

Abstract
Ceramide kinase (CERK) is the mammalian lipid kinase from which the bioactive sphingolipid, ceramide-1-phosphate (C1P), is derived. CERK has been implicated in several pro-malignant phenotypes with little known as to mechanistic underpinnings. In this study, the mechanism of how CERK inhibition decreases cell survival in mutant (Mut) KRAS non-small cell lung cancer (NSCLC), a major lung cancer subtype, was revealed. Specifically, NSCLC cells possessing a KRAS mutation were more responsive to inhibition, downregulation, and genetic ablation of CERK compared to those with wild-type (WT) KRAS regarding a reduction in cell survival. Inhibition of CERK induced ferroptosis in Mut KRAS NSCLC cells, which required elevating VDAC-regulated mitochondria membrane potential (MMP) and the generation of cellular reactive oxygen species (ROS). Importantly, through modulation of VDAC, CERK inhibition synergized with the first-line NSCLC treatment, cisplatin, in reducing cell survival and in vivo tumor growth. Further mechanistic studies indicated that CERK inhibition impacted MMP and cell survival by limiting AKT activation and translocation to mitochondria, and thus, blocking VDAC phosphorylation and tubulin recruitment.
Implications:
Our findings depict how CERK inhibition may serve as a new key point in combination therapeutic strategy for NSCLC, specifically precision therapeutics targeting NSCLC possessing a KRAS mutation.
Keywords: ceramide kinase, ceramide-1-phosphate, ferroptosis, cisplatin, KRAS, lung cancer, cell survival, AKT, VDAC, mitochondria
INTRODUCTION
Sphingolipids have emerged in recent years as mediators of a number of cancer-related processes, including survival, proliferation, migration and metastasis. Regarding cell survival, ceramide (Cer) and its metabolites, such as sphingosine-1-phosphate (S1P) and ceramide-1-phosphate (C1P), have been implicated in different cell-death pathways in tumor cells, including apoptosis, autophagy and necrosis, in response to chemo-, radio- or targeted- therapy (1-7). Specifically, Cer is generally considered as anti-apoptotic, anti-survival or anti-proliferation factor whereas S1P and C1P have been reported to exert pro-apoptotic or pro-survival signaling (8,9). Cer and Cer-derivatives also contribute to therapeutic drug-resistance in cancer cells (1,3,10,11).
Despite numerous findings on mechanistic function of Cer and S1P in different aspects of cancer cells, little is known regarding the pro-surviving or pro-malignant properties of C1P. C1P is generated by ceramide kinase (CERK), an enzyme localized to the plasma membrane, Golgi apparatus, and mitochondria (12,13,14). C1P is also transported to different cell compartments by C1P-transport protein (CPTP), which can impact function (15,16). Potential targets of C1P for augmenting tumor cell migration and invasion have been implicated such as PI3K/AKT, mTOR, and RhoA (17,18).
In this study, we identified a new molecular mechanism for the action of CERK in the cell survival of NSCLC. Specifically, we demonstrate that NSCLC cells with an oncogenic KRAS mutation (Mut KRAS) are addicted to CERK-derived C1P by stabilizing VDAC-mediated mitochondria function and suppressing the activation of ferroptotic cell death. CERK inhibition synergized with cisplatin, the first-line therapeutic drugs for NSCLC treatment, on reducing cell survival and tumor growth. In Mut KRAS cells, CERK modulated VDAC-regulated mitochondria function via AKT translocation to mitochondria for VDAC phosphorylation and subsequently stabilizing the tubulin-VDAC interaction. Overall, our findings provide the first mechanistic underpinnings of CERK-derived C1P in modulating cell survival pathways, and our study further shows CERK as a crucial component in therapeutics precisely targeting NSCLC with Mut KRAS.
