier, and compared by log-rank test. A value of P < 0.05 was regarded as statistically significant.
Supplementary Material
Acknowledgements
This work was supported by Natural Science Foundation of Jiangsu Province (BK20171216 to T.P.), the Heart and Stroke Foundation of Canada (G-17-0018361 to T.P.), and Natural Science Foundation of Zhejiang Province (LY14H020005 to J.L.).
Footnotes
Conflict of interest
None
References
- Abeyrathna P, Kovacs L, Han W, Su Y (2016) Calpain-2 activates Akt via TGF-beta1-mTORC2 pathway in pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol 311(1):C24–34 doi:ajpcell.00295.2015 [pii] 10.1152/ajpcell.00295.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briz V, Hsu YT, Li Y, Lee E, Bi X, Baudry M (2013) Calpain-2-mediated PTEN degradation contributes to BDNF-induced stimulation of dendritic protein synthesis. J Neurosci 33(10):4317–28 doi:33/10/4317 [pii] 10.1523/JNEUROSCI.4907-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H, Zhang L, Xu PT, et al. (2011a) Nuclear translocation of calpain-2 regulates propensity toward apoptosis in cardiomyocytes of tail-suspended rats. J Cell Biochem 112(2):571–80 doi: 10.1002/jcb.22947 [DOI] [PubMed] [Google Scholar]
- Chang WT, Li J, Haung HH, et al. (2011b) Baicalein protects against doxorubicin-induced cardiotoxicity by attenuation of mitochondrial oxidant injury and JNK activation. J Cell Biochem 112(10):2873–81 doi: 10.1002/jcb.23201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez AS, Diwan A, Odley AM, et al. (2007) Cardiomyocyte degeneration with calpain deficiency reveals a critical role in protein homeostasis. Circ Res 100(7):1071–8 doi:01.RES.0000261938.28365.11 [pii] 10.1161/01.RES.0000261938.28365.11 [DOI] [PubMed] [Google Scholar]
- Glading A, Bodnar RJ, Reynolds IJ, et al. (2004) Epidermal growth factor activates m-calpain (calpain II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol Cell Biol 24(6):2499–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goll DE, Thompson VF, Li H, Wei W, Cong J (2003) The calpain system. Physiol Rev 83(3):731–801 doi: 10.1152/physrev.00029.2002 83/3/731 [pii] [DOI] [PubMed] [Google Scholar]
- Ho WC, Pikor L, Gao Y, Elliott BE, Greer PA (2012) Calpain 2 regulates Akt-FoxO-p27(Kip1) protein signaling pathway in mammary carcinoma. J Biol Chem 287(19):15458–65 doi:M112.349308 [pii] 10.1074/jbc.M112.349308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MY, Zhang Y, Matkovich SJ, Diwan A, Chishti AH, Dorn GW 2nd (2010) Receptor-independent cardiac protein kinase Calpha activation by calpain-mediated truncation of regulatory domains. Circ Res 107(7):903–12 doi:CIRCRESAHA.110.220772 [pii] 10.1161/CIRCRESAHA.110.220772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwano Y, Gorospe M (2008) Protecting the stress response, guarding the MKP-1 mRNA. Cell Cycle 7(17):2640–2 doi:6534 [pii] 10.4161/cc.7.17.6534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhu H, Shen E, Wan L, Arnold JM, Peng T (2010) Deficiency of rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes 59(8):2033–42 doi:db09-1800 [pii] 10.2337/db09-1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Zhang L, Ni R, et al. (2016) Disruption of calpain reduces lipotoxicity-induced cardiac injury by preventing endoplasmic reticulum stress. Biochim Biophys Acta 1862(11):2023–2033 doi:S0925-4439(16)30194-6 [pii] 10.1016/j.bbadis.2016.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Ma J, Zhu H, et al. (2011) Targeted inhibition of calpain reduces myocardial hypertrophy and fibrosis in mouse models of type 1 diabetes. Diabetes 60(11):2985–94 doi:db10-1333 [pii] 10.2337/db10-1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Shepherd EG, Nelin LD (2007) MAPK phosphatases--regulating the immune response. Nat Rev Immunol 7(3):202–12 doi:nri2035 [pii] 10.1038/nri2035 [DOI] [PubMed] [Google Scholar]
