

Abstract
Background/objectives:
Sickle cell disease (SCD) is an important, hidden cause of childhood mortality worldwide. It is most prevalent in sub-Saharan Africa where national newborn screening programs remain unavailable and most children in rural areas are never diagnosed. We conducted a study at a rural district hospital in northern Tanzania to determine the birth prevalence and community awareness of SCD and to determine the feasibility of using point-of-care testing to enroll newborns in a new SCD clinic for ongoing treatment.
Design/methods:
We screened infants at Shirati KMT hospital for SCD using HemoTypeSC, an inexpensive point-of-care test. Infants who screened positive were enrolled in the SCD clinic and instructed to return at 6–12 weeks for confirmatory testing, counseling, and preventive care.
Results:
A total of 999 newborns were screened from February to September 2019. Among these, 31.6% (315/999) had sickle cell trait and 3.9% (39/999) had SCD. No hemoglobin C was detected. Very few parents knew their own sickle cell status (0.3%). At 5 months after completion, 12 infants from the screening study and 30 additional children had been seen at the SCD clinic for ongoing counseling and care.
Conclusions:
Birth prevalence of SCD in rural Tanzania is extremely high and community awareness is low. Newborn point-of-care testing enhances case finding and enables early enrollment in preventive care for SCD, even in rural sub-Saharan Africa with minimal laboratory capacity. SCD-specific clinical services implemented at the district hospital level could expand access to many children and significantly reduce early SCD morbidity and mortality.
Keywords: linkage-to-care, newborn screening, prevalence, sickle cell disease, sub-Saharan Africa
1 |. INTRODUCTION
Sickle cell disease (SCD) is an extremely important cause of childhood mortality in low-resource settings, where lack of diagnosis obscures its significant contribution to under-5 years mortality.1 Sub-Saharan Africa is estimated to account for 75% of the approximately 400 000 infants born globally with SCD each year.2,3 Unfortunately, most children with SCD in sub-Saharan Africa will die before the age of 5,4 making SCD responsible for at least 6–16% of under-5 mortality in Africa.5,6
Adoption of universal newborn screening for SCD in many high-resource countries enables early diagnosis and enrollment in care. Preventive measures such penicillin prophylaxis and pneumococcal vacci-nation and disease-modifying therapy such as hydroxyurea and blood transfusion protocols have steadily increased life expectancy for SCD in high-resource countries over the last several decades,7 reaching 58 years within the United States.8 Similar gains in low-resource countries have been inhibited by lack of access to SCD diagnostic and treatment services.
Universal newborn screening is thought to be highly cost-effective,9 but it is challenging to implement in low-resource settings because it requires specialized equipment, adequate reagents, and laboratory staff. Large referral hospitals in Africa have demonstrated significant improvement in outcomes with the introduction of local screening and implementation of a basic health care package, but services have seldom expanded to more rural areas.10 Until a national newborn strategy is available, equipping district hospitals to diagnose and care for SCD could substantially expand services and decrease morbidity and mortality from SCD.11
Therefore, we conducted a study to further understand the extent of SCD and the feasibility of expanding services into a rural part of northern Tanzania. Our primary objective was to determine the birth prevalence and community awareness of SCD in the town of Shirati. Secondary objectives were to determine the feasibility of using point-of-care (POC) testing in rural Tanzania and the ability to follow up newly diagnosed children in a local SCD clinic. This information is essential for improving access to diagnosis and treatment, reducing morbidity, and increasing survival among children with SCD in sub-Saharan Africa.
2 |. METHODS
2.1 |. Study area
This prospective cohort study was conducted among newborns in Shirati KMT Hospital located in northern Tanzania. The hospital was established in 1935 and currently contains 100 beds. Specialist services are available sporadically when consultants supported by the African Medical and Research Foundation (AMREF) visit four to six times per year. The outpatient department sees 100–150 patients per day. The maternity ward has approximately 1500 deliveries annually.
