

Abstract
Inhibitors targeting the antiapoptotic molecule BCL-2 have therapeutic potential for the treatment of acute myeloid leukaemia (AML); however, BCL-2 inhibitors such as venetoclax exhibit limited monotherapy efficacy in relapsed or refractory human AML. PI3Kδ/AKT signalling has been shown to be constitutively active in AML patients. Here, we demonstrate that the combination of BCL-2 and PI3Kδ inhibitors exerts synergistic antitumour effects both in vitro and in vivo in AML. Cotreatment with venetoclax and the specific PI3Kδ inhibitor idelalisib significantly enhanced antiproliferative effects and induced caspase-dependent apoptosis in a panel of AML cell lines. The synergistic effects were mechanistically based on the inactivation of AKT/4E-BP-1 signalling and the reduction of MCL-1 expression, which diminished the binding of Bim to MCL-1. Notably, compared with the parental FLT3-ITD-positive MV-4-11, the acquired FLT3 inhibitor quizartinib-resistant xenograft model carrying the F691L mutation, exhibited a markedly higher sensitivity to venetoclax. Furthermore, venetoclax combined with idelalisib led to tumour regression in all animals in this quizartinib-resistant AML model. Thus, these data indicate that combined inhibition of BCL-2 and PI3Kδ may be a promising strategy in AML, especially for patients with FLT3-ITD and/or FLT3-TKD mutations.
Keywords: Acute myeloid leukaemia, BCL-2, PI3Kδ, FLT3, synergistic lethality
Introduction
Acute myeloid leukaemia (AML) is an aggressive heterogeneous clonal disease of haemopoietic progenitor cells [1-3]. In adults, AML is the most common form of acute leukaemia [4]. Until recently, therapy options for AML patients have been limited, it’s largely due to the high heterogeneity and dynamics of AML, which is characterized by driver gene mutations acquired by multiple somatic cells, coexisting competitive cloning, and disease development over time [5]. Although many molecular targeted drugs have exhibited improved survival rates [6], due to the genome instability of AML cells and the rapid emergence of drug resistance mutation, durable response with monotherapy for AML patients is often rarely seen [7]. Therefore, novel therapeutic strategies against specific targets have become an intensely important research area, and rational combinations are believed to be additional effective therapeutic options for AML patients.
The B-cell lymphoma 2 (BCL-2) protein is a critical regulator in the mitochondrial apoptotic pathway. Previous researches have shown that the expression of BCL-2 family proteins is increased in AML mother cells, and the survival of most AML stem cells depends on the aberrantly high levels of BCL-2 [8,9]. Abnormal overexpression of BCL-2 is related to tumorigenesis and increased chemotherapy resistance in multiple malignancies, including AML [8]. Venetoclax is a highly selective BCL-2 inhibitor and has been approved for use in chronic lymphocytic leukaemia [10]. However, venetoclax has limited and transient single-agent activity against AML in both preclinical and clinical investigations [11,12]. Venetoclax-based combinations are under exploring, for example, cotreatment with intensive chemotherapy drugs [13] or other targeted inhibitors [14,15]. In October 2020, the FDA has granted full approval for venetoclax in combination with azacytidine, decitabine or low-dose cytarabine for treating newly diagnosed AML [16,17].
The class IA phosphoinositide 3-kinase (PI3K)/AKT pathway has been reported to be extensively hyperactive in AML cells, which prompts cell survival and proliferation [18,19]. The catalytic subunits of class IA PI3K are subdivided into 4 isoforms: p110α, p110β, p110δ and p110γ [20]. The expression and activity of the p110δ isoform of PI3K are consistently high in AML, and this isoform plays a critical role in disease progression, which makes it a potential therapeutic target [21]. However, monotherapy with idelalisib, a selective PI3Kδ inhibitor that has been approved for use in relapsed chronic lymphocytic leukaemia and indolent lymphoma [22], exhibited limited effect against AML in a phase I dose-escalation study [23]. We previously reported that co-inhibition of PI3Kδ and FMS-like tyrosine kinase-3 (FLT3) signalling exerted synergistic antitumour efficacy and overcame acquired drug resistance in FLT3-activated AML [24]. Although synergy between targeting BCL-2 and MCL-1 has been previously reported in AML, prior MCL-1 inhibitors have not been suitable for clinical development [25,26]. Previous studies have shown that PI3K/mTOR pathway inhibition significantly diminishes MCL-1 protein level [27]. Furthermore, the acquired resistance of BCL-2 inhibitor develops due to increased activation of the PI3K/AKT/mTOR signalling pathway and upregulation of MCL-1 and BCL-XL that sequester Bim [27]. Collectively, PI3Kδ inhibitor-based combinations may be a promising option for AML.
FLT3 mutations are the most common molecular alterations in AML, seen in approximately 30% of patients with newly diagnosed AML [28,29]. FLT3-internal tandem duplication (ITD) mutation, which is the most common genetic alteration in AML [29]. In the venetoclax monotherapy trial, a small subset of FLT3-ITD+ patients exerted no response and some patients relapsed with novel FLT3-ITD mutation [28]. Recently, many FLT3 inhibitors, including midostaurin and gilteritinib, have been developed and presented impressive preclinical and clinical activity in AML. However, drug resistance rapidly ensues following treating AML patients with FLT3 inhibitor, leading to modest durable responses and poor clinical therapeutic efficacy [30]. A potential strategy for overcoming acquired drug resistance in many tumour types is the usage of combination regimens [31]. In this study, we demonstrate that the combination of BCL-2 and PI3Kδ inhibitors is synergistically lethal against AML, which is mediated by enhanced inhibition of AKT/4E-binding protein (4E-BP) and MCL-1. Importantly, venetoclax alone or in combination with idelalisib is highly potent in FLT3-ITD+ AML carrying the F691L mutation, which acquires resistance to quizartinib. These findings provide the potential for combining venetoclax with PI3Kδ inhibitors to lower the dosage of venetoclax and enhance its applicability in venetoclax- or quizartinib-resistant AML.
Materials and methods
Materials
Venetoclax, quizartinib, crenolanib and gilteritinib were purchased from MedChemExpress (New Jersey, USA). Idelalisib, sunitinib, sorafenib and paclitaxel were purchased from Melone Pharmaceutical (Dalian, China).
Antibodies specific for PI3Kinase p110δ, phospho-FLT3 (Tyr589/591), FLT3, STAT5, phospho-STAT5 (Tyr694), AKT, phospho-AKT (Ser473), phospho-4E-BP-1 (Thr37/46), phospho-ERK1/2 (Thr202/Tyr204), ERK1/2, phospho-rpS6 (Ser235/236), rpS6, caspase-8, caspase-9, caspase-3, PARP, MCL-1, BCL-XL, BCL-2, Bim, β-Actin and GAPDH were all purchased from Cell Signaling Technology (Danvers, MA).
