

Abstract
We and others have reported that calpain-1 was increased in myocardial mitochondria from various animal models of heart disease. This study investigated whether constitutive up-regulation of calpain-1 restricted to mitochondria induced myocardial injury and heart failure, and if so, whether these phenotypes could be rescued by selective inhibition of mitochondrial superoxide production. Transgenic mice with human CAPN1 up-regulation restricted to mitochondria in cardiomyocytes (Tg-mtCapn1/tTA) were generated and characterized with low and high over-expression of transgenic human CAPN1 restricted to mitochondria, respectively. Transgenic up-regulation of mitochondria-targeted CAPN1 dose-dependently induced cardiac cell death, adverse myocardial remodelling, heart failure, and early death in mice, the changes of which were associated with mitochondrial dysfunction and mitochondrial superoxide generation. Importantly, a daily injection of mitochondria-targeted superoxide dismutase mimetics mito-TEMPO for one month starting from age 2 months attenuated cardiac cell death, adverse myocardial remodelling and heart failure, and reduced mortality in Tg-mtCapn1/tTA mice. In contrast, administration of TEMPO did not achieve similar cardiac protection in transgenic mice. Furthermore, transgenic up-regulation of mitochondria-targeted CAPN1 induced a reduction of ATP5A1 protein and ATP synthase activity in hearts. In cultured cardiomyocytes, increased calpain-1 in mitochondria promoted mitochondrial permeability transition pore (mPTP) opening and induced cell death, which were prevented by over-expression of ATP5A1, mito-TEMPO or cyclosporin A, an inhibitor of mPTP opening. In conclusion, this study has provided direct evidence demonstrating that increased mitochondrial calpain-1 is an important mechanism contributing to myocardial injury and heart failure by disrupting ATP synthase, and promoting mitochondrial superoxide generation and mPTP opening.
Keywords: Calpain, Heart failure, Mitochondria, Superoxide onion, ATP synthase
1. Introduction
Calpains belong to a 15 member-family of calcium-dependent proteases. Among them, calpain-1 and calpain-2 are ubiquitously expressed. Both consist of a large catalytic subunit CAPN1 or CAPN2 encoded by capn1 or capn2, respectively, and a small regulatory subunit CAPNS1 encoded by capns1 [14]. Activation of calpains has been implicated in a variety of cardiac diseases. Both pharmacological and genetic inhibition of calpains prevent cardiac injury and adverse myocardial remodelling under several pathological conditions including ischemia, diabetes, sepsis [31] [5, 19-22, 28, 29, 35, 38]. These studies underscore a possible role of calpain in the progression of heart failure. This was indeed supported by experimental evidence showing that cardiomyocyte-specific over-expression of calpain-1 sufficiently induced dilated heart failure and early death in transgenic mice [13]. Thus, calpain may represent a promising therapeutic target for cardiac diseases.
While it is well-known that calpains are cytoplasmic proteases, calpains are also present in other organelles including mitochondria [17]. In addition, recent studies from our lab and others have shown that the protein levels and activities of calpain-1 and/or calpain-2 are increased in cardiomyocyte mitochondria under various stresses including ischemia/reperfusion, diabetic conditions, and sepsis [4, 5, 28, 29, 35, 38]. Importantly, increased mitochondrial calpain may target and disrupt respiratory chain complex I[35, 38] and V or ATP synthase in cardiomyocytes through proteolysis of respiratory chain complex subunits, such as ATP synthase subunit alpha (ATP5A1) [28, 29]. This leads to mitochondrial dysfunction and subsequent reactive oxygen species (ROS) production, e.g. superoxide onion [28, 29], which has been shown to contribute to cardiac cell death and hypertrophy [7, 9, 27], two main features of adverse myocardial remodelling. Mitochondrial ROS may serve as a signalling molecule triggering pro-inflammatory responses in cardiomyocytes [41] and it also impairs mitochondrial respiration in ischemic heart disease [16]. Thus, selective inhibition of mitochondrial ROS reduced cardiac disease in animal models [7-9, 27]. These previous findings raise an intriguing possibility that increased mitochondrial calpain may be a common mechanism contributing to mitochondrial ROS generation, cardiac injury and heart failure. However, direct evidence is lacking about whether selectively increasing calpains in mitochondria can sufficiently induce cardiac injury and heart failure.
In this study, we generated a novel line of transgenic mice with over-expression of CAPN1 restricted to mitochondria and investigated whether constitutive up-regulation of calpain-1 restricted to mitochondria induced myocardial injury and heart failure; if so, we aimed to determine whether these phenotypes were associated with disruption of ATP synthase and could be reversed by the selective inhibition of mitochondrial superoxide production.
2. Methods
2.1. Animals
This investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication, 8th Edition, 2011). All experimental procedures were approved by the Animal Use Subcommittee at Soochow University in China. Breeding pairs of C57BL/6 mice were purchased from the Jackson Laboratory.
The transgenic vector containing full-length human capn1 gene (coding region) with mitochondrial signal peptide (MTS) and myc-tag under the tetracycline transactivator (tTA) inducible mouse alpha-myosin heavy chain (α-MHC) promoter was constructed, and transgenic mice containing the mouse α-MHC promoter along with the MTS-capn1-myc-tag cDNA (Tg-mtCapn1) were created as previously described [32]. The Tg-mtcapn1 mice were crossed with transgenic mice with cardiomyocyte-specific over-expression of tTA (Tg-tTA) to produce wild-type, Tg-tTA, Tg-mtCapn1 and Tg-Capn1/tTA mice, which were identified by polymerase chain reaction (PCR) using both tTA and human capn1 primers (Supplementary Figure 1A). The beta-2-microglobulin (B2GM) gene was simultaneously amplified by PCR as a control. Both Tg-tTA mice and the transgenic vector containing the tTA inducible mouse α-MHC promoter were kindly provided by Dr. Jeffrey Robbins [32].
All animals were housed in a temperature- and humidity-controlled facility with 12-hour light and dark cycles with water and food ad libitum.
2.2. Experimental protocol
In an effort to distinguish the role of mitochondrial superoxide versus global cellular superoxide, male mice received a daily injection of mitochondrial-targeted 2,2,6,6-Tetramethyl-1-piperidinyloxy (mito-TEMPO, 0.7 mg/kg/day, i.p.), a superoxide dismutase mimetics selectively targeting and scavenging mitochondrial superoxide, or the same dose of TEMPO which scavenges global cellular superoxide as a control starting from age 2 months for a total of 30 days. This dose of mito-TEMPO was based on our recent report [27]. There were 5-10 mice in each group.
