

Abstract
Quantum chemical calculations are performed to explore if the reactivity of diarylethene switches toward photocyclization can be controlled by the excited-state aromaticity of their bridging π-linker. Using an archetypal diarylethene with a non-aromatic π-linker as a reference, completely different outcomes are found when the π-linker is allowed to become either aromatic (no reaction) or antiaromatic (fast reaction) upon photoexcitation. The results demonstrate a possibility to use the excited-state aromaticity concept for actual modulation of photochemical reactivity.
Introduction
The use of the excited-state aromaticity (ESA) and antiaromaticity (ESAA) concepts1 to describe cyclic, conjugated organic molecules in their electronically excited states has in the past decade or so witnessed a resurgence,2 helping explain and predict both photophysical properties and photochemical reactivity of such compounds in a variety of contexts. For example, as for photophysical properties, these concepts have been employed to explain differences in Stokes shifts of benzoxazole fluorophores,3 to design chromophores with singlet-triplet energy gaps amenable to singlet-fission photovoltaics,4 and to design red-light fluorophores based on a simple benzene core.5 Regarding photochemical reactivity, in turn, the concepts have proven very useful in finding ways to improve the quantum yields of E/Z photoisomerizations that power light-driven molecular motors,6 in identifying the driving force for excited-state proton transfer7 and conformational planarization8 reactions, and in understanding the mechanisms of electron-catalyzed photodissociation processes.9
As for the impact of aromaticity on other types of photochemical reactions, which has been less investigated, electrocyclic reactions of molecular photoswitches are a curious case in that the presence of an aromatic moiety can have both positive10 and negative11 consequences for these transformations. As an example of the former situation, a study of diarylethene switches12 recently presented both experimental and computational evidence indicating that the insertion of benzene as the bridging π-linker between the two aryl units (in the form of thienyl groups) facilitates the photocyclization of the ring-open form of the corresponding dithienylbenzene into its ring-closed form.10 Specifically, it was found that the photocyclization is driven by the complete loss of aromaticity in the benzene moiety upon photoexcitation, which creates a reactive, antiaromatic excited state in the Franck–Condon (FC) region that allows ring-closing to occur in an energetically downhill fashion through the subsequent relief of this antiaromaticity.10 Contrarily, in a study of the photoinduced conversion of a benzannulated dihydroazulene switch into the corresponding vinylheptafulvene isomer, it was noted that the loss of aromaticity in the benzene motif from the initial electronic excitation is only partial, whereby the relief of the remaining aromaticity during the subsequent excited-state evolution introduces a barrier for the reaction.11
In light of these findings that photoinduced changes in aromaticity can influence electrocyclic reactions of different photoswitches so differently, it becomes pertinent to explore the extent to which it is possible to control these reactions by modulating the ESA among a specific type of switches. The present work reports the first investigation of this important but hitherto unresolved problem. To this end, we here model the photocyclization reactions of the three diarylethenes shown in their ring-open (1o/2o/3o) and ring-closed (1c/2c/3c) isomeric forms in Scheme 1, by performing both quantum chemical calculations and non-adiabatic molecular dynamics (NAMD) simulations.131o is the aforementioned dithienylbenzene switch featuring an aromatic benzene bridge that becomes antiaromatic in the FC region of the lowest singlet excited state (S1) populated by UV absorption.102o, in turn, is a dithienylcyclobutadiene compound where the benzene bridge is replaced by an antiaromatic cyclobutadiene motif that, conversely, turns aromatic upon UV absorption (this will be demonstrated below). 3o, finally, is a dithienylcyclohexene compound where benzene instead is replaced by a non-aromatic cyclohexene motif that remains non-aromatic after UV absorption (this will also be demonstrated below).
Scheme 1. Photocyclization Reactions Studied in This Work.

With these systems, our work encompasses all three relevant scenarios for the character of the photoactive excited state in the FC region, ranging from antiaromatic in 1o to non-aromatic in 3o and aromatic in 2o. By comparing the results for 1o and 2o, the calculations will reveal whether the photocyclization reactivity is indeed tunable by ESA. Furthermore, by comparing the results for 1o/2o with those for 3o, whose non-aromatic cyclohexene bridge best represents available diarylethene switches12 (including systems with a perfluorocyclohexene bridge14) considered for applications in, for example, molecular electronics15 and photopharmacology,16 the calculations will also assess whether the photocyclization reactivity achieved by ESA-tuned diarylethenes parallels that achieved by typical diarylethenes.
Results and Discussion
The full technical details of the modeling, carried out at levels of theory previously benchmarked against experimental UV–vis absorption data,10 are given in the Supporting Information.
Static Modeling of Photocyclization Reactions
First, the photocyclization reactions of 1o/2o/3o into 1c/2c/3c were modeled by performing quantum chemical calculations using the B3LYP hybrid density functional in combination with the cc-pVTZ basis set and the SMD continuum solvation approach17 to model an acetonitrile solvent, which was used in the aforementioned experimental studies of 1o.10 In these calculations, which are summarized in Figure 1, the S1 state of the different species was described in the framework of time-dependent density functional theory (TD-DFT).18 Besides modeling the photocyclization processes, the reaction free energies and free energy barriers for the corresponding thermal electrocyclization and cycloreversion (i.e., ring-opening) processes in the ground S0 state were also calculated (at the same level of theory). These results are presented in Figure S1 and Table S1 in the Supporting Information.