Cell lines
MV-4-11, THP-1, RS4;11, U937, and TF-1 cell lines were purchased from the ATCC (Manassas, VA). EOL-1, MOLM-13. SET-2 and HEL cell lines were purchased from DSMZ (Braunschweig, Germany). Cell authentication was conducted by Geneing Biotechnologies Inc. (Shanghai, China). MV-4-11/quizartinib was generated by continuous incubating with gradually increased concentration of quizartinib.
Cell proliferation assay
MTT (Sigma-Aldrich, St. Louis, MO) assay was used to measure the cytotoxicity after cells were incubated with gradient diluted concentrations of drugs for 72 h, and 50% inhibitory concentration (IC50) values were calculated using Prism 5 (GraphPad Software, San Diego, CA) curve-fitting software. The interaction between two drugs was evaluated via calculating the combination index (CI) values conducted in Calcusyn (Calcusyn, Inc., Paramus, NJ). Synergistic effects: CI < 1, addictive effects: CI = 1, antagonistic effects: CI > 1.
Western blot and co-immunoprecipitation (IP)
Whole-cell lysates were prepared in SDS sample loading buffer (Melone, Dalian, China) after treatment, and then separated by SDS-PAGE and electroblotted onto polyvinylidene difluoride membranes (Millipore, Bedford, MA). After membrane closure and antibody incubation, the chemiluminescence intensity was determined by Tanon 4600 (Tanon, Shanghai, China).
For co-IP, whole-cell lysates were incubated with anti-Bim antibody and protein G agarose (Cell Signaling Technology, Danvers, MA), and the levels of immunoprecipitated proteins were subsequently analysed by Western blotting.
Survival and correlation analyses
The survival analyses of PI3Kδ and BCL-2 for AML patients and the correlation analyses of these two genes in the TCGA-LAML were performed via the Gene Expression Profiling Interactive Analysis web service (GEPIA, http://gepia.cancer-pku.cn/index.html).
Annexin V-fluorescein isothiocyanate (annexin V-FITC)/propidium iodide (PI) flow cytometry assay
After treatment with venetoclax and/or idelalisib for 24 h, AML cells were collected and then using the Annexin V-FITC/PI Apoptosis Kit (BD Biosciences, San Jose, CA) to quantify cell apoptosis according to the manufacturer’s instructions.
Analysis of mitochondrial membrane potential
Cells were collected at the end of treatment and their viability and membrane potential were determined using the Mitochondrial Membrane Potential Detection Kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions. Fluorescence was acquired using CytoFLEXTM Flow Cytometer (Beckman Coulter, Inc., CA, USA). And the samples were visualized using Zeiss Axio Observer A1 fluorescence microscope (ZEISS, Oberkochen, Germany).
In vivo study
5-6 weeks old female athymic BALB/c nude mice were purchased from Charles River Laboratories (Beijing, China). Nude mice were inoculated subcutaneously with cells to establish MV-4-11 and MV-4-11/quizartinib tumour xenografts. Mice were randomly assigned to the vehicle and treatment groups (n = 5-6/group) when tumours grew to a volume of 100-200 mm3. The treatment groups were administrated by oral venetoclax, idelalisib, or their combination daily. Tumour volumes were monitored by callipers and calculated as (length×width2)/2. Tumours were dissected 3 h following the final dose and analysed by Western blotting or immunohistochemistry (IHC). All the animal-related procedures were canonically conducted according to the Institutional Animal Care and Use Committee guidelines of the Shanghai Institute of Materia Medica Chinese Academy of Sciences.
Statistical analysis
Means, SD, and SEM were analysed using GraphPad Prism 5.0. Statistical differences were calculated by the Student’s t-test between two groups or one-way ANOVA among three or more groups and considered significant if P values < 0.05.
Results
The combination of BCL-2 and PI3Kδ inhibitors exerts synergistic antiproliferative activity in AML cell lines
First, we assessed the single-agent activity of the BCL-2 inhibitor venetoclax and the PI3Kδ inhibitor idelalisib on the proliferation in a panel of AML cell lines with different expression levels of the BCL-2 family and PI3Kδ proteins. Consistent with previous reports [12], overall, the level of BCL-2 correlated with sensitivity to venetoclax. However, the level of BCL-XL inversely correlated with venetoclax sensitivity (Figure 1A, 1C). MV-4-11, EOL-1, THP-1, RS4;11 and MOLM13 cells sensitive to venetoclax with IC50 values ranging from 16 nM to 102 nM, exhibited high protein level of BCL-2 and low protein level of BCL-XL; in contrast, minimal cytotoxicity was noted in AML cells with low or absent protein level of BCL-2, such as U937, SET-2, HEL and TF-1. Idelalisib exhibited limited antiproliferative effects in all tested AML cells, even though PI3Kδ was ubiquitously expressed (Figure 1B, 1C).
Figure 1.
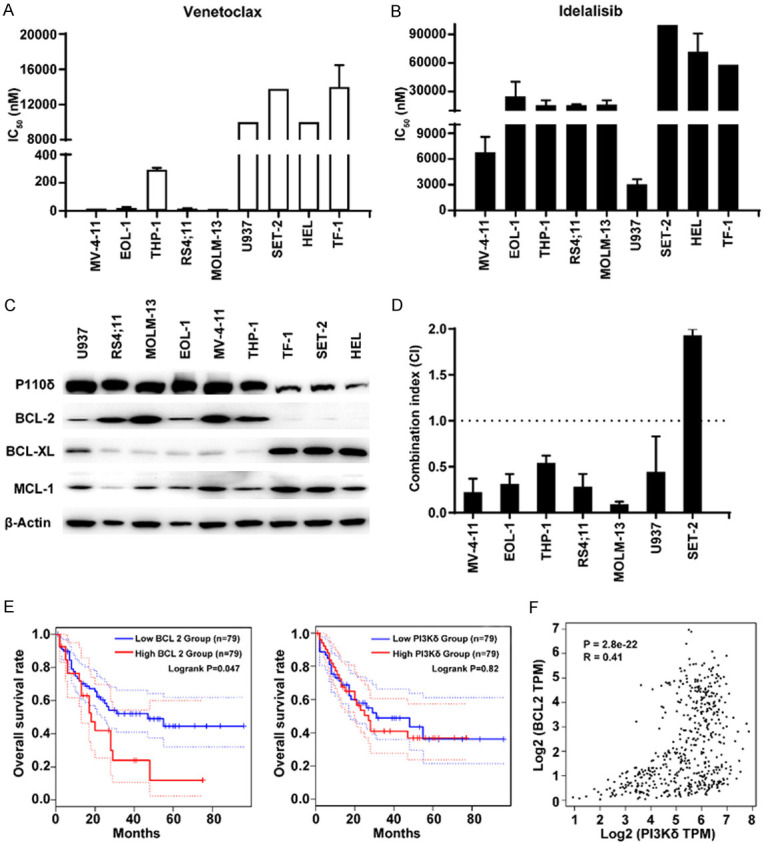
Antiproliferative activity of the co-inhibition of BCL-2 and PI3Kδ in AML cell lines. (A, B) Antiproliferative activity of venetoclax (A) or idelalisib (B) in a panel of AML cell lines. After cells were treated with venetoclax or idelalisib for 72 h, MTT assay was used to assess cell viability. (C) Protein levels of PI3Kδ, BCL-2, BCL-XL, and MCL-1 in a panel of AML cell lines were assessed by Western blotting. (D) Synergistic antiproliferative effects in vitro. After AML cells were treated with venetoclax and/or idelalisib for 72 h, MTT assay was used to assess cell viability. CI was calculated with CalcuSyn DemoVersion 2.0 software. Synergistic efficacy: CI < 1. (E) The differences in survival related to BCL-2 or PI3Kδ mRNA expression were compared in each group (log-rank test). The dotted line indicated the 95% confidence interval. (F) Scatter plot showed the correlation between BCL-2 and PI3Kδ mRNA levels in AML. Statistical analysis was performed using a two-tailed Student’s t-test. R, Spearman’s correlation coefficient.