Shirati KMT Hospital is the designated district hospital for Rorya district along the eastern edge of Lake Victoria (Figure 1 inset). Rorya has a population of approximately 265 000,12 and Shirati is the largest town. Approximately 95% of the population lives in a rural setting, and 19% of the population is < 5 years old. The nearest regional hospital, Musoma Hospital, is in Musoma, 2 h away by car, and the nearest zonal hospital, Bugando Medical Centre (BMC), is in Mwanza, 4.5 h away by car (see Figure 1).
FIGURE 1.
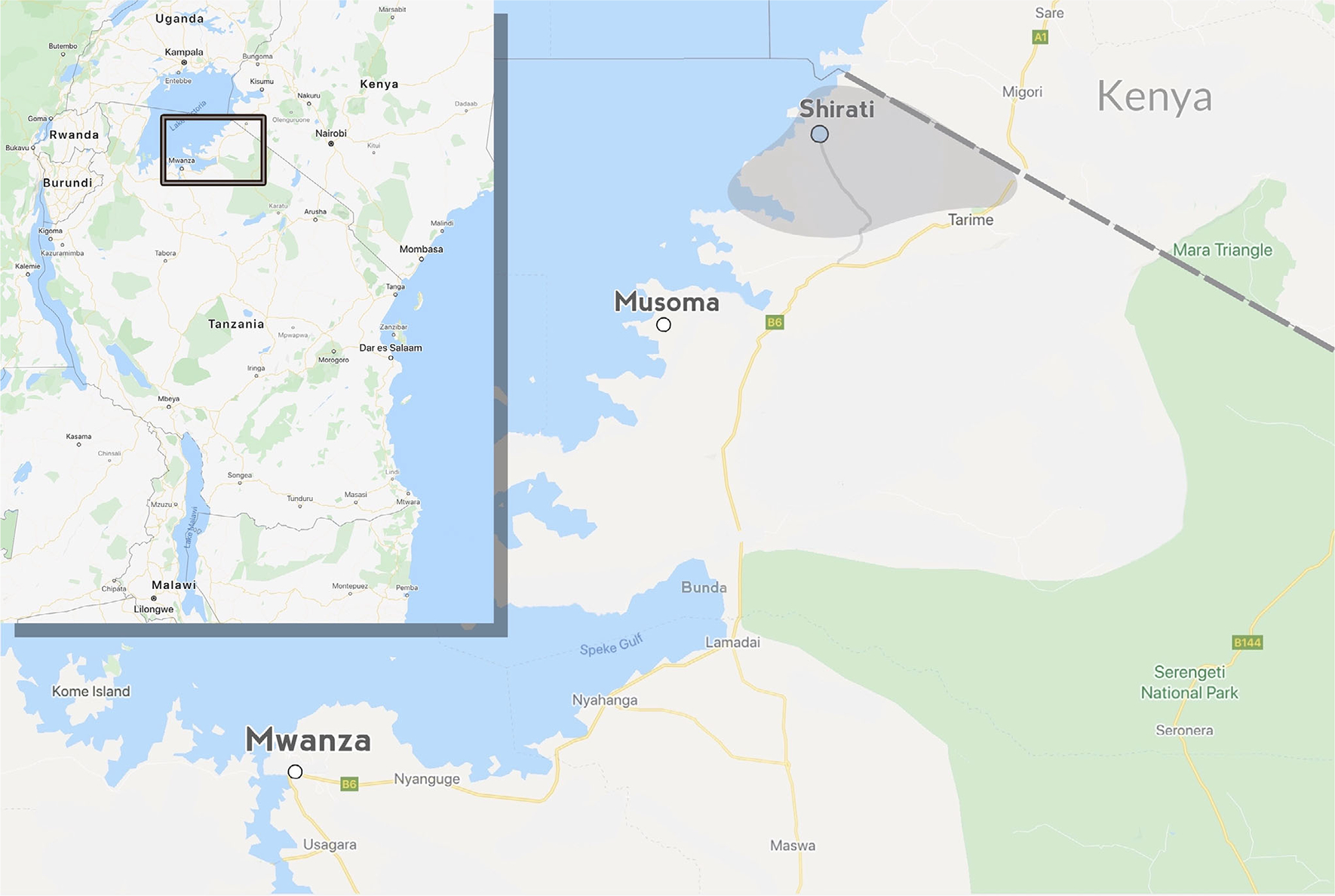
Map of northern Tanzania showing the relationship and distance between Shirati (light blue dot), Musoma, and Mwanza (white dots). Shaded area represents region served by Shirati Hospital. Inset showing relationship of highlighted region within Tanzania
2.2 |. Recruitment/enrollment
All infants born at Shirati KMT Hospital were eligible for enrollment from birth until the day of discharge. Infants who received a blood transfusion before enrollment were supposed to be excluded per the protocol, but none of the infants born at Shirati Hospital received a blood transfusion during the study period. Mothers are hospitalized for 2 days following the birth of an infant. Enrollment occurred 6 days per week (Monday through Saturday), enabling the parents of every neonate born at Shirati Hospital during the study period to be approached for enrollment prior to discharge. The study was briefly explained in Swahili to all families in the maternity ward each morning by one of the study personnel (Brian Dee Akungo). Swahili written informed consent was obtained from all those who wished to have their infant tested.
