

Abstract
The ascites ecosystem in ovarian cancer is inhabited by complex cell types and is bathed in an environment rich in cytokines, chemokines, and growth factors that directly and indirectly impact metabolism of cancer cells and tumor associated cells. This milieu of malignant ascites, provides a ‘rich’ environment for the disease to thrive, contributing to every aspect of advanced ovarian cancer, a devastating gynecological cancer with a significant gap in targeted therapeutics. In this perspective we focus our discussions on the ‘acellular’ constituents of this liquid malignant tumor microenvironment, and how they influence metabolic pathways. Growth factors, chemokines and cytokines are known modulators of metabolism and have been shown to impact nutrient uptake and metabolic flexibility of tumors, yet few studies have explored how their enrichment in malignant ascites of ovarian cancer patients contributes to the metabolic requirements of ascites-resident cells. We focus here on TGF-βs, VEGF and ILs, which are frequently elevated in ovarian cancer ascites and have all been described to have direct or indirect effects on metabolism, often through gene regulation of metabolic enzymes. We summarize what is known, describe gaps in knowledge, and provide examples from other tumor types to infer potential unexplored roles and mechanisms for ovarian cancer. The distribution and variation in acellular ascites components between patients poses both a challenge and opportunity to further understand how the ascites may contribute to disease heterogeneity. The review also highlights opportunities for studies on ascites-derived factors in regulating the ascites metabolic environment that could act as a unique signature in aiding clinical decisions in the future.
Keywords: Growth factors, metabolism, ascites, ovarian cancer, TGF-β, VEGF
Introduction
A major clinical feature of tumors with peritoneal carcinomatosis is fluid accumulation in the peritoneal cavity. Ovarian cancer (OC) is the most common cancer associated with development of malignant ascites1. The American Cancer Society estimated approximately 21,410 new OC cases and 13,770 OC-related deaths for 2021. Roughly 90% of these OCs are epithelial ovarian cancers (EOC), classified histologically as either Type 1 (endometroid, mucinous, clear cell, low grade serous) or Type 2 (poorly differentiated, carcinosarcoma, and high grade serous: HGSOC)2 3. Recent copy number analysis at single cell resolution reveals heterogeneity between high-grade and low-grade tumors as anticipated, but less heterogeneity in metastases as compared to primary tumors4. Additional molecular subtype signature-based classifications have also emerged for high grade serous cancers (HGSOC) from single-cell (sc) RNA-seq and drop-Seq studies indicating the presence of an epithelial to mesenchymal (EMT)- high subtype that can identify with poor prognosis5. Regardless of the classification, most ovarian cancers lead to ascites6. However much of our information comes from HGSOC, primarily due to the highest incidence of this subtype in the patient population. Recurrent HGSOC is incurable, and the presence of ascites is a common feature. Over 70% of patients will have recurrent disease with the likelihood being 90–95% in patients with stage 4 disease.
Ascites accumulation in patients frequently correlates with the extent of metastatic disease, and both ascitic volume and components in ascites can be used to grade, stage and predict survival and chemoresistance outcomes, which have been proposed as independent prognostic factors7–9. In some instances, ascites accumulation can lead to infections (bacterial peritonitis) and hernia, due to increased abdominal pressure, and can directly contribute to morbidity due to complications associated with gastrointestinal problems. Much like edema (capillary permeability and hydraulic and oncotic pressure gradients), arterial vasodilation and venous obstruction are the main physiological causes of ascites accumulation10. In advanced EOC, increased vascular permeability of vessels lining the peritoneum and/or lymphatic obstruction by disease burden leads to altered lymph drainage and angiogenesis and thereby ascites accumulation1,11,12. While in most cases treating the underlying disease will reduce ascites, untreatable ascites can be a recurrent and frequent problem, requiring drainage and paracentesis.
The malignant ascites environment is comprised of diverse cell types (reviewed in detail recently in13 14 and summarized in Fig 1). A subset of these include macrophages, fibroblasts, endothelial cells, lymphocytes and mesothelial cells and a small fraction of tumor cells15,16. In depth scRNA-seq has confirmed and identified 18 distinct cell clusters including epithelial, macrophage, cancer associated fibroblasts (CAF), dendritic cells, B cells, T cells and erythrocytes with immune cells being the most abundant (~65%). Layered on the complexity of these cell types, is the additional patient by patient variability in the percentage of these cells present, as described recently16. Notably, malignant cells exhibited an inflammatory transcriptional program16.
Figure 1.
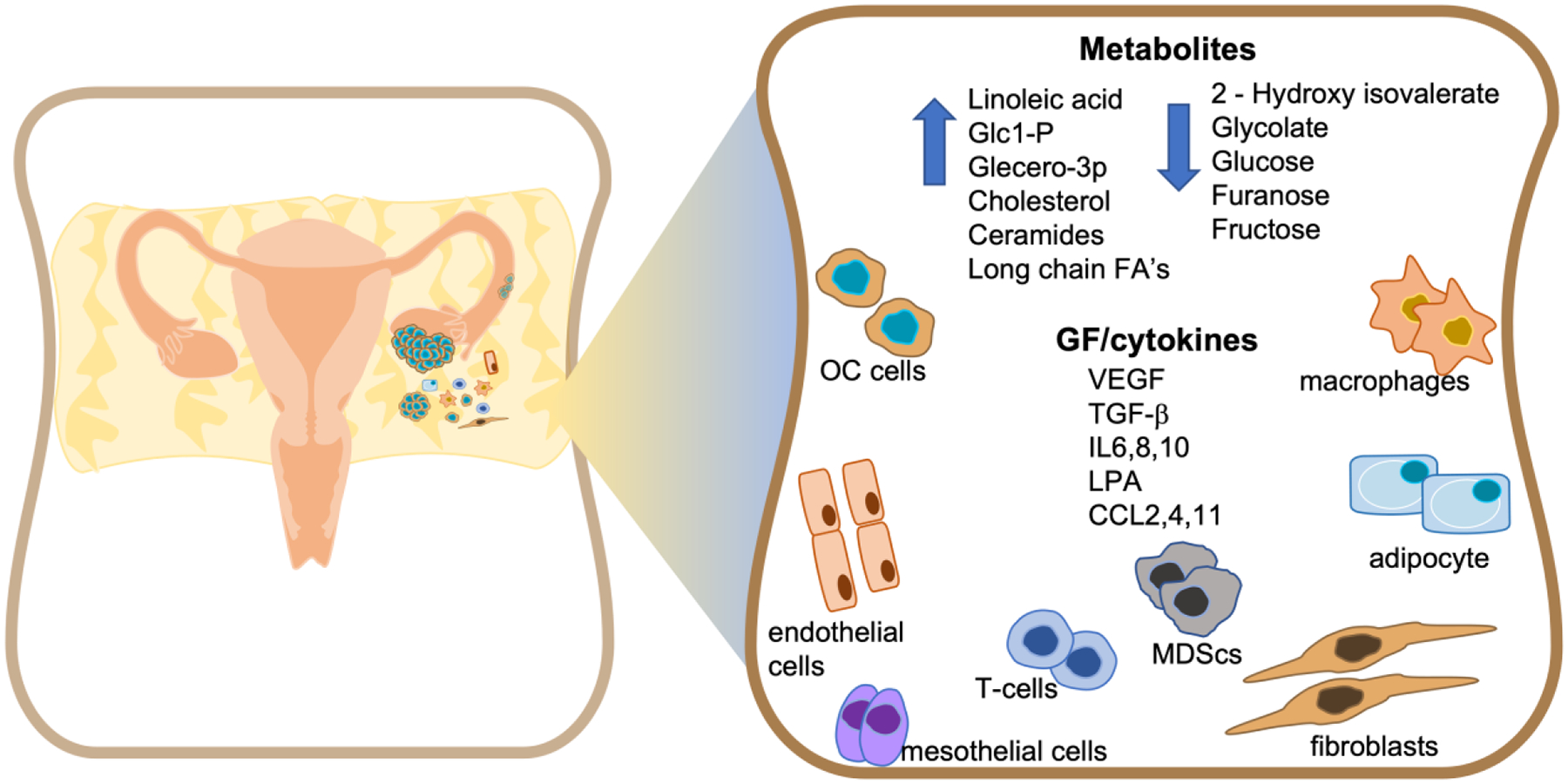
Overview of the major cellular components, metabolites, cytokines and growth factors (GF) found in the ovarian cancer ascites (OC) environment.
In addition to the cellular make-up of ascites, significant variability exists in the acellular components found in the ascitic fluid, including growth factors, cytokines and metabolites that result from secretion by, and metabolic activity of, the cell types found in the ascites (Fig 1). Much has been discussed about the signaling pathways associated with ascites-derived growth factors and cytokines and their effects on cellular programs driving ovarian cancer progression17 18 19 20. However, limited integration of information exists on the link between growth factors and the metabolite environment of the ascites fluid, and regulation of the metabolism specifically of ascites resident cells. Undoubtedly, changes to cell signaling by growth factors in the ascites impacts nutrient uptake and energy production required for tumor cell survival and the interaction between the different cell types in the ascites. While an in-depth analysis of every cytokine and growth factor is beyond the scope of the review, we have discussed those that have established direct roles in ascites development and have been explored as treatment options for ovarian cancer in the clinic, particularly focusing on VEGF and TGF-β, and a subset of interleukins (IL). We focus on their relationship to metabolism and discuss the impact on nutrient balance in subsets of cell types found within the OC ascites, drawing inferences from other cancer types.
1. Acellular Components in Malignant Ascites
1.1. Growth factors, cytokines and chemokines
Ascitic fluid is a reservoir of soluble factors that directly contributes to transcoelomic, hematogenous and lymphatic metastasis of OC21,22. Thus, characterization of these soluble factors is essential to understand how the ascites tumor environment affects OC progression23–27, and as a resource for staging, diagnosis and prognostic biomarker development in the clinic. Several studies have implicated the presence of multiple cytokines, chemokines and growth factors within ascites with OC etiology (Table 1), with significant variation seen between studies in part due to sample handling28 and sensitivity of the detection method used (Table 1). Several of these factors are elevated specifically in malignant ascites in comparison with benign ascites or in normal or diseased sera indicating OC-dependent increases29–31. Chemokines including CCL2, −3, −4, −5, −8 and −22 as well as chemokine receptors CCR1, −2a, −3, −4, −5, and −8 are also found in OC ascites with studies indicating CCL2 as being the most abundant32. Among the pro-tumor cytokines, IL-4, −5, −6, −8, −10, −13, and TGF-β exhibit strong immune modulatory and angiogenetic functions33 34 35 36. Increased levels of IL6, IL8, IL10, CCL2 (MCP-1), IFNγ, and TNF-α have been linked to poor prognosis and IL6 has been proposed as an independent factor for predicting overall survival (OS) in advanced ovarian cancer20. Specifically, IL6 has been linked to paclitaxel and cisplatin resistance in multiple in vitro OC studies37. Further, neo adjuvant chemotherapy can lead to lowered IL6 and IL8 levels and improve survival outcomes9,38–43. Comparisons of IL6 levels in matching specimens indicates an almost 100 times increase within peritoneal fluid as compared to plasma levels in the circulation44, suggesting that OC ascites is a highly pro-metastatic and inflammatory environment. Indeed, incorporation of IL6 levels as a biomarker improves the Risk of Malignancy Index (RMI)45, an algorithm used widely for assessing ovarian cancer risk along with CA125 and HE4. Notably, IL6 was most effective at distinguishing advanced malignant disease46. IL10, is also present at higher concentrations in ascites and is linked to shorter progression free survival (PFS) in OC patients40,43. IL-6 and −10 have also been shown to stimulate CA-125 expression47, and enhance ascites accumulation in OC patients48. However, a recent cell-free and concentrated ascites reinfusion therapy (CART) study indicated that higher levels of IL10 after CART, correlated with longer survival in response to CART49, suggesting some variability in outcomes associated with IL10 levels.
