

Abstract
Macrophage polarization determines the production of pro‐ or anti‐inflammatory cytokines in response to various bacterial and virus infections. Here, we report that pro‐inflammatory macrophage polarization induced by lipopolysaccharide (LPS) skews the TRIM21‐SIRT5 interplay toward TRIM21 activation and SIRT5 degradation, resulting in an enhancement of interleukin (IL)‐1β production in vitro and in vivo. Mechanistically, LPS challenge enhances the interaction between TRIM21 and SIRT5 to promote SIRT5 ubiquitination and degradation, while reducing the binding of SIRT5 to HAUSP, a deubiquitinating enzyme that stabilizes SIRT5. In a feedback loop, SIRT5 degradation sustains the acetylation of TRIM21 at Lys351, thereby increasing its E3 ligase activity in LPS‐activated macrophages. Thus, we identify a functional balance between TRIM21 and SIRT5 that is tilted toward SIRT5 suppression in response to LPS stimulation, thereby enhancing IL‐1β production during inflammation.
Keywords: colitis, IL‐1β, SIRT5, TRIM21, ubiquitination
Subject Categories: Immunology, Molecular Biology of Disease, Post-translational Modifications & Proteolysis
The competitive binding of the E3 ligase TRIM21 and the DUB HAUSP is skewed in favor of TRIM21 upon LPS stimulation, resulting in SIRT5 degradation and enhanced pro‐inflammatory cytokine production in macrophages.
Introduction
Tripartite motif‐containing 21 (TRIM21; also known as Ro52) is a ubiquitously expressed cytosolic antibody receptor predominantly involved in innate immunity (Rhodes & Isenberg, 2017). Acting as a major autoantigen, TRIM21 has been shown to be directly linked to rheumatoid arthritis, systemic lupus erythematosus, and Sjögren's syndrome (Keeble et al, 2008). TRIM21 can directly neutralize infection by targeting antibody‐opsonized viruses and bacteria for proteasomal degradation. For instance, during pathogen infection, TRIM21 detects and binds to antibody‐bound pathogens in infected cells via its carboxy (C)‐terminal PRYSPRY domain (McEwan et al, 2013) and quickly undergoes degradation dependent of proteasome (Mallery et al, 2010) and AAA ATPase valosin‐containing protein (VCP) (Hauler et al, 2012). Recently, TRIM21 has been shown to cooperate with the DNA sensor cGAS to intercept antibody‐coated viruses which escape into the cytosol (Labzin et al, 2019). Having E3 ligase activity, TRIM21 plays a role in regulating inflammatory signaling in part through ubiquitination of various downstream signaling factors. To restrain the innate immune response to intracellular double‐stranded DNA, TRIM21 promotes Lys48 (K48)‐linked ubiquitination and degradation of DDX41 in dendritic cells, macrophages, and neutrophils (Zhang et al, 2013). Yet, upon RNA viral infection, TRIM21 activates the innate immune response through Lys27 (K27)‐linked polyubiquitination of MAVS (Xue et al, 2018). In addition, TRIM21 interacts and degrades sterile alpha motif and histidine–aspartic acid domain‐containing protein 1 (SAMHD1) through the proteasomal pathway upon enterovirus 71 (EV71) infection and HIV‐1 infection (Li et al, 2020). Moreover, cytosolic antibody receptor and ubiquitin ligase TRIM21 promotes antigen presentation and T‐cell activation by targeting immune complexes for efficient proteasomal degradation (Albecka et al, 2021; Caddy et al, 2021). TRIM21 also plays a role in redox regulation by directly binding with p62 and ubiquitylating p62 via K63‐linkage to suppress protein sequestration, which is its inherent cellular function (Pan et al, 2016). A recent study reported that genetic ablation of TRIM21 in mice conferred protection from LPS‐induced inflammation and dextran sulfate sodium‐induced colitis through suppressing Gasdermin D activity (Gao et al, 2022). Although activation of innate immune‐signaling pathways by recognition of antibody‐coated pathogens may endow TRIM21 with some ability to upregulate cytokine expression through TAK1‐dependent signaling (McEwan et al, 2013), the role of TRIM21 in regulating inflammatory cytokine production and how the E3 ligase activity of TRIM21 is regulated still remain unclear.
Macrophages are believed to be key mediators of inflammatory responses and dysregulation of macrophage activation may lead to the development of various inflammatory diseases including atherosclerosis and arthritis. When exposed to classical activators such as TLR4‐ligand lipopolysaccharide (LPS) and/or IFN‐γ, macrophages are polarized into M1 macrophages, produce pro‐inflammatory cytokines, such as IL‐1β, and infiltrate into injured tissues (Arnold et al, 2007). M2 or anti‐inflammatory macrophages are usually residentially polarized by alternative activators such as interleukin (IL)‐4 or IL‐13 (Martinez & Gordon, 2014) and appear at late stages of injured tissue repair and remodeling (Biswas & Mantovani, 2012). It has recently been reported that, SIRT5, a mitochondria NAD+‐dependent lysine deacetylase, is functionally involved in IL‐1β production in LPS‐activated macrophages by desuccinylating and activating pyruvate kinase M2 (PKM2) and thereby preventing DSS‐induced colitis (Wang et al, 2017). Otherwise, unlike the other members of SIRT family, the role of SIRT5 in shaping immunity, metabolism, and bioenergetics during inflammation remains unknown (Vachharajani et al, 2016). In the past few years, SIRT5 has attracted considerable attention in metabolic regulation. Initially, SIRT5 was shown to deacetylate carbamoyl phosphate synthetase 1 (CPS1) to promote urea cycle for ammonia detoxification and disposal (Nakagawa et al, 2009). Moreover, SIRT5 serves as a global regulator of lysine succinylation in mitochondria (Park et al, 2013) and desuccinylates the rate‐limiting ketogenic enzyme 3‐hydroxy‐3‐methylglutaryl‐CoA synthase 2 (HMGCS2) to inhibit ketogenesis (Rardin et al, 2013). In addition, SIRT5‐mediated lysine desuccinylation impacts amino acid degradation, the tricarboxylic acid cycle (TCA) cycle, and fatty acid metabolism (Park et al, 2013). Although it has been recently implicated in inhibition of IL‐1β production in LPS‐activated macrophages (Wang et al, 2017), how SIRT5 is regulated and the function of SIRT5 in inflammation still remains largely unknown.
In this study, we report that TRIM21 as an E3 ligase directly binds to and promotes SIRT5 degradation through K48‐linkage ubiquitination. Reciprocally, SIRT5 suppresses TRIM21 E3 ligase activity through deacetylating TRIM21 at Lys351. Moreover, HAUSP as a deubiquitinating enzyme competes with TRIM21 to interact with SIRT5, thereby stabilizing SIRT5. Notably, in LPS‐activated macrophages, SIRT5 is prone to form complex with TRIM21, but not HAUSP, which leads to SIRT5 degradation and IL‐1β production. Furthermore, SIRT5 knockout aggravates DSS‐induced colitis in mice, while knockout of TRIM21 in SIRT5‐deficiency mice confers protection against colitis. Thus, our findings reveal that LPS stimulation changes the balance of the TRIM21‐SIRT5 interplay toward SIRT5 degradation so that favoriting pro‐inflammatory cytokine production in macrophages.
Results
TRIM21 interacts with SIRT5 and promotes SIRT5 degradation
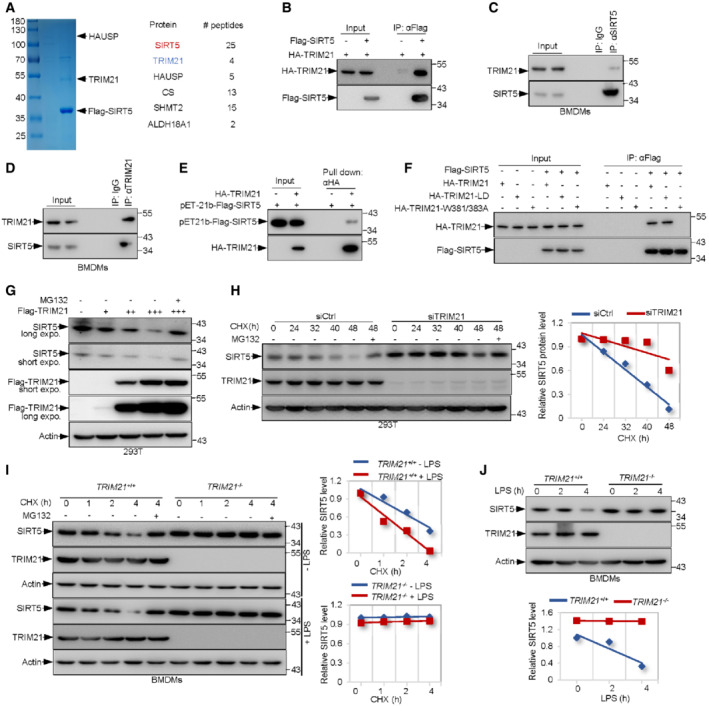
The deacetylase SIRT5 is a multiple function enzyme involved in metabolic regulation. To explore how SIRT5 is controlled physiologically, we sought to investigate whether it associates with some regulatory factors that can modulate its activity directly by immunoprecipitation assays followed by mass spectrometry analysis. Interestingly, we found that TRIM21, a RlNG‐containing E3 ubiquitin‐ligase of the TRIM protein family, was strongly pulled down by SIRT5 (Fig 1A). This result was strengthened by the observation that several known SIRT5‐binding partners including CS and SHMT2 were also identified concomitantly (Fig 1A). Moreover, immunofluorescence assays indicated these two proteins mainly localized in cytoplasm, providing the spatial possibility for their interaction (Fig EV1A). Furthermore, IP assay using subcellular fractionation from BMDMs showed that SIRT5 interacts with TRIM21 mainly in the cytoplasm (Fig EV1B). Thus, it is likely that TRIM21 can potentially form complex with SIRT5. In keeping with this, results from immunoprecipitation assays in 293T cells by overexpressing these two proteins with different tags revealed a noticeable interaction between them (Figs 1B and EV1C). Notably, SIRT5 could bind to TRIM21 at endogenous level in mouse bone marrow‐derived macrophages (BMDMs) (Fig 1C and D), and in vitro binding assays further suggest that SIRT5‐TRIM21 interaction occurs directly (Figs 1E and EV1D).
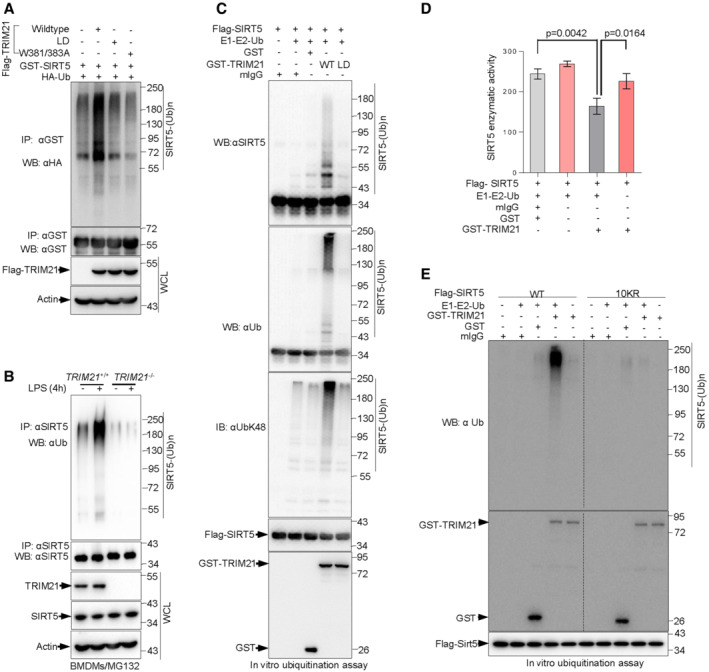
Figure 1. TRIM21 directly binds to SIRT5 and promotes its degradation.
