

Summary
Hereditary Hemorrhagic Telangiectasia or Rendu-Osler-Weber Disease is a rare fibrovascular dysplasia that makes vascular walls vulnerable to trauma and rupture, causing skin and mucosa bleeding. It is of dominant autosomal inheritance, characterized by recurrent epistaxis and telangiectasia on the face, hands and oral cavity; visceral arteriovenous malformations and positive family history. Epistaxis is often the first and foremost manifestation. It's associated to arteriovenous malformations in several organs. There are possible hematologic, neurologic, pulmonary, dermatologic and gastrointestinal complications. Treatment is supportive and helps prevent complications. This study is a case report of a patient with this syndrome who came to the ENT Outpatient Ward of the Faculdade de Medicina de Marília; and we have done a bibliographic review of the disease's etiopathogenesis, clinical manifestations and clinical-surgical treatment options.
Keywords: epistaxis, rendu-osler-weber syndrome, hereditary hemorrhagic telangiectasia
INTRODUCTION
The Rendu-Osler-Weber syndrome or Hereditary Hemorrhagic Telangiectasia is a rare systemic fibrovascular dysplasia which bears, as basic defect, an alteration in the elastic and muscle layers of vessel walls, making them more vulnerable to spontaneous ruptures and injuries1, 2.
The disease is autosomal dominant, although in about 20% of the cases, there is no family history. The incidence in the general population is of 1-2/100,000 and has a homogeneous race and gender distribution3.
Diagnosis is made according to the Curaçao Criteria: telangiectasia on the face, hands and oral cavity; recurrent epistaxis; arteriovenous malformations with visceral involvement; family history. Diagnosis is confirmed upon the presence of at least 3 of these manifestations4.
Otorhinolaryngological manifestations are the most frequent, and recurrent epistaxis is the main complaint. Blood vessels from other regions may also be involved, especially those from the lungs, brain, skin and gastro-intestinal tract1, 4, 5, 6.
Treatment is of palliative nature only; there still is no consensus on the best treatment option. The important thing here is to control the disease for as long as possible. Options vary between anterior and posterior nasal packing and chemical or laser cauterization of the lesions, all the way to surgical treatment, such as dermoseptoplasty and nasal cavity obliteration through Young's approach5, 6. With the progress in diagnostic image tests, today it is possible to embolize the Maxillary, Ethmoidal or Sphenoidal arteries, as well as ligation of the Maxillary or Palatine arteries in recurrent cases, in an attempt to contain bleeding and improve the patient's anemia5.
Because of this lack of consensus regarding the best treatment for these cases, all cases should be described so as to provide a better understanding of this disease. This paper also aims at alerting otolaryngologists that, when facing a patient with recurrent epistaxis, one must suspect of Rendu-Osler-Weber syndrome, and perform a nasal fibroscopy to better diagnose this syndrome.
The present investigation reports the case of one patient with Rendu-Osler-Weber syndrome; seen at the ENT Ward at Faculdade de Medicina de Marília and we review the literature and discuss available treatment options.
CASE PRESENTATION
C.L.S., 53 years old, came to the Medical School of Marília Department of Otorhinolaryngology, complaining of mild recurrent epistaxis for 15 years, with worsening of the clinical Picture in the past 3 months, having had bleeding for more than one hour three times a day, always after physical exercise or sneezes. So far the patient had not had the need for nasal packing or other treatment.
For about 30 years he had hemangiomas spread throughout his body, especially in the chest, lower lip (Figure 1), tongue, cheek mucosa, face, fingers and nail bed. (Figure 2). He was also hypertensive and used captopril 25mg bid and propanolol 40mg bid. He did not use salicylate-based drugs. His father had repetition epistaxis, however milder and with telangiectasia lesions on his skin and mucosa, without visceral lesions; and he had an 8 year old grandson with mild epistaxis.
Figure 1.