Table 1.
Published reports of growth factor, cytokine, and chemokine levels in ovarian cancer
| Growth factors and cytokines | Levels in circulation (Healthy) [pg/ml] | Levels in circulation (Ovarian Cancer) [pg/ml] Unless indicated | Levels in OC ascites fluid (AF) [pg/ml] Unless indicated | Additional information |
|---|---|---|---|---|
| IL6 | 5.186156 | 29.9743 | 14845.2843 391.09157 2955(median)40 641938 |
Increased in malignant AF compared with benign9,157 |
| IL8 | 10.9 (median)158 29.3 (median)159 |
28.0543 100.4 (median)160 |
140838 202.17 in HGSOC and 456.03 in Mucinous and endometroid157 48.7 (median)160 |
Increased in non-serous OC AF compared with benign peritoneal effusion157 Increased in malignant AF compared with benign160 |
| IL10 | 12.6 (median)159 | 17.6043 17.3 (median)160 |
24 (median)40 137.8543 23.5 (median)160 |
Increased in AF compared with plasma43,161 No change in malignant AF compared with benign160 |
| IL2 | 14 (median)159 | 14.9743 | 3.7443 | Reduced in AF of OC compared with plasma level. No difference between AF from malignant vs benign162 |
| VEGF | 61.6 (median)159 | 96.6543 9470163 |
10670.3543 10250163 2330 (median)164 |
Increased in AF compared with plasma43 Increased in malignant AF compared with benign163 |
| TGF β1 | 1.985 ng/ml165 1.000 ng/mL166 |
4–20 ng/ml28,167,168 | 14.435 ng/ml162 3.5 – 47.734 ng/mL57 388 pg/mL31 |
Blood level higher in all OC tumors (benign and malignant) compared with healthy controls162 Elevated in AF from serous and endometroid OC compared with benign fluid31 |
| TGF β2 | <0.2 ng/mL168 | 8.1 ng/mL169 | <0.1–4 ng/mL57 | Higher serum level was reported to corelate with improved survival169 |
| TNF-α | 0.06170 | 11.0143 8.5170 |
16.6343 210170 |
Increased serum level in OC patients compared with healthy individuals170 Increased in AF of OC compared with peritoneal lavage of healthy individuals170 |
| IFN-γ | 12.99171 | 284.5143 | 91.5543 2.76 (median)172 |
No difference between AF of OC and benign tumors162 Decreased in AF of OC compared to plasma level43 |
| PDGF-BB | 8473 (median)173 | 960.1943 11422(median)173 |
31.3143 | Reduced level in OC, AF compared with plasma43 Increased level in serum OC compared to normal individuals173 |
| FGF2 | 41.7 (median)159 33 (median)173 |
53 (median)173 | Increased in serum of OC compared with normal individuals | |
| RANTES | 5839 (median)159 | 13269.1343 | 228.5543 | Decreased in OC AF compared with serum43 Elevated serum level in OC compared with benign ovarian cyst174 |
| CCL2 (MCP-1) | 41.5 (median)159 230175 |
490175 108.4543 |
4280175 1456.2143 |
Increased serum level in OC compared with healthy individuals43,175 |
| CCL3 (MIP-1α) | 7.1 (median)159 | 13.4043 | 15.5543 | No change detected between OC serum and AF43 |
| CCL4 (MIP-1β) | 70176 | 145.0743 24.1 (median)177 |
729.7843 22.5 (median)177 |
Elevated in OC AF compared with serum43 No changes reported in177 |
| CCL11 (Eotaxin) | 242178 | 243.8 (median)177 | 104.1 (median)177 | Reduced level in AF compared with blood |
| CXCL1 | 58.6179 | 32131 | Elevated in OC AF compared with benign fluid31 | |
| CXCL5 | 162 (median)180 | 91331 | Elevated in OC AF compared with benign fluid31 | |
| CXCL10 | 576.2 (median)159 | 726.2 (median)177 | 10,001 (median)177 | Elevated in AF compared with blood from OC177 |
The significance of VEGF in the ascites is exemplified by the use of bevacizumab as standard of care for homologous recombination (HR) proficient patients, and the ability of such anti-VEGF therapies to reduce ascites burden and prolong the time between paracentesis50. Findings from the 2014 AURELIA trial have also led to bevacizumab use in platinum resistant patients51,52, where progression free survival (PFS) was improved, and ascites control observed. It remains unclear whether the improved PFS was due primarily to ascites control, or if a reduction in ascites was the result of delayed recurrence. Adverse side effects such as gastrointestinal events, hypertension, bleeding, thromboembolism and delayed wound healing remain with anti-VEGF therapies53, and identification of additional non-toxic angiogenic targets remains and an active area of investigation to combat OC progression and ascites formation.
In addition to VEGF, which is arguably the most dominant driver of angiogenesis in cancer, TGF-β, IL6 and IL8 are considered angiostimulatory and are negatively associated with progression free survival54 17 55. Consistently, TGF-β2 and particularly TGFβ1 are elevated in the ascites of EOC patients56 57 (Table 1). Not all TGF-β superfamily members are elevated however, as a subset of BMP’s can be present below the levels found in normal circulation57 and caution must be used in treating all TGF-β superfamily members equally. The significance of the presence of high TGF-β and engagement of its signaling machinery in ascites-resident cells17 is underscored by TGF-β pathway blockade strategies that not only abolish ascites formation in preclinical models but also suppress VEGF production and improve ascites drainage by lymphatic vessels58 36,58. In addition to potential direct contributions to ascites development via the vasculature, TGF-βs are also key drivers of epithelial mesenchymal plasticity in OC cells and in multicellular spheroid aggregates59,60 61. In addition, TGF-βs regulate mesenchymal cells and the immune cell population62, which together can increase tumor burden and the formation of malignant ascites.
The source of cytokines and growth factors is as varied as the growth factors themselves and includes secretions by tumor cells, immune cells, stromal cells, as well as contribution from the diseased host tissue, including the omentum and peritoneal organs. For instance, besides tumor cells, CD163 positive macrophages enhance tumor angiogenesis and metastasis by inducing secretion of VEGF, IL6 and IL10, in the ovarian tumor microenvironment63. What is evident from cytokine screens from different studies is that significant heterogeneity and variability exists in the levels of these soluble factors between patients (Table 1). The types of growth factors, cytokines and chemokines detected often differ between patients and between studies. The relative levels detected can also be highly variable. This underscores the need for a standardized screening method if these soluble factors are to be used as bio- and prognostic markers in future.
1.2. Metabolites
Although less well studied than the growth factor and cytokine components of ascites, metabolites are an important subset of the acellular components in this liquid tumor environment. Metabolomic, lipidomic and proteomic studies of EOC patient ascites and in vivo tumor models have highlighted the presence and importance of some key signature metabolites in the ascites fluid, which are integral to regulating the metabolism of tumor and tumor associated cells in the ascites64–67 (Fig 1). Malignant ascites display a distinct metabolite pool relative to benign ascites. For example, principal component analysis can distinguish OC ascites from cirrhosis derived ascites by the presence of amides of linoleic acid and oleic acid, and high glucose-1-phosphate, glycero-3-phosphate, cholesterol and ceramide levels. Conversely, 2-hydroxyisovalerate, glycolate, glucose, furanose, and fructose were less abundant in malignant ascites relative to cirrhosis-derived ascites65 (Fig 1). Several glucose metabolism intermediates are altered in a way that suggest increased consumption of glucose by ascites resident cells. For example, levels of glucose, furanose, glycolate and fructose are significantly reduced in the malignant ascites, whereas glycero-3-phosphate (G3P), and glucose-1-phosphate (G1P) are significantly elevated65,68 (Fig 1).
Lipid accumulation is also a common feature of malignant ascites65,66, and has been linked to both increased lipogenesis and fatty acid oxidation by ascites resident cells. Lipid species detected include long chain fatty acids that are derived from lipid biosynthesis in tumor and adipocytes in the peritoneal tissues, as well as signaling lipids. The types of long chain fatty acids also appear to be specific to ascites. For example, lipidomic profiling found a higher accumulation of unsaturated fatty acids (UFA) compared to saturated fatty acids (SFA) in patient ascites66. In addition, increased ratios of UFAs to SFAs were found in OC cell lines cultured in conditioned ascites or omentum-derived media66. The higher UFA levels in ascites likely reflect increased biosynthesis of long chain fatty acids, which are both synthesized, stored and used as fuels for beta-oxidation by tumor cells. Examples of PUFAs enriched in ascites are prostanoids, hydroxy eicosatetraenoic acids, leukotrienes, linoleic acid, docosahexaenoic acid and prostaglandin E2.
Phospholipids are also detected in the malignant EOC ascites69. Levels of the bioactive lipid molecule LPA (lysophosphatidic acid) and sphingosine1-phosphate (S1P) are elevated in malignant EOC ascites fluid compared to benign ascites27, and high levels of LPA are associated with adverse outcomes70. Furthermore, concurrent with elevated levels of LPA, human EOC ascites also had high levels of cytosolic and calcium-independent Phospholipase 2 (PLA2) activity64. PLA2 is critical for breakdown of phosphatidylcholine (PtdCho), the major membrane lipid, to form lyso- phosphatidylcholine (lyso-PtdCho) which is present at higher levels in EOC ascites compared to benign ascites71.
Other metabolic intermediates have also been noted to be significantly altered in ascites. For example, presence of BHB (β-hydroxybutyrate), a ketone body formed from acetyl-CoA and two other metabolites of the TCA cycle, citrate and maleic acid, is elevated in ascites in in-vivo tumor studies72. In contrast, 2-hydroxyisovalerate is one of the most depleted metabolites in malignant ascites compared to benign liver cirrhosis ascites. Even though, the cause of this decrease in 2-hydroxyisovalerate is not clearly understood, it might be the result of increased amino acid catabolism as 2-hydroxyisovalerate is produced upon breakdown of branched amino acids68. Many of the causes and consequences of changes in metabolites such as amino acids still require further investigation and analysis.
2. Linking growth factors and metabolism in malignant ascites
Metabolic flexibility is an important hallmark of OC to allow cells to survive under fluctuations in nutrient availability in response to changing tumor microenvironments. Several growth factors and cytokines that are elevated in ascites have also been implicated in regulating proliferation and metastatic progression by impacting nutrient uptake and metabolic flexibility in tumors. TGF-βs, VEGF and ILs have all been described to have direct or indirect effects either by gene expression of enzymes involved in metabolic reactions in autocrine or paracrine fashions. Additionally, targeting these growth factors, particularly VEGF, can lead to metabolic alterations and impact therapeutic response and resistance mechanisms.
2.1. Glycolysis & Pentose phosphate pathway
Malignant ascites of EOC patients have lower levels of glucose and a higher level of G1P and G3P as compared to benign ascites (Fig 1). This decrease in glucose levels is thought to be reflective of enhanced consumption of glucose by ascites resident cells65 (Fig 2). Increased consumption of glucose in the tumor microenvironment (TME) is frequently associated with enhanced glycolytic activity by tumor cells, shifting mitochondrial ATP production to macromolecular biosynthesis pathways to support proliferation (Fig 2). Of the 2000 proteins and peptides identified in metastatic EOC ascites73,74 two glycolytic enzymes pyruvate kinase isozymes M1/M2 (PKM1/2) and glyceraldehyde phosphate dehydrogenase (GAPDH) showed significant difference between patient and control ascites and serum samples. In addition to promoting glucose metabolism, PKM2 has been implicated with platinum-resistance in ovarian cancer cell lines75. While an increase in aerobic glycolysis, otherwise known as the Warburg effect, is a common phenotype of cancer cells, the fate of glucose in tumor cells is cell state dependent, as highlighted in subsequent sections, and the depletion of ascites glucose could have consequences on rewiring towards utilization of alternate fuel sources. Moreover, increased glucose utilization by tumor cells decreases glucose availability to other tumor associated cells, such as effector T-cells76 (Fig. 2). Thus, tumor cells and T-cells (and potentially other immune cells) have distinct metabolic profiles as a consequence of glucose competition within ascites, which has also been observed in other solid tumor tissues77.