-
AMass spectrometry (MS) assay of indicated electrophoretic bands in Coomassie blue staining (left panel), and proteins pulled down by Flag‐tagged SIRT5 from 293T cells were indicated on the right.
-
BLysates from 293T cells transfected with HA‐TRIM21 and Flag‐SIRT5 or vector control (−) as indicated were immunoprecipitated (IP) using anti‐Flag antibody, and bound proteins were analyzed by Western blotting (WB).
-
C, DLysates from BMDMs were immunoprecipitated with anti‐SIRT5 (C), anti‐TRIM21 (D) or control IgG antibodies as indicated and then analyzed by Western blotting.
-
EBacterially expressed and purified Flag‐SIRT5 were incubated with purified HA‐TRIM21 proteins prior to pull down with anti‐HA antibody followed by Western blot analysis.
-
FImmunoblot (IB) analysis of whole‐cell lysates (WCLs) and immunoprecipitates (IPs) derived from 293T cells transfected with Flag‐SIRT5 together with HA‐TRIM21 or indicated HA‐tagged mutant TRIM21 constructs. Cells were treated with 10 μM MG132 for 4 h before harvesting. HA, hemagglutinin.
-
GIB analysis of WCLs derived from 293T cells transfected with increasing amounts of Flag‐TRIM21 construct. Where indicated, 10 μM MG132 was added for 4 h before harvesting.
-
H293T cells transfected with control or TRIM21 siRNA for 48 h were then treated with 100 ng/ml CHX for the indicated time period before harvesting. Protein expression was analyzed by Western blotting (left panel). The SIRT5 protein abundance was quantified by ImageJ and plotted as indicated (right panel).
-
IIB analysis of WCLs derived from TRIM21 +/+ and TRIM21 −/− BMDMs treated with 100 ng/ml CHX for the indicated time period before harvesting (left panel). The SIRT5 protein abundance was quantified (right panel).
-
JIB analysis of WCLs derived from TRIM21 +/+ and TRIM21 −/− BMDMs treated with 100 ng/ml LPS for the indicated time period before harvesting (top panel). The SIRT5 protein abundance was quantified (bottom panel).
Data information: All IP and WB data in this work otherwise indicated are representative of at least three independent experiments.
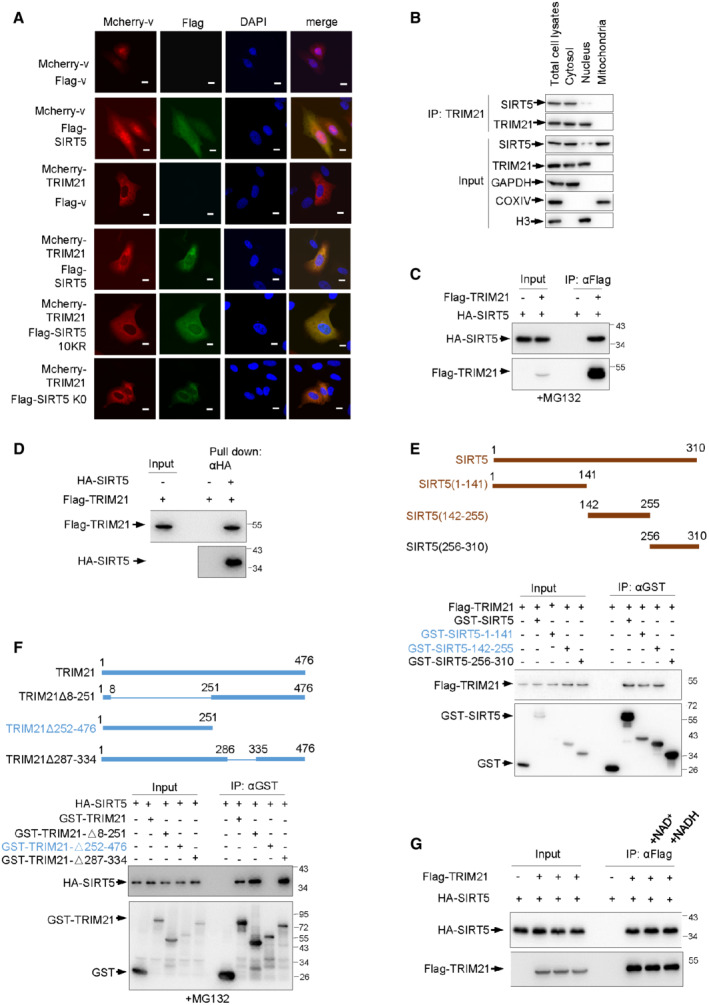
Figure EV1. TRIM21 is a binding partner for SIRT5.
-
AIndirect immunofluorescence assay of 293T cells transfected with indicated constructs. Scale bar: 10 μm.
-
BBMDM cells were fractionated. Total cell lysates from BMDM cells, and cytoplasmic, mitochondria and nuclear protein extracts were used for IP assay with anti‐TRIM21 antibody and analyzed by Western blot using indicated antibodies. GAPDH, COXIV and total H3 were used as fraction and loading controls respectively.
-
CImmunoblot (IB) analysis of whole‐cell lysates (WCLs) and immunoprecipitates (IPs) derived from 293T cells transfected with Flag‐TRIM21 together with HA‐SIRT5 constructs. Cells were treated with 10 μM MG132 for 4 h before harvesting.
-
DPurified HA‐SIRT5 protein were incubated with purified recombinant Flag‐TRIM21 proteins as indicated for 90 min at 30°C, followed by pull down with anti‐HA antibody and immunoblot analysis.
-
E(top panel) Scheme for generation of the expression constructs encoding the serial deletions of SIRT5 as indicated. (bottom panel) IB analysis of WCLs and IPs derived from 293T cells transfected with Flag‐TRIM21 together with the indicated SIRT5 deletion constructs. Cells were treated with 10 μM MG132 for 4 h before harvesting.
-
F(top panel) Scheme for generation of the expression constructs encoding the serial deletions of TRIM21 as indicated. (bottom panel) IB analysis of WCLs and IPs derived from 293T cells transfected with HA‐SIRT5 together with the indicated TRIM21 deletion constructs. Cells were treated with 10 μM MG132 for 4 h prior to harvesting.
-
GIB analysis of WCLs and immunoprecipitates derived from 293T cells transfected with Flag‐TRIM21 together with HA‐SIRT5 constructs in the presence or absence of 3 mM NAD+ or NADH as indicated. Cells were treated with 10 μM MG132 for 4 h before harvesting.
Data information: All IP and WB data in this work otherwise indicated are representative of at least three independent experiments.
We next mapped the interaction domains of SIRT5 and TRIM21 by constructing series of deletion mutants and showed that the residues 1–255 of SIRT5 and 335–476 of TRIM21 were responsible for their binding (Fig EV1E and F). Specifically, a two‐point mutation (W381A/W383A) within the interaction domain of TRIM21 totally abolished the interaction between TRIM21 and SIRT5 (Fig 1F). Interestingly, a ligase‐dead (LD) mutant TRIM21 (C16A, C31A, and H33W) showed no defect in binding to SIRT5, suggesting that the E3 ligase activity is not required for the assembly of these two proteins (Fig 1F). SIRT5 is a NAD‐dependent protein lysine deacetylase. Yet, supplying cells with NAD+ or NADH had no effect on the formation of TRIM21‐SIRT5 complex (Fig EV1G). These data together suggest that TRIM21 can directly bind to SIRT5.
Next, we wanted to know the physiological outcome(s) of the TRIM21‐SIRT5 interaction. Interestingly, co‐expression of TRIM21 led to a decrease in SIRT5 protein expression levels (Fig EV2A). Moreover, a decrease in endogenous SIRT5 levels was observed with increasing expression of TRIM21 (Fig 1G). By contrast, we did not observe significant changes in SIRT5 mRNA levels in BMDMs derived from TRIM21 −/− mice (Fig EV2B). Together, these data suggest that TRIM21 may have a role in regulating SIRT5 degradation, not its transcription. In support of this, treatment with MG132, a proteasome inhibitor, restored the protein levels of SIRT5 in cells overexpressing TRIM21 (Fig 1G). In addition, a cycloheximide (CHX) pulse‐chase assay was performed to examine the function of TRIM21 on SIRT5 stability. Remarkably, SIRT5 was stabilized when TRIM21 was silent (Fig 1H). Similarly, TRIM21 knockout prevented SIRT5 degradation in BMDMs (Fig 1I). LPS polarizes macrophages to M1 pro‐inflammatory macrophages. Surprisingly, administration of LPS to BMDMs reduced the stability of SIRT5 in TRIM21 +/+ cells, but not in TRIM21 −/− cells (Fig 1I). Moreover, supplying cells with LPS led to decreased SIRT5 levels in a time‐dependent manner (Fig 1J). These findings indicate that M1 polarization may promote TRIM21‐mediated degradation of SIRT5.
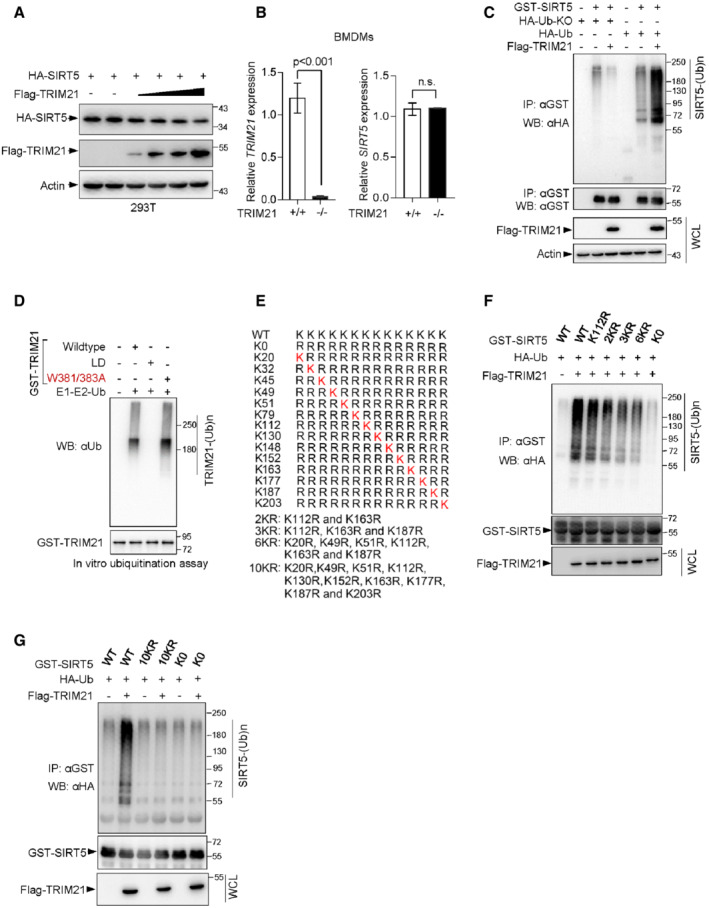
Figure EV2. Identification of the lysine residues in SIRT5 that are responsible for TRIM21 ubiquitination.
-
AIB analysis of WCLs from 293T cells transfected with HA‐SIRT5 together with increasing amounts of Flag‐TRIM21 constructs.
-
BmRNA levels of TRIM21 and SIRT5 in TRIM21 +/+ and TRIM21 −/− BMDMs were analyzed by qRT‐PCR. Data are means ± SD. n = 3 biological replicates. P‐values were determined by unpaired two‐tailed Student's t‐tests. ns, not significant.
-
CIB analysis of WCLs and anti‐GST immunoprecipitated products derived from 293T cells transfected with indicated constructs. Cells were treated with 10 μM MG132 for 4 h before harvesting.
-
DPurified TRIM21 and its mutant proteins as indicated were incubated with purified E1, E2 (UBE2D2) and ubiquitin at 30°C for 90 min. TRIM21 ubiquitination and protein levels were analyzed by Western blotting.
-
EMutants of human SIRT5 in which various lysine residues (K) are substituted with arginine (R).