2.3. Echocardiography
Animals were lightly anaesthetized with inhaled isoflurane (1%) and imaged on a warm handling platform using a 40 MHz linear array transducer attached to a preclinical ultrasound system (Vevo 2100, FUJIFILM Visual Sonics, Canada) with nominal in-plane spatial resolution of 40 μm (axial) × 80 μm (lateral) as we described recently [28]. Left ventricular (LV) end-systolic inner diameter (LVIDs), LV end-diastolic inner diameter (LVIDd), and fractional shortening (FS) were analyzed. The pulsed wave Doppler measurements of maximal early (E) and late (A) transmitral velocities in diastole were performed in the apical view with the cursor at mitral valve inflow. At the end of each experiment, mice were euthanized by cervical dislocation under anaesthesia with inhaled isoflurane (3-5%) after ensuring that they did not respond to needle-punch to the skin.
2.4. Cell membrane permeability to evans blue dye (EBD)
EBD was dissolved in saline and injected into mice (100 mg/kg body weight, i.p.). Twenty-four hours later, mice were euthanized by cervical dislocation under anaesthesia with a mixture of ketamine (100 mg/kg) /zylaxine (5 mg/kg, i.p.) after ensuring that they did not respond to needle-punch to the skin. The heart was harvested and embedded in optimal cutting temperature (OCT) compound (Sakura), snap frozen in liquid nitrogen, and cut into 5-μm cryosections. EBD uptakes (red) were visualized under fluorescence microscopy. The EBD positive cells were counted in whole section and they were presented in each high power field (HPF).
2.5. Histological examination
After being fixed in 10% formalin, heart tissues were routinely processed, embedded, and sectioned. Haematoxylin and eosin staining was performed in heart tissue sections. Cardiomyocyte cross-sectional areas and collagen deposition of heart sections were measured as we previously described [18].
2.6. Measurement of ROS generation in freshly isolated mitochondria
Mitochondria were isolated from freshly harvested hearts as described previously [28]. The isolated mitochondria were further purified using Percoll density gradient centrifugation. Mitochondrial ROS generation was determined on addition of pyruvate/malate by using Amplex Red and horseradish peroxidise (Invitrogen, USA) according to the manufacturer’s instructions.
2.7. Measurement of OCR
Mitochondrial oxygen consumption rate (OCR) was measured by the Version 3.6 with Instech Viewer 2.51 (Instech, USA). Fifty μg of freshly isolated mitochondria were mixed with 160 μL respiration buffer (225 mM mannitol,75 mM sucrose, 10 mM KCl, 10 mM Tris-HCl, 5mM KH2PO4), which was saturated with oxygen at 30°C and added with ADP (125 nmol). Next, the complex I substrates including malate (5 μM) and pyruvate (5 μM) were added. Respiration in the presence of ADP is called State 3; when the ADP is exhausted, respiration turns to a resting state or State 4. OCR was presented for State 3 and State 4 respiration.
2.8. Determination of malondialdehyde (MDA)
The levels of MDA in freshly heart tissue lysates were measured using a TBARS assay kit (Cayman Chemical Company, USA) following the manufacturer’s instructions.
2.9. ATP synthase activity
ATP synthase activity was measured in heart tissue lysates using an assay coupled with pyruvate kinase, which converts ADP to ATP and produces pyruvate from phosphoenolpyruvate, as described previously [28].
2.10. Real-time reverse transcriptase-PCR
Total RNA was extracted from heart tissues using the Trizol Reagent (Sigma-Aldrich, USA) following the manufacturer's instructions. Real-time reverse transcriptase-PCR was performed for analyzing mRNA levels of ANP, β-MHC, Collagen-I, Collagen-III and GAPDH as previously described [18].
2.11. Western blot analysis
The protein levels of CAPN1, CAPN2, ATP5A1, ATP synthase β subunit, VDAC1 and GAPDH were analyzed in heart tissue lysates by western blot using their specific antibodies from Cell Signaling Technology, USA (1:1000 for CAPN1, CAPN2 and VDAC1), Abcam, Canada (1:1000 for ATP5A1 and ATP synthase β subunit) or Santa Cruz Biotechnology, USA (1:5000 for GAPDH).
2.12. Isolation and cultures of adult mouse cardiomyocytes
Adult mice were killed by neck dislocation. Hearts were isolated and perfused after ensuring that mice did not respond to needle-punch to the skin. Mouse ventricle cardiomyocytes were isolated and cultured according to methods we described previously [27-29].
2.13. Infection with adenoviral vectors
Cultured cells were infected with adenoviral vectors containing mitochondria-targeted capn1 and atp5a1 gene (Ad-mtcapn1, Ad-ATP5A1, SignaGen, USA) or hemagglutinin (Ad-HA, SignaGen, USA) as a control at a multiplicity of infection of 100 plaque forming units/cell. Twenty-four hours late, cells were subjected to various treatments.
2.14. Determination of cell death
Adult cardiomyocyte death was detected by Annexin-V and nuclei were stained using Hoechst 33324 as described previously [27, 29]. At least 200 adult cardiomyocytes were examined for each sample.
2.15. Assessment of the mitochondrial permeability transition pore (mPTP) opening in H9c2 cells
The rat myoblast H9c2 cell line was purchased from ATCC (USA). The mPTP opening was determined using calcein AM following the manufacturer’s instructions. Briefly, the cells were loaded with calcein AM and a cytosolic calcein fluorescence quencher (CoCl2) for 30 minutes at 37 °C [39]. Mitochondrial calcein fluorescence images were acquired with a fluorescence microscope. The nuclei were stained using Hoechst 33342.
2.16. Statistical analysis
Data were given as Mean ± SD. Student’s t test was employed for comparisons within two groups. ANOVA followed by Newman-Keuls test was performed for multi-group comparisons. Survival curves were created by the method of Kaplan and Meier, and compared by log-rank test. A value of P < 0.05 was regarded as statistically significant.