Figure 2. Potential GF-metabolic pathway interactions in the OC ascites.
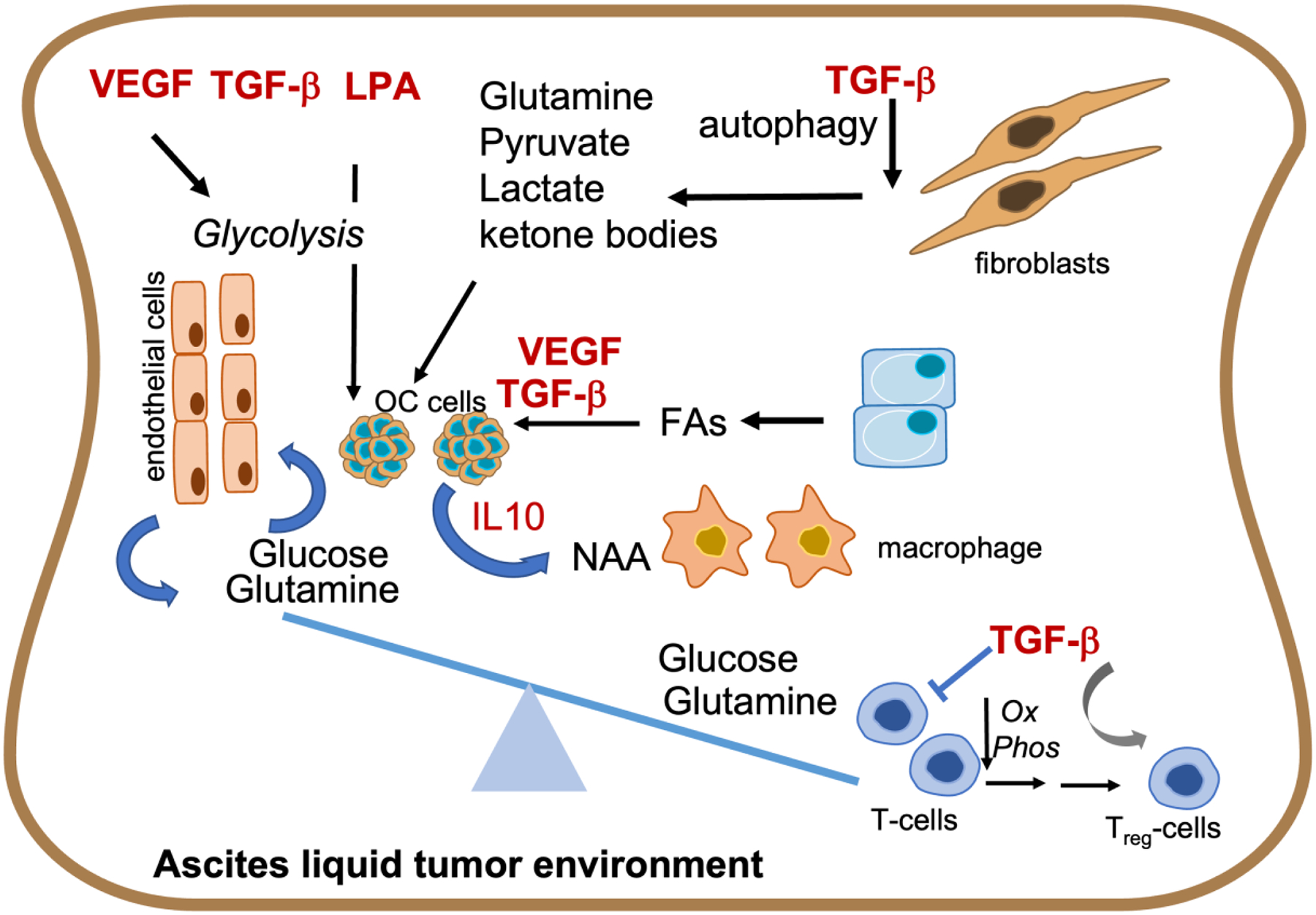
TGF-β, VEGF and LPA are known regulators of tumor cell metabolism, including increasing glycolysis and fatty acid (FA) uptake, and glutamine production from fibroblasts. Increased use of metabolites, such as glucose and glutamine by OC tumor cells alters the ascites metabolite environment. This can lead to depletion of the same metabolites for T-cell metabolism resulting in reduced Oxphos and increase in differentiation and expansion of the regulatory T cell population (Treg) that can be enhanced by TGF-β. Addiction of OCs to glutamine can also increase NAA to fuel M2 macrophages thus skewing the ascites metabolite pool to create a tumor permissive immune environment.
PKM2 is also a downstream target of TGF-β during epithelial to mesenchymal transition (EMT) and this is accompanied by increases in glucose transporters78 (Fig 2). Notably, several lines of investigation in non-ovarian models link TGF-β during the process of EMT to metabolic changes associated with a shift to aerobic glycolysis79 80 81. This glycolytic reprogramming is also partially required for TGF-β-induced EMT82. A reciprocal relationship between glucose metabolism and the TGF-β’s exists (Fig 3), wherein glucose can also stimulate TGF-β signaling by increasing TGF-β cell surface receptor levels and activating latent TGF-β in the extracellular space83. Energy production is an accepted requirement for EMT as metabolic changes accompany EMT. However, it is currently difficult to distinguish if metabolic changes are by products or drivers of the process. Moreover, with the significant plasticity of the process of EMT itself, it is likely that the metabolic flexibility mirrors the EMT process84.
Figure 3. GF regulation of the metabolic pathways active in ascites resident OC cells.
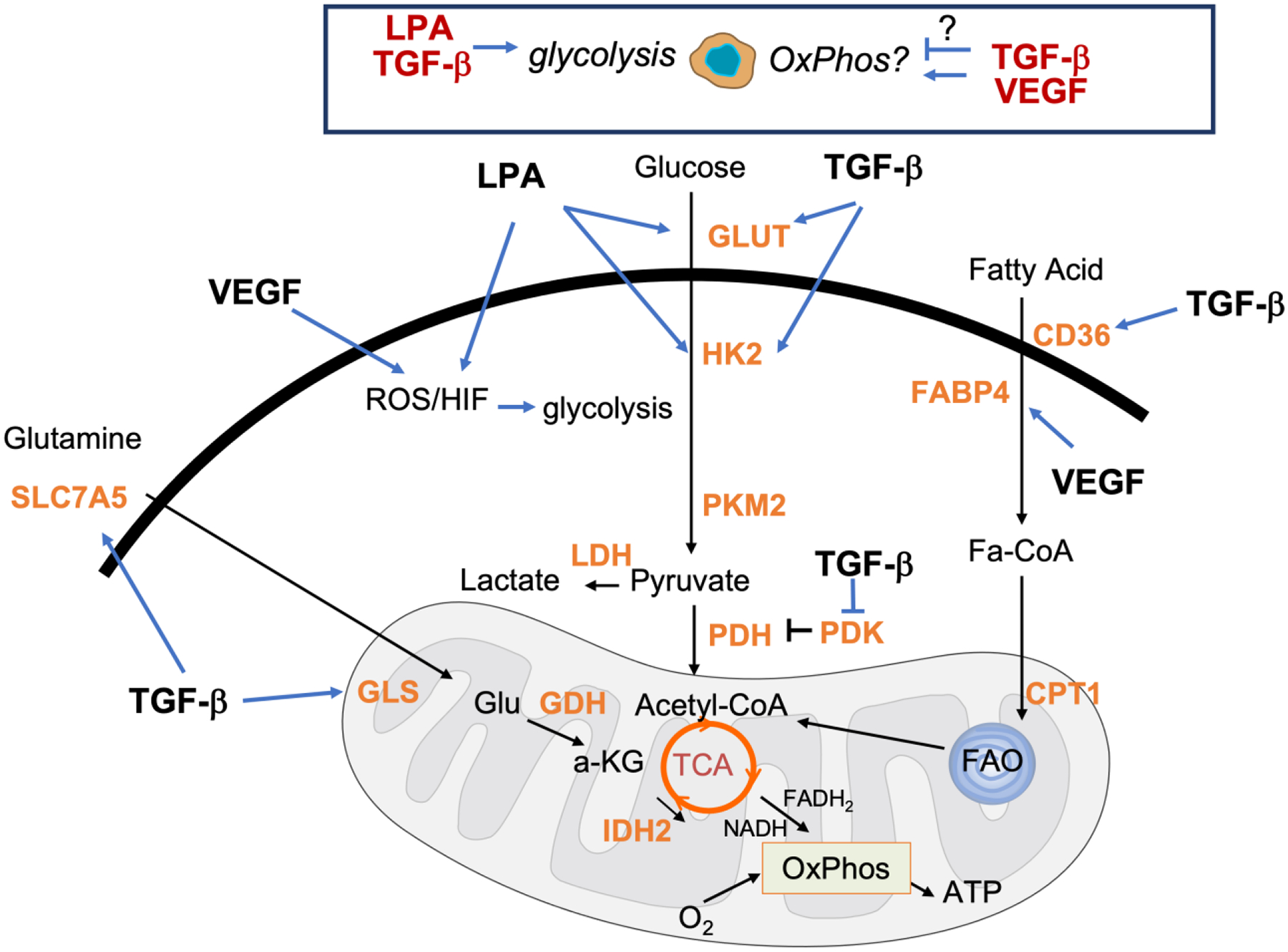
Ascites resident OC cells exhibit both glycolysis and oxidative phosphorylation with GF’s been shown to regulate both processes in other cell types. TGF-β can increase expression of genes involved in glucose, glutamine and FA uptake and metabolism (GLUT, CD36 and SLCA5) and (HK2, GLS1, and FABP4) respectively. A mutual relationship exists between glucose and TGF-β wherein glucose can stimulate TGF-β signaling. LPA induces pseudohypoxia by increasing HIF1α and likely increased expression of GLUT1 and HK2, leading to an LPA-mediated glycolytic shift in OC cells. VEGF can regulate FABP4 and increase ROS production.
Epithelial to Mesenchymal Transition (EMT) is an important reversible transcriptional program process during metastasis that shares gene expression signatures with cancer stem cells85. EOC subpopulations isolated from malignant ascites that are enriched in stem like cells86,87 may be prone towards a rewiring of metabolism towards glycolysis. Specifically, an ALDH+/CD44+ cancer stem cell subpopulation has been shown to be reliant on aerobic glycolysis, which is dependent on their upregulation of pyruvate dehydrogenase kinase 4 (PDK4)88. Interestingly, the increased activity of PDK4 in ALDH+/CD44+ ascites-resident cancer stem cells also increases IL-8 expression88, suggesting that metabolic changes lead to alterations in ascites cytokine levels. Whether or not cytokines like IL-8 and IL-6 also conversely contribute to metabolic reprograming of cells within malignant ascites remains to be further explored. However, evidence from other tumor types suggests that this interplay is possible. In pancreatic cancer, stromal IL6 increases glycolytic flux in tumor cells, leading to lactate efflux and selection of cancer stem cells89. Whether IL6 in the ascites could favor EOC stem cell enrichment via metabolic changes is speculative, yet intriguing (Fig 3).
