

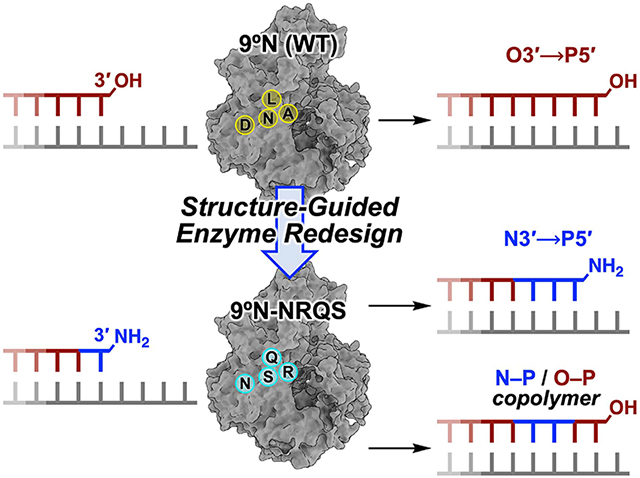
Abstract
DNA polymerases have evolved to feature a highly conserved activity across the tree of life: formation of, without exception, internucleotidyl O─P linkages. Can this linkage selectivity be overcome by design to produce xenonucleic acids? Here, we report that the structure-guided redesign of an archaeal DNA polymerase, 9°N, exhibits a new activity undetectable in the wild-type enzyme: catalyzing the formation of internucleotidyl N─P linkages using 3′-NH2-ddNTPs. Replacing a metal-binding aspartate in the 9°N active site with asparagine was key to the emergence of this unnatural enzyme activity. MD simulations provided insights into how a single substitution enhances the productive positioning of a 3′-amino nucleophile in the active site. Further remodeling of the protein–nucleic acid interface in the finger subdomain yielded a quadruple-mutant variant (9°N-NRQS) displaying DNA-dependent NP-DNA polymerase activity. In addition, the engineered promiscuity of 9°N-NRQS was leveraged for one-pot synthesis of DNA—NP-DNA copolymers. This work sheds light on the molecular basis of substrate fidelity and latent promiscuity in enzymes.
Graphical Abstract
INTRODUCTION
Transfer of genetic information is orchestrated by protein enzymes in all extant organisms on Earth. DNA replication, the process of producing copies of DNA, is essential for information transfer and catalyzed by diverse DNA polymerases. Despite large differences in composition, catalytic activity, and replication fidelity, all known DNA polymerases have evolved to feature a stringent internucleotidyl linkage selectivity in generating the repeating sugar-phosphate backbone of DNA.1 They catalyze the formation of the O3′ → P5′ phosphodiester (or simply O─P) bond between the 3′-OH of a DNA primer and the 5′-α-phosphate of a 2′-deoxynucleoside triphosphate (dNTP) monomer. This transformation requires the presence of divalent metal cations, typically Mg2+, which activate the 3′-OH nucleophile and thus reduce the activation energy to reach the phosphorylation transition state.2,3
Despite exquisite preference in utilizing hydroxyl nucleophiles, certain DNA polymerases can display low-level promiscuous activities. For example, they can accept non-canonical nucleotides with modifications in the nucleobase4,5 or sugar framework6-10 and synthesize non-natural nucleic acids, also known as xenonucleic acids (XNAs).11,12 However, enzymatic sugar-phosphate connection between atoms other than oxygen and phosphorus has rarely been observed, suggesting that the molecular machinery essential for genetic information transfer has evolved to favor, with high stringency, O─P bond formation.
One distinct nucleic acid that contains a non-O─P internucleotidyl linkage is 3′-nitrogen-5′-phosphorus-linked DNA (NP-DNA), where each 3′-O atom is replaced by a 3′-NH, thereby possessing N3′ → P5′ phosphoramidate (or simply N─P) linkages. NP-DNA mimics RNA in that its duplex structure adopts an RNA-like A-form helical geometry in solution and in crystallo.13 Notably, reverse transcriptases can recognize templates made of NP-DNA and synthesize a complementary DNA strand.14 In addition, it can form duplexes with both ssDNA and ssRNA that exhibit higher thermal stability than isosequential duplexes made of DNA and RNA.13 Furthermore, NP-DNA has been proposed as a potential inhibitor of RNAs and RNA-binding proteins.15,16 It exhibits resistance against nucleolytic degradation17 and can inhibit gene expression both in vitro and in vivo by acting as a steric block in translation without the need to promote endogenous RNase H activity.18 Therefore, NP-DNA has chemical and structural features that make it a unique polymer with significant potential in synthetic biology and biotechnology.
Enzymatic synthesis of NP-DNA is fundamentally challenging because forming the internucleotidyl N─P bond is not a primary function of known wild-type (WT) polymerases. NP-DNA can be synthesized nonenzymatically from 3′-amino-nucleotides with a reactive 5′-leaving group.19-23 The N─P bonds in NP-DNA can form under reaction conditions where divalent metal ions are not required for the 3′-amino group to function as a nucleophile.20,21 In the absence of divalent metal ions, nonenzymatic primer extension is substantially faster with a 3′-amino nucleophile compared to 3′-hyrdoxyl nucleophile.21 In contrast, at least two divalent metal ions are needed in polymerase-catalyzed, canonical, O─P bond formation (Figure 1D).2,3 However, there is no established evidence to suggest that a similar metal ion-dependent mechanism could be employed by a polymerase to form N─P bonds. Therefore, we postulated that a WT DNA polymerase with full metal occupancy may have little-to-no ability to catalyze internucleotidyl N─P bond formation and may be redesigned with reduced metal occupancy to exhibit this novel function. Indeed, a WT DNA polymerase from Bacillus stearothermophilus (Bst) with reduced active-site metal occupancy has recently been reported to display low-level promiscuous N─P bond-forming activity.24 Structural characterization indicated single-metal ion coordination to the incoming nucleoside triphosphate (in the B-site) but no coordination to the primer 3′-amino group (in the A-site), despite the presence of two canonical Asp residues in the active site.24 Although certain residues in the active site of WT DNA polymerases can be modified without major impact on their activity, metal-binding Asp residues are considered immutable without complete loss of function.25 Consequently, we reasoned that substituting these metal-binding Asp residues could result in an extreme trade-off between the WT and novel internucleotidyl linkage selectivities, which is rare in enzyme evolution but not uncommon in designed biocatalysts.26,27
Figure 1.

Structure-guided redesign of 9°N DNA polymerase to catalyze the formation of internucleotidyl N-P bonds. (A) Model of 9°N bound to a primer/template complex. (B,C) Insets show the positioning of the primer and the incoming nucleoside triphosphate within (B) the WT 9°N (PDB accession codes: 5OMV and 5OMQ) and (C) its quadruple-mutant variant 9°N-NRQS (Rosetta based model). Sites of substitutions in 9°N-NRQS are highlighted by red spheres. Active site metal ions are shown: (B) Mg2+ in green and (C) Mn2+ in purple. Residues are drawn as sticks with color by atom (carbon, white; nitrogen, blue; oxygen, red; phosphorus, orange). (D,E) Template-directed primer-extension reactions catalyzed by (D) WT 9°N and (E) 9°N-NRQS. Chemical structure of (D) natural phosphodiester-linked DNA and (E) N3′ → P5′ phosphoramidate-linked DNA (NP-DNA).
In this work (Figure 1), using structure-guided computational design, we engineered variants of 9°N, an archaeal B-family replicative DNA polymerase, that can form the internucleotidyl N─P bond using 3′-amino-2′,3′-dideoxynucleoside 5′-triphosphates (3′-NH2-ddNTPs). Replacing metal-binding Asp-404 in the 9°N active site (Figures 1A,B) with asparagine (Asn) (Figure 1C) drastically decreased its DNA polymerase activity. Strikingly, however, this substitution provided the desired novel function, which is undetectable in the WT enzyme: catalyzing internucleotidyl N─P bond formation (Figure 1E). Computational modeling revealed that productive nucleophile positioning in the active site, in the absence of an A-site metal ion, was key to the emergence of this new enzyme activity. The polymerase processivity was enhanced via three amino acid substitutions in the distal subdomains of the polymerase (Figure 1C). The resulting quadruple-mutant variant (9°N-NRQS) displayed DNA-dependent NP-DNA polymerase activity with modest rates and fidelity (Figure 1E) and enabled the synthesis of short stretches of NP-DNA. This work, to our knowledge, presents the first example of an otherwise inactive DNA polymerase to catalyze the formation of internucleotidyl N─P bonds upon substituting one of its metal-binding amino acids in the active site, endowing archaeal B-family DNA polymerases with a novel function.
RESULTS
Polymerase Active-Site Remodeling Allows for the New Enzyme Function: Catalyzing N─P Bond Formation.
