

Abstract
Lysosomes are degradative organelles and play vital roles in a variety of cellular processes. Ion channels on the lysosomal membrane are key regulators of lysosomal function. TMEM175 has been identified as a lysosomal potassium channel, but its modulation and physiological functions remain unclear. Here, we show that the apoptotic regulator Bcl‐2 binds to and inhibits TMEM175 activity. Accordingly, Bcl‐2 inhibitors activate the channel in a caspase‐independent way. Increased TMEM175 function inhibits mitophagy, disrupts mitochondrial homeostasis, and increases production of reactive oxygen species (ROS). ROS further activates TMEM175 and thus forms a positive feedback loop to augment apoptosis. In a 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) mouse model of Parkinson's disease (PD), knockout (KO) of TMEM175 mitigated motor impairment and dopaminergic (DA) neuron loss, suggesting that TMEM175‐mediated apoptosis plays an important role in Parkinson's disease (PD). Overall, our study reveals that TMEM175 is an important regulatory site in the apoptotic signaling pathway and a potential therapeutic target for Parkinson's disease (PD).
Keywords: apoptosis, lysosome, Parkinson's disease, potassium channel, TMEM175
Subject Categories: Autophagy & Cell Death, Membranes & Trafficking, Neuroscience
The anti‐apoptotic mitochondrial protein Bcl‐2 regulates lysosomal K+ channel TMEM175. TMEM175 forms a positive feedback loop with ROS to augment apoptosis and promote the death of DA neurons in Parkinson's disease.
Introduction
Lysosomes are acidic vesicular organelles involved in the degradation of most cellular macromolecules and metabolites in eukaryotic cells (Saftig & Klumperman, 2009). In addition to their recycling function, lysosomes have also been found to engage in plasma membrane (PM) repair, immune responses, nutrient sensing, signal transduction, vesicle trafficking, cell death, and various other biological processes (Guicciardi et al, 2004; Settembre et al, 2013; Lawrence & Zoncu, 2019). Lysosomal dysfunction is associated with a variety of diseases, such as lysosomal storage diseases (LSDs), neurodegenerative diseases, and cancers (Platt et al, 2012; Fraldi et al, 2016; Fennelly & Amaravadi, 2017; Tomala & Gabryel, 2017).
Normally, mature lysosomes contain more than 60 hydrolases for degradation. Genetic defects in these enzymes lead to aberrant degradation and excessive accumulation of undegraded intralysosomal substrates, which in turn initiate cell damage and death (Platt et al, 2018). Many lysosomal enzymes are acidic hydrolases whose activity requires a low lysosomal pH (Mindell, 2012). Moreover, lysosomes are important calcium storage sites in cells (Morgan et al, 2011). Therefore, lysosomal ion transport and ion homeostasis are critical for the functions of lysosomes. Transporters and ion channel proteins in lysosomal membranes play fundamental roles in maintaining lysosomal ion homeostasis (Xu & Ren, 2015; Xiong & Zhu, 2016; Sterea et al, 2018). A variety of lysosomal ion channels have been identified, and the dysfunction of several ion channels has been shown to cause certain diseases, such as type IV mucolipidosis, osteopetrosis, and neurodegeneration (Kornak et al, 2001; Kasper et al, 2005; Dong et al, 2008).
Potassium ions are the most abundant cations inside cells and are critical for cellular physiological processes. Lysosomes are immersed in cytosol containing a high concentration of potassium, so the potassium permeability of lysosomal membranes also significantly affects the physiological states of the lysosomes. TMEM175 is a newly identified lysosomal potassium channel with a unique structure enabling ion selection (Cang et al, 2015; Lee et al, 2017). A previous study revealed the important roles of TMEM175 in regulating lysosomal membrane potential, lysosomal pH, and lysosome–autophagosome fusion (Cang et al, 2015); however, information on the organismal functions of this protein and its importance to health and diseases is still lacking. The activity of other lysosomal ion channels has been reported to be regulated by multiple intracellular molecules and signals, including mammalian target of rapamycin (mTOR), adenosine triphosphate (ATP), phosphatidylinositol 4,5‐bisphosphate (PIP2), Mg2+, and p38 (Dong et al, 2010; Cang et al, 2013; Jha et al, 2014), yet the intracellular regulation of TMEM175 is still unclear.
In this study, we discovered an interaction between TMEM175 and apoptosis regulator Bcl‐2, and revealed an important role of TMEM175 in apoptosis. Bcl‐2 binds to and inhibits TMEM175. Bcl‐2 inhibitors and reactive oxygen species (ROS) activated TMEM175, and TMEM175 activation promoted ROS production and apoptosis. In addition, TMEM175 knockout (KO) showed a neuroprotective effect in a 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP)‐induced mouse model of Parkinson's disease (PD), suggesting that TMEM175 plays an important role in neuronal cell death in PD.
Results
Bcl‐2 family‐specific inhibitors activate TMEM175
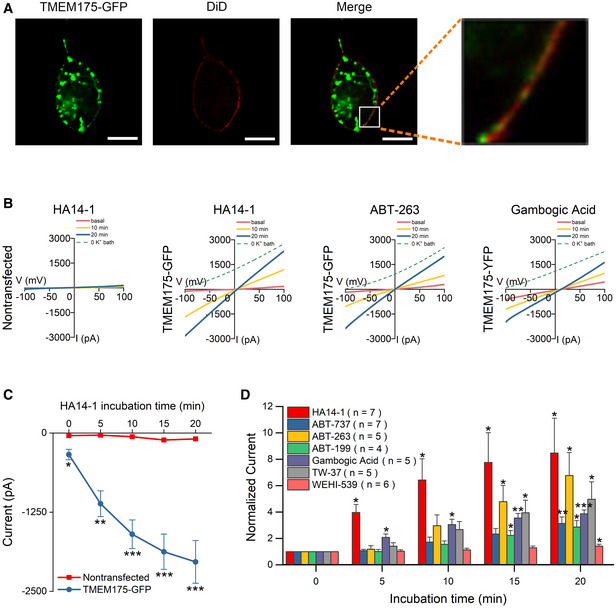
TMEM175 was first identified as an endolysosomal potassium channel using the endolysosomal patch‐clamp technique, which is an ideal methodology for investigation of the electrophysiological properties of organellar ion channels. In this technique, endolysosomes are extracted from cells and recorded. Since the isolated endolysosomes have been removed from the normal intracellular physiological environment, this method is not suitable for exploration of the intracellular signals acting on TMEM175. One feasible way to address this issue is to redirect TMEM175 to the PM and test its activity in intact cells (Vergarajauregui & Puertollano, 2006; Dong et al, 2009). In this study, we noticed that HEK293T cells overexpressing green fluorescent protein (GFP)‐tagged TMEM175 exhibited visible GFP signals on their PMs (Fig 1A). Whole‐cell patch‐clamp detected a linear current under symmetrical K+ conditions in GFP‐tagged and nontagged TMEM175‐transfected cells but not in nontransfected cells, indicating that functional TMEM175 channels existed on the PMs (Figs 1B and C, and EV1A and B).
Figure 1. Inhibitors selective for Bcl‐2 family proteins activate TMEM175 currents.
-
ACo‐localization of green fluorescent protein (GFP)‐tagged TMEM175 (TMEM175‐GFP) and the plasma membrane (PM) marker DiD in HEK293T cells. Scale bars = 10 μm.
-
BRepresentative whole‐cell currents recorded at different time points after application of the Bcl‐2‐selective inhibitors HA14‐1 in nontransfected HEK293T cells and HA14‐1, ABT‐263, and gambogic acid in TMEM175‐GFP‐transfected HEK293T cells. The currents were recorded with ramp protocols (−100 to +100 mV in 1 s, every 10 s, Vh = 0 mV).
-
CCurrent amplitudes measured at −100 mV in the ramp protocol used in (B). n = 5 for nontransfected group. n = 7 for TMEM175‐GFP group.
-
DCurrent amplitudes (at −100 mV) measured at different time points after the application of Bcl‐2‐targeting agents normalized to the basal value (0 min). The HA14‐1 data are replotted from (C) for comparison.
Data information: The data are presented as the mean ± SEM. Statistical significance was analyzed using post hoc tests with analysis of variance (ANOVA) (C) or calculated with two‐sided Student's t‐tests (D), and is indicated with * for P < 0.05, ** for P < 0.01, and *** for P < 0.001. n value means the number of biological replicates made for each data point.
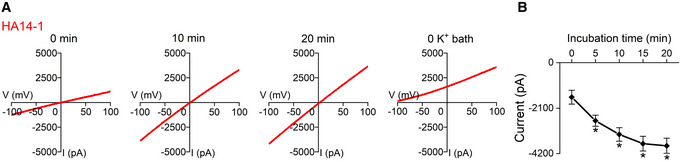
Figure EV1. HA14‐1 activates nontagged TMEM175 current.
-
ARepresentative whole‐cell currents recorded in HEK293T cells expressing nontagged TMEM175.
-
BCurrent amplitudes measured at −100 mV in the ramp protocol used in (A). n = 6.
Data information: The data are presented as the mean ± SEM. Statistical significance was analyzed with two‐sided Student's t‐tests, and is indicated with * for P < 0.05. n value means the number of biological replicates made for each data point.
We next used whole‐cell patch‐clamp to test the effect of numerous compounds known to target intracellular signaling molecules on the current of TMEM175, and found that the Bcl‐2 inhibitor HA14‐1 strongly activated TMEM175. HA14‐1 at a concentration of 10 μM continuously increased the current, which progressed from −339.3 ± 82.0 to −2034.6 ± 336.9 pA in 20 min (Fig 1C). This increase was detected only in TMEM175‐transfected cells, suggesting that activated TMEM175 was responsible rather than endogenously expressed potassium channels (Fig 1B and C). We detected similar current increases in cells expressing nontagged TMEM175, excluding the possibility that the effect of HA14‐1 was due to the GFP tag (Fig EV1A and B).
The linear TMEM175 current could be easily contaminated by a leak current due to the instability of the seal between the pipette tip and the PM. Therefore, we performed a quality control step at the end of each recording by replacing all the K+ in the bath solution (producing a 0 K+ bath) with N‐methyl‐d‐glucamine (NMDG+), a large cation that cannot pass through TMEM175 channels. NMDG+ replacement should eliminate the vast majority of the inward current; the persistence of the inward current indicates a leaky seal. Recordings for which leaky seals were detected were discarded.
HA14‐1 is a selective inhibitor of antiapoptotic Bcl‐2 proteins. It induces apoptosis by antagonizing Bcl‐2 and translocating proapoptotic proteins from the cytosol to the mitochondria. HA14‐1 may activate TMEM175 by directly binding to the channel protein or by inhibiting Bcl‐2. To verify the mechanism, we tested other specific inhibitors of antiapoptotic Bcl‐2 proteins, including ABT‐737 (10 μM), ABT‐263 (1 μM), ABT‐199 (1 μM), gambogic acid (1 μM), TW‐37 (10 μM), and WEHI‐539 (10 μM). All these inhibitors activated TMEM175 currents, indicating the involvement of the Bcl‐2 family in the regulation of TMEM175 (Fig 1D).
Bcl‐2 binds to TMEM175 and regulates its function through caspase‐independent way
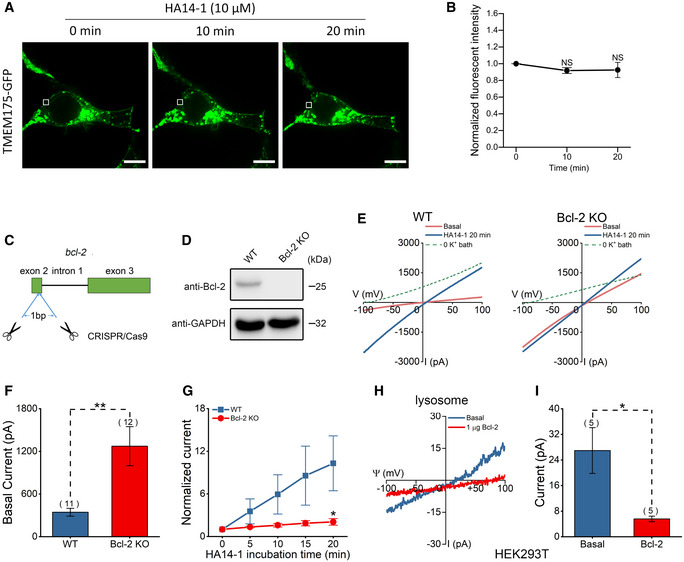
A whole‐cell current indicates an overall movement of K+ ions across the PM through all opened channels. Changes in whole‐cell currents may result from changes in total channel numbers or channel activity. In TMEM175‐overexpressing cells, only a small portion of channel proteins were localized to the PM. Upon monitoring the GFP fluorescence density on the PM, we did not detect a considerable PM translocation of TMEM175 after HA14‐1 application (Fig EV2A and B). This result suggested that HA14‐1 potentiated TMEM175 current by increasing the channel activity.
Figure EV2. HA14‐1 activates TMEM175 through Bcl‐2 without inducing translocation of TMEM175 to the plasma membrane (PM).
-
ALocalization of C‐terminally green fluorescent protein (GFP)‐tagged TMEM175 before (0 min) and after incubation with HA14‐1; 10 and 20 min are the incubation times. The white boxes indicate TMEM175‐expressing PM regions. Scale bars = 10 μm.
-
BFluorescence intensities of selected PM regions indicating the expression of TMEM175. n = 4 (four PM regions selected from different cells in the same experiment).