MATERIALS AND METHODS
Cell culture
NSCLC cell lines (A549, H838, H1792, H1299, H358, H460) were cultured as previously described and used within 6 passages (19-26) from receipt and thawing. Cell lines were tested every two months for mycoplasma (universal mycoplasma detection kit, ATCC) throughout the study starting two months after thawing cells received from ATCC. Parental cell lines were authenticated by STR (short tandem repeat) profiling. For the comparison studies among NSCLC cell lines, cells were plated in regular growth medium for each cell line, which gave 40% confluency. The following day, medium was changed to serum-free RPMI for 16 hours prior to being harvested for analysis. All treatments (Sup. Tbl.S1 for list of drugs/reagents) were undertaken in serum-free RPMI medium.
Cell survival assays
NSCLC (4-6x102) cells were plated in 35 mm dishes (6-well plates) in normal growth medium, incubated for 6-12 days under SIC, washed, fixed with 10% formalin, and stained with 0.05% crystal violet solution. For clonogenic survival assays, the colonies are then counted and plotted as number of colonies/plate (≥40 cells per colony) as described (19,20,21). For a spectrophotometer survival assays, crystal violet dye was solubilized in 10% acetic acid solution, diluted in water, and measured by absorbance at 595nM. The absorbance value is proportional to cell biomass attached to the plate or to cell viability level.
siRNA transfection
NSCLC cells were transfected with ON-TARGET plus Non-targeting Control Pool siRNA or CERK/VDAC siRNA (Horizon) using Dharmafect 1 transfection reagent (Horizon). Cells (2-3 x 105) were plated in each well of 6-well tissue culture dishes in regular growth medium. After 24 hrs, cells were transfected with 50 nM of siRNA following manufacturer instructions. After 8 hrs, medium was changed to regular growth medium. For double siRNA treatment, 24 hrs post-transfection with the first siRNA, cells were transfected with second siRNA following the same procedure. 48 hrs after siRNA transfection, cells were used for further analyses.
Generation of CERK+/− (Het) cells by CRISPR/Cas9 technology
NSCLC cells were transduced with both Cas-9 and CERK sgRNA CRISPR adenovirus (Abm, cat# K0435821) for 24 hours. Then cells underwent clonal selection to select cell clones with at least 50% decrease in CERK mRNA expression examined by quantitative RT-PCR (CERK Het cells). Control CERK WT cells were randomly selected from colonies yielded from clonal selection of cells transfected with both Cas9 and null control virus.
TMRM assay for MMP
TMRM assay was performed using TMRM Assay Kit (Abcam) following manufacturer’s protocol and visualized by using Keyence all-in-one florescence microscopy BZ-X710 (Ex/Em = 548/573). Fluorescence intensity in each image were measured by using Fuji software, subtracted to background signal, and normalized to total cell number in the image. Fluorescence intensity value in each well is the average of normalized background-subtracted florescence intensity of 3 random areas captured from each well.
Cellular ROS assay
The Cellular ROS assay was performed using DCFDA Cellular ROS Detection Assay Kit (Abcam) following manufacturer’s protocol and visualized by using Keyence all-in-one fluorescence microscopy BZ-X710 (Ex/Em = 485/535). Fluorescence intensity in each image were measured by using Fuji software, subtracted to background signal and normalized to total cell number in the image. Fluorescence intensity value in each well is the average of normalized background-subtracted fluorescence intensity of 3 random areas captured from each well.
Lipid peroxidation (MDA) assay
The lipid peroxidation assay was performed using the Lipid Peroxidation (MDA) Assay Kit (Sigma-Aldrich) following manufacturer’s protocol. The amount of malondialdehyde (MDA), one of the end products of lipid peroxidation, was measured and normalized to the amount of total protein from each cell sample (2x106 cells).
Quantitative RT-PCR
Quantitative RT-PCR (RT-qPCR) were performed as previously described (22-26). The relative mRNA expression of each gene was normalized to 18S RNA.
Mitochondria and cytoplasmic extraction
Cytoplasmic and mitochondria extracts were separated and prepared from cells using Mitochondria Isolation Kit for Cultured Cells (Thermo scientific) following manufacture’s protocol. Mitochondria pellets were resuspended in 1X cell lysis buffer for Western immunoblotting or in 1X coIP buffer for coIP or IP assays.