2.3 |. Point-of-care testing
After written informed consent was obtained from the parents of infants, demographic and clinical information was collected using a standardized questionnaire. A blood sample was collected using a heel stick procedure. Three dried blood spots (DBS) were collected from each infant on Whatman 1001–125 filter paper. One DBS was used for the HemoTypeSC test, and the two remaining DBS were stored for repeat testing when required and confirmatory testing at a later date.
Blood samples were processed according to the instructions provided in the package insert for HemoTypeSC. Using a dropper pipette, six drops of water were added to a small vial. A 3/16-inch circular punch from a DBS was placed in the vial and completely submerged for 10 min. A test strip was then inserted into the vial and the distal end submerged for 10 min. The test strip was then removed and interpreted using the guide provided by the manufacturer (Figure 2). The absence of a line indicated the presence of the hemoglobin variant. Results were interpreted as HbAA, HbAS, HbAC, HbSS, HbSC, HbCC, or invalid. The result was written on each infant’s questionnaire at the time of interpreting the result. A photo of each test strip including the study ID of the infant was taken as a backup. The filter paper with remaining DBS was stored at −20°C in a ziploc bag with desiccant.
FIGURE 2.
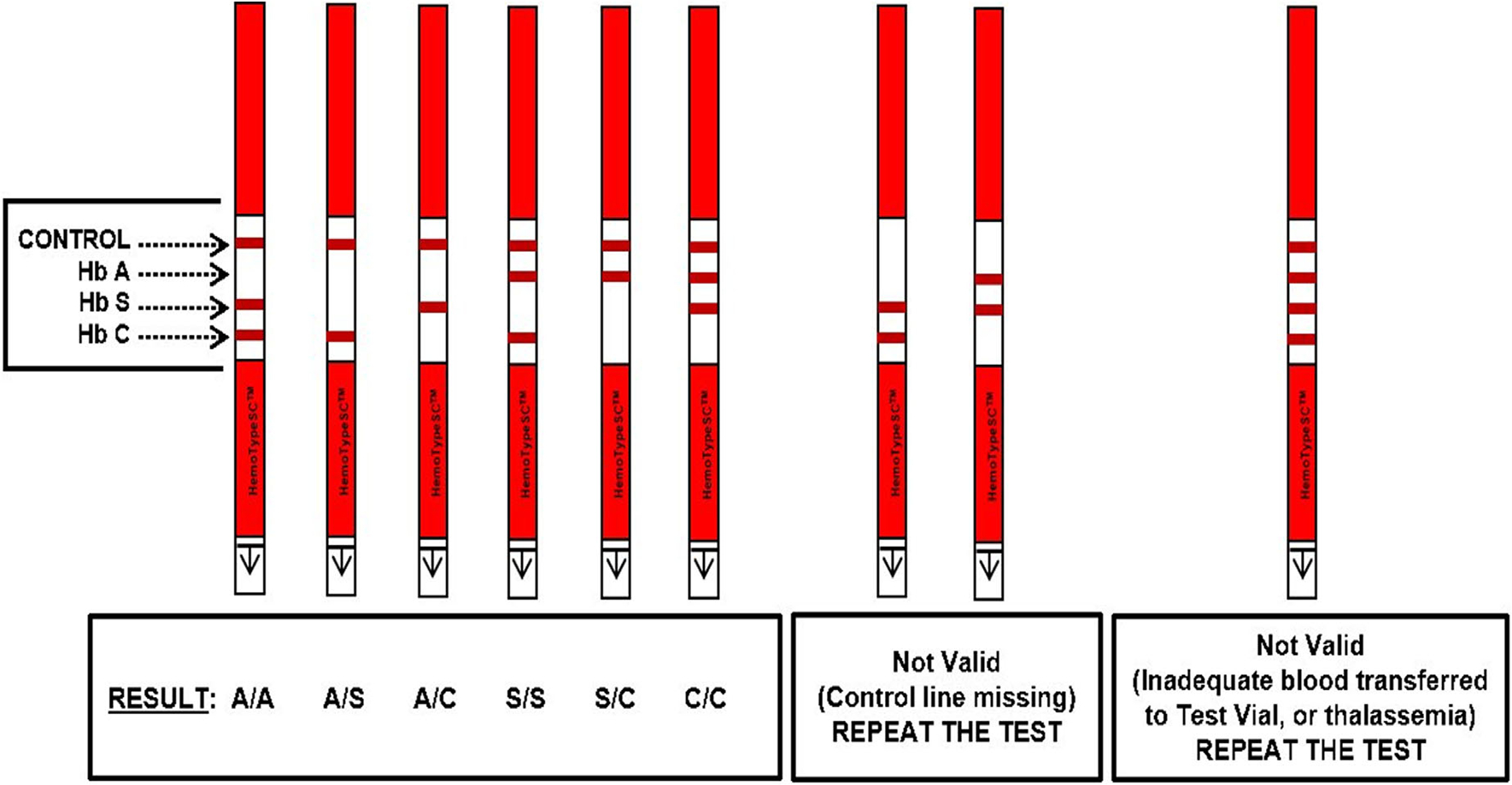
HemoTypeSC point-of-care interpretation
2.4 |. Counseling and referral
POC test results for each neonate were immediately disclosed to his/her caregiver at the conclusion of the test. The infants with a HemoTypeSC result consistent with either SCD or sickle cell trait (SCT) were provided general counseling about SCD and how it is inherited. All infants with SCD were also given an educational handout. Infants with SCD were enrolled in a new SCD clinic at Shirati KMT Hospital that was established to provide follow-up care for infants diagnosed during this study. The SCD clinic sees follow-up visits 2 days per week. Acute care on other days is provided at the