

Abstract
Coenzyme Q10 (CoQ10) is necessary for mitochondrial electron transport. Mutations in CoQ10 biosynthetic genes cause primary CoQ10 deficiency (PCoQD) and manifest as mitochondrial disorders. It is often stated that PCoQD patients can be treated by oral CoQ10 supplementation. To test this, we compiled all studies describing PCoQD patients up to May 2022. We excluded studies with no data on CoQ10 treatment, or with insufficient description of effectiveness. Out of 303 PCoQD patients identified, we retained 89 cases, of which 24 reported improvements after CoQ10 treatment (27.0%). In five cases, the patient's condition was reported to deteriorate after halting of CoQ10 treatment. 12 cases reported improvement in the severity of ataxia and 5 cases in the severity of proteinuria. Only a subjective description of improvement was reported for 4 patients described as responding. All reported responses were partial improvements of only some symptoms. For PCoQD patients, CoQ10 supplementation is replacement therapy. Yet, there is only very weak evidence for the efficacy of the treatment. Our findings, thus, suggest a need for caution when seeking to justify the widespread use of CoQ10 for the treatment of any disease or as dietary supplement.
Keywords: coenzyme Q, CoQ biosynthesis, CoQ10 supplementation, mitochondrial disorders, primary CoQ10 deficiency, ubiquinone
1. INTRODUCTION
Coenzyme Q10 (CoQ10), also known as ubiquinone (UQ10), is composed of a redox active aromatic ring and a ten‐repeat long polyprenyl sidechain. CoQ10 is an essential component of the mitochondrial respiratory chain, where it functions as a mobile carrier for the transfer of electrons from respiratory complexes I and II to complex III, and as cofactor in complex III function. In addition, it feeds electrons into the respiratory chain from other entry points, including the electron transfer flavoprotein, sulphide‐quinone reductase and dihydroorotate dehydrogenase. 1 , 2 , 3 , 4 CoQ10 is known to have antioxidant properties and to be involved in several other cellular functions outside of mitochondria. 5 , 6 As far as is known, all cells rely exclusively on endogenous CoQ synthesis. So far, 11COQ genes whose products participate in CoQ10 biosynthesis have been identified in humans. Some of them function as enzymes and others as structural components of the CoQ biosynthetic complex (Figure 1A). 7 , 8 , 9 , 10 , 11 Mutations in COQ genes cause primary CoQ10 deficiency (PCoQD), a clinically heterogeneous and rare disorder. 12 , 13 Symptoms often resemble those of typical inborn mitochondrial respiratory chain disorders (Figure 1B), including early onset, multi‐organ involvement and with prevalent neurological and muscular manifestations. It some cases the symptoms predominantly affect a particular organ or tissue (e.g. kidney‐ or cerebellum‐limited phenotypes). 8 , 14 , 15 , 16 Secondary CoQ10 deficiency refers to all the conditions in which the etiology of a CoQ10 deficiency is not a molecular lesion in the CoQ10 biosynthetic pathway. 17 , 18 In fact, a variety of conditions have been found to be associated with CoQ10 deficiency. Statins were shown to reduce serum and muscle CoQ10 levels. 19 , 20 Mutations in the electron transfer flavoprotein dehydrogenase (ETFDH) and mitochondrial DNA (mtDNA) lesions, including low mtDNA copy number, were also shown to lower steady state level of CoQ10. 21 , 22 , 23 , 24 , 25 , 26 The mechanisms leading to deficiency in these cases are unknown, except for the effect of statins, which inhibit the synthesis of mevalonate, the molecular precursors of the CoQ10 sidechain.
FIGURE 1.
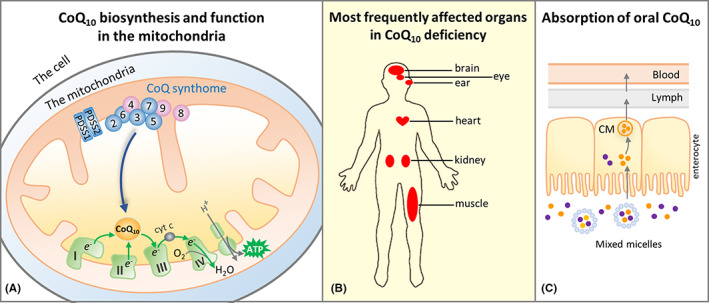
CoQ10 in the mitochondria, pathology of CoQ10 deficiency and oral supplementation. (A) The final steps of CoQ10 biosynthesis are carried out in the inner mitochondrial membrane. The CoQ10 biosynthetic pathway includes both enzymes (in blue) and structural or regulatory components (in purple). Only the numbers in their names are shown for COQ proteins (COQ2‐7, COQ8A, COQ8B and COQ9). They are known to form a large complex, the CoQ biosynthetic complex or CoQ‐synthome. COQ10A and COQ10B whose functions are uncertain and not known to be part of the complex are not shown. The most essential function of CoQ10 is to transport electrons in the mitochondrial respiratory chain. Although CoQ10 is found in the mitochondrial membrane, in the figure this is not shown for clarity. (B) Primary CoQ10 deficiency predominantly manifests as mitochondrial disorder, with organs with high energy needs being most often affected. (C) Intestinal absorption of CoQ10 is thought to occur through the formation of mixed micelles with other dietary lipids. Once inside the enterocytes, CoQ10 is incorporated into chylomicrons (CM), which are transported via the lymphatics to the blood circulation. Because of its extreme hydrophobicity and its relatively large size, the absorption of orally administered CoQ10 has been reported to be poor
In tissue samples or cultured cells from patients, CoQ10 deficiency can be diagnosed by measuring CoQ10 levels, which can be complemented by the observation of impaired CoQ10‐dependent respiratory chain activities (Complex I‐III and Complex II‐III). In the last few decades, with the increasing availability and affordability of genomic sequencing technology, whole genome or exome sequencing is increasingly becoming the first‐line diagnostic test for patients suspected of having genetic disorders, including PCoQD. This has accelerated the discovery of novel PCoQD disease variants. 27 Disease‐causing mutations have been reported for 9 out of 11 COQ genes required for CoQ biosynthesis. Below we report that at least 303 PCoQD patients have been reported so far. CoQ10 supplementation is frequently initiated immediately after diagnosis (Figure 1C), and the majority of the literature on CoQ10 deficiency states that CoQ10 deficiency is treatable by supplementation with exogenous CoQ10. 28 , 29 , 30 , 31 , 32 , 33 However, there is lack of clear evidence for such a claim.