-
F, GIn vivo ubiquitination assay analysis of WCLs and anti‐GST immunoprecipitated products derived from 293T cells transfected with the indicated plasmids. 10 μM MG132 was added for 4 h before harvesting.
Data information: All IP and WB data in this work otherwise indicated are representative of at least three independent experiments.
TRIM21 is an E3 ligase for SIRT5 promoting its ubiquitination, which is potentiated in LPS‐activated macrophages
To investigate the mechanism for the destabilization of SIRT5 by TRIM21, we tested whether TRIM21 promotes SIRT5 ubiquitination by acting as an E3 ligase. Indeed, overexpression of TRIM21 led to increased polyubiquitination of SIRT5, while this effect disappeared when cells expressed a mutant ubiquitin (Ub‐KO, all lysine residues replaced by arginine residues) (Figs 2A and EV2C). By contrast, a ligase‐dead TRIM21 mutant (LD) had no effect on SIRT5 ubiquitination (Fig 2A). Notably, the TRIM21(W381/383A) mutant (Fig EV2D), which lost the ability to bind SIRT5 but still possessed E3 ligase activity, failed to promote the ubiquitination of SIRT5, suggesting that direct interaction is required for TRIM21 to ubiquitinate SIRT5 (Fig 2A). In keeping with the findings that M1 polarization promoted TRIM21‐dependent degradation of SIRT5 (Fig 1H–J), LPS supplementation strongly triggered SIRT5 ubiquitination in TRIM21 +/+ BMDMs, but not in TRIM21 −/− BMDMs (Fig 2B).
Figure 2. TRIM21 is an E3 ligase of SIRT5 promoting its ubiquitination, which is enhanced by LPS challenge.
-
AImmunoblot analysis of WCLs and anti‐GST immunoprecipitates derived from 293T cells transfected with the indicated plasmids for 48 h, cells were then treated with the proteasome inhibitor MG132 for another 4 h prior to harvesting.
-
BWCLs from TRIM21 +/+ and TRIM21 −/− BMDMs treated with 100 ng/ml LPS for 4 h were immunoprecipitated with anti‐SIRT5 antibody and analyzed by Western blotting. 10 μM MG132 was added for 4 h before harvesting.
-
CTRIM21 ubiquitinates SIRT5 in vitro. Immunopurified SIRT5 were incubated with purified recombinant TRIM21 or mutant TRIM21 proteins, E1 and E2 (UBE2D2), and ubiquitin as indicated in 30°C for 90 min. The ubiquitination reaction products were resolved by SDS‐PAGE and probed with the indicated antibodies.
-
DPurified SIRT5 protein were incubated with purified recombinant TRIM21, E1 and E2 (UBE2D2), and ubiquitin in the presence or absence of control mIgG or GST proteins as indicated for 90 min at 30°C. SIRT5 enzymatic activity was measured. Data are from n = 3 biological replicates. Data are the mean ± SD. Statistical significance was determined by unpaired two‐tailed Student's t‐tests.
-
EPurified SIRT5 and mutant SIRT5‐10KR proteins were incubated with purified E1 and E2 (UBE2D2), ubiquitin and/or purified recombinant TRIM21 or control mIgG or GST proteins as indicated at 30°C for 90 min. SIRT5 ubiquitination and TRIM21 protein levels were analyzed by Western blotting with the indicated antibodies.
Data information: All IP and WB data in this work otherwise indicated are representative of at least three independent experiments.
To elucidate whether TRIM21 can directly ubiquitinate SIRT5, we performed an in vitro ubiquitination assay using purified proteins. In line with the cell transfection data (Figs 2A and EV2C), TRIM21 strongly triggered SIRT5 ubiquitination, while TRIM21‐LD did not (Fig 2C). Significantly, ubiquitination modification may block SIRT5 activation. Induction of SIRT5 ubiquitination by adding TRIM21 proteins and other ubiquitination components (E1, E2, and ubiquitin) led to a sharp reduction in SIRT5 enzymatic activity, and this effect could be inhibited when E1, E2 and ubiquitin were absent (Fig 2D). These data suggest that TRIM21 inhibits SIRT5 enzymatic activity through inducing the degradation of SIRT5.
Next, we wanted to identify the lysine residues that are responsible for TRIM21‐mediated ubiquitination. SIRT5 has 14 lysine residues, and we first mutated all lysine residues of SIRT5 to arginine (SIRT5‐K0), and then reintroduced individual lysine residues into SIRT5‐K0 to generate the single‐lysine mutants (Fig EV2E). Results from co‐immunoprecipitation experiment showed that SIRT5 ubiquitination gradually declined with an increase in the number of lysine‐to‐arginine substitutions compared with that of wild‐type SIRT5 (WT) (Fig EV2F). Notably, the level of ubiquitinated SIRT5‐10KR mutant was similar to that of SIRT5‐K0 mutant (Fig EV2G). This finding was further confirmed in vitro ubiquitination assay. While TRIM21 readily induced the polyubiquitination of wild‐type SIRT5(WT), it showed no capability to ubiquitinate SIRT5‐10KR (Fig 2E). Together, these findings suggest that TRIM21 is an E3 ligase for SIRT5 and catalyzes the polyubiquitination of SIRT5 during M1 macrophage polarization.
TRIM21‐mediated ubiquitination of SIRT5 is K48‐linked
To investigate the type of polyubiquitin chains on SIRT5 formed by TRIM21, we generated ubiquitin mutants (K48R) and (K63R), in which either lysine 48 or lysine 63 was substituted with arginine. Specifically, TRIM21 catalyzed polyubiquitination of SIRT5 in the presence of HA‐tagged wild‐type ubiquitin (HA‐Ub) and HA‐Ub‐K63R, but not in the presence of HA–Ub‐K48R (Fig 3A). Alternatively, we also generated ubiquitin mutants (K48) and (K63), in which all lysine residues were substituted with arginine except the lysine at positions 48 and 63, respectively. Consistently, upon overexpression of ubiquitin K48 (Ub‐K48), not Ub‐K63, the ubiquitination of SIRT5 was observed in the presence of TRIM21 (Fig 3B).
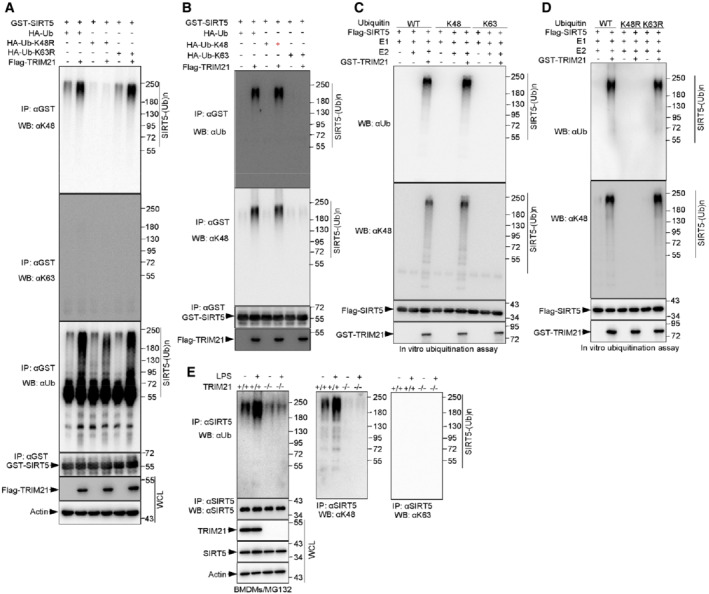
Figure 3. TRIM21 catalyzes Lys48‐linked ubiquitination of SIRT5.
-
AIn vivo ubiquitination assay analysis of WCLs and GST‐tagged immunoprecipitated products derived from 293T cells transfected with the indicated plasmids. 10 μM MG132 was added for 4 h before harvesting.
-
BIB analysis of WCLs and anti‐HA immunoprecipitates of 293T cells transfected with the indicated plasmids. 48 h post‐transfection, cells were treated with 10 μM MG132 for 4 h before harvesting.
-
CImmunopurified SIRT5 proteins were incubated with purified recombinant E1 and ubiquitin (Ub) or mutant Ub‐K48 or Ub‐K63 in the presence or absence of purified E2 (UBE2D2) and/or TRIM21 proteins as indicated in 30°C for 90 min. The ubiquitination reaction products were resolved by SDS‐PAGE and probed with the indicated antibodies.
-
DImmunopurified SIRT5 proteins were incubated with purified recombinant TRIM21, E1 and E2 (UBE2D2), and ubiquitin (Ub) or mutant Ub‐K48R or Ub‐K63R proteins as indicated for 45 min at 30°C. The ubiquitination reaction products were resolved by SDS‐PAGE and probed with the indicated antibodies.
-
EWCLs from TRIM21 +/+ and TRIM21 −/− BMDMs treated with 100 ng/ml LPS for 4 h were immunoprecipitated with anti‐SIRT5 antibody and analyzed by Western blotting using antibodies specifically against Ub‐K48 and Ub‐K63. 10 μM MG132 was added for 4 h before harvesting.
Data information: All IP and WB data in this work otherwise indicated are representative of at least three independent experiments.
To further validate these findings, we performed in vitro ubiquitination assays. TRIM21 was able to trigger the ubiquitination of SIRT5 in the presence of wild‐type Ub or Ub‐K48, but not Ub‐K63 (Fig 3C). Moreover, using the mutant Ub‐K48R and Ub‐K63R, we found that TRIM21 could induce the ubiquitination of SIRT5 in the presence of wild‐type Ub or Ub‐K63R, but not Ub‐K48R (Fig 3D).
To confirm these findings in vivo, we measured the polyubiquitination of SIRT5 in LPS‐treated primary macrophages BMDMs which prepared from TRIM21 +/+ and TRIM21 −/− mice. In accordance with in vitro data, the endogenous SIRT5 was robustly ubiquitinated with K48‐linked, but not K63‐linked chains in TRIM21 +/+ macrophages, but not in TRIM21 −/− macrophages (Fig 3E). Moreover, LPS treatment increased the ubiquitination level of SIRT5 in TRIM21 +/+ macrophages (Fig 3E). Collectively, these data indicate that TRIM21 catalyzes the Lys48‐dependent ubiquitination of SIRT5.
HAUSP binds to and stabilizes SIRT5, which is inhibited by LPS challenge
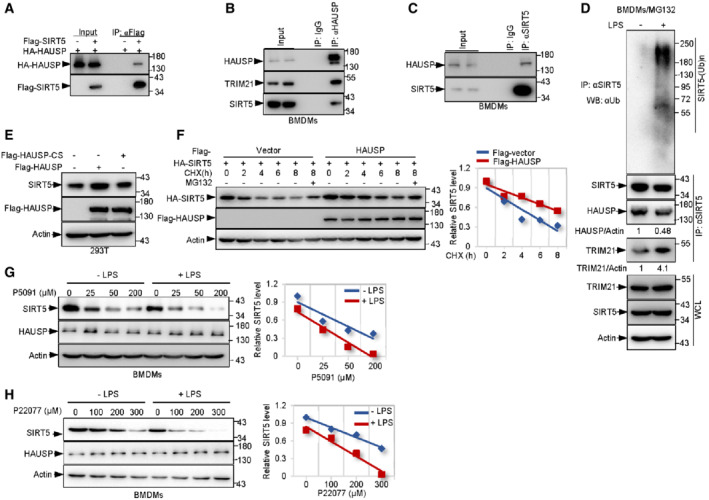
To investigate how exactly is SIRT5 regulated, we next wanted to identify the deubiquitinating enzyme responsible for deubiquitinating SIRT5. As shown in Fig 1A, mass spectrometry analysis revealed five unique peptides identical to HAUSP (herpesvirus‐associated ubiquitin‐specific protease). To confirm this, we overexpressed HAUSP and SIRT5 in 293T cells. Co‐immunoprecipitation assay showed that HAUSP bound to SIRT5 (Figs 4A and EV3A). Moreover, an endogenous association between HAUSP and SIRT5 was detected in BMDMs (Fig 4B and C). The direct interaction between HAUSP and SIRT5 was confirmed in vitro with purified proteins (Fig EV3B). Moreover, C‐terminal (residues 256–310) of SIRT5 failed to form complex with HAUSP, confirming that N‐terminal (residues 1–255) of SIRT5 is required for its binding to HAUSP (Fig EV3C). Together with the finding that TRIM21 also binds to this region (Fig EV1E), it is likely that HAUSP and TRIM21 compete for binding to SIRT5, which is quite understandable as they usually have opposite functions in regulating protein ubiquitination and degradation. In support of this, during polarization induced by LPS, SIRT5 showed increased binding intensity to TRIM21, while the binding between SIRT5 and HAUSP noticeably declined (Fig 4D).