hospital emergency department or the pediatric outpatient department. Children seen in the clinic are started on penicillin V prophylaxis 125 mg twice daily and daily folate supplementation at no cost to the family. The clinic also ensures vaccinations are up to date and administers catch up vaccinations as needed. The cost of vaccinations are covered by the Tanzania Ministry of Health.
2.5 |. Confirmatory testing
All DBS interpreted as HbSS were shipped to BMC in one batch after enrollment was completed to attempt confirmatory testing. BMC performs isoelectric focusing (IEF) with the RESOLVE hemoglobin kit and JB-2 staining system (PerkinElmer, Inc., Waltham, MA, USA) with control specimens containing HbA, HbF, HbS, and HbC. Results were interpreted by two individuals independently and differences were resolved through discussion. The presence of HbA, HbF, HbS, HbC, or variant bands were identified, and the results were reported as normal (FA), sickle cell trait (FSA or FAS), disease (FS), variant, or uninterpretable. Uninterpretable results were repeated a second time.
2.6 |. Feasibility
The study set out to assess three measures of feasibility: (a) willingness of families to undergo testing; (b) ability of families to follow up in clinic; and (c) successful completion of confirmatory testing. It was hypothesized that > 80% of families would be willing to undergo testing, > 50% of families would return to clinic, and > 90% of confirmatory test results could be communicated back to Shirati.
2.7 |. Data collection/analysis
Data was collected using paper case report forms and then entered in a REDCap online electronic database.
2.8 |. Ethical issues
The research protocol was approved by the Shirati Hospital review board and the Shirati Health, Education and Development (SHED) Foundation, a nongovernmental organization, as well as the institutional review board of the University of Rochester. Written informed consent was obtained from the parent of each infant. All parents were informed of the POC test results and instructed to return to clinic for confirmatory testing.