Figure 1.

Calculated photocyclization paths in the S1 state of 1o (a), 2o (b), and 3o (c) with energies (ΔE) in each case given relative to the most stable species in the S0 state (1o, 2c, and 3o, respectively). Also shown are the S0 energies at the S1 geometries along the photocyclization paths. Complementary calculations performed using other (than B3LYP) quantum chemical methods that are discussed in the Supporting Information (see Tables S2 and S3) corroborate the shapes of these paths.
From Figure 1, wherein the reaction coordinate starting from the vertically excited S1 FC point is taken to be the C1–C1′ distance (the atom numbering is given in Scheme 1), it can first be seen that the calculated photocyclization paths of 1o and 2o are distinctly different. For 1o, the reaction is completely barrierless and enables the S1 state to gradually come closer and closer to the S0 state in a region of a presumed S1/S0 conical intersection (CI) seam that connects the photocyclization path to 1c. Notably, reinforcing our previous results on 1o obtained using a different density functional than B3LYP (ωB97X-D),10 the motion toward this seam is found to have an appreciable driving force, as revealed by the fact that the S1 energy at C1–C1′ = 1.78 Å (where the S1/S0 gap is the smallest) lies 1.55 eV below the FC point at 3.38 Å. For 2o, on the other hand, there is a minimum at 3.55 Å along the photocyclization path, in the near vicinity of the FC point at 3.61 Å, and any further motion toward shorter C1–C1′ distances is impeded by a continuous increase in the S1 energy. Accordingly, from this comparison of 1o and 2o, it does appear that the photocyclization reactivity of diarylethene switches can be tuned through the aromatic character of the π-linker between the two aryl units. Moreover, since the calculated photocyclization path of 1o has the same favorable features as that of 3o (see Figure 1), which represents a typical diarylethene with a non-aromatic π-linker,12 it seems possible for diarylethenes that are tuned in this way to be fully competitive in terms of their intrinsic photochemical performance.
Regarding the thermal electrocyclization reactions, in turn, the calculated free energies are markedly different for 1o and 2o (see Table S1), with 1o having a very large barrier (181 kJ mol–1) and a distinctly positive reaction energy (112 kJ mol–1), and 2o showing a much smaller barrier (104 kJ mol–1) and a negative reaction energy (−24 kJ mol–1). This difference reflects that the π-linker loses aromaticity in the reaction of 1o and antiaromaticity in the reaction of 2o. Furthermore, it is also of interest to compare the +112 kJ mol–1 reaction energy of 1o with the +46 kJ mol–1 reaction energy of 3o, which has a non-aromatic π-linker. This comparison reveals that diarylethenes such as 1o/1c are an interesting prospect for applications in molecular solar thermal energy storage,19 because of the high energy content of their ring-closed forms. At the same time, such applications would require the ring-closed forms to be stable toward thermal cycloreversion, which could be a challenge. Indeed, the calculations predict that 1c is more susceptible to this process than 3c (cf. barriers of 70 and 142 kJ mol–1, respectively, in Table S1). A similar trade-off has previously been documented for related diarylethenes.20
NAMD-Based Modeling of Photocyclization Reactions
Although Figure 1 offers a clear picture of the differences between 1o and 2o and of the similarities between 1o and 3o as to their photocyclization reactivity, it should be noted that the underlying calculations are based on a predefined C1–C1′ reaction coordinate, whereby any influence of competing side reactions is neglected. Moreover, because of their static nature, calculations of this type cannot predict the time scales over which the photocyclizations occur. Therefore, in order to circumvent these limitations, NAMD simulations of 1o–3o were performed for maximally 300 fs at the B3LYP/cc-pVDZ level of theory using Tully’s fewest switches algorithm,21 as further described in the Supporting Information. These simulations extend the scope of our previous NAMD-based study of the photocyclization dynamics of 1o,10 both by now considering diarylethenes with different excited-state character and by running many more trajectories (25 instead of 10), whereby the reaction efficiency can be assessed with a higher degree of statistical certainty.
The results from the NAMD simulations, launched at the respective S1 FC point, are summarized in Figure 2 and Figures S3 and S4 in the Supporting Information. Starting with the 2o system, Figure S3b shows that the S1 and S0 states approach degeneracy almost instantly in the simulations, in keeping with the small magnitudes of the S1/S0 energy gaps along the corresponding photocyclization path in Figure 1b. However, as revealed by Figure 2b, none of the 2o trajectories evolve toward shorter C1–C1′ distances, which is consistent with the photocyclization path being energetically unfavorable. Combined with the fact that all 25 trajectories decay to the S0 state already within 25 fs (see Figure S4b), this means that there is no dynamical preference for ring-closing to occur. Thus, rather than producing the 2c photoproduct, the excited-state dynamics reforms the initial 2o species.