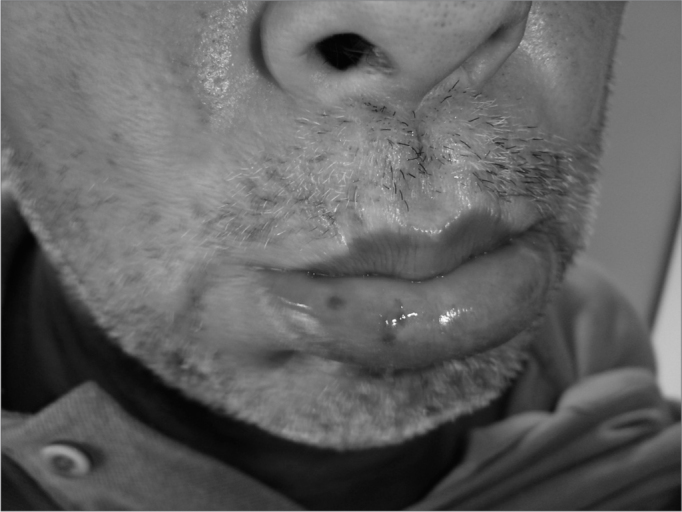
Lower lip Telangiectasia.
Figure 2.
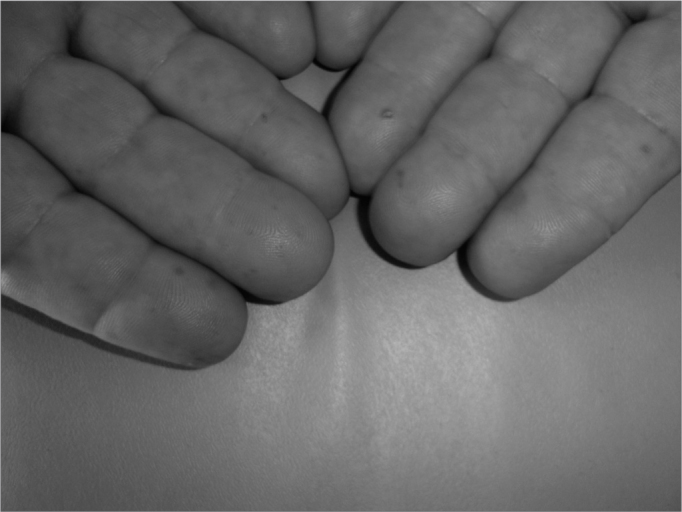
Fingers Telangiectasia.
During his physical exam, we noticed telangiectasia lesions spread throughout his chest, nails, fingers, lower lip, oropharynx, tongue and face. Doing anterior rhinoscopy, we noticed hematic points on his septal mucosa and his left middle turbinate. There were also hematic crusts on his right septal mucosa.
His chest x-ray, abdominal echography and skull and abdomen CT scan did not show arteriovenous malformations. Blood tests showed Hb 8.3g/dl, Ht 26.6%.
As first treatment, we packed his nose and admitted him to the hospital for blood transfusion. After we removed the nasal packing, the epistaxis episodes relapsed, and we then decided to cauterize the nasal lesions through nasal endoscopy. Finally, we carried out right side maxillary artery embolization with an improvement in the intensity of the epistaxis episodes, however without complete remission. The patient remains under follow up in an outpatient basis and has been referred to Hematology, Gastroenterology and Thoracic Surgery for evaluation and they found no pulmonary and gastrointestinal malformations. He is still being followed up in the hematology ward treating anemia and using aminocaproic acid since then. His last blood test showed Hb 12.4g/dL and Ht 37.4%.
BIBLIOGRAPHIC REVIEW AND DISCUSSION
Background
This disorder was first described in 1864, by Henry Gawen Sutton (1836 - 91), who reported on a disorder that caused epistaxis, skin telangiectasia and internal bleeding7. One year afterwards, Benjamin Guy Babington (1794 - 1866) described repetition epistaxis in 5 generations of a family, and he was a Pioneer in describing the hereditary characteristic of this disease4. In 1876, John Wickham Legg (1843 - 1921) also described this disease, but just like Chiari in 1887 and Chaufffadin, in 1896, he was not able to differentiate this disease from hemophilia2, 4.
In was only in 1896, that Louis Marie Rendu (1844 - 1902) published a description of a 52-year-old man with recurrent epistaxis and telangiectasia on his face and torso, and lesions on his lips and soft palate. He was the first to suspect of nasal lesions causing epistaxis. He also noticed the presence of nasal bleeding in the patient's mother and brother4.
In 1901, Wiliam Osler (1849 - 1919) reported three cases in which he described the disease's hereditary characteristic; he was also the first to report that viscera could be affected4, 5, 6.
In 1907, Frederick Parkes Weber (1863 - 1962) reported a series of patients in whom he noticed lesions on their fingers, especially under the nails. It was in 1909 that Hanes coined the term Hereditary Hemorrhagic Telangiectasia, however, the disease is known today by the eponym of Rendu-Osler-Weber's syndrome4.