3. Results
3.1. Characterization of cardiomyocyte-specific and mitochondria-targeted capn1 expression in Tg-mtCapn1/tTA double-transgenic mice
Two different lines of Tg-mtCapn1/tTA mice were generated. Western blot analysis revealed that low and high levels of transgenicCAPN1 protein were detected in mitochondria but not cytosolic fractions from heart tissues of two lines of Tg-mtCapn1/tTA, respectively (Supplementary Figure 1B). Accordingly, these two lines of transgenic mice were designated as Tg-mtCapn1/tTAlow (low over-expression of transgenic CAPN1 or TG54) and Tg-mtCapn1/tTAhigh (high over-expression of transgenic CAPN1 or TG38). In contrast, transgenic expression of CAPN1 protein was not detected in lung and skeletal muscle tissues from Tg-mtCapn1/tTA mice (Supplementary Figure 1C). These results verify cardiac-specific and mitochondria-targeted expression of transgenic CAPN1 in Tg-mtCapn1/tTA mice. All Tg-mtCapn1 and Tg-tTA mice grew normal without any cardiac phenotypes (also see Supplementary Table-1) and thus, a mixture of wild-type, Tg-mtCapn1 and Tg-tTA served as wild-type littermates for the following studies.
To manipulate inducible transgenic expression of CAPN1, we fed the breeding pairs of Tg-mtCapn1/tTAhigh mice with doxycycline in drinking water during gestation and immediately postpartum. After weaning, the offspring were continually given doxycycline in drinking water for another 2 months. The expression of transgenic CAPN1 protein was not detected in Tg-mtCapn1/tTAhigh mouse hearts in the presence of doxycycline in drinking water (Supplementary Figure 2A). These results confirmed that the tTA system was effective in inducing the expression of transgenic CAPN1 protein in Tg-mtCapn1/tTA mice.
3.2. Transgenic over-expression of mitochondria-targeted CAPN1 sufficiently induces dilated heart failure and early death in Tg-mtCapn1/tTAhigh mice
Echocardiographic analysis revealed that LV chamber diameter was greater and myocardial function decreased in Tg-mtCapn1/tTAhigh mice compared with their wild-type littermates at age 2 months (Figure 1A-1D). The LV chamber diameter was further increased and myocardial function was progressively decreased in Tg-mtCapn1/tTAhigh mice at age 3 months. The heart weight was much greater in Tg-mtCapn1/tTAhigh mice compared with their wild-type littermates at age 3 months (Supplementary Table-1). Histological and anatomic analyses demonstrated dilated cardiomyopathic changes in Tg-mtCapn1/tTAhigh mice including cardiac cell death and resultant fibrosis, increased cross-sectional cardiomyocyte areas and collagen deposition, and increased heart size (Figure 1E-1G). Evans blue staining of heart tissues confirmed an increase in cardiac cell death in Tg-mtCapn1/tTAhigh mice (Supplementary Figure 3). Histological analysis of the lungs demonstrated interstitial edema and pronounced peri-bronchial inflammatory processes in Tg-mtCapn1/tTAhigh mice at age 3 months, providing additional evidence supporting the occurrence of heart failure (Figure 1E). As a consequence, Tg-mtCapn1/tTAhigh mice began to die at age 1.5 months, and by age 3 months the mortality was around 30% (Figure 1H). To confirm that the cardiac phenotypes were due to up-regulation of mitochondria-targeted CAPN1 in Tg-mtCapn1/tTAhigh mice, we administered doxycycline through their drinking water during gestation and 3 months postpartum. Echocardiography showed normal LV chamber diameter and myocardial function in Tg-mtCapn1/tTAhigh mice fed with doxycycline in drinking water at age 3 months, with normal cardiomyocyte size and collagen deposition by histological analysis (Supplementary Figure 2B-2E).
Figure 1. (A, B, C, D) Echocardiographic analysis.

Echocardiography was conducted on transgenic mice (TG54 and TG38) and their wild-type littermates (a mixture of wild-type, Tg-mtCapn1 and Tg-tTA). Each mouse was scanned twice at age 2 and 3 months, respectively. (A) E/A ratio. (B) Fractional shortening. (C) Left ventricle diastolic diameter (LVDD). (D) Left ventricle systolic diameter (LVSD). Data are mean ± SD, n = 5. *P< 0.05 versus WT (2 months), †P< 0.05 versus WT (3 months), ‡P< 0.05 versus TG54 (2 months), and #P< 0.05 versus TG54 (3 months) (two-way ANOVA followed by Newman-Keuls test).
(E, F, G, H) Histological and anatomic analyses and mortality. Heart and lung tissues were collected and processed for histological analysis from transgenic mice (TG54 and TG38) and their wild-type littermates (WT) at age 3 months. (E) A representative histological picture of H&E staining for heart and lung, wheat germ agglutinin (WGA) staining, Sirius red staining and whole heart. (F) Quantification for cardiomyocyte cross-sectional areas. (G) Quantification for collagen deposition. Data are mean ± SD, n = 4-6. *P< 0.05 versus WT, †P< 0.05 versus TG54 (one-way ANOVA followed by Newman-Keuls test). (H) Survival.
Although no death was observed in Tg-mtCapn1/tTAlow mice by age 3 months, these animals displayed myocardial dysfunction and increases in cross-sectional cardiomyocyte areas and collagen deposition (Figure 1A-1G). However, the degree of these changes was much less in Tg-mtCapn1/tTAlow compared with Tg-mtCapn1/tTAhigh mice at the same ages. Taken together, these results demonstrate that transgenic up-regulation of mitochondria-targeted CAPN1 dose-dependently induces dilated cardiomyopathy and heart failure in mice.
3.3. Transgenic over-expression of mitochondria-targeted CAPN1 induces mitochondrial ROS generation in hearts
We have recently reported that the selective inhibition of mitochondrial calpain prevents mitochondrial superoxide generation in cardiomyocytes under diabetic and septic conditions [28, 29], suggesting an important role for mitochondrial calpain in mitochondrial superoxide generation. To provide direct evidence to support this prior finding, we analyzed OCR and mitochondrial ROS generation in isolated mitochondria from Tg-mtCapn1/tTA mouse hearts. As shown in Figure 2A and 2B, the State 3 OCR and the ratio of State 3 OCR over State 4 OCR were much lower in Tg-mtCapn1/tTA mice compared with their wild-type littermates, indicative of mitochondrial dysfunction. Transgenic over-expression of mitochondrial-targeted CAPN1 increased mitochondrial ROS generation in a dose-dependent manner, which was inhibited by mito-TEMPO but not TEMPO, validating mitochondrial ROS production (Figure 2C). Mitochondrial ROS generation correlated well with the elevation of MDA levels in Tg-mtCapn1/tTA mouse hearts (Figure 2D). Thus, transgenic expression of mitochondrial-targeted CAPN1 is sufficient to induce mitochondrial ROS generation in hearts.
