

An easy synthetic route towards ethyl 1-(3-tosylquinolin-4-yl)piperidine-4-carboxylate was found. Its molecular and crystal structures are described as well and the biological activity is also predicted using molecular docking studies.
Keywords: ethyl 1-(3-tosylquinolin-4-yl)piperidine-4-carboxylate, antibacterial properties, molecular and crystal structure, Hirshfeld surface analysis, pairwise interaction energy, molecular docking
Abstract
The title compound, C24H26N2O4S, can be obtained via two synthetic routes. According to our investigations, the most suitable way is by the reaction of ethyl 2-bromoacetate with sodium tosylsulfinate in dry DMF. It was crystallized from methanol into the monoclinic P21/n space group with a single molecule in the asymmetric unit. Hirshfeld surface analysis was performed to define the hydrogen bonds and analysis of the two-dimensional fingerprint plots was used to distinguish the different types of interactions. Two very weak non-classical C—H⋯O hydrogen bonds were found and the contributions of short contacts to the Hirshfeld surface were determined. Molecules form an isotropic network of intermolecular interactions according to an analysis of the pairwise interaction energies. A molecular docking study evaluated the interactions in the title compound with the active centers of macromolecules of bacterial targets (Staphylococcus aureus DNA Gyrase PDB ID: 2XCR, Mycobacterium tuberculosis topoisomerase II PDB ID: 5BTL, Streptococcus pneumoniae topoisomerase IV PDB ID: 4KPF) and revealed high affinity towards them that exceeded the reference antibiotics of the fluoroquinolone group.
1. Chemical context
Quinolone-based compounds have become strikingly conspicuous in recent years. Generally, quinolone derivatives can possess antibacterial, antiparasitic and antiviral (including malaria, hepatitis, HIV, herpes), anticancer and immunosuppressant activities. They can be used in the treatment of obesity, diabetes and neurodegenerative diseases (Horta et al., 2017 ▸). Thus, in this work, we decided to broaden the scope of the quinolone scaffolds utilized in our previous works (Bylov et al., 1999 ▸; Silin et al., 2004 ▸; Savchenko et al., 2007 ▸; Hryhoriv et al., 2021 ▸) toward a promising new class of arylsulfonylquinolin derivatives, namely ethyl 1-(3-tosylquinolin-4-yl)piperidine-4-carboxylate.
Effective synthetic approaches toward these compounds are versatile. The most notable among them are green chemistry methods and microwave-assisted synthesis (Dhiman et al., 2019 ▸; Atechian et al. 2007 ▸). However, to date, very few data are available for arylsulfonylquinolins. Kang et al. (2016 ▸) described a straightforward and mild one-pot method to synthesize 3-(phenylsulfonyl)-2,3-dihydro-4(1H)quinolinones via a Cu-catalyzed aza-Michael addition/base-mediated cyclization reaction. Other researchers (Ivachtchenko et al. 2012a
▸,b
▸) have reported new 3-(phenylsulfonyl)quinoline derivatives as serotonin 5-HT receptor antagonists, performed molecular docking studies, and proposed them for preventing and treating central nervous system (CNS) diseases such as psychiatric disorders, schizophrenia, anxiety disorders, and obesity. The preparation method for 3-methanesulfonylquinolines such as GABA-B enhancers was patented by Malherbe et al. (2006 ▸). In vivo investigations of 4-amino-3-arylsulfoquinolin derivatives as metabotropic glutamate 5(mGlu) receptor negative allosteric modulators have shown efficacy for treating anxiety and depression (Galambos et al., 2017 ▸).

In the present paper, we study an optimal synthetic route for ethyl 1-(3-tosylquinolin-4-yl)piperidine-4-carboxylate and report its molecular and crystal structures as well as potential biological properties.
2. Structural commentary
The asymmetric unit contains one molecule of ethyl 1-(3-tosylquinolin-4-yl)piperidine-4-carboxylate (Fig. 1 ▸). The presence of two bulky substituents in vicinal positions at the pyridine ring results in a rotation of the piperidine ring with respect to the bicyclic fragment [the dihedral angle between their mean planes is 76.83 (13)°]. The piperidine ring adopts a chair conformation with puckering parameters (Zefirov et al., 1990 ▸) S = 1.16 (1), Θ = 0.6 (1)°, Ψ = 66.2 (12)°. The atoms N2 and C19 deviate from the mean plane of the other ring atoms by −0.640 (2) and 0.675 (3) Å, respectively. The atom N2 has a pyramidal configuration with a bond-angle sum of 345.4°. The ethyl ester group is located in an equatorial position with respect to the piperidine ring [the C17—C18—C19—C22 torsion angle is 179.8 (2)°]. It is disordered over the two positions (A and B) due to rotation around the C19—C22 bond with an occupancy ratio of 0.562 (12):0.438 (12). The ethyl group is almost orthogonal to the carboxylic fragment in conformer A and is located in the intermediate position between +ac and ap in conformer B [the C22—O4—C23—C24 torsion angle is −98.1 (14) and 150 (2)° in conformers A and B, respectively]. The tolyl substituent is located in a -sc position relative to the endocyclic C7—C8 bond [C7—C8—S1—C10 = −71.5 (3)°] and rotated about the C8—S1 bond [C8—S1—C10—C11 = 124.9 (2)°].
Figure 1.

Molecular structure of the title compound. Displacement ellipsoids are shown at the 50% probability level.
3. Supramolecular features
Regarding the van der Waals radii proposed in Bondi (1964 ▸) for all atoms except for the hydrogens (Rowland & Taylor, 1996 ▸), the analysis of intermolecular interactions revealed two very weak non-classical hydrogen bonds, C4—H4⋯O3A and C5—H5⋯O2 (Table 1 ▸). The first is formed by an oxygen atom of the carboxylic group and a hydrogen atom of the benzene ring (Fig. 2 ▸
a). An oxygen atom of the sulfonyl group is involved in the second hydrogen bond, similarly with a hydrogen atom of the benzene ring (Fig. 2 ▸
b). Connected with the initial molecule by the symmetry operations x −
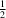 , −y +
, −y +
 , z +
and x +
, −y +
, z +
, these hydrogen bonds are affected by both twofold screw axes <010> and glide plane family {010}. On their own, these hydrogen bonds form the chains in the [10
, z +
and x +
, −y +
, z +
, these hydrogen bonds are affected by both twofold screw axes <010> and glide plane family {010}. On their own, these hydrogen bonds form the chains in the [10
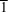 ] and [101] directions, respectively.
] and [101] directions, respectively.
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C4—H4⋯O3A i | 0.93 | 2.52 | 3.421 (16) | 163 |
| C5—H5⋯O2ii | 0.93 | 2.58 | 3.415 (4) | 149 |
Symmetry codes: (i)
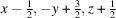 ; (ii)
; (ii)
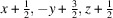 .
.
Figure 2.

Crystal packing of the chains built with the hydrogen bonds C4—H4⋯O3A (a) and C5—H5⋯O2 (b) in cyan. Projection in the [010] direction.