-
CSchematic diagram showing the knockout (KO) strategy of Bcl‐2 in HEK293T cells.
-
DThe knockout (KO) of Bcl‐2 protein was detected by Western blotting.
-
ERepresentative whole‐cell currents recorded in wild‐type (WT) (left) and Bcl‐2 KO (right) HEK293T cells transfected with TMEM175 at 0 min (basal) and 20 min after bath application of 10 μM HA14‐1. The currents were recorded with ramp protocols (−100 to +100 mV in 1 s, every 10 s, Vh = 0 mV).
-
FAmplitudes of basal currents measured at −100 mV in the ramp protocol used in (E).
-
GCurrent amplitudes measured at different time points after the application of HA14‐1 normalized to the basal values (0 min).
-
HRepresentative endogenous TMEM175 lysosomal currents recorded in HEK293T cells with or without 1 μg Bcl‐2 application in bath solution.
-
IStatistics of the currents in (H).
Data information: The data are presented as the mean ± SEM. Statistical significance was analyzed with two‐sided Student's t‐tests, and is indicated with NS for not significant (P > 0.05), and * for P < 0.05, ** for P < 0.01. In panel (B), n value means the number of technical replicates. In other panels, n value means the number of biological replicates made for each data point.
Inhibition of antiapoptotic Bcl‐2 proteins should activate the caspase cascade and induce apoptosis. However, the pancaspase inhibitor Z‐VAD‐FMK (zVAD; carbobenzoxy‐valyl‐alanyl‐aspartyl‐[O‐methyl]‐fluoromethylketone) failed to prevent TMEM175 from being activated by HA14‐1 in HEK293T cells (Fig 2A and B), indicating that a noncaspase mechanism was involved. To further validate this point, we generated Bax/BAK1 double knockout (dKO) HEK293T cells using the CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR‐associated protein 9) technique (Fig 2C and D). Whole‐cell recordings revealed that 10 μM HA14‐1 could still activate TMEM175 channels in Bax/BAK1 dKO cells overexpressing TMEM175 (Fig 2E and F).
Figure 2. Bcl‐2 binds to and regulates TMEM175 through caspase‐independent way.
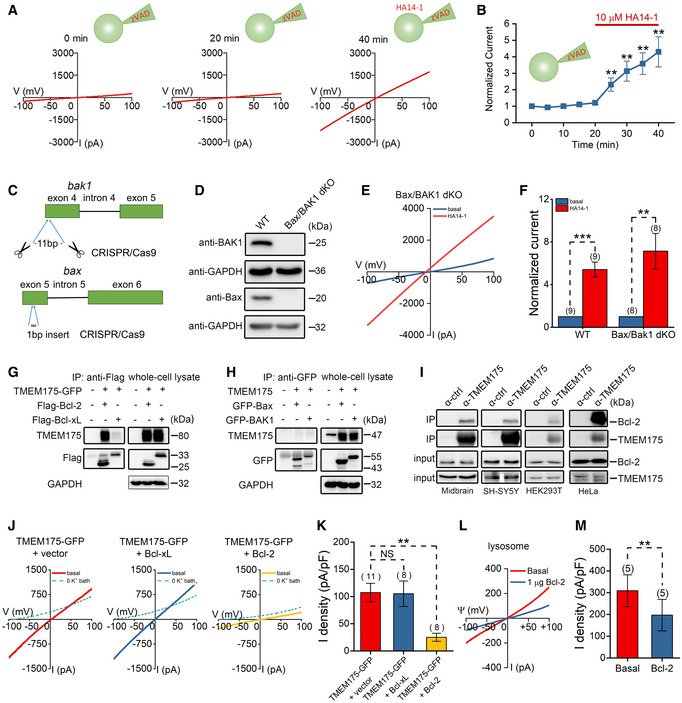
-
ARepresentative current traces recorded with the pan‐caspase inhibitor Z‐VAD‐FMK (zVAD; carbobenzoxy‐valyl‐alanyl‐aspartyl‐[O‐methyl]‐fluoromethylketone) at a concentration of 10 μM in pipette solution. The basal currents were recorded for 20 min, and then 10 μM HA14‐1 was added to the bath solution.
-
BCurrent amplitudes (at −100 mV) normalized to the basal values (0 min). The red solid line indicates the time frame for HA14‐1 application. n = 8.
-
CSchematic diagram showing the double knockout (dKO) strategy of Bax/BAK1 in HEK293T cells.
-
DBax/BAK1 protein levels were detected by Western blotting.
-
ERepresentative TMEM175 whole‐cell currents recorded in Bax/BAK1 double knockout (dKO) cells overexpressing TMEM175 with 10 μM HA14‐1 bath application.
-
FCurrent amplitudes measured at −100 mV at 20 min after application of HA14‐1 normalized to the basal value (0 min).
-
G, HResults of immunoblotting with immunoprecipitates (IPs) or whole‐cell lysates from HEK293T cells transfected with different constructs as indicated.
-
IImmublotting results with IP or input (whole‐cell lysates) from mouse midbrain, SH‐SY5Y, HEK293T, or HeLa cells as indicated. The α‐ctrl represents that the magnetic beads used in IP were not preincubated with any antibody.
-
JRepresentative TMEM175 whole‐cell currents recorded in HEK293T cells expressing TMEM175 co‐transfected or not co‐transfected with Bcl‐2.
-
KStatistics of the current densities in (J).
-
LRepresentative TMEM175 lysosomal currents recorded in HEK293T cells expressing TMEM175 with or without Bcl‐2 application in bath solution.
-
MStatistics of the current densities in (L).
Data information: The data are presented as the mean ± SEM. Statistical significance was calculated with two‐sided Student's t‐test, and is indicated with NS for not significant (P > 0.05), ** for P < 0.01, and *** for P < 0.001. n value means the number of biological replicates made for each data point.
Another possibility is that Bcl‐2 proteins regulate TMEM175 activity by directly binding to the channel. Although Bcl‐2 family proteins are primarily located in mitochondria, they can still physically contact lysosomal membrane proteins (Wong et al, 2018). In our coimmunoprecipitation (co‐IP) assay, we observed significant binding between TMEM175 and Bcl‐2, and a very weak binding between TMEM175 and Bcl‐xL; however, we observed no binding between TMEM175 and the proapoptotic protein Bax or BAK1 (Fig 2G and H). In cells co‐expressing mCherry‐tagged TMEM175 and cyan fluorescent protein (CFP)‐tagged Bcl‐2 or Bcl‐xL, we also observed a direct contact between mCherry and CFP signals (Appendix Fig S1), indicating that the two proteins may interact at mitochondria‐lysosome contact sites. Although the interaction between TMEM175 and Bcl‐xL was barely detectable, the Bcl‐xL‐specific inhibitor WEHI‐539 at 10 μM effectively activated TMEM175, probably through nonspecific inhibition of Bcl‐2. The IC50 of WEHI‐539 to Bcl‐xL is 1.1 nM. Its selectivity for Bcl‐xL is 500‐fold over Bcl‐2, which means that the IC50 for Bcl‐2 is also at the sub‐micromolar level (Lessene et al, 2013). We also found that 10 nM WEHI‐539 failed to activate TMEM175 (Appendix Fig S2), indicating that it does not act through Bcl‐xL. Endogenous binding of TMEM175 and Bcl‐2 can also be detected in mouse midbrain, SH‐SY5Y, HEK293T, and HeLa cells (Fig 2I). Next, we transfected Bcl‐2 or Bcl‐xL into HEK293T cells and recorded whole‐cell TMEM175 currents. Interestingly, overexpression of Bcl‐2, but not Bcl‐xL, suppressed the TMEM175 currents (Fig 2J and K). We then knocked out Bcl‐2 protein in HEK293T cells using the CRISPR/Cas9 technique (Fig EV2C and D) and overexpressed TMEM175 in wild‐type (WT) or Bcl‐2 KO cells. The basal whole‐cell current of TMEM175 was higher in Bcl‐2 KO cells (Fig EV2E and F), indicating a release of inhibition on TMEM175. Furthermore, the activation of TMEM175 by 10 μM HA14‐1 was greatly reduced in Bcl‐2 KO cells (Fig EV2G). Nonetheless, HA14‐1 still induced approximately 2‐fold activation in Bcl‐2 KO cells, indicating that HA14‐1 activates TMEM175 through both Bcl‐2‐dependent and Bcl‐2‐independent mechanisms. This may also be the reason why HA14‐1 works better than other Bcl‐2 inhibitors in activating TMEM175. Moreover, using lysosomal patch‐clamp performed in HEK293T cells overexpressing TMEM175, we found that application of Bcl‐2 protein to the bath solution also inhibited the lysosomal TMEM175 currents significantly (Fig 2L and M). In nontransfected HEK293T cells, Bcl‐2 protein decreased endogenous lysosomal TMEM175 currents likewise (Fig EV2H and I).
Bcl‐2 regulates TMEM175 through physical contact
Based on the previously reported crystal structure of Bcl‐2 and cryo‐EM structure of TMEM175 (Birkinshaw et al, 2019; Oh et al, 2020), we predicted the binding interface of the two proteins using docking software ZDOCK. Two main binding modes were revealed by analyzing top three predicted complexes of Bcl‐2 bound with open‐ or closed‐state TMEM175, respectively. Binding mode I was found in complexes containing open‐ (Fig EV3) or closed‐state TMEM175 (Fig 3A, left). Hydrogen bond between sidechain R377 of TMEM175 and E135 of Bcl‐2 contributes the main interaction. Binding mode II was only found in complexes with closed‐state TMEM175 (Fig 3A, right). Backbone V145 of TMEME175 and sidechain N172 of Bcl‐2 form hydrogen bond instead.
Figure EV3. Predicted binding mode I for Bcl‐2 bound with open‐state TMEM175.
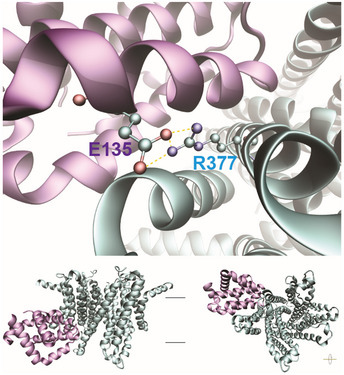
The protein backbone is shown as cartoon and colored in cyan for TMEM175 and purple for Bcl‐2. Pairs of residues forming hydrogen bond are displayed in sticks and colored by atom type. Hydrogen bonds are displayed in yellow dashed lines. Lower: the overhead and lateral views of the bound complexes.
Figure 3. Bcl‐2 regulates TMEM175 through physical contact.
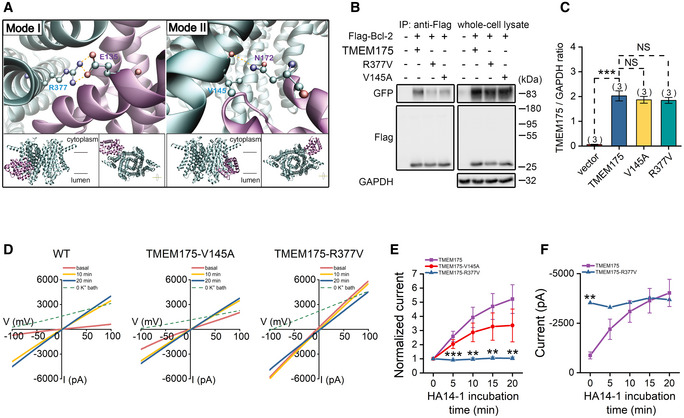
-
APredicted binding mode I (left) and mode II (right) for Bcl‐2 (PDB ID: 6O0K) bound with TMEM175 (PDB ID: 6WC9). The insert shows the overhead and lateral views of the bound complexes. The protein backbone is shown as cartoon and colored in cyan for TMEM175 and purple for Bcl‐2. Pairs of residues forming hydrogen bond are displayed in sticks and colored by atom type. Hydrogen bonds are displayed in yellow dashed lines.
-
BResults of immunoblotting with immunoprecipitates (IPs) or whole‐cell lysates from cells transfected with Bcl‐2 and TMEM175 or its mutants TMEM175‐V145A, TMEM175‐R377V, as indicated.
-
CStatistics of the relative TMEM175 protein levels normalized to GAPDH (glyceraldehyde‐3‐phosphate dehydrogenase) in vector, TMEM175, TMEM175‐V145A, or TMEM175‐R377V overexpressing HEK293T cells.
-
DRepresentative whole‐cell currents recorded at different time points after application of the Bcl‐2‐selective inhibitors HA14‐1 in TMEM175, TMEM175‐V145A, or TMEM175‐R377V overexpressing HEK293T cells.
-
ENormalized currents (at −100 mV) to the basal value (0 min) in the ramp protocol used in (D). n = 5 for TMEM175 group. n = 6 for TMEM175‐V145A group. n = 6 for TMEM175‐R377V group.
-
FCurrent amplitudes of TMEM175 and TMEM175‐R377V‐transfected HEK293T cells measured at −100 mV in the ramp protocol used in (D). n = 5 for TMEM175 group. n = 6 for TMEM175‐R377V group.
Data information: The data are presented as the mean ± SEM. Statistical significance was calculated with two‐sided Student's t‐tests (C and F) or analyzed using post hoc tests with analysis of variance (ANOVA) (E), and is indicated with NS for not significant (P > 0.05), ** for P < 0.01, and *** for P < 0.001. n value means the number of biological replicates made for each data point.