High glucose consumption by tumor cells in malignant ascites is likely also driven by a need for macromolecular biosynthesis pathways. Glucose is frequently wired into the Pentose phosphate pathway (PPP), which provides anabolic substrates for ribonucleotide synthesis to support cancer growth, and the production of NADPH (Fig 4), which is necessary for maintenance of a reduced glutathione (GSH) pool20 The PPP thus represents an important mechanism for tumor cells to manage oxidative stress. For example, cancer cells are able to cope with the oxidative stress associated with loss of matrix attachment by reroute glucose metabolites into the PPP90 91 (Fig 4). This metabolic adaptation is important for anoikis resistance of breast cancer cells90, and was demonstrated in OCs cultured in anchorage independent spheroid conditions, which were shown to exhibit increased glycolytic flux and NADPH generation91. Moreover, enhanced activity of 6-phosphogluconate dehydrogenase (6PGD), an important enzyme for NADPH synthesis in the PPP has been associated with cisplatin resistance in OC92. Whether ascites fluid or specific growth factor cooperate to regulate PPP enzymes in OCs or tumor associated cells remains to be explored. However, a link to VEGF signaling has been made in endothelial cells, wherein G6PD positively regulates VEGFR2 signaling and VEGF-induced EC proliferation, migration, and tube formation93.
Figure 4. Metabolic pathways active in ascites resident OC cells.
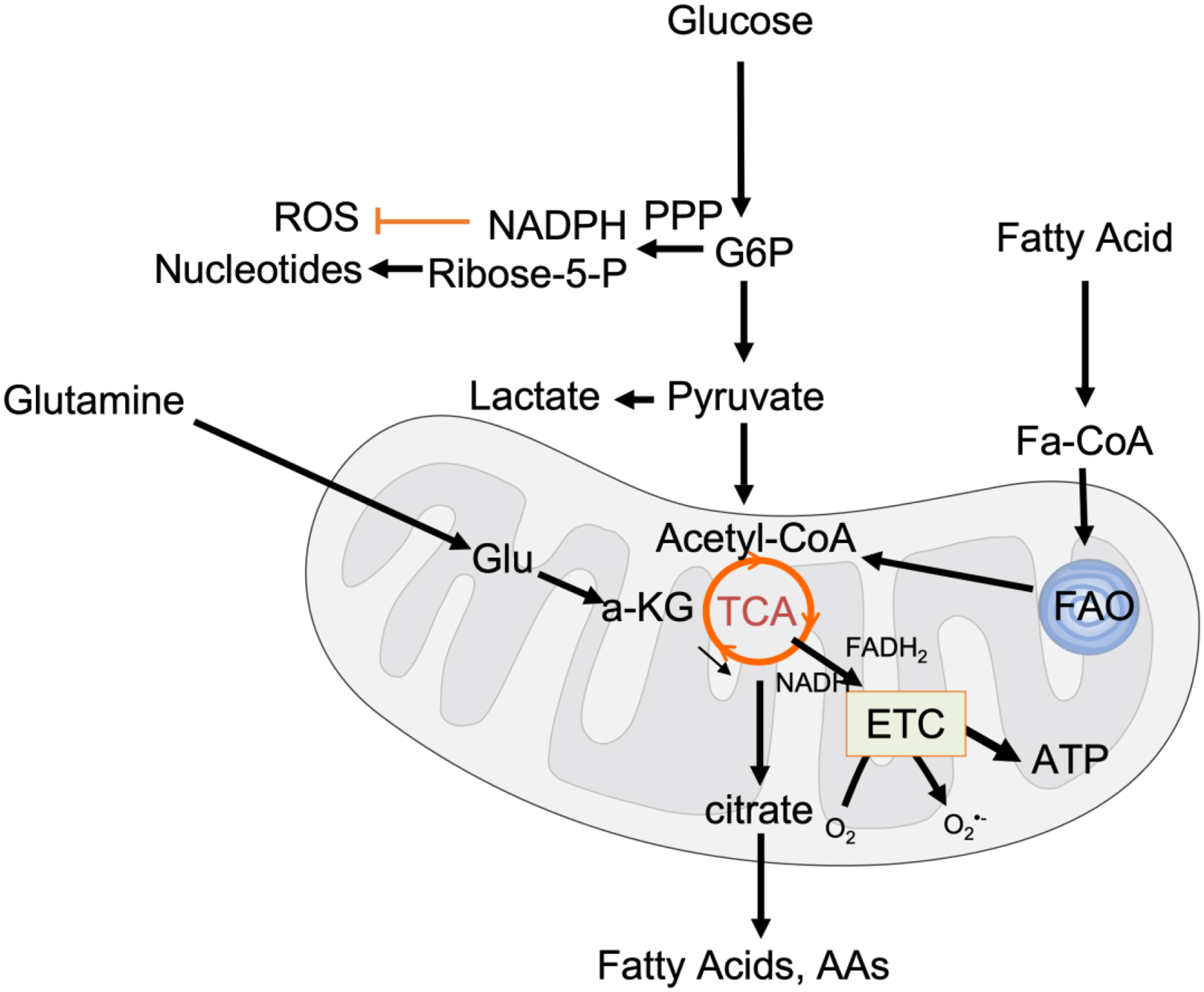
OC tumor cells demonstrate metabolic flexibility by their ability to utilize glucose, glutamine and fatty acids as fuel sources. Glucose-6-phosphate (G6P) can be routed into the Pentose phosphate pathway (PPP) for generation of NADPH and ribose 5-phosphate, that aids in ROS scavenging and nucleotide synthesis, respectively. Fatty acid oxidation (FAO) and glutaminolysis provide alternate carbon sources for the tricarboxylic acid cycle (TCA). The TCA provides metabolic substrates for macromolecular synthesis necessary for tumor cell proliferation and reducing equivalents FADH2 and NADH for Oxidative Phosphorylation and ATP production at the electron transport chain (ETC). A consequence of increased oxygen flow thought the ETC is superoxide production (O2·−) which can lead to mitochondrial redox signaling used by tumor cells.
Like other cancers, OCs require increased supplies of nutrients and thereby depend on angiogenesis, which concomitantly contributes to ascites formation. VEGF is the predominant driver of angiogenic changes in cancers including OC, and much of VEGF’s role in metabolism has been delineated in endothelial cells. Similar to tumor cells, the tumor vasculature prefers glycolysis, as 2-Deoxy-glucose was shown to be highly toxic to endothelial cells94, likely due to the need to preserve oxygen for blood supply to the tumor/tissues. Endothelial cells are also highly plastic and are poised to proliferate in response to tumor secreted VEGF (angiogenic switch), and during such bursts, can significantly increase their VEGF-dependent glycolytic flux95 (Fig 3). However, VEGF can influence additional cells in the ascites, including tumor cells, stromal/fibroblasts and immune cells. Approximately 85% of human ovarian tumors express VEGFR2 receptor, with most ovarian cancer cells expressing functional VEGFR296. Notably short-term pharmacological inhibition of VEGFR2 signaling in EOC cells is a strong suppressor of survival in suspension cultures97. It is possible that VEGF may also contribute to metabolic reprograming of ascites resident EOCs, yet this remains to be clearly defined (Fig 3).
The mildly hypoxic environment of the ascites and hypoxic micro-environments found within the core of multicellular tumor spheroids aggregates suspended within ascites can stimulate VEGF synthesis98–100, likely further contributing to a re-wiring of metabolism, in addition to increases in factors, such as hypoxia inducible factors (HIF). HIF activation can also be driven by LPA, (Fig 3) which is highly abundant in malignant ascites and has been shown to be secreted by both tumor cells and Tumor associated macrophages (TAMs). LPA induces pseudohypoxia in OC cells with the resultant expression of HIF1α via Gαi2, Rac1, NOX2 and the generation of reactive oxygens species (ROS)101 (Fig 3). This may lead to increased expression of glucose transporter-1 (GLUT1/SLC2A1) and the glycolytic enzyme hexokinase-2 (HK2), leading to an LPA-mediated glycolytic shift in EOC cells70. Like the effect of LPA, both GLUT1 and HK2 are also established targets of TGF-β in non- ovarian cancer models, and corelate strongly with EMT and glucose uptake during TGF-β-induced EMT78 (Fig 3).
Aside from tumor cells, TGF-β are well-known regulators of the stromal environment driving fibroblast reprogramming102, which mimics the EMT process of tumor cells. TGF-β pathways increase oxidative stress, autophagy/mitophagy and glycolysis in fibroblasts in part via caveolin regulation103. TGF-β can enhance autophagy in cancer associated fibroblasts, which further fuels metabolism of tumor cells by providing metabolites such as pyruvate, lactate and ketone bodies104 105 (Fig 2).
Another scenario that presents in the clinic, is with the use of anti-angiogenics (anti-VEGF) to manage ovarian cancer, that induce vessel pruning and leads to starvation of tumor cells. It is widely appreciated that such starvation can intensify the hypoxic/acidic environment106 107 and may explain the dramatic depletion of glucose and ATP in tumors seen. These metabolic changes are accompanied by partial tumor regression in highly glycolytic ovarian tumors108, with poorly glycolytic ovarian tumors being more growth arrested by anti-VEGF, and highly glycolytic ones becoming resistant more rapidly108. Thus, the metabolic profile may impact response to anti-VEGF and potentially other targeted therapies
2.2. Mitochondrial Oxidative Phosphorylation (OxPhos)
While the Warburg effect is a major pathway for the consumption of glucose by tumor cells, OC cells do not exclusively rely on glycolysis as a metabolic pathway (Fig 3,4). This is supported by OC cells exhibiting both oxidative phosphorylation and glycolysis and an increased ability to survive under anchorage-independent conditions109 (Fig 3). Similarly, CD44+/CD117+ ovarian cancer stem cell spheroids isolated from patients’ ascitic fluid exhibited not just enhanced glucose uptake as described in the previous section, but also had heightened OxPhos110. OC tumor initiating cells have been shown to have increased flexibility in mitochondrial function in response to mitochondrial uncouplers72. Chemo resistant OC cell lines are also less sensitive to glucose deprivation and display increased ability to switch from glycolysis to OxPhos111. Given the ability to upregulate both glycolysis and OxPhos, it appears that OC cells are highly metabolically flexible, which may provide survival adaptations under conditions of nutrient depletion. Interestingly, TGF-β present in ascites fluid can promote anchorage independent survival via increasing the stemness gene SOX257 112. Whether this also contributes to a shift in metabolism towards OxPhos remains untested but anticipated. As pointed out above, EMT is frequently associated with a shift toward aerobic glycolysis. However, in non-small cell lung cancer, TGF-β treatment leads to increased OxPhos and a shift away from glycolysis, via the repression of repression of pyruvate dehydrogenase kinase 482. Conversely, TGF-β may have negative effects on mitochondrial function of tumor associated cells, which contributes to its immunomodulatory function. TGF-β blocks mitochondrial OxPhos in CD4+ T cells, leading to repression of IFN-gamma production113. Thus, it is possible that the role of ascites-derived TGF-β on mitochondrial function is cell type dependent.
It should be noted that an increase in OxPhos presents challenges to tumor cells due to increased generation of mitochondria-produced reactive oxygen species, derived from electron leakage at the electron transport chain (Fig 4). OC cells can overcome this in anchorage-independent conditions by upregulating mitochondrial antioxidant defense mechanisms114. Coping with lethal oxidative stress is a necessary adaptation for tumor cell survival, as similarly illustrated above through increased pentose phosphate pathway - dependent NADPH generation. However, sublethal levels of ROS can conversely also be drivers of tumor progression. An example being NOX-derived ROS in stimulating pseudohypoxia via the regulation of HIF, as mentioned above. Growth factors can also manipulate ROS generation via the mitochondria. For instance, VEGF is widely reported to stimulate ROS production in endothelial cells115 (Fig 4), and can induce the production of mitochondrial ROS by enhancing mitochondrial function116. Mitochondria-derived ROS can have a number of consequences on endothelial cell function (reviewed in117), including enhanced migration by ROS-mediated Rac1 activation in response to VEGF116. VEGF-dependent regulation of OxPhos and subsequent mitochondrial ROS generation might thus be contributors to tumor angiogenesis. ROS can also increase VEGFR2 phosphorylation demonstrating a reciprocal link between redox and VEGF signaling117. Whether the same mechanisms utilized by endothelial cells are also seen in OC or other cells in the ascites that also express VEGF receptors is currently unclear (Fig 3). It is worth mentioning that cancers with HR defects, as is the case for a proportion of HGSOCs’, are sensitive to OxPhos inhibition, as a consequence of their high need for NAD+ and ATP, substrates required for PARP-dependent DNA repair118. Thus, the current therapeutic regimen of anti- angiogenic strategies (VEGF) along with PARP inhibitors being evaluated as a combination strategy in the clinic is highly timely119 120, and metabolic links to their mechanistic synergy should be further explored.