Figure 2.

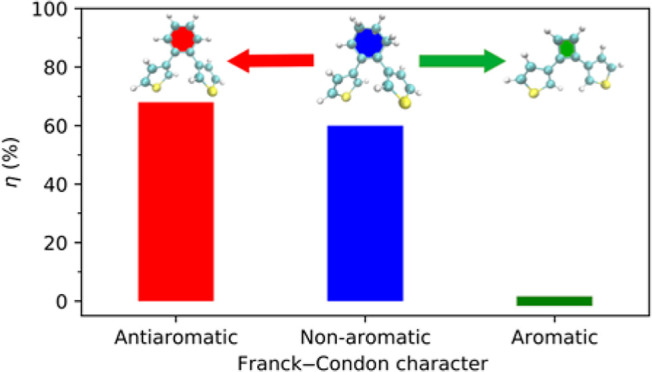
Changes in C1–C1′ distances during the 25 NAMD trajectories run for each of 1o (a), 2o (b), and 3o (c). As described in the NAMD section of the Supporting Information, the trajectories are separated into successful and failed ones that, respectively, complete and do not complete photocyclization within 300 fs. Percentage values refer to the proportions of successful trajectories.
Continuing with the 1o and 3o systems, the situation is markedly different. Here, the fast (<100 fs) approaching of degeneracy observed in Figure S3a,c is accompanied by pronounced decreases in the C1–C1′ distances (see Figure 2a,c), which conform to the barrierless shapes of the corresponding photocyclization paths (see Figure 1a,c) and show that, for both systems, ring-closing is a major component of the excited-state dynamics. In fact, after 200 fs, almost all of the two sets of trajectories reside in the S0 state (see Figure S4a,c) and more than half oscillate around a C1–C1′ distance of 1.5 Å (see Figure 2a,c). In other words, within 200 fs, more than half of the trajectories complete the photocyclizations by producing the 1c and 3c photoproducts, seemingly without any strong competition from side reactions. Qualitatively, this time scale agrees with experimental measurements of the ring-closing dynamics of diarylethenes switches in different solvents.22 At the end of the simulations, 68 and 60% of the trajectories have formed the 1c and 3c photoproducts, respectively, with the two hydrogen atoms at the C1 and C1′ carbons consistently adopting an anti conformation (see Scheme 1) as a result of the ring-closing occurring in a conrotatory fashion, in accordance with the Woodward–Hoffmann rules. Although the slightly lower efficiency of the reaction of 3o is compensated for by it being somewhat faster (the average photocyclization time, calculated as described in the NAMD section of the Supporting Information, is ∼30 fs shorter for 3o than for 1o), these differences between the two systems are too small to allow for any quantitative investigation of their origin by the present simulations.
Calculated Aromaticity Indices
Having found by both static quantum chemical calculations and NAMD simulations that 1o and 2o are very different with respect to their photocyclization reactivity, it remains to establish whether this can be understood in terms of a difference in the aromatic character of their π-linkers following photoexcitation. In fact, such a difference can be anticipated from the observation in Figure 1 that the photon energy required to excite 2o is only half of that needed to excite 1o. Accordingly, for 2o, the photon energy may be insufficient to overcome any barrier for the photocyclization.
To this end, the complete active space self-consistent field (CASSCF) method23 was used to calculate nucleus-independent chemical shift (NICS) indices24 for the π-linkers of 1o and 2o (as well as 3o) at key points along the corresponding photocyclization paths, as further detailed in the Supporting Information. Briefly, providing a magnetic measure of aromaticity, these indices were calculated through a NICS-scan procedure25 to obtain NICSzz values24b at distances 1.50/1.60/1.70/1.80/1.90/2.00 Å above the geometric centers of the π-linkers. This procedure was applied equally to the S0 and S1 states and was adopted to avoid having to choose (more arbitrarily) one specific distance at which to calculate a single NICSzz value.25 Furthermore, the rationale for choosing the current scan interval is to simultaneously minimize both the contributions from σ-electrons to the induced magnetic field25b and the offset between the center of the field and the normal axis passing through the geometric center of the π-linker.25d,25e For ease of analysis, the resulting values were averaged over the different distances to yield mean NICSzz values (<NICSzz>) that are presented in Figure 3. The full set of NICSzz values are given in Table S4 in the Supporting Information. Moreover, the Supporting Information also includes a discussion of results (summarized in Figures S5 and S6) from calculations of electronic aromaticity indices that support the NICS-based analysis below.
Figure 3.

<NICSzz> values for the benzene/cyclobutadiene/cyclohexene π-linkers of 1o/2o/3o calculated at key points along the corresponding photocyclization paths. The points are presented from left to right in the order in which they appear along the paths (see Table S4 for the corresponding C1–C1′ distances).