In addition to patients with documented CoQ10 deficiency and/or COQ mutations, CoQ10 is frequently recommended to mitochondrial disease patients. 34 , 35 In fact, it is a component of the so‐called mitochondrial cocktail, which is a collection of high‐dose nutraceuticals with the potential to support mitochondrial functioning. 36 Moreover, although there is no consistent scientific evidence for beneficial effects, CoQ10 is often recommended for treating a wide range of other conditions (e.g. heart failure and neurodegenerative diseases) and it is widely available over the counter as an anti‐ageing dietary supplement. 32 By estimation, the global market size of CoQ10 amounts to close to 600 M USD a year.
This review aims to summarize and evaluate the available evidence for the effectiveness of CoQ10 supplementation for the treatment of PCoQD. Patients with PCoQD should be the most amenable to CoQ10 treatment because their CoQ10 deficiency is the only cause of all their symptoms, and therefore, CoQ10 treatment is simple replacement therapy. Thus, examining outcomes of CoQ10 treatment for these patients is the first key step to address the effectiveness of any CoQ10 therapy and to promote a rational use of CoQ10 for disease treatment or as a health supplement.
2. MATERIALS AND METHODS
2.1. Search strategy and selection criteria
A literature search was performed in PubMed for studies that described PCoQD patients, up until May 01, 2022. The PubMed query used is given in Supporting Information. The references cited in the articles identified were manually screened for any additional relevant study. We imposed no publication status or language restrictions. We considered any type of study regardless of research design.
The following information was sought in each paper: descriptive characteristics of PCoQD patients including sex, age of onset, major symptoms, age at the last reported exam or death, molecular lesions in COQ genes or proteins, severity of CoQ10 deficit, respiratory chain complex (RCC) activities, CoQ10 treatment received and clinical outcomes and laboratory tests known to be relevant to mitochondrial disease. CoQ10 levels and RCC activities are most often reported in patient‐derived skin fibroblasts or muscle biopsies. Study data were extracted by one reviewer (YW) and verified by another reviewer (SH) for accuracy, narrative summaries and interpretation. When data were reported more than once for the same patients, which was exceedingly rare, the data that were included were those from the most recent comprehensive report. If no data on patient treatment with CoQ10 were provided in a study, or if patients were treated but outcome data were not reported, or the reported effects were contradictory or ambiguous, the study was excluded from the final data synthesis (Figure 2).
FIGURE 2.

Flow diagram for identification and selection of primary CoQ10 deficiency patients
2.2. Data analysis
We synthesized data using tabulations that include narrative summaries. The effect of CoQ10 treatment on clinical outcomes is considered as positive (responding) if one of the following criteria is satisfied: a) a positive effect on a quantifiable clinical measure was reported; b) some improvement was noted after CoQ10 treatment and stopping/halting the treatment resulted in deterioration of a patient's condition; and/or c) no quantifiable clinical evidence was provided but at least two symptoms were described to be improved following CoQ10 treatment. Fulfilling any one of the first two criteria is defined as responding with an objective description of the response. Whereas if symptom improvement was described without relying on any quantifiable measure, we categorize it as a subjective description of the response to CoQ10 therapy. Patients counted as not responding include cases where no significant effect was noted after CoQ10 treatment, or the reported effect(s) were minimal, or when, though some clinical improvement was noted, the patient's condition had deteriorated (e.g. developed new symptoms) while on CoQ10 therapy. No restriction on CoQ10 dosage (dose, formulation, dose frequency), time of initial treatment, duration of treatment or concurrent treatments was made. The two authors independently assigned the patient cases to the categories. Disagreements were resolved by discussion and consensus.
2.3. Statistical analysis
Violin graphs were plotted and analysed by using GraphPad Prism 9 (GraphPad Software, Inc.). Differences between groups were tested using Student's t‐test.
3. RESULTS
The literature search yielded 78 published studies, from which a total of 303 patients with PCoQD were identified. Their characteristics are summarized in Table 1, and details are available in Table S1. Of the 303 PCoQD patients, 142 [46.7%] were reported to receive oral supplement of CoQ10. The dosage was reported as mg/day in some studies and as mg/kg/day in others. Doses ranged from 60 mg/day to 2100 mg/day or from 5 mg/kg/day to 100 mg/kg/day, and the reported duration of treatment was from 1 month to 8 years. 37 , 38 , 39 , 40 Following the exclusion criteria, 53 treated patients were removed from the final analysis (Table S2). Among the excluded patients, 16 were excluded because the reported follow‐up findings were judged to be ambiguous or inadequate to judge treatment efficiency, for example reports that mention symptom stabilization or CoQ10 treatment combined with other simultaneous treatments. All other exclusions were because no treatment outcome was reported.
TABLE 1.