Figure 4. HAUSP binds to and stabilizes SIRT5, which is repressed in LPS‐activated macrophages.
-
ALysates from 293T cells transfected with HA‐HAUSP and Flag‐SIRT5 or vector control (−) as indicated were immunoprecipitated (IP) using anti‐Flag antibody, and bound proteins were analyzed by Western blotting (WB).
-
B, CLysates from BMDMs were immunoprecipitated with anti‐HAUSP (B), anti‐SIRT5 (C) or control IgG antibodies as indicated and then analyzed by Western blotting.
-
DIB analysis of WCLs and anti‐SIRT5 immunoprecipitates of BMDMs treated with 100 ng/ml LPS for 4 h. Cells were treated with 10 μM MG132 for 4 h before harvesting.
-
E293T cells were transfected with Flag‐tagged HAUSP, mutant HAUSP‐CS (C223S) or vector control as indicated for 48 h. protein expression was determined by Western blot analysis using indicated antibodies.
-
FIB analysis of WCLs derived from 293T cells transfected with HA‐SIRT5 together with Flag‐HAUSP or vector control constructs (left panel). Cells were treated with 100 ng/ml CHX for the indicated time period prior to harvesting. The SIRT5 protein abundance was quantified (right panel).
-
G, HWCLs from BMDMs treated with increasing amounts of P5091(G) or P22077 (H) in the presence or absence of 100 ng/ml LPS were subjected to IB analysis using indicated antibodies (left panel). The SIRT5 protein abundance was quantified (right panel).
Data information: All IP and WB data in this work otherwise indicated are representative of at least three independent experiments.
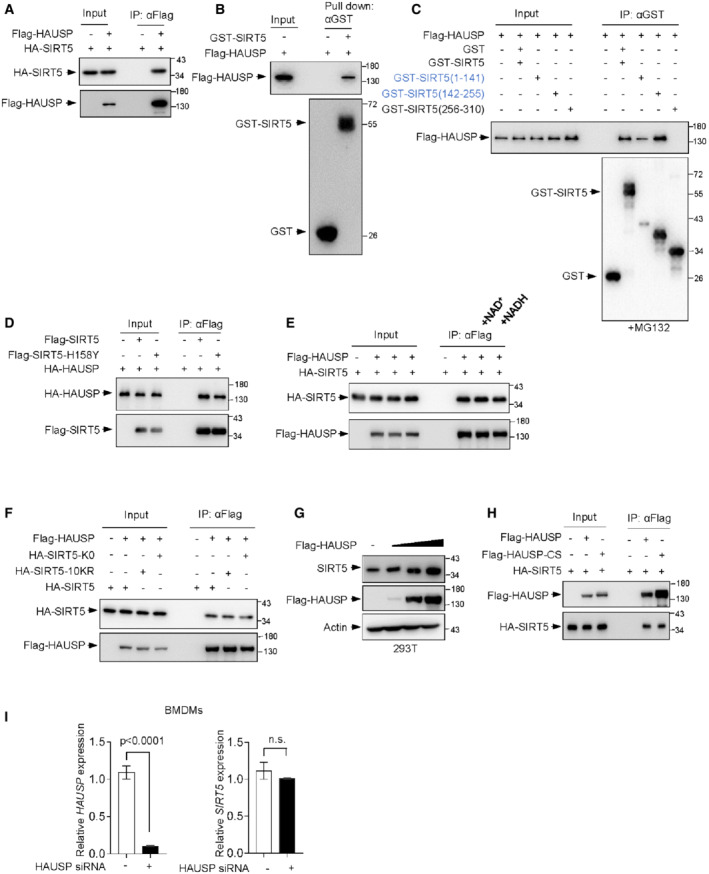
Figure EV3. HAUSP physically interacts with SIRT5.
-
AIB analysis of WCLs and immunoprecipitates derived from 293T cells transfected with Flag‐HAUSP together with HA‐SIRT5 constructs. Cells were treated with 10 μM MG132 for 4 h before harvesting.
-
BPurified GST‐SIRT5 proteins were incubated with purified recombinant Flag‐HAUSP proteins as indicated for 90 min at 30°C, followed by pull down with anti‐GST antibody and Western blot analysis.
-
CIB analysis of WCLs and IPs derived from 293T cells transfected with Flag‐HAUSP together with the indicated SIRT5 deletion constructs. Cells were treated with 10 μM MG132 for 4 h before harvesting.
-
DIB analysis of WCLs and immunoprecipitates derived from 293T cells transfected with indicated constructs. Cells were treated with 10 μM MG132 for 4 h before harvesting.
-
EIB analysis of WCLs and immunoprecipitates derived from 293T cells transfected with Flag‐HAUSP together with HA‐SIRT5 constructs in the presence or absence of 3 mM NAD+ or NADH as indicated. Cells were treated with 10 μM MG132 for 4 h before harvesting.
-
FIB analysis of WCLs and immunoprecipitates derived from 293T cells transfected with Flag‐HAUSP together with HA‐SIRT5, HA‐SIRT5‐K0, HA‐SIRT5‐10KR as indicated. Cells were treated with 10 μM MG132 for 4 h before harvesting.
-
G293T cells were transfected with increasing amounts of Flag‐HAUSP construct and endogenous SIRT5 expression was analyzed by Western blotting.
-
HIB analysis of WCLs and immunoprecipitates derived from 293T cells transfected with indicated constructs. Cells were treated with 10 μM MG132 for 4 h before harvesting.
-
ImRNA levels of HAUSP and SIRT5 in BMDMs transfected with control siRNA (−) or HAUSP siRNA for 48 h were measured by qRT‐PCR. Data are means ± SD. n = 3 biological replicates. P‐values were determined by unpaired two‐tailed Student's t‐tests. ns, not significant.
Data information: All IP and WB data in this work otherwise indicated are representative of at least three independent experiments.
Furthermore, changes in SIRT5 activity through either direct mutation (H158Y) or addition of NAD+ or NADH did not affect the interaction between HAUSP and SIRT5 (Fig EV3D and E). Intriguingly, HAUSP was still able to interact with the mutant SIRT5 (K0 or 10KR), which cannot be ubiquitinated by TRIM21 (Fig EV3F). These findings together suggest that HAUSP is a binding partner for SIRT5.
We next examined if HAUSP can influence the stability of SIRT5. Notably, enforced expression of wild‐type HAUSP led to elevated protein levels of SIRT5 (Figs 4E and EV3G). By contrast, although the catalytically inactive mutant of HAUSP (HAUSP‐CS(C223S)) still retained the ability to interact with SIRT5 (Fig EV3H), it failed to enhance the protein levels of SIRT5 (Fig 4E). In addition, unlike protein expression, the mRNA levels of SIRT5 were not changed upon HAUSP ablation in BMDMs (Fig EV3I). Thus, these findings suggest that HAUSP may have a role in the improvement of SIRT5 stability. To further confirm this, we performed CHX pulse‐chase analysis. The protein of HA‐SIRT5 was much more stable in Flag‐HAUSP expressing cells than in vector control cells (Fig 4F). Moreover, treatment of MG132 blocked the degradation of SIRT5 in both situations (Fig 4F), suggesting a role for HAUSP in stabilizing SIRT5. In keeping with the findings that M1 macrophage polarization promoted degradation of SIRT5 (Fig 1I and J) and TRIM21 reduced SIRT5 stability (Fig 1H and I), supplying cells with LPS to trigger M1 polarization resulted in a decrease in the interaction between SIRT5 and HAUSP, and increased the binding of SIRT5 to TRIM21 (Fig 4D). Moreover, suppression of HAUSP using the selective and cell‐permeable HAUSP inhibitor P5091 accelerated the degradation of SIRT5 induced by LPS (Fig 4G). Similar results were obtained using another potent and selective HAUSP inhibitor P22077 (Fig 4H). Taken together, these data suggest that HAUSP has a protective role in regulating SIRT5 stability, which is restricted during M1 polarization.
HAUSP erases SIRT5 ubiquitination by acting as a deubiquitinating enzyme and LPS treatment suppresses this effect in macrophages
To explore whether HAUSP enhances SIRT5 stability through deubiquitinating SIRT5, we examined the effect of HAUSP on SIRT5 ubiquitination. Overexpression of HAUSP led to a decrease in ubiquitination levels of SIRT5 (Fig 5A and B), and this effect could be largely reversed by P22077‐mediated HAUSP inhibition (Fig 5B). The deubiquitination of SIRT5 by HAUSP seems very robust, as it could almost totally remove the ubiquitin chains from SIRT5 (Fig 5C). In support of this, we found that HAUSP overexpression resulted in a complete block to the ubiquitination of SIRT5 even in the presence of TRIM21 (Fig 5C). In line with the protein expression data (Fig 4E), a catalytically inactive mutation of HAUSP (HAUSP‐CS) failed to deubiquitinate SIRT5, suggesting HAUSP‐mediated SIRT5 deubiquitination requires the deubiquitinating enzymatic activity of HAUSP (Fig 5D). In addition, the specificity of HAUSP for SIRT5 was demonstrated by an in vitro deubiquitination assay. Ubiquitinated SIRT5 was pulled down from cells co‐transfected with HA‐Ub, and then treated with purified wild‐type HAUSP or the catalytically inactive HAUSP mutant (HAUSP‐CS). Remarkably, HAUSP addition reduced the ubiquitination levels of SIRT5, whereas the HAUSP‐CS mutant showed no effect (Fig 5E).
Figure 5. HAUSP deubiquitinates SIRT5, and LPS treatment suppresses this effect in macrophages.
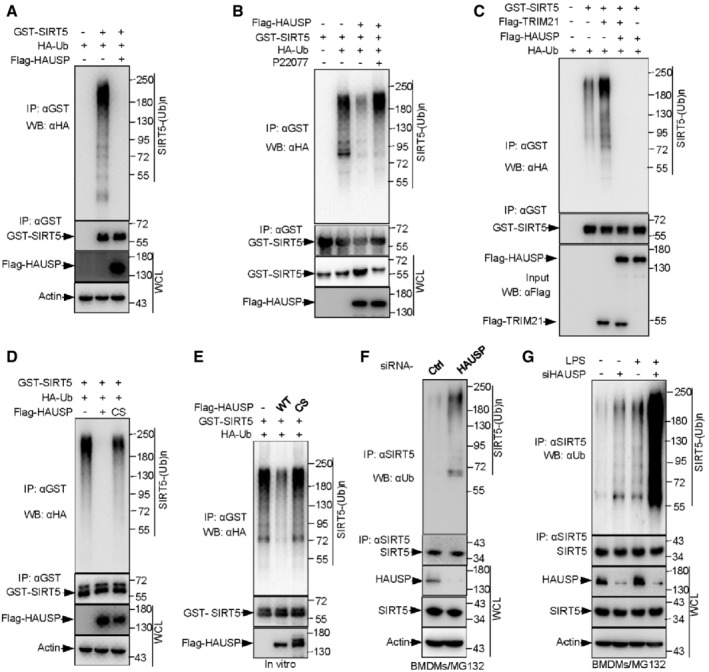
-
AIn vivo ubiquitination assay analysis of WCLs and GST‐tagged immunoprecipitated products derived from 293T cells transfected with the indicated plasmids. 10 μM MG132 was added for 4 h before harvesting.