Genetic Heritage
It is a hereditary disorder, with dominant autosomal transmission, despite the fact that about 20% of the cases do not have a family history - they could represent sporadic mutations7.
It occurs in heterozygotes with incomplete expression in some cases. The homozygote form is not compatible with life6. Many genes have been identified and implicated with the disease's pathogenesis; however, the most important ones so far are the Endoglin (ENG) in chromosome 9 and the type II A Activin - Como I (ACVRL-1) receptor, being the former associated with a variant with pulmonary manifestations, known as THH1 and the latter with a more subtle phenotype of late onset8. Both genes code a membrane glycoprotein that is specially expressed in endothelial tissue cells and make up the surface receptor for the β growth factor (TGF-β), which will mediate vascular remodeling by affecting the extracellular matrix production8, 9. ENG, ACVRL - 1 and TGF-β function are essential for angiogenesis8, 9.
Histopathology
The histological findings once described by Jahnke in 1970 were an increase of submucosal vessels in intact endothelium, dilations of capillary, post-capillary and collector-type venules, with large elongated clusters of erythrocytes with fibrin channels in the connective tissue, red cells spread in the interstice around affected vessels, endothelial discontinuity and degeneration2, 5, 10. The first to describe these alterations was Menefee et al., in 1975, after a study with electron microscopy10.
The endothelial cells described were of three types: Normal - 90% of them; Degenerative - 7%, with a dense cytoplasm and less organelles present. They are less adherent to adjacent cells and develop slots between them; Cuboids - 3% found at random10. The basal lamina of these cells were intact, sometimes doubled, even where the cells were degenerated or there were slots between them. Associated with a reduplication of the basal lamina, we observed frequent microfibrillae in the connective tissue, without other clear alterations2.
In 1990, Braverman, studied the development of skin telangiectasia. In a normal skin, the arterioles are connected to the venules through multiple capillaries in the papillary dermis region. These vessels come from other larger vessels at the skin-fat tissue junction. The post-capillary venule's normal ultra-structure includes the lumen (L), endothelial cells and two or three layers of adventitious cells. At first, the simple venules become dilated; however they are still connected to the arterioles by means of one or more capillaries. There is a perivascular lymphocytic infiltrate. In a more advanced stage, the venules and their branches are markedly dilated, elongated and rolled in the dermis. The connection arterioles are also dilated and such dilation makes the capillaries disappear after some time, causing the arteriovenous malformations (shunts and fistulas). The perivascular lymphocytic infiltrate is still present. The vessel walls loose their elastic fibers, while the endothelial layer, the basal membrane and the smooth muscles remain intact11.
Diagnosis
Clinical diagnosis is made based on the Curaçao Criteria, established by the Scientific Division of the Hereditary Hemorrhagic Telangiectasia International Foundation. It is based on:
-
1-
epistaxis - spontaneous and recurrent nasal bleeding;
-
2-
telangiectasia - multiple and in characteristic sites (lips, oropharynx, fingers and nose)
-
3-
visceral lesions - such as gastrointestinal telangiectasia with or without bleeding; pulmonary, hepatic, brain and spinal arteriovenous malformations;
-
4-
family history - one first degree relative with Hereditary Hemorrhagic Telangiectasia4.
The multiple clinical manifestations associated with Hereditary Hemorrhagic Telangiectasia involve nasal, skin, pulmonary, cerebral and gastrointestinal tract vascular abnormalities5, 6.
Clinical Manifestations
Nasal
Epistaxis caused by spontaneous bleeding of nasal mucosa telangiectasia is the most common manifestation of this disease; however, it does not happen to all patients, about 80% of them have recurrent epistaxis5. Disease severity varies from severe epistaxis that requires multiple blood transfusions and oral iron supplementation to such mild manifestations that the disease is never suspected6. Bleeding starts around 10 year of age in some patients and before 21 years of age in almost all; however, they become more severe in later decades in two-thirds of the patients2.
Skin
The most characteristic lesion is the macular telangiectasia, with about 2 millimeters in diameter, which affect the face, lips, nose, tongue, ears, hands, upper body and feet2, 5. They usually manifest later than epistaxis, and are detected around the third decade of life6. These lesions may bleed, however the bleeding is not clinically relevant.