Primary CoQ10 deficiency patients reported in the literature
| Gene | No. of pathogenic variants | No. of patients | No. of families | Range of age of onset (years) | CoQ10 level (% of control) [No. of patients examined] | Common clinical manifestations a | No. of CoQ10‐ treated patients b | |
|---|---|---|---|---|---|---|---|---|
| Skin fibroblasts | Muscle | |||||||
| PDSS1 | 1 | 2 | 1 | 1–3 | <5% [2] | ND | Encephalopathy, deafness | 0 |
| PDSS2 | 5 | 4 | 4 | infancy‐2 | 12% [1] | 14% [1] | NS, encephalopathy | 2 |
| COQ2 | 20 | 25 | 17 | infancy‐68 | 9–36% [7] | 3–38% [5] | NS, encephalopathy | 10 |
| COQ4 | 27 | 32 | 25 | infancy‐9 | 22–98% [8] | 2–63% [6] | Encephalopathy, cardiac pathology | 21 |
| COQ5 | 1 | 3 | 1 | childhood | ND | 57% [1] | cerebellar ataxia, encephalopathy | 3 |
| COQ6 | 15 | 28 | 20 | infancy‐6.4 | ND | ND | NS, SND | 52 |
| COQ7 | 7 | 6 | 5 | infancy‐childhood | 10–70% [4] | 10% [1] | Spasticity, motor difficulties | 3 |
| COQ8A/ADCK3 | 79 | 112 | 88 | infancy‐42 | 35–100% [8] | 2–100% [13] | Cerebellar ataxia, exercise intolerance | 592 |
| COQ8B/ADCK4 | 36 | 88 | 51 | infancy‐32 | 27% [2] | ND | NS | 33 |
| COQ9 | 3 | 3 | 3 | infancy | 11–18% [2] | 15% [1] | Encephalopathy | 2 |
Abbreviations: ND, not determined; NS, nephrotic syndrome; SND, sensorineural deafness.
The most common symptoms among the reported patients are listed.
Including treatment with ubiquinol (reduced form of CoQ); in addition, the number of patients treated with the CoQ analogue idebenone are indicated as superscripts.
In the final analysis, we included and assessed a total of 89 patients. The results are shown in Table 2. Details, including total count of patients treated and numbers of exclusions for each gene, can be found in Table S3. We classified 65 out of the 89 patients (73.0%) as not responding to CoQ10 treatment according to the evaluation criteria (Table S4). Among those, there are nine cases in which patients showed infantile onset and multisystem involvement. Such cases may be more challenging to treat, but this is only speculation. Of the 24 cases (27.0%) that were identified as responders, 20 were found to provide objective descriptions of responses and four are considered to be responders because they meet the criterion of having a subjective description of responses to CoQ10 therapy (Table S5). Note, however, that all responses were partial, and responses are frequently only observed with a single symptom. Table 3 highlights the five cases in which a worsening of patient conditions after stopping/halting of CoQ10 treatment or regimen change was reported. Four out of the five also reported recovery to some extent following treatment resumption. These cases potentially provide the most tantalizing evidence for a partial efficacy of CoQ10 treatment for CoQ10 deficiency. We should note, however, that the possibility of placebo effects cannot be excluded. Furthermore, in one of the cases the patient's condition was reported to worsen after replacement of CoQ10 with idebenone and thus it is impossible to distinguish between the effects of stopping CoQ10 and potential idebenone toxicity. Of the other 15 cases of responses with objective description, four cases reported a decrease of proteinuria after CoQ10 treatment as an indication of kidney function improvement and ten reported a reduction in a severity score of ataxia or another motor performance test at a follow‐up. However, five of the patients classified as responders because of an amelioration of proteinuria had only kidney symptoms and in two cases only proteinuria.
TABLE 2.
Reported partial effects of CoQ10 treatment in primary CoQ10 deficiency patients
| Gene | No. of patients included in the analysis | Responding | Not responding | |
|---|---|---|---|---|
| Objective description | Subjective description | |||
| PDSS1 | 0 | – | – | – |
| PDSS2 | 2 | 0 | 0 | 2 |
| COQ2 | 7 | 1 | 0 | 6 |
| COQ4 | 19 | 3 | 2 | 14 |
| COQ5 | 3 | 3 | 0 | 0 |
| COQ6 | 3 | 1 | 0 | 2 |
| COQ7 | 3 | 0 | 0 | 3 |
| COQ8A/ADCK3 | 41 | 9 | 2 | 30 |
| COQ8B/ADCK4 | 9 | 3 | 0 | 6 |
| COQ9 | 2 | 0 | 0 | 2 |
Note: Treatment effects established by quantitative or semi‐quantitative measures to describe the response to CoQ10 treatment were counted as responding with objective description, while descriptions of positive effects but without relying on quantitative or semi‐quantitative measures were counted as responding with subjective description. ‘Not responding’ include the patients who were reported not to respond to CoQ10 treatment or whose responses we consider lacking a convincing demonstration of a response to CoQ10 supplementation.
TABLE 3.
Therapeutic efficacy of CoQ10 suggested by the effects of treatment interruptions
| COQ gene | Mutation [patient ID#] a | Effects of CoQ10 treatment | Strength of Evidence | Ref. |
|---|---|---|---|---|
| COQ4 | monoallelic deletion (HET) | Improvement in physical status and social function. Conditions worsened (weakness and diffuse myalgia) after formulation change and dosage reduction. Remission of symptoms within a week after reverting to the original dosage. | Weak evidence: small benefits and a possible placebo effect and/or observer bias | [78] |
| COQ6 | A353D (HOM) [A1072‐22] | The patient was diagnosed with SRNS at the age of 2.5 years and SND at 4 years old. CoQ10 treatment was started at age 5.5 years when the subject was in partial remission from cyclosporine treatment, which was discontinued at 5.8 years. A decrease of proteinuria was observed (from 117 to 76 mg/day) at 2 months into treatment and remission was maintained at the end of the study period. No hearing improvement was observed. Proteinuria reoccurred at a level of 1100 mg/day after temporary cessation of CoQ10 treatment and decreased again to 188 mg/day following reinstitution of CoQ10 treatment. |
Weak evidence: the effect observed after the first 2 months of treatment is confounded by the presence of another intervention; unclear cause for the surge of proteinuria after interruption of CoQ10 |
[16] |
| COQ8A | G272D/Q605GfsX125 (CH) | CoQ10 and L‐carnitine were initiated at age 5, improved exercise tolerance and fewer vomiting episodes were noted after 3 months of therapy. Blood lactate level also decreased but without totally normalizing. The patient continued to develop new neurologic symptoms, for example cerebellar syndrome with tremors, with increasing age. CoQ10 was replaced with idebenone at the age of 9 years, and within the following 4 months, severe exercise intolerance reappeared with numerous episodes of vomiting. The clinical deterioration was accompanied by an elevation of lactatemia. Reverting back to the initial CoQ10 treatment resulted in returns to the previous clinical status within 3 months. |
Weak evidence: partial improvement of only a few symptoms; confounding effects from another intervention; worsening of the patient's condition after stopping CoQ10 is also consistent with idebenone toxicity |
[79] |
| COQ8A | T584delACC/P502R (CH) | CoQ10 was initiated at 5 years of age with partial improvement in motor skills, balance and strength. After 6 years, the patient gradually stopped taking CoQ10 and her condition deteriorated including severe psychiatric involvement. |
Weak evidence: partial improvement; vague description of effects; possible placebo effect and/or observer bias |
[80] |
| COQ8A | S616LfsX114/R301Q (CH) | Self‐reported fatigue and exercise tolerance improvement after 2 weeks of therapy. After 2 years of therapy, ataxia and head tremor diminished. SARA total score improved from 13 to 8. When the treatment was stopped for a month, the patient's condition deteriorated, rendering him to resume taking CoQ10. |
Weak evidence: placebo effect and improvements as a result of the natural course of the illness, could not be ruled out |
[81] |
Abbreviations: CH, compound heterozygous; HET, heterozygous; HOM, homozygous; SARA, Scale for the Assessment and Rating of Ataxia; SND, sensorineural deafness; SRNS, steroid‐resistant nephrotic syndrome.