In order to confirm these predicted binding modes, we introduced point mutations V145A and R377V to TMEM175, respectively. In our co‐IP assay, weakened bindings between TMEM175 mutants and Bcl‐2 were expectedly detected (Fig 3B). The decrease in co‐IP signals was not caused by impairment of protein level, because the expression of WT TMEM175 and mutants was basically the same (Fig 3C). Both TMEM175 mutants could mediate whole‐cell potassium current, but their sensitivity to the Bcl‐2 inhibitor HA14‐1 is significantly different from that of wild‐type TMEM175. The V145A mutant was less activated by HA14‐1, while the R377V mutant was not activated by HA14‐1 at all (Fig 3D and E). Interestingly, the basal current of TMEM175–R377V was much larger than that of wild‐type TMEM175, and was comparable to the current of TMEM175 after 20 min treatment of 10 μM HA14‐1 (Fig 3F). These results indicate that Bcl‐2 dominantly inhibits TMEM175 by physical binding. The R377V mutation of TMEM175 relieves the binding and consequently eliminated the inhibition.
ROS regulate TMEM175 through positive feedback
EM20‐25 is an analog of HA14‐1 that disrupts Bcl‐2/Bax interaction and activates caspase‐9 in cells overexpressing Bcl‐2. Unlike HA14‐1, EM20‐25 does not affect mitochondrial respiration (Milanesi et al, 2006). In HEK293T cells, after 20 μM EM20‐25 was applied, the whole‐cell TMEM175 current exhibited only a slight tendency to increase (Fig 4A), in contrast to the strong effect exhibited after HA14‐1 treatment (Fig 1C and D). A serious consequence of mitochondrial dysfunction is a dramatic increase in ROS, therefore we assumed that ROS also contribute to the activation of TMEM175. In accordance with this hypothesis, we found that perfusion with 200 μM H2O2 directly increased the TMEM175 current by ~2.8 times in 20 min (Fig 4B). Adding 1 mM antioxidant GSH (reduced glutathione) into pipette solution largely reduced the activation effect of HA14‐1 (Fig 4C and D), further indicating that the activation of TMEM175 is related to the increase of ROS levels. 1‐Methyl‐4‐phenylpyridinium (MPP+) and rotenone are both classic compounds that cause ROS and apoptosis. Changes in the interaction and localization of Bcl‐2 family proteins occur during mitochondrial apoptosis, which may also affect the regulation of TMEM175. Therefore, we induced apoptosis with these compounds and tested their effects on TMEM175 currents. Both compounds, MPP+ and rotenone, significantly potentiated TMEM175 currents (Fig 4E and F, and EV4A and B). The activation mediated by MPP+ could not be abolished by the pancaspase inhibitor Z‐VAD‐FMK (zVAD) but was largely abolished by GSH (Fig 4E and F). Moreover, bath application of H2O2 also significantly activated lysosomal TMEM175 currents (Fig 4G and H). These results confirmed that ROS play critical roles in the activation of TMEM175. Upon using CM‐H2DCFDA (5‐(and‐6)‐chloromethyl‐2'‐7'‐dichlorodihydrofluorescein diacetate) as a fluorescent ROS indicator, we also observed an interesting phenomenon in which overexpression of TMEM175 increased ROS levels (Fig 4I and J, and EV4C and D). Thus, TMEM175 and ROS regulate each other through positive feedback.
Figure 4. Reactive oxygen species (ROS) regulate TMEM175 through positive feedback.
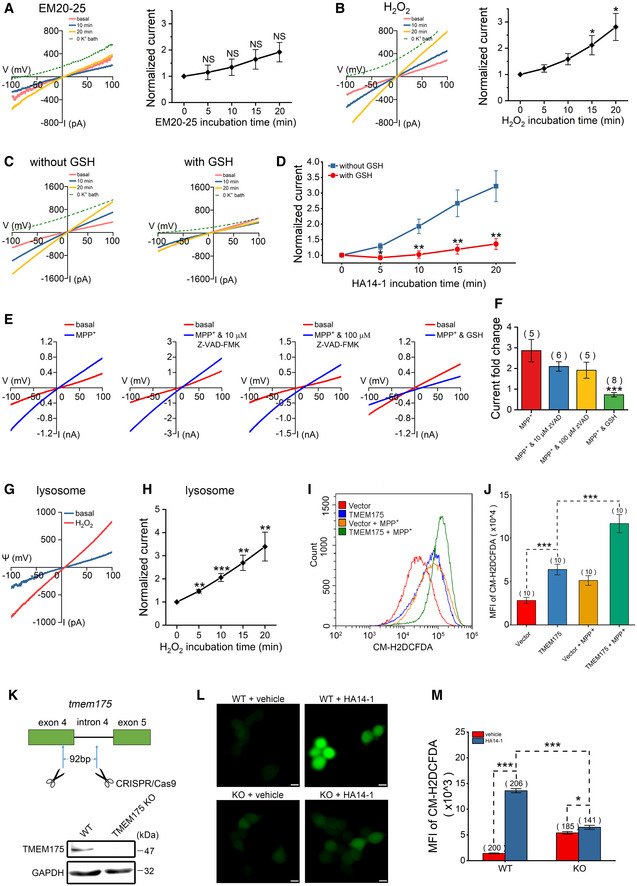
-
A–FWhole‐cell recording was performed in HEK293T cells transfected with TMEM175‐GFP (green fluorescent protein). (A) (Left) Representative current traces showing the effects of 20 μM EM20‐25 in the bath solution. (Right) Statistics of the currents normalized to the basal values (0 min). n = 6. (B) (Left) Representative current traces showing the effects of 200 μM H2O2 in the bath solution. (Right) Statistics of the currents normalized to the basal values (0 min). n = 5. (C) Representative current traces showing the effects of GSH (reduced glutathione; 1 mM in pipette solution) on HA14‐1 (1 μM added to bath)‐induced activation of TMEM175 currents. (D) Statistics of the currents in (C) normalized to the basal values (0 min). n = 6 for each group. (E) Representative currents recorded with pipette solutions containing 100 μM MPP+ and 10 μM Z‐VAD‐FMK (zVAD), 100 μM Z‐VAD‐FMK (zVAD) or 1 mM GSH, as indicated. The red traces indicate the basal currents. The blue traces indicate the currents recorded 20 min after the initiation of whole‐cell mode. (F) Quantification of the current fold changes in (E) normalized to the basal currents.
-
GRepresentative TMEM175 lysosomal current traces showing the effects of 500 μM H2O2 in the bath solution. The blue trace indicates basal current. The red trace indicates the current recorded 20 min after application of H2O2.
-
HStatistics of current fold changes in (G) normalized to basal currents (0 min). n = 7.
-
I, JROS levels in HEK293T cells overexpressing TMEM175 or empty vectors pretreated or not pretreated with 500 μM MPP+ (1‐methyl‐4‐phenylpyridinium) for 1 h. ROS were detected using the CM‐H2DCFDA (5‐(and‐6)‐chloromethyl‐2'‐7'‐dichlorodihydrofluorescein diacetate) dye by flow cytometry (I), and the quantified results are presented in (J).
-
K(Upper) schematic diagram showing the strategy of TMEM175 KO in HEK293T cells. As much as 92 bp in exon 4 and intron 4 were removed from the tmem175 locus. (Lower) TMEM175 levels in the WT or TMEM175 KO cells were detected by Western blotting.
-
LCM‐H2DCFDA (green) indicating cellular ROS levels of WT or TMEM175 KO HEK293T cells pretreatment with vehicle or 10 μM HA14‐1 for 20 min. Scale bars = 10 μm.
-
MStatistics of CM‐H2DCFDA fluorescence intensity of (L). The n values represent the cell numbers counted within the same experiment.
Data information: The data are presented as the mean ± SEM. Statistical significance was calculated with two‐sided Student's t‐tests (A, B, F, H, J, M) or analyzed using post hoc tests with analysis of variance (ANOVA) (D), and is indicated with NS for not significant (P > 0.05), * for P < 0.05, ** for P < 0.01, and *** for P < 0.001. In panel (M), n value means the number of technical replicates. In other panels, n value means the number of biological replicates made for each data point.
Figure EV4. Activation of TMEM175 by rotenone.
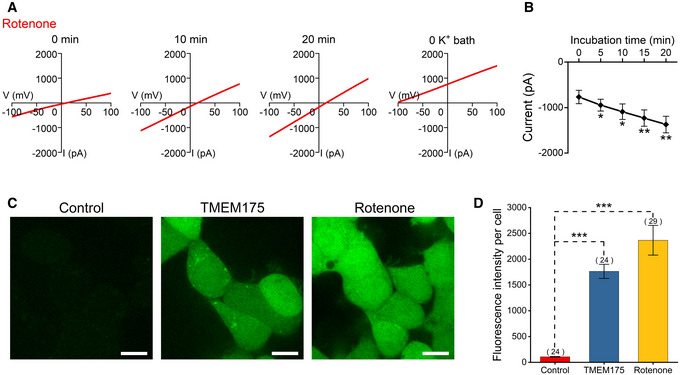
-
ARepresentative current traces showing the effects of 10 μM rotenone in the bath solution.
-
BQuantification of the current amplitudes measured at −100 mV in the ramp protocol used in (A). n = 8.
-
CCM‐H2DCFDA (5‐(and‐6)‐chloromethyl‐2'‐7'‐dichlorodihydrofluorescein diacetate) (green) indicating cellular reactive oxygen species (ROS) levels of HEK293T cells overexpressing TMEM175 or pretreatment of 10 μM rotenone for 1 h. Scale bars = 10 μm.
-
DStatistics of CM‐H2DCFDA fluorescence intensity of (C). The n values represent the cell number counted within the same experiment.
Data information: The data are presented as the mean ± SEM. Statistical significance was analyzed with two‐sided Student's t‐tests, and is indicated with * for P < 0.05, ** for P < 0.01, and *** for P < 0.001. In panel (B), n value means the number of biological replicates made for each data point. In panel (D), n value means the number of technical replicates.
To further verify the relationship between TMEM175 and increasing ROS, we generated TMEM175 knockout (KO) HEK293T cell line by using the CRISPR/Cas9 technique (Fig 4K). Bath application of HA14‐1 (10 μM, 20 min) drastically increased ROS level in WT cells, but only showed marginal effect in TMEM175 KO cells (Fig 4L and M). This result further indicated that TMEM175 plays an important role in ROS regulation.
TMEM175 overexpression impairs mitochondrial function
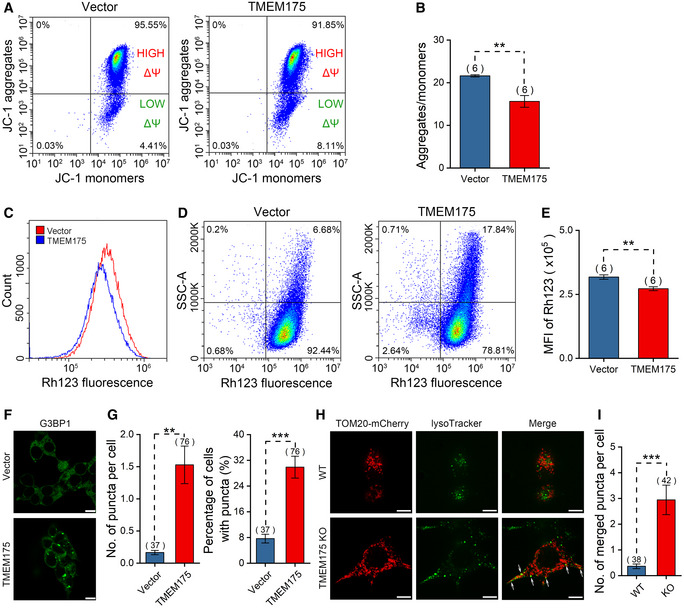
Mitochondria are the major source of ROS. Excessive generation of ROS caused by upregulated TMEM175 implies changes in mitochondrial function. JC‐1 and Rhodamine 123 dyes are frequently used for monitoring mitochondrial membrane potential (ΔΨ) to assess mitochondrial function. In TMEM175‐overexpressing HEK293T cells, we observed a decreased ratio of red fluorescent JC‐1 aggregates to green fluorescent JC‐1 monomers (Fig 5A and B), as well as a decreased Rhodamine 123 fluorescence intensity (Fig 5C–E). These results suggested that overexpression of TMEM175 caused depolarization of mitochondrial membrane potential. In addition, we found that the side scatter signal (SSC) of TMEM175‐overexpressing cells increased, indicating a higher intracellular complexity. Intracellular granular structure is an important contributor to the intracellular complexity. TMEM175‐induced ROS generation may promote the generation of stress granules (SGs). Thus, we labeled SGs with antibody against a SG‐specific marker protein G3BP1 (Ras‐GTPase‐activating protein (GAP)‐binding protein 1) and found more SGs in TMEM175‐overexpressing cells (Fig 5F and G).
Figure 5. TMEM175 overexpression impairs mitochondrial membrane potential and mitophagy in HEK293T cells.
-
AFlow cytometric analysis of JC‐1 staining in HEK293T cells transfected with mock vector or TMEM175. Horizontal axis and vertical axis represent fluorescence intensities of JC‐1 monomers and JC‐1 aggregates, respectively.
-
BStatistics of the ratios of JC‐1 aggregates/monomers in (A).
-
CFlow cytometric analysis of Rhodamine 123 staining in HEK293T cells transfected with mock vector or TMEM175.
-
DRhodamine 123 fluorescence of cells in (C) is plotted against side scatter signal (SSC).