Noting that negative/positive NICS values reflect aromaticity/antiaromaticity,24aFigure 3 clearly indicates that the difference in photocyclization reactivity between 1o and 2o can be explained from the aromatic character of their π-linkers following photoexcitation. Specifically, while photoexcitation of 1o generates a reactive excited state through the conversion of its benzene π-linker into an antiaromatic moiety in the S1 FC region (the <NICSzz> value changes from −20 to 43 ppm), photoexcitation of 2o conversely produces a non-reactive excited state via the transformation of its cyclobutadiene π-linker into an aromatic motif in the S1 FC region (the <NICSzz> value changes from 11 to −15 ppm). At the nearby minimum along the photocyclization path of 2o (see Figure 1b), this aromaticity is further strengthened (the <NICSzz> value is lowered to −26 ppm), which explains why any additional ring-closing motion beyond the minimum is energetically unfavorable.
In sharp contrast to the distinct photoinduced changes in aromaticity shown by 1o and 2o, the cyclohexene π-linker of 3o maintains its non-aromatic character throughout the photocyclization process, consistently exhibiting <NICSzz> values close to zero (see Figure 3). Pleasingly, the stark differences among 1o–3o documented in Figure 3 are further reinforced by complementary NICSzz calculations, as summarized in Figure S7 in the Supporting Information, in which the aforementioned 1.50–2.00 Å scan interval was extended to 1.00–2.50 Å. In this regard, it may be noted that it is not uncommon to consider even shorter distances than 1.00 Å for such calculations.25c However, as alluded to above, this will typically exaggerate predictions of aromatic/antiaromatic character, because of the ensuing contamination of the induced magnetic field by σ-electrons.25b At the same time, at short distances, this effect is somewhat countered by the offset between the center of the field and the geometric ring center, which tends to lead to an underestimation of the strength of the field.25d,25e
Besides photoinduced aromaticity, another potential reason for the non-reactivity of 2o is ring strain in the four-membered π-linker. In order to assess this possibility, a photocyclization path was also calculated for a diarylethene switch 4o in which the antiaromatic cyclobutadiene π-linker of 2o is replaced by another four-membered motif (cyclobutene, see Figure S8a in the Supporting Information) that is non-aromatic and remains non-aromatic during the reaction. The resulting path is shown in Figure S8b in the Supporting Information. Notably, the path is completely barrierless. Hence, it seems unlikely that ring strain is a reason for the non-reactivity of 2o. Rather, the only discernible impediment to the photocyclization of 2o is the photoinduced aromaticity of the cyclobutadiene π-linker.
As indicated above and discussed in the Supporting Information, the scenarios predicted by the NICS data in Figure 3 are corroborated by the calculation of electronic aromaticity indices. Particularly, focusing on Shannon aromaticity (SA) indices,26 the −log10(SA) values for the different π-linkers in Figure S6 confirm that the photoexcited S1 state of 1o/2o experiences a marked loss/gain in aromaticity relative to the S0 state (the values decrease/increase by more than 1 unit), whereas the character of the S1 state of 3o is altered to a lesser extent, seemingly becoming somewhat more aromatic (the values increase by ∼0.5 units). At the same time, even these relatively minor increases in the −log10(SA) values may be too large to support quantitatively the prediction by the NICS data that the cyclohexene π-linker of 3o remains, in an absolute sense, non-aromatic in S1. In this regard, it is worth noting that, especially for excited states, there are no well-established boundaries between aromatic/non-aromatic/antiaromatic systems in terms of their typical −log10(SA) values.
Conclusions
In summary, based on quantum chemical calculations and NAMD simulations, we have discovered that it is possible to tune the photocyclization reactivity of diarylethene switches by modulating the ESA of the π-linker between the two aryl units. Furthermore, we have found that this tuning can be implemented without taking away from the reactivity achieved by archetypal diarylethenes12 featuring a non-aromatic π-linker. Given the many possible areas of application for these switches,12,15,16 a natural goal of future research is to also find ways to maximize their reactivity through this approach.
Computational Methods
The full computational details are given in the Supporting Information. Briefly, the photocyclization reactions of 1o/2o/3o into 1c/2c/3c were modeled with DFT and TD-DFT18 by performing static quantum chemical calculations and NAMD simulations at the B3LYP/cc-pVTZ/SMD and B3LYP/cc-pVDZ levels of theory, respectively. NICS indices at key points along the respective photocyclization path were calculated using CASSCF/cc-pVDZ and (12,12)/(12,12)/(10,10) active spaces for the reactions of 1o/2o/3o. The corresponding SA indices, in turn, were calculated at the B3LYP/cc-pVTZ/SMD level of theory. Static DFT and TD-DFT calculations were performed using Gaussian 16.27 NAMD simulations were performed using TURBOMOLE 7.4.28 NICS indices were calculated using Dalton 2016.2.29 SA indices were calculated using Multiwfn 3.7.30
Acknowledgments
This work was supported by the Olle Engkvist Foundation (grant 204-0183), the Swedish Research Council (grant 2019-03664), ÅForsk (grant 20-570), and the Carl Trygger Foundation (grant CTS 20:102). The calculations were enabled by resources provided by (a) the Swedish National Infrastructure for Computing at the National Supercomputer Centre partially funded by the Swedish Research Council (grant 2018-05973) and (b) the National Supercomputer Centre funded by Linköping University.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.2c01172.