- Liu ZF, Ji JJ, Zheng D, Su L, Peng T (2018) Calpain-2 protects against heat stress-induced cardiomyocyte apoptosis and heart dysfunction by blocking p38 mitogen-activated protein kinase activation. J Cell Physiol doi: 10.1002/jcp.27750 [DOI] [PubMed] [Google Scholar]
- Lou H, Danelisen I, Singal PK (2005) Involvement of mitogen-activated protein kinases in adriamycin-induced cardiomyopathy. Am J Physiol Heart Circ Physiol 288(4):H1925–30 doi:288/4/H1925 [pii] 10.1152/ajpheart.01054.2004 [DOI] [PubMed] [Google Scholar]
- Ma J, Wei M, Wang Q, et al. (2012) Deficiency of Capn4 gene inhibits nuclear factor-kappaB (NF-kappaB) protein signaling/inflammation and reduces remodeling after myocardial infarction. J Biol Chem 287(33):27480–9 doi:M112.358929 [pii] 10.1074/jbc.M112.358929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min K, Kwon OS, Smuder AJ, et al. (2015) Increased mitochondrial emission of reactive oxygen species and calpain activation are required for doxorubicin-induced cardiac and skeletal muscle myopathy. J Physiol 593(8):2017–36 doi: 10.1113/jphysiol.2014.286518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni R, Zheng D, Wang Q, et al. (2015) Deletion of capn4 Protects the Heart Against Endotoxemic Injury by Preventing ATP Synthase Disruption and Inhibiting Mitochondrial Superoxide Generation. Circ Heart Fail 8(5):988–96 doi:CIRCHEARTFAILURE.115.002383 [pii] 10.1161/CIRCHEARTFAILURE.115.002383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng T, Lu X, Lei M, Feng Q (2003) Endothelial nitric-oxide synthase enhances lipopolysaccharide-stimulated tumor necrosis factor-alpha expression via cAMP-mediated p38 MAPK pathway in cardiomyocytes. J Biol Chem 278(10):8099–105 doi: 10.1074/jbc.M207288200 M207288200 [pii] [DOI] [PubMed] [Google Scholar]
- Rastogi R, Jiang Z, Ahmad N, et al. (2013) Rapamycin induces mitogen-activated protein (MAP) kinase phosphatase-1 (MKP-1) expression through activation of protein kinase B and mitogen-activated protein kinase kinase pathways. J Biol Chem 288(47):33966–77 doi:M113.492702 [pii] 10.1074/jbc.M113.492702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanbe A, Gulick J, Hanks MC, Liang Q, Osinska H, Robbins J (2003) Reengineering inducible cardiac-specific transgenesis with an attenuated myosin heavy chain promoter. Circ Res 92(6):609–16 doi: 10.1161/01.RES.0000065442.64694.9F 01.RES.0000065442.64694.9F [pii] [DOI] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, et al. (2004) Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117(3):399–412 doi:S0092867404004003 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson L, Parsons R (2001) PTEN: life as a tumor suppressor. Exp Cell Res 264(1):29–41 doi: 10.1006/excr.2000.5130 S0014-4827(00)95130-9 [pii] [DOI] [PubMed] [Google Scholar]
- Sugden PH (2003) Ras, Akt, and mechanotransduction in the cardiac myocyte. Circ Res 93(12):1179–92 doi: 10.1161/01.RES.0000106132.04301.F5 93/12/1179 [pii] [DOI] [PubMed] [Google Scholar]
- Takemura G, Fujiwara H (2007) Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog Cardiovasc Dis 49(5):330–52 doi:S0033-0620(06)00120-4 [pii] 10.1016/j.pcad.2006.10.002 [DOI] [PubMed] [Google Scholar]