-
BIB analysis of WCLs and anti‐GST immunoprecipitates of 293T cells transfected with the indicated plasmids in the presence or absence of 100 μM P22077 for 24 h. Cells were treated with 10 μM MG132 for 4 h before harvesting.
-
C, DIB analysis of WCLs and GST‐tagged immunoprecipitated products derived from 293T cells transfected with the indicated plasmids. 10 μM MG132 was added for 4 h before harvesting.
-
EIn vitro SIRT5 deubiquitination assay. Ubiquitinated SIRT5 was pulled down from 293T cells cotransfected HA‐Ub, and then incubated with purified wild‐type HAUSP or the catalytically inactive HAUSP mutant (HAUSP‐CS) 30°C for 90 min. The reaction products were resolved by SDS‐PAGE and probed with the indicated antibodies.
-
FIB analysis of WCLs and anti‐SIRT5 immunoprecipitated products derived from BMDMs transfected with control or HAUSP siRNA for 48 h. 10 μM MG132 was added for 6 h before harvesting.
-
GBMDMs transfected with control (−) or HAUSP siRNA for 48 h were then treated with 100 ng/ml mM LPS for 4 h, and 10 μM MG132 was added for 4 h before harvesting. WCLs and anti‐SIRT5 immunoprecipitated products were prepared and subjected to IB analysis using indicated antibodies.
Data information: All IP and WB data in this work otherwise indicated are representative of at least three independent experiments.
To confirm if HAUSP‐mediated erasure of SIRT5 ubiquitination occurs at endogenous level, we knocked down HAUSP in BMDMs and found that HAUSP ablation led to a strong increase in the ubiquitination of SIRT5 (Fig 5F and G). More importantly, LPS stimulation promoted SIRT5 ubiquitination, and this effect was exacerbated when HAUSP was silent (Fig 5G). Collectively, these findings reveal a role for HAUSP in deubiquitinating SIRT5 and preventing its degradation, for instance, during macrophage M1 polarization.
SIRT5 reciprocally modulates TRIM21 activity by deacetylation
Many important biological functions of SIRT5 have been revealed due to its deacetylase activity. The direct assembly of TRIM21‐SIRT5 complex further prompted us to investigate whether acetylation modification modulates TRIM21 activity. It was reported that the demalonylase or desuccinylase activity of SIRT5 is higher than its deacetylase activity (Du et al, 2011; Rardin et al, 2013). To determine which type of modification of TRIM21 is regulated by SIRT5, we analyzed the effect of SIRT5 on lysine acetylation, glutarylation, succinylation, and molonylation of TRIM21, respectively. Only acetylation levels of TRIM21 were reduced in SIRT5 overexpressed cells (Fig 6A). Conversely, the levels of TRIM21 acetylation were increased in SIRT5 knockdown cells (Fig 6B). These data suggest that SIRT5 may catalyze TRIM21 deacetylation. Next, we investigate how TRIM21 is acetylated. Coimmunoprecipitation assays revealed that TRIM21 was able to form complex with two major acetyltransferases, histone acetyl transferase 1 (HAT1) (Fig EV4A) and N‐acetyltransferase 8 (NAT8) (Fig EV4B), which was further confirmed by an in vitro binding assay using purified proteins (Fig EV4C). The interaction between TRIM21 and these two acetyltransferases raised a possibility that HA‐NAT8 or HA‐HAT1 may promote TRIM21 acetylation. Indeed, cells expressing either HA‐NAT8 or HA‐HAT1 displayed increased acetylation levels of TRIM21 (Fig EV4D). Together, these data indicate a role for the acetyltransferases NAT8 and HAT1 in positively regulating TRIM21 acetylation.
Figure 6. SIRT5 suppresses TRIM21 activity via deacetylation of TRIM21 at Lys351.
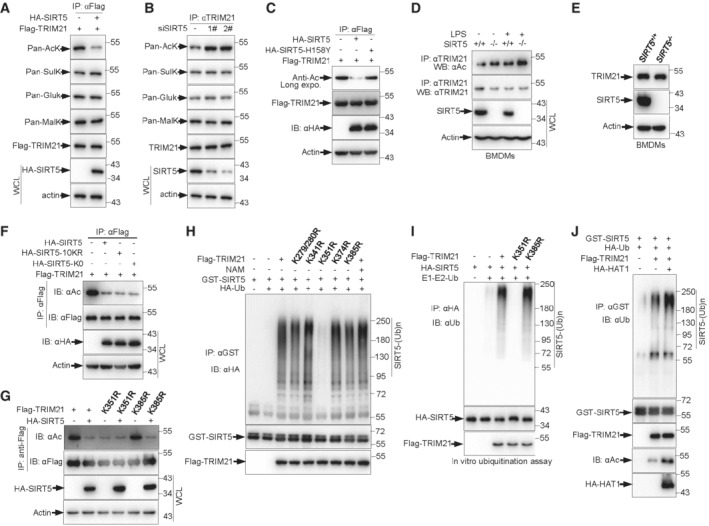
-
AIB analysis of WCLs and anti‐Flag immunoprecipitated products derived from 293T cells transfected with the indicated plasmids using anti‐Flag, anti‐Acetyllysine, anti‐Succinyllysine, anti‐Glutaryllysine, and anti‐Malonyllysine antibodies.
-
BIB analysis of WCLs and anti‐TRIM21 immunoprecipitated products derived from 293T cells transfected with control or SIRT5 siRNA using anti‐Flag, anti‐acetyl Lysine (AcK), anti‐succinyl Lysine (SulK), anti‐glutaryl Lysine (GluK), and anti‐malonyl Lysine (MalK) antibodies.
-
CIB analysis of WCLs and anti‐Flag immunoprecipitated products derived from 293T cells transfected with the indicated plasmids. 10 μM MG132 was added for 4 h before harvesting.
-
DIB analysis of WCLs and anti‐TRIM21 immunoprecipitated products derived from BMDMs treated 100 ng/ml LPS or left untreated for 4 h. 10 μM MG132 was added for 4 h before harvesting.
-
EProtein expression in SIRT5+/+ and SIRT5−/− BMDMs was determined by Western blot analysis.
-
F, GIB analysis of WCLs and anti‐Flag immunoprecipitated products derived from 293T cells transfected with the indicated plasmids. 10 μM MG132 was added for 4 h before harvesting.
-
HIB analysis of WCLs and anti‐GST immunoprecipitated products derived from 293T cells transfected with the indicated plasmids in the presence or absence of 3 mM NAM for 24 h. 10 μM MG132 was added for 4 h before harvesting.
-
IPurified TRIM21, TRIM21‐K351R and TRIM21‐K385R proteins were incubated with purified E1 and E2 (UBE2D2), ubiquitin and SIRT5 proteins as indicated at 30°C for 90 min. The ubiquitination reaction products were resolved by SDS‐PAGE and probed with the indicated antibodies.
-
JIB analysis of WCLs and anti‐GST immunoprecipitated products derived from 293T cells transfected with the indicated plasmids. 10 μM MG132 was added for 4 h before harvesting.
Data information: All IP and WB data in this work otherwise indicated are representative of at least three independent experiments.
Figure EV4. Both HAT1 and NAT8 can bind to TRIM21.
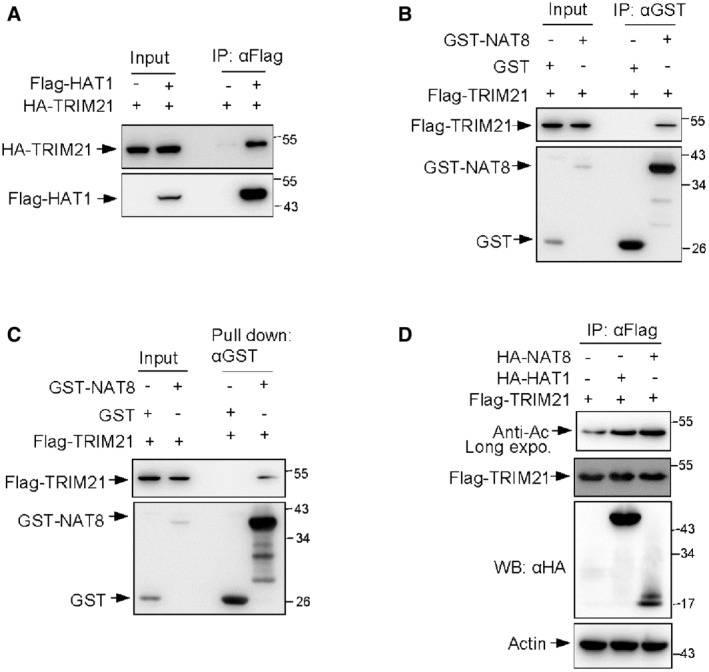
-
AIB analysis of WCLs and immunoprecipitates derived from 293T cells transfected with Flag‐HAT1 together with Flag‐TRIM21 constructs.
-
BIB analysis of WCLs and anti‐GST immunoprecipitated products derived from 293T cells transfected with indicated constructs.
-
CPurified Flag‐TRIM21 proteins were incubated with purified GST or GST‐NAT8 proteins as indicated for 90 min at 30°C, followed by pull down with anti‐GST antibody and Western blot analysis.
-
DIB analysis of WCLs and anti‐Flag immunoprecipitated products derived from 293T cells transfected with the indicated plasmids. 10 μM MG132 was added for 4 h before harvesting.
Data information: All IP and WB data in this work otherwise indicated are representative of at least three independent experiments.
We next examined whether SIRT5 catalyzes TRIM21 deacetylation. Consistent with the previous finding (Fig 6A and B), the acetylation levels of TRIM21 were noticeably reduced upon SIRT5 overexpression in 293T cells (Fig 6C). Conversely, SIRT5 knockout led to increased acetylation levels of TRIM21 in BMDMs, especially when cells were exposed to LPS treatment (Fig 6D). Additionally, knockout of SIRT5 had no effect on the total levels of TRIM21 (Fig 6E). Notably, the deacetylation of TRIM21 relies on the deacetylase activity of SIRT5, as transfection of an enzyme‐deficient mutant SIRT5 (SIRT5‐H158Y) did not affect TRIM21 acetylation (Fig 6C). Interestingly, ubiquitination of SIRT5 induced by TRIM21 seems unable to impair the deacetylase activity of SIRT5, and still, introduction of either SIRT5‐K0 or 10KR resulted in a strong decrease in TRIM21 acetylation (Fig 6F). To identify the site(s) of SIRT5‐mediated deacetylation, we generated a series of single–lysine mutant constructs of TRIM21 by replacing lysine with arginine. Specifically, substitution of the lysine 351 with arginine led to reduced acetylation levels of TRIM21 and completely abolished the deacetylation effect of SIRT5 on TRIM21 (Fig 6G). In sum, these findings demonstrate that SIRT5 deacetylates TRIM21 at lysine 351.
Of note, upon LPS stimulation, we found an explicit elevation of TRIM21 acetylation in BMDMs, suggesting that acetylation may enhance TRIM21 activity during macrophage M1 polarization (Fig 6D). Consistent with this, overexpression of the mutant TRIM21‐K351R, which had low level of acetylation and could not be further deacetylated by SIRT5 (Fig 6G), showed no capability to ubiquitinate SIRT5 in 293T cells (Fig 6H). This finding was further confirmed by an in vitro ubiquitination assay. In the presence of E1, E2, and ubiquitin, the addition of wild‐type TRIM21 or a control mutant (TRIM21‐K385R) catalyzed the ubiquitination of SIRT5; however, TRIM21‐K351R supplementation failed to induce SIRT5 ubiquitination (Fig 6I). To further confirm our observations that acetylation of TRIM21 enhances its E3 ligase activity, we co‐transfected 293T cells with GST‐SIRT5 together with Flag‐TRIM21 in the presence or absence of HA‐HAT1. GST‐SIRT5 was immunoprecipitated and its ubiquitination was examined with anti‐Ubiquitin antibody. Again, TRIM21 promoted SIRT5 ubiquitination, and HAT1 enhanced this effect (Fig 6J). Collectively, acetylation increases TRIM21 activity, which is inhibited by SIRT5. Physiologically, in macrophages, LPS‐mediated M1 polarization increases the binding of TRIM21 to SIRT5, as well as enhances the acetylation of TRIM21, thereby leading to SIRT5 ubiquitination and degradation. Reciprocally, more SIRT5 degradation leads to even higher activity of TRIM21 in a feedback loop manner.