Next, we explored the effects of cotreatment with idelalisib and venetoclax in seven AML cell lines which displayed different sensitivities to idelalisib or venetoclax in monotherapy. As shown in Figure 1D, the combination clearly increased antiproliferative effects in both venetoclax-sensitive and venetoclax-insensitive cell lines, with CI values ranging from 0.09 to 0.54, except that antagonism was seen in the BCL-2-negative SET-2 cell line. Furthermore, the combination of venetoclax and idelalisib reached a strong synergism (CI < 0.3) in FLT3-ITD+ MV-4-11 and MOLM-13 cells.
Additionally, we studied the roles of the BCL-2 and PI3Kδ genes in the survival of patients with AML using the GEPIA database. The data suggested that increased BCL-2 was related to poor overall survival of AML (Figure 1E, P = 0.047 < 0.05). Although the expression of PI3Kδ was not associated with patient survival, the BCL-2 level was positively correlated with PI3Kδ by correlation analysis (Figure 1F, R = 0.41, P < 0.05). These results may provide some evidence for the potential clinical relevance of combining BCL-2 and PI3Kδ inhibitors in AML treatment.
The combination of BCL-2 and PI3Kδ inhibitors synergistically induces caspase-dependent apoptosis in AML cell lines
To measure the cell apoptosis induced by idelalisib and/or venetoclax in AML cell lines, the annexin V/PI assay was performed. Cotreatment produced significantly enhanced cell apoptosis compared with single drug treatment (Figure 2A), which was accompanied by the increased cleaved caspase 9, caspase 8, caspase 3 and PARP levels in both MV-4-11 (FLT3-ITD+) cells and EOL-1 (FLT3-wild type activated) cells (Figure 2B). To further examine the effects of venetoclax and idelalisib on mitochondrial function, MitoTracker Red CMXRos assays were conducted. After pretreatment of MV-4-11 with the pan-caspase inhibitor Z-VAD-FMK, the amount of green fluorescent (Annexin V-FITC) was significantly decreased, together with increased red fluorescence, compared with venetoclax and idelalisib cotreatment, indicating that the caspase inhibitor rescued cell viability and increased the stability of the mitochondrial membrane potential (Figure 2C, 2D). In summary, these results suggested that the synergistic antiproliferative effect induced by co-inhibition of BCL-2 and PI3Kδ mechanistically involved caspase-dependent apoptosis.
Figure 2.
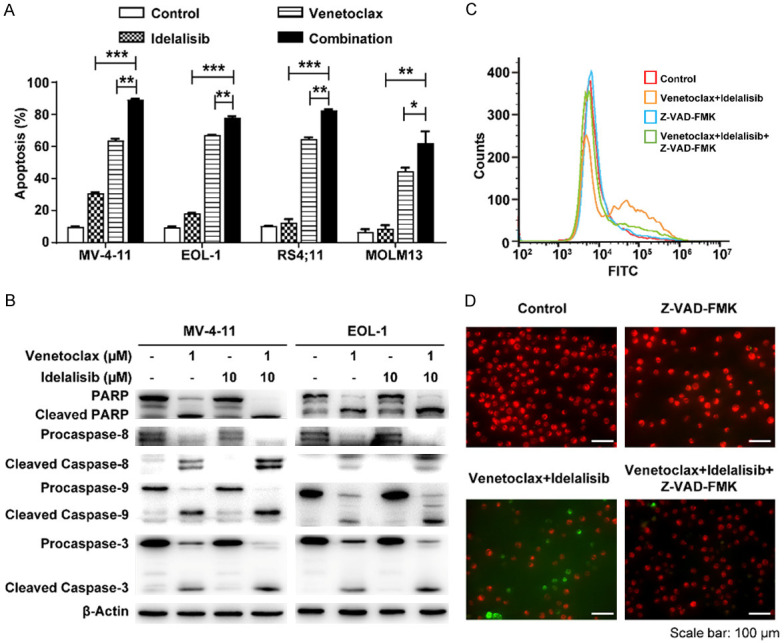
The combination of BCL-2 and PI3Kδ inhibitors synergistically induces caspase-dependent apoptosis in AML cell lines. (A) AML cell lines were treated with idelalisib and/or venetoclax for 24 h, and then cell apoptosis was assessed by annexin V-FITC/PI staining and FACS analysis. The concentrations used in this study were 10 µM idelalisib, 50 nM, 25 nM, 50 nM, and 10 nM venetoclax for MV-4-11, EOL-1, RS4;11 and MOLM13 cells, respectively. (B) Protein levels of PARP, Caspase 8, Caspase-9, Caspase-3 in MV-4-11 and EOL-1 cells were determined by Western blotting after treatment with idelalisib and/or venetoclax for 24 h. (C, D) MV-4-11 cells were treated with drugs for 24 h and then cell viability and mitochondrial membrane potential were measured by annexin V-FITC/MitoTracker Red CMXRos staining. FITC histograms were obtained by flow cytometry (C), and samples were visualized under a fluorescence microscope (D). The concentrations used in this study were 50 µM Z-VAD-FMK, 5 nM venetoclax, and 10 µM idelalisib. *P < 0.05, **P < 0.01, ***P < 0.001.
Combination of BCL-2 and PI3Kδ inhibitors downregulates the expression of MCL-1 mediated by AKT/4E-BP-1 inactivation
Given that venetoclax monotherapy exhibits limited single-agent activity against AML with FLT3-ITD mutation in both preclinical and clinical investigations [11,12], MV-4-11 and EOL-1, these two FLT3-activated AML cell lines were chosen to further identify the mechanism of synergistic apoptosis induced by the combination of venetoclax and idelalisib. Compared with either drug alone, venetoclax combined with idelalisib exhibited greater inhibition of MCL-1, but not BCL-2 or BCL-XL (Figure 3A). To determine the role of MCL-1 in the apoptosis induced by the combination, we performed co-IP experiment by Bim antibody and the interaction between MCL-1 and Bim was analysed. The combination of venetoclax and idelalisib markedly decreased the protein levels of MCL-1 bound to Bim in both MV-4-11 and EOL-1 cells (Figure 3B).