Computational details, Cartesian coordinates and electronic energies of different geometries of 1–3, and additional results (stationary points and free energies for thermal electrocyclization and cycloreversion reactions, vertical excitation energies and FC relaxation energies, molecular orbitals, analyses of NAMD trajectories, aromaticity indices, and photocyclization path of 4o) (PDF)
Animation of a representative trajectory from the NAMD simulations of 1o (MP4)
Animation of a representative trajectory from the NAMD simulations of 2o (MP4)
Animation of a representative trajectory from the NAMD simulations of 3o (MP4)
The authors declare no competing financial interest.
Supplementary Material
References
- a Baird N. C. Quantum Organic Photochemistry. II. Resonance and Aromaticity in the Lowest 3ππ* State of Cyclic Hydrocarbons. J. Am. Chem. Soc. 1972, 94, 4941–4948. 10.1021/ja00769a025. [DOI] [Google Scholar]; b Karadakov P. B. Ground- and Excited-State Aromaticity and Antiaromaticity in Benzene and Cyclobutadiene. J. Phys. Chem. A 2008, 112, 7303–7309. 10.1021/jp8037335. [DOI] [PubMed] [Google Scholar]; c Ottosson H. Exciting Excited-State Aromaticity. Nat. Chem. 2012, 4, 969–971. 10.1038/nchem.1518. [DOI] [PubMed] [Google Scholar]
- a Rosenberg M.; Dahlstrand C.; Kilså K.; Ottosson H. Excited State Aromaticity and Antiaromaticity: Opportunities for Photophysical and Photochemical Rationalizations. Chem. Rev. 2014, 114, 5379–5425. 10.1021/cr300471v. [DOI] [PubMed] [Google Scholar]; b Kim J.; Oh J.; Osuka A.; Kim D. Porphyrinoids, a Unique Platform for Exploring Excited-State Aromaticity. Chem. Soc. Rev. 2022, 51, 268–292. 10.1039/d1cs00742d. [DOI] [PubMed] [Google Scholar]
- Lampkin B. J.; Nguyen Y. H.; Karadakov P. B.; VanVeller B. Demonstration of Baird’s Rule Complementarity in the Singlet State with Implications for Excited-State Intramolecular Proton Transfer. Phys. Chem. Chem. Phys. 2019, 21, 11608–11614. 10.1039/c9cp02050k. [DOI] [PubMed] [Google Scholar]
- a Fallon K. J.; Budden P.; Salvadori E.; Ganose A. M.; Savory C. N.; Eyre L.; Dowland S.; Ai Q.; Goodlett S.; Risko C.; Scanlon D. O.; Kay C. W. M.; Rao A.; Friend R. H.; Musser A. J.; Bronstein H. Exploiting Excited-State Aromaticity to Design Highly Stable Singlet Fission Materials. J. Am. Chem. Soc. 2019, 141, 13867–13876. 10.1021/jacs.9b06346. [DOI] [PubMed] [Google Scholar]; b El Bakouri O.; Smith J. R.; Ottosson H. Strategies for Design of Potential Singlet Fission Chromophores Utilizing a Combination of Ground-State and Excited-State Aromaticity Rules. J. Am. Chem. Soc. 2020, 142, 5602–5617. 10.1021/jacs.9b12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.; Park W.; Kim Y.; Filatov M.; Choi C. H.; Lee D. Relief of Excited-State Antiaromaticity Enables the Smallest Red Emitter. Nat. Commun. 2021, 12, 5409. 10.1038/s41467-021-25677-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Oruganti B.; Wang J.; Durbeej B. Excited-State Aromaticity Improves Molecular Motors: A Computational Analysis. Org. Lett. 2017, 19, 4818–4821. 10.1021/acs.orglett.7b02257. [DOI] [PubMed] [Google Scholar]; b Durbeej B.; Wang J.; Oruganti B. Molecular Photoswitching Aided by Excited-State Aromaticity. ChemPlusChem 2018, 83, 958–967. 10.1002/cplu.201800307. [DOI] [PubMed] [Google Scholar]; c Wang J.; Oruganti B.; Durbeej B. A Straightforward Route to Aromatic Excited States in Molecular Motors that Improves Photochemical Efficiency. ChemPhotoChem 2019, 3, 450–460. 10.1002/cptc.201800268. [DOI] [Google Scholar]