4. Hirshfeld surface analysis
The complementation of the Hirshfeld surface, i.e. the surface splitting the regions of crystal into molecular domains within the ratio of promolecular to procrystal electronic density, with geometric parameters, especially the normalized contact distance (d
norm), implemented in CrystalExplorer17 (Spackman et al., 2021 ▸) allowed us to distinguish the intermolecular interactions in a more thorough way. The standard ‘high’ surface resolution was used. Two regions with d
norm significantly lower than the van der Waals contact length (in red) emerge on the surface (Fig. 3 ▸
a). Both of them concern the C4—H4⋯O3A hydrogen bond and show it to be the sole directed interaction in the crystal. The chains built up by these hydrogen bonds are parallel to the [
01] direction. However, they cannot be considered as a structural motif because the aforementioned hydrogen bonds are very weak, exist solely for conformer A and one of them was not revealed for conformer B. At the same time, the short contact C24B⋯O1 appears just for conformer B (Fig. 3 ▸
b). Differences in the distribution of d
norm for the two conformers and so the short contacts and hydrogen bonds can be easily be seen from the two projections (top and bottom) shown in Fig. 3 ▸.
Figure 3.

Distribution of the value d norm onto the Hirshfeld surfaces of the conformers A (a) and B (b).
In addition to the Hirshfeld surface analysis, the 2D fingerprint plots were computed for ethyl 1-(3-tosylquinolin-4-yl)piperidine-4-carboxylate. The contributions of the three types of intermolecular contacts get to areas with the values of the internal and external distances (d i and d e) below the van der Waals radii of the corresponding atoms (Fig. 4 ▸). These contributions belong to the short O⋯H, C⋯H and H⋯H contacts. They are 20.2% and 19.9% of the Hirshfeld surface area for H⋯O/O⋯H for the disorder components A and B, respectively, 16.7% and 17.7%, respectively, for C⋯H/H⋯C and 54.3% for H⋯H. The differences for the disordered positions A and B can be explained by the rearrangement of the interactions network described above.
Figure 4.

Contributions of the O⋯H/H⋯O (a), C⋯H/H⋯C (b) and H⋯H (c) contacts to the fingerprint plots built using the Hirsfeld surfaces of conformer A.
5. Analysis of the pairwise interaction energies
The strength of the non-classical C—H⋯O hydrogen bonds is often underestimated, as mentioned in Sutor (1962 ▸) and Desiraju (1996 ▸, 2005 ▸). Thus, to extend the knowledge of the supramolecular structure of the title compound and to prove the small contribution of these interactions to the structure, analysis of the pairwise interaction energies was performed as proposed by Konovalova et al. (2010 ▸) and Shishkin et al. (2012 ▸). The procedure was implied in a very similar way to the one described in detail in Vaksler et al. (2021 ▸). The single molecule was considered as a building unit. The interactions in the molecular pairs containing the aforementioned hydrogen bonds are −7.4 and −10.5 kcal mol−1 for conformer A and −3.2 and −11.7 kcal mol−1 for conformer B (data given for the C24B⋯O1 short contact and the C5—H5⋯O2 hydrogen bond). These values are comparable to those for the non-directed interactions in other pairs of neighboring molecules. In addition to this, the interaction energy decomposition was performed using an ‘accurate’ energy model in CrystalExplorer17 for the molecular pairs with C—H⋯O hydrogen bonds. It showed that the sum of electrostatic and polarization components is rather low in comparison with the dispersion and repulsion terms (−1.0 versus −7.5 and 2.6 kcal mol−1 for the C4—H4⋯O3A hydrogen bond, −4.6 versus −9.7 and 4.2 kcal mol−1 for C5—H5⋯O2) implying minimal contributions of hydrogen bonds in general bonding. Despite the apparent layering (Fig. 5 ▸ a) parallel to the (010) plane, the energetic structure of the title compound can be considered isotropic, which can easily be seen from the energy vector diagrams (Fig. 5 ▸ b). The total energy of interaction between a basic molecule and its first coordination sphere is −95.8 and −95.5 kcal mol−1 for conformers A and B, respectively.
Figure 5.

Crystal packing of the molecules (a) and energy vector diagrams (b). Projection in the [100] direction.
6. Database survey
A search of the Cambridge Structural Database [Version 5.42, update of November 2020; Groom et al., 2016 ▸] shows no similarities between the title compound and 4-(piperidin-1-yl)-3-sulfone-quinoline derivatives.
7. Molecular docking
A molecular docking study was performed in order to estimate the application efficiency of ethyl 1-(3-tosylquinolin-4-yl)piperidine-4-carboxylate in terms of medicinal chemistry as antimicrobials. For receptor-oriented flexible docking, the Autodock 4.2 software package (Morris et al., 2009 ▸) was used. Ligands were prepared using the MGL Tools 1.5.6 (Sanner, 1999 ▸) and optimized within the Avogadro (Hanwell et al., 2012 ▸) (United Force Field with the steepest descent algorithm). The biotargets were chosen on the basis of structural similarity between the title compound and known antibacterial agents from the group of fluoroquinolones. The active centers of macromolecules of bacterial targets [Staphylococcus aureus DNA Gyrase PDB ID: 2XCR (Bax et al., 2010 ▸); Mycobacterium tuberculosis topoisomerase II PDB ID: 5BTL (Blower et al., 2016 ▸); Streptococcus pneumoniae topoisomerase IV PDB ID: 4KPF (Laponogov et al., 2016 ▸)] from the Protein Data Bank (PDB) were used for docking.
The receptor maps were made with MGL Tools and AutoGrid (Sanner, 1999 ▸). The docking parameters were defined closely to the ones mentioned in Syniugin et al. (2016 ▸; see supporting information). These parameters were chosen to bring the formation of a complex between the tested molecule and the receptor as close as possible to the conditions that exist in biological systems.
Inhibitory activity against bacterial targets can be realized by the formation of their complexes with ligands [as ethyl 1-(3-tosylquinolin-4-yl)piperidine-4-carboxylate]. In turn, the stability of complexes can be estimated from the strength of the intermolecular interactions. The scoring function indicating the enthalpy contribution to the value of the free binding energy (affinity DG), the values of the free binding energy and binding constants [E Doc (kcal mol−1) and Ki (μM)] are represented for the most profitable conformation positions (Table 2 ▸). All the parameters show that the title compound is superior to the reference medicines of the same type.
Table 2. The values of affinity DG, free binding energy, and binding coefficients for the best conformational positions of the title compound in combination with biotargets (PDB ID: 2XCR, 5BTL, 4KPF). Values are also given for reference compounds.
| Molecule | Affinity DG (kcal mol−1) | E Doc (kcal mol−1) | Ki (υM) |
|---|---|---|---|
| PDB ID: 2XCR | |||
| Title compound | −7.5 | −5.62 | 76.15 |
| Ciprofloxacin | −7.2 | −5.10 | 183.79 |
| Norfloxacin | −7.2 | −4.30 | 708.28 |
| PDB ID: 5BTL | |||
| Title compound | −8.2 | −5.64 | 73.02 |
| Ciprofloxacin | −7.5 | −5.51 | 91.69 |
| Norfloxacin | −7.8 | −5.25 | 142.92 |
| PDB ID: 4KPF | |||
| Title compound | −8.1 | −6.13 | 31.90 |
| Ciprofloxacin | −7.4 | −5.38 | 113.52 |
| Norfloxacin | −7.4 | −4.78 | 315.73 |
8. Synthesis and crystallization
The starting compounds were obtained from commercial sources and were used without further purification.