2.3. Glutamine
Glutamine is an important carbon source for the TCA cycle and is frequently used by tumor cells, stromal cells and immune cells in the tumor environment as an alternate fuel source (Fig 4). OC cells require glutamine for optimal proliferation, survival and metastasis as evidenced by highly invasive cells exhibiting higher glutamine dependency121,122. Such glutamine dependency may be regulated by growth factors like TGF-β that can increase glutamine uptake in cancer cells, including driving expression of the glutamine transporter SLC7A5123. During TGF-β induced EMT genes involved in glutamine metabolism, particularly Glutaminase (GLS), the first enzyme of glutaminolysis necessary for shuttling of glutamine to the TCA cycle are also increased123 124. TGF-β’s effects on glutamine uptake by tumor cells could be indirect as well, where TGF-β dependent fibroblast reprogramming leads to production of glutamine that can then be utilized by neighboring cancer cells103 (Fig 2). Whether TGF-β in the ascites and in OC models plays similar roles needs to be determined and may depend on the immune environment as well, since both TGF-β and glutamine are immunosuppressive. Specifically, in an OC model, immunosuppressive CD11b+Gr1+ myeloid cells are primed by exposure to ID8 mouse ovarian tumor cells to increase their glutamine metabolism to fuel mitochondrial respiration125. Together with myeloid-derived suppressor cells (MDSCs), Tregs contribute to the immunosuppressive environment of ovarian and other cancers62. In contrast to MDSCs glutamine deprivation has been shown to promote TGF-β-mediated conversion of CD4+T cells into Foxp3 T regs126, implicating this axis in immune tolerance. Decreased glucose uptake by ascites-resident T-cells, potentially as a consequence of tumor cell depletion of ascites glucose, may lead to the reduced glutamine uptake seen by T-cells, that inhibits mitochondrial OxPhos and decreases anti-tumor immunity76 (Fig 2). This is consistent with findings that glutamine depletion blocks T effector cell proliferation127, much like the effects of TGF-β on effector Tcells62. However, the effect of glutamine in Treg homeostasis could be cancer model dependent and remains to be clarified in the OC ascites environment. Given the direct, established role of TGF-β in promoting expansion of Tregs, it is possible that glutamine uptake as impacted by the competition for ascites metabolites also plays a direct role in immunosuppression in OC.
In addition to effects on the adaptive immune system, exposure to tumor cell metabolites and alterations in macrophage metabolism within the ascites environment regulate macrophage polarization128–130. For example, addiction to extracellular glutamine by OC cells results in increased glutaminolysis and secretion of N-acetylaspartate (NAA), which is abundantly found in ovarian cysts and ascitic fluids131. NAA treatment can increase glutamine synthetase (GLS) expression in macrophages, a marker upregulated in M2 macrophages, leading to and enhancing M2 reprogramming. NAA may also work synergistically with cytokines in the ascites including IL10132 (Fig 2).
Glutamine metabolism is also crucial for VEGF dependent angiogenesis as endothelial cells were found to consume glutamine more than any other amino acid133. However, endothelial cells may differ from tumor cells in their response to glutamine deprivation in their ability to use asparagine instead. Indeed, asparagine suppresses glutamine deprivation-induced death, but not the proliferation defect of cancer cells134.
The diverse dependencies on glutamine are being explored for therapeutic intervention124 by ways of inhibiting the enzyme GLS1 which is required for incorporation of glutamine into the central carbon metabolism. Most current trials are exploring the use of GLS1 inhibitors in a broad spectrum of OCs including clear cell carcinomas with ARID1A mutations, which demonstrate increased GLS1 expression135. Glutamine metabolism has also been associated with OC chemoresistance136 137 and as such GLS1 inhibitors are currently in Phase I trials alongside PARP inhibitors (Niraparib) for platinum resistant homologous recombination (HR) proficient OC patients as well.
2.4. Fatty acid metabolism
The interaction between ascites and the adipocyte rich omentum and peritoneum allows exchange and supply of free fatty acids to the tumor cells in the ascites fluid. Importantly, migration and proliferation of human OC cells are significantly increased when cultured with omentum adipocytes, or their conditioned media138 139. In these studies, adipocytes increase the lipolysis of triglycerides to produce free fatty acids. Lipolysis and lipogenesis are important in maintaining high ATP production in OC cells, necessary for proliferation, increased lipid metabolism and increased lipid synthesis by OC cells in response to ascites66. The lipolysis also likely contributes to the high free fatty acid content in ascitic fluid (Fig 1) that may contribute to the metabolic reprograming of OC cells from aerobic glycolysis to fatty acid β-oxidation (Fig 4). It has been shown that OC cells utilize free fatty acids from ascites or omentum-conditioned media and in response alter expression of genes involved in fatty acid oxidation and lipogenesis66. Anchorage-independent cancer cells, which are commonly enriched in cancer stem cells can also switch metabolism to utilization of fatty acids for beta-oxidation as an alternate fuel source including the production of ATP and NADPH71,140. Multicellular aggregates found in ascites are also frequently hypoxic, and hypoxic cells have elevated FA uptake141.
FA uptake is mediated primarily by the FA transport family protein CD36, which is corelated with poor prognosis in OC and drives metastasis142. Interestingly, both free fatty acids and CD36 exacerbate TGF-β driven EMT143. Besides CD36, FABP4, frequently overexpressed in OC cells is important in the uptake of FAs138. FABP4 is also regulated by VEGF to facilitate free fatty acid transport across the endothelium144 (Fig 3). However, the contribution of fatty acid oxidation (FAO) to energy production in endothelial cells can be variable. Given the use of anti- VEGF therapies as front line treatment for ovarian cancers, it is worth noting that OC cells can switch their lipid metabolism and storage in response to anti-VEGF leading to alterations in the lipidomic profile that has been shown to confer antiangiogenic drug resistance145. Notably, anti- angiogenic treatment mediated oxygen deprivation can lead to tumor cells switching from glycolysis to FAO metabolism upon treatment in non-ovarian cancers, particularly in tissues proximal to adipose deposits146.
Fatty acid metabolism is also important for tumor associated cells. For example, tumor cell line derived high molecular weight hyaluronic acid can alter macrophage membrane composition by significantly decreasing membrane cholesterol content. Consequently, this was found to alter macrophage activation by promoting IL-4-mediated pro-tumor reprogramming147. Peritoneal resident macrophages also exhibited tumor cell induced increases in fatty-acid oxidation and production of itaconic acid, which increased OxPhos mediated ROS generation in macrophages and tumor cells148. Poly unsaturated fatty acids (PUFA), such as prostaglandin E2, can also exert immune suppressor functions by inhibiting expression of TH1 cytokines TNFα, IFNγ and IL-2, and increasing expression of TH2 cytokines IL-4, IL-10 and IL-6 (reviewed in149). PUFAs have also been implicated in mediating T-cell suppression by OC ascites150
LPA, which is abundant in ascites can increase de novo lipid synthesis in OC to promote proliferation71,101. Both LPA and S1P stimulate expression of IL8 in OC cells151, thus contributing to the high cytokine levels in ascites. It is widely accepted that obesity and adipocytes recapitulate persistent inflammation that is marked by increased cytokines including IL6, IL8 and VEGF152 which are all elevated in ascites and contribute to the reciprocal crosstalk. Such interactions provide a rationale for targeting the availability of free fatty acids for ovarian cancer management.
3. Summary and Future Perspectives
VEGF and TGF-β represent two highly abundant growth factors in the OC ascites and in combination with other abundant factors including LPA, IL6, IL10 and additional chemokines, are likely contributors to metabolic alterations in ascites resident cells, and thereby the metabolite environment of the ascites. We highlight here the existence of metabolic plasticity in OC cells and the tumor associated cells in the ascites environment. This flexibility confers a higher level of ‘cellular fitness’ especially to tumor cells that can switch between glycolysis, fatty acid oxidation, glutaminolysis, pentose phosphate pathways and OXPHOS. The flexibility is likely a feature of OC cells that evade anoikis and survive under anchorage independent stress, leading to shifts in the metabolite pools in the ascites. As a consequence of the metabolic flexibility, a possible outcome is also immune evasion by eliciting metabolic changes in the immune cells and the creation of a tolerogenic environment.
The described examples also highlight potential links to reprograming of metabolism, particularly glycolysis in the ascites tumor environment that maybe influenced by the growth factors and cytokines enriched in the ascites. No discussion on metabolism and signaling is complete without appreciating the significance of nutrient sensing mechanisms likely to be active in all the cell types found in the ascites. Rewiring and changing metabolism in cells is controlled by the key transcription factors HIF, SREBP and ATF4 and the signaling pathways including mTORC1 and AMPK. It is also widely appreciated that mTORC1 activation is mediated by both growth factors and amino acids153 154. The processes regulated by these mechanisms include scavenging, autophagy, mitophagy and pinocytosis. All these mechanisms can in turn be regulated by growth factors as well, to balance proliferation and survival155 154. Since nutrient availability is challenging to analyze, growth factor measurements may be more feasible in a clinical setting. Hence defining the relationship between the two is central to understanding the metabolic outcomes of ascites accumulation and in defining treatment modalities that impact it.
At present it also is unclear how heterogeneity and individual variations in patient ascites metabolite pools and cytokine and growth factor levels as described here influence, tumor progression, response to therapy and patient survival. The lack of studies on the interplay between metabolites and GFs, exposes a knowledge gap and presents an important opportunity for future investigations. Together, the advances in standardization in measuring growth factors and metabolites, which are greatly influenced by sample handling and detection methods, and understanding of this link, could in the future lead to the utilization of the composition of ascites as a significant individualistic parameter to consider for personalized medicine approaches.
Acknowledgements
This work was supported in part by NIH grant R01CA230628 to Nadine Hempel and Mythreye Karthikeyan and NIH grants R01CA219495 to Mythreye Karthikeyan, and R01CA242021 to Nadine Hempel.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Footnote: “The co-first authors may list their name first on publication lists, such as CVs to demonstrate their equal contribution.”
Declaration of Competing Interest
Authors have no conflicts to declare.