Patient IDs are provided when more than one individual was described in the original patient reports.
As shown in Figure 3 and S1, there is no significant differences in treatment dosage and duration of treatment between the non‐responding and responding patients. The highest reported dosage is 2100 mg/day. No substantial adverse effects have been reported for the CoQ10‐treated PCoQD patients. However, an adverse reaction has been reported in one case of treatment with the synthetic CoQ analogue idebenone, which has a hydroxydecyl instead of a decaprenyl side chain and higher solubility than CoQ10. 41
FIGURE 3.

Violin plots of CoQ10 treatment dose and duration. (A) Two graphs are shown for dosage comparisons because CoQ10 treatment dosages were reported in 2 different units (mg/kg/day and mg/day). (B) Comparison of CoQ10 treatment duration. (C) Disease durations before CoQ10 treatment. ns: not significant (Student's t‐test). Sample sizes are indicated on the graphs. Note that the measures plotted in these graphs only include the patients for which the information was provided in the case reports, which is why the sample sizes are different
4. DISCUSSION
In humans, mutations have so far been reported in all the genes required for CoQ10 biosynthesis, except COQ3. COQ3 is an O‐methyltransferase and it is the only COQ protein that is required for more than one step in the CoQ biosynthetic pathway. 42 Thus, one possible explanation for the lack of reports of COQ3 patients is that, because it is required for two enzymatic steps, pathogenic mutations in COQ3 are more detrimental to CoQ production and thus are more likely to be lethal. Among the reported PCoQD patients, 37.0% (112/303) carry a mutation in the COQ8A gene and 29.0% (88/303) carry a mutation in the COQ8B gene. The reason for the higher COQ8A and COQ8B patient counts is most likely because genetic screening studies were performed for COQ8A and COQ8B on a relative larger scale. Two studies reported screening for COQ8A mutations in patients with ataxic symptoms, resulting in the identification of 69 patients carrying rare biallelic variants. 15 , 39 Screens for COQ8B mutations in patients with renal disorders, including nephrotic syndrome and chronic renal failure, were described in three studies, which in total reported the identification of 63 COQ8B patients. 43 , 44 , 45 COQ8A and COQ8B are orthologues of yeast Coq8p, which plays a regulatory role in CoQ biosynthesis. 40 , 43 , 46 COQ8A is expressed in most tissues, but there is a relative enrichment of COQ8B in podocytes. 43 , 47 Consistently, COQ8B patients were described to have a less severe clinical course and manifest largely kidney‐limited phenotypes. 43 , 48 In mice, the Coq8a −/− model was shown to develop ataxia accompanied by minor neurological and muscle phenotypes. 47 More interestingly, unlike for other Coq genes, including Coq8b −/− , which are embryonically lethal, Coq8a −/− mice are viable and maintain a moderate level of residual CoQ. 47 , 49 , 50 , 51 , 52 , 53 , 54 , 55 Thus, the mutation frequency observed for a given COQ gene is likely influenced by the role it plays in CoQ10 biosynthesis and its tissue expression pattern. With increasing affordability and accessibility of genome or exome sequencing, 56 more and more PCoQD patients are being reported, and a more accurate picture of PCoQD patients' frequency should soon emerge.
Often CoQ10 deficiency patients are started on oral CoQ10 supplementation immediately after diagnosis. Various oral formulations of CoQ10 are available. 57 The scientific literature as well as the general media mostly state that oral CoQ10 supplementation is effective and thus that CoQ10 deficiency is treatable. 33 However, to the best of our knowledge, there is no other evidence that could support such a belief than the set of studies reviewed here. The final step of our analysis is based on published studies on 89 PCoQD patients for which we consider there to be sufficient information available to estimate the clinical effectiveness of the CoQ10 treatment. Of them, 65 cases fit our criteria for not responding, including patients with age of onset ranging from neonatal to 42 years of age and that present with multisystem symptoms or primarily one organ‐specific manifestation (e.g. cerebellar ataxia or nephrotic syndrome). Among the 24 cases identified as responsive, 12 cases reported improvement of an ataxia rating score and 7 out of them are patients with COQ8A mutations for whom ataxia is often the most prominent symptom. Five cases reported proteinuria improvement at a post‐treatment follow‐up, and in all five of them renal dysfunction was the only manifestation. However, many PCoQD patients with ataxia or kidney symptoms were reported to show no response or the condition continued to deteriorate after CoQ10 treatment (Table S4). Therefore, the observed relative prevalence of positive effects on ataxia or proteinuria does not indicate that the kidneys and cerebellum are more sensitive to supplemental treatment with CoQ10. Of note, none of the studied that reported symptomatic improvement found a profound rescue of the patients' conditions. Furthermore, in patients with multisystem manifestations, effects were reported only for a few symptoms and most of the other symptoms still persisted after CoQ10 treatment. Detrimental effects of treatment interruption were noted in five cases, which potentially constitute the best evidence for some effectiveness of CoQ10 therapy. However, as these are not blinded studies, the possibility of placebo effects remains of concern.