Two ways were proposed for the synthesis of ethyl 1-(3-tosylquinolin-4-yl)piperidine-4-carboxylate: the classical one and an alternative one with a lower number of steps and higher yield of the final product:
Classical synthesis. In the first stage, the addition of methyl propiolate 2 to aniline 1 produces labile cis–trans mixtures of enamine 3. Thermal cyclization of enamine provides a synthesis of 4(1H)-quinolone 4 (Gray et al., 1951 ▸).

Conversion of 4-hydroxyquinoline 4 to 4-chloroquinoline 6 can be carried out by a known halogenation method with POCI3 5, or other suitable reagents (e.g. SOCI2, PCI5, POBr3, PBr3). The obtained 4-chloroquinoline 6 can be converted to 4-aminoquinoline derivative 8 by an aromatic nucleophilic substitution reaction with secondary amine 7. Standard bromination of quinoline 8 gives the product 10. 4-Amino-3-bromoquinoline 10 can be substituted by the sodium salt of thiophenol 11 to provide compound 12. Oxidation of 4-amino-3-arylsulfanylquinoline 12 can be accomplished by known methods, preferably in a suitable acid (e.g. acetic acid) at 273–278 K with potassium permanganate 15 to give 4-amino-3-arylsulfinylquinolines 14 or with aqueous hydrogen peroxide 13 in a suitable acid (e.g. acetic acid or trifluoroacetic acid). To obtain the title compound 16, further oxidation of compound 14 is required. The reaction can be carried out by known methods, preferably in a suitable acid (e.g. acetic acid) at 273–278 K with potassium permanganate 15 (Keserü et al., 2007 ▸). The yield of the title compound is 46.0%.
Alternative synthesis. Ethyl 2-tosylacetate 19 is obtained by the reaction of ethyl 2-bromoacetate 18 with sodium tosylsulfinate 17 in dry DMF. Compound 21 can be obtained by the condensation reaction of compound 19 with N,N-dimethylformamide dimethylacetal 20 without using a solvent or in a minimum amount of dioxane. Compound 21, upon reaction with aniline 22 in isopropanol/AcOH medium, produces an E/Z isomer mixture of enamine 23, which is converted to 3-tosylquinoline-4-(1H)-one 24 by thermal cyclization in diphenyl ether. Chlorination of compound 24 is carried out according to a known method with phosphorus oxychloride 5. The final product 16 is obtained by the reaction of aromatic nucleophilic substitution of 4-chloro-3-tosylquinoline 25 with ethyl piperidine-4-carboxylate 7 in a dry DMF medium using a base (triethylamine, DBU), or excess of secondary amine (Keserü et al., 2007 ▸). The yield of the title compound is 73.6%. Recrystallization by slow evaporation of a solution in acetonitrile produced block-like colorless crystals suitable for X-ray diffraction analysis. The advantages of this synthesis make it seem preferable to the common one.

9. NMR characterization
The NMR spectra were recorded on a Varian MR-400 spectrometer with standard pulse sequences operating at 400 MHz for 1H NMR, 101 MHz for 13C NMR. For the NMR spectra, DMSO-d 6 was used as a solvent. Chemical shift values are referenced to residual protons (δ 2.49 ppm) and carbons (δ 39.6 ppm) of the solvent as an internal standard. LC/MS spectra were recorded on a ELSD Alltech 3300 liquid chromatograph equipped with a UV detector (λmax 254 nm), API-150EX mass-spectrometer using a Zorbax SB-C18 column, Phenomenex (100 × 4 mm) Rapid Resolution HT Cartridge 4.6 × 30mm, 1.8-Micron. Elution started with an 0.1 M solution of HCOOH in water and ended with an 0.1 M solution of HCOOH in acetonitrile using a linear gradient at a flow rate of 0.15 ml min−1 and an analysis cycle time of 25 min.
Characteristics of the title molecule:
1H NMR (400 MHz, DMSO-d 6) δ 9.37 (s, 1H), 8.28 (d, J = 8.6 Hz, 1H), 8.17 (d, J = 8.4 Hz, 1H), 7.93 (t, J = 7.7 Hz, 1H), 7.72 (t, J = 8.0 Hz, 3H), 7.39 (d, J = 8.0 Hz, 2H), 4.10 (q, J = 7.1 Hz, 2H), 3.37 (d, J = 19.2 Hz, 1H), 3.31–3.27 (m, 1H), 2.92 (d, J = 11.2 Hz, 2H), 2.55 (d, J = 9.2 Hz, 1H), 2.38 (s, 3H), 1.64 (dd, J = 13.2, 3.8 Hz, 2H), 1.41–1.36 (m, 1H), 1.35 (s, 1H), 1.22 (t, J = 7.1 Hz, 3H).
13C NMR (101 MHz, DMSO-d 6) δ 174.17, 158.14, 151.56, 149.44, 143.64, 139.23, 132.37, 130.31, 129.66, 127.43, 126.19, 125.86, 59.85, 50.56, 27.26, 21.02, 14.11.
10. Refinement
Crystal data, data collection and structure refinement details are summarized in Table 3 ▸. All hydrogen atoms were positioned geometrically (C–H= 0.93–0.97 Å) and refined using a riding model with U iso(H) = nU eq of the carrier atom (n = 1.5 for methyl groups and n = 1.2 for other hydrogen atoms). During the refinement the distances between the atoms of the disordered part were restrained to the following values: 1.497 Å for the bond C19—C22, 1.196 Å for O3—C22, 1.336 Å for O4—C22, 1.452 Å for O4—C23 and 1.513 Å for C23—C24 according to the mean values in Dunitz & Bürgi (1994 ▸). The estimated standard deviation was set at 0.005 Å for all the bonds.