SIRT5 and TRIM21 coordinate LPS responses
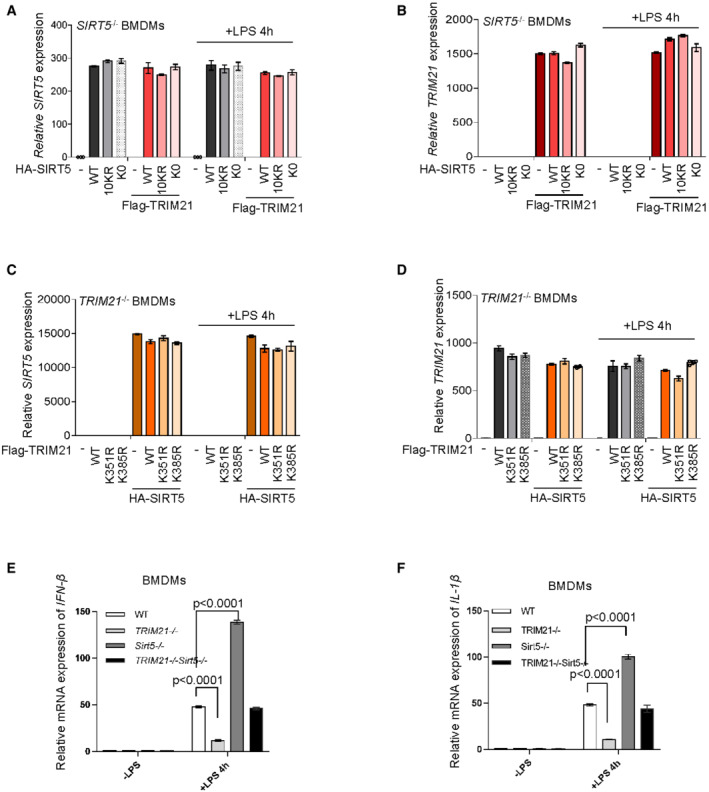
To elucidate the physiological impact(s) of the interplay between TRIM21 and SIRT5, we examined the production of pro‐inflammation cytokines in LPS‐activated macrophages. SIRT5 has been shown to negatively modulate IL‐1β production in LPS‐activated macrophages (Wang et al, 2017). Consistent with this, we found that macrophages stably overexpressing wild‐type SIRT5, as well as the mutants SIRT5‐K0 and SIRT5‐10KR, produced much less IFN‐β and IL‐1β, compared with vector control cells upon LPS treatment (Figs 7A and B, and EV5A and B). In keeping with the finding that TRIM21 mediates SIRT5 ubiquitination and degradation, the suppressive effect of wild‐type SIRT5 on IFN‐β and IL‐1β could be totally reversed by enforced expression of Flag‐TRIM21 (Fig 7A and B), suggesting a critical role for SIRT5 in TRIM21‐mediated pro‐inflammatory cytokine generation. More importantly, introduction of TRIM21 could not restore the production of these pro‐inflammatory cytokines in LPS‐activated cells expressing SIRT5‐K0 or SIRT5‐10KR (Fig 7A and B). These data together indicate that TRIM21 may promote LPS‐induced macrophage inflammation through ubiquitination of SIRT5. Reciprocally, we also wanted to know the effect of SIRT5‐mediated acetylation of TRIM21 on the production of pro‐inflammatory cytokines in LPS‐treated macrophages. The expression of IFN‐β and IL‐1β was higher in cells transfected with wild‐type TRIM21 or the nonfunctional mutant TRIM21‐K385R (Figs 7C and D, and EV5C and D). In comparison, the acetylation‐ and activity‐defective mutant TRIM21‐K351R restrained its ability to promote IFN‐β and IL‐1β expressions (Fig 7C and D), suggesting that the acetylation of TRIM21 is crucial for its activity in LPS‐mediated cytokine production in macrophages. Moreover, SIRT5 overexpression could largely reduce the expression IFN‐β and IL‐1β in these cells, especially in TRIM21‐ and TRIM21‐K385R‐expressing cells in response to LPS treatment (Fig 7C and D). Together, these findings suggest a feedback regulation of LPS response in macrophages by TRIM21 and SIRT5.
Figure 7. Mutual regulation of SIRT5 and TRIM21 influences IL‐1β production in LPS‐activated macrophages and DSS‐induced colitis in mice.
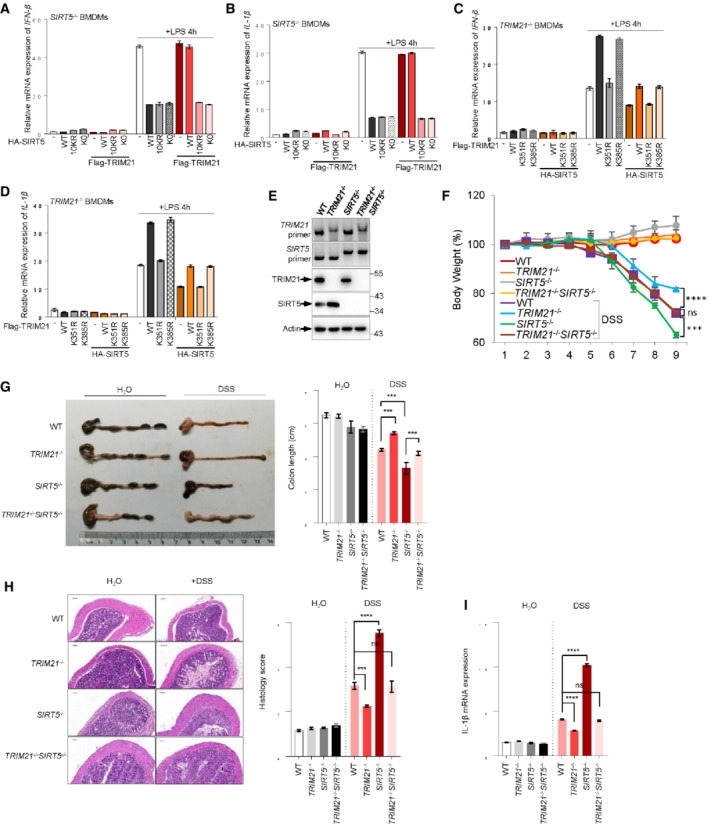
-
A, BSIRT5 −/− macrophages (BMDMs) were transfected with HA‐SIRT5 WT or mutants in the absence or presence of Flag‐TRIM21 as indicated. Macrophages were stimulation with 100 ng/ml LPS for 4 h. mRNA expression of IFN‐β (A) and IL‐1β (B) was determined by qRT‐PCR analysis.
-
C, DTRIM21 −/− macrophages (BMDMs) were transfected with Flag‐TRIM21 WT or mutants in the absence or presence of HA‐SIRT5 as indicated. Macrophages were stimulation with 100 ng/ml LPS for 4 h. mRNA expression of IFN‐β (C) and IL‐1β (D) was determined by qRT‐PCR analysis.
-
EGenotyping of TRIM21 −/−, SIRT5 −/− and TRIM21 −/− SIRT5 −/− mice. DNA derived from the tail of wild‐type and knockout animals was analyzed by PCR. Protein expression of TRIM21 and SIRT5 in BMDMs derived from mice were also analyzed by Western blotting.
-
F–IWild‐type (WT, n = 5), TRIM21 −/− (n = 5), SIRT5 −/−(n = 5), and TRIM21 −/− SIRT5 −/− (n = 5) mice were orally administrated with 2.5% (w/v) DSS in drinking water for 7 days and regular drinking water for another 2 days. Mice body weight (F) and Colon‐length (G) changes were measured. The colon tissues were examined microscopically for pathological changes (H, left panel, scale bar: 100 μm), which were further semiquantitatively scored (H, right panel). (I) IL‐1β mRNA expression in colon tissues determined by qRT‐PCR.
Data information: Data are means ± SD. In (A–D), n = 3 biological replicates, in (F–I), n = 5 biological replicates. Western blots in (E) represent three independent experiments. P‐values were determined by unpaired two‐tailed Student's t‐tests. ns, not significant; ***P < 0.001, ****P < 0.0001.
Figure EV5. Effect of SIRT5 and TRIM21 mutual regulation on macrophage pro‐inflammatory cytokine production.
-
A, BmRNA expression of SIRT5 (A) and TRIM21 (B) in LPS‐treated and untreated SIRT5 −/− BMDMs expressing Flag‐TRIM21 together with HA‐SIRT5 or various HA‐tagged SIRT5 mutants as indicated was determined by qRT‐PCR analysis and normalized to β‐actin. Macrophages were stimulation with 100 ng/ml LPS for 4 h.
-
C, DmRNA expression of SIRT5 (C) and TRIM21 (D) in LPS‐treated and untreated TRIM21 −/− BMDMs stably expressing HA‐SIRT5 together with Flag‐TRIM21 or various Flag‐tagged TRIM21 mutants as indicated was determined by qRT‐PCR analysis and normalized to β‐actin. Macrophages were stimulation with 100 ng/ml LPS for 4 h.
-
E, FmRNA expression of IFN‐β (E) and IL‐1β (F) in LPS‐treated and untreated BMDMs derived from wild‐type (WT), TRIM21 −/−, SIRT5 −/− and TRIM21 −/− SIRT5 −/− mice. Macrophages were stimulation with 100 ng/ml LPS for 4 h.
Data information: Data are means ± SD. In (A–F), n = 3 biological replicates. P‐values were determined by unpaired two‐tailed Student's t‐tests. ns, not significant.
To further validate the influence of the interplay between TRIM21 and SIRT5 on macrophage inflammation response in vivo, we generated TRIM21 and SIRT5 double knockout mice (Fig 7E). In agreement with the above findings (Fig 7A–D), knockout of TRIM21 strongly impaired, while SIRT5 knockout increased the expression of IFN‐β and IL‐1β in LPS‐activated macrophages derived from TRIM21 −/− and SIRT5 −/− mice, respectively (Fig EV5E and F). Notably, simultaneous knockout of TRIM21 and SIRT5 (TRIM21 −/− SIRT5 −/−) brought the cytokine expression back to the level of that observed in wild‐type control cells (Fig EV5E and F). DSS‐induced colitis is driven by activated intestinal macrophages that release pro‐inflammatory cytokines and chemokines causing tissue damage in the colon. To better understand the role of TRIM21‐SIRT5 interplay in inflammation responses, we conducted DSS‐induced colitis in vivo model. After 9 days of oral administration of DSS, SIRT5 −/− mice showed severe body‐weight loss, and further loss of TRIM21 significantly improved the body weight (Fig 7F). Another important evaluation parameter in DSS‐induced colitis model is the length of colon (Okayasu et al, 1990). Consistent with previous findings (Wang et al, 2017), the colons of SIRT5 −/− mice were about 25% shorter than that of wild‐type mice (Fig 7G). Notably, TRIM21 knockout could largely restore the colon length in DSS‐treated SIRT5 −/− mice (Fig 7G). Moreover, representative colon sections from DSS‐treated mice showed that, compared with that of wild‐type mice, the colonic sections of SIRT5 −/− mice displayed noticeable histopathological changes as characterized by severe transmural inflammation with focal areas of extensive ulceration and necrotic lesions (Fig 7H). Consistently, upon DSS treatment, the expression of IL‐1β was significantly higher in colons of SIRT5 −/− mice, and robustly reduced when TRIM21 was knocked out in these mice (Fig 7I). Taken together, these data suggest mutual regulation of SIRT5 and TRIM21 functionally dictates inflammation in DSS‐induced colitis.