Overall, most descriptions of the effects of CoQ10 treatment have incomplete information and lack a complete clinical picture. Doctors and patients are aware of the treatments (i.e. no blinding). There can of course be no ‘no‐treatment’ control group of patients. For these reasons, we consider the cases where a minimal effect only was reported as not responding to treatment. It has been hypothesized that CoQ10 treatment cannot reverse severe tissue damage due to PCoQD when the disease has already progressed too far before therapy is initiated. 30 , 58 However, animal studies with an unnatural CoQ biosynthetic precursor suggest that most phenotypes due to severe CoQ deficiency can be completely rescued by a partial replenishment of CoQ levels. 59 , 60 , 61 It remains to be seen how various disease symptoms due to CoQ10 deficiency can be effectively treated in human patients by sufficient restoration of CoQ10 levels. It is likely that it would be more challenging for symptoms associated with severe cell loss, such as neuronal loss in the central nervous system. Nevertheless, if the remaining cells and neurons can be made to function more efficiently by alleviating their CoQ10 deficiency, significant partial functional recovery might be possible. In addition, patients with late onset of symptoms would be expected to have sustained less irretrievable damage and could benefit substantially. Overall, it seems reasonable to hope that worthwhile clinical benefits are possible even in severely impaired PCoQD patients if in fact a significant amount of CoQ10 were absorbed and could reach affected tissues.
The results from our analysis indicate that most PCoQD patients treated with CoQ10 showed little or no response, and, in the cases of positive reports, the overall clinical benefit was only very limited. This strongly suggests a lack of efficacy of CoQ10 treatment. It is noteworthy that clinical trials have been conducted to assess the potential benefit of CoQ10 in the treatment of patients with secondary CoQ10 deficiency or mitochondrial disease. CoQ10 supplementation was shown to elicit no benefit to the patients with statin‐induced myalgia. 62 To date, only few double‐blind and randomized clinical trials evaluating CoQ10 in the treatment of mitochondrial disorders have been completed. There were reports of minor effects for improved muscle strength and attenuation of lactate rise post‐exercise. However, the overall conclusion remained that CoQ10 is ineffective for the treatment of patients with mitochondrial disorder, or at least there is no solid evidence to suggest otherwise. 63 , 64
CoQ10 is extremely lipophilic and practically insoluble in water; therefore, to develop pharmaceutical CoQ10 preparations, a number of formulation strategies for insoluble compounds have been tried, such as oil solution, emulsion, cyclodextrin complexation and liposomal nanoencapsulation. 65 Presently, all currently marketed formulations of CoQ10 are for oral administration only. Like all dietary lipids, orally administered CoQ10 is absorbed in the enterocytes, packaged into chylomicrons (large lipoprotein particles) and then transported via the lymphatics to the circulation (Figure 1C) where CoQ10 is mostly packaged into lipoproteins. 66 In humans, the level of total plasma CoQ10 is less than 2 μg/ml. Increases several‐fold above normal plasma level has been reported after CoQ10 treatment. 66 , 67 , 68 However, it is not known how blood CoQ10 concentration is related to effectiveness in relieving symptoms. Moreover, the mechanism of tissue uptake of CoQ10 is still poorly understood. In rodents, after oral CoQ10 supplementation high concentrations of CoQ10 were reported for several tissues including the liver, ovaries, brown adipocytes and spleen, but not for the heart, kidney, muscle and brain, the main affected tissues in PCoQD. 59 , 69 , 70 , 71 , 72 , 73 Key factors that influence the tissue or cellular uptake of CoQ10 await future studies.
There have been discussions on the possible merits of using the reduced form of CoQ10, also known as ubiquinol, to enhance the bioavailability of CoQ10. 74 However, this is not yet strongly supported by all studies, and ubiquinol's claimed to superior bioavailability is still in question. 66 Out of the 89 cases included in our final analysis, 6 were reported to be treated with ubiquinol (Table S4 and S5). Two met our criteria of responding and 4 did not. Thus, these data also do not point to better bioavailability of ubiquinol over regular CoQ10 in PCoQD patients.
In sum, the results of the present review suggest the need to develop alternative strategies of providing CoQ10 for treating PCoQD. For example, our recent study suggests the possibility of intravenously administering CoQ10 solubilized with the fungicide caspofungin to achieve much higher plasma concentration and thus more effective CoQ10 therapy. 75 Moreover, modified precursors of the quinone ring of CoQ10, for example, DHB, have been considered as potential alternative treatment option for some types of PCoQD. 59 , 60 , 61 , 76 , 77 Future work is warranted to further explore these possibilities and unleash the full potential of CoQ10 therapy. Another implication of our study is that better empirical and clinical documentation of the effects of CoQ10 treatments is needed. Our study also stresses the need for caution when seeking to justify the widespread use of CoQ10 for disease treatment or as a dietary supplement. Oral CoQ10 could benefit conditions that affect the few tissues where it readily accumulates. However, so far no such indications have been identified.
AUTHOR CONTRIBUTIONS
YING WANG: Data curation (equal); formal analysis (equal); investigation (equal); methodology (equal); visualization (equal); writing – original draft (equal); writing – review and editing (equal). Siegfried Hekimi: Conceptualization (equal); data curation (equal); formal analysis (equal); funding acquisition (equal); investigation (equal); methodology (equal); supervision (equal); validation (equal); writing – original draft (equal); writing – review and editing (equal).
CONFLICT OF INTEREST
SH and YW have received royalty payment from Clarus Therapeutics Holdings. SH also consults for Clarus Therapeutics Holdings.
Supporting information
Appendix S1
ACKNOWLEDGEMENTS
Research in the laboratory of SH is funded by a Foundation grant from the Canadian Institutes of Health Research: FDN‐159916. SH is Campbell Chair of Developmental Biology.