Table 3. Experimental details.
| Crystal data | |
| Chemical formula | C24H26N2O4S |
| M r | 438.53 |
| Crystal system, space group | Monoclinic, P21/n |
| Temperature (K) | 298 |
| a, b, c (Å) | 8.2608 (3), 17.9433 (7), 15.2470 (7) |
| β (°) | 100.626 (4) |
| V (Å3) | 2221.25 (16) |
| Z | 4 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.18 |
| Crystal size (mm) | 0.3 × 0.2 × 0.1 |
| Data collection | |
| Diffractometer | Xcalibur, Sapphire3 |
| Absorption correction | Multi-scan (CrysAlis PRO; Rigaku OD, 2018 ▸) |
| T min, T max | 0.400, 1.000 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 22680, 6477, 3049 |
| R int | 0.104 |
| (sin θ/λ)max (Å−1) | 0.703 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.077, 0.253, 1.04 |
| No. of reflections | 6477 |
| No. of parameters | 321 |
| No. of restraints | 9 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.33, −0.52 |
Supplementary Material
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989022007691/dx2047sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989022007691/dx2047Isup2.hkl
The parameters used for molecular docking. DOI: 10.1107/S2056989022007691/dx2047sup3.pdf
Supporting information file. DOI: 10.1107/S2056989022007691/dx2047Isup4.cml
CCDC reference: 2193735
Additional supporting information: crystallographic information; 3D view; checkCIF report
supplementary crystallographic information
Crystal data
| C24H26N2O4S | F(000) = 928 |
| Mr = 438.53 | Dx = 1.311 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| a = 8.2608 (3) Å | Cell parameters from 2249 reflections |
| b = 17.9433 (7) Å | θ = 3.5–22.9° |
| c = 15.2470 (7) Å | µ = 0.18 mm−1 |
| β = 100.626 (4)° | T = 298 K |
| V = 2221.25 (16) Å3 | Block, colourless |
| Z = 4 | 0.3 × 0.2 × 0.1 mm |
Data collection
| Xcalibur, Sapphire3 diffractometer | 6477 independent reflections |
| Radiation source: fine-focus sealed X-ray tube, Enhance (Mo) X-ray Source | 3049 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.104 |
| Detector resolution: 16.1827 pixels mm-1 | θmax = 30.0°, θmin = 3.0° |
| ω scans | h = −11→11 |
| Absorption correction: multi-scan (CrysAlisPro; Rigaku OD, 2018) | k = −25→24 |
| Tmin = 0.400, Tmax = 1.000 | l = −21→20 |
| 22680 measured reflections |
Refinement
| Refinement on F2 | Hydrogen site location: inferred from neighbouring sites |
| Least-squares matrix: full | H-atom parameters constrained |
| R[F2 > 2σ(F2)] = 0.077 | w = 1/[σ2(Fo2) + (0.106P)2] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.253 | (Δ/σ)max < 0.001 |
| S = 1.04 | Δρmax = 0.33 e Å−3 |
| 6477 reflections | Δρmin = −0.52 e Å−3 |
| 321 parameters | Extinction correction: SHELXL-2017/1 (Sheldrick 2015b), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 9 restraints | Extinction coefficient: 0.0057 (16) |
| Primary atom site location: structure-invariant direct methods |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | Occ. (<1) | |
| S1 | 0.12710 (8) | 0.79643 (4) | 0.44018 (5) | 0.0610 (3) | |
| O1 | 0.1623 (3) | 0.71849 (11) | 0.44202 (15) | 0.0728 (6) | |
| O2 | −0.0150 (2) | 0.82165 (14) | 0.37816 (14) | 0.0790 (6) | |
| N1 | −0.0721 (3) | 0.90921 (14) | 0.61830 (19) | 0.0700 (7) | |
| N2 | 0.3448 (3) | 0.76877 (12) | 0.62148 (16) | 0.0557 (6) | |
| C1 | 0.0279 (4) | 0.89220 (16) | 0.6975 (2) | 0.0608 (7) | |
| C2 | −0.0099 (4) | 0.92616 (19) | 0.7745 (2) | 0.0765 (9) | |
| H2 | −0.095862 | 0.960273 | 0.769311 | 0.092* | |
| C3 | 0.0792 (5) | 0.9093 (2) | 0.8568 (3) | 0.0852 (10) | |
| H3 | 0.055013 | 0.932402 | 0.907452 | 0.102* | |
| C4 | 0.2055 (5) | 0.8577 (2) | 0.8645 (2) | 0.0811 (10) | |
| H4 | 0.262181 | 0.844427 | 0.920871 | 0.097* | |
| C5 | 0.2481 (4) | 0.82599 (18) | 0.7907 (2) | 0.0690 (8) | |
| H5 | 0.335630 | 0.792592 | 0.797560 | 0.083* | |
| C6 | 0.1626 (3) | 0.84274 (15) | 0.70451 (19) | 0.0571 (6) | |
| C7 | 0.2030 (3) | 0.81265 (14) | 0.62430 (19) | 0.0529 (6) | |
| C8 | 0.0973 (3) | 0.82935 (15) | 0.54556 (19) | 0.0571 (7) | |
| C9 | −0.0392 (3) | 0.87693 (18) | 0.5469 (2) | 0.0669 (8) | |
| H9 | −0.110214 | 0.885782 | 0.493097 | 0.080* | |
| C10 | 0.2968 (3) | 0.84632 (15) | 0.41666 (17) | 0.0549 (6) | |
| C11 | 0.4327 (3) | 0.80964 (17) | 0.3971 (2) | 0.0649 (8) | |
| H11 | 0.438815 | 0.757924 | 0.399994 | 0.078* | |