Immunoprecipitation (IP) and coimmunoprecipitation (coIP)
For VDAC1 coimmunoprecipitation (coIP), cells were lysed in 1X coIP buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 1mM EDTA pH 8.0, 0.5% NP-40 and 1X phosphatase/protease inhibitor cocktail), and VDAC1 immunoprecipitated using a VDAC1 antibody (Cell Signaling) as describedxx followed by purification with protein A/G magnetic beads (MedChemExpress), solubilization is Laemmli buffer, and the supernatant subjected to SDS-PAGE/immunoblotting for tubulin, VDAC1 or AKT. VDAC1 IP was done as above except cells were lysed in 1X coIP buffer with addition of 1% SDS followed by heating at 92°C for 2 minutes and subjected to SDS-PAGE/immunoblotting for Phospho-AKT substrate or VDAC1.
VDAC-tubulin co-localization assay
A549 cells (3x104) were plated in each chamber of the 8-chamber culture slide. The following day, cells were treated with NVP-231 (400 nM) for 24 hours. Then cells were washed with 1X PBS, fixed in 4% PFA for 5 minutes, washed gently with 1X PBS, permeabilized in 0.25% Triton X-100 for 5 minutes, washed with 1X PBS, blocked with 10% goat serum for 30 minutes, washed with 1X PBS, incubated with primary antibody (anti-VDAC1 antibody (ab154856, Abcam) and anti-Tubulin antibody (ab7291, Abcam)), washed, incubated in secondary antibody (Goat Anti-Rabbit IgG H&L (Alexa Fluor® 488, ab150077, Abcam) and Goat Anti-Mouse IgG H&L (Alexa Fluor® 594, ab150116, Abcam)). After washing, the coverslip is added with DAPI-containing mounting media and sealed. Images were taken using Keyence all-in-one florescence microscopy BZ-X710 (VDAC1, Tubulin and DAPI channel). Spearman’s rank correlation value for colocalization was calculated using coloc 2 plugin in ImageJ from the raw merged image of VDAC1 and Tubulin channel and the correlation value representative for each chamber was the average of correlation values from 3 areas randomly selected from each chamber.
Adenovirus transduction
Adenovirus control or adenovirus expressing constitutively active AKT2 were used at ~10 MOI for transduction as previously described (27). Cas9 and CERK sgRNA CRISPR adenovirus (Abm) were used at ~ 5 MOI each for transfection.
Mouse tumor xenograft models
Five-week-old female nude (Foxn1nu/nu) mice were received subcutaneous injections of NSCLC cells (106, CERK Het or WT cells) mixed in matrigel solution (50%) into the hind flanks. Tumor size was measured 2-3 times per week by a vernier caliper. When tumors reached approximately 100 mm3, mice were randomly enrolled onto vehicle control (PBS) or cisplatin (3 mg/kg, i.p., twice/week) group. After 2 weeks of treatment (total 4 treatments), mice were euthanized and tumors were excised for weight measurement as well as histological analysis. All animals were maintained according to the University of South Florida (8230R) and James A. Haley VAH (#4433V) approved IACUC protocols.
Ultra-Performance Liquid Chromatography Mass Spectrometry
Sphingolipids were extracted from cell pellets using a modified Bligh Dyer Extraction and analyzed via mass spectrometry utilizing electrospray ionization and a Sciex Triple Quad 5500 Mass Spectrometer as previously described (28-32).
Histology and immunohistochemistry
Tumors were fixed in 10% formalin and embedded in paraffin for H&E (hematoxylin and eosin) and immunohistochemical analyses. Briefly, following a heat-induced epitope retrieval step (0.01 M citrate buffer, pH 6.0), sections were incubated with Ki67 antibody (Cell signaling technology) at 1:100 dilution or TFR1 antibody (CD71, 3B8 2A1, Santa Cruz Biotechnology) at 1:50 dilution, followed by biotinylated secondary antibody (Vector labs) and avidin-biotin-alkaline phosphatase (Vector labs). Then slides were visualized with Vulcan Fast Red Chromogen (Biocarta) along with hematoxylin counterstain.