Wang Y, Hekimi S. The efficacy of coenzyme Q10 treatment in alleviating the symptoms of primary coenzyme Q10 deficiency: A systematic review. J Cell Mol Med. 2022;26:4635‐4644. doi: 10.1111/jcmm.17488
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Wang Y, Hekimi S. Understanding ubiquinone. Trends Cell Biol. 2016;26(5):367‐378. [DOI] [PubMed] [Google Scholar]
- 2. Quinzii CM, Luna‐Sanchez M, Ziosi M, Hidalgo‐Gutierrez A, Kleiner G, Lopez LC. The role of sulfide oxidation impairment in the pathogenesis of primary CoQ deficiency. Front Physiol. 2017;8:525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lenaz G, Genova ML. Structure and organization of mitochondrial respiratory complexes: a new understanding of an old subject. Antioxid Redox Signal. 2010;12(8):961‐1008. [DOI] [PubMed] [Google Scholar]
- 4. Crane FL. Biochemical functions of coenzyme Q10. J Am Coll Nutr. 2001;20(6):591‐598. [DOI] [PubMed] [Google Scholar]
- 5. Morre DJ, Morre DM. Non‐mitochondrial coenzyme Q. Biofactors. 2011;37(5):355‐360. [DOI] [PubMed] [Google Scholar]
- 6. Bentinger M, Brismar K, Dallner G. The antioxidant role of coenzyme Q. Mitochondrion. 2007;7:S41‐S50. [DOI] [PubMed] [Google Scholar]
- 7. Tran UC, Clarke CF. Endogenous synthesis of coenzyme Q in eukaryotes. Mitochondrion. 2007;7:S62‐S71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang Y, Hekimi S. Molecular genetics of ubiquinone biosynthesis in animals. Crit Rev Biochem Mol Biol. 2013;48(1):69‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stefely JA, Pagliarini DJ. Biochemistry of mitochondrial coenzyme Q biosynthesis. Trends Biochem Sci. 2017;42(10):824‐843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang Y, Hekimi S. The complexity of making ubiquinone. Trends Endocrinol Metab. 2019;30(12):929‐943. [DOI] [PubMed] [Google Scholar]
- 11. Tsui HS, Clarke CF. Ubiquinone biosynthetic complexes in prokaryotes and eukaryotes. Cell Chem Biol. 2019;26(4):465‐467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ogasahara S, Engel AG, Frens D, Mack D. Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc Natl Acad Sci U S A. 1989;86(7):2379‐2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hughes BG, Harrison PM, Hekimi S. Estimating the occurrence of primary ubiquinone deficiency by analysis of large‐scale sequencing data. Sci Rep. 2017;7(1):17744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Doimo M, Desbats MA, Cerqua C, Cassina M, Trevisson E, Salviati L. Genetics of coenzyme q10 deficiency. Mol Syndromol. 2014;5(3–4):156‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Traschutz A, Schirinzi T, Laugwitz L, et al. Clinico‐genetic, imaging and molecular delineation of COQ8A‐ataxia: a multicenter study of 59 patients. Ann Neurol. 2020;88(2):251‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Heeringa SF, Chernin G, Chaki M, et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Invest. 2011;121(5):2013‐2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Quinzii CM, Hirano M. Primary and secondary CoQ(10) deficiencies in humans. Biofactors. 2011;37(5):361‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gueguen N, Baris O, Lenaers G, Reynier P, Spinazzi M. Secondary coenzyme Q deficiency in neurological disorders. Free Radic Biol Med. 2021;165:203‐218. [DOI] [PubMed] [Google Scholar]
- 19. Deichmann R, Lavie C, Andrews S. Coenzyme q10 and statin‐induced mitochondrial dysfunction. Ochsner J. 2010;10(1):16‐21. [PMC free article] [PubMed] [Google Scholar]
- 20. Folkers K, Langsjoen P, Willis R, et al. Lovastatin decreases coenzyme Q levels in humans. Proc Natl Acad Sci U S A. 1990;87(22):8931‐8934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Montero R, Grazina M, Lopez‐Gallardo E, et al. Coenzyme Q(1)(0) deficiency in mitochondrial DNA depletion syndromes. Mitochondrion. 2013;13(4):337‐341. [DOI] [PubMed] [Google Scholar]
- 22. Gempel K, Topaloglu H, Talim B, et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron‐transferring‐flavoprotein dehydrogenase (ETFDH) gene. Brain. 2007;130(Pt 8):2037‐2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sacconi S, Trevisson E, Salviati L, et al. Coenzyme Q10 is frequently reduced in muscle of patients with mitochondrial myopathy. Neuromuscul Disord. 2010;20(1):44‐48. [DOI] [PubMed] [Google Scholar]
- 24. Woerner AC, Vockley J. Mitochondrial disease and coenzyme Q10 deficiency: commentary. J Pediatr. 2021;228:14‐15.e1. [DOI] [PubMed] [Google Scholar]
- 25. Kuhl I, Miranda M, Atanassov I, et al. Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. Elife. 2017;6:e30952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yubero D, Montero R, Martin MA, et al. Secondary coenzyme Q10 deficiencies in oxidative phosphorylation (OXPHOS) and non‐OXPHOS disorders. Mitochondrion. 2016;30:51‐58. [DOI] [PubMed] [Google Scholar]
- 27. Berardo A, Quinzii CM. Redefining infantile‐onset multisystem phenotypes of coenzyme Q10‐deficiency in the next‐generation sequencing era. J Transl Genet Genom. 2020;4:22‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Diomedi‐Camassei F, Di Giandomenico S, Santorelli FM, et al. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol. 2007;18(10):2773‐2780. [DOI] [PubMed] [Google Scholar]
- 29. Trevisson E, DiMauro S, Navas P, Salviati L. Coenzyme Q deficiency in muscle. Curr Opin Neurol. 2011;24(5):449‐456. [DOI] [PubMed] [Google Scholar]