| C12 | 0.5601 (4) | 0.85057 (18) | 0.3730 (2) | 0.0699 (8) | |
| H12 | 0.651240 | 0.825780 | 0.359412 | 0.084* | |
| C13 | 0.5542 (4) | 0.92746 (17) | 0.3688 (2) | 0.0676 (8) | |
| C14 | 0.4163 (4) | 0.96262 (17) | 0.3886 (2) | 0.0755 (9) | |
| H14 | 0.410682 | 1.014372 | 0.386521 | 0.091* | |
| C15 | 0.2875 (4) | 0.92320 (17) | 0.4111 (2) | 0.0708 (8) | |
| H15 | 0.194724 | 0.947991 | 0.422600 | 0.085* | |
| C16 | 0.6935 (4) | 0.9711 (2) | 0.3433 (3) | 0.0968 (12) | |
| H16A | 0.758596 | 0.992709 | 0.395828 | 0.145* | |
| H16B | 0.650002 | 1.009876 | 0.302399 | 0.145* | |
| H16C | 0.761151 | 0.938456 | 0.315502 | 0.145* | |
| C17 | 0.3276 (3) | 0.68933 (15) | 0.6411 (2) | 0.0591 (7) | |
| H17A | 0.221619 | 0.671292 | 0.610173 | 0.071* | |
| H17B | 0.332639 | 0.682536 | 0.704646 | 0.071* | |
| C18 | 0.4648 (3) | 0.64532 (16) | 0.6112 (2) | 0.0629 (7) | |
| H18A | 0.452600 | 0.648235 | 0.546801 | 0.076* | |
| H18B | 0.456382 | 0.593353 | 0.627312 | 0.076* | |
| C19 | 0.6334 (3) | 0.67519 (16) | 0.65413 (19) | 0.0610 (7) | |
| H19 | 0.643943 | 0.669921 | 0.718917 | 0.073* | |
| C20 | 0.6433 (4) | 0.75735 (18) | 0.6337 (2) | 0.0705 (8) | |
| H20A | 0.636820 | 0.764020 | 0.570004 | 0.085* | |
| H20B | 0.748421 | 0.776840 | 0.663831 | 0.085* | |
| C21 | 0.5054 (4) | 0.80045 (17) | 0.6634 (3) | 0.0726 (9) | |
| H21A | 0.517450 | 0.797924 | 0.727861 | 0.087* | |
| H21B | 0.510842 | 0.852386 | 0.646572 | 0.087* | |
| C22 | 0.7719 (4) | 0.63318 (19) | 0.6266 (2) | 0.0780 (10) | |
| O3A | 0.8692 (12) | 0.6593 (9) | 0.5867 (10) | 0.110 (4) | 0.562 (12) |
| O4A | 0.7720 (9) | 0.5613 (2) | 0.6503 (6) | 0.083 (2) | 0.562 (12) |
| C23A | 0.8975 (13) | 0.5082 (7) | 0.6336 (6) | 0.102 (4) | 0.562 (12) |
| H23A | 0.925138 | 0.473765 | 0.683210 | 0.123* | 0.562 (12) |
| H23B | 0.996805 | 0.534136 | 0.625668 | 0.123* | 0.562 (12) |
| C24A | 0.8233 (12) | 0.4672 (7) | 0.5499 (7) | 0.112 (3) | 0.562 (12) |
| H24A | 0.812699 | 0.500471 | 0.499914 | 0.169* | 0.562 (12) |
| H24B | 0.716630 | 0.448644 | 0.555390 | 0.169* | 0.562 (12) |
| H24C | 0.893277 | 0.426205 | 0.541008 | 0.169* | 0.562 (12) |
| O3B | 0.9009 (14) | 0.6554 (14) | 0.6125 (15) | 0.132 (7) | 0.438 (12) |
| O4B | 0.7400 (14) | 0.5610 (4) | 0.6108 (11) | 0.135 (6) | 0.438 (12) |
| C23B | 0.888 (2) | 0.5271 (7) | 0.5881 (13) | 0.122 (6) | 0.438 (12) |
| H23C | 0.986565 | 0.552319 | 0.618191 | 0.147* | 0.438 (12) |
| H23D | 0.884938 | 0.528434 | 0.524241 | 0.147* | 0.438 (12) |
| C24B | 0.8811 (16) | 0.4482 (5) | 0.6209 (14) | 0.124 (6) | 0.438 (12) |
| H24D | 0.782857 | 0.424517 | 0.589912 | 0.187* | 0.438 (12) |
| H24E | 0.880177 | 0.448590 | 0.683798 | 0.187* | 0.438 (12) |
| H24F | 0.975808 | 0.421342 | 0.609985 | 0.187* | 0.438 (12) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.0576 (4) | 0.0605 (5) | 0.0624 (5) | −0.0034 (3) | 0.0045 (3) | −0.0067 (3) |
| O1 | 0.0859 (15) | 0.0524 (12) | 0.0821 (16) | −0.0119 (10) | 0.0206 (12) | −0.0138 (10) |
| O2 | 0.0629 (13) | 0.1024 (18) | 0.0651 (13) | 0.0003 (11) | −0.0052 (10) | −0.0052 (12) |
| N1 | 0.0643 (15) | 0.0667 (16) | 0.0800 (18) | 0.0106 (12) | 0.0159 (13) | −0.0014 (13) |
| N2 | 0.0525 (13) | 0.0452 (12) | 0.0690 (15) | −0.0003 (9) | 0.0105 (11) | −0.0004 (10) |
| C1 | 0.0627 (16) | 0.0499 (15) | 0.0722 (19) | 0.0008 (12) | 0.0184 (14) | −0.0022 (13) |
| C2 | 0.079 (2) | 0.073 (2) | 0.083 (2) | 0.0051 (16) | 0.0285 (18) | −0.0110 (17) |
| C3 | 0.100 (3) | 0.085 (2) | 0.078 (2) | −0.007 (2) | 0.035 (2) | −0.0181 (19) |
| C4 | 0.106 (3) | 0.075 (2) | 0.063 (2) | −0.003 (2) | 0.0190 (18) | −0.0012 (16) |
| C5 | 0.079 (2) | 0.0652 (18) | 0.0626 (19) | 0.0032 (15) | 0.0113 (15) | 0.0011 (15) |
| C6 | 0.0622 (16) | 0.0500 (15) | 0.0603 (17) | −0.0040 (12) | 0.0143 (12) | −0.0008 (12) |
| C7 | 0.0490 (14) | 0.0428 (13) | 0.0662 (17) | −0.0034 (10) | 0.0088 (12) | −0.0001 (12) |
| C8 | 0.0551 (15) | 0.0489 (15) | 0.0658 (17) | 0.0006 (11) | 0.0071 (12) | −0.0013 (12) |
| C9 | 0.0555 (16) | 0.0664 (19) | 0.076 (2) | 0.0085 (13) | 0.0058 (13) | 0.0055 (15) |
| C10 | 0.0566 (15) | 0.0512 (15) | 0.0548 (15) | 0.0017 (11) | 0.0049 (11) | −0.0003 (12) |
| C11 | 0.0641 (18) | 0.0492 (15) | 0.081 (2) | 0.0081 (13) | 0.0120 (15) | 0.0012 (14) |