3 |. RESULTS
3.1 |. Demographic and clinical characteristics
Between February 2019 and October 2019, 999 children were born at Shirati Hospital. All families were consented, and all infants were enrolled and tested. The median age was 1 day (interquartile range, 1–1 day) (Table 1). There were 504 (50.5%) male and 495 (49.5%) female infants, and 48 (4.8%) were born prematurely (< 37 weeks gestational age). Most parents had attended some primary school (77.8% mothers, 70.2% fathers). Fewer parents had any secondary education (17.8% mothers, 22.1% fathers). Very few parents knew their sickle cell status (0.3%). Only six (0.6%) of the infants had a sibling death with confirmed SCD. A sibling with more than or equal to three blood transfusions was reported in 32 (3.2%), three of whom had HbSS. Recurrent jaundice in a sibling was reported in 254 (25.4%) of all infants, and eight of the 254 had HbSS. A sibling with recurrent swelling of fingers or toes was only reported in 13 (1.3%) of the families, and none of the infants had HbSS. Family history of known SCD and sibling history of transfusion, recurrent jaundice, or dactylitis were not associated with SCD in the infant screened (2.9% prevalence of SCD in newborns with positive family history or clinical symptoms vs 4.3% prevalence in newborns without positive family history or siblings with clinical symptoms; P = .34).
TABLE 1.
Baseline characteristics of 999 neonates screened for hemoglobinopathies at Shirati KMT Hospital from February to September 2019
| Characteristic | Number (%)or median (interquartile range) |
|---|---|
| Age (days) | 1 (1–1) |
| Gender | |
| Male | 504 (50.5%) |
| Female | 495 (49.5%) |
| Premature birth (< 37 weeks gestation) | |
| Yes | 48 (4.8%) |
| No | 947 (94.8%) |
| Mother’s education | |
| Never attended | 37 (3.7%) |
| Attended primary | 777 (77.8%) |
| Attended secondary | 178 (17.8%) |
| Attended university | 6 (0.6%) |
| Father’s education | |
| Never attended | 27 (2.7%) |
| Attended primary | 701 (70.2%) |
| Attended secondary | 221 (22.1%) |
| Attended university | 14 (1.4%) |
| Maternal sickle cell status | |
| Unknown | 997 (99.8%) |
| Sickle cell disease | 2 (0.2%) |
| Sickle cell trait | 0 (0.0%) |
| Paternal sickle cell status | |
| Unknown | 994 (99.5%) |
| Sickle cell disease | 5 (0.5%) |
| Sickle cell trait | 0 (0.0%) |
| Sibling history | |
| Sibling death from sickle cell disease | 6 (0.6%) |
| Sibling ≥ three blood transfusions | 32 (3.2%) |
| Sibling with recurrent/frequent jaundice | 254 (25.4%) |
| Sibling with recurrent finger/toe swelling | 13 (1.3%) |
3.2 |. Prevalence of hemoglobinopathies
The distribution of hemoglobin types identified by HemoTypeSC among the study participants is shown in Figure 3. An abnormal hemoglobin was detected in 354 of 999 (35.4%) infants. Of the 999 screened infants, 315 (31.6%) had SCT, 39 (3.9%) had SCD, which was in Hardy-Weinberg equilibrium. No HbC was detected.
FIGURE 3.