Figure 2. Mitochondrial function and ROS measurement.
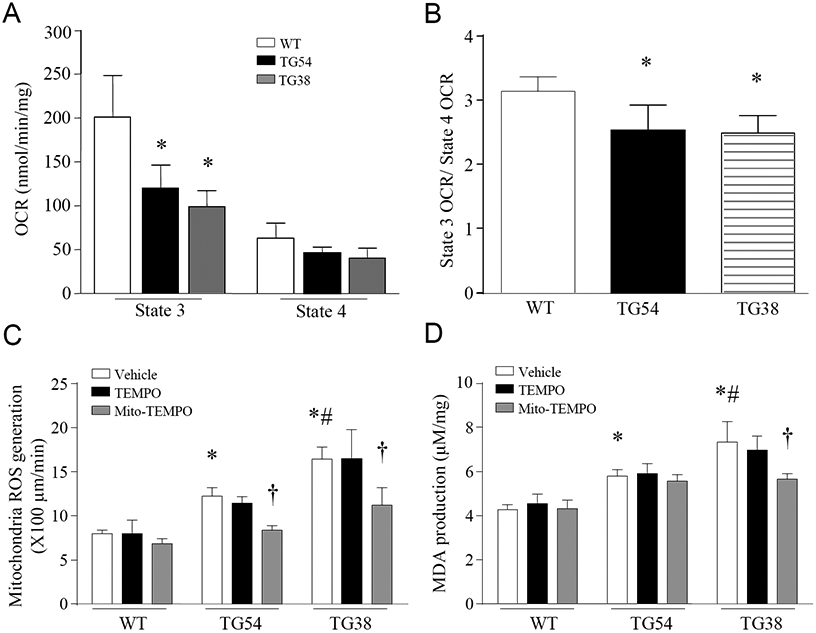
(A, B, C) Mitochondria were isolated from transgenic (TG54 and TG38) and wild-type (WT) mouse hearts at age 3 months. Oxygen consumption rate in state 3 and state 4 (OCR, A), the ratio of state 3/state 4 OCR (B) and mitochondrial ROS generation (C) were measured. (D) Malondialdehyde (MDA) in whole heart lysates. Data are mean ± SD, n = 4-6. *P< 0.05 versus WT (Vehicle), †P< 0.05 versus TG54 (TEMPO) or TG38 (TEMPO) and #P< 0.05 versus TG54 (Vehicle). One-way ANOVA followed by Newman-Keuls test was conducted for (A) and (B), and two-way ANOVA followed by Newman-Keuls test for (C) and (D).
3.4. Administration of mitochondrial-targeted antioxidant mito-TEMPO halts the progression of dilated heart failure in Tg-mtCapn1/tTA mice
Excessive mitochondrial ROS promotes mitochondrial dysfunction, cell death and myocardial hypertrophy, all of which contribute to myocardial dysfunction and heart failure [7, 9, 27]. Indeed, selectively scavenging mitochondrial superoxide using mitochondrial-targeted superoxide dismutase mimetics mito-TEMPO reduced heart weight and LV chamber size (Supplementary Table-1), improved survival, attenuated cardiac cell death and resultant fibrosis, and mitigated myocardial dysfunction in Tg-mtCapn1/tTA mice (Figure 3A-3D). Administration of mito-TEMPO also attenuated myocardial hypertrophic changes and collagen deposition in Tg-mtCapn1/tTAhigh mice (Figure 4). As a control, TEMPO which scavenges global cellular superoxide production did not provide cardio-protective effects in Tg-mtCapn1/tTA mice (Figure 3 and 4). These results clearly indicate that mitochondrial superoxide generation but not global cellular superoxide production is an important mechanism contributing to cardiomyopathic changes in Tg-mtCapn1/tTA mice.
Figure 3. The survival, histological analysis and functional assessment.

Echocardiography was conducted on transgenic mice (TG54 and TG38) and their wild-type (WT) littermates at age 2 months. These animals were then given a daily injection of mito-TEMPO or TEMPO (i.p.) for one month. At age 3 months, they were scanned again by echocardiography (A) Survival rate. (B) A representative H&E staining of heart tissues. (C and D) Assessment of myocardial function by echocardiography. Data are mean ± SD, n = 4-6. *P< 0.05 versus WT (before treatment), #P< 0.05 versus TG54 (before treatment), †P< 0.05 versus TG54 (after treatment), $P< 0.05 versus TG38 (before treatment) and ‡P< 0.05 versus TG54 (before treatment). Two-way ANOVA followed by Newman-Keuls test was conducted for (C) and (D).
Figure 4. Histological analyses of heart tissues.

Transgenic mice (TG54 and TG38) and their wild-type littermates (WT) were given mito-TEMPO or TEMPO (i.p.) starting from age of 2 months for one month. Heart tissues were then collected and processed for histological analyses in mice at age 3 months. (A) A representative histological picture of wheat germ agglutinin (WGA) staining. (B) A representative histological picture of sirius red staining. (C) Quantification for cardiomyocyte cross-sectional areas. (D) Quantification for collagen deposition. (E-H) Real-time RT-PCR for mRNA expression of ANP (E), β-MHC (G), collagen- I (F) and collagen-III (H) in heart tissues. Data are mean ± SD, n = 4-6. *P< 0.05 versus WT (Vehicle), †P< 0.05 versus TG54 (TEMPO), #P< 0.05 versus TG54 (Vehicle) and ‡P< 0.05 versus TG38 (TEMPO). Two-way ANOVA followed by Newman-Keuls test was conducted for (C)-(H).
3.5. Transgenic over-expression of mitochondria-targeted CAPN1 is associated with disruption of ATP synthase in Tg-mtCapn1/tTA mice
Our recent studies have demonstrated that calpain-1 targets and cleaves ATP5A1, the alpha-subunit of ATP synthase, thereby disrupting ATP synthase activity in the mitochondria of cardiomyocytes [28, 29]. In support of these previous findings, transgenic up-regulation of mitochondria-targeted CAPN1 decreased protein levels of ATP5A1 but not of ATP synthase β-subunit in Tg-mtCapn1/tTAhigh mouse hearts (Figure 5A and 5B). Down-regulation of ATP5A1 protein correlated with a reduction of ATP synthase activity (Figure 5C). The ATP synthase activity was also lower in Tg-matCapn1/tTAlow compared with their wild-type littermate mouse hearts (Figure 5D).