9°N displays intrinsic thermostability,28 can tolerate amino acid mutations,29 and can accommodate non-canonical nucleoside triphosphates as monomers in synthesis of RNA-like nucleic acids.6,7 Given these versatile characteristics, we selected 9°N as the starting point in our investigation of the requirements for enzymatic internucleotidyl N3′ → P5′ phosphoramidate bond formation.
To test our hypothesis that reduced metal ion occupancy of the A-site of the polymerase would enable N─P bond formation, we first explored single-point substitutions of metal-coordinating Asp residues. 9°N possesses two conserved Asp residues in the active site,30 Asp-404 and Asp-542, each coordinating one or more divalent metal cations (Figure 1B). Activation of the pyrophosphate leaving group is expected to be dependent on the metal ion occupancy of the B-site.2
Therefore, we synthesized a total of five 9°N variants (Tables S1 and S2), each constituting a variation in side chain polarity at either of the two Asp positions:31 Asn, Ala, Ser, His, and Thr. In particular, Asn is isosteric with Asp but is expected to only weakly coordinate a single metal ion compared to two-metal ion coordination by WT Asp residues. For one of the five variants, Asp-404 was substituted with Asn (9°N-N), and for the other four, Asp-542 was substituted with either Ala (9°N-A′), Ser (9°N-S), Thr (9°N-T), or His (9°N-H). Rosetta-based models of these variants showed steric compatibility with the WT enzyme conformation, suggesting that all of the variants should be sufficiently stable. Indeed, we were able to produce all of these proteins by recombinant expression (Figure S1A). To assay WT 9°N and the designed 9°N variants, we used 3′-NH2-ddTTP (100 μM) as the monomer and a primer/template complex composed of a 5′-FAM-labeled 15 nt DNA primer ending with a 3′-NH2-G (1 μM) and a 22 nt complementary DNA template containing a 5′-C2A5 overhang (1 μM) (Table S3). We set the pH of the primer-extension reaction mixtures in this work as 8.8. At this pH, a substantial population of the 3′-amino nucleophile is expected to be neutral, as the pKa of the 3′-ammonium group of 2′,3′-dideoxynucleosides is ~7.5–7.7.19,21 Divalent metal cations can affect the rate and fidelity of nucleic acid synthesis catalyzed by DNA polymerases.32 Therefore, these assays were conducted in the presence of a panel of divalent metal cations, including Mg2+, Ca2+, Mn2+, Zn2+, Ni2+, Co2+, Fe2+, and Cu2+. Denaturing gel analysis indicated that the control sample lacking 9°N gave no primer-extension product and 9°N (1 μM) failed to extend the 3′-amino primer at 55 °C with any of these divalent metals (Figure S2). Notably, we only observed N3′ → P5′ phosphoramidate bond formation (+1 product) for the polymerase-metal ion combination of 9°N-N and Mn2+ (Figure S2). These results indicate that low-level N─P bond formation is a latent promiscuous activity of the polymerase that can be accessed by changing merely a single non-hydrogen atom (oxygen to nitrogen) in a side chain group.
Molecular Dynamics Simulations Shed Mechanistic Light on the Consequences of Asp-to-Asn Substitution and Role of Mn2+.
To gain a better understanding of the influence of D404N substitution, A-site metal ion, and the identity of the primer 3′-nucleophile on the structure and dynamics of the active site of 9°N, we performed exhaustive molecular dynamics (MD) simulations of 9°N with various active site configurations. In total, we carried out ~0.5 μs-long, independent MD simulations for 8 different active site configurations of 9°N, specifically the WT and D404N variant, in the presence of either a 3′-OH or 3′-NH2 primer in the presence and absence of the A-site metal ion. The results from these MD simulations are summarized in Figure 2A as two-dimensional scatter plots of the in-line attack angle (τ, the angle between O3′, scissile phosphorus atom, and O5′) and the distance between the 3′-nucleophile and scissile phosphorus atom (r), created from the corresponding MD configurations. The configurations are classified as active (A) or non-active (NA), where active configurations are defined as those in which the active site possesses in-line fitness (τ > 150°) and in which the 3′-nucleophile is favorably positioned for catalysis (distance between 3′-nucleophile and P ≤ 3.5 Å).
Figure 2.

Analysis of 3′-nucleophile positioning using molecular dynamics (MD) simulations. (A) 2D scatter plots of the in-line attack angle (τ, the angle between the 3′-nucleophile, scissile phosphorus atom, and O5′) and the distance between the 3′-nucleophile and scissile phosphorus atom (r), obtained from MD simulations of 9°N in various active site configurations. In total, eight different active site scenarios are explored: the four panels correspond to simulations in which residue 404 is Asp and the A-site metal ion is present (top left) or absent (top right), residue 404 is Asn and the A-site metal ion is present (bottom left) or absent (bottom right). In each panel, the red and blue colors correspond to the primer being 3′-OH (red) or 3′-NH2 (blue). The data points are classified as active (A) or non-active (NA), where active configurations are defined as those in which the active site possesses in-line fitness (τ > 150°) and in which the 3′-nucleophile is favorably positioned for catalysis (distance between 3′-nucleophile and P ≤ 3.5 Å). The inset bars represent percentage of favorable configurations of the 3′-nucleophile for catalysis via in-line attack. (B) MD snapshot of the 9°N-N active site showing a representative configuration with high in-line fitness (τ ~ 173°, solid black line). The distance between the 3′-amino nucleophile and D542 is 3.0 Å, suggesting that D542 may serve as a base. Observed hydrogen bond and charge interactions in the active site are highlighted (residues in sticks).
WT Polymerase Simulations.
In the WT polymerase ensemble, 3′-OH is favorably positioned for catalysis in >90% of Asp-404 configurations when Mn2+ is present (Figure 2A, top left, red), compared to a 10-fold decrease (9%) in active configurations when Mn2+ is absent (Figure 2A, top right, red), suggesting that the presence of divalent metals is crucial to the reactivity of primer 3′-OH in 9°N. Similarly, primer 3′-NH2 is favorably positioned for catalysis in >90% of Asp-404 configurations when Mn2+ is present, with a corresponding 3-fold decrease (35%) in active configurations when Mn2+ is absent (Figure 2A, top left, blue), indicating that there is still a significant fraction of active Asp-404 configurations without Mn2+.
D404N Variant Polymerase Simulations.
3′-OH was favorably positioned in 28% of Asn-404 configurations when Mn2+ was present (Figure 2A, bottom left, red), compared to a 14-fold decrease (2%) in active configurations when Mn2+ was absent (Figure 2A, bottom right, red). In contrast, 3′-NH2 was favorably positioned for catalysis in 10% of Asn-404 configurations when Mn2+ was present (Figure 2A, bottom left, blue), compared to an ~2-fold increase (22%) when Mn2+ was absent (Figure 2A, bottom right, blue).
Taken together, these simulations provide a mechanistic scenario in which (i) positioning of both 3′-OH and 3′-NH2 is influenced by both the identity of the 404 residue and the presence of Mn2+, (ii) D404N substitution is key to low metal occupancy of the A-site, (iii) N─P bond formation may occur via an in-line attack of 3′-NH2 on the 5′-α-phosphate of the incoming nucleotide (Figure 2B), (iv) the primer 3′-NH2 may act as a more potent nucleophile in the absence of Mn2+ in the A-site, and (v) D542 may act as a base (Figure 2B) facilitating the deprotonation of 3′-N upon the N─P bond formation step. Consistent with this mechanistic scenario, D542N (9°N-N′) and D542N + D404N (9°N-NN′) showed no enzyme activity in catalyzing N─P bond formation. However, it is possible that D542N reduces metal occupancy in the B-site and thus diminishes activity. While ascertaining the identity of the general base requires further mechanistic investigations, the presence of an Asp residue that is sufficiently proximal for hydrogen bonding to the 3′-amino group to potentially act as a base is consistent with the proposed mechanism for N─P bond formation catalyzed by Bst.24
Structure-Guided Mutagenesis Gives Insights into the Role of Active Site and Finger Domains in Affecting the Rate of N─P Bond Formation.
Using Rosetta-based modeling,33 we explored additional substitutions beyond D404N throughout 9°N (Figure 3A). First, we sought to reduce divalent metal occupancy in the active site by altering the hydrogen bond network: Y402H (9°N-NH) (Figure 3B) and E580D (9°N-ND) (Figure 3C). Further remodeling was conducted to increase local positive charge: Y402K (9°N-NK) and E580Q (9°N-NQ**). We also investigated making a stronger hydrogen bond with the primer 3′-amino group: T541S (9°N-NS*).
Figure 3.