Figure 3.
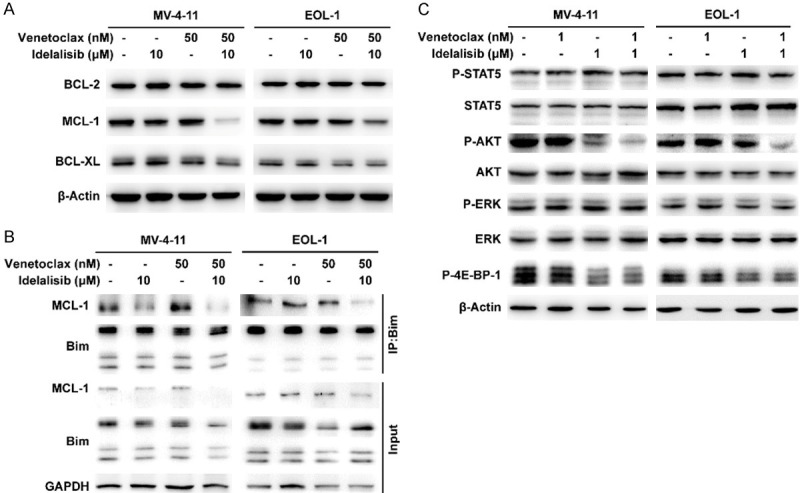
The combination of BCL-2 and PI3Kδ inhibitors inhibits the AKT pathway and downregulates MCL-1, resulting in reduced binding of MCL-1 to Bim. (A) Protein levels of BCL-2, MCL-1 and BCL-XL in MV-4-11 and EOL-1 cells were determined via Western blotting after treatment with idelalisib and/or venetoclax for 3 h. (B) MCL-1/Bim interaction in MV-4-11 and EOL-1 cells was measured by co-IP experiment using Bim antibody after 3 h treatment with idelalisib and/or venetoclax, and the corresponding MCL-1 and Bim were analysed by Western blotting. (C) Phosphorylation levels of STAT5, AKT, ERK and 4E-BP-1 in MV-4-11 and EOL-1 cells were analysed by Western blotting after treatment with venetoclax and/or idelalisib for 3 h.
It has been shown that the PI3K/AKT/mTOR pathway regulates MCL-1 expression by 4E-BP-1/mTORC1-dependent translation in AML [32]. Next, we assessed the effect of cotreatment on the JAK/STAT, PI3K/AKT, and MAPK/ERK pathways in MV-4-11 and EOL-1 cells. Combination markedly inhibited the phosphorylation of AKT and 4E-BP-1 compared with single drug treatment (Figure 3C). Collectively, these results indicated that combined inhibition of PI3Kδ and BCL-2 leads to synergistic apoptosis through the inactivation of AKT/4E-BP-1 and the downregulation of MCL-1, which may diminish the binding of Bim to MCL-1 and lead to the activation of Bak/Bax.
The combination of BCL-2 and PI3Kδ inhibitors overcomes acquired resistance to FLT3 inhibitors in AML cells
Although many inhibitors for FLT3 have been explored and demonstrated impressive responses in AML, resistance often developed in most AML patients after prolonged FLT3 inhibitor monotreatment [30]. To examine whether the combination of BCL-2 and PI3Kδ inhibitors could also overcome FLT3 inhibitors resistance, MV-4-11/quizartinib, the acquired quizartinib-resistant cell line was generated by continuous incubating with gradually increasing concentration of the FLT3 inhibitor quizartinib. MV-4-11/quizartinib cells exhibited disparate sensitivities to multiple FLT3 inhibitors with resistance factors (RFs) of 1009.4, 1536.8 for quizartinib and sorafenib, whereas they showed less resistance to the type I FLT3 inhibitors crenolanib, gilteritinib and sunitinib, with RFs of 6.6, 31.8 and 5.6, respectively (Table 1). Additionally, MV-4-11/quizartinib-resistant cells retained sensitivity to both venetoclax and idelalisib, as well as the cytotoxic drug paclitaxel (Figure 4A; Table 1).
Table 1.
Antiproliferative effects of compounds against MV-4-11 and MV-4-11/quizartinib cells
| Compound | IC50 (nM, Mean ± SD) | RF | |
|---|---|---|---|
|
| |||
| MV-4-11 | MV-4-11/quizartinib | ||
| Quizartinib | 0.2 ± 0.1 | 181.8 ± 15.5 | 1009.4 |
| Crenolanib | 2.1 ± 0.1 | 13.7 ± 0.9 | 6.6 |
| Gilteritinib | 1.5 ± 0.6 | 47.0 ± 28.0 | 31.8 |
| Sunitinib | 4.1 ± 0.1 | 23.3 ± 0.8 | 5.6 |
| Sorafenib | 0.7 ± 0.1 | 1116.0 ± 152.7 | 1536.8 |
| Paclitaxel | 0.4 ± 0.1 | 0.2 ± 0.1 | 0.5 |
Notes: MV-4-11 and MV-4-11/quizartinib cells were treated with different concentrations of compounds for 72 h. Cell survival was analysed by MTT assay. Data are presented as the means ± SD of three independent experiments. RF, resistance factor.
Figure 4.
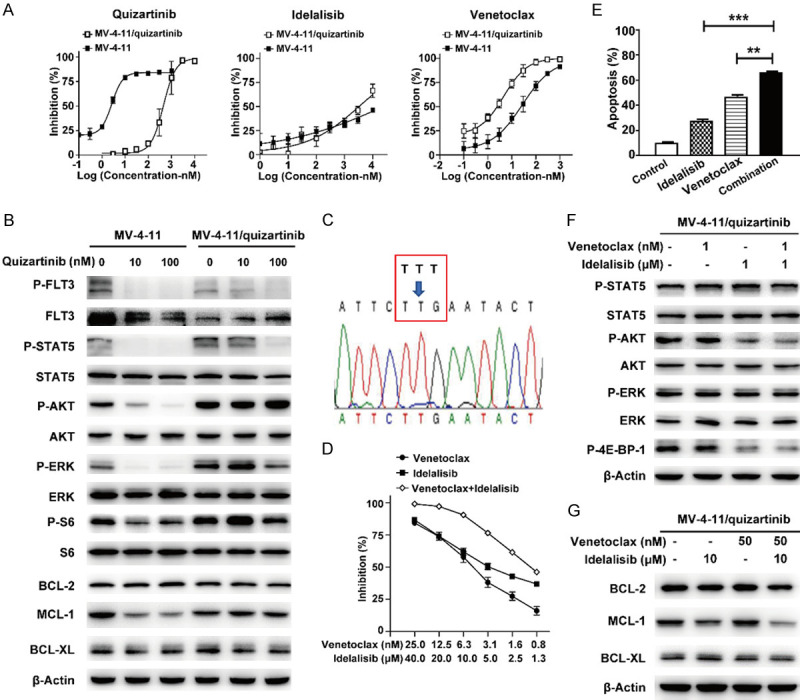
The combination of BCL-2 and PI3Kδ inhibitors overcomes acquired resistance to FLT3 inhibitors in AML cells. (A) After MV-4-11 and MV-4-11/quizartinib cell lines were treated with quizartinib, idelalisib or venetoclax for 72 h, MTT assay was used to assess cell viability. (B) After 3 h treatment with quizartinib in MV-4-11 and MV-4-11/quizartinib cell lines, the phosphorylation status of FLT3 signalling protein and expression of BCL-2 family members were determined by Western blotting. (C) Sequencing of FLT3 in MV-4-11/quizartinib. The “T” to “G” replacement at the codon 691 led to a Phe-to-Leu exchange (F691L). (D) After MV-4-11/quizartinib cells were treated with venetoclax, idelalisib, or a combination for 72 h, MTT assay was used to assess cell viability. (E) Cells were treated with idelalisib (10 µM), venetoclax (50 nM), or their combination for 24 h, and cell apoptosis was measured by annexin V-FITC/PI staining. (F, G) After MV-4-11/quizartinib cells were treated with venetoclax and/or idelalisib for 3 h, the lysates were detected by Western blot with the indicated antibodies. **P < 0.01, ***P < 0.001.