- a Gutiérrez-Arzaluz L.; Cortés-Guzmán F.; Rocha-Rinza T.; Peón J. Ultrafast Excited State Hydrogen Atom Transfer in Salicylideneaniline Driven By Changes in Aromaticity. Phys. Chem. Chem. Phys. 2015, 17, 31608–31612. 10.1039/c5cp03699b. [DOI] [PubMed] [Google Scholar]; b Nishina N.; Mutai T.; Aihara J. Excited-State Intramolecular Proton Transfer and Global Aromaticity. J. Phys. Chem. A 2017, 121, 151–161. 10.1021/acs.jpca.6b11684. [DOI] [PubMed] [Google Scholar]; c Wu C.-H.; Karas L. J.; Ottosson H.; Wu J. I. Excited-State Proton Transfer Relieves Antiaromaticity in Molecules. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 20303–20308. 10.1073/pnas.1908516116. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Karas L. J.; Wu C.-H.; Ottosson H.; Wu J. I. Electron-Driven Proton Transfer Relieves Excited-State Antiaromaticity in Photoexcited DNA Base Pairs. Chem. Sci. 2020, 11, 10071–10077. 10.1039/d0sc02294b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hada M.; Saito S.; Tanaka S.; Sato R.; Yoshimura M.; Mouri K.; Matsuo K.; Yamaguchi S.; Hara M.; Hayashi Y.; Röhricht F.; Herges R.; Shigeta Y.; Onda K.; Miller R. J. D. Structural Monitoring of the Onset of Excited-State Aromaticity in a Liquid Crystal Phase. J. Am. Chem. Soc. 2017, 139, 15792–15800. 10.1021/jacs.7b08021. [DOI] [PubMed] [Google Scholar]; b Yamakado T.; Takahashi S.; Watanabe K.; Matsumoto Y.; Osuka A.; Saito S. Conformational Planarization versus Singlet Fission: Distinct Excited-State Dynamics of Cyclooctatetraene-Fused Acene Dimers. Angew. Chem., Int. Ed. 2018, 57, 5438–5443. 10.1002/anie.201802185. [DOI] [PubMed] [Google Scholar]; c Toldo J.; El Bakouri O.; Solà M.; Norrby P.-O.; Ottosson H. Is Excited-State Aromaticity a Driving Force for Planarization of Dibenzannelated 8π-Electron Heterocycles?. ChemPlusChem 2019, 84, 712–721. 10.1002/cplu.201900066. [DOI] [PubMed] [Google Scholar]; d Kotani R.; Liu L.; Kumar P.; Kuramochi H.; Tahara T.; Liu P.; Osuka A.; Karadakov P. B.; Saito S. Controlling the S1 Energy Profile by Tuning Excited-State Aromaticity. J. Am. Chem. Soc. 2020, 142, 14985–14992. 10.1021/jacs.0c05611. [DOI] [PubMed] [Google Scholar]
- Banerjee A.; Halder D.; Ganguly G.; Paul A. Deciphering the Cryptic Role of a Catalytic Electron in a Photochemical Bond Dissociation Using Excited State Aromaticity Markers. Phys. Chem. Chem. Phys. 2016, 18, 25308–25314. 10.1039/c6cp03789e. [DOI] [PubMed] [Google Scholar]
- Oruganti B.; Pál Kalapos P. P.; Bhargav V.; London G.; Durbeej B. Photoinduced Changes in Aromaticity Facilitate Electrocyclization of Dithienylbenzene Switches. J. Am. Chem. Soc. 2020, 142, 13941–13953. 10.1021/jacs.0c06327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skov A. B.; Ree N.; Gertsen A. S.; Chabera P.; Uhlig J.; Lissau J. S.; Nucci L.; Pullerits T.; Mikkelsen K. V.; Nielsen M. B.; Sølling T. I.; Hansen T. Excited-State Topology Modifications of the Dihydroazulene Photoswitch Through Aromaticity. ChemPhotoChem 2019, 3, 619–629. 10.1002/cptc.201900183. [DOI] [Google Scholar]
- a Irie M. Diarylethenes for Memories and Switches. Chem. Rev. 2000, 100, 1685–1716. 10.1021/cr980069d. [DOI] [PubMed] [Google Scholar]; b Irie M.; Fukaminato T.; Matsuda K.; Kobatake S. Photochromism of Diarylethene Molecules and Crystals: Memories, Switches, and Actuators. Chem. Rev. 2014, 114, 12174–12277. 10.1021/cr500249p. [DOI] [PubMed] [Google Scholar]
- a Barbatti M. Nonadiabatic Dynamics with Trajectory Surface Hopping Method. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1, 620–633. 10.1002/wcms.64. [DOI] [Google Scholar]; b Tapavicza E.; Bellchambers G. D.; Vincent J. C.; Furche F. Ab Initio Non-Adiabatic Molecular Dynamics. Phys. Chem. Chem. Phys. 2013, 15, 18336–18348. 10.1039/c3cp51514a. [DOI] [PubMed] [Google Scholar]