Rosetta modeling-guided double and combinatorial mutagenesis of 9°N-N. (A) Classification of variants based on the site of amino acid substitution and combinatorial status. Substitutions leading to an increase in NP-DNA polymerase activity are highlighted in green, the variant with the highest catalytic rate is highlighted in blue. (B–G) Rosetta-based structural models of selected key substitutions, highlighting the second-shell effects on the catalytic site (B,C) and the distal finger subdomain (D–G). Active site metal ion Mn2+ is in purple. Residues are drawn as sticks with color by atom (carbon, white; nitrogen, blue; oxygen, red; phosphorus, orange). Dashed yellow line indicates the second shell interaction.
With these 9°N variants (Table S1 and Figure S1B), we conducted a comparative polymerase activity analysis for the extension of the same 3′-amino primer on a 5′-A2C5 DNA template with 3′-NH2-ddGTP at 55 °C (Figure S3). We measured the observed rate constant, kobs, for the primer extension by following the depletion of the primer over 24 h (Table S4). The substitutions in 9°N-NH and 9°N-ND resulted in an ~2.5-fold primer-extension rate enhancement (Table S4); however, for the other three variants, the rates were slower than that for 9°N-N by ~20%.
Certain processive mutations in KOD, a related B-family DNA polymerase, have been reported to increase the polymerization rate of threose nucleic acid,34 which is another XNA that adopts a helical geometry similar to that of A-form RNA. We therefore sought to transfer some of these mutations to 9°N-N to facilitate a faster NP-DNA extension. To this end, we modeled four double-mutant variants with substitutions on the distal subdomains: A485R (9°N-NR) (Figures 3D,G), N491S (9°N-NS) (Figure 3E), L489Q (9°N-NQ) (Figures 3F,G), and E664I (9°N-NI). In our models, the finger subdomain substitutions A485R and L489Q formed additional hydrogen bonds with the residues S348 and T349, which are at the N-terminus of a helix (residues 349–359). This N-terminal segment in turn makes hydrogen bonding and electrostatic interactions (via its helix dipole) with the downstream 5′-phosphate of the DNA template. These interactions may thus indirectly help stabilize the template in an orientation productive for the enzymatic catalysis. Furthermore, position 491 is located on the active-site-facing side of the helix, and in our model, S491 is poised to make hydrogen-bonding interactions with both the pyrophosphate and 3′-amino group of the incoming monomer. Based on such possible favorable interactions, we synthesized these double-mutant variants (Table S1 and Figure S1B). These variants resulted in 2.6 to 3.3-fold primer-extension rate enhancements (Table S4).
Active-site and distal substitutions that led to an increase in catalytic activity were then investigated for synergistic effects through the synthesis of triple-mutant variants (9°N-NRQ, 9°N-NRS, 9°N-NQI, 9°N-NQS, 9°N-NSI, 9°N-NRI, and 9°N-RQS) (Table S1 and Figure S1C) and quadruple-mutant variants (9°N-NQSI, 9°N-NRQS, 9°N-NRSI, and 9°N-NRQI) (Table S1 and Figure S1D). Among the triple-mutant variants, 9°N-NRS and 9°N-NQS provided significantly (~10-fold) larger kobs. The majority of the quadruple-mutant variants had a similar activity to 9°N-NRS and 9°N-NQS, while 9°N-NRQS catalyzed N3′ → P5′ phosphoramidate bond formation with the fastest rate (kobs = 0.13 h−1) (Table S4). Of note, compared to the extension kinetics for double-mutant variants, kobs values for 9°N-NRS and 9°N-NQS were ~4-fold faster, whereas kobs for 9°N-NRQI was 1.4-fold slower. These findings suggest that N491S substitution, in combination with A485R and L489Q substitutions, can synergistically improve the polymerase activity.
Assessing NP-DNA and DNA Polymerase Activities.
To shed light on the factors governing polymerase activity, we examined the enzyme dependence on the divalent metal ion and 3′-nucleophile (Figure 4A). For these studies, we focused on 9°N-N and 9°N-NRQS, as the former variant constitutes the amino acid substitution essential for the desired unnatural activity and the latter produces internucleotidyl phosphoramidate linkages at the fastest rates among all 9°N variants we screened. For both 9°N-N and 9°N-NRQS, the primer extensions, whether the DNA primer has a 3′-OH or 3′-NH2 nucleophile, were substantially faster with Mn2+ than Mg2+ (Figure 4B). For 9°N-N, kobs of the 3′-amino-primer extension with 3′-NH2-ddGTP in the presence of Mn2+ was 0.013 ± 0.008 h−1 (Figure 4B, entry 8). 9°N-NRQS catalyzed the formation of phosphoramidate bonds with ~11-fold enhanced kinetics at 55 °C compared to 9°N-N (entry 12). Further systematic investigations on polymerase activity supported the observations that substituting Asp-404 with Asn-404 (D404N) was necessary for catalyzing the formation of N3′ → P5′ phosphoramidate bonds: 9°N-RQS (Table S1 and Figure S1C), which is the close analog of 9°N-NRQS with Asp-404 intact, lost this unnatural catalytic function completely.
Figure 4.

Enzymatic DNA and NP-DNA primer-extensions. (A) Primer-extension reaction conditions: 1 μM 3′-amino-G primer, 1 μM DNA template, 100 μM 3′-NH2-ddNTP, 1 mM M2+ (Mg2+ or Mn2+), 1 μM polymerase (9°N, 9°N-N, or 9°N-NRQS) in 1× ThermoPol buffer at pH 8.8 at 55 °C. 5′-end of the primer strand is labeled with 6-carboxyfluorescein (FAM), represented as the green sphere. (B) Summary of primer-extension reaction rate, kobs (h−1). For entries 3 and 4, an upper bound of the kobs is provided. a: N.D.: not determined. Product formation is complete in less than 1 min. For 9°N-catalyzed dGTP addition in the presence of Mg2+, kpol was measured as 183,600 h−1 (see ref 35); b: reaction temperature is 65 °C. For entries 5–13, R2 values for the fits are 0.942, 0.991, 0.967, 0.991, 0.913, 0.997, 0.992, 0.994, and 0.977. (C) Substrate concentration-dependent kobs vs 3′-NH2-ddGTP concentration for 9°N-N (red) and 9°N-NRQS (dark gray) at 55 °C. Comparison of maximum rates of 3′-amino-G incorporation, kpol, and dissociation constants, Kd, for both 9°N-N and 9°N-NRQS.
Given the thermophilic character of 9°N, we sought to explore how reaction temperature affects the NP-DNA polymerase activity of 9°N-NRQS. Reaction mixtures with identical contents (3′-amino-G primer/5′-A2C5 DNA template, 3′-NH2-ddGTP, Mn2+) were incubated for 12 h at temperatures ranging from 45 to 75 °C with 5 °C intervals (Figure S4). The combined yields of the extension products were the lowest (32–34% for the total of +1 and +2 products) for the samples incubated at temperatures below 55 °C. A similar outcome was observed for the sample incubated at 75 °C (52% for the total of +1, +2, and +3 products), likely due to the irreversible destabilization of the primer/template complex or inactivation of the polymerase. Efficiency of the N─P bond formation was highest at 65 and 70 °C, with the +3 product fraction reaching up to ~17% for both conditions. Due to the higher risk of inactivation of the polymerase for longer incubations at 70 °C, we elected 65 °C as the suitable reaction temperature. At 65 °C, kobs of 3′-amino-G primer extension with 3′-NH2-ddGTP was 0.27 ± 0.04 h−1 (Figure 4B, entry 13), a 90% enhancement of the kinetics observed for the 55 °C condition. This rate was similar to that we measured for a longer primer/template complex. A 21 nt primer on full length complementary DNA with the same overhang (5′-A2C5) displayed kobs of 0.25 ± 0.02 h−1, suggesting that the 15-bp primer/template duplex does not suffer from thermal destabilization at 65 °C.
We compared the NP-DNA polymerase activities of 9°N-N and 9°N-NRQS by determining the maximum rate of 3′-amino-G incorporation, kpol, and dissociation constant, Kd (Figure 4C) at 55 °C. For 9°N-N and 9°N-NRQS, kpol values were 0.014 and 0.15 ± h−1, respectively, which revealed a 10-fold difference in catalytic efficiency. Kd values of 3′-NH2-ddGTP with 9°N-N and 9°N-NRQS were 18 and 20 μM, respectively, suggesting a similar binding affinity of the 3′-NH2-ddGTP monomer with either enzyme.
As the 9°N active-site mutations enable enzymatic NP-DNA synthesis, we sought to gain insight into how these mutations affect DNA polymerase activity. Both 9°N-N and 9°N-NRQS displayed significantly reduced activity for extending the canonical 3′-hydroxyl DNA primer with dGTP. D404N substitution, not surprisingly, appeared to be the major cause of the reduced DNA extension rate. When 9°N-N was used, kobs values were measured as 1.2 ± 0.1 h−1 for Mg2+ (Figure 4B, entry 5) and 14.2 ± 1.2 h−1 for Mn2+ (entry 6). DNA extension was the slowest (kobs = 0.12 ± 0.01 h−1) with 9°N-NRQS and Mg2+ at 55 °C (entry 9).