Figure 5. Determination of ATP5A1 and ATP synthase β subunit protein expression as well ATP synthase activity.
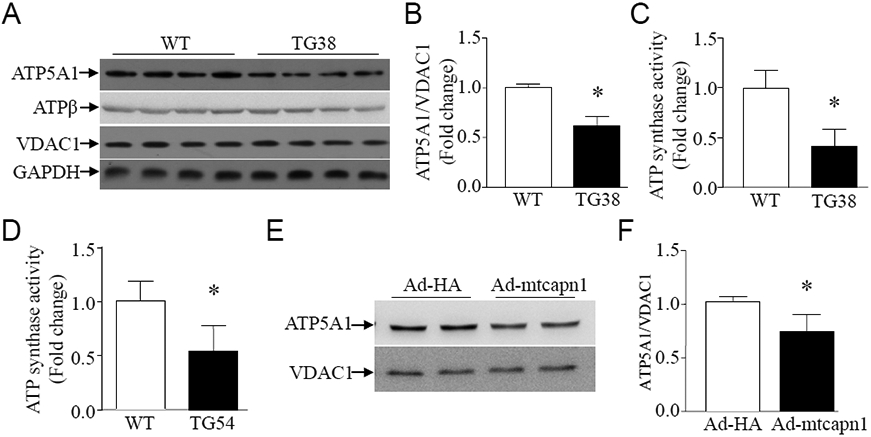
(A, B, C, D) Heart tissues were collected from transgenic mice (TG38 and TG54) and their wild-type littermates (WT) at age 3 months. (A) A representative western blot for ATP5A1 and ATP synthase β subunit (ATPβ) from 4 different hearts in each group. (B) Quantification of ATP5A1 relative to VDAC1. (C and D) ATP synthase activity (n = 5 in each group). (E, F) Cultured adult mouse cardiomyocytes were infected with Ad-mtcapn1 or Ad-HA. Twenty-four hours later, western blot analysis was conducted for ATP5A1 and VDAC1. (E) A representative western blot from 3 independent cell cultures for ATP5A1 and VDAC1. (F) Quantification for ATP5A1 relative to VDAC1. Data are mean ± SD, n = 3-5. *P< 0.05 versus WT and Ad-HA. T test was conducted for (B), (C), (D) and (F).
To provide further evidence supporting that the reduction of ATP5A1 was due to increased mitochondrial calpain-1 in hearts, we infected adult mouse cardiomyocytes with Ad-mtcapn1 and Ad-HA as a control. In line with the in vivo observation (Figure 5A and 5B), infection with Ad-mtcapn1 induced a reduction of ATP5A1 in cardiomyocytes (Figure 5E and 5F).
3.6. Increased mitochondria-targeted CAPN1 promotes the mPTP opening, which is prevented by over-expression of ATP5A1 and mito-TEMPO in cardiomyocytes
Given that mitochondrial ROS [33] and ATP synthase [11] play an important role in regulating the mPTP opening, an increase of which promotes cell death [12], we determined whether increased mitochondrial calpain-1 induced mPTP opening through mitochondrial superoxide generation and ATP5A1 reduction. To address this, we infected H9c2 cells with Ad-mtcapn1 or Ad-HA, and then with Ad-ATP5A1. After adenoviral infection, cardiomyocytes were incubated with mito-TEMPO, cyclosporine A (a specific inhibitor for the mPTP opening) [6] or vehicle for 24 hours. The mPTP opening was determined using calcein AM. Infection with Ad-mtcapn1 resulted in a reduction of calcein AM signals (Figure 6A-6C). However, incubation with cyclosporine A increased calcein AM signals in Ad-mtcapn1 infected H9c2 cells (Figure 6A), indicating that increased mitochondrial calpain-1 promoted the mPTP opening. Similarly, co-infection with Ad-ATP5A1 or incubation with mito-TEMPO attenuated the mPTP opening induced by increased mitochondrial calpain-1 (Figure 6B and 6C).
Figure 6. Assessment of the mPTP opening in H9c2 cells.

The H9c2 cells were infected with Ad-mtcapn1 or Ad-HA and then Ad-ATP5A1. After adenoviral infection, the cells were incubated with cyclosporine A, mito-TEMPO or vehicle for 24 hours. The mPTP opening was determined using calcein AM (green) and nuclei were staining by Hoechst 33342 (blue). (A, B, C) Representative pictures for the mPTP opening were presented from 3 independent cell cultures.
3.7. Up-regulation of mitochondria-targeted CAPN1 induces cell death, which is prevented by over-expression of ATP5A1, mito-TEMPO, and inhibition of the mPTP opening in cardiomyocytes
Adult cardiomyocytes were infected with Ad-mtcapn1 or Ad-HA, and then with Ad-ATP5A1. After adenoviral infection, cardiomyocytes were incubated with mito-TEMPO, cyclosporine A or vehicle for 24 hours. Cell death was determined by annexin V staining. Infection with Ad-mtcapn1 sufficiently induced cell death. However, co-infection with Ad-ATP5A1, incubation with mito-TEMPO or cyclosporine A attenuated cell death in Ad-mtcapn1 infected cardiomyocytes (Figure 7A-7F).
Figure 7. Determination of cell death in cardiomyocytes by annexin V staining.

Adult cardiomyocytes were isolated from mice. After attachment, cardiomyocytes were infected with Ad-mtcapn1 or Ad-HA and then Ad-ATP5A1. After adenoviral infection, the cells were incubated with cyclosporine A, mito-TEMPO or vehicle for 24 hours. Cell death was determined by annexin V staining (green). Nuclei were stained with Hoechst 33342 (blue). (A, C, E) Representative pictures from 3 independent cell cultures. (B, D, F) Percentages of annexin V positive cells. Data are mean ± SD, n = 3. *P < 0.05 versus Ad-HA in the same category and †P < 0.05 versus Ad-mtcapn1 in the same category (two-way ANOVA followed by Newman-Keuls test).