| C12 | 0.0590 (17) | 0.0669 (19) | 0.085 (2) | 0.0080 (14) | 0.0157 (15) | 0.0062 (16) |
| C13 | 0.0655 (18) | 0.0618 (18) | 0.071 (2) | 0.0006 (14) | 0.0020 (14) | 0.0111 (14) |
| C14 | 0.083 (2) | 0.0476 (16) | 0.098 (2) | 0.0051 (15) | 0.0211 (18) | 0.0097 (15) |
| C15 | 0.0688 (19) | 0.0560 (18) | 0.090 (2) | 0.0118 (14) | 0.0196 (16) | 0.0005 (15) |
| C16 | 0.077 (2) | 0.090 (3) | 0.122 (3) | −0.0133 (19) | 0.014 (2) | 0.031 (2) |
| C17 | 0.0572 (16) | 0.0496 (15) | 0.0722 (18) | 0.0004 (12) | 0.0166 (13) | 0.0017 (13) |
| C18 | 0.0643 (17) | 0.0509 (15) | 0.0748 (19) | 0.0046 (12) | 0.0159 (14) | −0.0026 (14) |
| C19 | 0.0553 (15) | 0.0706 (18) | 0.0576 (16) | 0.0056 (13) | 0.0120 (12) | 0.0058 (14) |
| C20 | 0.0494 (16) | 0.076 (2) | 0.085 (2) | −0.0058 (14) | 0.0087 (15) | 0.0061 (16) |
| C21 | 0.0569 (17) | 0.0540 (17) | 0.104 (3) | −0.0059 (13) | 0.0070 (16) | −0.0066 (16) |
| C22 | 0.064 (2) | 0.095 (3) | 0.078 (2) | 0.0186 (19) | 0.0220 (17) | 0.002 (2) |
| O3A | 0.078 (5) | 0.159 (8) | 0.101 (5) | 0.047 (5) | 0.037 (4) | 0.034 (5) |
| O4A | 0.068 (3) | 0.064 (3) | 0.122 (7) | 0.012 (2) | 0.031 (3) | −0.019 (3) |
| C23A | 0.091 (5) | 0.111 (9) | 0.104 (7) | 0.014 (6) | 0.015 (5) | −0.006 (6) |
| C24A | 0.093 (6) | 0.091 (7) | 0.153 (9) | −0.007 (5) | 0.021 (6) | −0.010 (7) |
| O3B | 0.058 (5) | 0.167 (11) | 0.177 (17) | 0.009 (6) | 0.039 (8) | 0.054 (10) |
| O4B | 0.129 (9) | 0.149 (9) | 0.141 (12) | 0.072 (7) | 0.062 (8) | 0.008 (6) |
| C23B | 0.107 (9) | 0.134 (12) | 0.144 (14) | 0.056 (9) | 0.068 (10) | 0.002 (11) |
| C24B | 0.093 (8) | 0.059 (6) | 0.229 (19) | 0.025 (5) | 0.050 (10) | −0.024 (8) |
Geometric parameters (Å, º)
| S1—O1 | 1.428 (2) | C16—H16B | 0.9600 |
| S1—O2 | 1.438 (2) | C16—H16C | 0.9600 |
| S1—C8 | 1.771 (3) | C17—H17A | 0.9700 |
| S1—C10 | 1.755 (3) | C17—H17B | 0.9700 |
| N1—C1 | 1.366 (4) | C17—C18 | 1.518 (4) |
| N1—C9 | 1.306 (4) | C18—H18A | 0.9700 |
| N2—C7 | 1.419 (3) | C18—H18B | 0.9700 |
| N2—C17 | 1.469 (3) | C18—C19 | 1.523 (4) |
| N2—C21 | 1.476 (3) | C19—H19 | 0.9800 |
| C1—C2 | 1.407 (4) | C19—C20 | 1.512 (4) |
| C1—C6 | 1.412 (4) | C19—C22 | 1.494 (3) |
| C2—H2 | 0.9300 | C20—H20A | 0.9700 |
| C2—C3 | 1.367 (5) | C20—H20B | 0.9700 |
| C3—H3 | 0.9300 | C20—C21 | 1.514 (4) |
| C3—C4 | 1.384 (5) | C21—H21A | 0.9700 |
| C4—H4 | 0.9300 | C21—H21B | 0.9700 |
| C4—C5 | 1.364 (4) | C22—O3A | 1.191 (5) |
| C5—H5 | 0.9300 | C22—O4A | 1.340 (4) |
| C5—C6 | 1.405 (4) | C22—O3B | 1.194 (5) |
| C6—C7 | 1.431 (4) | C22—O4B | 1.334 (5) |
| C7—C8 | 1.381 (4) | O4A—C23A | 1.464 (5) |
| C8—C9 | 1.417 (4) | C23A—H23A | 0.9700 |
| C9—H9 | 0.9300 | C23A—H23B | 0.9700 |
| C10—C11 | 1.381 (4) | C23A—C24A | 1.502 (5) |
| C10—C15 | 1.383 (4) | C24A—H24A | 0.9600 |
| C11—H11 | 0.9300 | C24A—H24B | 0.9600 |
| C11—C12 | 1.387 (4) | C24A—H24C | 0.9600 |
| C12—H12 | 0.9300 | O4B—C23B | 1.462 (5) |
| C12—C13 | 1.382 (4) | C23B—H23C | 0.9700 |
| C13—C14 | 1.383 (4) | C23B—H23D | 0.9700 |
| C13—C16 | 1.501 (4) | C23B—C24B | 1.506 (5) |
| C14—H14 | 0.9300 | C24B—H24D | 0.9600 |
| C14—C15 | 1.373 (4) | C24B—H24E | 0.9600 |
| C15—H15 | 0.9300 | C24B—H24F | 0.9600 |
| C16—H16A | 0.9600 | ||
| O1—S1—O2 | 117.38 (14) | N2—C17—H17B | 109.7 |
| O1—S1—C8 | 111.76 (13) | N2—C17—C18 | 109.8 (2) |
| O1—S1—C10 | 109.63 (13) | H17A—C17—H17B | 108.2 |
| O2—S1—C8 | 104.97 (13) | C18—C17—H17A | 109.7 |
| O2—S1—C10 | 106.93 (13) | C18—C17—H17B | 109.7 |
| C10—S1—C8 | 105.39 (13) | C17—C18—H18A | 109.4 |
| C9—N1—C1 | 116.9 (3) | C17—C18—H18B | 109.4 |
| C7—N2—C17 | 114.9 (2) | C17—C18—C19 | 111.2 (2) |
| C7—N2—C21 | 117.0 (2) | H18A—C18—H18B | 108.0 |
| C17—N2—C21 | 113.5 (2) | C19—C18—H18A | 109.4 |
| N1—C1—C2 | 116.7 (3) | C19—C18—H18B | 109.4 |
| N1—C1—C6 | 123.1 (3) | C18—C19—H19 | 107.7 |
| C2—C1—C6 | 120.1 (3) | C20—C19—C18 | 109.5 (2) |
| C1—C2—H2 | 119.8 | C20—C19—H19 | 107.7 |
| C3—C2—C1 | 120.4 (3) | C22—C19—C18 | 112.9 (3) |
| C3—C2—H2 | 119.8 | C22—C19—H19 | 107.7 |
| C2—C3—H3 | 120.1 | C22—C19—C20 | 111.1 (2) |
| C2—C3—C4 | 119.8 (3) | C19—C20—H20A | 109.4 |
| C4—C3—H3 | 120.1 | C19—C20—H20B | 109.4 |
| C3—C4—H4 | 119.5 | C19—C20—C21 | 111.3 (2) |
| C5—C4—C3 | 120.9 (3) | H20A—C20—H20B | 108.0 |
| C5—C4—H4 | 119.5 | C21—C20—H20A | 109.4 |
| C4—C5—H5 | 119.3 | C21—C20—H20B | 109.4 |