Primer-Extension Kinetics with Homopolymeric DNA Templates.
We compared the 3′-amino-primer extensions on homopolymeric DNA templates with each matching 3′-NH2-ddNTP (Figure 5A). Monomers 3′-NH2-ddATP, 3′-NH2-ddGTP, 3′-NH2-ddTTP, and 3′-NH2-ddCTP were incubated separately with mixtures containing DNA templates 5′-C2T5, 5′-A2C5, 5′-C2A5, and 5′-T2G5, respectively (Figure 5B and Table S1). The rate of primer extension with 3′-NH2-ddATP on 5′-C2T5 (kobs = 0.204 ± 0.013 h−1) was similar to that with 3′-NH2-ddGTP on 5′-A2C5 (kobs = 0.27 ± 0.04 h−1) (Figure 5C). The rates of primer extension with 3′-NH2-ddTTP on 5′-C2A5 and with 3′-NH2-ddCTP on 5′-T2G5 were slower (kobs = 0.160 ± 0.014 and 0.149 ± 0.005 h−1, respectively). Despite nearly 90% or more of the primer being consumed within 24 h, the +3 product band intensities for the reactions involving 3′-NH2-ddCTP and 3′-NH2-ddTTP plateaued at 23% and 5%, respectively. The extension reactions involving 3′-NH2-ddGTP and 3′-NH2-ddATP proved to be faster: the +3 products were observed within 3 h, with the band intensities reaching 37% and 12%, respectively, after 24 h. Within this timeframe, the primer extension with 3′-NH2-ddGTP provided the +4 product, albeit at 5% yield. This finding suggests that the primer-extension rate is substantially influenced by factors other than canonical nucleobase hydrogen bonding between the 3′-NH2-ddNTPs and the template within the 9°N-NRQS binding site. Such factors, including base stacking and sterics, have been recognized to play critical roles in governing enzymatic nucleic acid polymerization.36
Figure 5.

9°N-NRQS-catalyzed extensions of the 3′-amino-terminated primer on homopolymeric DNA templates. (A) Primer-extension reaction condition. Four different reaction mixtures were prepared using a specific 3′-NH2-ddNTP (N: base incorporated to the primer). Y: template base; q: overhang. (B) Denaturing SDS-PAGE analyses of the extension reaction mixtures containing 1 μM 3′-amino-G primer, 1 μM DNA template, 100 μM 3′-NH2-ddNTP, 1 mM Mn2+, and 1 μM 9°N-NRQS in 1× ThermoPol buffer at pH 8.8; for each reaction, samples were quenched at the designated time points. Imaging performed at FAM excitation (epi-blue, 460–490 nm). (C) Plot of the percentage of the unreacted primer strand versus incubation time. The observed rate constants, kobs (h−1), was calculated using a single exponential decay fit. Error estimates were calculated from the standard mean error of three replicates. R2 values for the fits are 0.998 (3′-NH2-ddTTP), 0.995 (3′-NH2-ddATP), 0.993 (3′-NH2-ddCTP), and 0.977 (3′-NH2-ddGTP).
9°N-NRQS Displays Template-Directed Polymerase Activity.
The DNA template-dependent activity of 9°N-NRQS was assessed using polymerase end point assays. We investigated nucleotide incorporation fidelity by comparing the primer-extension reactions for a total of sixteen different 3′-NH2-ddNTP–template combinations at both 55 and 65 °C (Figure 6A). The buffered mixtures of 9°N-NRQS, Mn2+, the 3′-amino-G primer, and a DNA template selected from 5′-C2A5, 5′-C2T5, 5′-T2G5, and 5′-A2C5 were incubated with a given 3′-NH2-ddNTP. Mixtures were quenched after 24 h, and percentages of the primer and the extension product fractions were measured after denaturing PAGE separation. For each unique nucleotide-template combination, the nucleotide incorporation fidelity was expressed as the selectivity ratio (SR), which is defined as [total band intensity of the extension products for a given 3′-NH2-ddNTP]/[total band intensity of all extension products combined]. In this analysis, a higher SR represents a more favorable nucleotide selectivity. For the samples incubated at either 55 or 65 °C, SRs for the matching nucleotide were higher than those for the mismatches. 9°N-NRQS mostly displayed at least an order of magnitude selectivity toward the matching 3′-NH2-ddNTP over the mismatch A, G, or C monomer. The degree of mismatch was the highest for 3′-NH2-ddTTP at either 55 or 65 °C: The SRs were determined to be 12.3 or 3.9 for 5′-C2T5, 15.2 or 2.2 for 5′-T2G5, and 47.9 or 5.1 for 5′-A2C5, respective of the reaction temperature. The observation that misincorporation aptitude is high for T is consistent with the reported low selectivity of Therminator polymerase, an engineered 9°N variant, for dTTP.37 Furthermore, the presence of Mn2+ has been associated with a reduced DNA polymerase specificity.38-40 It should be noted that the outcomes in Figure 6a represent the upper bound of misincorporation kinetics, as the mismatch conditions use a single type of 3′-NH2-ddNTP.
Figure 6.

Effect of template on 9°N-NRQS-catalyzed extensions of the 3′-amino-terminated primer. (A) Investigation of the NP-DNA polymerase activity using the nucleotide-template mismatch assays at 55 °C (top) and 65 °C (bottom) after 24 h of incubation. Each reaction contained 1 μM 3′-amino-G primer, 1 μM DNA template, 100 μM 3′-NH2-ddNTP, 1 mM Mn2+, and 1 μM 9°N-NRQS in 1× ThermoPol buffer at pH 8.8. Selectivity ratio (SR) for each template-nucleotide combination was calculated based on the ratio of [total band intensity of the extension products for a given 3′-NH2-ddNTP]/[total band intensity of all extension products combined]. Heatmaps range from red (minimum) to blue (maximum), with values normalized to the lowest SR as 1.0 in each row separately. (B) Investigation of the polymerase activity of 9°N-NRQS on templates with mixed sequences. Reaction mixtures (i–iv) contained 1 μM 3′-amino-G primer, 1 μM DNA template, 200 μM each of 3′-NH2-ddNTP or dNTP, 1 mM Mn2+, and 5 μM 9°N-NRQS in 1× ThermoPol buffer at pH 8.8. Reaction temperature: 65 °C.
We investigated NP-DNA polymerase activity of 9°N-NRQS with DNA templates containing mixed sequences (Figure 6B, conditions i and ii). The extensions of the 15 nt 3′-amino-G primer on 5′-ATGCATGC (i) and 5′-AATGCTGC (ii) in the presence of all 3′-NH2-ddNTPs were examined. The comparison of these two templates was intended to gain insight into the effect of addition of T on the primer-extension efficiency. Within 24 h of incubation at 65 °C, the +3 and +4 products were observed for the mixtures including templates 5′-ATGCATGC and 5′-AATGCTGC, respectively. At the 48 h mark, the relative abundance of these extension products reached up to 34% and 15%. Replacing A with T on the template resulted in a greater extension efficiency, thus providing a relatively longer (+4) extension product. The absence of ≥ +5 products suggests that either 9°N-NRQS loses catalytic activity after 24 h of incubation or the emerging structure of the extending NP-DNA primer on a DNA template stalls 9°N-NRQS. Although either proposition may be correct, no major extension beyond four consecutive N3′ → P5′ phosphoramidate linkages was observed using a 16 nt primer containing an N─P bond between the G15 and G16 residues (5′-FAM-AGCGTGACTGACTGGNPG-NH2).
9°N-NRQS Can Synthesize Chimeric DNA–NP-DNA Copolymers in One Pot.
In contrast to 9°N, 9°N-NRQS displayed polymerase activity for extending both the DNA and NP-DNA primers (Figure 4B). Therefore, we reasoned that 9°N-NRQS can be utilized to generate relatively long polymers containing both NP-DNA and DNA residues from a mixture of diverse nucleotide building blocks in a one-pot reaction (Figure 6B, conditions iii and iv). Toward this end, a mixture of 3′-NH2-ddNTP and dNTP monomers was used for extending the 15 nt 3′-amino-G primer on 5′-AATGCTGC. The reaction mixture containing 3′-NH2-ddGTP, 3′-NH2-ddCTP, dATP, and dTTP (iii) generated the +4 product within 1 h. Gel analysis showed that the +2 product, formed via addition of C from 3′-NH2-ddCTP, was rapidly converted into +3 with the addition of A from dATP. Indeed, when dATP was excluded from the monomer pool, primarily the +2 product was observed (Figure S5A). With all nucleotide monomers present, traces of the full length (+8) product was observed within 24 h. The yield of the +8 product was further enhanced when the reaction mixture contained 3′-NH2-ddGTP, dCTP, dATP, and dTTP (iv). The +4 and +8 products were observed within 10 min and 2 h, respectively. Their relative abundance reached up to 18% and 9% after a total incubation time of 3 h. When 3′-NH2-ddGTP was excluded from the monomer pool, no extension product was observed (Figure S5B), indicating that the initial N─P bond formation step with 3′-NH2-ddGTP on C-template was necessary for subsequent additions on dNTPs. Taken together, these results show the unique versatility of 9°N-NRQS in synthesizing nucleic acid copolymers on mixed DNA templates in a sequence-specific manner.