To investigate the mechanism of resistance to quizartinib in MV-4-11/quizartinib cells, the FLT3 signalling in parental and resistant cells was profiled. In MV-4-11/quizartinib, quizartinib still potently decreased the phosphorylation of FLT3 and STAT5, but to a lesser extent compared with parental MV-4-11 cells (Figure 4B). Nevertheless, it significantly suppressed the phosphorylation of AKT, ERK and S6 only in parental cells (Figure 4B). Additionally, quizartinib only suppressed the level of MCL-1 in parental but not in MV-4-11/quizartinib cells without affecting other antiapoptotic proteins of the BCL-2 family, including BCL-2 or BCL-XL. Next-generation sequencing of FLT3 in MV-4-11/quizartinib cells revealed substitution of F691L within the FLT3 tyrosine kinase domain (TKD) (Figure 4C), which was previously identified in AML patients who relapsed upon treatment with quizartinib [33].
Furthermore, the combination of idelalisib and venetoclax showed strong synergistic antiproliferative effects with a CI value of 0.08 and increased the quantity of apoptosis in MV-4-11/quizartinib cells (Figure 4D, 4E). Additionally, cotreatment greatly decreased the phosphorylation of AKT and 4E-BP-1 and the level of MCL-1 (Figure 4F, 4G). Together, these findings indicated the potency of the BCL-2 and PI3Kδ inhibitor combination in refractory AML with FLT3-ITD/F691L dual mutations based on decreasing MCL-1 expression through the inactivation of AKT/4E-BP-1 signalling.
The combination of BCL-2 and PI3Kδ inhibitors has synergistic antitumour activity in AML xenografts
Next, we investigated the in vivo antitumour efficacy of cotreatment with PI3Kδ and BCL-2 inhibitors. Consistent with previous data [28], although MV-4-11 cells were sensitive to venetoclax in vitro, inherent resistance to venetoclax in MV-4-11 xenografts was observed. Idelalisib (180 mg/kg) or venetoclax (75 mg/kg) alone showed weak antitumour activities with tumour growth inhibition (TGI) values of 38.3% and 31.4%, respectively, whereas cotreatment produced an enhanced antitumour efficacy with a TGI of 73.5% (Figure 5A). No obvious body weight loss was found when treated with drug alone or in combination (Figure 5B). Furthermore, compared with either drug alone, decreased expression of MCL-1 and an increased level of cleaved caspase 3 in tumour tissues from cotreatment groups were observed by Western blotting and IHC assays (Figure 5C, 5D).
Figure 5.

The combination of BCL-2 and FLT3 inhibitors exerts enhanced antitumour activity in mice xenografted with human AML. (A-D) MV-4-11 xenograft mice received vehicle, idelalisib (180 mg/kg), venetoclax (75 mg/kg), their combination or quizartinib (1 mg/kg). Tumour volumes were determined (A), and mouse body weights were measured (B). The mice were euthanized 3 h following the final dose, and then tumours were excised, lysed, and analysed by Western blotting (C) or IHC (D). (E-G) MV-4-11/quizartinib tumour-bearing mice were treated with vehicle or the indicated dosages of idelalisib, venetoclax, their combination or quizartinib. Tumour volumes were measured (E), relative tumour volumes were indicated as percent change from baseline at Day 18 (F), and body weights were shown as the means ± SD (G). **P < 0.01, ***P < 0.001, versus vehicle; ##P < 0.01 versus either single agent alone.
Consistent with the in vitro data, MV4-11/quizartinib xenografts were resistant to quizartinib with a TGI of 12.0% at a dosage of 1 mg/kg, compared with 88.1% in MV-4-11 xenografts. Unexpectedly, MV-4-11/quizartinib xenografts displayed preferable sensitivity to venetoclax with a TGI of 85.4% at a dosage of 75 mg/kg. Moreover, in comparison to each single agent, the combination led to further reduced tumour growth in the MV-4-11/quizartinib xenograft model (P < 0.01) (Figure 5E). Importantly, tumour regression was observed in all mice from the cotreatment group, while no regression of tumours was observed when treated with single agent (Figure 5F). Furthermore, complete tumour regression was found in one of five mice in the cotreatment group. The combination treatment was generally well-tolerated, and no obvious loss of body weight was found in all groups (Figure 5G). In summary, these results indicated that venetoclax combined with idelalisib exhibits synergistic benefits in AML with FLT-3-ITD and/or acquired TKD mutation.
Discussion
BCL-2 overexpression is related to poor therapy response in AML, which makes it a theoretically promising treatment target. However, monotherapy with the BCL-2 specific inhibitor venetoclax shows limited efficacy at the relapse setting in AML [9]. Thus, there is a clinical rationale for exploring venetoclax-based combination regimens that maximize efficacy while minimizing toxicity. PI3Kδ, which is constitutively activated in AML blast cells [21,34], appears to be a suitable target of combination treatments against AML. In our study, we demonstrated that co-inhibition of PI3Kδ and BCL-2 exerted synergistic lethality and overcame FLT3 inhibitors acquired resistance in AML through AKT/4E-BP-1 inactivation and MCL-1 downregulation.
It has been shown that the combination of venetoclax and PI3K/mTOR inhibitors exhibits a synergistic effect. As PI3Kδ is uniformly expressed and constitutively activated in AML, it may be more suitable for combination with a higher selectivity and lower toxicity. In the current study, the combination of venetoclax and a selective PI3Kδ inhibitor idelalisib exhibited a synergistic antiproliferative effect and induced caspase-dependent apoptosis both in venetoclax-sensitive and venetoclax-resistant AML cells. Since the complex interactions among BCL-2 family members, individual measurements of the various antiapoptotic proteins may be insufficient to provide comprehensive data on the evaluation of antiapoptotic dependencies in AML. FLT3-ITD mutation, which is one of the most common genetic alterations in AML, emerged at relapse after venetoclax monotherapy and a small subset of FLT3-ITD+ patients showed no responses to venetoclax [12,35]. Although FLT3-ITD+ MV-4-11 cells have high expression of BCL-2 and are sensitive to venetoclax in vitro, as seen in our and others’ previous studies [28], venetoclax exhibited weak in vivo antitumour activity in this xenograft model. However, the combination of venetoclax and idelalisib was synergistically lethal and exhibited significantly enhanced antitumour activity in FLT3-ITD+ AML xenografts.