-
EStatistics of Rhodamine123 mean fluorescence intensities in (C).
-
FFluorescence of G3BP1 (Ras‐GTPase‐activating protein (GAP)‐binding protein 1) excited with 488 nm in control (upper) and TMEM175‐overexpressing (lower) cells. Scale bars = 10 μm.
-
GStatistics of the numbers of puncta per cell (left) and the percentage of cells with puncta in (F). We counted 547 cells in Vector group from 37 different fields of view and 332 cells in TMEM175 group from 76 different fields of view. n value represents the number of different fields of view randomly selected in the same imaging experiment.
-
HCo‐localization of mitochondrial indicator TOM20‐mCherry and lysosomal indicator LysoTracker in WT (upper row) or TMEM175 KO (lower row) HEK293T cells. Co‐localization of mitochondrion and lysosome is observed as a yellow color in the merged image (marked by white arrows). Scale bars = 10 μm.
-
IThe number of merged puncta per cell in (H). We counted 139 cells in wild‐type (WT) group from 38 different fields of view and 148 cells in TMEM175 KO group from 42 different fields of view. n value represents the number of different fields of view randomly selected in the same imaging experiment.
Data information: The data are presented as the mean ± SEM. Statistical significance was calculated with two‐sided Student's t‐test, and is indicated with ** for P < 0.01, and *** for P < 0.001. In panels (G and I), n value means the number of technical replicates. In other panels, n value means the number of biological replicates made for each data point.
TMEM175 has previously been shown to inhibit autophagic flow (Cang et al, 2015), therefore impaired mitophagy may be the cause of mitochondrial damage and increased intracellular complexity. To verify this idea, we labeled mitochondria and lysosomes with Tom20‐mCherry and LysoTracker green, respectively. The co‐localization of Tom20 and lysosomes represented autophagic mitochondria delivered to the lysosome for degradation. We observed more co‐localization in TMEM175 KO cells, suggesting that TMEM175 inhibits mitophagy (Fig 5H and I).
TMEM175 regulates apoptosis
Both Bcl‐2 and ROS are key regulators of apoptosis. Their interaction with TMEM175 incorporates this lysosomal ion channel into the complex network of apoptosis. To verify this, we incubated WT and TMEM175 KO cells with 2 mM MPP+ for 24 h. The results showed that MPP+ induced apoptosis in WT cells, but not in KO cells (Fig 6A and B). Conversely, overexpression of TMEM175 significantly increased the proportion of apoptotic cells. Application of MPP+ or rotenone in TMEM175‐overexpressing cells did not further increase the apoptosis levels (Figs 6C and D, and EV5A–E), indicating that MPP+/rotenone and TMEM175 may act through the same downstream ROS pathway. Meanwhile, our patch‐clamp data showed that extracellular application of 2 mM MPP+ could activate TMEM175 current (Fig EV5F and G).
Figure 6. TMEM175 regulates apoptosis.
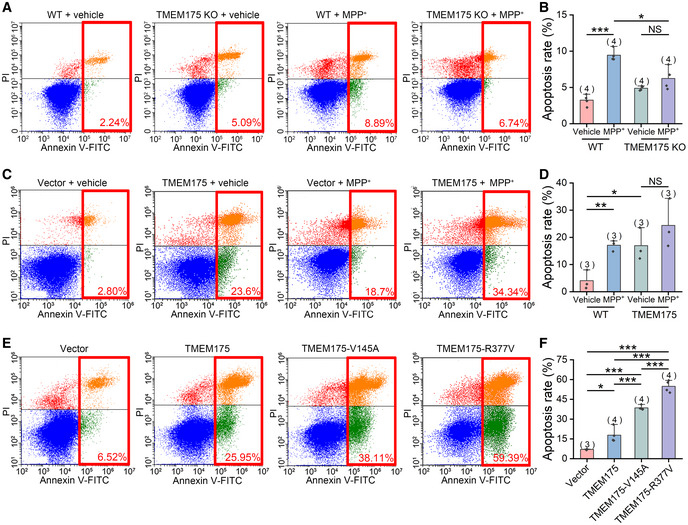
-
AWild‐type (WT) or TMEM175 KO HEK293T cells were treated with vehicle or 2 mM MPP+ (1‐methyl‐4‐phenylpyridinium) for 24 h. Apoptotic cell death was detected by Annexin V–fluorescein isothiocyanate (FITC)/propidium iodide (PI) double staining followed by flow cytometric analysis.
-
BStatistics of the apoptosis rates in (A). Four independent experiments were performed.
-
CApoptosis detection of HEK293T cells transfected with empty vectors or nontagged TMEM175 treated with vehicle or 2 mM MPP+.
-
DStatistics of the apoptosis rates in (C). Three independent experiments were performed.
-
EApoptosis detection of vector, TMEM175, TMEM175‐V145A, or TMEM175‐R377V‐transfected HEK293T cells.
-
FStatistics of the apoptosis rates in (E). Four independent experiments were performed.
Data information: The data are presented as the mean ± SEM. Statistical significance was analyzed with two‐sided Student's t‐tests, and is indicated with NS for not significant (P > 0.05), * for P < 0.05, ** for P < 0.01, and *** for P < 0.001.
Figure EV5. Rotenone and MPP+ (1‐methyl‐4‐phenylpyridinium) regulate TMEM175 and apoptosis.
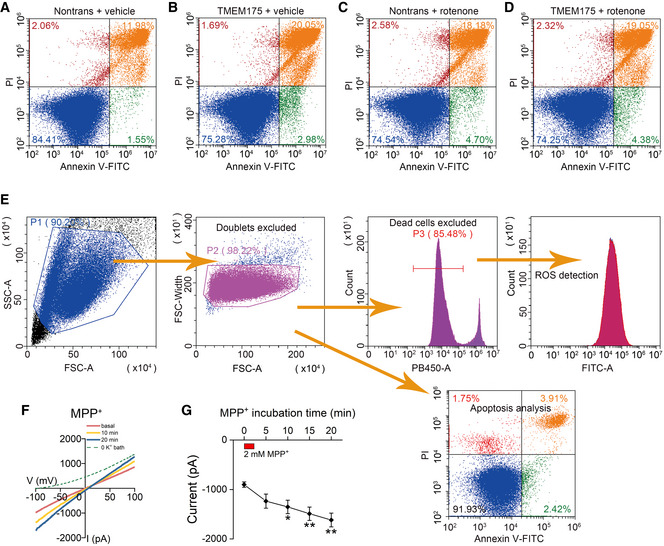
-
A–DNontransfected (A and C) and TMEM175 transfected (B and D) HEK293T cells were treated with vehicle (A and B) or 10 μM rotenone (C and D) for 24 h. The apoptotic cell death was detected by Annexin V–fluorescein isothiocyanate (FITC)/propidium iodide (PI) double staining followed by flow cytometric analysis.
-
EGating strategy of (A–D) and flow cytometry of Figs 4I, 5A, C and D, and 6A, C and E. The initial cell population gating (forward scatter (FSC)‐Area vs. side scatter (SSC)‐Area) was adopted to remove cell debris. Secondary cell population gating (FSC‐Area vs. FSC‐Width) was applied to exclude doublets. For the detection of intracellular reactive oxygen species (ROS), 4',6‐diamidino‐2‐phenylindole (DAPI) staining was used to exclude dead cells. The same gating strategy was applied to all samples analyzed simultaneously.
-
FRepresentative current traces recorded at different time points after transient bath application (2 min) of 2 mM MPP+.
-
GQuantification of the current amplitudes in (F) (measured at −100 mV). The red rectangle indicates the time frame for MPP+ application. n = 7.
Data information: The data are presented as the mean ± SEM. Statistical significance was analyzed with two‐sided Student's t‐tests, and is indicated with * for P < 0.05, and ** for P < 0.01. n value means the number of biological replicates made for each data point.
Previous results showed that V145A and R377V mutations in TMEM175 attenuated the binding between TMEM175 and Bcl‐2, and increased TMEM175 activity (Fig 3E and F). In our apoptosis detection, we found that in comparison with WT TMEM175, the V145A and R377V mutations intensified the degree of apoptosis, of which TMEM175‐R377V has greater effect (Fig 6E and F).
TMEM175 KO shows neuroprotective effects in an MPTP‐induced PD mouse model
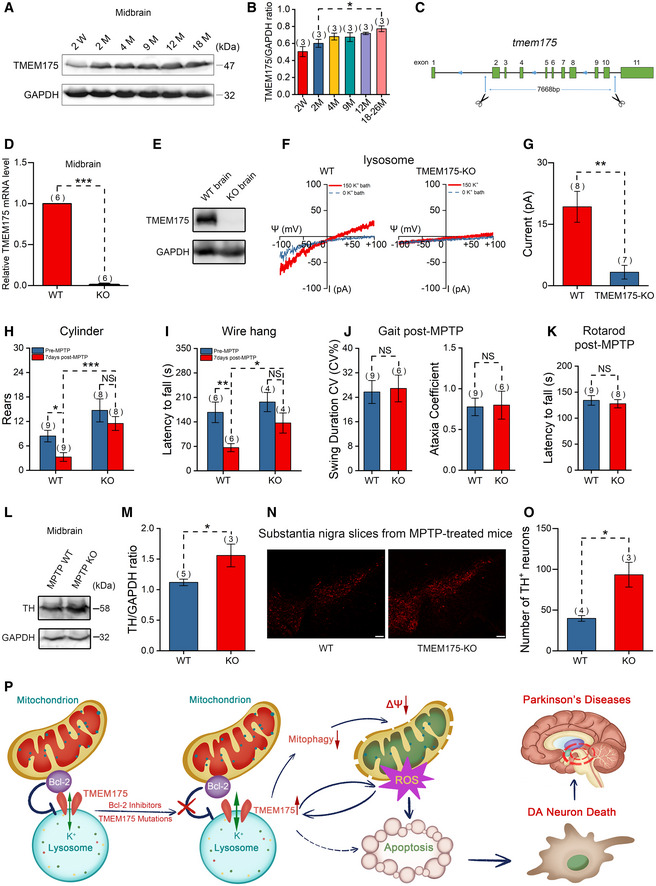
TMEM175 is widely expressed in many eukaryotic cell types. Apoptotic cell death resulting from increased TMEM175 activity or protein levels could occur in many tissues and organs but will cause the most serious consequences in tissues lacking sufficient regenerative ability, e.g., the nervous system. In the mouse midbrain, TMEM175 expression increased during aging (Fig 7A and B), suggesting that TMEM175 may play an important role in brain aging or neurodegeneration. To verify this possibility, we generated TMEM175 KO mice by using the CRISPR/Cas9 technique. Two single guide RNAs (sgRNAs) targeting the first and tenth introns were used, resulting in deletion of a large genome segment including exons 2–10 (Fig 7C). In KO mice brain, the messenger RNA (mRNA) and protein level of TMEM175 was eliminated (Fig 7D and E, and Appendix Fig S3). Moreover, the lysosomal potassium current was greatly reduced in bone marrow macrophages from KO mice (Fig 7F and G). These data proved a successful knockout of TMEM175. Surprisingly, the TMEM175 KO mice were viable and fertile and had no obvious morphological defects.
Figure 7. TMEM175 knockout (KO) mitigates 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP)‐induced Parkinson's disease (PD) symptoms in mice.
-
ARepresentative immunoblots for TMEM175 expression levels in the midbrains of mice of different ages (W: weeks; M: months).
-
BStatistics of the relative TMEM175 protein levels normalized to GAPDH (glyceraldehyde‐3‐phosphate dehydrogenase) in (A).
-
CSchematic diagram showing the KO strategy of TMEM175 in mice. Introns 2–10 were removed from the TMEM175 locus.
-
DTMEM175 messenger RNA (mRNA) levels in the midbrains of wild‐type (WT) or TMEM175 KO mice were detected by reverse transcription–quantitative polymerase chain reaction (RT‐qPCR) using SYBR Green PCR Master Mix.
-
EThe knockout of TMEM175 in mouse was verified by Western blot of brain tissues.
-
FRepresentative TMEM175 basal lysosomal currents recorded in macrophages of WT or TMEM175 KO mice. Ψ is the lysosomal membrane potential (defined as Vcytosol – Vlumen). The currents were elicited with ramp voltage protocols (−100 to +100 mV in 1 s, Vh = 0 mV).
-
GStatistics of the current amplitudes of (F).
-
H, IQuantification of rears in the cylinder (H) and the latency to fall from a wire hang apparatus (I) pre‐ and post‐MPTP treatment.
-
JSwing duration coefficient of variation (CV) (CV%) (left panel) and Ataxia coefficient (right panel) in Gait tests 7 days post‐MPTP.
-
KLatency to fall in rotarod tests 7 days post‐MPTP.
-
LImmunoblots of the tyrosine hydroxylase (TH) levels post‐MPTP treatment in WT and TMEM175 KO mouse midbrains.
-
MQuantification of the relative TH levels normalized to GAPDH post‐MPTP.
-
NTH immunofluorescent labeling on substantia nigra slices of WT or TMEM175 KO mice post‐MPTP treatment. Scale bars = 100 μm.
-
OStatistics of the TH‐positive neurons in (N).
-
PAn overview model illustrating the presumed roles of TMEM175 in apoptosis and PD. Antiapoptotic mitochondrial protein Bcl‐2 binds directly to and regulates lysosomal potassium channel TMEM175. Bcl‐2 inhibitors and mutations of the binding sites release the inhibition and thus activate TMEM175. Activation of TMEM175 inhibits mitophagy, impairs mitochondrial function, and consequently increases reactive oxygen species (ROS) production. ROS activates TMEM175 through a positive feedback way to augment apoptosis. TMEM175‐mediated apoptosis promotes the death of DA neurons in PD.