4. Discussion
Calpains play essential roles in pathophysiological processes and may represent useful targets for therapy [30]. Increased calpains have been implicated in a variety of cardiac diseases, and these disease processes were reduced when calpains were blocked [3, 19-22, 28, 29, 35, 38, 40]. Interestingly, the immune-activity of calpain-1 has been detected in both the inner membrane and matrix of cardiac mitochondria [4, 5, 28, 29, 35, 38]. An early study has shown that mitochondrial calpain-1 is involved in the cleavage of apoptosis-inducing factor (AIF) to truncated AIF, which is released into the intermembrane space in hearts following ischemia and reperfusion [5]. It has also been reported that calpain-1 in the matrix of cardiac mitochondria may target and disrupt respiratory chain complex-I, which contributes to ischemia/reperfusion injury [38]. In mouse models of diabetes and sepsis, we have recently reported that the protein levels and activities of calpain-1 were increased in mouse heart mitochondria [28, 29].These previous studies suggest that increased mitochondrial calpain-1 may be a common mechanism contributing to cardiac diseases. Our present study set up low and high levels of cardiac-specific and mitochondria-targeted expression of CAPN1 in transgenic Tg-mtCapn1/tTA mice. Based on these two models, we demonstrate that increased mitochondrial calpain-1 dose-dependently induces classic changes of dilated cardiomyopathy including enlarged LV chamber diameter, increased heart size and weight, cardiomyocyte death and resultant fibrosis, and cardiac hypertrophy in Tg-mtCapn1/tTA mice. Importantly, in Tg-mtCapn1/tTAhigh mice, the cardiomyopathic changes resulted in dilated heart failure and early death. Thus, this study provides direct evidence in support of the role of increased mitochondrial calpain-1 in myocardial injury and heart failure. Nevertheless, future studies are needed to determine whether calpain-1 is increased in human failing hearts.
Subsequently, we explored the mechanism of mitochondria-targeted CAPN1 in inducing dilated heart disease. Mitochondria are important ATP-generating organelles, and ROS is a byproduct of the respiration chain [26], In simple terms, ROS are reactive molecules containing oxygen, such as hydroxyl radical (HO•), peroxynitrite (OONO─), superoxide (O2·─), and hydrogen peroxide (H2O2) [2]. There are at least ten sites of mitochondrial ROS production, and these are not limited to the respiration chain [15], The damaging effects of ROS have been studied for decades, and include DNA mutagenesis, inflammation, and multiple cell death pathways [23]. In relation to the heart, cardiomyocyte is rich in mitochondria, and mitochondrial dysfunction is implicated in numerous cardiac diseases [1, 10]. ROS is also a key player in cardiac cell death, myocardial infarction, and reperfusion injury [34, 36, 37]. In our study, the over-expression of mitochondria-targeted CAPN1 influenced the function of mitochondria by inducing a lower OCR and increased superoxide generation. Previous studies have demonstrated that mitochondria-targeted enzymatic antioxidants have cardioprotective effects [7, 9, 27]. Thus, in the present study, we used mito-TEMPO, a mitochondrial-targeted superoxide dismutase mimetics, to specifically scavenge mitochondrial superoxide production. The results showed that the progression of dilated heart failure by the over-expression of mitochondria-targeted CAPN1 was halted successfully through the use of mito-TEMPO. This finding underscores a critical role of mitochondrial superoxide generation as global cellular superoxide dismutase mimetics TEMPO failed to provide cardio-protection in mice with over-expression of mitochondria-targeted CAPN. The discrepant effects of mito-TEMPO versus TEMPO can be explained by the following two facts: firstly, the concentration of TEMPO in mitochondria was much lower relative to mito-TEMPO, which may be not high enough to effectively scavenge mitochondrial superoxide production in cardiomyocytes, and secondly, superoxide in mitochondria can’t diffuse into the cytosol though some forms of ROS such as H2O2 can freely diffuse between organelles within cells.
Multiple mechanisms may be involved in mitochondrial calpain-1 induced superoxide generation in cardiomyocytes. In addition to impairment of complex I in the respiratory chain [38], which induces superoxide generation, in our recent studies [28, 29], we reported that mitochondrial calpain-1 directly targets and negatively regulates ATP5A1 protein, leading to ATP synthase disruption in cardiomyocytes under diabetic and septic conditions. This is also verified in our in vivo study with a decreased ATP5A1 level and a normal ATP synthase β-subunit level in Tg-mtCapn1/tTAhigh mouse hearts and in our in vitro study with a reduction of ATP5A1 in cultured cardiomyocytes with over-expression of mitochondrial targeted calpain-1. Disruption of ATP synthase may cause “back-up” of electron within the respiratory chain thereby inducing superoxide generation. Thus, our data support a mode that increased mitochondrial calpain-1 targets and cleaves ATP5A1, which disrupts ATP synthase, leading to superoxide generation from the respiratory chain in cardiomyocytes. Additionally, disruption of ATP synthase impairs ATP production thereby directly compromising myocardial function [11].
Dilated heart failure is the irreversible endpoint of dilated cardiomyopathy, and is characterized by left ventricle dilation and reduced systolic function [24, 25]. Without heart transplantation, the final outcome for these patients is death. In our study, we have shown that mitochondria-targeted calpain-1 played essential roles in inducing dilated heart failure and early death. Moreover, the damages to the heart caused by mitochondria-targeted CAPN1 were related to the generation of mitochondrial superoxide. The addition of mito-TEMPO, a mitochondrial-targeted superoxide dismutase mimetics, inhibited this progression of dilated heart failure in Tg-mtCapn1/tTA mice. These findings further suggest an important role of increased mitochondrial calpain-1 and subsequent superoxide generation in myocardial injury and heart failure. This is also supported by our in vitro study in cultured cardiomyocytes, which shows that over-expression of mitochondria calpain-1 induced cell death was prevented by over-expression of ATP5A1 and mito-TEMPO.
Increased mitochondrial ROS has been reported to promote cardiomyocyte death, myocardial hypertrophy and inflammatory response, all of which contribute to heart failure. However, the exact molecular mechanisms by which mitochondrial superoxide generation induces myocardial injury and dilated heart failure remain to be elucidated. It is well known that mitochondrial oxidative damage induces the mPTP opening, a phenomenon observed in cardiomyocytes under various pathological conditions, which promotes cell death [33]. In the present study, over-expression of mitochondria targeted calpain-1 led to an increase in the mPTP opening in cultured cardiomyocytes, which was prevented by incubation with mito-TEMPO or up-regulation of ATP5A1. Importantly, incubation with mito-TEMPO, up-regulation of ATP5A1 or inhibition of the mPTP opening attenuated cell death induced by increased mitochondrial calpain-1. Thus, we argue the mode that increased mitochondrial calpain-1 induces cell death via a reduction of ATP5A1 and subsequent increases in mitochondrial ROS generation and the mPTP opening in cardiomyocytes.