In addition to BCL-2, MCL-1 is frequently overexpressed and plays an important prosurvival role in AML [36]. Upregulated MCL-1 has appeared to be a common determinant of venetoclax resistance [37]. SiRNA knockdown of MCL-1 in venetoclax-resistant cells resulted in improved efficacy of venetoclax [38], and ectopic overexpression of MCL-1 attenuated apoptosis induced by venetoclax [35]. A previous study showed that the PI3K/AKT/mTOR pathway regulated the expression of MCL-1 by 4E-BP-1/mTORC1-dependent translation in AML [32]. Another recent study suggested that MCL-1 disrupted intramolecular PH/KD interactions by its PEST domain with AKT at the PH domain, resulting in AKT activation in cancer cells [39]. In the present study, compared with either drug alone, venetoclax combined with idelalisib exhibited greater inhibition of AKT/4E-BP-1 phosphorylation and MCL-1 expression in venetoclax-sensitive AML cells. Additionally, the combination greatly diminished the level of MCL-1 protein bound to Bim both in MV-4-11 and EOL-1 cells. It is known that released Bim binding to MCL-1 is one of the mechanisms accounting for the intrinsic resistance to venetoclax [40]. Thus, PI3Kδ inhibition, via downregulating AKT, would lead to reduced MCL-1 expression, and the combination with the BCL-2 selective inhibitor venetoclax would prevent BCL-2 from sequestering free Bim, thereby activating Bim and allowing induction of apoptosis. Certainly, the specific mechanism still requires more detailed experimental demonstrations.
FLT3 inhibitors have shown limited effect in AML due to the emergence of resistance, even the next-generation FLT3 inhibitor quizartinib [33,41,42]. Our results demonstrated that quizartinib-resistant MV-4-11 cells exhibited disparate sensitivities to multiple FLT3 inhibitors, with high-level resistance to both quizartinib and sorafenib, whereas they showed less resistance to the type I FLT3 inhibitors crenolanib, gilteritinib and sunitinib. Sequencing of FLT3 revealed the gatekeeper mutation of F691L within the TKD in quizartinib-resistant MV-4-11 cells. Accordingly, a previous study showed that AML patients with acquired point mutation at F691L developed resistance to the type II kinase inhibitor quizartinib but remained sensitive to the type I inhibitor [33]. Quizartinib markedly downregulated the phosphorylation of AKT and ERK, as well as MCL-1, only in parental MV-4-11 cells. However, the combination of venetoclax and idelalisib exhibited enhanced inhibition of the phosphorylation of AKT and expression of MCL-1 and synergistic efficacy in MV-4-11/quizartinib cells, as it did in the parental cells. Thus, MCL-1 may be one of the mechanisms contributing to intrinsic resistance to venetoclax and acquired resistance to quizartinib, which can be overcome by the combination of venetoclax and idelalisib.
It has been reported that FLT3-ITD+ cells become BCL-2-dependent following quizartinib treatment [28]. Accordingly, in the present study, venetoclax alone was considerably more active against FLT3 inhibitor-resistant cell line MV-4-11/quizartinib than parental cell line MV-4-11 both in vitro and in vivo. The TGI values for venetoclax at a dose of 75 mg/kg were 31.4% and 85.4% in parental and quizartinib-resistant MV-4-11 xenografts, respectively. Although these results suggest that the shift of dependence to different BCL-2 family members may contribute to the potential antitumour effects of venetoclax in MV-4-11/quizartinib AML with the F691L mutation, it remains important to understand the mechanism underlying this regulation in the future. Moreover, the combination of venetoclax and idelalisib further improved the reduction of tumour burden in the MV-4-11/quizartinib xenograft than drug treatment alone (P < 0.01) and induced tumour regression in all mice from the cotreatment group. In addition, the cotreatment was generally well-tolerated, and no obvious loss in body weight was found during the experiments in all groups. Thus, co-inhibition of BCL-2 and PI3Kδ may provide a new option against AML with higher selectivity and lower toxic effects and may also prevent the emergence of FLT3-mutated, venetoclax-resistant subclones in patients.
In summary, cotreatment with venetoclax and idelalisib exerts synergistic lethality against AML cells co-expressing BCL-2 and PI3Kδ and overrides acquired resistance to quizartinib in FLT3-ITD+ AML, both in vitro and in vivo, which is mediated by the greater attenuation of P-AKT and MCL-1 levels. Given the encouraging preliminary clinical results of venetoclax monotherapy in AML, our findings suggest that the combination of venetoclax and PI3Kδ inhibitors is a promising strategy for the treatment of AML, particularly for FLT3-ITD+ patients who develop quizartinib resistance because of FLT3-TKD mutations.
Acknowledgements
This research was supported by grants from the Natural Science Foundation of Shanghai (19ZR1467700), the Lingang Laboratory (LG202101-01-06) and the Major Research Plan of the National Natural Science Foundation of China (91953203).
Disclosure of conflict of interest
None.