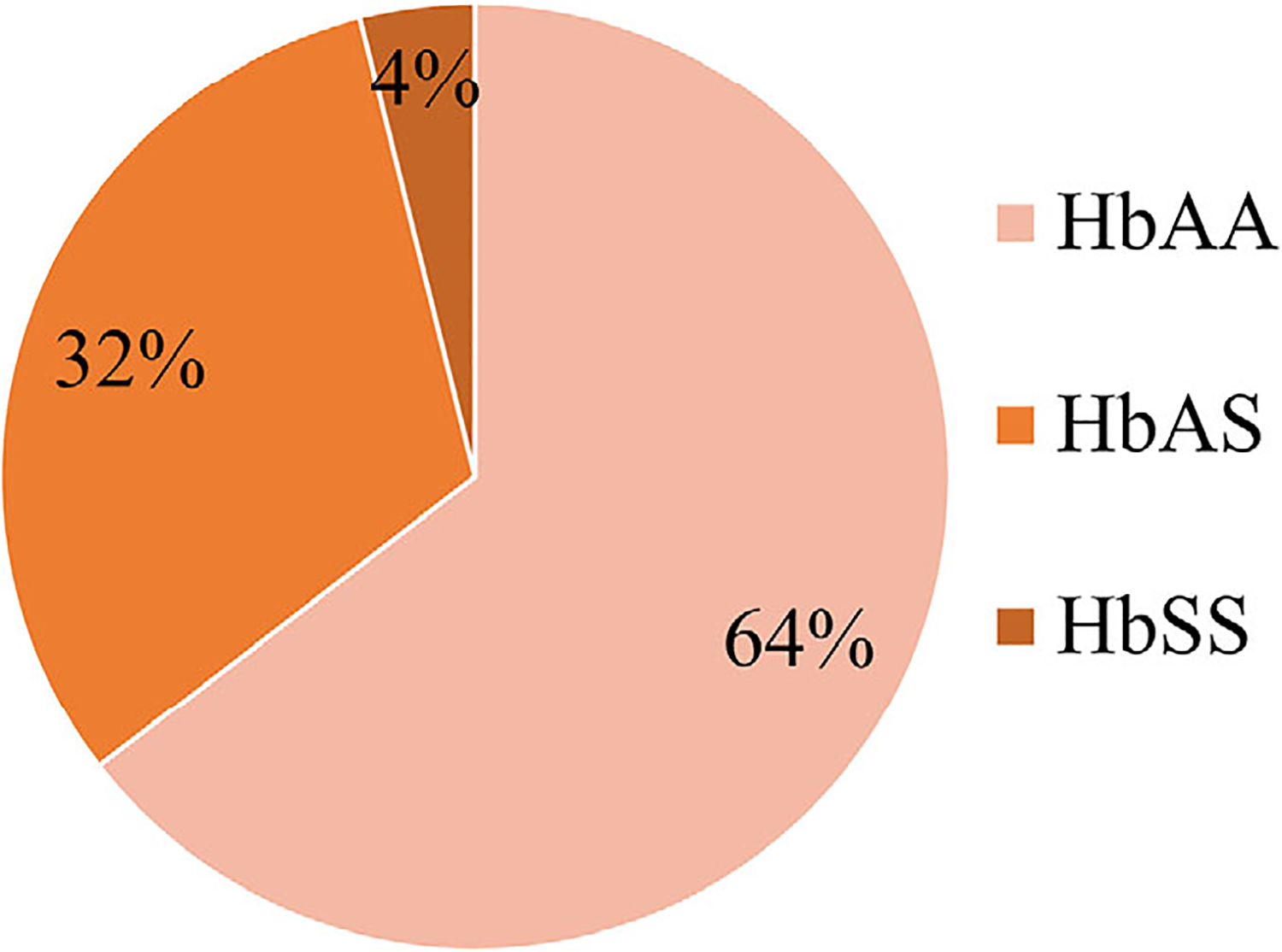
Prevalence of hemoglobinopathies in 999 infants screened from February to September 2019 at Shirati KMT Hospital, Shirati, Tanzania
3.3 |. Follow up
At 5 months after completion of the study, 12 of the 39 newborns (31%) had been seen in the SCD follow-up clinic. Another 30 children under 5 years of age from the community had been referred and seen in clinic, mostly older siblings and relatives of newborns screened in the study. All are receiving prophylactic penicillin, folic acid, and immunizations.
3.4 |. Confirmatory results of the HemoTypeSC
Stored DBS from the 39 infants who had SCD by the POC testing were later shipped and the conclusion of the enrollment to the referral hospital for confirmatory testing using IEF. Of the 39 that were identified as having SCD, 30 were confirmed by IEF as having SCD. Of the remaining nine that were not confirmed as having SCD, two were identified as normal and seven as having SCT.