Data information: The data are presented as the mean ± SEM. Statistical significance was analyzed with two‐sided Student's t‐tests, and is indicated with NS for not significant (P > 0.05), * for P < 0.05, ** for P < 0.01, and *** for P < 0.001. n value means the number of biological replicates made for each data point.
Source data are available online for this figure.
Our previous data showed that MPP+ and rotenone could activate TMEM175. Both rotenone and the prodrug of MPP+, MPTP, have long been used to induce symptoms of PD in rodent models. Here, we also used an MPTP‐induced PD model to evaluate the function of TMEM175 at the animal level. In a cylinder test, systemic administration of MPTP caused significant reductions in rear times in wild‐type (WT) mice but not in KO mice (Fig 7H). TMEM175 KO mice also performed better in the wire hang test than WT mice after MPTP induction (Fig 7I). However, no difference was detected between WT and TMEM175 KO mice in gait and rotarod tests post‐MPTP application (Fig 7J and K). There were fewer dopaminergic (DA) neurons in the midbrain in WT mice than in KO mice after MPTP treatment, as shown by analysis of the protein levels of tyrosine hydroxylase (TH; Fig 7L and M) and TH‐positive DA neurons in the substantia nigra (Fig 7N and O). Thus, KO of TMEM175 exerted a neuroprotective effect, reflecting an important role of TMEM175 in PD.
Discussion
We have discovered novel intracellular regulatory mechanisms of the lysosomal potassium channel TMEM175. The apoptosis regulatory factor Bcl‐2 directly binds to and inhibits TMEM175, while Bcl‐2‐specific inhibitors can enhance the activity of TMEM175. Exposure to the apoptotic reagent MPP+ or rotenone increases generation of ROS, which can activate TMEM175 through positive feedback and in turn promote apoptosis (Fig 7P). This study, together with previous reports (Dong et al, 2010; Cang et al, 2013; Jha et al, 2014; Cao et al, 2015; Wie et al, 2021), has led us to a certain understanding of the intracellular regulatory mechanisms of the identified major lysosomal ion channels.
Using structure‐based protein–protein binding algorithm and site‐specific mutagenesis, we identified two amino acid residues of TMEM175 that bind to Bcl‐2. Mutations of these two residues, especially the R377V, disrupted the binding between TMEM175 and Bcl‐2, caused increases in channel currents, prevented HA14‐1 from activating the TMEM175 currents, and promoted apoptosis dramatically. These results suggest that Bcl‐2 dominantly inhibits TMEM175 by directly binding to this channel. Relieving this inhibition will unleash the proapoptotic effect of TMEM175 and promote cell death. These findings revealed a new way for Bcl‐2 to regulate apoptosis.
The interaction between Bcl‐2 and TMEM175 suggests that the expression level of Bcl‐2 may affect the function of TMEM175. In fact, we did observe that overexpression of Bcl‐2 significantly reduced TMEM175 currents (Fig 2J and K). Changes in Bcl‐2 expression have been shown to be associated with various diseases. Increased expression of the antiapoptotic Bcl‐2 family members and/or reduced expression of proapoptotic members are common features in B cell lymphomas (Ashkenazi et al, 2017). Decreased Bcl‐2 expression and increased apoptosis were found in polycystic kidney disease 1 (PKD1)‐mutant‐induced polycystic kidney disease (PKD; Duplomb et al, 2017). In Alzheimer's disease, amyloid‐β was found to provoke a rapid and sustained downregulation of Bcl‐2 and subsequently promote cell death (Paradis et al, 1996). Abnormal Bcl‐2 expression in these diseases may be accompanied by changes in TMEM175 activity. Verification of this conjecture is expected to reveal the role of TMEM175 in these diseases and provide new insights into the pathogenesis.
As key regulators of mitochondrial apoptosis, Bcl‐2 proteins mainly localize to and act on mitochondria. How do they gain opportunities to bind to a lysosomal protein? There are two possibilities. First, binding may happen at the sites where mitochondria and lysosomes contact each other. Wong et al (2018) revealed that these two organelles physically contact each other using superresolution microscopy and discovered that a lysosomal protein, Rab7, mediates this contact. Lysosomes are also found in close proximity to mitochondria in cardiac ventricular myocytes (Aston et al, 2017). The physical contact between mitochondria and lysosomes may facilitate their crosstalk. An interesting example is that transient receptor potential mucolipin 1 (TRPML1) was previously found to mediate direct lysosome–mitochondrial calcium transfer at the contact site (Peng et al, 2020). It is likely that at the contact sites of the two organelles, Bcl‐2 tethers mitochondria with its membrane‐spanning C terminus, and binds to TMEM175 with its cytosolic portion containing the predicted binding sites (E135 and N172). This provides a new anchoring mechanism for the contact of the two organelles. Second, Bcl‐2 family proteins have been located on organelles other than mitochondria, including the endoplasmic reticulum, nuclear membrane, Golgi apparatus, peroxisomes, and lysosomes (Krajewski et al, 1993; Dumitru et al, 2012; Guan et al, 2015; Hosoi et al, 2017). Thus, it is possible that lysosomal Bcl‐2s bind to TMEM175 directly.
The interactions between mitochondria and lysosomes, two key organelles involved in cellular metabolism and homeostasis, play important roles in regulating cellular physiological status and function. Lysosomes participate in the execution and regulation of mitophagy, a key process in mitochondrial quality control. In this process, damaged and aged mitochondria are transported to lysosomes for degradation through macroautophagy and thus prevent cell damage caused by the accumulation of dysfunctional mitochondria (Raimundo et al, 2016). The direct physical contact of the two organelles also presents possibilities for previously unidentified interaction patterns. The lysosomal membrane protein Rab7 has been found to regulate mitochondrial fission at the contact site, thereby affecting the dynamics of the mitochondrial network. Conversely, the mitochondrial protein TBC (Tre‐2, Bub2 and Cdc16) domain family member 15 (TBC1D15) regulates lysosomal transport, fusion, and maturation by regulating the hydrolysis of RAS‐related protein (Rab7; Wong et al, 2018, 2019). Lysosomes are also involved in the regulation of the mitochondrial apoptotic pathway (Repnik & Turk, 2010). Abnormalities in the interactions between mitochondria and lysosomes are associated with multiple diseases, such as neurodegenerative diseases. For example, multiple PD‐associated genes are involved in endosomal lysosomal trafficking and mitochondrial function, and mutations in these genes disrupt mitochondrial autophagy and impair mitochondrial function (Plotegher & Duchen, 2017; Audano et al, 2018). Our study revealed interactions among Bcl‐2, TMEM175, and ROS as well as their roles in apoptosis regulation, thus revealing a new mitochondrial lysosomal interaction mechanism.
ROS are normal byproducts of cellular aerobic metabolism, and mitochondria are major generators of ROS. These organelles produce significant amounts of ROS at respiratory chain complexes I and III (Brand, 2010). ROS play different roles under different physiological conditions. Proper/moderate ROS concentrations help maintain normal cellular physiological functions, while excessive ROS result in oxidative stress, which in turn causes cell damage (Valko et al, 2007). Mitochondria themselves are also targets of ROS. Constant exposure to high levels of ROS triggers downstream apoptotic signaling cascades (Murphy, 2009). Damaged mitochondria in turn produce more ROS through ROS‐induced ROS release (RIRR), thus forming a positive feedback loop to augment apoptosis (Zorov et al, 2000, 2014). Interestingly, our data showed that ROS and TMEM175 form another positive feedback loop. TMEM175 has previously been reported to be a suppressor of autophagic flux (Cang et al, 2015). In this study, we found that overexpression of TMEM175 inhibited mitophagy and depolarized mitochondrial membrane potential, leading to mitochondrial damage. The activation of TMEM175 by ROS may inhibit the clearance of damaged mitochondria through mitophagy and consequently increase the production of ROS. Therefore, TMEM175 could act as a relay station between ROS and mitochondria. In contrast, ROS scavengers can decrease apoptosis levels (Tapeinos et al, 2018) and were found to prevent TMEM175 activation by the apoptosis inducer MPP+ (Fig 4E and F). Another lysosomal channel, TRPML1, has been identified as a ROS sensor that orchestrates an autophagy‐dependent negative feedback program to alleviate oxidative stress in cells (Zhang et al, 2016). Therefore, TRPML1 initiates the ROS removal process, while the newly identified ROS sensor TMEM175 promotes the cell death process. This difference suggests a complex mechanism by which lysosomes sense and regulate intracellular ROS.
Previous genome‐wide association studies (GWASs) have discovered the potential relevance of TMEM175 genetic variation to PD. Two single‐nucleotide polymorphisms (SNPs) of TMEM175 have been identified. One of them (rs34311866) has been reported to decrease the age of onset in PD patients and to produce a partial phenocopy of TMEM175 KO in a cellular model (Nalls et al, 2014; Lill et al, 2015; Jinn et al, 2019). Another study has further suggested that TMEM175 may play a role in PD. Researchers reported that knockout of TMEM175 in SH‐SY5Y cells induces pathological features of PD (Jinn et al, 2017). A recent study also reported that TMEM175 deficiency led to accelerated spreading of pathogenic α‐synuclein in cultured neurons and impaired motor function in mice (Wie et al, 2021). Surprisingly, we found that TMEM175 KO mice did not show behavioral deficit in the cylinder and wire hang tests compared with WT mice. More importantly, after MPTP induction, the KO mice performed even better than WT mice. And we detected higher midbrain TH level and more TH‐positive neurons in substantia nigra from KO mice. Our findings indicate that KO of TMEM175 exerted a neuroprotective effect, which is inconsistent with the results of the previous study on SH‐SY5Y cells and culture neurons (Jinn et al, 2017; Wie et al, 2021). The different PD models we adopted may underlie the different results in these studies. The two previous studies used exogenous α‐synuclein fibrils to stimulate the cells in vitro, while we injected MPTP into mice to induce symptoms in vivo. In addition, in the behavioral experiments testing the effect of TMEM175 deletion on motor function, Wie et al used heterozygote TMEM175 KO mice without any exogenous drug treatment, which is more similar to the PD induced by genetic factors, such as familial PD. In these heterozygous mice, the function of TMEM175 was reduced by about 50%, an extent close to that caused by the M393T variant. Although this variant increases the risk of PD in human, the overall incidence of PD in people who carry this variant remains very low. However, Wie et al found that the heterozygous mice displayed significant impairments in motor function, indicating that human and mouse have quite different susceptibility to partial reduction of TMEM175 function. In our animal behavioral experiments, we used homozygous TMEM175 KO mice and MPTP‐induced model of PD. MPTP has been shown to cause permanent Parkinsonism in human (Langston et al, 1983), so the MPTP‐induced mouse model is widely used in PD research. This model can better mimic the PD caused by environmental factors in the human population, which accounts for the vast majority of PD. We found that after administration of MPTP, TMEM175 KO mice performed better in motor skills than WT mice. So far, no cases of complete loss of TMEM175 function in human have been identified, so it is unclear whether this complete loss of function causes consistent phenotypes in humans and mice. Wie et al and our study suggest that TMEM175 may play a dual role in different types of PD. Moreover, different degrees of TMEM175 deletion may also lead to different results. Therefore, it may be important to maintain TMEM175 function within an appropriate range.
Bcl‐2 proteins have also been shown to be closely related to PD. The imbalance of autophagy and apoptosis due to changes in expression and activity of Bcl‐2 may be an important cause of neuronal death in PD (Liu et al, 2019). Together with the previous study (Cang et al, 2015), we have revealed regulatory roles of TMEM175 in both autophagy and apoptosis. Thus, this lysosomal ion channel links the two vital cellular processes together. Increase in the expression or activity of TMEM175 will inhibit autophagy and promote apoptosis, and is likely to promote neuronal death in PD. In this study, we found that KO of TMEM175 induced remarkable neuronal protection in MPTP mouse PD model but did not result in significant defects, suggesting that targeting TMEM175 could be a promising and safe therapeutic strategy for PD.
In summary, we have identified TMEM175 as a key molecule that participates in the most notable cellular process, apoptosis, by acting on ROS through positive feedback. The proapoptotic function of TMEM175 may be involved in the death of neurons in neurodegenerative diseases. Further research using genetically manipulated animal models will reveal more organismal functions of this new ion channel. TMEM175 is also expected to be a new target for autophagy, apoptosis, and related diseases. Development of specific channel blockers and activators will be an important direction for future research.
Materials and Methods
Animals
The animal procedures were approved by the Animal Care and Use Committee of the University of Science and Technology of China. WT and TMEM175 KO C57BL/6N male mice at 3–4 months of age were used for behavioral tests. No specific randomization procedures were used. The mice were housed at a density of no more than 5 per cage under constant temperature (23 ± 1°C) with a 12‐h light/dark cycle and were given food and water ad libitum under specific pathogen‐free conditions. WT C57BL/6N mice were purchased from Beijing Vital River Laboratory Animal Technology. The TMEM175 KO mouse model was generated by Beijing Biocytogen Co., Ltd. To introduce a large‐fragment deletion into the TMEM175 gene, zygotes derived from WT C57BL/6N mice were coinjected with Cas9 mRNA and two sgRNAs: one targeting the first intron, and one targeting the tenth intron (as shown in Fig 7C). Therefore, successful CRISPR/Cas9 editing resulted in the deletion of 9 exons (exons 2–10). Finally, a founder strain with a 7668‐base pair deletion in the TMEM175 gene locus was used for breeding. Heterozygous mice were backcrossed to C57BL/6N mice for more than five generations before being used to generate homozygous KOs. No specific randomization procedures were used.