Pulmonary
These are arteriovenous malformations causing direct communication between the pulmonary vein and artery by means of a thin-walled aneurism2. They are multiple and appear in both lungs, having a predilection for the lower pulmonary lobes. It is estimated that about 60% of the persons with pulmonary arteriovenous malformations have Hereditary Hemorrhagic Telangiectasia; however, between 15 and 33% of the persons with such disease have pulmonary arteriovenous malformations, although such incidence vary according to the specific gene involved9.
They produce a right-to-left shunt6. Symptoms start around the third or fourth decade of life and patients may, depending on shunt severity, present with profound dyspnea, fatigue, cyanosis or polycytemia6. Nonetheless, initial manifestations usually are sequela of neurologic lesions such as ischemic stroke and cerebral abscess.
They may be found as a holosystolic extracardiac murmur during deep inspiration or by means of image exams6. Chest X-Ray shows a typical mass with artery and vein enlargement, however, the lesions may be found in the posteroinferior lung region2, 5. Chest CT-scan must be used and pulmonary angiography should be used only for radiological or surgical treatment planning6.
Cerebral
Results from pulmonary arteriovenous fistulas in about 60% of the cases, cerebral malformations in about 28%, spinal chord malformation in 8% and portasystemic encephalopathy in 3%5, 6.
They happen in between 8 and 12% of the patients and symptoms include: headaches, vertigo, syncope, visual and hearing disorders, focal and generalized crisis, obnubilation and paraparesias6.
Brain abscesses, Ischemic Strokes, Bacterial encephalitis happen only in patients with right-to-left shunt and pulmonary arteriovenous malformations, which facilitate the passage of septic emboli to the brain circulation2.
Gastrointestinal Tract
In about 10% of the cases there is recurrent and small high and low gastrointestinal tract bleeding1. They happen mainly between the fifth or sixth decades of life and are caused by mucosal telangiectasia similar to those that happen in the oral and nasal mucosa1, 4, 5. In half of the patients, the bleeding cause may be located in the stomach or duodenum5. There may be larger telangiectasia, angiodysplasia or less commonly, arteriovenous malformations2. There are reports of a greater incidence of duodenal ulcer; however, without clinical correlation with the disease12.
Liver involvement is rare. An arteriovenous malformation between the hepatic vein and artery may produce a left-to-right shunt, which may cause congestive heart failure2, 5. A shunt between the hepatic and porta vein may cause hepatic encephalopathy and gastrointestinal bleeding. Malformations between the hepatic artery and the porta vein may cause portal hypertension with esophageal varicose veins. This may be suspected when there is hepatomegalia or a noise over the liver. Other findings include a mild increase in alkaline phosphatase, γ-GT, and cholestasis. Ultrasound and CT scan may be used for diagnostic purposes 6.
Another type of liver involvement is hereditary hemorrhagic telangiectasia cirrhosis, characterized by dilated vessels surrounded by a variable amount of stroma and randomly distributed throughout the liver. Within these structures we have normal liver tissue6.
Treatment
Treatment is rarely necessary for skin lesions. However, when the telangiectasia cause cosmetic changes or bleed frequently, laser ablation or topic agents may be used5.
In pulmonary malformation, the classical treatment is surgery, however it is invasive and must remove as little as possible from the healthy pulmonary tissue around the malformation. The current treatment of choice is embolization of the vessels found, although there are still no studies regarding the long term follow up of these patients9.
The treatment of small gastrointestinal bleedings may be carried out by a small combined dose of estrogen and progesterone that, according to Carney13, has reduced the need for transfusion even for some months after the end of treatment. Results with endoscopic laser and bipolar coagulation are disheartening, because the lesions, especially those in the small bowel, are not easily found13. Aminocaproic acid has been suggested as a treatment option; however its efficacy has been questioned13.
Treatment of hepatic malformations is conservative14. Experience with surgical ligation is limited and data on embolization, although limited, show mortality in about 25% of the cases14.
Regarding malformations in the nervous system, there is no consensus as to the best treatment option - conservative or surgical, thus the decision depend on surgical risks and lethal bleeding, as well as surgeon's experience and lesion location. Different treatment modes such as embolization and estereotactic radiosurgery with γ rays may be used alone or in combination6.
And finally, this study highlights epistaxis control. There is no standard treatment1, 15. The multiple modes vary: anterior and posterior nasal packing, electric and/or chemical cauterization, vessel ligation, dermoseptoplasty, estrogen, hormonal therapy and, more recently, laser treatment16. However, no treatment has obtained full success and often times it is necessary to use more than one type of treatment to control the epistaxis1, 2, 16.