In addition to calpain-1, calpain-2 was also reported to be increased in heart mitochondria following ischemia/reperfusion, and increased mitochondrial calpain-2 induced the mPTP opening thereby promoting myocardial ischemia/reperfusion injury [35]. Future studies may further investigate other calpains, including calpain-2, in the pathophysiological processes of cardiac diseases. Together with previous findings [4, 5, 28, 29, 35, 38], our study opens new avenues for the research, prevention, and treatment of clinical dilated heart failure through the selective inhibition of mitochondrial calpain.
Supplementary Material
5. Acknowledgments
This study was supported by operating grants from the National Natural Science Foundation of China (81470499 to T.P., 81521001 and 31570904 to R.C.), Natural Science Foundation of Jiangsu Province (BK20171216 to T.P.) and the Canadian Institutes of Health Research (MOP-133657 to T.P.).
Footnotes
Conflict of interest:
None declared
References:
- 1.Barth E, Stammler G, Speiser B, Schaper J (1992) Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol 24:669–681 doi: 10.1016/0022-2828(92)93381-S [DOI] [PubMed] [Google Scholar]
- 2.Brown DI, Griendling KK (2015) Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res 116:531–549 doi: 10.1161/CIRCRESAHA.116.303584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bukowska A, Lendeckel U, Bode-Boger SM, Goette A (2012) Physiologic and pathophysiologic role of calpain: implications for the occurrence of atrial fibrillation. Cardiovasc Ther 30:e115–127 doi: 10.1111/j.1755-5922.2010.00245.x [DOI] [PubMed] [Google Scholar]
- 4.Chen Q, Lesnefsky EJ (2015) Heart mitochondria and calpain 1: Location, function, and targets. Biochim Biophys Acta 1852:2372–2378 doi: 10.1016/j.bbadis.2015.08.004 [DOI] [PubMed] [Google Scholar]
- 5.Chen Q, Paillard M, Gomez L, Ross T, Hu Y, Xu A, Lesnefsky EJ (2011) Activation of mitochondrial mucalpain increases AIF cleavage in cardiac mitochondria during ischemia-reperfusion. Biochem Biophys Res Commun 415:533–538 doi: 10.1016/j.bbrc.2011.10.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crompton M, Ellinger H, Costi A (1988) Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem J 255:357–360 doi: 10.2042/bj2550357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai DF, Chen T, Szeto H, Nieves-Cintron M, Kutyavin V, Santana LF, Rabinovitch PS (2011) Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol 58:73–82 doi: 10.1016/j.jacc.2010.12.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dai DF, Hsieh EJ, Chen T, Menendez LG, Basisty NB, Tsai L, Beyer RP, Crispin DA, Shulman NJ, Szeto HH, Tian R, MacCoss MJ, Rabinovitch PS (2013) Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ Heart Fail 6:1067–1076 doi: 10.1161/CIRCHEARTFAILURE.113.000406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintron M, Chen T, Marcinek DJ, Dorn GW 2nd, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS (2011) Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res 108:837–846 doi: 10.1161/CIRCRESAHA.110.232306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dromparis P, Michelakis ED (2013) Mitochondria in vascular health and disease. Annu Rev Physiol 75:95–126 doi: 10.1146/annurev-physiol-030212-183804 [DOI] [PubMed] [Google Scholar]
- 11.Fernandez-Sanz C, Ruiz-Meana M, Castellano J, Miro-Casas E, Nunez E, Inserte J, Vazquez J, Garcia-Dorado D (2015) Altered FoF1 ATP synthase and susceptibility to mitochondrial permeability transition pore during ischaemia and reperfusion in aging cardiomyocytes. Thromb Haemost 113:441–451 doi: 10.1160/TH14-10-0901 [DOI] [PubMed] [Google Scholar]
- 12.Gadicherla AK, Wang N, Bulic M, Agullo-Pascual E, Lissoni A, De Smet M, Delmar M, Bultynck G, Krysko DV, Camara A, Schluter KD, Schulz R, Kwok WM, Leybaert L (2017) Mitochondrial Cx43 hemichannels contribute to mitochondrial calcium entry and cell death in the heart. Basic Res Cardiol 112:27 doi: 10.1007/s00395-017-0618-1 [DOI] [PubMed] [Google Scholar]
- 13.Galvez AS, Diwan A, Odley AM, Hahn HS, Osinska H, Melendez JG, Robbins J, Lynch RA, Marreez Y, Dorn GW 2nd (2007) Cardiomyocyte degeneration with calpain deficiency reveals a critical role in protein homeostasis. Circ Res 100:1071–1078 doi: 10.1161/01.RES.0000261938.28365.11 [DOI] [PubMed] [Google Scholar]
- 14.Goll DE, Thompson VF, Li H, Wei W, Cong J (2003) The calpain system. Physiol Rev 83:731–801 doi: 10.1152/physrev.00029.2002 [DOI] [PubMed] [Google Scholar]
- 15.Goncalves RL, Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Brand MD (2015) Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J Biol Chem 290:209–227 doi: 10.1074/jbc.M114.619072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang PT, Chen CL, Lin P, Chilian WM, Chen YR (2017) Impairment of pH gradient and membrane potential mediates redox dysfunction in the mitochondria of the post-ischemic heart. Basic Res Cardiol 112:36 doi: 10.1007/s00395-017-0626-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kar P, Samanta K, Shaikh S, Chowdhury A, Chakraborti T, Chakraborti S (2010) Mitochondrial calpain system: an overview. Arch Biochem Biophys 495:1–7 doi: 10.1016/j.abb.2009.12.020 [DOI] [PubMed] [Google Scholar]