Cell culture
HEK293T and HeLa cells were cultured in Dulbecco's modified Eagle's Medium (DMEM; Gibco) supplemented with GlutaMAX (Gibco), penicillin/streptomycin (Biosharp), and 10% fetal bovine serum (FBS; Biological Industries). SH‐SY5Y cells were maintained in DMEM/Nutrient Mixture F12 supplemented with glutagro (Corning), penicillin/streptomycin, and 20% FBS. All cells were cultured at 37°C in a humidified CO2 incubator. The cells used for the patch‐clamp recordings were plated on poly‐L‐lysine‐coated round coverslips 12 h before the experiment and then transferred to a patch‐clamp recording chamber before the experiment. Macrophages were cultured from mouse bone marrow. Briefly, the femurs were dissected from sacrificed mice. Remaining tissue on the bone was removed. Bone marrow was extracted by injection and rinsing the bone marrow cavity with ice‐cold phosphate‐buffered saline (PBS). The obtained cells were cultured in DMEM supplemented with 20% FBS and penicillin/streptomycin. As much as 10 ng/ml granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) was added to the culture 7 days before patch‐clamp recordings.
CRISPR knockout of TMEM175, Bcl‐2, or Bax/BAK1 in HEK293T cells
The CRISPR/Cas9 technique was used to knock out TMEM75, Bcl‐2, or Bax/BAK1 in HEK293T cells. To knock out TMEM175, the targeted hTMEM175 sequence, namely TTCTCATCGTGACAGTGGCC [TGG] (with TGG serving as the protospacer adjacent motif (PAM)), was targeted using the plentiCRISPR‐V2 vector. To knock out Bcl‐2, hBcl‐2 sequence ACATCTCGGCGAAGTCGCGG[CGG] (with CGG serving as the PAM) was targeted. To knockout Bax and BAK1, the hBax sequence GGCTGGATCCAAGACCAGGG[TGG] (with TGG serving as the PAM) and the hBAK1 sequence GGAACTCTGAGTCATAGCGT[CGG] (with CGG serving as the PAM) were targeted using the plentiCRISPR‐V2 vector.
Lentiviruses were generated by co‐transfecting HEK293T cells with plentiCRISPR‐V2, lentiviral‐packing (psPAX2), and envelope (pMD2.G) plasmids via Lipofectamine 3000 (Invitrogen) transfection reagent. At 48 h after transfection, the culture medium containing lentiviral particles was collected and added to fresh HEK293T cells cultured in antibiotic‐free medium containing 8 μg/ml of polybrene. Starting at 24 h after infection, the cells were selected with 3 μg/ml of puromycin for 48 h. Single‐cell clones were established using a limiting dilution method and were then sequenced for the knockout confirmation.
Protein‐binding site prediction
Structures of Bcl‐2 (PDB ID: 6O0K), TMEM175 in closed state (PDB ID: 6WC9) and open state (PDB ID: 6WCA) were obtained from the Protein Data Bank (PDB) database and prepared as follows: all hydrogen atoms were added with H‐bonding orientation; all ligands and water were removed; related missing sidechains were repaired; atom bumps were removed with 100 cycles energy minimized. Bcl‐2 protein was docked with TMEM175 in both states using ZDOCK 3.0.2 software (Pierce et al, 2014). None of the residues were selected to restrict the docking site manually. Top three predicting bound complexes were extracted and further analyzed. All interface residues and hydrogen bonds between interacting proteins were analyzed using Edu PyMoL (v 1.70) software (Schrödinger, Inc. 2016). All figures were mapped using Visual Molecular Dynamics (VMD) software (Humphrey et al, 1996).
cDNA constructs and transfection
DNA constructs encoding human TMEM175 were gifts from Dejian Ren. For GFP‐tagged TMEM175, the complementary DNA (cDNA) of TMEM175 was subcloned into the NheI/KpnI sites of pEGFP‐N1. For nontagged TMEM175, the cDNA of TMEM175 was subcloned into the pdECFP Gateway destination vector, and the extended connectivity fingerprint (ECFP) was removed. TMEM175‐V145A and TMEM175‐R377V mutants were gnerated using polymerase chain reaction (PCR) from phTMEM175‐GFP and were confirmed by sequencing. DNA constructs of human Bcl‐2, Bcl‐xL, Bax, and BAK1 were purchased from Sino Biological. The cDNA sequences of human Bcl‐2 and Bcl‐xL with N‐terminal Flag tags were subcloned into the KpnI/XbaI sites of pCMV3‐N‐FLAG vectors, while the cDNA sequences of Bax and BAK1 with N‐terminal GFPSpark tags were subcloned into the KpnI/XbaI sites of pCMV3‐N‐GFPSpark vectors. For mCherry‐tagged TOM20, the cDNA sequence of TOM20 was subcloned into mCherry2‐N1. DNA transfection was performed with PolyJet (SignaGen Laboratories) according to the manufacturer's protocol.
Electrophysiology
Patch‐clamp recordings were performed using a Multiclamp 700B or Axopatch 200B patch‐clamp amplifier, a Digidata 1550B data acquisition system, and pClamp software (Molecular Devices). Recording pipettes were made from borosilicate glass tubes using a P‐1000 puller (Sutter Instrument). For whole‐cell recordings, the pipette solution contained 150 mM K‐methanesulfonate, 2.5 mM MgCl2, 5 mM EGTA (ethylene glycol‐bis(β‐aminoethyl ether)‐N,N,N′,N′‐tetraacetic acid), and 10 mM HEPES (4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid) (pH 7.2). The filled pipettes had resistances of 3–6 MΩ. The bath solution contained 150 mM K‐methanesulfonate, 10 mM triethanolamine (TEA), 3 mM HCl, 1 mM MgCl2, 10 mM glucose, and 10 mM HEPES (pH 7.4). Whole‐lysosome patch‐clamp recordings followed previously described methods (Cang et al, 2013; Chen et al, 2017). Lysosomes were enlarged by treatment with 1 μM vacuolin‐1 for 3–5 h (macrophage) or overnight (HEK293T; Dong et al, 2010), and dissected out with glass pipette before recordings. Pipette solution (luminal) used for lysosomal recordings contained 150 mM K‐methanesulfonate, 2.5 mM MgCl2, 10 mM HEPES, and 10 mM MES (2‐(N‐morpholino)ethanesulfonic acid; pH 4.6). The filled pipettes had resistances of 5–8 MΩ. Bath solution (cytosolic) contained 150 mM K‐methanesulfonate, 2.5 mM MgCl2, 5 mM EGTA, and 10 mM HEPES (pH 7.2). When measuring the leakage of the seal, K‐methanesulfonate in the bath solution was substituted with NMDG‐methanesulfonate. The liquid junction potential was corrected online.
Confocal imaging
All images were taken using a confocal system consist of a Nikon Eclipse Ti inverted microscope, a Confocal Scanner Unit CSU‐X1 Spinning Disk Unit (Yokogawa), a DU‐897U EMCCD camera (Andor), a laser controlling module (Andor), and iQ3 imaging software (Andor). The 488‐, 594‐, and 445‐nm lasers were used to excite GFP/CM‐H2DCFDA/LysoTracker, DiD/mCherry, and CFP, and the corresponding emission light was passed through 525/50‐, 617/73‐, and 483/32‐nm filters (Semrock), respectively. The statistics of fluorescence intensity and image processing were performed in ImageJ software. Cells were plated on poly‐L‐lysine‐coated chamber (NEST, 801001) for 16 h before imaging. For DiD staining (Fig 1A), cells were incubated with 5 mg/ml DiD (a gift from Dr. Yucai Wang) for 20 min in CO2 incubator and washed twice with PBS before imaging.
ROS detection
Intracellular ROS levels were determined with CM‐H2DCFDA (Invitrogen) using a Nikon Ti2‐E fluorescence microscope (Fig 4L and M), flow cytometry (Fig 4I and J), and confocal imaging (Fig EV4C and D). HEK293T cells were transfected with empty vector or TMEM175 for 24 h before experiments. For the data in Fig 4L and M, the cells were treated with vehicle or 10 μM HA14‐1 for 20 min and then incubated with 2.5 μM CM‐H2DCFDA in culture medium for 30 min. Before imaging, the cells were washed twice with prewarmed PBS. For the flow cytometric analysis, the cells were treated with vehicle or 500 μM MPP+ for 1 h and then incubated with 2.5 μM CM‐H2DCFDA in PBS for 30 min. The cells were then washed twice with ice‐cold PBS and resuspended in 200 μl of PBS. Before testing, 1 μg/ml 4′,6‐diamidino‐2‐phenylindole (DAPI) was added to exclude dead cells. The ROS and DAPI signals were analyzed on the 525/40 BP and 450/45 BP channels of a CytoFLEX cytometer (Beckman Coulter), respectively. For the data in Fig EV4C and D, the cells were treated with vehicle or 10 μM rotenone for 1 h and incubated with 2.5 μM CM‐H2DCFDA for 30 min. The cells were then washed twice with PBS prewarmed to 37°C.
Mitochondrial membrane potential measurement
JC‐1 (MedChemExpress) and Rhodamine 123 (Invitrogen) were used to detect mitochondrial membrane potential. HEK293T cells were cultured in 35‐mm Petri dishes and transfected with TMEM175 or empty vector when the cells reached 50% confluence. Twenty‐four hours later, the cells were digested with trypsin/EDTA (ethylenediaminetetraacetic acid) and dissociated by pipetting up and down. As many as 5 × 105 cells were incubated in 500 μl of culture medium containing 2 μg/ml JC‐1 or 10 μM Rhodamine 123 for 15 min, and washed twice with prewarmed PBS. After centrifugation at 800 × g for 5 min, the cells were resuspended in 300 μl PBS and analyzed by a flow cytometer (CytoFLEX, Beckman Coulter). The fluorescence signals of JC‐1 monomers and aggregates were collected through 525/40 BP and 585/42 BP channels, respectively. The fluorescence signals of Rhodamine 123 were collected through 525/40 BP channel. Both dyes were excited at 488 nm.
Mitophagy detection
TOM20‐mCherry was transfected into WT or TMEM175 KO HEK293T cells for 24 h. The cells were stained with 50 nM LysoTracker Green for 20 min before imaging. The fluorescent labeling of organelles was observed using a confocal system. As many as 488‐ and 594‐nm lasers were used to excite LysoTracker Green and TOM20‐mCherry, respectively. The corresponding emission light was passed through 525/50 and 617/73 nm filters, respectively.
Annexin V/PI double staining for apoptosis
Annexin V/PI double staining was used to measure apoptosis. HEK293T cells were cultured in 35‐mm Petri dishes and divided into groups with different interventions. Before apoptosis detection, the cells were detached with trypsin at 37°C for 2 min, harvested, and washed twice with ice‐cold PBS. Thereafter, the cells were resuspended in 500 μl of annexin‐binding buffer, filtered through a 300‐mesh screen (Suoqiao Bio), and adjusted to a density of 3–5 × 105 cells/ml. Three microliters (μl) of Alexa Fluor 488‐labeled annexin V and 1 μl of propidium iodide (PI) were added to 100 μl of cell suspension, and the mixture was incubated for 15 min at room temperature. The samples were detected by flow cytometry as soon as possible. The annexin V–fluorescein isothiocyanate (FITC) and PI signals were analyzed on the 525/40 BP and 585/42 BP channels, respectively.
Antibodies
Rabbit polyclonal anti‐TMEM175 antibodies (19925‐1‐AP), mouse monoclonal GFP‐tagged antibodies (66002‐1‐IG), rabbit polyclonal GFP‐tagged antibodies (50430‐2‐AP), mouse monoclonal anti‐GAPDH (glyceraldehyde‐3‐phosphate dehydrogenase) antibodies (60004‐1‐IG), horseradish peroxidase (HRP)‐conjugated goat anti‐mouse immunoglobulin G (IgG; H+L; SA00001‐1), and HRP‐conjugated goat anti‐rat IgG (H+L; SA00001‐2) were purchased from Proteintech. Mouse monoclonal Flag‐tagged antibodies (T0003), rabbit polyclonal Flag‐tagged antibodies (T0053), and rabbit polyclonal HA‐tagged antibodies (T0050) were purchased from Affinity. Rabbit polyclonal anti‐TH antibodies (ab112) were purchased from Abcam. Alexa Fluor 594‐conjugated goat anti‐rabbit IgG (H+L; E‐AB‐1060) was purchased from Elabscience. Mouse monoclonal Bcl‐2 antibodies (sc‐7382) and mouse monoclonal anti‐Bax antibodies (sc‐20067) were purchased from Santa Cruz. Rabbit polyclonal anti‐BAK antibodies (12105) were purchased from Cell Signaling Technology. IPKine HRP, goat Anti‐Mouse IgG HCS (human chorionic somatomammotropin) (A25112) and IPKine HRP, mouse Anti‐Rabbit IgG LCS (A25022) were obtained from Abbkine.