Discussion
Our findings reveal that TRIM21 and SIRT5 functionally interact, which plays a role in modulating inflammatory cytokines including IL‐1β production in LPS‐activated macrophages. Vice versa, macrophage M1 polarization induced by LPS skews the TRIM21‐SIRT5 interplay toward elevation of TRIM21 E3 ligase activity, and leads to SIRT5 degradation. It has been well‐recognized that TRIM21 expression is upregulated by type I interferons for more potent neutralization. Thus, it could be imagined that through functional interaction with TRIM21, SIRT5 might be linked to IFNs‐mediated antiviral immunity, and testing the potential involvement of SIRT5 in host immune protection and pathology during infection would be of great interest. TRIM21 can be acetylated at multiple lysine reduces, and as we have identified here, two acetyltransferases HAT1 and NAT8 catalyze the acetylation of TRIM21, and specifically, SIRT5 suppresses TRIM21 E3 ligase activity by deacetylating it at Lys351, but not Lys385. Interestingly, recent work suggests that TRIM21 is also a substrate of histone deacetylase 6 (HDAC6) and deacetylation of TRIM21 at Lys385 and Lys387 by HDAC6 causes its homodimerization, thereby leading to increased ubiquitination activity and binding to antibody‐coated virus complex for degradation (Xie et al, 2020). However, in LPS‐activated macrophages, we did not find any activity of TRIM21‐K385R toward SIRT5 ubiquitination. Based on these observations, it is likely that acetylation at different lysine residues may have different impacts on TRIM21 activity, and orchestration by deacetylating of these lysine acetylation states may enable cells to response to different stress stimulations.
Although a previous study reported that SIRT5 +/+ and SIRT5 −/− BMDMs secreted similar levels of TNF and IL‐6 in response to 10 ng/ml LPS stimulation (Heinonen et al, 2018), in the present study, we found that when BMDMs were treated with 100 ng/ml LPS, compared with WT BMDMs, SIRT5 −/− BMDMs showed a significant increase in the levels of IFN‐β and IL‐1β. This finding is consistent with the work by Wang et al (2017) in which the BMDMs were treated with 100 ng/ml LPS. The different responses of SIRT5 −/− BMDMs to LPS may be due to the different concentration of LPS treatment. That is, SIRT5 has an inhibitory effect on IL‐1b production when high doses of LPS‐treated macrophages. Interestingly, LPS‐activated macrophages show suppressed SIRT5 expression level and NAD+ level, which in turn increases the succinylation of several key glycolytic enzymes (e.g., PKM2) and IL‐1β production (Tannahill et al, 2013; Palsson‐McDermott et al, 2015). Together with these findings, it appears that in LPS‐induced pro‐inflammatory M1 macrophages, SIRT5 is repressed at different levels. On the one hand, LPS stimulation decreases the expression and NAD+‐dependent enzymatic activity of SIRT5, which may lead to enhancement of TRIM21 E3 ligase activity by sustaining its acetylation at Lys351. On the contrary, LPS treatment also increases the binding of TRIM21 to SIRT5 for ubiquitination and degradation, while reducing HAUSP‐mediated deubiquitination and stabilization of SIRT5. Thus, through TRIM21, LPS induces a positive feedback loop to control SIRT5 and keep it at a low level. Moreover, the tight control of SIRT5 involving TRIM21 strongly demonstrate the importance of TRIM21‐SIRT5 interplay in LPS‐mediated IL‐1β production and inflammatory response.
Metabolic reprogramming, for instance, glycolytic remodeling, is essential for antimicrobial inflammatory response. Over the past several years, extensively studies have been focused on the regulation of metabolism by SIRT5, especially the impressive role of SIRT5 in modulating mitochondrial biogenesis. Given the strong and multiple‐level control of SIRT5, whether metabolic changes in LPS‐activated macrophages contribute to SIRT5‐mediated macrophages inflammatory status and effectiveness is of great interest and requires further investigation. Nevertheless, although the role of SIRT5 and TRIM21, as well as their effect on other inflammatory responses remains unclear, our findings may highlight a potential therapeutic approach by changing the balance of TRIM21‐SIRT5 interplay for the treatment of inflammatory diseases.
Materials and Methods
Cell culture, reagents, and antibodies
HEK293T (also named 293T in the manuscript) cells were purchased from ATCC (Maryland, USA). The cells were maintained in Dulbecco's modified Eagle medium (Thermo Scientific) with 10% fetal bovine serum. Cells were tested negative for mycoplasma contamination. The following reagents were used: MG132 (Merck), LPS (Sigma‐Aldrich), CHX (Sigma‐Aldrich), NAD+ (Sigma‐Aldrich), NADH (Sigma‐Aldrich), DSS (MW 36–50 kDa) (MP Biomedicals), GST Beads (GE Healthcare), Flag‐M2 beads (Bimake), HA beads (thermo), DAPI (thermo), SIRT5 inhibitor 1 (MedChemExpress), SIRT5 enzymatic activity assay kit (Enzo Life Sciences), E1 (R&D Systems), E2 (R&D Systems), His6‐Ub (R&D Systems), Ub‐K48R (R&D Systems), Ub‐K63R (R&D Systems), Ub‐K48 (R&D Systems), Ub‐K63 (R&D Systems), Normal mouse IgG (Santa Cruz), Normal rabbit IgG (Cell Signaling Technology), SIRT5 antibody (Cell Signaling Technology), TRIM21 antibody (Cell Signaling Technology), Ubiquitin antibody (Cell Signaling Technology), Ubiquitin‐K48 antibody (Cell Signaling Technology), Ubiquitin‐K63 antibody (Cell Signaling Technology), HSUSP antibody (Cell Signaling Technology), Acetyl antibody (Thermo), actin antibody (EASYBIO), GST antibody (EASYBIO), Flag antibody (EASYBIO), HA antibody (EASYBIO), P5049 (Selleck), P22077 (Selleck), SYBR Green Q‐PCR Mix (GenStar), RevertAid First Strand cDNA Synthesis Kit (Thermo), GM‐CSF (Peprotech).
siRNAs
The following siRNAs were purchased from GenePharma Company (Suzhou, China): human TRIM21: 5’‐GCAUGGUCUCCUUCUACAA‐3′; human SIRT5‐1#: 5’‐GCAUCCCAGUUGAGAAACU‐3′; human SIRT5‐2#: 5’‐GAGAUCCAUGGUAGCUUAUUU‐3′; Mouse HAUSP: 5’‐GCGUAGCUUUAUUUGAGUCUU‐3′.
Mice
Wild‐type and TRIM21 tm1Hm (010724, TRIM21 knockout mice) mice were purchased from The Jackson Laboratory. C57BL/6‐SIRT5 tm1cyagen mice (SIRT5 knockout mice) were purchased from Cyagen Biosciences. TRIM21 tm1Hm were crossed with SIRT5 tm1cyagen mice to obtain TRIM21 −/− SIRT5 −/− mice. All animals were used at 6–8 weeks of age, and only male mice were used. All mice were maintained under specific pathogen‐free conditions, and used in accordance with protocols approved by the Institutional Animal Care and Use Committees of Tsinghua University for animal welfare. All procedures performed in this study were approved by the Tsinghua University Animal Care and Use Committee (TUACUC).
Mouse bone marrow‐derived macrophages (BMDMs) isolation and transfection
Mouse macrophages were isolated from mouse bones and then cultured in M‐CSF‐supplied DMEM medium containing 10% FBS for 7 days for BMDM differentiation. On day 8, the cells were re‐seeded and used for subsequent experiments.
Briefly, BMDMs were seeded into a six‐well or 60‐mm dish and cultured in DMEM containing 10% FBS for 24 h. Then, 40–50% confluent cells were changed with fresh medium. siRNA and plasmids were transfected into BMDMs using Lipofectamine 3000 (L3000150, Thermo Fisher Scientific) regents according to the manufacturer's instructions. 4 μl Lipofectamine 3000 reagent was diluted with 100 μl Opti‐MEM (31985070, Life Technologies, Invitrogen), and 100 pmol siRNA was diluted with 100 μl Opti‐MEM medium at room temperature for 15 min, respectively. These two transfection complexes were subsequently gently mixed together and incubated for another 15 min at room temperature, and then added into cells in a drop‐wise manner and shaken well. Fresh medium was replaced 24 h after transfection and BMDM cells were collected 48 h after transfection for RT‐qPCR and Western blotting analysis.
Induction of DSS‐induced acute colitis
Wild‐type (WT), TRIM21 −/−, SIRT5 −/−, TRIM21 −/− SIRT5 −/− mice were induced with 2.5% (w/v) DSS (MW 36–50 kDa) dissolved in sterile, distilled water. Six weeks old of male mice had free access to the DSS‐treated water for 1–7 days, which was replaced by normal drinking water until the end of the experiment on day 8. DSS water was prepared fresh on day 3 and day 5. Control mice were provided the same drinking water without DSS for 9 days. Mice were sacrificed on day 9. Body weight were daily measured and colon length were measured when mice were sacrificed.
In vivo ubiquitination assay
Bone marrow‐derived macrophages (BMDMs) from WT, TRIM21 −/− mice and 293T cells were treated with 10 μM MG132 for 4 h before harvesting. Cells were lysed by IP lysis buffer. Cell lysates were precipitated with indicated antibodies and A&G beads overnight. Then, beads were collected and eluted by acidic elution buffer. Neutralization buffer was adding to the eluted fraction to neutralize the low pH and boiled 10 min and analyzed by SDS‐PAGE followed by immunoblotting.
In vivo deubiquitination assay
BMDMs were transfected with control siRNA or HAUSP siRNA for 48 h and were treated with 10 μM MG132 for 4 h before harvesting. For cell‐based deubiquitination assays, GST‐SIRT5 or mutations, HA‐Ubiquitin or HA‐Ubiquitin‐K48R, K63R, K48, K63 were cotransfected with Flag vector, Flag‐HAUSP (WT or C223S) and were treated with 10 μM MG132 for 4 h before harvesting. Cells were lysed in IP lysis buffer for 1 h and GST beads or protein A/G beads were added to the lysates at 4°C overnight with gentle rotation. Then beads were collected and eluted by acidic elution buffer and boiled 10 min after adding neutralization buffer and 5× SDS‐loading buffer.
In vitro ubiquitination assay
GST or GST‐TRIM21 purified from 293T were incubated with 100 nM E1, 1 μM E2 (UBE2D2) (Wada & Kamitani, 2006; Pan et al, 2016; Zhu et al, 2022), His‐ubiquitin or K48R, K63R, K48, K63 (2 μg), and Sirt5 bounded on beads in 40 μl ubiquitination assay buffer (50 mM Tris–HCl, 2.5 mM MgCl2, 0.5 mM DTT and 2 mM ATP) for 2 h at 30°C. Then stopped by adding 5× SDS‐loading buffer and boiled for 10 min.
In vitro deubiquitination assay
Flag‐HAUSP (WT or C223S) were overexpressed in 293T cells, immunoprecipitated by anti‐Flag M2 beads, and eluted by Flag peptides (50 mg/ml) in the deubiquitination buffer (50 mM Tris–HCl pH 8.0, 50 mM NaCl, 1 mM EDTA, 10 mM DTT, 5% glycerol). GST‐Sirt5 and HA‐Ubiquitin were overexpressed in 293T cells for 48 h, and purified by GST beads. Purified Flag‐HAUSP or mutation were incubated with Sirt5 bounded on GST beads in deubiquitination buffer for 2 h at 30°C. Then stopped by adding 5× SDS‐loading buffer, boiled for 10 min and analyzed by Western blotting.
Quantitative real‐time PCR
Total RNA was extracted from the BMDMs using the TRIzol reagent (Invitrogen) according to the manufacturer's instructions, followed by cDNA preparation using the revertaid first strand cDNA synthesis kit (Thermo Scientific). Quantitative real‐time PCR were performed using SYBR Green mix (GenStar) with the CFX96 Real‐Time PCR Detection System (Bio‐Rad). The primer pairs for human genes were as follows: β‐actin, 5’‐GACCTGACTGACTACCTCATGAAGAT‐3′ and 5′‐GTCACACTTCATGATGGAGTTGAAGG‐3′; SIRT5, 5′‐TATTAGAAAGCAGCCGTGGAGA‐3′ and 5′‐CGCATCAGGGTTTGTCTGTAG‐3′; TRIM21, CAGTGCATGGAGAGAGACT and 5′‐AGTTCCCCTAATGCCACCTG‐3′.