DISCUSSION
The formation of the N3′ → P5′ phosphoramidate bond was observed when Asp-404, one of the two metal-binding Asp residues in the active site, was replaced with Asn (9°N-N). As anticipated, an active-site residue with little affinity toward a divalent metal but more polar than aliphatic or aromatic counterparts could thus allow phosphorylation of a primer 3′-amino group. Further metal-binding-site remodeling aimed at altering the electrostatics, hydrogen bonding, and hydrophobicity of the catalytic site by additional single substitutions resulted in additional, albeit marginal, increases in activity. These results highlight the possibilities as well as limits on repurposing a highly evolved active site for a promiscuous activity by solely focusing on screening mutations in the catalytic site. As enzyme activity and selectivity likely emerge by the collective action of many protein structural elements, subsequent exploration of substitutions was focused on the distal subdomains of the polymerase, which were expected to be key elements underlying processivity of 9°N based on previous studies with KOD.34 Substitutions A485R, L489Q, and N491S provided 9°N-NRQS as the most active variant (among all 26 variants expressed), with up to ~13-fold enhanced kinetics at 55 °C compared to 9°N-N. Investigation of the effect of temperature on 3′-amino primer extension via 9°N-NRQS indicated that the extension rate at 65 °C was faster than that at 55 °C. At 65 °C, 9°N-NRQS produced internucleotidyl N─P bonds with a kobs further enhanced by nearly 90%, which corresponds to an ~21-fold enhanced kinetics compared to that measured for 9°N-N at 55 °C. The kobs values were measured as 0.27 ± 0.04 and 0.204 ± 0.013 h−1 for the primer extensions on 5′-A2C5 and 5′-C2T5 DNA templates with 3′-NH2-ddGTP and 3′-NH2-ddATP, respectively.
It is crucial to note that 9°N-NRQS has substantially reduced polymerase activity for extending the canonical 3′-hydroxyl DNA primer. DNA extension from dGTP using 9°N-NRQS is the slowest in the presence of Mg2+ at 55 °C. As a consequence, this variant displays an unnatural polymerase activity (catalyzing NP-DNA extension) similar to what was a natural activity (catalyzing DNA extension) prior to the active-site mutations. It is interesting to note that unlike examples of evolutionary trajectories in natural enzymes,26 there is a clear trade-off between the original and designed promiscuous activities of 9°N. Such a precipitous trade-off was previously observed between deaminase (original) and organophosphate hydrolase (designed) activities in another redesigned metalloenzyme.27
WT DNA polymerases have evolved to catalyze the formation of internucleotidyl O─P linkages in genetic polymers. 9°N, an archaeal B-family DNA polymerase, can accommodate non-canonical deoxynucleoside triphosphate monomers for extending primers but does not catalyze the formation of internucleotidyl N─P linkages. We hypothesized that the 3′-amino group of a primer could serve as a nucleophile in an engineered 9°N active site with altered metal-binding properties while maintaining the sterics required to accommodate the nucleophile. Through a mechanism-guided, structure-based design approach, we performed systematic active-site mutagenesis to transform 9°N into a catalyst that can slowly extend 3′-amino terminated primers with 3′-NH2-ddNTP monomers.
Our experimental results show that 9°N-NRQS can extend 3′-amino-G primer on DNA templates at rates similar to those measured for nonenzymatic template-directed extensions with the chemically activated nucleotide building blocks, such as 3′-amino-nucleoside phosphoroimidazolides.21 In addition, the results suggest that catalytic-site distal elements may also play a key role in the proper positioning of substrates leading to a significant enhancement of the promiscuous activity of the enzyme. Further improvement in the 3′-amino primer-extension rate is necessary to make 9°N, KOD, or other ortholog polymerases viable biocatalysts for the synthesis of long NP-DNA sequences, which may be achieved through directed evolution. Further processivity-enhancing polymerase mutations discovered in other studies, such as those reported for Tgo,41 can conceivably be incorporated into these designs. More detailed mechanistic investigations using computationally intensive methods, such as QM/MM, may shed further light on the phosphoramidate bond formation in the active site of these bespoke enzymes.
9°N-NRQS possesses a unique capability of template-dependent synthesis of diverse nucleic acid polymer types, including DNA, NP-DNA, and DNA/NP-DNA chimeras. Copolymers of DNA and its analogs with non-natural internucleotidyl linkages may provide access to unique structural and functional properties. For example, DNA/NP-DNA chimeras could adopt complex, potentially DNA/RNA-like, tertiary structures while exhibiting higher levels of resistance against nucleolytic degradation. However, these properties of chimeric informational polymers with non-natural backbones remain underexplored. Future studies capitalizing on the engineered promiscuity of 9°N-NRQS would allow systematic investigations of nucleic acid copolymers obtained enzymatically from easily accessible nucleoside triphosphate building blocks.
The work described here provides insights on the catalytic mechanisms and latent promiscuity in metalloenzymes. Specifically, we demonstrated that the highly tuned environment in the metal-binding enzyme active site can also be highly susceptible to substitution of metal-chelating residues and can provide a general strategy to explore novel activities in metalloenzymes by redesign. One underexplored yet captivating novel enzyme activity is to generate XNAs, which are emerging molecular tools with major potential in synthetic biology, biotechnology, and molecular medicine.42-45 Recent reports showed that these nucleic acid analogs, whether in the homopolymeric or hybrid form, can store and transfer information11,46-48 as well as evolve to bind a molecular target49-52 or catalyze a reaction.53 Accordingly, we expect that novel polymerase functions, combined with the expansion of genetic information space, will continue to push the boundaries of designer biological systems.
METHODS
Computational Modeling Using Rosetta.
PyRosetta54 was used to analyze the enzyme structure and perform in silico mutagenesis. To explore substitutions that were expected to alter the microenvironment of Asp404, we substituted all neighboring residues that are non-catalytic to all 19 amino acids. All residue pairs with Cα─Cα distance <5.5 Å were considered neighbors, and residue pairs with Cα─Cα distance <11 Å were also considered neighbors if their Cα─Cβ vectors were at an angle <75°.55 Structural models for each variant were computed by replacing the reference side chain atomic coordinates in the starting model generated from the crystal structures of 9°N polymerase (PDB codes: 5OMV and 5OMQ) with those of the substituted amino acid(s) and performing three rounds of Monte Carlo optimization of rotamers for all side chains (except those involved in catalysis) falling within an 8 Å radius of the substitution(s) followed by gradient-based energy minimization of the entire structure, with atom positional restraints on the backbone, incoming monomer, and the primer–template complex to limit significant changes to backbone geometry.56 Computed structural model optimizations were performed with the REF2015.57 Energetic consequences of amino acid substitutions were determined by performing identical side chain optimization and energy minimization on both wild-type and substituted models thrice and subtracting the total energy of the lowest-scoring wild-type model from that of the lowest-scoring substituted model.58 Energy changes within 3 Rosetta Energy Units for each residue position were considered acceptable, and the resulting models were visually inspected to choose substitutions for experimental characterization.
MD Simulations.
MD simulations were performed in truncated octahedral boxes containing TIP4P-Ew59 water molecules, having a buffer distance of 12 Å with the solute. Monovalent ions (Na+ and Cl−) were added to neutralize the boxes and provide them with ionic strengths of 150 mM. Simulations were run with full periodic boundary conditions. The smooth particle mesh Ewald (PME) method60 with a 12 Å cutoff was used to calculate long range electrostatic interactions, and the SHAKE61 algorithm was used to constrain all bonds involving hydrogen. Langevin dynamics with a collision frequency (γ) of 5.0 was employed to maintain constant temperature during simulations. For constant pressure and temperature simulations, the Berendsen barostat62 with a pressure relaxation time of 1 ps and isotropic pressure scaling was used along with Langevin dynamics (γ = 5.0). After equilibrating the system through several stages that held either pressure or volume constant and varied temperature,63 production trajectories of 400–600 ns were computed at 298 K in the canonical ensemble (i.e., constant NVT) with a 1 fs time step. All simulations were performed with the AMBER18 software package,64 using the pmemd.cuda program65 and employing the Amber ff14SB force field66,67 for proteins and bsc168 force field for DNAs.
Protein Expression and Purification.
Site-Directed Mutagenesis (SDM) and Transformation.