References
- 1.Hackl H, Astanina K, Wieser R. Molecular and genetic alterations associated with therapy resistance and relapse of acute myeloid leukemia. J Hematol Oncol. 2017;10:51. doi: 10.1186/s13045-017-0416-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Short NJ, Konopleva M, Kadia TM, Borthakur G, Ravandi F, DiNardo CD, Daver N. Advances in the treatment of acute myeloid leukemia: new drugs and new challenges. Cancer Discov. 2020;10:506–525. doi: 10.1158/2159-8290.CD-19-1011. [DOI] [PubMed] [Google Scholar]
- 3.Khwaja A, Bjorkholm M, Gale RE, Levine RL, Jordan CT, Ehninger G, Bloomfield CD, Estey E, Burnett A, Cornelissen JJ, Scheinberg DA, Bouscary D, Linch DC. Acute myeloid leukaemia. Nat Rev Dis Primers. 2016;2:16010. doi: 10.1038/nrdp.2016.10. [DOI] [PubMed] [Google Scholar]
- 4.De Kouchkovsky I, Abdul-Hay M. ‘Acute myeloid leukemia: a comprehensive review and 2016 update’. Blood Cancer J. 2016;6:e441. doi: 10.1038/bcj.2016.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, Dombret H, Ebert BL, Fenaux P, Larson RA, Levine RL, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz M, Sierra J, Tallman MS, Tien HF, Wei AH, Löwenberg B, Bloomfield CD. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–447. doi: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu H. Emerging agents and regimens for AML. J Hematol Oncol. 2021;14:49. doi: 10.1186/s13045-021-01062-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, Perl AE, Travers KJ, Wang S, Hunt JP, Zarrinkar PP, Schadt EE, Kasarskis A, Kuriyan J, Shah NP. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485:260–263. doi: 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campos L, Rouault JP, Sabido O, Oriol P, Roubi N, Vasselon C, Archimbaud E, Magaud JP, Guyotat D. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood. 1993;81:3091–3096. [PubMed] [Google Scholar]
- 9.Konopleva M, Letai A. BCL-2 inhibition in AML: an unexpected bonus? Blood. 2018;132:1007–1012. doi: 10.1182/blood-2018-03-828269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L, Wong S, Dunbar M, Zhu M, Desai MB, Cerri E, Heitner Enschede S, Humerickhouse RA, Wierda WG, Seymour JF. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:311–322. doi: 10.1056/NEJMoa1513257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, McKeegan E, Salem AH, Zhu M, Ricker JL, Blum W, DiNardo CD, Kadia T, Dunbar M, Kirby R, Falotico N, Leverson J, Humerickhouse R, Mabry M, Stone R, Kantarjian H, Letai A. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016;6:1106–1117. doi: 10.1158/2159-8290.CD-16-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pan RQ, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, Cortes J, DeAngelo DJ, Debose L, Mu H, Dohner H, Gaidzik VI, Galinsky I, Golfman LS, Haferlach T, Harutyunyan KG, Hu JH, Leverson JD, Marcucci G, Muschen M, Newman R, Park E, Ruvolo PP, Ruvolo V, Ryan J, Schindela S, Zweidler-McKay P, Stone RM, Kantarjian H, Andreeff M, Konopleva M, Letai AG. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014;4:362–375. doi: 10.1158/2159-8290.CD-13-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shahswar R, Beutel G, Klement P, Rehberg A, Gabdoulline R, Koenecke C, Markel D, Eggers H, Eder M, Stadler M, Hambach L, Ehrlich S, Göhring G, Schlegelberger B, Dammann E, Reuter M, Wichmann M, Neziri B, Ganser A, Thol F, Heuser M. FLA-IDA salvage chemotherapy combined with a seven-day course of venetoclax (FLAVIDA) in patients with relapsed/refractory acute leukaemia. Br J Haematol. 2020;188:e11–e15. doi: 10.1111/bjh.16268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maiti A, Franquiz MJ, Ravandi F, Cortes JE, Jabbour EJ, Sasaki K, Marx K, Daver NG, Kadia TM, Konopleva MY, Masarova L, Borthakur G, DiNardo CD, Naqvi K, Pierce S, Kantarjian HM, Short NJ. Venetoclax and BCR-ABL tyrosine kinase inhibitor combinations: outcome in patients with philadelphia chromosome-positive advanced myeloid leukemias. Acta Haematol. 2020;143:567–573. doi: 10.1159/000506346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma J, Zhao SJ, Qiao XN, Knight T, Edwards H, Polin L, Kushner J, Dzinic SH, White K, Wang G, Zhao LJ, Lin H, Wang Y, Taub JW, Ge YB. Inhibition of Bcl-2 synergistically enhances the antileukemic activity of midostaurin and gilteritinib in preclinical models of FLT3-mutated acute myeloid leukemia. Clin Cancer Res. 2019;25:6815–6826. doi: 10.1158/1078-0432.CCR-19-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei AH, Montesinos P, Ivanov V, DiNardo CD, Novak J, Laribi K, Kim I, Stevens DA, Fiedler W, Pagoni M, Samoilova O, Hu Y, Anagnostopoulos A, Bergeron J, Hou JZ, Murthy V, Yamauchi T, McDonald A, Chyla B, Gopalakrishnan S, Jiang Q, Mendes W, Hayslip J, Panayiotidis P. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: a phase 3 randomized placebo-controlled trial. Blood. 2020;135:2137–2145. doi: 10.1182/blood.2020004856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DiNardo CD, Jonas BA, Pullarkat V, Thirman MJ, Garcia JS, Wei AH, Konopleva M, Döhner H, Letai A, Fenaux P, Koller E, Havelange V, Leber B, Esteve J, Wang J, Pejsa V, Hájek R, Porkka K, Illés Á, Lavie D, Lemoli RM, Yamamoto K, Yoon SS, Jang JH, Yeh SP, Turgut M, Hong WJ, Zhou Y, Potluri J, Pratz KW. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383:617–629. doi: 10.1056/NEJMoa2012971. [DOI] [PubMed] [Google Scholar]
- 18.Tamburini J, Elie C, Bardet V, Chapuis N, Park S, Broet P, Cornillet-Lefebvre P, Lioure B, Ugo V, Blanchet O, Ifrah N, Witz F, Dreyfus F, Mayeux P, Lacombe C, Bouscary D. Constitutive phosphoinositide 3-kinase/Akt activation represents a favorable prognostic factor in de novo acute myelogenous leukemia patients. Blood. 2007;110:1025–1028. doi: 10.1182/blood-2006-12-061283. [DOI] [PubMed] [Google Scholar]
- 19.Foukas LC, Berenjeno IM, Gray A, Khwaja A, Vanhaesebroeck B. Activity of any class IA PI3K isoform can sustain cell proliferation and survival. Proc Natl Acad Sci U S A. 2010;107:11381–11386. doi: 10.1073/pnas.0906461107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Ding J, Meng LH. PI3K isoform-selective inhibitors: next-generation targeted cancer therapies. Acta Pharmacol Sin. 2015;36:1170–1176. doi: 10.1038/aps.2015.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sujobert P, Bardet V, Cornillet-Lefebvre P, Hayflick JS, Prie N, Verdier F, Vanhaesebroeck B, Muller O, Pesce F, Ifrah N, Hunault-Berger M, Berthou C, Villemagne B, Jourdan E, Audhuy B, Solary E, Witz B, Harousseau JL, Himberlin C, Lamy T, Lioure B, Cahn JY, Dreyfus F, Mayeux P, Lacombe C, Bouscary D. Essential role for the p110delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood. 2005;106:1063–1066. doi: 10.1182/blood-2004-08-3225. [DOI] [PubMed] [Google Scholar]