| C4—C5—C6 | 121.4 (3) | N2—C21—C20 | 109.8 (2) |
| C6—C5—H5 | 119.3 | N2—C21—H21A | 109.7 |
| C1—C6—C7 | 118.5 (3) | N2—C21—H21B | 109.7 |
| C5—C6—C1 | 117.3 (3) | C20—C21—H21A | 109.7 |
| C5—C6—C7 | 124.2 (3) | C20—C21—H21B | 109.7 |
| N2—C7—C6 | 124.0 (2) | H21A—C21—H21B | 108.2 |
| C8—C7—N2 | 119.2 (2) | O3A—C22—C19 | 124.7 (8) |
| C8—C7—C6 | 116.8 (2) | O3A—C22—O4A | 123.3 (9) |
| C7—C8—S1 | 123.0 (2) | O4A—C22—C19 | 111.9 (4) |
| C7—C8—C9 | 119.7 (3) | O3B—C22—C19 | 129.6 (13) |
| C9—C8—S1 | 117.3 (2) | O3B—C22—O4B | 116.4 (14) |
| N1—C9—C8 | 124.6 (3) | O4B—C22—C19 | 113.9 (5) |
| N1—C9—H9 | 117.7 | C22—O4A—C23A | 123.1 (7) |
| C8—C9—H9 | 117.7 | O4A—C23A—H23A | 110.5 |
| C11—C10—S1 | 120.9 (2) | O4A—C23A—H23B | 110.5 |
| C11—C10—C15 | 120.0 (3) | O4A—C23A—C24A | 106.0 (8) |
| C15—C10—S1 | 118.9 (2) | H23A—C23A—H23B | 108.7 |
| C10—C11—H11 | 120.3 | C24A—C23A—H23A | 110.5 |
| C10—C11—C12 | 119.5 (3) | C24A—C23A—H23B | 110.5 |
| C12—C11—H11 | 120.3 | C23A—C24A—H24A | 109.5 |
| C11—C12—H12 | 119.4 | C23A—C24A—H24B | 109.5 |
| C13—C12—C11 | 121.3 (3) | C23A—C24A—H24C | 109.5 |
| C13—C12—H12 | 119.4 | H24A—C24A—H24B | 109.5 |
| C12—C13—C14 | 118.0 (3) | H24A—C24A—H24C | 109.5 |
| C12—C13—C16 | 120.7 (3) | H24B—C24A—H24C | 109.5 |
| C14—C13—C16 | 121.4 (3) | C22—O4B—C23B | 107.5 (9) |
| C13—C14—H14 | 119.1 | O4B—C23B—H23C | 111.1 |
| C15—C14—C13 | 121.8 (3) | O4B—C23B—H23D | 111.1 |
| C15—C14—H14 | 119.1 | O4B—C23B—C24B | 103.2 (8) |
| C10—C15—H15 | 120.3 | H23C—C23B—H23D | 109.1 |
| C14—C15—C10 | 119.4 (3) | C24B—C23B—H23C | 111.1 |
| C14—C15—H15 | 120.3 | C24B—C23B—H23D | 111.1 |
| C13—C16—H16A | 109.5 | C23B—C24B—H24D | 109.5 |
| C13—C16—H16B | 109.5 | C23B—C24B—H24E | 109.5 |
| C13—C16—H16C | 109.5 | C23B—C24B—H24F | 109.5 |
| H16A—C16—H16B | 109.5 | H24D—C24B—H24E | 109.5 |
| H16A—C16—H16C | 109.5 | H24D—C24B—H24F | 109.5 |
| H16B—C16—H16C | 109.5 | H24E—C24B—H24F | 109.5 |
| N2—C17—H17A | 109.7 | ||
| S1—C8—C9—N1 | −175.8 (2) | C9—N1—C1—C2 | −179.0 (3) |
| S1—C10—C11—C12 | 175.7 (2) | C9—N1—C1—C6 | 0.5 (4) |
| S1—C10—C15—C14 | −176.9 (3) | C10—S1—C8—C7 | −71.5 (3) |
| O1—S1—C8—C7 | 47.6 (3) | C10—S1—C8—C9 | 106.4 (2) |
| O1—S1—C8—C9 | −134.6 (2) | C10—C11—C12—C13 | 0.5 (5) |
| O1—S1—C10—C11 | 4.5 (3) | C11—C10—C15—C14 | −1.9 (5) |
| O1—S1—C10—C15 | 179.5 (2) | C11—C12—C13—C14 | −0.6 (5) |
| O2—S1—C8—C7 | 175.8 (2) | C11—C12—C13—C16 | 179.5 (3) |
| O2—S1—C8—C9 | −6.4 (3) | C12—C13—C14—C15 | −0.5 (5) |
| O2—S1—C10—C11 | −123.7 (2) | C13—C14—C15—C10 | 1.7 (5) |
| O2—S1—C10—C15 | 51.3 (3) | C15—C10—C11—C12 | 0.8 (4) |
| N1—C1—C2—C3 | 176.8 (3) | C16—C13—C14—C15 | 179.4 (3) |
| N1—C1—C6—C5 | −175.5 (3) | C17—N2—C7—C6 | 83.1 (3) |
| N1—C1—C6—C7 | 4.2 (4) | C17—N2—C7—C8 | −98.2 (3) |
| N2—C7—C8—S1 | 1.7 (4) | C17—N2—C21—C20 | 57.5 (3) |
| N2—C7—C8—C9 | −176.1 (2) | C17—C18—C19—C20 | −55.7 (3) |
| N2—C17—C18—C19 | 55.9 (3) | C17—C18—C19—C22 | 179.8 (2) |
| C1—N1—C9—C8 | −3.7 (5) | C18—C19—C20—C21 | 55.9 (3) |
| C1—C2—C3—C4 | −1.0 (5) | C18—C19—C22—O3A | 117.3 (10) |
| C1—C6—C7—N2 | 173.2 (2) | C18—C19—C22—O4A | −61.1 (6) |
| C1—C6—C7—C8 | −5.5 (4) | C18—C19—C22—O3B | 143.1 (14) |
| C2—C1—C6—C5 | 3.9 (4) | C18—C19—C22—O4B | −32.5 (9) |
| C2—C1—C6—C7 | −176.4 (3) | C19—C20—C21—N2 | −56.2 (4) |
| C2—C3—C4—C5 | 3.3 (5) | C19—C22—O4A—C23A | −178.2 (7) |
| C3—C4—C5—C6 | −1.9 (5) | C19—C22—O4B—C23B | −177.9 (9) |
| C4—C5—C6—C1 | −1.7 (5) | C20—C19—C22—O3A | −6.2 (11) |
| C4—C5—C6—C7 | 178.7 (3) | C20—C19—C22—O4A | 175.4 (5) |
| C5—C6—C7—N2 | −7.1 (4) | C20—C19—C22—O3B | 19.6 (15) |
| C5—C6—C7—C8 | 174.1 (3) | C20—C19—C22—O4B | −156.0 (9) |
| C6—C1—C2—C3 | −2.6 (5) | C21—N2—C7—C6 | −53.7 (4) |
| C6—C7—C8—S1 | −179.56 (19) | C21—N2—C7—C8 | 124.9 (3) |
| C6—C7—C8—C9 | 2.7 (4) | C21—N2—C17—C18 | −57.4 (3) |
| C7—N2—C17—C18 | 164.2 (2) | C22—C19—C20—C21 | −178.7 (3) |
| C7—N2—C21—C20 | −165.0 (2) | C22—O4A—C23A—C24A | −98.1 (14) |
| C7—C8—C9—N1 | 2.1 (5) | C22—O4B—C23B—C24B | 150 (2) |
| C8—S1—C10—C11 | 124.9 (2) | O3A—C22—O4A—C23A | 3.4 (14) |
| C8—S1—C10—C15 | −60.1 (3) | O3B—C22—O4B—C23B | 5.9 (17) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C4—H4···O3Ai | 0.93 | 2.52 | 3.421 (16) | 163 |
| C5—H5···O2ii | 0.93 | 2.58 | 3.415 (4) | 149 |
Symmetry codes: (i) x−1/2, −y+3/2, z+1/2; (ii) x+1/2, −y+3/2, z+1/2.