REFERENCES
- 1.Kipps E, Tan DS & Kaye SB Meeting the challenge of ascites in ovarian cancer: new avenues for therapy and research. Nat Rev Cancer 13, 273–282, doi: 10.1038/nrc3432 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kurman RJ & Shih Ie M The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol 34, 433–443, doi: 10.1097/PAS.0b013e3181cf3d79 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lisio MA, Fu L, Goyeneche A, Gao ZH & Telleria C High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int J Mol Sci 20, doi: 10.3390/ijms20040952 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar M, Bowers RR & Delaney JR Single-cell analysis of copy-number alterations in serous ovarian cancer reveals substantial heterogeneity in both low- and high-grade tumors. Cell Cycle 19, 3154–3166, doi: 10.1080/15384101.2020.1836439 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu Z et al. The Repertoire of Serous Ovarian Cancer Non-genetic Heterogeneity Revealed by Single-Cell Sequencing of Normal Fallopian Tube Epithelial Cells. Cancer Cell 37, 226–242 e227, doi: 10.1016/j.ccell.2020.01.003 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Szender JB et al. Impact of ascites volume on clinical outcomes in ovarian cancer: A cohort study. Gynecol Oncol 146, 491–497, doi: 10.1016/j.ygyno.2017.06.008 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feigenberg T et al. Molecular profiling and clinical outcome of high-grade serous ovarian cancer presenting with low- versus high-volume ascites. Biomed Res Int 2014, 367103, doi: 10.1155/2014/367103 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ford CE, Werner B, Hacker NF & Warton K The untapped potential of ascites in ovarian cancer research and treatment. Br J Cancer 123, 9–16, doi: 10.1038/s41416-020-0875-x (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lane D et al. Inflammation-regulating factors in ascites as predictive biomarkers of drug resistance and progression-free survival in serous epithelial ovarian cancers. BMC cancer 15, 1–11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garrison RN, Galloway RH & Heuser LS Mechanisms of malignant ascites production. J Surg Res 42, 126–132, doi: 10.1016/0022-4804(87)90109-0 (1987). [DOI] [PubMed] [Google Scholar]
- 11.Nagy JA, Herzberg KT, Dvorak JM & Dvorak HF Pathogenesis of malignant ascites formation: initiating events that lead to fluid accumulation. Cancer Res 53, 2631–2643 (1993). [PubMed] [Google Scholar]
- 12.Tamsma J The pathogenesis of malignant ascites. Cancer Treat Res 134, 109–118, doi: 10.1007/978-0-387-48993-3_6 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Rickard BP et al. Malignant Ascites in Ovarian Cancer: Cellular, Acellular, and Biophysical Determinants of Molecular Characteristics and Therapy Response. Cancers (Basel) 13, doi: 10.3390/cancers13174318 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmed N & Stenvers KL Getting to know ovarian cancer ascites: opportunities for targeted therapy-based translational research. Front Oncol 3, 256, doi: 10.3389/fonc.2013.00256 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheid B Angiogenic effects of macrophages isolated from ascitic fluid aspirated from women with advanced ovarian cancer. Cancer Lett 62, 153–158, doi: 10.1016/0304-3835(92)90186-y (1992). [DOI] [PubMed] [Google Scholar]
- 16.Izar B et al. A single-cell landscape of high-grade serous ovarian cancer. Nat Med 26, 1271–1279, doi: 10.1038/s41591-020-0926-0 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumari A et al. TGFbeta signaling networks in ovarian cancer progression and plasticity. Clin Exp Metastasis 38, 139–161, doi: 10.1007/s10585-021-10077-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pogge von Strandmann E, Reinartz S, Wager U & Muller R Tumor-Host Cell Interactions in Ovarian Cancer: Pathways to Therapy Failure. Trends Cancer 3, 137–148, doi: 10.1016/j.trecan.2016.12.005 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Smolle E et al. Targeting signaling pathways in epithelial ovarian cancer. Int J Mol Sci 14, 9536–9555, doi: 10.3390/ijms14059536 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Browning L, Patel MR, Horvath EB, Tawara K & Jorcyk CL IL-6 and ovarian cancer: inflammatory cytokines in promotion of metastasis. Cancer Manag Res 10, 6685–6693, doi: 10.2147/CMAR.S179189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weidle UH, Birzele F, Kollmorgen G & Rueger R Mechanisms and Targets Involved in Dissemination of Ovarian Cancer. Cancer Genomics Proteomics 13, 407–423, doi: 10.21873/cgp.20004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Judson PL et al. Preoperative detection of peripherally circulating cancer cells and its prognostic significance in ovarian cancer. Gynecol Oncol 91, 389–394, doi: 10.1016/j.ygyno.2003.08.004 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Rickard BP et al. Malignant Ascites in Ovarian Cancer: Cellular, Acellular, and Biophysical Determinants of Molecular Characteristics and Therapy Response. Cancers 13, 4318 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahmed N & Stenvers K Getting to know ovarian cancer ascites: opportunities for targeted therapy-based translational research. Frontiers in oncology 3, 256 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Auer K et al. Role of the immune system in the peritoneal tumor spread of high grade serous ovarian cancer. Oncotarget 7, 61336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buas MF et al. Quantitative global lipidomics analysis of patients with ovarian cancer versus benign adnexal mass. Scientific reports 11, 1–12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Worzfeld T et al. The unique molecular and cellular microenvironment of ovarian cancer. Frontiers in oncology 7, 24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao L et al. The influence of the blood handling process on the measurement of circulating TGF-beta1. Eur Cytokine Netw 23, 1–6, doi: 10.1684/ecn.2012.0298 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Uruski P et al. Malignant Ascites Promote Adhesion of Ovarian Cancer Cells to Peritoneal Mesothelium and Fibroblasts. International Journal of Molecular Sciences 22, 4222 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rudlowski C et al. Prognostic significance of vascular endothelial growth factor expression in ovarian cancer patients: a long-term follow-up. International Journal of Gynecologic Cancer 16 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Mikuła-Pietrasik J et al. Biochemical composition of malignant ascites determines high aggressiveness of undifferentiated ovarian tumors. Medical Oncology 33, 1–4 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Milliken D, Scotton C, Raju S, Balkwill F & Wilson J Analysis of chemokines and chemokine receptor expression in ovarian cancer ascites. Clin Cancer Res 8, 1108–1114 (2002). [PubMed] [Google Scholar]
- 33.Nicholas C & Lesinski GB Immunomodulatory cytokines as therapeutic agents for melanoma. Immunotherapy 3, 673–690, doi: 10.2217/imt.11.45 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neufeld G & Kessler O Pro-angiogenic cytokines and their role in tumor angiogenesis. Cancer Metastasis Rev 25, 373–385, doi: 10.1007/s10555-006-9011-5 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Goumans MJ, Liu Z & ten Dijke P TGF-beta signaling in vascular biology and dysfunction. Cell Res 19, 116–127, doi: 10.1038/cr.2008.326 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Goel S et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev 91, 1071–1121, doi: 10.1152/physrev.00038.2010 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Izabela S-K, Michal K, Marek N & Magdalena K The implication of IL-6 in the invasiveness and chemoresistance of ovarian cancer cells. Systematic review of its potential role as a biomarker in ovarian cancer patients. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer, 188639 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Lane D, Matte I, Rancourt C & Piche A Prognostic significance of IL-6 and IL-8 ascites levels in ovarian cancer patients. BMC Cancer 11, 210, doi: 10.1186/1471-2407-11-210 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodrigues ISS et al. IL-6 and IL-8 as prognostic factors in peritoneal fluid of ovarian Cancer. Immunological investigations 49, 510–521 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Matte I, Lane D, Laplante C, Rancourt C & Piché A Profiling of cytokines in human epithelial ovarian cancer ascites. American journal of cancer research 2, 566 (2012). [PMC free article] [PubMed] [Google Scholar]
- 41.Fahmi MN, Pradjatmo H, Astuti I & Nindrea RD Cytokines as Prognostic Biomarkers of Epithelial Ovarian Cancer (EOC): A Systematic Review and Meta-Analysis. Asian Pacific Journal of Cancer Prevention: APJCP 22, 315 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coosemans A et al. Increased immunosuppression is related to increased amounts of ascites and inferior prognosis in ovarian cancer. Anticancer research 39, 5953–5962 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Giuntoli RL et al. Ovarian cancer-associated ascites demonstrates altered immune environment: implications for antitumor immunity. Anticancer research 29, 2875–2884 (2009). [PubMed] [Google Scholar]
- 44.Hina Amer, K. a. AER P. M Elevated Interleukin-6 Levels in the Circulation and Peritoneal Fluid of Patients with Ovarian Cancer as a Potential Diagnostic Biomarker: A Systematic Review and Meta-Analysis. Journal of Personalized Medicine 11, doi: 10.3390/jpm11121335 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jacobs I et al. A risk of malignancy index incorporating CA 125, ultrasound and menopausal status for the accurate preoperative diagnosis of ovarian cancer. Br J Obstet Gynaecol 97, 922–929, doi: 10.1111/j.1471-0528.1990.tb02448.x (1990). [DOI] [PubMed] [Google Scholar]
- 46.Kampan NC et al. Pre-operative sera interleukin-6 in the diagnosis of high-grade serous ovarian cancer. Sci Rep 10, 2213, doi: 10.1038/s41598-020-59009-z (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matte I, Garde-Granger P, Bessette P & Piche A Ascites from ovarian cancer patients stimulates MUC16 mucin expression and secretion in human peritoneal mesothelial cells through an Akt-dependent pathway. BMC Cancer 19, 406, doi: 10.1186/s12885-019-5611-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mesiano S, Ferrara N & Jaffe RB Role of vascular endothelial growth factor in ovarian cancer: inhibition of ascites formation by immunoneutralization. Am J Pathol 153, 1249–1256, doi: 10.1016/S0002-9440(10)65669-6 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ito T et al. Ascitic IL‐10 concentration predicts prognosis of patients undergoing cell‐free and concentrated ascites reinfusion therapy. Therapeutic Apheresis and Dialysis 24, 90–95 (2020). [DOI] [PubMed] [Google Scholar]
- 50.Smolle E, Taucher V & Haybaeck J Malignant ascites in ovarian cancer and the role of targeted therapeutics. Anticancer Res 34, 1553–1561 (2014). [PubMed] [Google Scholar]
- 51.Pujade-Lauraine E et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: The AURELIA open-label randomized phase III trial. J Clin Oncol 32, 1302–1308, doi: 10.1200/JCO.2013.51.4489 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Sehouli J et al. Bevacizumab Combined with Platinum-Taxane Chemotherapy as First-Line Treatment for Advanced Ovarian Cancer: Results of the NOGGO Non-Interventional Study (OTILIA) in 824 Patients. Cancers (Basel) 13, doi: 10.3390/cancers13194739 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rossi L et al. Bevacizumab in ovarian cancer: A critical review of phase III studies. Oncotarget 8, 12389–12405, doi: 10.18632/oncotarget.13310 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dalal V et al. Biomarker potential of IL-6 and VEGF-A in ascitic fluid of epithelial ovarian cancer patients. Clin Chim Acta 482, 27–32, doi: 10.1016/j.cca.2018.03.019 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Yeung TL et al. Systematic Identification of Druggable Epithelial-Stromal Crosstalk Signaling Networks in Ovarian Cancer. J Natl Cancer Inst 111, 272–282, doi: 10.1093/jnci/djy097 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santin AD et al. Increased levels of interleukin-10 and transforming growth factor-beta in the plasma and ascitic fluid of patients with advanced ovarian cancer. BJOG 108, 804–808, doi: 10.1111/j.1471-0528.2001.00206.x (2001). [DOI] [PubMed] [Google Scholar]
- 57.SHONIBARE Z et al. Reciprocal epigenetic Sox2 regulation by SMAD1-SMAD3 is critical for anoikis resistance and metastasis in cancer. bioRxiv, 2022.2001.2011.475900, doi: 10.1101/2022.01.11.475900 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liao S et al. TGF-beta blockade controls ascites by preventing abnormalization of lymphatic vessels in orthotopic human ovarian carcinoma models. Clin Cancer Res 17, 1415–1424, doi: 10.1158/1078-0432.CCR-10-2429 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rafehi S et al. TGFbeta signaling regulates epithelial-mesenchymal plasticity in ovarian cancer ascites-derived spheroids. Endocr Relat Cancer 23, 147–159, doi: 10.1530/ERC-15-0383 (2016). [DOI] [PubMed] [Google Scholar]