- a Yuan K.; Boixel J.; Le Bozec H.; Boucekkine A.; Doucet H.; Guerchais V.; Jacquemin D. Perfluorocyclohexene Bridges in Inverse Diarylethenes: Synthesis through Pd-Catalyzed C–H Bond Activation, Experimental and Theoretical Studies on Their Photoreactivity. Chem. Commun. 2013, 49, 7896–7898. 10.1039/c3cc43754j. [DOI] [PubMed] [Google Scholar]; b Hatano E.; Morimoto M.; Hyodo K.; Yasuda N.; Yokojima S.; Nakamura S.; Uchida K. Photosalient Effect of a Diarylethene with a Perfluorocyclohexene Ring. Chem.—Eur. J. 2016, 22, 12680–12683. 10.1002/chem.201603020. [DOI] [PubMed] [Google Scholar]; c Hatano E.; Morimoto M.; Imai T.; Hyodo K.; Fujimoto A.; Nishimura R.; Sekine A.; Yasuda N.; Yokojima S.; Nakamura S.; Uchida K. Photosalient Phenomena that Mimic Impatiens Are Observed in Hollow Crystals of Diarylethene with a Perfluorocyclohexene Ring. Angew. Chem., Int. Ed. 2017, 56, 12576–12580. 10.1002/anie.201706684. [DOI] [PubMed] [Google Scholar]
- a Orgiu E.; Crivillers N.; Herder M.; Grubert L.; Pätzel M.; Frisch J.; Pavlica E.; Duong D. T.; Bratina G.; Salleo A.; Koch N.; Hecht S.; Samorì P. Optically Switchable Transistor via Energy-Level Phototuning in a Bicomponent Organic Semiconductor. Nat. Chem. 2012, 4, 675–679. 10.1038/nchem.1384. [DOI] [PubMed] [Google Scholar]; b El Gemayel M.; Börjesson K.; Herder M.; Duong D. T.; Hutchison J. A.; Ruzié C.; Schweicher G.; Salleo A.; Geerts Y.; Hecht S.; Orgiu E.; Samorì P. Optically Switchable Transistors by Simple Incorporation of Photochromic Systems into Small-Molecule Semiconducting Matrices. Nat. Commun. 2015, 6, 6330. 10.1038/ncomms7330. [DOI] [PubMed] [Google Scholar]
- Lerch M. M.; Hansen M. J.; van Dam G. M.; Szymanski W.; Feringa B. L. Emerging Targets in Photopharmacology. Angew. Chem., Int. Ed. 2016, 55, 10978–10999. 10.1002/anie.201601931. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Casida M. E.; Huix-Rotllant M. Progress in Time-Dependent Density-Functional Theory. Annu. Rev. Phys. Chem. 2012, 63, 287–323. 10.1146/annurev-physchem-032511-143803. [DOI] [PubMed] [Google Scholar]
- a Cacciarini M.; Skov A. B.; Jevric M.; Hansen A. S.; Elm J.; Kjaergaard H. G.; Mikkelsen K. V.; Nielsen M. B. Towards Solar Energy Storage in the Photochromic Dihydroazulene-Vinylheptafulvene System. Chem.—Eur. J. 2015, 21, 7454–7461. 10.1002/chem.201500100. [DOI] [PubMed] [Google Scholar]; b Sun C.-L.; Wang C.; Boulatov R. Applications of Photoswitches in the Storage of Solar Energy. ChemPhotoChem 2019, 3, 268–283. 10.1002/cptc.201900030. [DOI] [Google Scholar]
- a Kitagawa D.; Nakahama T.; Nakai Y.; Kobatake S. 1,2-Diarylbenzene as Fast T-Type Photochromic Switch. J. Mater. Chem. C 2019, 7, 2865–2870. 10.1039/c8tc05357j. [DOI] [Google Scholar]; b Nakahama T.; Kitagawa D.; Kobatake S. Tuning of Optical Properties and Thermal Cycloreversion Reactivity of Photochromic Diarylbenzene by Introducing Electron-Donating Substituents. J. Phys. Chem. C 2019, 123, 31212–31218. 10.1021/acs.jpcc.9b09953. [DOI] [Google Scholar]; c Hamatani S.; Kitagawa D.; Kobatake S. Fast T-Type Photochromic Crystals of Diarylbenzene. J. Phys. Chem. C 2021, 125, 4588–4594. 10.1021/acs.jpcc.0c10988. [DOI] [Google Scholar]
- a Tully J. C. Molecular Dynamics with Electronic Transitions. J. Chem. Phys. 1990, 93, 1061–1071. 10.1063/1.459170. [DOI] [Google Scholar]; b Tapavicza E.; Meyer A. M.; Furche F. Unravelling the Details of Vitamin D Photosynthesis by Non-Adiabatic Molecular Dynamics Simulations. Phys. Chem. Chem. Phys. 2011, 13, 20986–20998. 10.1039/c1cp21292c. [DOI] [PubMed] [Google Scholar]
- a Bertarelli C.; Gallazzi M. C.; Stellacci F.; Zerbi G.; Stagira S.; Nisoli M.; De Silvestri S. Ultrafast Photoinduced Ring-Closure Dynamics of a Diarylethene Polymer. Chem. Phys. Lett. 2002, 359, 278–282. 10.1016/s0009-2614(02)00706-6. [DOI] [Google Scholar]; b Ishibashi Y.; Fujiwara M.; Umesato T.; Saito H.; Kobatake S.; Irie M.; Miyasaka H. Cyclization Reaction Dynamics of a Photochromic Diarylethene Derivative as Revealed by Femtosecond to Microsecond Time-Resolved Spectroscopy. J. Phys. Chem. C 2011, 115, 4265–4272. 10.1021/jp112370a. [DOI] [Google Scholar]