- 22.Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, Barrientos JC, Zelenetz AD, Kipps TJ, Flinn I, Ghia P, Eradat H, Ervin T, Lamanna N, Coiffier B, Pettitt AR, Ma S, Stilgenbauer S, Cramer P, Aiello M, Johnson DM, Miller LL, Li D, Jahn TM, Dansey RD, Hallek M, O’Brien SM. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370:997–1007. doi: 10.1056/NEJMoa1315226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lannutti BJ, Meadows SA, Herman SEM, Kashishian A, Steiner B, Johnson AJ, Byrd JC, Tyner JW, Loriaux MM, Deininger M, Druker BJ, Puri KD, Ulrich RG, Giese NA. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117:591–594. doi: 10.1182/blood-2010-03-275305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He Y, Sun LP, Xu YP, Fu L, Li Y, Bao XB, Fu HY, Xie CY, Lou LG. Combined inhibition of PI3K delta and FLT3 signaling exerts synergistic antitumor activity and overcomes acquired drug resistance in FLT3-activated acute myeloid leukemia. Cancer Lett. 2018;420:49–59. doi: 10.1016/j.canlet.2018.01.071. [DOI] [PubMed] [Google Scholar]
- 25.Moujalled DM, Pomilio G, Ghiurau C, Ivey A, Salmon J, Rijal S, Macraild S, Zhang L, Teh TC, Tiong IS, Lan P, Chanrion M, Claperon A, Rocchetti F, Zichi A, Kraus-Berthier L, Wang YZ, Hamovic E, Morris E, Colland F, Segal D, Huang D, Roberts AW, Maragno AL, Lessene G, Geneste O, Wei AH. Combining BH3-mimetics to target both BCL-2 and MCL1 has potent activity in pre-clinical models of acute myeloid leukemia. Leukemia. 2019;33:905–917. doi: 10.1038/s41375-018-0261-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei AH, Roberts AW, Spencer A, Rosenberg AS, Siegel D, Walter RB, Caenepeel S, Hughes P, McIver Z, Mezzi K, Morrow PK, Stein A. Targeting MCL-1 in hematologic malignancies: rationale and progress. Blood Rev. 2020;44:100672. doi: 10.1016/j.blre.2020.100672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rahmani M, Aust MM, Attkisson E, Williams DC Jr, Ferreira-Gonzalez A, Grant S. Dual inhibition of Bcl-2 and Bcl-xL strikingly enhances PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and Bim-dependent mechanism. Cancer Res. 2013;73:1340–1351. doi: 10.1158/0008-5472.CAN-12-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raghuveer Singh M, Qi Z, RosaAnna D, Antonio C, Vinitha Mary K, Jayant R, Vidhi M, Edna FC, Monique D, Neil PS, Marina K, Deepak S, Elisabeth AL. Venetoclax combines synergistically with FLT3 inhibition to effectively target leukemic cells in FLT3-ITD+ acute myeloid leukemia models. Haematologica. 2020;106:1034–1046. doi: 10.3324/haematol.2019.244020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33:299–312. doi: 10.1038/s41375-018-0357-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Z, Cai J, Cheng J, Yang W, Zhu Y, Li H, Lu T, Chen Y, Lu S. FLT3 Inhibitors in acute myeloid leukemia: challenges and recent developments in overcoming resistance. J Med Chem. 2021;64:2878–2900. doi: 10.1021/acs.jmedchem.0c01851. [DOI] [PubMed] [Google Scholar]
- 31.Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. 2019;575:299–309. doi: 10.1038/s41586-019-1730-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rahmani M, Aust MM, Hawkins E, Parker RE, Ross M, Kmieciak M, Reshko LB, Rizzo KA, Dumur CI, Ferreira-Gonzalez A, Grant S. Co-administration of the mTORC1/TORC2 inhibitor INK128 and the Bcl-2/Bcl-xL antagonist ABT-737 kills human myeloid leukemia cells through Mcl-1 down-regulation and AKT inactivation. Haematologica. 2015;100:1553–1563. doi: 10.3324/haematol.2015.130351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Albers C, Leischner H, Verbeek M, Yu C, Illert AL, Peschel C, von Bubnoff N, Duyster J. The secondary FLT3-ITD F691L mutation induces resistance to AC220 in FLT3-ITD+ AML but retains in vitro sensitivity to PKC412 and Sunitinib. Leukemia. 2013;27:1416–1418. doi: 10.1038/leu.2013.14. [DOI] [PubMed] [Google Scholar]
- 34.Billottet C, Grandage VL, Gale RE, Quattropani A, Rommel C, Vanhaesebroeck B, Khwaja A. A selective inhibitor of the p110delta isoform of PI 3-kinase inhibits AML cell proliferation and survival and increases the cytotoxic effects of VP16. Oncogene. 2006;25:6648–6659. doi: 10.1038/sj.onc.1209670. [DOI] [PubMed] [Google Scholar]
- 35.Niu X, Wang G, Wang Y, Caldwell JT, Edwards H, Xie C, Taub JW, Li C, Lin H, Ge Y. Acute myeloid leukemia cells harboring MLL fusion genes or with the acute promyelocytic leukemia phenotype are sensitive to the Bcl-2-selective inhibitor ABT-199. Leukemia. 2014;28:1557–1560. doi: 10.1038/leu.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD, Izon DJ, Zuber J, Rappaport AR, Herold MJ, Alexander WS, Lowe SW, Robb L, Strasser A. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012;26:120–125. doi: 10.1101/gad.182980.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tahir SK, Smith ML, Hessler P, Rapp LR, Idler KB, Park CH, Leverson JD, Lam LT. Potential mechanisms of resistance to venetoclax and strategies to circumvent it. BMC Cancer. 2017;17:399. doi: 10.1186/s12885-017-3383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bogenberger JM, Kornblau SM, Pierceall WE, Lena R, Chow D, Shi CX, Mantei J, Ahmann G, Gonzales IM, Choudhary A, Valdez R, Camoriano J, Fauble V, Tiedemann RE, Qiu YH, Coombes KR, Cardone M, Braggio E, Yin H, Azorsa DO, Mesa RA, Stewart AK, Tibes R. BCL-2 family proteins as 5-Azacytidine-sensitizing targets and determinants of response in myeloid malignancies. Leukemia. 2014;28:1657–1665. doi: 10.1038/leu.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen G, Park D, Magis AT, Behera M, Ramalingam SS, Owonikoko TK, Sica GL, Ye KQ, Zhang C, Chen ZJ, Curran WJ, Deng XM. Mcl-1 interacts with akt to promote lung cancer progression. Cancer Res. 2019;79:6126–6138. doi: 10.1158/0008-5472.CAN-19-0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Niu X, Zhao J, Ma J, Xie C, Edwards H, Wang G, Caldwell JT, Xiang S, Zhang X, Chu R, Wang ZJ, Lin H, Taub JW, Ge Y. Binding of released Bim to Mcl-1 is a mechanism of intrinsic resistance to ABT-199 which can be overcome by combination with daunorubicin or cytarabine in AML cells. Clin Cancer Res. 2016;22:4440–4451. doi: 10.1158/1078-0432.CCR-15-3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, Belli B, Karaman MW, Pratz KW, Pallares G, Chao Q, Sprankle KG, Patel HK, Levis M, Armstrong RC, James J, Bhagwat SS. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML) Blood. 2009;114:2984–2992. doi: 10.1182/blood-2009-05-222034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith CC, Paguirigan A, Jeschke GR, Lin KC, Massi E, Tarver T, Chin CS, Asthana S, Olshen A, Travers KJ, Wang S, Levis MJ, Perl AE, Radich JP, Shah NP. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood. 2017;130:48–58. doi: 10.1182/blood-2016-04-711820. [DOI] [PMC free article] [PubMed] [Google Scholar]