All the mutations in the plasmid were incorporated by SDM (see Tables S1 and S2) and were sequence-confirmed by commercial Sanger sequencing (Genwiz). Plasmids containing the N-terminal His tag and Amp resistance gene were transformed into BL21(DE3) competent cells using the heat shock method and were selected on the LB plate with Amp (100 μg/mL).
Starter Culture and IPTG Induction.
5 mL of LB containing Amp (100 μg/mL) was inoculated with a single isolated colony and was grown at 37 °C overnight. The next day, the overnight culture was used to inoculate the 500 mL of LB with Amp (100 μg/mL) and grown at 37 °C until the optical density (OD600) of the culture reached 0.4–0.6. Protein expression was induced by the addition of IPTG to a final concentration of 500 μM. The sample was shaken at 18 °C for a minimum of 18 h.
Protein Purification.
Purification of the 9°N variants was performed based on a previously reported protocol69 with modifications. The cells were harvested by centrifugation (4000g, 30 min, 4 °C) and resuspended in 30 mL of lysis buffer. Cells were lysed by sonication for 10 min on ice. Cell lysate was incubated first in a 80 °C water bath for 60 min to denature the endogenous E. coli proteins and then on ice for 30 min. Cell debris and endogenous proteins were removed by centrifugation (50,000g, 4 °C, 30 min). To remove the nucleic acids, 10% PEI was added to a final concentration of 0.5% PEI. The solution was incubated on ice for 15 min and centrifuged (50,000g, 4 °C, 30 min). To remove the excess PEI and precipitate the protein, ammonium sulfate was added to a final concentration of 60%. Protein was precipitated (50,000g, 4 °C, 30 min), and the pellet was resuspended in wash buffer and filtered through a 0.22 μm syringe filter. The supernatant was then added to a Ni-NTA gravity column, which was pre-equilibrated with the wash buffer and manually eluted with the elution buffer (~15 mL). The eluted protein was dialyzed overnight into the buffer containing 0.5 mM EDTA to remove divalent metal ions to get apo-protein. The dialyzed protein buffer was exchanged with the polymerase storage buffer using a PD10 column, and the isolated fraction was concentrated to 10 μM using a 30 kDa cutoff Amicon centrifugal filter (Millipore). The protein expression was validated by SDS-PAGE gel analysis (see Figure S1).
Synthesis, Isolation and Purification of 3′-Amino-Primers.
The 15 nt 3′-amino primer was synthesized by the following protocol: The 14 nt primer precursor (5 μM), 5′-TTGGGGGCCAGTCAGTCACGCT (also referred to as 5′-T2G5 template) (5 μM), 3′-NH2-ddGTP (100 μM), Mg+2 (1 mM), and 9°N-WT (5 μM) were incubated in 1× ThermoPol buffer at 12 °C for 10 min. The 16 nt 3′-amino primer was synthesized by the following protocol: The 14-nt primer precursor (5 μM), 5′-AAAAACCAGTCAGTCACGCT (also referred to as 5′-A5C2 template) (5 μM), 3′-NH2-ddGTP (100 μM), Mg+2 (1 mM), and 9°N-WT (5 μM) were incubated in 1× ThermoPol buffer at 65 °C for 3 h. For each protocol, the reaction was quenched by addition of the stop buffer. Reaction contents were loaded on a 1.5 mm-thick 20% polyacrylamide gel, and the presence of the +1 product (15 nt 3′-amino primer) or +2 product (16 nt 3′-amino primer) was verified by gel imaging. The 3′-amino primer band was excised using a clean scalpel, isolated gel slab was finely crushed, and the resulting particulates were transferred to a 15 mL tube. The 3′-amino primer was then eluted from the gel particulates using a previously reported freeze–thaw method.70 After the elution process, the isolated filtrate was lyophilized overnight. The dried pellet was resuspended in 1 mL of nuclease-free water, to which 100 μL of sodium acetate (3 M, pH 5.5) followed by 2.2 mL of ethanol was added. The resulting mixture was chilled at −20 °C overnight and then centrifuged (20,000 rcf, 4 °C, 1 h) to precipitate the 3′-amino primer. The supernatant was removed, and the resulting pellet was washed with 70% ethanol and centrifuged (20,000 rcf, 4 °C, 15 min). The supernatant was discarded, and the pellet was resuspended in nuclease-free water. The oligonucleotide concentration was determined by UV (NanoDrop) measurements. Product purity was determined as 95% based on 20% denaturing PAGE analysis.
Primer Extension Assays and Kinetics.
Prior to each primer-extension assay, the primer and template strands (1 μM each) and nucleoside triphosphate (100 μM) were dissolved in the ThermoPol buffer. The primer/template complex was formed by heating the mixture at 90 °C for 2 min and subsequently cooling to 4 °C with a ramp rate of 0.1 °C/s using a thermocycler. The polymerase (1 μM) was then added to the cooled mixture. The primer-extension reaction was initiated by the addition of the divalent metal salt (1 mM). Reaction aliquots were taken at specific time intervals and quenched by adding the stop buffer. The resulting mixtures were then treated with formamide (50% v/v), heated at 80 °C for 2 min, and subsequently separated by 20% denaturing PAGE with 7 M urea. Gels were analyzed on a BioRad Chemidoc MP gel imager, and bands were quantified using Image Lab software. The plots for enzyme kinetics were prepared using GraphPad Prism 8 software.
Measurements of kobs, kpol, and Kd.
The pseudo-first-order reaction kinetics for primer-extension reactions were calculated by plotting the percentage of the unreacted primer strand versus incubation time, and observed rate constant, kobs (h−1), was calculated using a single exponential decay fit.71 For kpol and Kd measurement,72 primer-extension assays were performed at various concentrations of 3′-NH2-ddGTP (1, 10, 20, 50, 100, 250, 500, and 1000 μM) with the 3′-amino-G primer (1 μM), 5′-A2C5 DNA template (1 μM), Mn2+ (1 mM), and 9°N-NRQS (1 μM) in 1× ThermoPol buffer at 55 °C. Calculated kobs values for each of these reactions were then plotted against the concentration of 3′–NH2–ddGTP and fitted according to the following equation:
where kpol represents the maximum rate of the nucleotide incorporation and Kd represents the dissociation constant.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. Chaput and his coworkers, A. Ngor and A. Nikoomanzar, for the donation of the 9°N-WT plasmid.
Funding
This work was supported by the American Cancer Society, Institutional Research Grant Early Investigator Award and the Rutgers Cancer Institute of New Jersey NCI Cancer Center Support Grant P30CA072720 and US National Institutes of Health (NIH)/NIBIB Trailblazer Award EB029548 (to E.C.I.), National Science Foundation (NSF) grant CBET-1929237 and NIH grant GM132565 (to S.D.K.), and NIH grant GM107485 (to D.M.Y.). Computational resources were provided by the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF grant ACI-1548562 (COMET and EXPANSE at SDSC through allocation TG-CHE190067, and Frontera at TACC through allocation CHE20002).
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.2c00373.
Materials, chemicals, and biologics; classification and SDS-PAGE analysis of 9°N variants; list of oligonucleotides used in primer extension assays and site directed mutagenesis; divalent metal screen assay; comparative polymerase activity analysis; effect of temperature on 9°N-NRQS activity (PDF)
Contributor Information
Tushar Aggarwal, Department of Chemistry and Chemical Biology, Rutgers University, New Brunswick, New Jersey 08854, United States.
William A. Hansen, Institute for Quantitative Biomedicine, Rutgers University, New Brunswick, New Jersey 08854, United States
Jonathan Hong, Department of Chemistry and Chemical Biology, Rutgers University, New Brunswick, New Jersey 08854, United States.
Abir Ganguly, Institute for Quantitative Biomedicine and Laboratory for Biomolecular Simulation Research, Rutgers University, New Brunswick, New Jersey 08854, United States.
Darrin M. York, Department of Chemistry and Chemical Biology, Institute for Quantitative Biomedicine, and Laboratory for Biomolecular Simulation Research, Rutgers University, New Brunswick, New Jersey 08854, United States; Cancer Institute of New Jersey, Rutgers University, New Brunswick, New Jersey 08901, United States.
Sagar D. Khare, Department of Chemistry and Chemical Biology and Institute for Quantitative Biomedicine, Rutgers University, New Brunswick, New Jersey 08854, United States; Cancer Institute of New Jersey, Rutgers University, New Brunswick, New Jersey 08901, United States
Enver Cagri Izgu, Department of Chemistry and Chemical Biology, Rutgers University, New Brunswick, New Jersey 08854, United States; Cancer Institute of New Jersey and Rutgers Center for Lipid Research and New Jersey Institute for Food, Nutrition, and Health, Rutgers University, New Brunswick, New Jersey 08901, United States.