- Roos B. O.; Taylor P. R.; Siegbahn P. E. M. A Complete Active Space SCF Method (CASSCF) Using a Density Matrix Formulated Super-CI Approach. Chem. Phys. 1980, 48, 157–173. 10.1016/0301-0104(80)80045-0. [DOI] [Google Scholar]
- a Schleyer P. v. R.; Maerker C.; Dransfeld A.; Jiao H.; van Eikema Hommes N. J. R. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. 10.1021/ja960582d. [DOI] [PubMed] [Google Scholar]; b Fallah-Bagher-Shaidaei H.; Wannere C. S.; Corminboeuf C.; Puchta R.; Schleyer P. v. R. Which NICS Aromaticity Index for Planar π Rings Is Best?. Org. Lett. 2006, 8, 863–866. 10.1021/ol0529546. [DOI] [PubMed] [Google Scholar]
- a Stanger A. Nucleus-Independent Chemical Shifts (NICS): Distance Dependence and Revised Criteria for Aromaticity and Antiaromaticity. J. Org. Chem. 2006, 71, 883–893. 10.1021/jo051746o. [DOI] [PubMed] [Google Scholar]; b Gershoni-Poranne R.; Stanger A. The NICS-XY-Scan: Identification of Local and Global Ring Currents in Multi-Ring Systems. Chem. - Eur. J. 2014, 20, 5673–5688. 10.1002/chem.201304307. [DOI] [PubMed] [Google Scholar]; c Yuan B.; Zhuang J.; Kirmess K. M.; Bridgmohan C. N.; Whalley A. C.; Wang L.; Plunkett K. N. Pentaleno[1,2-a:4,5’]diacenaphthylenes: Uniquely Stabilized Pentalene Derivatives. J. Org. Chem. 2016, 81, 8312–8318. 10.1021/acs.joc.6b01480. [DOI] [PubMed] [Google Scholar]; d Stanger A. Reexamination of NICSπ,zz: Height Dependence, Off-Center Values, and Integration. J. Phys. Chem. A 2019, 123, 3922–3927. 10.1021/acs.jpca.9b02083. [DOI] [PubMed] [Google Scholar]; e Stanger A. NICS—Past and Present. Eur. J. Org. Chem. 2020, 2020, 3120–3127. 10.1002/ejoc.201901829. [DOI] [Google Scholar]
- Noorizadeh S.; Shakerzadeh E. Shannon Entropy as a New Measure of Aromaticity, Shannon Aromaticity. Phys. Chem. Chem. Phys. 2010, 12, 4742–4749. 10.1039/b916509f. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford CT, 2016.
- a Furche F.; Ahlrichs R.; Hättig C.; Klopper W.; Sierka M.; Weigend F. Turbomole. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2014, 4, 91–100. 10.1002/wcms.1162. [DOI] [Google Scholar]; b TURBOMOLE, V7.4 2019, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007; TURBOMOLE GmbH, 2007. http://www.turbomole.com (accessed April 25, 2022).
- a Aidas K.; Angeli C.; Bak K. L.; Bakken V.; Bast R.; Boman L.; Christiansen O.; Cimiraglia R.; Coriani S.; Dahle P.; Dalskov E. K.; Ekström U.; Enevoldsen T.; Eriksen J. J.; Ettenhuber P.; Fernández B.; Ferrighi L.; Fliegl H.; Frediani L.; Hald K.; Halkier A.; Hättig C.; Heiberg H.; Helgaker T.; Hennum A. C.; Hettema H.; Hjertenaes E.; Høst S.; Høyvik I.-M.; Iozzi M. F.; Jansík B.; Jensen H. J. A.; Jonsson D.; Jørgensen P.; Kauczor J.; Kirpekar S.; Kjaergaard T.; Klopper W.; Knecht S.; Kobayashi R.; Koch H.; Kongsted J.; Krapp A.; Kristensen K.; Ligabue A.; Lutnaes O. B.; Melo J. I.; Mikkelsen K. V.; Myhre R. H.; Neiss C.; Nielsen C. B.; Norman P.; Olsen J.; Olsen J. M. H.; Osted A.; Packer M. J.; Pawlowski F.; Pedersen T. B.; Provasi P. F.; Reine S.; Rinkevicius Z.; Ruden T. A.; Ruud K.; Rybkin V. V.; Sałek P.; Samson C. C. M.; de Merás A. S.; Saue T.; Sauer S. P. A.; Schimmelpfennig B.; Sneskov K.; Steindal A. H.; Sylvester-Hvid K. O.; Taylor P. R.; Teale A. M.; Tellgren E. I.; Tew D. P.; Thorvaldsen A. J.; Thøgersen L.; Vahtras O.; Watson M. A.; Wilson D. J. D.; Ziolkowski M.; Ågren H. The Dalton Quantum Chemistry Program System. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2014, 4, 269–284. 10.1002/wcms.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dalton, A Molecular Electronic Structure Program, Release Dalton 2016.2. 2016, http://daltonprogram.org (accessed July 25, 2022).
- Lu T.; Chen F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.