4 |. DISCUSSION
The birth prevalence of SCD is extraordinarily high in this rural area of northern Tanzania, with 3.9% of newborns having SCD, and 31.6% having SCT by POC testing. If discordant IEF results are recategorized, the prevalence of SCD is 3.0% and the prevalence of SCT is 32.2%.This is the highest prevalence ever reported in Tanzania and among the highest recorded in sub-Saharan Africa. The Tanzanian Sickle Surveillance Study (TS3), the largest study to date in Tanzania, documented an SCD prevalence of 1.2% across Rorya district. TS3 was restricted to specimens collected from HIV-exposed infants, and < 500 samples were collected within Rorya district. Our study tested twice as many newborns, but our population was restricted to a single hospital. Likely the prevalence of SCD across the district is around 1.2% and the prevalence in the community immediately around the hospital is much higher. The high prevalence of SCD, negligible knowledge about the disease, and lack of diagnostic testing in rural northern Tanzania undoubtedly contributes to the high under-5 and childhood mortality rates in Rorya district where Shirati KMT Hospital is located. Rorya has an under-5 mortality rate of 113.3 deaths/1000 live births, almost double the national average of 66.5 deaths/1000 live births, and the highest in the country.12 The under-5 mortality rate in Rorya is 20 times higher than the under-5 mortality rate in the United States, which was 5.8 deaths/100 000 live births in 2017.13
We attempted to confirm the POC test results at the local referral/zonal hospital with IEF, but seven of 39 (18%) were interpreted as SCT by IEF, and two of 39 (5%) were interpreted as normal. Although this could reflect the accuracy of the HemoTypeSC assay, it seems more likely that it is the consequence of poor preservation and transportation of DBS. The DBS in our study were not collected on traditional Whatman 903 cards, and many were not tested until > 6 months after initial collection. There may have been degradation of hemoglobin during storage and transport prior to IEF testing, making individual bands harder to distinguish. HemoTypeSC was both sensitive (93.4–100%) and specific (99.9–100%) during prior field testing in both a multicenter study conducted in Ghana, Martinique, and the USA, and a single-center study conducted in Nigeria.14,15 Therefore, we did not test all specimens to investigate the sensitivity and specificity of HemoTypeSC. Only a small number of discordant results were discovered in both prior studies, but the confirmatory testing modality and adjudication methods differed between these studies, making the results difficult to interpret.14,15
Another possible reason for the discrepant results in our study may be the age of the infants tested. All children were tested on day 1–2 of life when HbF is at its highest and HbS and HbA are very low. In prior studies, a minority of children tested with HemoTypeSC were < 30 days old, but no greater discrepancies were identified in younger age groups. Ultimately, all children with SCD results by HemoTypeSC in our study were instructed to return to the SCD clinic at Shirati so that confirmatory testing could be performed at 6–12 weeks of life, when HbF will have waned. These challenges highlight the need to perform confirmatory testing on more than one diagnostic platform, the need for proper storage and timely testing, and the need for central highly qualified laboratories that can investigate questionable results.
We established a follow-up clinic at Shirati so that infants diagnosed with SCD could be enrolled in care, receive confirmatory testing, counseling, and standard preventive therapy. Approximately one-third of infants returned to the clinic, and efforts to improve linkage-to-care after discharge from the maternity ward are ongoing. Education regarding SCD symptoms are a key aspect of follow-up care since almost none (0.3%) of the mothers knew their sickle cell status. Although we did not meet our goal of 50% of children returning to clinic, as a result of family education an additional 30 children under 5 years old have been diagnosed and referred to the SCD clinic, mostly older siblings or relatives of newborns screened.
Implementation of any case-finding strategies must be followed by linkage-to-care and provision of a basic health care package. Improving services at the district hospital level should be a priority of public health efforts to improve childhood survival for SCD. Urban SCD programs in sub-Saharan Africa have improved childhood survival of SCD with early diagnosis.10 We provide an example of how diagnosis and treatment for SCD can be expanded to rural district hospitals who have a collaborative relationship with their nearby referral center. This model could be implemented by individual district hospitals until a national newborn screening program becomes available.