- 60.Jeon S et al. Shift of EMT gradient in 3D spheroid MSCs for activation of mesenchymal niche function. Sci Rep 7, 6859, doi: 10.1038/s41598-017-07049-3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hao Y, Baker D & Ten Dijke P TGF-beta-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int J Mol Sci 20, doi: 10.3390/ijms20112767 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Batlle E & Massague J Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 50, 924–940, doi: 10.1016/j.immuni.2019.03.024 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baci D et al. The ovarian cancer tumor immune microenvironment (TIME) as target for therapy: a focus on innate immunity cells as therapeutic effectors. International journal of molecular sciences 21, 3125 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bharti SK et al. Metabolomic characterization of experimental ovarian cancer ascitic fluid. Metabolomics 13, doi: 10.1007/s11306-017-1254-3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shender VO et al. Proteome-metabolome profiling of ovarian cancer ascites reveals novel components involved in intercellular communication. Mol Cell Proteomics 13, 3558–3571, doi: 10.1074/mcp.M114.041194 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen RR et al. Targeting of lipid metabolism with a metabolic inhibitor cocktail eradicates peritoneal metastases in ovarian cancer cells. Commun Biol 2, 281, doi: 10.1038/s42003-019-0508-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Han Q et al. Tumor cellfibroblast heterotypic aggregates in malignant ascites of patients with ovarian cancer. Int J Mol Med 44, 2245–2255, doi: 10.3892/ijmm.2019.4361 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gong Y, Yang J, Wang Y, Xue L & Wang J Metabolic factors contribute to T-cell inhibition in the ovarian cancer ascites. Int J Cancer 147, 1768–1777, doi: 10.1002/ijc.32990 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bachmayr-Heyda A et al. Integrative Systemic and Local Metabolomics with Impact on Survival in High-Grade Serous Ovarian Cancer. Clin Cancer Res 23, 2081–2092, doi: 10.1158/1078-0432.CCR-16-1647 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Radhakrishnan R et al. Ovarian cancer cell-derived lysophosphatidic acid induces glycolytic shift and cancer-associated fibroblast-phenotype in normal and peritumoral fibroblasts. Cancer Lett 442, 464–474, doi: 10.1016/j.canlet.2018.11.023 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Penet MF et al. Ascites Volumes and the Ovarian Cancer Microenvironment. Front Oncol 8, 595, doi: 10.3389/fonc.2018.00595 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anderson AS, Roberts PC, Frisard MI, Hulver MW & Schmelz EM Ovarian tumor-initiating cells display a flexible metabolism. Exp Cell Res 328, 44–57, doi: 10.1016/j.yexcr.2014.08.028 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Elschenbroich S et al. In-depth proteomics of ovarian cancer ascites: combining shotgun proteomics and selected reaction monitoring mass spectrometry. J Proteome Res 10, 2286–2299, doi: 10.1021/pr1011087 (2011). [DOI] [PubMed] [Google Scholar]
- 74.Bery A, Leung F, Smith CR, Diamandis EP & Kulasingam V Deciphering the ovarian cancer ascites fluid peptidome. Clin Proteomics 11, 13, doi: 10.1186/1559-0275-11-13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hjerpe E et al. Metabolic markers GAPDH, PKM2, ATP5B and BEC-index in advanced serous ovarian cancer. BMC Clin Pathol 13, 30, doi: 10.1186/1472-6890-13-30 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Song M et al. IRE1alpha-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature 562, 423–428, doi: 10.1038/s41586-018-0597-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chang CH et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 162, 1229–1241, doi: 10.1016/j.cell.2015.08.016 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hua W, Ten Dijke P, Kostidis S, Giera M & Hornsveld M TGFbeta-induced metabolic reprogramming during epithelial-to-mesenchymal transition in cancer. Cell Mol Life Sci 77, 2103–2123, doi: 10.1007/s00018-019-03398-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Masin M et al. GLUT3 is induced during epithelial-mesenchymal transition and promotes tumor cell proliferation in non-small cell lung cancer. Cancer Metab 2, 11, doi: 10.1186/2049-3002-2-11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shiraishi T et al. Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget 6, 130–143, doi: 10.18632/oncotarget.2766 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dong C et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 23, 316–331, doi: 10.1016/j.ccr.2013.01.022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sun Y et al. Metabolic and transcriptional profiling reveals pyruvate dehydrogenase kinase 4 as a mediator of epithelial-mesenchymal transition and drug resistance in tumor cells. Cancer Metab 2, 20, doi: 10.1186/2049-3002-2-20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu L & Derynck R Essential role of TGF-beta signaling in glucose-induced cell hypertrophy. Dev Cell 17, 35–48, doi: 10.1016/j.devcel.2009.05.010 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Muralidharan S et al. Quantifying the patterns of metabolic plasticity and heterogeneity along the epithelial-hybrid-mesenchymal spectrum in cancer. bioRxiv, 2021.2012.2018.473275, doi: 10.1101/2021.12.18.473275 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Singh A & Settleman J EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29, 4741–4751, doi: 10.1038/onc.2010.215 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Latifi A et al. Isolation and characterization of tumor cells from the ascites of ovarian cancer patients: molecular phenotype of chemoresistant ovarian tumors. PLoS One 7, e46858, doi: 10.1371/journal.pone.0046858 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gening SO et al. Stem-like tumor cells and proinflammatory cytokines in the ascitic fluid of ovarian cancer patients. Klin Lab Diagn 66, 297–303, doi: 10.51620/0869-2084-2021-66-5-297-303 (2021). [DOI] [PubMed] [Google Scholar]
- 88.Jiang YX et al. Ascites-derived ALDH+CD44+ tumour cell subsets endow stemness, metastasis and metabolic switch via PDK4-mediated STAT3/AKT/NF-kappaB/IL-8 signalling in ovarian cancer. Br J Cancer 123, 275–287, doi: 10.1038/s41416-020-0865-z (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kesh K et al. Stroma secreted IL6 selects for “stem-like” population and alters pancreatic tumor microenvironment by reprogramming metabolic pathways. Cell Death Dis 11, 967, doi: 10.1038/s41419-020-03168-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schafer ZT et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113, doi: 10.1038/nature08268 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liao J et al. Ovarian cancer spheroid cells with stem cell-like properties contribute to tumor generation, metastasis and chemotherapy resistance through hypoxia-resistant metabolism. PLoS One 9, e84941, doi: 10.1371/journal.pone.0084941 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zheng W et al. Inhibition of 6-phosphogluconate Dehydrogenase Reverses Cisplatin Resistance in Ovarian and Lung Cancer. Front Pharmacol 8, 421, doi: 10.3389/fphar.2017.00421 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Leopold JA et al. Glucose-6-phosphate dehydrogenase modulates vascular endothelial growth factor-mediated angiogenesis. J Biol Chem 278, 32100–32106, doi: 10.1074/jbc.M301293200 (2003). [DOI] [PubMed] [Google Scholar]
- 94.Merchan JR et al. Antiangiogenic activity of 2-deoxy-D-glucose. PLoS One 5, e13699, doi: 10.1371/journal.pone.0013699 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.De Bock K et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154, 651–663, doi: 10.1016/j.cell.2013.06.037 (2013). [DOI] [PubMed] [Google Scholar]
- 96.Spannuth WA et al. Functional significance of VEGFR-2 on ovarian cancer cells. Int J Cancer 124, 1045–1053, doi: 10.1002/ijc.24028 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sher I, Adham SA, Petrik J & Coomber BL Autocrine VEGF-A/KDR loop protects epithelial ovarian carcinoma cells from anoikis. Int J Cancer 124, 553–561, doi: 10.1002/ijc.23963 (2009). [DOI] [PubMed] [Google Scholar]
- 98.Galdiero MR et al. Tumor associated macrophages and neutrophils in cancer. Immunobiology 218, 1402–1410, doi: 10.1016/j.imbio.2013.06.003 (2013). [DOI] [PubMed] [Google Scholar]
- 99.Lai YS, Wahyuningtyas R, Aui SP & Chang KT Autocrine VEGF signalling on M2 macrophages regulates PD-L1 expression for immunomodulation of T cells. J Cell Mol Med 23, 1257–1267, doi: 10.1111/jcmm.14027 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Santin AD et al. Secretion of vascular endothelial growth factor in ovarian cancer. Eur J Gynaecol Oncol 20, 177–181 (1999). [PubMed] [Google Scholar]
- 101.Ha JH et al. LPA Induces Metabolic Reprogramming in Ovarian Cancer via a Pseudohypoxic Response. Cancer Res 78, 1923–1934, doi: 10.1158/0008-5472.CAN-17-1624 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wu F et al. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct Target Ther 6, 218, doi: 10.1038/s41392-021-00641-0 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Guido C et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-beta drives tumor growth: connecting TGF-beta signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle 11, 3019–3035, doi: 10.4161/cc.21384 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Capparelli C et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle 11, 3599–3610, doi: 10.4161/cc.21884 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Capparelli C et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle 11, 2285–2302, doi: 10.4161/cc.20718 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ebos JM et al. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15, 232–239, doi: 10.1016/j.ccr.2009.01.021 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Paez-Ribes M et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 15, 220–231, doi: 10.1016/j.ccr.2009.01.027 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nardo G et al. Glycolytic phenotype and AMP kinase modify the pathologic response of tumor xenografts to VEGF neutralization. Cancer Res 71, 4214–4225, doi: 10.1158/0008-5472.CAN-11-0242 (2011). [DOI] [PubMed] [Google Scholar]
- 109.Dier U, Shin DH, Hemachandra LP, Uusitalo LM & Hempel N Bioenergetic analysis of ovarian cancer cell lines: profiling of histological subtypes and identification of a mitochondria-defective cell line. PLoS One 9, e98479, doi: 10.1371/journal.pone.0098479 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pasto A et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 5, 4305–4319, doi: 10.18632/oncotarget.2010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dar S et al. Bioenergetic Adaptations in Chemoresistant Ovarian Cancer Cells. Sci Rep 7, 8760, doi: 10.1038/s41598-017-09206-0 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Katsuno Y et al. Chronic TGF-beta exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci Signal 12, doi: 10.1126/scisignal.aau8544 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dimeloe S et al. Tumor-derived TGF-beta inhibits mitochondrial respiration to suppress IFN-gamma production by human CD4(+) T cells. Sci Signal 12, doi: 10.1126/scisignal.aav3334 (2019). [DOI] [PubMed] [Google Scholar]
- 114.Kim YS et al. Context-dependent activation of SIRT3 is necessary for anchorage-independent survival and metastasis of ovarian cancer cells. Oncogene 39, 1619–1633, doi: 10.1038/s41388-019-1097-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wright GL et al. VEGF stimulation of mitochondrial biogenesis: requirement of AKT3 kinase. FASEB J 22, 3264–3275, doi: 10.1096/fj.08-106468 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang Y et al. Regulation of VEGF-induced endothelial cell migration by mitochondrial reactive oxygen species. Am J Physiol Cell Physiol 301, C695–704, doi: 10.1152/ajpcell.00322.2010 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang DX & Gutterman DD Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am J Physiol Heart Circ Physiol 292, H2023–2031, doi: 10.1152/ajpheart.01283.2006 (2007). [DOI] [PubMed] [Google Scholar]