Treatment aims at reducing the number of bleeding episodes, as well as their intensity16. It tries to reduce the need for blood transfusions and hospitalizations1, 2. It also aims at improving the quality of life for those patients, who already suffer from constant bleeding episodes1, 2.
In general, treatment with cauterization, local pressure or packing is only palliative, and must be used in acute spells only1, 2. Arterial embolization, estrogen therapy or laser, Young's procedure and epithelial slice cultures are used as definitive treatment options in mild cases, bearing in mind that there is important bleeding recurrence and often times many treatment modalities are carried out16. Dermoseptoplasty and local microvascularized flaps are saved for the most severe cases16.
Hormonal treatment is carried out with high doses of estrogen, inducing nasal mucosa metaplasia towards a thick layer of keratinized squamous epithelium, which cover eventual nasal lesions, thus protecting them from local trauma2. Although of proven efficacy, hormonal therapy has not been broadly used because of its potential side effects - nausea, mammary sensitivity, weight gain and coronary diseases6. Moreover, male patients may develop gynecomastia, testicular atrophy and loss of libido1, 2.
Surgical treatment for this disease include: dermoseptoplasty and nostril closure by Young's technique16. Dermoseptoplasty or Saunders's procedure was described by the former in 1962, and used a thin skin layer to cover the anterior area of the nose. Skin grafts may be harvested from the thigh or arm, labial mucosa or amniotic membrane. Surgery consists of removing the affected mucosa and placing the flat over it, covering the nasal septum and inferior turbinates, bilaterally17. It is considered gold standard in the treatment1, 2, 6, 13. Nostril closure according to Young's procedure is carried out through an incision in the mucocutaneous junction of the nasal vestibule, which is taken backwards. It brings about severe side effects, such as oral breathing, and it is considered a last resort in very well selected cases17.
Microembolization is carried out using gelfoam or sclerosing substances from the maxillary artery under visualization of angiographic films16. Brachytherapy and fibrin glue injection in the nasal septum and inferior turbinates submucosa are efficacious, however they bring about only temporary symptom improvement18. Treatment with aminocaproic acid is not very much used; however it brings about significant symptoms improvement. Its side effects are: nausea, cramps, diarrhea, hypotension, dizziness, skin rash, myopathy, fatigue, rabdomyolysis, renal dysfunction and thrombosis16.
Laser is saved for acute bleeding spells, in an attempt to contain those. In the literature we have reports on CO2, o Nd-YAG, Argon, KTP and pulsed-dye lasers. It is difficult to compare these lasers because of a non-homogeneous classification in the current world medical literature1, 2, 16.
FINAL COMMENTS
Hereditary Hemorrhagic Telangiectasia is a multisystemic disease, first manifested by repetition epistaxis. Thus, it is fundamentally important that the otorhinolaryngologist be up-to-date in relation to the disease's etiopathogenesis and treatment options, so as to perform correct diagnosis as well as to prevent systemic complications of this disease.
Thus, this paper aimed at providing an update on the disease, as well as report a clinical case from the ENT Ward of the Medical School of Marília - Faculdade de Medicina de Marília, since there are treatment options, but we still lack a totally satisfactory treatment modality. It is very important to record all cases found in order to compare clinical manifestations and treatment approaches so that in the near future we may create Standards with effective results and life quality improvement for the patients.
Footnotes
This paper was submitted to the RBORL-SGP (Publishing Manager System) on 3 May 2005. code 293.