Our study had several limitations. Confirmatory testing was not performed on all participants. The confirmatory testing that was completed was delayed until the end of the study resulting in some low-quality specimens. Furthermore, we were unable to genotype the specimens to resolve the discrepancies between HemoTypeSC and IEF. However, children did receive follow-up instructions to return to the SCD clinic where they could obtain counseling and retesting when needed. Because we did not trace participants who missed a follow-up appointment, we were unable to identify barriers to follow up. Developing a plan to ensure reliable storage and transportation of DBS and to enhance linkage-to-care will be crucial for future success and should be considered by all those starting NBS programs in remote settings. This will be assessed in future work.
5 |. CONCLUSION
The birth prevalence of SCD around the town of Shirati in northern Tanzania is among the highest ever reported, and represents a focus of extremely high disease burden within the country. POC testing for SCD and introduction of SCD-specific clinical services is feasible in rural district hospitals who are supported by a referral center. District hospitals within Tanzania and other countries in sub-Saharan Africa could significantly expand diagnostic services using similar POC tests until universal newborn screening is available and provide life-saving treatment of SCD.
ACKNOWLEDGMENTS
We would like to thank the patients, their families, the Shirati Health Education and Development Foundation, the laboratory at Bugando Medical Centre, and the Shirati KMT Hospital administration and staff.
Abbreviations:
- BMC
Bugando Medical Centre
- DBS
dried blood spot
- IEF
isoelectric focusing
- POC
point-of-care
- SCD
sickle cell disease
- SCT
sickle cell trait
Footnotes
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
REFERENCES
- 1.Ware RE. Is sickle cell anemia a neglected tropical disease?. PLoS Negl Trop Dis. 2013;7(5):e2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Piel FB, Patil AP, Howes RE, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet. 2013;381(9861):142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piel FB, Howes RE, Patil AP, et al. The distribution of haemoglobin C and its prevalence in newborns in Africa. Sci Rep. 2013;3:1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, Williams TN. Sickle cell disease in Africa: a neglected cause of early childhood mortality. Am J Prev Med. 2011;41(6 Suppl 4):S398–S405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.World Health Assembly, 59. Sickle-Cell Anaemia: report by the Secretariat. World Health Organization; 2006. https://apps.who.int/iris/handle/10665/20890 [Google Scholar]
- 7.Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood. 2010;115(17):3447–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elmariah H, Garrett ME, De Castro LM, et al. Factors associated with survival in a contemporary adult sickle cell disease cohort. Am J Hematol. 2014;89(5):530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rajbhandari R, McMahon DE, Rhatigan JJ, Farmer PE. The neglected hospital - the district hospital’s central role in global health care delivery. N Engl J Med. 2020;382(5):397–400. [DOI] [PubMed] [Google Scholar]
- 10.Rahimy MC, Gangbo A, Ahouignan G, et al. Effect of a comprehensive clinical care program on disease course in severely ill children with sickle cell anemia in a sub-Saharan African setting. Blood. 2003;102(3):834–838. [DOI] [PubMed] [Google Scholar]
- 11.Kuznik A, Habib AG, Munube D, Lamorde M. Newborn screening and prophylactic interventions for sickle cell disease in 47 countries in sub-Saharan Africa: a cost-effectiveness analysis. BMC Health Serv Res. 2016;16:304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.National Bureau of Statistics. Mortality and Health. National Bureau of Statistics. The United Republic of Tanzania. Published July, 2015. https://www.nbs.go.tz/nbs/takwimu/census2012/Mortality_and_Health_Monograph.pdf. Accessed March 31, 2018.
- 13.CDC. Infant Mortality. Centers for Disease Control and Prevention. Updated March 27, 2019. https://www.cdc.gov/reproductivehealth/maternalinfanthealth/infantmortality.htm. Accessed March 19, 2020.
- 14.Steele C, Sinski A, Asibey J, et al. Point-of-care screening for sickle cell disease in low-resource settings: a multi-center evaluation of HemoTypeSC, a novel rapid test. Am J Hematol. 2019;94(1):39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nnodu O, Isa H, Nwegbu M, et al. HemoTypeSC, a low-cost point-of-care testing device for sickle cell disease: promises and challenges. Blood Cells Mol Dis. 2019;78:22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]