Western blotting
For the experiments testing, the TMEM175 protein expression levels in the midbrains of mice of different ages, WT male C57BL/6N mice aged 2 weeks, 2 months, 4 months, 9 months, 12 months, and 18–26 months were deeply anesthetized with isoflurane. The midbrains were quickly dissected on ice and stored frozen at −80°C. On the day of the experiment, each sample was immersed in an appropriate volume of prechilled radio‐immunoprecipitation assay (RIPA) lysis buffer (Biosharp) containing a working concentration of the protease inhibitor (Thermo Fisher, A32953) and homogenized for 10 s with a tissue homogenizer three times. The samples were placed on ice for 10 min after each homogenization to avoid damage to the proteins. Thereafter, the samples were centrifuged at 14,000 × g at 4°C for 5 min, and the supernatants were collected. For the verification of TMEM175 knockout (KO) in mouse (Fig 7E), WT and TMEM175 KO mice were deeply anesthetized with isoflurane. The whole brains were quickly dissected on ice and minced into small pieces. Each sample was immersed in 2 ml of freshly prepared RIPA lysis buffer (Biosharp) containing a working concentration of the protease inhibitor (Thermo Fisher, A32953), 10 mM sodium fluoride, and 1 mM sodium orthovanadate, and stroked 8–12 times to homogenize the tissue using a precooled glass Dounce homogenizer. The samples were then placed on ice for 30 min and thereafter centrifuged at 18,506 × g at 4°C for 10 min to obtain the supernatants. After quantification, the whole‐cell lysates were loaded, subjected to 10% sodium dodecyl sulfate–polyacrylamide get electrophoresis (SDS–PAGE), and electrophoretically transferred onto Immobilon‐P polyvinylidene difluoride (PVDF) membranes (Millipore) using a vertical electrophoresis system and a Trans‐Blot electrophoretic transfer system (Cavoy). The membranes were blocked for 2 h at room temperature with 5% nonfat dry milk in Tris‐buffered saline with Tween 20 (TBST) buffer containing 20 mM Tris‐HCl (pH 7.4), 137 mM NaCl, and 0.1% Tween 20. After incubating the membranes overnight at 4°C with primary antibodies against TMEM175 (1:1,000) or GAPDH (1:3,000), the blots were washed three times for 10 min each in TBST and then incubated with HRP‐labeled secondary antibodies for 2 h at room temperature. After the reaction, the blots were rewashed three times for 10 min in TBST. The immunostaining was revealed with WesternBright Sirius HRP substrate (Advansta, K‐12043‐D20) with a gel imaging system (ChemiDoc‐It510, UVP).
For the experiments testing the TH protein expression levels in the midbrains of WT and TMEM175 KO mice, the midbrains were quickly dissected on ice and stored frozen at −80°C after all behavioral tests. Whole‐cell lysate preparation and immunostaining detection were performed as described above with anti‐TH (1:1,500) and anti‐GAPDH (1:3,000) primary antibodies. For the experiments verifying the double knockout (KO) of Bax/BAK1, the knockout of Bcl‐2 and verifying the TMEM175 expression level of WT and mutant TMEM175 in HEK293T cells, cells from 35‐mm dishes were lysed with Nonidet P‐40 (NP‐40) lysis buffer (Biosharp) supplemented with protease inhibitor (Thermo Fisher Scientific) and incubated on ice for 30 min. After centrifugation at 18,500 × g for 10 min, the supernatants were collected and heated for 15 min at 72°C and were then performed as described above with anti‐Bax (1:500), anti‐BAK1 (1:1,000), anti‐Bcl‐2 (1:500), anti‐TMEM175 (1:1,000), and anti‐GAPDH (1:3,000) primary antibodies.
Coimmunoprecipitation analysis
For the experiments testing the interactions between TMEM175 and Bcl‐2 family proteins (Bcl‐2, Bcl‐xL, Bax, and BAK1; Fig 2G and H), transfected HEK293T cells were lysed in CHAPS (3‐[(3‐Cholamidopropyl)dimethyl‐ammonio]‐1‐propane sulfonate) buffer (1% CHAPS in Tris‐buffered saline [19.98 mM Tris, 136 mM NaCl, pH 7.4]) and one protease inhibitor mini tablet (Thermo Fisher, A32953) dissolved in 10 ml of lysate.
For the experiments testing the bindings between Bcl‐2 and TMEM175‐V145A or TMEM175‐R377V (Fig 3B), transfected HEK293T cells were lysed in ice‐cold lysis buffer containing 150 mM NaCl, 10 mM Tris–HCl (pH 7.4), 1% Triton X‐100, and one protease inhibitor mini tablet (Thermo Fisher, A32953) dissolved in 10 ml of lysate. After centrifugation at 18,500 × g for 10 min, the supernatants were collected and divided into two parts: one was used as the whole‐cell lysate input, and the other was used for immunoprecipitation. First, monoclonal anti‐Flag antibodies or monoclonal anti‐GFP antibodies were incubated with protein G‐coupled magnetic beads (Bio‐Rad, 1614823) at 4°C for 1 h. Then, the beads were washed three times with ice‐cold 0.02% phosphate‐buffered saline with Tween 20 (PBST), incubated with the whole‐cell lysates at 4°C for 2 h, subjected to additional three washes with 0.02% PBST, and eluted with 1% Triton X‐100 lysis buffer and 1× loading buffer (Servicebio, G2013). The samples were then heated for 15 min at 72°C and separated by 10% SDS–PAGE. The proteins were transferred onto PVDF membranes. The membranes were then blocked for 2 h in TBST with 5% nonfat dry milk and analyzed by immunoblotting using polyclonal anti‐TMEM175 (1:1,000), polyclonal anti‐Flag (1:2,000), polyclonal anti‐GFP (1:2,000), or monoclonal anti‐GAPDH (1:3,000).
For the experiment testing the interactions between endogenous TMEM175 and Bcl‐2 (Fig 2I), mouse midbrain, SH‐SY5Y, HEK293T, or HeLa cells in 60‐mm dishes were lysed in ice‐cold lysis buffer containing 50 mM Tris–HCl (pH 7.4), 150 mM NaCl, and 0.1% NP‐40 supplemented with one protease inhibitor mini tablet (Thermo Fisher, A32963) dissolved in 50 ml of lysate. After centrifugation at 20,000 × g for 30 min, we collected the supernatants and performed the subsequent immunoprecipitation experiment as described above. Polyclonal anti‐TMEM175 (1:1,000) and monoclonal anti‐Bcl‐2 (1:500) were used for the detection. The α‐ctrl in Fig 2I represents that the magnetic beads used in IP were not preincubated with any antibody.
Immunohistochemistry
After all behavioral tests, 3 TMEM175 KO and 4 WT mice were deeply anesthetized with isoflurane and then perfused with chilled PBS followed by 4% paraformaldehyde (PFA) in PBS. After decapitation, the brains were removed and submerged into a series of sucrose solutions (10, 20, and 30% sucrose) until they sank at 4°C. Then, the brains were sectioned with a freezing cryostat (Leica, CM1860) at 40‐μm thickness and collected in a 24‐well plate. The free‐floating coronal brain slices were washed in PBS for 5 min and blocked in 6% goat serum (v/v), 1% bovine serum albumin (BSA; w/v), and 0.2% Triton X‐100 in 0.01 M PBS overnight at 4°C. For labeling of DA neurons, the brain slices were incubated with polyclonal anti‐TH antibodies (1:2,000) at 4°C in medium containing 3% normal goat serum (v/v), 0.2% Triton X‐100, and 0.5% BSA (w/v) in 0.01 M PBS for 3 days. After washing three times with PBS (10 min each), the binding sites of the primary antibodies were revealed by incubation with Alexa Fluor 594‐conjugated goat anti‐rabbit IgG (H+L; 1:2,000) for 2 h at 4°C. The immunostained brain slices were imaged with an Olympus IX73 inverted microscope. The TH‐positive neurons were counted with ImageJ software.
RT‐qPCR analysis
Total RNA of tissues was extracted with RNA isolater (Vazyme Biotech, R401‐01) and lysed using an electric homogenizer. The purity of RNA was measured by a spectrophotometer (NanoDrop 2000, Thermo Fisher). The cDNA was acquired according to manufacturer's direction (Vazyme, R223‐01) and applied to reverse transcription–quantitative polymerase chain reaction (RT‐qPCR) detection (Vazyme, Q111‐02). RT‐qPCR was performed using the following primers: 5′‐AGCAACTAGGATCGCTGTCT‐3′ and 5′‐CCAACAACCTGGAACAATCTGG‐3′ for TMEM175, 5′‐GTCGCCGAGGCGATTCC‐3′ and 5′‐AGGGAAAAGACAGCCGCAAC‐3′ for TMEM175, 5′‐TGATGGGTGTGAACCACGAG‐3′ and 5′‐GCCCTTCCACAATGCCAAAG‐3′ for GAPDH. The expression levels of all target genes were normalized to the expression level of GAPDH. In order to ensure the correctness of the experimental results, six biological replicates (midbrain tissues from 6 WT and 6 KO mice) were made for each experiment. Based on the Livak and Schmittgen hypothesis, the method was used to calculate the relative mRNA expression levels of target genes.
Mouse behavioral procedures
For cylinder test, before the MPTP administration, each mouse was placed in a 15 cm × 30 cm × 5 mm (inner diameter × height × wall thickness) transparent acrylic cylinder with a 30 cm × 30 cm transparent acrylic bottom. The mouse was videotaped from below for 3 min and then placed back into its home cage. Each mouse underwent three trials of training with 15 min intertrial intervals. After each trial, the apparatus was cleaned with 75% ethanol. The rears made by each mouse in the cylinder were counted. A rear was defined as a movement in which the mouse stood on only its hind limbs, with both forelimbs off the ground (Fleming et al, 2013).
The wire hang test was also commonly used to evaluate the motor function and deficit in the MPTP mouse model of PD. The experiment was performed as described previously with modification (Li et al, 2013). Before MPTP administration, each mouse was place on the framed cage top which was then inverted and suspended about 50 cm above the home cage with soft bedding in it. The latency to when the mouse fell was recorded. Each test was performed with four trials with a 15‐min intertrial interval. The performance was presented as the average of the four trials. The mouse with a score of more than 60 s latency to fall was allowed to undergo post‐MPTP testing.
Beginning on the day after training, each mouse received an intraperitoneal injection of 30 mg/kg MPTP daily for 8 consecutive days, with a total cumulative dose of 240 mg/kg MPTP. Each mouse underwent a cylinder test and a wire hang test 7 days after MPTP administration. The procedure for testing was the same as that for training. The tests were recorded using digital video cameras. The videos were scored by an experimenter blinded to the genotype and treatment condition within the experiment.
The gait dynamics of mouse was analyzed with MSI DigiGait (RWD). Each mouse was pretrained 1 day pre‐MPTP administration and tested 7 days post‐MPTP treatment for one trail. The mouse was placed in a gait apparatus and walked voluntarily on a motorized transparent treadmill belt moving at a constant speed of 20 m/s. Ventral plane imaging for the manner of walking was recorded by a high‐speed digital camera. The swing duration coefficient of variation (CV) and Ataxia coefficient automatically determined by DigiGaitTM Imaging System were used to measure the impaired mobility in mice.
For rotarod analysis, the protocol was adapted, based on previously described methods (Aartsma‐Rus & van Putten, 2014). Mice were pretrained 4 consecutive days before the test on a rotating rotarod apparatus which was set to two accelerating mode steps: from 0 to 4 rpm (revolutions per minute) in 2 s for step 1; from 4 to 60 rpm in 300 s for step 2. The time (in seconds) for the mouse to fall off the rotating rod was recorded. And each mouse performed three consecutive trials (separated by 15 min intertrial intervals) which were then averaged. Seven days after MPTP administration, mice were tested using the same procedure.
Data processing and statistics
The data were analyzed using ImageJ, Clampfit (Molecular Devices), Origin (OriginLab), and Excel (Microsoft). Numeric data are shown as the mean ± SEM. Statistical significance was calculated with two‐sided Student's t‐tests or analysis of variance (ANOVA) with post hoc tests, and is indicated with * (* for P < 0.05, ** for P < 0.01, and *** for P < 0.001).
Author contributions
Lili Qu: Conceptualization; data curation; formal analysis; supervision; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Bingqian Lin: Conceptualization; data curation; formal analysis; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Wenping Zeng: Data curation; formal analysis; supervision; investigation; methodology. Chunhong Fan: Investigation. Haotian Wu: Investigation. Yushu Ge: Software; formal analysis; investigation; methodology. Qianqian Li: Investigation. Canjun Li: Investigation. Yanan Wei: Validation; investigation. Jing Xin: Investigation. Xingbing Wang: Formal analysis; supervision; investigation; methodology. Dan Liu: Formal analysis; supervision; investigation; methodology. Chunlei Cang: Conceptualization; resources; data curation; formal analysis; supervision; funding acquisition; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
CC and LQ conceived the project. LQ and BL performed patch‐clamp recordings, ROS, mitochondrial membrane potential, mitophagy and apoptosis detection, protein chemistry, and behavioral experiments. WZ and CL performed confocal imaging. CF contributed ROS and mitochondrial membrane potential detection. HW and QL contributed patch‐clamp recordings. YG and DL contributed binding site prediction of Bcl‐2 and TMEM175; XW contributed apoptosis analysis; CF, HW, and JX developed reagents. YW constructed Bcl‐2 KO and Bax/BAK1 dKO cell lines. CC and LQ wrote the paper. All authors reviewed the manuscript.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Figure 7
Source Data for Appendix
Acknowledgements
We thank Dr. Dejian Ren for the use of DNA constructs encoding human TMEM175 and Dr. Yucai Wang for the use of DiD. Thanks for supercomputing resources from the Bioinformatics Center of the University of Science and Technology of China, School of Life Sciences. This work was supported by funding from the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB39020400), the National Natural Science Foundation of China (31722018, 31570836, and 31770896), the Fundamental Research Funds for the Central Universities, and K.C. Wong Education Foundation.