The primer pairs for mouse genes were as follows: HPRT, 5′‐CAGTCCCAGCGTCGTGATTAG‐3′ and 5′‐AAACACTTTTTCCAAATCCTCGG‐3′; IFNβ, 5′‐TCCGAGCAGAGATCTTCAGGAA‐3′ and 5′‐TGCAACCACCACTCATTCTGAG ‐3′; IL‐1β 5′‐ACCTTCCAGGATGAGGACATGA −3′ and 5′‐CTAATGGGAACGTCACACACCA ‐3′; SIRT5, 5′‐CTCCGGGCCGATTCATTTC‐3′ and 5′‐GCGTTCGCAAAACACTTCCG‐3′; TRIM21, 5′‐TTCGTGGTTCAGAGCTGGAGC‐3′ and 5′‐GTCCTCAGCATCTTCTTCCGC‐3′.
Immunofluorescence confocal microscopy
Cells grown on glass coverslips were fixed with 4% paraformaldehyde in PBS for 30 min, permeabilized with 0.1% Triton X‐100, and blocked with 1% bovine serum albumin. Then, the cells were stained with the indicated primary antibodies followed by washing with phosphate‐buffered saline (PBS) twice, then incubating with fluorescent‐dye‐conjugated secondary antibodies. Nuclei were counterstained with DAPI (Sigma‐Aldrich). Then, the cells were subjected to confocal microscopy analysis.
Immunoprecipitation
293T cells were cotransfected with Flag‐SIRT5 and HA‐TRIM21 for 48 h, and then cells were lysed in IP lysis buffer for 1 h. Anti‐FLAG M2 affinity gels were adding to supernatants and incubated for 8 h. After incubation, beads were washed three times with lysis buffer and eluted by Flag peptide. Protein samples were boiled in 5× loading buffer for 10 min. As for sequential immunoprecipitation, 293T cells were cotransfected with Flag‐SIRT5, GST‐HAUSP, and HA‐TRIM21 for 48 h. Anti‐Flag M2 affinity beads and glutathione beads were used in the first round IP and the second round IP, respectively.
SIRT5 enzymatic activity assay
Briefly, GST, GST‐TRIM21 and Flag‐SIRT5 were overexpressed in HEK293T cells, and were purified with GST and Flag beads, respectively. GST‐TRIM21 and Flag‐SIRT5 proteins were incubated with E1 (100 nM), E2 (UBE2D2) (1 μM), His‐ubiquitin (2 μg) in 40 μl ubiquitination assay buffer (50 mM Tris–HCl, 2.5 mM MgCl2, 0.5 mM DTT and 2 mM ATP) for 2 h at 30°C, and then SIRT5 enzymatic activity assay was measured with SIRT5 enzymatic activity assay kit according to the manufacturer's instructions.
Histological analysis
The colon tissues were fixed with 4% paraformaldehyde at 4°C for more than 4 h, paraffin embedded, and sectioned (5 μm). The sections were deparaffinized and rehydrated before staining with hematoxylin and eosin (H&E). The histological scores of H&E stained sections were used to assess the colon pathology, based on assigned scores of tissue damage (0 = normal; 1 = irregular crypts; 2 = mild‐to‐moderate crypt loss (10–50%); 3 = severe crypt loss (50–90%); 4 = complete crypt loss) and inflammation (0 = absent, 1 = mucosal, 2 = submucosal, 3 = transmural extending into muscularis and serosa and 4 = diffuse). More than six different locations on each sample were used to count the score for each mouse. The scoring was done in a blinded manner.
Statistical analysis
Statistical parameters including the exact sample sizes and error bars are defined in the figures and corresponding legends. Unless otherwise stated, all figures are representative of at least three independent experiments. No statistical methods were used to predetermine sample size. For animal experiments, the authors who did the experiments were blinded to group allocation during data collection and/or analysis. The investigators were not blinded to all other experiments. All statistical analysis was performed and P‐values were obtained using the GraphPad Prism software 5.0 (GraphPad Software, Inc. USA). To assess statistical significance, comparisons between two groups were performed using an unpaired, two‐tailed Student's t‐test. Results are presented as the mean ± SD. P‐values <0.05 were considered statistically significant. Statistical significance is shown as *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Author contributions
Pengbo Yao: Data curation; formal analysis; investigation; methodology. Taiqi Chen: Methodology. Peng Jiang: Resources; project administration. Li Li: Methodology. Wenjing Du: Conceptualization; supervision.
In addition to the CRediT author contributions listed above, the contributions in detail are:
PY designed the experiments, collected, and analyzed the data. PY and TC performed most of the experiments. LL provided technical assistance. PJ provided the reagents. WD supervised the research and wrote the manuscript. All authors commented on the manuscript.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Acknowledgements
We thank Dr. Zhengfan Jiang at Peking University for materials. We thank Dr. Charles David at Tsinghua University for help with manuscript preparation. We thank Ye Liu at Tsinghua University for help with tissue slicing, and Youxiang Mao, Wenjun Xia and Di Shi for help with the mouse maintenance and injection, and all the Du laboratory members for discussion and technical assistance. This research was supported by CAMS Innovation Fund for Medical Sciences (CIFMS) (2021‐I2M‐1‐016), Haihe Laboratory of Cell Ecosystem Innovation Fund (HH22KYZX0011), National Key Research and Development Program of China (2019YFA0802600), CAMS Basic Research Fund (2019‐RC‐HL‐007) and State Key Laboratory Special Fund (2060204) to WD; National Natural Science Foundation of China (81902820) to PY.
EMBO reports (2022) 23: e54391
Data availability
This study includes no data deposited in external repositories.
References
- Albecka A, Clift D, Vaysburd M, Rhinesmith T, Caddy SL, Favara DM, Baxendale HE, James LC (2021) A functional assay for serum detection of antibodies against SARS‐CoV‐2 nucleoprotein. EMBO J 40: e108588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold L, Henry A, Poron F, Baba‐Amer Y, van Rooijen N, Plonquet A, Gherardi RK, Chazaud B (2007) Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med 204: 1057–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SK, Mantovani A (2012) Orchestration of metabolism by macrophages. Cell Metab 15: 432–437 [DOI] [PubMed] [Google Scholar]
- Caddy SL, Vaysburd M, Papa G, Wing M, O'Connell K, Stoycheva D, Foss S, Terje Andersen J, Oxenius A, James LC (2021) Viral nucleoprotein antibodies activate TRIM21 and induce T cell immunity. EMBO J 40: e106228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH et al (2011) Sirt5 is a NAD‐dependent protein lysine demalonylase and desuccinylase. Science 334: 806–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W, Li Y, Liu X, Wang S, Mei P, Chen Z, Liu K, Li S, Xu XW, Gan J et al (2022) TRIM21 regulates pyroptotic cell death by promoting Gasdermin D oligomerization. Cell Death Differ 29: 439–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauler F, Mallery DL, McEwan WA, Bidgood SR, James LC (2012) AAA ATPase p97/VCP is essential for TRIM21‐mediated virus neutralization. Proc Natl Acad Sci USA 109: 19733–19738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinonen T, Ciarlo E, Theroude C, Pelekanou A, Herderschee J, Le Roy D, Roger T (2018) Sirtuin 5 deficiency does not compromise innate immune responses to bacterial infections. Front Immunol 9: 2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeble AH, Khan Z, Forster A, James LC (2008) TRIM21 is an IgG receptor that is structurally, thermodynamically, and kinetically conserved. Proc Natl Acad Sci USA 105: 6045–6050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labzin LI, Bottermann M, Rodriguez‐Silvestre P, Foss S, Andersen JT, Vaysburd M, Clift D, James LC (2019) Antibody and DNA sensing pathways converge to activate the inflammasome during primary human macrophage infection. EMBO J 38: e101365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Huan C, Wang H, Liu Y, Liu X, Su X, Yu J, Zhao Z, Yu XF, Zheng B et al (2020) TRIM21‐mediated proteasomal degradation of SAMHD1 regulates its antiviral activity. EMBO Rep 21: e47528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallery DL, McEwan WA, Bidgood SR, Towers GJ, Johnson CM, James LC (2010) Antibodies mediate intracellular immunity through tripartite motif‐containing 21 (TRIM21). Proc Natl Acad Sci USA 107: 19985–19990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, Gordon S (2014) The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep 6: 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan WA, Tam JC, Watkinson RE, Bidgood SR, Mallery DL, James LC (2013) Intracellular antibody‐bound pathogens stimulate immune signaling via the Fc receptor TRIM21. Nat Immunol 14: 327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Lomb DJ, Haigis MC, Guarente L (2009) SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 137: 560–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R (1990) A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 98: 694–702 [DOI] [PubMed] [Google Scholar]
- Palsson‐McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, van den Bosch MW, Quinn SR, Domingo‐Fernandez R, Johnston DG et al (2015) Pyruvate kinase M2 regulates Hif‐1alpha activity and IL‐1beta induction and is a critical determinant of the warburg effect in LPS‐activated macrophages. Cell Metab 21: 65–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan JA, Sun Y, Jiang YP, Bott AJ, Jaber N, Dou Z, Yang B, Chen JS, Catanzaro JM, Du C et al (2016) TRIM21 ubiquitylates SQSTM1/p62 and suppresses protein sequestration to regulate redox homeostasis. Mol Cell 61: 720–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, Xie Z, Zhang Y, Zwaans BM, Skinner ME et al (2013) SIRT5‐mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell 50: 919–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, Guo A, Gut P, Sahu AK, Li B et al (2013) SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab 18: 920–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DA, Isenberg DA (2017) TRIM21 and the function of antibodies inside cells. Trends Immunol 38: 916–926 [DOI] [PubMed] [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, Palsson‐McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH et al (2013) Succinate is an inflammatory signal that induces IL‐1beta through HIF‐1alpha. Nature 496: 238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachharajani VT, Liu T, Wang X, Hoth JJ, Yoza BK, McCall CE (2016) Sirtuins link inflammation and metabolism. J Immunol Res 2016: 8167273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada K, Kamitani T (2006) Autoantigen Ro52 is an E3 ubiquitin ligase. Biochem Biophys Res Commun 339: 415–421 [DOI] [PubMed] [Google Scholar]
- Wang F, Wang K, Xu W, Zhao S, Ye D, Wang Y, Xu Y, Zhou L, Chu Y, Zhang C et al (2017) SIRT5 desuccinylates and activates pyruvate kinase M2 to block macrophage IL‐1beta production and to prevent DSS‐induced colitis in mice. Cell Rep 19: 2331–2344 [DOI] [PubMed] [Google Scholar]
- Xie S, Zhang L, Dong D, Ge R, He Q, Fan C, Xie W, Zhou J, Li D, Liu M (2020) HDAC6 regulates antibody‐dependent intracellular neutralization of viruses via deacetylation of TRIM21. J Biol Chem 295: 14343–14351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B, Li H, Guo M, Wang J, Xu Y, Zou X, Deng R, Li G, Zhu H (2018) TRIM21 promotes innate immune response to RNA viral infection through Lys27‐linked polyubiquitination of MAVS. J Virol 92: e00321‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Bao M, Lu N, Weng L, Yuan B, Liu YJ (2013) The E3 ubiquitin ligase TRIM21 negatively regulates the innate immune response to intracellular double‐stranded DNA. Nat Immunol 14: 172–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Xue J, Jiang X, Gong Y, Gao C, Cao T, Li Q, Bai L, Li Y, Xu G et al (2022) TRIM21 suppresses CHK1 activation by preferentially targeting CLASPIN for K63‐linked ubiquitination. Nucleic Acids Res 50: 1517–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]