Funding Statement
Funding for this research was provided by: Ministry of Health of Ukraine (grant No. 0121U109239).
References
- Atechian, S., Nock, N., Norcross, R. D., Ratni, H., Thomas, A. W., Verron, J. & Masciadri, R. (2007). Tetrahedron, 63, 2811–2823.
- Bax, B. D., Chan, P. F., Eggleston, D. S., Fosberry, A., Gentry, D. R., Gorrec, F., Giordano, I., Hann, M. M., Hennessy, A., Hibbs, M., Huang, J., Jones, E., Jones, J., Brown, K. K., Lewis, C. J., May, E. W., Saunders, M. R., Singh, O., Spitzfaden, C. E., Shen, C., Shillings, A., Theobald, A. J., Wohlkonig, A., Pearson, N. D. & Gwynn, M. N. (2010). Nature, 466, 935–940. [DOI] [PubMed]
- Blower, T. R., Williamson, B. H., Kerns, R. J. & Berger, J. M. (2016). PNAS 113, 7, 1706-1713. [DOI] [PMC free article] [PubMed]
- Bondi, A. (1964). J. Phys. Chem. 68, 3, 441–451.
- Bylov, I. E., Bilokin, Y. V. & Kovalenko, S. M. (1999). Heterocycl. Comm. 5, 3, 281–284.
- Desiraju, G. R. (1996). Acc. Chem. Res. 29, 441–449. [DOI] [PubMed]
- Desiraju, G. R. (2005). ChemComm, 24, 2995–3001.
- Dhiman, P., Arora, N., Thanikachalam, P. V. & Monga, V. (2019). Bioorg. Chem. 92, 103291. [DOI] [PubMed]
- Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2009). J. Appl. Cryst. 42, 339–341.
- Dunitz, J. D. & Bürgi, H.-B. (1994). Structure correlation, pp. 767–784. Weinheim: VCH.
- Galambos, J., Bielik, A., Wágner, G., Domány, G., Kóti, J., Béni, Z., Szigetvári, Á., Sánta, Z., Orgován, Z., Bobok, A., Kiss, B., Mikó-Bakk, M. L., Vastag, M., Sághy, K., Krasavin, M., Gál, K., Greiner, I., Szombathelyi, Z. & Keserű, G. M. (2017). Eur. J. Med. Chem. 133, 240–254. [DOI] [PubMed]
- Gray, F. W., Mosher, H. S., Whitmole, F. C. & Oakwood, T. S. (1951). J. Am. Chem. Soc. 73, 8, 3577–3578.
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed]
- Hanwell, M. D., Curtis, D. E., Lonie, D. C., Vandermeersch, T., Zurek, E. & Hutchison, G. R. (2012). J. Cheminform, 4, 17. [DOI] [PMC free article] [PubMed]
- Horta, P., Secrieru, A., Coninckx, A. & Cristiano, M. L. S. (2017). OMCIJ, 4, 91–93.
- Hryhoriv, H., Mariutsa, I., Kovalenko, S., Sidorenko, L., Perekhoda, L., Filimonova, N., Geyderikh, O. & Georgiyants, V. (2021). ScienceRise Pharm. Sci. 5, 33, 4-11.
- Ivachtchenko, A. V., Golovina, E. S., Kadieva, M. G., Kysil, V. M., Mitkin, O. D., Vorobiev, A. A. & Okun, I. (2012a). Bioorg. Med. Chem. Lett. 22, 4273–4280. [DOI] [PubMed]
- Ivachtchenko, A. V., Mitkin, O. D. & Kadieva, M. G. (2012b). Patent WO/2012/087182.
- Kang, S., Yoon, H. & Lee, Y. (2016). Chem. Lett. 45, 1356–1358.
- Keserü, G., Wéber, C., Bielik, A., Bobok, A. A., Gál, K., Meszlényiné Sipos, M., Molnár, L. & Vastag, M. (2007). Patent WO/2007/072093.
- Konovalova, I. S., Shishkina, S. V., Paponov, B. V. & Shishkin, O. V. (2010). CrystEngComm, 12, 3, 909–916.
- Laponogov, I., Pan, X.-S., Veselkov, D. A., Cirz, R. T., Wagman, A., Moser, H. E., Fisher, L. M. & Sanderson, M. R. (2016). Open Biol. 6, 160157. https://doi.org/10.1098/rsob.160157. [DOI] [PMC free article] [PubMed]
- Malherbe, P., Masciadri, R., Norcross, R. D. & Prinssen, E. (2006). Patent WO/2006/128802.
- Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S. & Olson, A. J. (2009). J. Comp. Chem. 30, 2785–2791. [DOI] [PMC free article] [PubMed]
- Rigaku OD (2018). CrysAlis PRO. Rigaku Oxford Diffraction, Yarnton, England.
- Rowland, R. S. & Taylor, R. (1996). J. Phys. Chem. 100, 18, 7384–7391.
- Sanner, M. F. (1999). J. Mol. Graph. Mod. 17, 57–61. [PubMed]
- Savchenko, T. I., Silin, O. V., Kovalenko, S. M., Musatov, V. I., Nikitchenko, V. M. & Ivachtchenko, A. V. (2007). Synth. Commun. 37, 8, 1321–1330.
- Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8.
- Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8.
- Shishkin, O. V., Dyakonenko, V. V. & Maleev, A. V. (2012). CrystEngComm, 14, 5, 1795–1804.
- Silin, O. V., Savchenko, T. I., Kovalenko, S. M., Nikitchenko, V. M. & Ivachtchenko, A. V. (2004). Heterocycles, 63, 8, 1883–1890.
- Spackman, P. R., Turner, M. J., McKinnon, J. J., Wolff, S. K., Grimwood, D. J., Jayatilaka, D. & Spackman, M. A. (2021). J. Appl. Cryst. 54, 1006–1011. [DOI] [PMC free article] [PubMed]
- Sutor, D. J. (1962). Nature, 195, 68–69.
- Syniugin, A. R., Ostrynska, O. V., Chekanov, M. O., Volynets, G. P., Starosyla, S. A., Bdzhola, V. G. & Yarmoluk, S. M. (2016). J. Enzyme Inhib. Med. Chem. 31, 160–169. [DOI] [PubMed]
- Vaksler, Ye. A., Idrissi, A., Urzhuntseva, V. V. & Shishkina, S. V. (2021). Cryst. Growth Des. 21, 4, 2176–2186.
- Zefirov, N. S., Palyulin, V. A. & Dashevskaya, E. E. (1990). J. Phys. Org. Chem. 3, 147–158.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989022007691/dx2047sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989022007691/dx2047Isup2.hkl
The parameters used for molecular docking. DOI: 10.1107/S2056989022007691/dx2047sup3.pdf
Supporting information file. DOI: 10.1107/S2056989022007691/dx2047Isup4.cml
CCDC reference: 2193735
Additional supporting information: crystallographic information; 3D view; checkCIF report