- Taneike M, Mizote I, Morita T, et al. (2011) Calpain protects the heart from hemodynamic stress. J Biol Chem 286(37):32170–7 doi:M111.248088 [pii] 10.1074/jbc.M111.248088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniyama Y, Walsh K (2002) Elevated myocardial Akt signaling ameliorates doxorubicin-induced congestive heart failure and promotes heart growth. J Mol Cell Cardiol 34(10):1241–7 doi:S0022282802920687 [pii] [DOI] [PubMed] [Google Scholar]
- Thandavarayan RA, Watanabe K, Sari FR, et al. (2010) Modulation of doxorubicin-induced cardiac dysfunction in dominant-negative p38alpha mitogen-activated protein kinase mice. Free Radic Biol Med 49(9):1422–31 doi:S0891-5849(10)00473-9 [pii] 10.1016/j.freeradbiomed.2010.08.005 [DOI] [PubMed] [Google Scholar]
- Wang Y, Lopez D, Davey PG, et al. (2016) Calpain-1 and calpain-2 play opposite roles in retinal ganglion cell degeneration induced by retinal ischemia/reperfusion injury. Neurobiol Dis 93:121–8 doi:S0969-9961(16)30101-2 [pii] 10.1016/j.nbd.2016.05.007 [DOI] [PubMed] [Google Scholar]
- Wang Y, Zheng D, Wei M, et al. (2013) Over-expression of calpastatin aggravates cardiotoxicity induced by doxorubicin. Cardiovasc Res 98(3):381–90 doi:cvt048 [pii] 10.1093/cvr/cvt048 [DOI] [PubMed] [Google Scholar]
- Weiss RB (1992) The anthracyclines: will we ever find a better doxorubicin? Semin Oncol 19(6):670–86 [PubMed] [Google Scholar]
- Xie P, Guo S, Fan Y, Zhang H, Gu D, Li H (2009) Atrogin-1/MAFbx enhances simulated ischemia/reperfusion-induced apoptosis in cardiomyocytes through degradation of MAPK phosphatase-1 and sustained JNK activation. J Biol Chem 284(9):5488–96 doi:M806487200 [pii] 10.1074/jbc.M806487200 [DOI] [PubMed] [Google Scholar]
- Yao Y, Xu X, Zhang G, Zhang Y, Qian W, Rui T (2012) Role of HMGB1 in doxorubicin-induced myocardial apoptosis and its regulation pathway. Basic Res Cardiol 107(3):267 doi: 10.1007/s00395-012-0267-3 [DOI] [PubMed] [Google Scholar]
- Yoshida T, Semprun-Prieto L, Sukhanov S, Delafontaine P (2010) IGF-1 prevents ANG II-induced skeletal muscle atrophy via Akt- and Foxo-dependent inhibition of the ubiquitin ligase atrogin-1 expression. Am J Physiol Heart Circ Physiol 298(5):H1565–70 doi:00146.2010 [pii] 10.1152/ajpheart.00146.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa T, Amo-Shiinoki K, Ishikura S, et al. (2016) Sequential cleavage of insulin receptor by calpain 2 and gamma-secretase impairs insulin signalling. Diabetologia 59(12):2711–2721 doi: 10.1007/s00125-016-4102-5 10.1007/s00125-016-4102-5 [pii] [DOI] [PubMed] [Google Scholar]
- Zhang S, Meng T, Liu J, Zhang X, Zhang J (2015a) Cardiac protective effects of dexrazoxane on animal cardiotoxicity model induced by anthracycline combined with trastuzumab is associated with upregulation of calpain-2. Medicine (Baltimore) 94(4):e445 doi: 10.1097/MD.0000000000000445 00005792-201501040-00020 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YY, Meng C, Zhang XM, et al. (2015b) Ophiopogonin D attenuates doxorubicin-induced autophagic cell death by relieving mitochondrial damage in vitro and in vivo. J Pharmacol Exp Ther 352(1):166–74 doi:jpet.114.219261 [pii] 10.1124/jpet.114.219261 [DOI] [PubMed] [Google Scholar]
- Zheng D, Wang G, Li S, Fan GC, Peng T (2015) Calpain-1 induces endoplasmic reticulum stress in promoting cardiomyocyte apoptosis following hypoxia/reoxygenation. Biochim Biophys Acta 1852(5):882–92 doi:S0925-4439(15)00051-4 [pii] 10.1016/j.bbadis.2015.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.