- 118.Lahiguera A et al. Tumors defective in homologous recombination rely on oxidative metabolism: relevance to treatments with PARP inhibitors. EMBO Mol Med 12, e11217, doi: 10.15252/emmm.201911217 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Boussios S et al. Combined Strategies with Poly (ADP-Ribose) Polymerase (PARP) Inhibitors for the Treatment of Ovarian Cancer: A Literature Review. Diagnostics (Basel) 9, doi: 10.3390/diagnostics9030087 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Alvarez Secord A, O’Malley DM, Sood AK, Westin SN & Liu JF Rationale for combination PARP inhibitor and antiangiogenic treatment in advanced epithelial ovarian cancer: A review. Gynecol Oncol 162, 482–495, doi: 10.1016/j.ygyno.2021.05.018 (2021). [DOI] [PubMed] [Google Scholar]
- 121.Yang L et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol Syst Biol 10, 728, doi: 10.1002/msb.20134892 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yuan L et al. Glutamine promotes ovarian cancer cell proliferation through the mTOR/S6 pathway. Endocr Relat Cancer 22, 577–591, doi: 10.1530/ERC-15-0192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Soukupova J et al. Role of the Transforming Growth Factor-beta in regulating hepatocellular carcinoma oxidative metabolism. Sci Rep 7, 12486, doi: 10.1038/s41598-017-12837-y (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Leone RD et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013–1021, doi: 10.1126/science.aav2588 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Udumula MP et al. Ovarian cancer modulates the immunosuppressive function of CD11b(+)Gr1(+) myeloid cells via glutamine metabolism. Mol Metab 53, 101272, doi: 10.1016/j.molmet.2021.101272 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Klysz D et al. Glutamine-dependent alpha-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal 8, ra97, doi: 10.1126/scisignal.aab2610 (2015). [DOI] [PubMed] [Google Scholar]
- 127.Carr EL et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol 185, 1037–1044, doi: 10.4049/jimmunol.0903586 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Galvan-Pena S & O’Neill LA Metabolic reprograming in macrophage polarization. Front Immunol 5, 420, doi: 10.3389/fimmu.2014.00420 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Thapa B & Lee K Metabolic influence on macrophage polarization and pathogenesis. BMB Rep 52, 360–372 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mehla K & Singh PK Metabolic Regulation of Macrophage Polarization in Cancer. Trends Cancer 5, 822–834, doi: 10.1016/j.trecan.2019.10.007 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kolwijck E et al. Ovarian cyst fluid of serous ovarian tumors contains large quantities of the brain amino acid N-acetylaspartate. PLoS One 5, e10293, doi: 10.1371/journal.pone.0010293 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Menga A et al. N-acetylaspartate release by glutaminolytic ovarian cancer cells sustains protumoral macrophages. EMBO Rep 22, e51981, doi: 10.15252/embr.202051981 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Huang H et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J 36, 2334–2352, doi: 10.15252/embj.201695518 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang J et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol Cell 56, 205–218, doi: 10.1016/j.molcel.2014.08.018 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wu S et al. Targeting glutamine dependence through GLS1 inhibition suppresses ARID1A-inactivated clear cell ovarian carcinoma. Nat Cancer 2, 189–200, doi: 10.1038/s43018-020-00160-x (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hudson CD et al. Altered glutamine metabolism in platinum resistant ovarian cancer. Oncotarget 7, 41637–41649, doi: 10.18632/oncotarget.9317 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Guo J et al. Reprogramming of glutamine metabolism via glutamine synthetase silencing induces cisplatin resistance in A2780 ovarian cancer cells. BMC Cancer 21, 174, doi: 10.1186/s12885-021-07879-5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nieman KM et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 17, 1498–1503, doi: 10.1038/nm.2492 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nieman KM, Romero IL, Van Houten B & Lengyel E Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim Biophys Acta 1831, 1533–1541, doi: 10.1016/j.bbalip.2013.02.010 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sawyer BT et al. Targeting Fatty Acid Oxidation to Promote Anoikis and Inhibit Ovarian Cancer Progression. Mol Cancer Res 18, 1088–1098, doi: 10.1158/1541-7786.MCR-19-1057 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bensaad K et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep 9, 349–365, doi: 10.1016/j.celrep.2014.08.056 (2014). [DOI] [PubMed] [Google Scholar]
- 142.Ladanyi A et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 37, 2285–2301, doi: 10.1038/s41388-017-0093-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nath A, Li I, Roberts LR & Chan C Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci Rep 5, 14752, doi: 10.1038/srep14752 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Elmasri H et al. Fatty acid binding protein 4 is a target of VEGF and a regulator of cell proliferation in endothelial cells. FASEB J 23, 3865–3873, doi: 10.1096/fj.09-134882 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Curtarello M et al. Rewiring of Lipid Metabolism and Storage in Ovarian Cancer Cells after Anti-VEGF Therapy. Cells 8, doi: 10.3390/cells8121601 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Iwamoto H et al. Cancer Lipid Metabolism Confers Antiangiogenic Drug Resistance. Cell Metab 28, 104–117 e105, doi: 10.1016/j.cmet.2018.05.005 (2018). [DOI] [PubMed] [Google Scholar]
- 147.Goossens P et al. Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression. Cell Metab 29, 1376–1389 e1374, doi: 10.1016/j.cmet.2019.02.016 (2019). [DOI] [PubMed] [Google Scholar]
- 148.Weiss JM et al. Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors. J Clin Invest 128, 3794–3805, doi: 10.1172/JCI99169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wang D & Dubois RN Eicosanoids and cancer. Nat Rev Cancer 10, 181–193, doi: 10.1038/nrc2809 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wefers C et al. Different Lipid Regulation in Ovarian Cancer: Inhibition of the Immune System. Int J Mol Sci 19, doi: 10.3390/ijms19010273 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wang D et al. S1P differentially regulates migration of human ovarian cancer and human ovarian surface epithelial cells. Mol Cancer Ther 7, 1993–2002, doi: 10.1158/1535-7163.MCT-08-0088 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Johnson AR, Milner JJ & Makowski L The inflammation highway: metabolism accelerates inflammatory traffic in obesity. Immunol Rev 249, 218–238, doi: 10.1111/j.1600-065X.2012.01151.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Torrence ME & Manning BD Nutrient Sensing in Cancer. Annual Review of Cancer Biology 2, 251–269, doi: 10.1146/annurev-cancerbio-030617-050329 (2018). [DOI] [Google Scholar]
- 154.Thompson CB & Bielska AA Growth factors stimulate anabolic metabolism by directing nutrient uptake. J Biol Chem 294, 17883–17888, doi: 10.1074/jbc.AW119.008146 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yuan HX, Xiong Y & Guan KL Nutrient sensing, metabolism, and cell growth control. Mol Cell 49, 379–387, doi: 10.1016/j.molcel.2013.01.019 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Said EA et al. Defining IL‐6 levels in healthy individuals: A meta‐analysis. Journal of Medical Virology 93, 3915–3924 (2021). [DOI] [PubMed] [Google Scholar]
- 157.Chudecka-Głaz AM et al. Assessment of selected cytokines, proteins, and growth factors in the peritoneal fluid of patients with ovarian cancer and benign gynecological conditions. OncoTargets and therapy 8, 471 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhang J & Bai C Elevated serum interleukin-8 level as a preferable biomarker for identifying uncontrolled asthma and glucocorticosteroid responsiveness. Tanaffos 16, 260 (2017). [PMC free article] [PubMed] [Google Scholar]
- 159.Kleiner G, Marcuzzi A, Zanin V, Monasta L & Zauli G Cytokine levels in the serum of healthy subjects. Mediators of inflammation 2013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Crispim PCA et al. IL6, IL8, and IL10 in the distinction of malignant ovarian neoplasms and endometriomas. American Journal of Reproductive Immunology 84, e13309 (2020). [DOI] [PubMed] [Google Scholar]
- 161.Tonetti CR et al. Ovarian Cancer-Associated Ascites Have High Proportions of Cytokine-Responsive CD56bright NK Cells. Cells 10, 1702 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Nowak M et al. Proinflammatory and immunosuppressive serum, ascites and cyst fluid cytokines in patients with early and advanced ovarian cancer and benign ovarian tumors. Neuroendocrinology Letters 31, 375–383 (2010). [PubMed] [Google Scholar]
- 163.Liang B, Guo Z, Li Y & Liu C Elevated VEGF concentrations in ascites and serum predict adverse prognosis in ovarian cancer. Scandinavian journal of clinical and laboratory investigation 73, 309–314 (2013). [DOI] [PubMed] [Google Scholar]
- 164.Yigit R et al. Cytokine analysis as a tool to understand tumour–host interaction in ovarian cancer. European Journal of Cancer 47, 1883–1889 (2011). [DOI] [PubMed] [Google Scholar]
- 165.Giles BM et al. Analytical characterization of an enzyme-linked immunosorbent assay for the measurement of transforming growth factor β1 in human plasma. The Journal of Applied Laboratory Medicine 3, 200–212 (2018). [DOI] [PubMed] [Google Scholar]
- 166.Mancini D et al. New methodologies to accurately assess circulating active transforming growth factor-β1 levels: implications for evaluating heart failure and the impact of left ventricular assist devices. Translational Research 192, 15–29 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Khan SA, Joyce J & Tsuda T Quantification of active and total transforming growth factor-beta levels in serum and solid organ tissues by bioassay. BMC Res Notes 5, 636, doi: 10.1186/1756-0500-5-636 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wakefield LM et al. Transforming growth factor-beta1 circulates in normal human plasma and is unchanged in advanced metastatic breast cancer. Clin Cancer Res 1, 129–136 (1995). [PubMed] [Google Scholar]
- 169.Bozas G et al. Prechemotherapy serum levels of CD105, transforming growth factor beta2, and vascular endothelial growth factor are associated with prognosis in patients with advanced epithelial ovarian cancer treated with cytoreductive surgery and platinum-based chemotherapy. Int J Gynecol Cancer 20, 248–254, doi: 10.1111/IGC.0b013e3181cc25c3 (2010). [DOI] [PubMed] [Google Scholar]
- 170.Moradi MM et al. Serum and ascitic fluid levels of interleukin‐1, interleukin‐6, and tumor necrosis factor‐alpha in patients with ovarian epithelial cancer. Cancer 72, 2433–2440 (1993). [DOI] [PubMed] [Google Scholar]
- 171.Hussain S et al. Level of interferon gamma in the blood of tuberculosis patients. Iranian Journal of Immunology 7, 240–246 (2010). [PubMed] [Google Scholar]
- 172.Chen Y-L et al. Interferon-gamma in ascites could be a predictive biomarker of outcome in ovarian carcinoma. Gynecologic oncology 131, 63–68 (2013). [DOI] [PubMed] [Google Scholar]
- 173.Madsen CV et al. Serum platelet-derived growth factor and fibroblast growth factor in patients with benign and malignant ovarian tumors. Anticancer research 32, 3817–3825 (2012). [PubMed] [Google Scholar]
- 174.Tsukishiro S, Suzumori N, Nishikawa H, Arakawa A & Suzumori K Elevated serum RANTES levels in patients with ovarian cancer correlate with the extent of the disorder. Gynecologic oncology 102, 542–545 (2006). [DOI] [PubMed] [Google Scholar]
- 175.Negus R et al. The detection and localization of monocyte chemoattractant protein-1 (MCP-1) in human ovarian cancer. The Journal of clinical investigation 95, 2391–2396 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sadeghi M et al. Serum levels of chemokines CCL4 and CCL5 in cirrhotic patients indicate the presence of hepatocellular carcinoma. British journal of cancer 113, 756–762 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rådestad E et al. Immune profiling and identification of prognostic immune-related risk factors in human ovarian cancer. Oncoimmunology 8, e1535730 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Syversen SW et al. A high serum level of eotaxin (CCL 11) is associated with less radiographic progression in early rheumatoid arthritis patients. Arthritis research & therapy 10, 1–4 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Cheng W-L et al. Overexpression of CXCL1 and its receptor CXCR2 promote tumor invasion in gastric cancer. Annals of oncology 22, 2267–2276 (2011). [DOI] [PubMed] [Google Scholar]
- 180.Dimberg J, Dienus O, Löfgren S, Hugander A & Wågsäter D Expression and gene polymorphisms of the chemokine CXCL5 in colorectal cancer patients. International journal of oncology 31, 97–102 (2007). [PubMed] [Google Scholar]