EMBO reports (2022) 23: e53234
Data availability
All data needed to evaluate the conclusions that are present in the paper and/or the Expanded View. No data have been deposited in a public database.
References
- Aartsma‐Rus A, van Putten M (2014) Assessing functional performance in the mdx mouse model. J Vis Exp: 51303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ (2017) From basic apoptosis discoveries to advanced selective BCL‐2 family inhibitors. Nat Rev Drug Discov 16: 273–284 [DOI] [PubMed] [Google Scholar]
- Aston D, Capel RA, Ford KL, Christian HC, Mirams GR, Rog‐Zielinska EA, Kohl P, Galione A, Burton RA, Terrar DA (2017) High resolution structural evidence suggests the Sarcoplasmic Reticulum forms microdomains with Acidic Stores (lysosomes) in the heart. Sci Rep 7: 40620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audano M, Schneider A, Mitro N (2018) Mitochondria, lysosomes, and dysfunction: their meaning in neurodegeneration. J Neurochem 147: 291–309 [DOI] [PubMed] [Google Scholar]
- Birkinshaw RW, Gong JN, Luo CS, Lio D, White CA, Anderson MA, Blombery P, Lessene G, Majewski IJ, Thijssen R et al (2019) Structures of BCL‐2 in complex with venetoclax reveal the molecular basis of resistance mutations. Nat Commun 10: 2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD (2010) The sites and topology of mitochondrial superoxide production. Exp Gerontol 45: 466–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang C, Aranda K, Seo YJ, Gasnier B, Ren D (2015) TMEM175 is an organelle K(+) channel regulating lysosomal function. Cell 162: 1101–1112 [DOI] [PubMed] [Google Scholar]
- Cang C, Zhou Y, Navarro B, Seo YJ, Aranda K, Shi L, Battaglia‐Hsu S, Nissim I, Clapham DE, Ren D (2013) mTOR regulates lysosomal ATP‐sensitive two‐pore Na(+) channels to adapt to metabolic state. Cell 152: 778–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Q, Zhong XZ, Zou Y, Zhang Z, Toro L, Dong XP (2015) BK channels alleviate lysosomal storage diseases by providing positive feedback regulation of lysosomal Ca2+ release. Dev Cell 33: 427–441 [DOI] [PubMed] [Google Scholar]
- Chen CC, Cang C, Fenske S, Butz E, Chao YK, Biel M, Ren D, Wahl‐Schott C, Grimm C (2017) Patch‐clamp technique to characterize ion channels in enlarged individual endolysosomes. Nat Protoc 12: 1639–1658 [DOI] [PubMed] [Google Scholar]
- Dong XP, Cheng X, Mills E, Delling M, Wang F, Kurz T, Xu H (2008) The type IV mucolipidosis‐associated protein TRPML1 is an endolysosomal iron release channel. Nature 455: 992–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong XP, Shen D, Wang X, Dawson T, Li X, Zhang Q, Cheng X, Zhang Y, Weisman LS, Delling M et al (2010) PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat Commun 1: 38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong XP, Wang X, Shen D, Chen S, Liu M, Wang Y, Mills E, Cheng X, Delling M, Xu H (2009) Activating mutations of the TRPML1 channel revealed by proline‐scanning mutagenesis. J Biol Chem 284: 32040–32052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitru R, Gama V, Fagan BM, Bower JJ, Swahari V, Pevny LH, Deshmukh M (2012) Human embryonic stem cells have constitutively active Bax at the Golgi and are primed to undergo rapid apoptosis. Mol Cell 46: 573–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duplomb L, Droin N, Bouchot O, Thauvin‐Robinet C, Bruel AL, Thevenon J, Callier P, Meurice G, Pata‐Merci N, Loffroy R et al (2017) A constitutive BCL2 down‐regulation aggravates the phenotype of PKD1‐mutant‐induced polycystic kidney disease. Hum Mol Genet 26: 4680–4688 [DOI] [PubMed] [Google Scholar]
- Fennelly C, Amaravadi RK (2017) Lysosomal biology in cancer. Methods Mol Biol 1594: 293–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SM, Ekhator OR, Ghisays V (2013) Assessment of sensorimotor function in mouse models of Parkinson's disease. J Vis Exp: 50303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraldi A, Klein AD, Medina DL, Settembre C (2016) Brain disorders due to lysosomal dysfunction. Annu Rev Neurosci 39: 277–295 [DOI] [PubMed] [Google Scholar]
- Guan JJ, Zhang XD, Sun W, Qi L, Wu JC, Qin ZH (2015) DRAM1 regulates apoptosis through increasing protein levels and lysosomal localization of BAX. Cell Death Dis 6: e1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guicciardi ME, Leist M, Gores GJ (2004) Lysosomes in cell death. Oncogene 23: 2881–2890 [DOI] [PubMed] [Google Scholar]
- Hosoi KI, Miyata N, Mukai S, Furuki S, Okumoto K, Cheng EH, Fujiki Y (2017) The VDAC2‐BAK axis regulates peroxisomal membrane permeability. J Cell Biol 216: 709–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14: 33–38, 27–28 [DOI] [PubMed] [Google Scholar]
- Jha A, Ahuja M, Patel S, Brailoiu E, Muallem S (2014) Convergent regulation of the lysosomal two‐pore channel‐2 by Mg(2)(+), NAADP, PI(3,5)P(2) and multiple protein kinases. EMBO J 33: 501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinn S, Blauwendraat C, Toolan D, Gretzula CA, Drolet RE, Smith S, Nalls MA, Marcus J, Singleton AB, Stone DJ (2019) Functionalization of the TMEM175 p.M393T variant as a risk factor for Parkinson disease. Hum Mol Genet 28: 3244–3254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinn S, Drolet RE, Cramer PE, Wong AH, Toolan DM, Gretzula CA, Voleti B, Vassileva G, Disa J, Tadin‐Strapps M et al (2017) TMEM175 deficiency impairs lysosomal and mitochondrial function and increases alpha‐synuclein aggregation. Proc Natl Acad Sci USA 114: 2389–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper D, Planells‐Cases R, Fuhrmann JC, Scheel O, Zeitz O, Ruether K, Schmitt A, Poet M, Steinfeld R, Schweizer M et al (2005) Loss of the chloride channel ClC‐7 leads to lysosomal storage disease and neurodegeneration. EMBO J 24: 1079–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornak U, Kasper D, Bosl MR, Kaiser E, Schweizer M, Schulz A, Friedrich W, Delling G, Jentsch TJ (2001) Loss of the ClC‐7 chloride channel leads to osteopetrosis in mice and man. Cell 104: 205–215 [DOI] [PubMed] [Google Scholar]
- Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC (1993) Investigation of the subcellular distribution of the bcl‐2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res 53: 4701–4714 [PubMed] [Google Scholar]
- Langston JW, Ballard P, Tetrud JW, Irwin I (1983) Chronic Parkinsonism in humans due to a product of meperidine‐analog synthesis. Science 219: 979–980 [DOI] [PubMed] [Google Scholar]
- Lawrence RE, Zoncu R (2019) The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol 21: 133–142 [DOI] [PubMed] [Google Scholar]
- Lee C, Guo J, Zeng W, Kim S, She J, Cang C, Ren D, Jiang Y (2017) The lysosomal potassium channel TMEM175 adopts a novel tetrameric architecture. Nature 547: 472–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessene G, Czabotar PE, Sleebs BE, Zobel K, Lowes KN, Adams JM, Baell JB, Colman PM, Deshayes K, Fairbrother WJ et al (2013) Structure‐guided design of a selective BCL‐X(L) inhibitor. Nat Chem Biol 9: 390–397 [DOI] [PubMed] [Google Scholar]
- Li S, Sun B, Nilsson MI, Bird A, Tarnopolsky MA, Thurberg BL, Bali D, Koeberl DD (2013) Adjunctive beta2‐agonists reverse neuromuscular involvement in murine Pompe disease. FASEB J 27: 34–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill CM, Hansen J, Olsen JH, Binder H, Ritz B, Bertram L (2015) Impact of Parkinson's disease risk loci on age at onset. Mov Disord 30: 847–850 [DOI] [PubMed] [Google Scholar]
- Liu J, Liu W, Yang H (2019) Balancing apoptosis and autophagy for Parkinson's disease therapy: targeting BCL‐2. ACS Chem Neurosci 10: 792–802 [DOI] [PubMed] [Google Scholar]
- Milanesi E, Costantini P, Gambalunga A, Colonna R, Petronilli V, Cabrelle A, Semenzato G, Cesura AM, Pinard E, Bernardi P (2006) The mitochondrial effects of small organic ligands of BCL‐2: sensitization of BCL‐2‐overexpressing cells to apoptosis by a pyrimidine‐2,4,6‐trione derivative. J Biol Chem 281: 10066–10072 [DOI] [PubMed] [Google Scholar]
- Mindell JA (2012) Lysosomal acidification mechanisms. Annu Rev Physiol 74: 69–86 [DOI] [PubMed] [Google Scholar]
- Morgan AJ, Platt FM, Lloyd‐Evans E, Galione A (2011) Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem J 439: 349–374 [DOI] [PubMed] [Google Scholar]
- Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M et al (2014) Large‐scale meta‐analysis of genome‐wide association data identifies six new risk loci for Parkinson's disease. Nat Genet 46: 989–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S, Paknejad N, Hite RK (2020) Gating and selectivity mechanisms for the lysosomal K(+) channel TMEM175. eLife 9: e53430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Douillard H, Koutroumanis M, Goodyer C, LeBlanc A (1996) Amyloid beta peptide of Alzheimer's disease downregulates Bcl‐2 and upregulates bax expression in human neurons. J Neurosci 16: 7533–7539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Wong YC, Krainc D (2020) Mitochondria‐lysosome contacts regulate mitochondrial Ca(2+) dynamics via lysosomal TRPML1. Proc Natl Acad Sci USA 117: 19266–19275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z (2014) ZDOCK server: interactive docking prediction of protein‐protein complexes and symmetric multimers. Bioinformatics 30: 1771–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt FM, Boland B, van der Spoel AC (2012) The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol 199: 723–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt FM, d'Azzo A, Davidson BL, Neufeld EF, Tifft CJ (2018) Lysosomal storage diseases. Nat Rev Dis Primers 4: 27 [DOI] [PubMed] [Google Scholar]
- Plotegher N, Duchen MR (2017) Crosstalk between lysosomes and mitochondria in Parkinson's disease. Front Cell Dev Biol 5: 110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimundo N, Fernandez‐Mosquera L, Yambire KF, Diogo CV (2016) Mechanisms of communication between mitochondria and lysosomes. Int J Biochem Cell Biol 79: 345–349 [DOI] [PubMed] [Google Scholar]
- Repnik U, Turk B (2010) Lysosomal‐mitochondrial cross‐talk during cell death. Mitochondrion 10: 662–669 [DOI] [PubMed] [Google Scholar]
- Saftig P, Klumperman J (2009) Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 10: 623–635 [DOI] [PubMed] [Google Scholar]
- Schrödinger, Inc. (2016) The PyMOL Molecular Graphics System, Version Educational v.1.79.
- Settembre C, Fraldi A, Medina DL, Ballabio A (2013) Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 14: 283–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterea AM, Almasi S, El Hiani Y (2018) The hidden potential of lysosomal ion channels: a new era of oncogenes. Cell Calcium 72: 91–103 [DOI] [PubMed] [Google Scholar]
- Tapeinos C, Larranaga A, Sarasua JR, Pandit A (2018) Functionalised collagen spheres reduce H2O2 mediated apoptosis by scavenging overexpressed ROS. Nanomedicine 14: 2397–2405 [DOI] [PubMed] [Google Scholar]
- Tomala K, Gabryel B (2017) Lysosomal dysfunction in neurodegenerative diseases. Postepy Hig Med Dosw (Online) 71: 291–306 [DOI] [PubMed] [Google Scholar]
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39: 44–84 [DOI] [PubMed] [Google Scholar]
- Vergarajauregui S, Puertollano R (2006) Two di‐leucine motifs regulate trafficking of mucolipin‐1 to lysosomes. Traffic 7: 337–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wie J, Liu Z, Song H, Tropea TF, Yang L, Wang H, Liang Y, Cang C, Aranda K, Lohmann J et al (2021) A growth‐factor‐activated lysosomal K(+) channel regulates Parkinson's pathology. Nature 591: 431–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Kim S, Peng W, Krainc D (2019) Regulation and function of mitochondria‐lysosome membrane contact sites in cellular homeostasis. Trends Cell Biol 29: 500–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Ysselstein D, Krainc D (2018) Mitochondria‐lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554: 382–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J, Zhu MX (2016) Regulation of lysosomal ion homeostasis by channels and transporters. Sci China Life Sci 59: 777–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Ren D (2015) Lysosomal physiology. Annu Rev Physiol 77: 57–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Cheng X, Yu L, Yang J, Calvo R, Patnaik S, Hu X, Gao Q, Yang M, Lawas M et al (2016) MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun 7: 12109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ (2000) Reactive oxygen species (ROS)‐induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 192: 1001–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Sollott SJ (2014) Mitochondrial reactive oxygen species (ROS) and ROS‐induced ROS release. Physiol Rev 94: 909–950 [DOI] [PMC free article] [PubMed] [Google Scholar]