- 30. Emmanuele V, Lopez LC, Berardo A, et al. Heterogeneity of coenzyme Q10 deficiency: patient study and literature review. Arch Neurol. 2012;69(8):978‐983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Duncan AJ, Bitner‐Glindzicz M, Meunier B, et al. A nonsense mutation in COQ9 causes autosomal‐recessive neonatal‐onset primary coenzyme Q10 deficiency: a potentially treatable form of mitochondrial disease. Am J Hum Genet. 2009;84(5):558‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hernandez‐Camacho JD, Bernier M, Lopez‐Lluch G, Navas P. Coenzyme Q10 supplementation in aging and disease. Front Physiol. 2018;9:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Acosta MJ, Vazquez Fonseca L, Desbats MA, et al. Coenzyme Q biosynthesis in health and disease. Biochim Biophys Acta. 2016;1857(8):1079‐1085. [DOI] [PubMed] [Google Scholar]
- 34. Parikh S, Saneto R, Falk MJ, et al. A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol. 2009;11(6):414‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hargreaves IP. Coenzyme Q10 as a therapy for mitochondrial disease. Int J Biochem Cell Biol. 2014;49:105‐111. [DOI] [PubMed] [Google Scholar]
- 36. Tarnopolsky MA. The mitochondrial cocktail: rationale for combined nutraceutical therapy in mitochondrial cytopathies. Adv Drug Deliv Rev. 2008;60(13–14):1561‐1567. [DOI] [PubMed] [Google Scholar]
- 37. Mero S, Salviati L, Leuzzi V, et al. New pathogenic variants in COQ4 cause ataxia and neurodevelopmental disorder without detectable CoQ10 deficiency in muscle or skin fibroblasts. J Neurol. 2021;268(9):3381‐3389. [DOI] [PubMed] [Google Scholar]
- 38. AbuMaziad AS, Thaker TM, Tomasiak TM, Chong CC, Galindo MK, Hoyme HE. The role of novel COQ8B mutations in glomerulopathy and related kidney defects. Am J Med Genet A. 2021;185(1):60‐67. [DOI] [PubMed] [Google Scholar]
- 39. Mignot C, Apartis E, Durr A, et al. Phenotypic variability in ARCA2 and identification of a core ataxic phenotype with slow progression. Orphanet J Rare Dis. 2013;8:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lagier‐Tourenne C, Tazir M, Lopez LC, et al. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am J Hum Genet. 2008;82(3):661‐672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mollet J, Delahodde A, Serre V, et al. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am J Hum Genet. 2008;82(3):623‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Clarke CF, Williams W, Teruya JH. Ubiquinone biosynthesis in Saccharomyces cerevisiae. Isolation and sequence of COQ3, the 3,4‐dihydroxy‐5‐hexaprenylbenzoate methyltransferase gene. J Biol Chem. 1991;266(25):16636‐16644. [PubMed] [Google Scholar]
- 43. Ashraf S, Gee HY, Woerner S, et al. ADCK4 mutations promote steroid‐resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest. 2013;123(12):5179‐5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Atmaca M, Gulhan B, Korkmaz E, et al. Follow‐up results of patients with ADCK4 mutations and the efficacy of CoQ10 treatment. Pediatr Nephrol. 2017;32(8):1369‐1375. [DOI] [PubMed] [Google Scholar]
- 45. Korkmaz E, Lipska‐Zietkiewicz BS, Boyer O, et al. ADCK4‐associated glomerulopathy causes adolescence‐onset FSGS. J Am Soc Nephrol. 2016;27(1):63‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xie LX, Hsieh EJ, Watanabe S, et al. Expression of the human atypical kinase ADCK3 rescues coenzyme Q biosynthesis and phosphorylation of coq polypeptides in yeast coq8 mutants. Biochim Biophys Acta. 2011;1811(5):348‐360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stefely JA, Licitra F, Laredj L, et al. Cerebellar ataxia and coenzyme Q deficiency through loss of unorthodox kinase activity. Mol Cell. 2016;63(4):608‐620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maeoka Y, Doi T, Aizawa M, et al. A case report of adult‐onset COQ8B nephropathy presenting focal segmental glomerulosclerosis with granular swollen podocytes. BMC Nephrol. 2020;21(1):376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Peng M, Falk MJ, Haase VH, et al. Primary coenzyme Q deficiency in Pdss2 mutant mice causes isolated renal disease. PLoS Genet. 2008;4(4):e1000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lu S, Lu LY, Liu MF, et al. Cerebellar defects in Pdss2 conditional knockout mice during embryonic development and in adulthood. Neurobiol Dis. 2012;45(1):219‐233. [DOI] [PubMed] [Google Scholar]
- 51. Lapointe J, Wang Y, Bigras E, Hekimi S. The submitochondrial distribution of ubiquinone affects respiration in long‐lived Mclk1+/− mice. J Cell Biol. 2012;199(2):215‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jiang N, Levavasseur F, McCright B, Shoubridge EA, Hekimi S. Mouse CLK‐1 is imported into mitochondria by an unusual process that requires a leader sequence but no membrane potential. J Biol Chem. 2001;276(31):29218‐29225. [DOI] [PubMed] [Google Scholar]
- 53. Nakai D, Yuasa S, Takahashi M, et al. Mouse homologue of coq7/clk‐1, longevity gene in Caenorhabditis elegans, is essential for coenzyme Q synthesis, maintenance of mitochondrial integrity, and neurogenesis. Biochem Biophys Res Commun. 2001;289(2):463‐471. [DOI] [PubMed] [Google Scholar]
- 54. Garcia‐Corzo L, Luna‐Sanchez M, Doerrier C, et al. Dysfunctional Coq9 protein causes predominant encephalomyopathy associated with CoQ deficiency. Hum Mol Genet. 2013;22(6):1233‐1248. [DOI] [PubMed] [Google Scholar]
- 55. EUCOMM . European Conditional Mouse Mutagenesis Program. https://www.mousephenotype.org/about‐impc/about‐ikmc/eucomm/ [DOI] [PubMed]