REFERENCES
- (1).Rothwell PJ; Waksman G Structure and Mechanism of DNA Polymerases. Adv. Protein Chem 2005, 71, 401–440. [DOI] [PubMed] [Google Scholar]
- (2).Yang W; Lee JY; Nowotny M Making and Breaking Nucleic Acids: Two-Mg2+-Ion Catalysis and Substrate Specificity. Mol. Cell 2006, 22, 5–13. [DOI] [PubMed] [Google Scholar]
- (3).Gao Y; Yang W Capture of a Third Mg2+ Is Essential for Catalyzing DNA Synthesis. Science 2016, 352, 1334–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Loakes D; Gallego J; Pinheiro VB; Kool ET; Holliger P Evolving a Polymerase for Hydrophobic Base Analogues. J. Am. Chem. Soc 2009, 131, 14827–14837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Li Q; Maola VA; Chim N; Hussain J; Lozoya-Colinas A; Chaput JC Synthesis and Polymerase Recognition of Threose Nucleic Acid Triphosphates Equipped with Diverse Chemical Functionalities. J. Am. Chem. Soc 2021, 143, 17761–17768. [DOI] [PubMed] [Google Scholar]
- (6).Kempeneers V; Vastmans K; Rozenski J; Herdewijn P Recognition of Threosyl Nucleotides by DNA and RNA Polymerases. Nucleic Acids Res. 2003, 31, 6221–6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ichida JK; Zou K; Horhota A; Yu B; McLaughlin LW; Szostak JW An in Vitro Selection System for TNA. J. Am. Chem. Soc 2005, 127, 2802–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Pinheiro VB; Taylor AI; Cozens C; Abramov M; Renders M; Zhang S; Chaput JC; Wengel J; Peak-Chew S-Y; McLaughlin SH; et al. Synthetic Genetic Polymers Capable of Heredity and Evolution. Science 2012, 336, 341–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Chen T; Hongdilokkul N; Liu Z; Adhikary R; Tsuen SS; Romesberg FE Evolution of Thermophilic DNA Polymerases for the Recognition and Amplification of C2′-Modified DNA. Nat. Chem 2016, 8, 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hoshino H; Kasahara Y; Kuwahara M; Obika S DNA Polymerase Variants with High Processivity and Accuracy for Encoding and Decoding Locked Nucleic Acid Sequences. J. Am. Chem. Soc 2020, 142, 21530–21537. [DOI] [PubMed] [Google Scholar]
- (11).Laos R; Thomson JM; Benner SA DNA Polymerases Engineered by Directed Evolution to Incorporate Non-Standard Nucleotides. Front. Microbiol 2014, 5, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Houlihan G; Arangundy-Franklin S; Holliger P Exploring the Chemistry of Genetic Information Storage and Propagation through Polymerase Engineering. Acc. Chem. Res 2017, 50, 1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tereshko V; Gryaznov S; Egli M Consequences of Replacing the DNA 3′-Oxygen by an Amino Group: High-Resolution Crystal Structure of a Fully Modified N3′→P5′ Phosphoramidate DNA Dodecamer Duplex. J. Am. Chem. Soc 1998, 120, 269–283. [Google Scholar]
- (14).Lelyveld VS; O’Flaherty DK; Zhou L; Izgu EC; Szostak JW DNA Polymerase Activity on Synthetic N3′→P5′ Phosphoramidate DNA Templates. Nucleic Acids Res. 2019, 47, 8941–8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Heidenreich O; Gryaznov S; Nerenberg M RNase H-Independent Antisense Activity of Oligonucleotide N3′→P5′ Phosphoramidates. Nucleic Acids Res. 1997, 25, 776–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Faria M; Spiller DG; Dubertret C; Nelson JS; White MRH; Scherman D; Hélène C; Giovannangeli C Phosphoramidate Oligonucleotides as Potent Antisense Molecules in Cells and in Vivo. Nat. Biotechnol 2001, 19, 40–44. [DOI] [PubMed] [Google Scholar]
- (17).Hanna RL; Gryaznov SM; Doudna JA A Phosphoramidate Substrate Analog Is a Competitive Inhibitor of the Tetrahymena Group I Ribozyme. Chem. Biol 2000, 7, 845–854. [DOI] [PubMed] [Google Scholar]
- (18).Rigl CT; Lloyd DH; Tsou DS; Gryaznov SM; Wilson WD Structural RNA Mimetics: N3′→P5′ Phosphoramidate DNA Analogs of HIV-1 RRE and TAR RNA Form A-Type Helices That Bind Specifically to Rev and Tat-Related Peptides. Biochemistry 1997, 36, 650–659. [DOI] [PubMed] [Google Scholar]
- (19).Kervio E; Hochgesand A; Steiner UE; Richert C Templating Efficiency of Naked DNA. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 12074–12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhang S; Blain JC; Zielinska D; Gryaznov SM; Szostak JW Fast and Accurate Nonenzymatic Copying of an RNA-like Synthetic Genetic Polymer. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 17732–17737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Izgu EC; Oh SS; Szostak JW Synthesis of Activated 3′-Amino-3′-Deoxy-2-Thio-Thymidine, a Superior Substrate for the Nonenzymatic Copying of Nucleic Acid Templates. Chem. Commun 2016, 52, 3684–3686. [DOI] [PubMed] [Google Scholar]
- (22).Hänle E; Richert C Enzyme-Free Replication with Two or Four Bases. Angew. Chem. Int. Ed 2018, 57, 8911–8915. [DOI] [PubMed] [Google Scholar]
- (23).O’Flaherty DK; Zhou L; Szostak JW Nonenzymatic Template-Directed Synthesis of Mixed-Sequence 3′-NP-DNA up to 25 Nucleotides Long Inside Model Protocells. J. Am. Chem. Soc 2019, 141, 10481–10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lelyveld VS; Zhang W; Szostak JW Synthesis of Phosphoramidate-Linked DNA by a Modified DNA Polymerase. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 7276–7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Patel PH; Loeb LA DNA Polymerase Active Site Is Highly Mutable: Evolutionary Consequences. Proc. Natl. Acad. Sci. U. S. A 2000, 97, 5095–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Khersonsky O; Tawfik DS Enzyme Promiscuity: A Mechanistic and Evolutionary Perspective. Annu. Rev. Biochem 2010, 79, 471–505. [DOI] [PubMed] [Google Scholar]
- (27).Khare SD; Kipnis Y; Greisen PJ; Takeuchi R; Ashani Y; Goldsmith M; Song Y; Gallaher JL; Silman I; Leader H; et al. Computational Redesign of a Mononuclear Zinc Metalloenzyme for Organophosphate Hydrolysis. Nat. Chem. Biol 2012, 8, 294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Southworth MW; Kong H; Kucera RB; Ware J; Jannasch HW; Perler FB Cloning of Thermostable DNA Polymerases from Hyperthermophilic Marine Archaea with Emphasis on Thermococcus Sp. 9 Degrees N-7 and Mutations Affecting 3′-5′ Exonuclease Activity. Proc. Natl. Acad. Sci. U. S. A 1996, 93, 5281–5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Duffy K; Arangundy-Franklin S; Holliger P Modified Nucleic Acids: Replication, Evolution, and next-Generation Therapeutics. BMC Biol. 2020, 18, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kropp HM; Betz K; Wirth J; Diederichs K; Marx A Crystal Structures of Ternary Complexes of Archaeal B-Family DNA Polymerases. PLoS One 2017, 12, No. e0188005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Song X An Inhomogeneous Model of Protein Dielectric Properties: Intrinsic Polarizabilities of Amino Acids. J. Chem. Phys 2002, 116, 9359–9363. [Google Scholar]
- (32).Vashishtha AK; Wang J; Konigsberg WH Different Divalent Cations Alter the Kinetics and Fidelity of DNA Polymerases. J. Biol. Chem 2016, 291, 20869–20875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Leaver-Fay A; Tyka M; Lewis SM; Lange OF; Thompson J; Jacak R; Kaufman KW; Renfrew PD; Smith CA; Sheffler W; et al. Chapter Nineteen Rosetta3 An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. Methods Enzymol. 2011, 487, 545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Nikoomanzar A; Vallejo D; Chaput JC Elucidating the Determinants of Polymerase Specificity by Microfluidic-Based Deep Mutational Scanning. ACS Synth. Biol 2019, 8, 1421–1429. [DOI] [PubMed] [Google Scholar]
- (35).LinWu S-W; Tu Y-H; Tsai T-Y; Maestre-Reyna M; Liu M-S; Wu W-J; Huang J-Y; Chi H-W; Chang W-H; Chiou C-F; et al. Thermococcus sp. 9°N DNA Polymerase Exhibits 3′-Esterase Activity that can be Harnessed for DNA Sequencing. Commun. Biol 2019, 2, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kool ET Hydrogen Bonding, Base Stacking, and Steric Effects in DNA Replication. Annu. Rev. Biophys. Biomol. Struct 2001, 30, 1–22. [DOI] [PubMed] [Google Scholar]