- 18.Li J, Zhu H, Shen E, Wan L, Arnold JM, Peng T (2010) Deficiency of rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes 59:2033–2042 doi: 10.2337/db09-1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li S, Zhang L, Ni R, Cao T, Zheng D, Xiong S, Greer PA, Fan GC, Peng T (2016) Disruption of calpain reduces lipotoxicity-induced cardiac injury by preventing endoplasmic reticulum stress. Biochim Biophys Acta 1862:2023–2033 doi: 10.1016/j.bbadis.2016.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Li Y, Shan L, Shen E, Chen R, Peng T (2009) Over-expression of calpastatin inhibits calpain activation and attenuates myocardial dysfunction during endotoxaemia. Cardiovasc Res 83:72–79 doi: 10.1093/cvr/cvp100 [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Ma J, Zhu H, Singh M, Hill D, Greer PA, Arnold JM, Abel ED, Peng T (2011) Targeted inhibition of calpain reduces myocardial hypertrophy and fibrosis in mouse models of type 1 diabetes. Diabetes 60:2985–2994 doi: 10.2337/db10-1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma J, Wei M, Wang Q, Li J, Wang H, Liu W, Lacefield JC, Greer PA, Karmazyn M, Fan GC, Peng T (2012) Deficiency of Capn4 gene inhibits nuclear factor-kappaB (NF-kappaB) protein signaling/inflammation and reduces remodeling after myocardial infarction. J Biol Chem 287:27480–27489 doi: 10.1074/jbc.M112.358929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mann PJ, Quastel JH (1946) Toxic effects of oxygen and of hydrogen peroxide on brain metabolism. Biochem J 40:139–144 doi: 10.1042/bj0400139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merlo M, Caiffa T, Gobbo M, Adamo L, Sinagra G (2018) Reverse remodeling in Dilated Cardiomyopathy: Insights and future perspectives. Int J Cardiol Heart Vasc 18:52–57 doi: 10.1016/j.ijcha.2018.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merlo M, Cannata A, Gobbo M, Stolfo D, Elliott PM, Sinagra G (2018) Evolving concepts in dilated cardiomyopathy. Eur J heart Fail 20:228–239 doi: 10.1002/ejhf.1103 [DOI] [PubMed] [Google Scholar]
- 26.Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417:1–13 doi: 10.1042/BJ20081386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ni R, Cao T, Xiong S, Ma J, Fan GC, Lacefield JC, Lu Y, Le Tissier S, Peng T (2016) Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic Biol Med 90:12–23 doi: 10.1016/j.freeradbiomed.2015.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ni R, Zheng D, Wang Q, Yu Y, Chen R, Sun T, Wang W, Fan GC, Greer PA, Gardiner RB, Peng T (2015) Deletion of capn4 Protects the Heart Against Endotoxemic Injury by Preventing ATP Synthase Disruption and Inhibiting Mitochondrial Superoxide Generation. Circ Heart Fail 8:988–996 doi: 10.1161/CIRCHEARTFAILURE.115.002383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ni R, Zheng D, Xiong S, Hill DJ, Sun T, Gardiner RB, Fan GC, Lu Y, Abel ED, Greer PA, Peng T (2016) Mitochondrial Calpain-1 Disrupts ATP Synthase and Induces Superoxide Generation in Type 1 Diabetic Hearts: A Novel Mechanism Contributing to Diabetic Cardiomyopathy. Diabetes 65:255–268 doi: 10.2337/db15-0963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ono Y, Saido TC, Sorimachi H (2016) Calpain research for drug discovery: challenges and potential. Nat Rev Drug Discov 15:854–876 doi: 10.1038/nrd.2016.212 [DOI] [PubMed] [Google Scholar]
- 31.Poncelas M, Inserte J, Aluja D, Hernando V, Vilardosa U, Garcia-Dorado D (2017) Delayed, oral pharmacological inhibition of calpains attenuates adverse post-infarction remodelling. Cardiovasc Res 113:950–961 doi: 10.1093/cvr/cvx073 [DOI] [PubMed] [Google Scholar]
- 32.Sanbe A, Gulick J, Hanks MC, Liang Q, Osinska H, Robbins J (2003) Reengineering inducible cardiac-specific transgenesis with an attenuated myosin heavy chain promoter. Circ Res 92:609–616 doi: 10.116/01.RES.0000065442.64694.9F [DOI] [PubMed] [Google Scholar]
- 33.Seidlmayer LK, Juettner VV, Kettlewell S, Pavlov EV, Blatter LA, Dedkova EN (2015) Distinct mPTP activation mechanisms in ischaemia-reperfusion: contributions of Ca2+, ROS, pH, and inorganic polyphosphate. Cardiovasc Res 106:237–248 doi: 10.1093/cvr/cvv097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sheeran FL, Pepe S (2006) Energy deficiency in the failing heart: linking increased reactive oxygen species and disruption of oxidative phosphorylation rate. Biochim Biophys Acta 1757:543–552 doi: 10.1016/j.bbabio.2006.03.008 [DOI] [PubMed] [Google Scholar]
- 35.Shintani-Ishida K, Yoshida K (2015) Mitochondrial m-calpain opens the mitochondrial permeability transition pore in ischemia-reperfusion. Int J Cardiol 197:26–32 doi: 10.1016/j.ijcard.2015.06.010 [DOI] [PubMed] [Google Scholar]
- 36.Sorescu D, Griendling KK (2002) Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. Congest Heart Fail 8:132–140 doi: 10.1111/j.1527-5299.2002.00717.x [DOI] [PubMed] [Google Scholar]
- 37.Teshima Y, Takahashi N, Nishio S, Saito S, Kondo H, Fukui A, Aoki K, Yufu K, Nakagawa M, Saikawa T (2014) Production of reactive oxygen species in the diabetic heart. Roles of mitochondria and NADPH oxidase. Circ J 78:300–306 doi: 10.1253/circj.CJ-13-1187 [DOI] [PubMed] [Google Scholar]
- 38.Thompson J, Hu Y, Lesnefsky EJ, Chen Q (2016) Activation of mitochondrial calpain and increased cardiac injury: beyond AIF release. Am J physiol Heart Circ Physiol 310:H376–384 doi: 10.1152/ajpheart.00748.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu T, Ding W, Ao X, Chu X, Wan Q, Wang Y, Xiao D, Yu W, Li M, Yu F, Wang J (2019) ARC regulates programmed necrosis and myocardial ischemia/reperfusion injury through the inhibition of mPTP opening. Redox Biol 20:414–426 doi: 10.1016/j.redox.2018.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang D, Ma S, Tan Y, Li D, Tang B, Zhang X, Sun M, Yang Y (2010) Increased expression of calpain and elevated activity of calcineurin in the myocardium of patients with congestive heart failure. Int J Mol Med 26:159–164 doi: 10.3892/ijmm_00000448 [DOI] [PubMed] [Google Scholar]
- 41.Zhu H, Shan L, Schiller PW, Mai A, Peng T (2010) Histone deacetylase-3 activation promotes tumor necrosis factor-alpha (TNF-alpha) expression in cardiomyocytes during lipopolysaccharide stimulation. J Biol Chem 285:9429–9436 doi: 10.1074/jbc.M109.071274 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.