- 56. Saneto RP. Mitochondrial diseases: expanding the diagnosis in the era of genetic testing. J Transl Genet Genom. 2020;4:384‐428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lopez‐Lluch G, Del Pozo‐Cruz J, Sanchez‐Cuesta A, Cortes‐Rodriguez AB, Navas P. Bioavailability of coenzyme Q10 supplements depends on carrier lipids and solubilization. Nutrition. 2019;57:133‐140. [DOI] [PubMed] [Google Scholar]
- 58. Chung WK, Martin K, Jalas C, et al. Mutations in COQ4, an essential component of coenzyme Q biosynthesis, cause lethal neonatal mitochondrial encephalomyopathy. J Med Genet. 2015;52(9):627‐635. [DOI] [PubMed] [Google Scholar]
- 59. Wang Y, Oxer D, Hekimi S. Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis. Nat Commun. 2015;6:6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hidalgo‐Gutierrez A, Barriocanal‐Casado E, Bakkali M, et al. beta‐RA reduces DMQ/CoQ ratio and rescues the encephalopathic phenotype in Coq9 (R239X) mice. EMBO Mol Med. 2019;11(1):e9466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hidalgo‐Gutierrez A, Barriocanal‐Casado E, Diaz‐Casado ME, et al. Beta‐RA targets mitochondrial metabolism and adipogenesis, leading to therapeutic benefits against CoQ deficiency and age‐related overweight. Biomedicine. 2021;9(10):1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Taylor BA, Lorson L, White CM, Thompson PD. A randomized trial of coenzyme Q10 in patients with confirmed statin myopathy. Atherosclerosis. 2015;238(2):329‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Glover EI, Martin J, Maher A, Thornhill RE, Moran GR, Tarnopolsky MA. A randomized trial of coenzyme Q10 in mitochondrial disorders. Muscle Nerve. 2010;42(5):739‐748. [DOI] [PubMed] [Google Scholar]
- 64. Phase III Trial of coenzyme Q10 in mitochondrial disease. In: https://ClinicalTrials.gov/show/NCT00432744.
- 65. Zaki NM. Strategies for oral delivery and mitochondrial targeting of CoQ10. Drug Deliv. 2016;23(6):1868‐1881. [DOI] [PubMed] [Google Scholar]
- 66. Mantle D, Dybring A. Bioavailability of coenzyme Q10: an overview of the absorption process and subsequent metabolism. Antioxidants (Basel). 2020;9(5):386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shults CW, Oakes D, Kieburtz K, et al. Effects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol. 2002;59(10):1541‐1550. [DOI] [PubMed] [Google Scholar]
- 68. Bhagavan HN, Chopra RK. Plasma coenzyme Q10 response to oral ingestion of coenzyme Q10 formulations. Mitochondrion. 2007;7:S78‐S88. [DOI] [PubMed] [Google Scholar]
- 69. Lass A, Forster MJ, Sohal RS. Effects of coenzyme Q10 and alpha‐tocopherol administration on their tissue levels in the mouse: elevation of mitochondrial alpha‐tocopherol by coenzyme Q10. Free Radic Biol Med. 1999;26(11–12):1375‐1382. [DOI] [PubMed] [Google Scholar]
- 70. Ben‐Meir A, Burstein E, Borrego‐Alvarez A, et al. Coenzyme Q10 restores oocyte mitochondrial function and fertility during reproductive aging. Aging Cell. 2015;14(5):887‐895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Anderson CM, Kazantzis M, Wang J, et al. Dependence of brown adipose tissue function on CD36‐mediated coenzyme Q uptake. Cell Rep. 2015;10(4):505‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Saiki R, Lunceford AL, Shi Y, et al. Coenzyme Q10 supplementation rescues renal disease in Pdss2kd/kd mice with mutations in prenyl diphosphate synthase subunit 2. Am J Physiol Renal Physiol. 2008;295(5):F1535‐F1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Garcia‐Corzo L, Luna‐Sanchez M, Doerrier C, et al. Ubiquinol‐10 ameliorates mitochondrial encephalopathy associated with CoQ deficiency. Biochim Biophys Acta. 2014;1842(7):893‐901. [DOI] [PubMed] [Google Scholar]
- 74. Bhagavan HN, Chopra RK, Craft NE, Chitchumroonchokchai C, Failla ML. Assessment of coenzyme Q10 absorption using an in vitro digestion‐Caco‐2 cell model. Int J Pharm. 2007;333(1–2):112‐117. [DOI] [PubMed] [Google Scholar]
- 75. Wang Y, Hekimi S. Micellization of coenzyme Q by the fungicide caspofungin allows for safe intravenous administration to reach extreme supraphysiological concentrations. Redox Biol. 2020;36:101680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Widmeier E, Airik M, Hugo H, et al. Treatment with 2,4‐dihydroxybenzoic acid prevents FSGS progression and renal fibrosis in podocyte‐specific Coq6 knockout mice. J Am Soc Nephrol. 2019;30(3):393‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Widmeier E, Yu S, Nag A, et al. ADCK4 deficiency destabilizes the coenzyme Q complex, which is rescued by 2,4‐dihydroxybenzoic acid treatment. J Am Soc Nephrol. 2020;31(6):1191‐1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Salviati L, Trevisson E, Rodriguez Hernandez MA, et al. Haploinsufficiency of COQ4 causes coenzyme Q10 deficiency. J Med Genet. 2012;49(3):187‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Aure K, Benoist JF, Ogier de Baulny H, Romero NB, Rigal O, Lombes A. Progression despite replacement of a myopathic form of coenzyme Q10 defect. Neurology. 2004;63(4):727‐729. [DOI] [PubMed] [Google Scholar]
- 80. Blumkin L, Leshinsky‐Silver E, Zerem A, Yosovich K, Lerman‐Sagie T, Lev D. Heterozygous mutations in the ADCK3 Gene in siblings with cerebellar atrophy and extreme phenotypic variability. JIMD Rep. 2014;12:103‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhang L, Ashizawa T, Peng D. Primary coenzyme Q10 deficiency due to COQ8A gene mutations. Mol Genet Genomic Med. 2020;8(10):e1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.