REFERENCES
- 1.Rapoport PG, Uvo IP, Costa KS, Cecatto SB, Garcia RID. Síndrome de Rendu-Osler-Weber: tratamento clínico e cirúrgico. Rev Bras Otorrinolaringol. 2003;694:577–580. [Google Scholar]
- 2.Maudonnet EN, Gomes CC, Sakano E. Telangiectasia Hemorrágica Hereditária (Doença de Rendu-Osler-Weber): um diagnóstico otorrinolaringológico. Rev Bras Otorrinolaringol. 2000;662:172–180. [Google Scholar]
- 3.Pau H, Carney AS, Murty GE. Hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): otorhinolaryngological manifestation. Clin Otolaryngol. 2001;26:93–98. doi: 10.1046/j.1365-2273.2001.00442.x. [DOI] [PubMed] [Google Scholar]
- 4.Fuchizaki U, Miyamori H, Kitagawa S, Kaneko S, Kobayashi K. Hereditary Haemorrhagic Telangiectasia (Rendu-Osler-Weber Disease) Lancet. 2003;362:1490–1494. doi: 10.1016/S0140-6736(03)14696-X. [DOI] [PubMed] [Google Scholar]
- 5.Guttmacher AE, Marchuk DA, White RL. Hereditary Haemorragic Telangiectasia. New England J Med. 1995;333:918–924. doi: 10.1056/NEJM199510053331407. [DOI] [PubMed] [Google Scholar]
- 6.Haitjema T, Westermann CJJ, Overtoom TTC, Timmer R, Disch F, Mauser H, Lammers JWJ. Hereditary Hemorrhagic Telangiectasia (Osler-weber-Rendu Disease) Arch Intern Med. 1996;56(8):714–719. [PubMed] [Google Scholar]
- 7.Carpes OLF, Moussalle MM, Ravanello R, Moraes VA, Swarowsky AM. Síndrome de Rendu-Osler-Weber: relato de caso e revisão bibliográfica. Rev Bras Otorrinolaringol. 1999;654:354–356. [Google Scholar]
- 8.Dakeishi M, Shioya T, Wada Y, Shindo T, Otaka K, Manabe M, Nozaki J, Inoue S, Koizume A. Genetic epidemiolology oh Hereditary hemorragic telangiectasia in a local communityin the northern part of Japan. 2002;19(2):140–148. doi: 10.1002/humu.10026. [DOI] [PubMed] [Google Scholar]
- 9.Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I, Simonneau G, Galie N, Loyd JE, Humbert M, Nichols WC, Morrel NW. Clinical and molecular genetic features of Pulmonary Hypertension in patients with Hereditary Hemorrhagic Telangiectasia. N Engl J Med. 2001;345(5):325–334. doi: 10.1056/NEJM200108023450503. [DOI] [PubMed] [Google Scholar]
- 10.Menefee MG, Flessa HC, Gluck SPH. Hereditary Hemorragic Telangiectasia (Osler Weber Disease) - An electron microscopic study of the vascular lesions before and after therapy with hormones. Arch Otolaryngol. 1975;101:246–251. doi: 10.1001/archotol.1975.00780330042011. [DOI] [PubMed] [Google Scholar]
- 11.Braverman IM, Keh A, Jacobson BS. Ultrastructure and three-dimensional organization of the telangiectases of hereditary hemorrhagic telangiectasia. J Invest Dermatol. 1990;95:422–427. doi: 10.1111/1523-1747.ep12555569. [DOI] [PubMed] [Google Scholar]
- 12.Perry WH. Clinical spectrum of Hereditary Hemorragic Telangiectasia (Osler Weber Disease) Am J Medicine. 1987;82:897–899. doi: 10.1016/0002-9343(87)90162-8. [DOI] [PubMed] [Google Scholar]
- 13.Carney PH, Murty GE. Hereditary haemorragic telangiectasia (OslerWeberRendu syndrome): otorhinolaringological manifestations. Clin Otolaryngol. 2001;26(2):93–98. doi: 10.1046/j.1365-2273.2001.00442.x. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Tsao G, Korzenik JR, Young L, Henderson KJ, Jain D, Byrd B, Pollack JS, White RI. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2000;343(13):931–936. doi: 10.1056/NEJM200009283431305. [DOI] [PubMed] [Google Scholar]
- 15.Martins RHG, Nakajima V, Dias NH, Sousa JC. Epistaxe associada a Síndrome de Rendu-Osler-Weber (Telangiectasia Hemorrágica Hereditária) Arquivos de Otorrinolaringologia. 1999:34. [Google Scholar]
- 16.Siegel MB, Keane WM, Atkins JP, Rosen MR. Control of epistaxis in patients with Herditary Hemorragic Telangiectasia. Otol Head Neck Surg. 1991;105:675–679. doi: 10.1177/019459989110500507. [DOI] [PubMed] [Google Scholar]
- 17.Lund VJ, Howard DJ. Closure of nasal cavities in the treatment of refractory Hereditary Haemorrhagic Telangiectasia. J Laryngol Otol. 1997;111:30–33. doi: 10.1017/s0022215100136369. [DOI] [PubMed] [Google Scholar]
- 18.Turcato G, Pizzi GB, Polico R, Antonello M, Busetto M. Epistaxis in Rendu-Weber-Osler Disease. The role of Brachytherapy. Acta Otorhinolaryngol Ita. 1996;16:513–516. [PubMed] [Google Scholar]