- (37).Litosh VA; Wu W; Stupi BP; Wang J; Morris SE; Hersh MN; Metzker ML Improved Nucleotide Selectivity and Termination of 3′-OH Unblocked Reversible Terminators by Molecular Tuning of 2-Nitrobenzyl Alkylated HOMedU Triphosphates. Nucleic Acids Res. 2011, 39, e39–e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Tabor S; Richardson CC Effect of Manganese Ions on the Incorporation of Dideoxynucleotides by Bacteriophage T7 DNA Polymerase and Escherichia Coli DNA Polymerase I. Proc. Natl. Acad. Sci. U. S. A 1989, 86, 4076–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Cadwell RC; Joyce GF Randomization of Genes by PCR Mutagenesis. Genome Res. 1992, 2, 28–33. [DOI] [PubMed] [Google Scholar]
- (40).Horhota A; Zou K; Ichida JK; Yu B; McLaughlin LW; Szostak JW; Chaput JC Kinetic Analysis of an Efficient DNA-Dependent TNA Polymerase. J. Am. Chem. Soc 2005, 127, 7427–7434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Liu C; Cozens C; Jaziri F; Rozenski J; Maréchal A; Dumbre S; Pezo V; Marlière P; Pinheiro VB; Groaz E; et al. Phosphonomethyl Oligonucleotides as Backbone-Modified Artificial Genetic Polymers. J. Am. Chem. Soc 2018, 140, 6690–6699. [DOI] [PubMed] [Google Scholar]
- (42).Herdewijn P; Marlière P Toward Safe Genetically Modified Organisms through the Chemical Diversification of Nucleic Acids. Chem. Biodiversity 2009, 6, 791–808. [DOI] [PubMed] [Google Scholar]
- (43).Taylor AI; Arangundy-Franklin S; Holliger P Towards Applications of Synthetic Genetic Polymers in Diagnosis and Therapy. Curr. Opin. Chem. Biol 2014, 22, 79–84. [DOI] [PubMed] [Google Scholar]
- (44).Chaput JC Redesigning the Genetic Polymers of Life. Acc. Chem. Res 2021, 54, 1056–1065. [DOI] [PubMed] [Google Scholar]
- (45).Wang Y; Nguyen K; Spitale RC; Chaput JC A Biologically Stable DNAzyme That Efficiently Silences Gene Expression in Cells. Nat. Chem 2021, 13, 319–326. [DOI] [PubMed] [Google Scholar]
- (46).Malyshev DA; Dhami K; Lavergne T; Chen T; Dai N; Foster JM; Corrêa IR; Romesberg FE A Semi-Synthetic Organism with an Expanded Genetic Alphabet. Nature 2014, 509, 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Chen Z; Lichtor PA; Berliner AP; Chen JC; Liu DR Evolution of Sequence-Defined Highly Functionalized Nucleic Acid Polymers. Nat. Chem 2018, 10, 420–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Hoshika S; Leal NA; Kim M-J; Kim M-S; Karalkar NB; Kim H-J; Bates AM; Watkins NE; SantaLucia HA; Meyer AJ; DasGupta S; Piccirilli JA; Ellington AD; SantaLucia J Jr.; Georgiadis MM; Benner SA Hachimoji DNA and RNA: A Genetic System with Eight Building Blocks. Science 2019, 363, 884–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).McCloskey CM; Li Q; Yik EJ; Chim N; Ngor AK; Medina E; Grubisic I; Keh LCT; Poplin R; Chaput JC Evolution of Functionally Enhanced A-l-Threofuranosyl Nucleic Acid Aptamers. ACS Synth. Biol 2021, 10, 3190–3199. [DOI] [PubMed] [Google Scholar]
- (50).Kong D; Yeung W; Hili R In Vitro Selection of Diversely Functionalized Aptamers. J. Am. Chem. Soc 2017, 139, 13977–13980. [DOI] [PubMed] [Google Scholar]
- (51).Kimoto M; Yamashige R; Matsunaga K; Yokoyama S; Hirao I Generation of High-Affinity DNA Aptamers Using an Expanded Genetic Alphabet. Nat. Biotechnol 2013, 31, 453–457. [DOI] [PubMed] [Google Scholar]
- (52).Yu H; Zhang S; Chaput JC Darwinian Evolution of an Alternative Genetic System Provides Support for TNA as an RNA Progenitor. Nat. Chem 2012, 4, 183–187. [DOI] [PubMed] [Google Scholar]
- (53).Taylor AI; Pinheiro VB; Smola MJ; Morgunov AS; Peak-Chew S; Cozens C; Weeks KM; Herdewijn P; Holliger P Catalysts from Synthetic Genetic Polymers. Nature 2015, 518, 427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Chaudhury S; Lyskov S; Gray JJ PyRosetta: A Script-Based Interface for Implementing Molecular Modeling Algorithms Using Rosetta. Bioinformatics 2010, 26, 689–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Lubin JH; Zardecki C; Dolan EM; Lu C; Shen Z; Dutta S; Westbrook JD; Hudson BP; Goodsell DS; Williams JK; et al. Evolution of the SARS-CoV-2 Proteome in Three Dimensions (3D) during the First 6 Months of the COVID-19 Pandemic. Proteins 2022, 90, 1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Nivón LG; Moretti R; Baker D A Pareto-Optimal Refinement Method for Protein Design Scaffolds. PLoS One 2013, 8, No. e59004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Alford RF; Leaver-Fay A; Jeliazkov JR; O’Meara MJ; DiMaio FP; Park H; Shapovalov MV; Renfrew PD; Mulligan VK; Kappel K; et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput 2017, 13, 3031–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Kellogg EH; Leaver-Fay A; Baker D Role of Conformational Sampling in Computing Mutation-induced Changes in Protein Structure and Stability. Proteins: Struct., Funct., Bioinf 2011, 79, 830–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Horn HW; Swope WC; Pitera JW Characterization of the TIP4P-Ew Water Model: Vapor Pressure and Boiling Point. J. Chem. Phys 2005, 123, 194504. [DOI] [PubMed] [Google Scholar]
- (60).Darden T; York D; Pedersen L Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- (61).Ryckaert J-P; Ciccotti G; Berendsen HJC Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
- (62).Berendsen HJC; Postma JPM; van Gunsteren WF; DiNola A; Haak JR Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys 1984, 81, 3684–3690. [Google Scholar]
- (63).Ganguly A; Weissman BP; Giese TJ; Li N-S; Hoshika S; Rao S; Benner SA; Piccirilli JA; York DM Confluence of Theory and Experiment Reveals the Catalytic Mechanism of the Varkud Satellite Ribozyme. Nat. Chem 2020, 12, 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Lee T-S; Cerutti DS; Mermelstein D; Lin C; LeGrand S; Giese TJ; Roitberg A; Case DA; Walker RC; York DM GPU-Accelerated Molecular Dynamics and Free Energy Methods in Amber18: Performance Enhancements and New Features. J. Chem. Inf. Model 2018, 58, 2043–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Salomon-Ferrer R; Götz AW; Poole D; Le Grand S; Walker RC Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput 2013, 9, 3878–3888. [DOI] [PubMed] [Google Scholar]
- (66).Cornell WD; Cieplak P; Bayly CI; Gould IR; Merz KM; Ferguson DM; Spellmeyer DC; Fox T; Caldwell JW; Kollman PA A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc 1995, 117, 5179–5197. [Google Scholar]
- (67).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Ivani I; Dans PD; Noy A; Pérez A; Faustino I; Hospital A; Walther J; Andrio P; Goñi R; Balaceanu A; et al. Parmbsc1: a refined force field for DNA simulations. Nat. Methods 2016, 13, 55–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Nikoomanzar A; Dunn MR; Chaput JC Engineered Polymerases with Altered Substrate Specificity: Expression and Purification. Curr. Protoc. Nucleic Acid Chem 2017, 69, 4.75.1–4.75.20. [DOI] [PubMed] [Google Scholar]
- (70).Chen Z; Ruffner DE Modified Crush-and-Soak Method for Recovering Oligodeoxynucleotides from Polyacrylamide Gel. Bio-Techniques 1996, 21, 820–822. [DOI] [PubMed] [Google Scholar]
- (71).Blain JC; Ricardo A; Szostak JW Synthesis and Nonenzymatic Template-Directed Polymerization of 2′-Amino-2′-deoxythreose Nucleotides. J. Am. Chem. Soc 2014, 136, 2033–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Gardner AF; Wang J; Wu W; Karouby J; Li H; Stupi BP; Jack WE; Hersh MN; Metzker ML Rapid Incorporation Kinetics and Improved Fidelity of a Novel Class of 3′-OH Unblocked Reversible Terminators. Nucleic Acids Res. 2012, 40, 7404–7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.