Western immunoblotting
Western immunoblotting was accomplished as described (25,26) using primary antibodies listed in the Sup Tbl.S2. Secondary antibodies were horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG antibodies (Cell Signaling Technology).
Gene expression analysis in lung cancer patients
Normalized Illumina HiSeq RNA-seq data and clinical patient data for lung adenocarcinoma was sourced from TCGA PanCancer Atlas (33-36) using cBioPortal for cancer genomics (37,38) as the download source. RNA-seq and clinical patient data were processed using the R programming language package Tidyverse (39). Outlier determination was performed on all genes of interest using the Prism ROUT method (40). Welch’s t-test was used to determine statistical significance in gene expression between groups.
Statistical analysis
Unless being noted, statistical differences between 2 groups were determined by a 2-tailed, unpaired Student’s t test and statistical differences among 3 and more groups were determined by ANOVA followed by Tukey HSD test. All tests were performed using R program. P values of less than 0.05 were considered significant.
RESULTS
Inhibition of CERK decreases the survival of NSCLC cells with KRAS mutation
CERK and C1P have been linked to cancer cell survival and cancer relapse (18,41), but the neither the susceptibility of NSCLC tumors or cells to CERK inhibition nor the mechanistic underpinnings as to a role of CERK and C1P in cancer cell signaling has been studied in depth. In this study, NSCLC cell lines with different oncogenic mutations and subtypes (lung adenocarcinoma (LUAD), bronchioloalveolar carcinoma (BAC) and large cell carcinoma (LCC)) (Sup. Tbl.S3) were treated with the well-characterized CERK inhibitor, NVP-231 (42), at varying concentrations reported to suppress intracellular C1P levels (≥400nM). Interestingly, only the NSCLC lines harboring activating mutations of KRAS (Mut KRAS) (e.g., A549 (LUAD), H358 (BAC), H460 (LCC), and H1792 (LUAD) cell lines) demonstrated significantly lower cell survival with NVP-231 versus wild-type (WT) KRAS NSCLC cells lines (H838 (LUAD) and H1299 (LCC) cell lines) (Fig.1A,B, Sup. Fig.S1A, Tbl.S4). This difference was not due to a difference in NSCLC subtype as H460s and H1299s, both large cell carcinomas (Sup. Tbl.S3), showed the same specificity for the mutant KRAS oncogenotype in regard to NVP-231 sensitivity as the lung adenocarcinoma cell lines (A549, H1792, and H838) (Fig.1A,B, Sup. Fig.S1A, Tbl.S4). The difference in cell survival was also not due to a lack of a differential inhibitory effect of NVP-231 as C1P levels were suppressed equally among the cell lines irrespective of KRAS mutation (Fig.1C). Importantly, NSCLC cells with Mut KRAS had a higher level of CERK expression (Fig.1D) and C1P levels (Fig.1E) versus WT KRAS NSCLC cell lines. Furthermore, human NSCLC (i.e., human lung adenocarcinomas (LUADs)) harboring KRASG12 mutations (either G12A, G12C or G12D), the most common KRAS mutations in NSCLC, has significantly higher CERK expression (Fig.1F; Sup. Fig.S2A) in contrast to common EGFR mutations observed in NSCLC (Supp Fig. 2B). NSCLC with Mut KRAS also demonstrated a lower survival outcome (Sup. Fig.S2C), which correlates with higher CERK expression. These data show that CERK-derived C1P is important for the survival of NSCLC with a Mut KRAS oncogenotype and suggestive of an addiction for cellular C1P in this NSCLC oncogenotype.
Figure 1: NSCLC cells with activating KRAS mutation are more responsive to CERK inhibition for effects on cell survival.
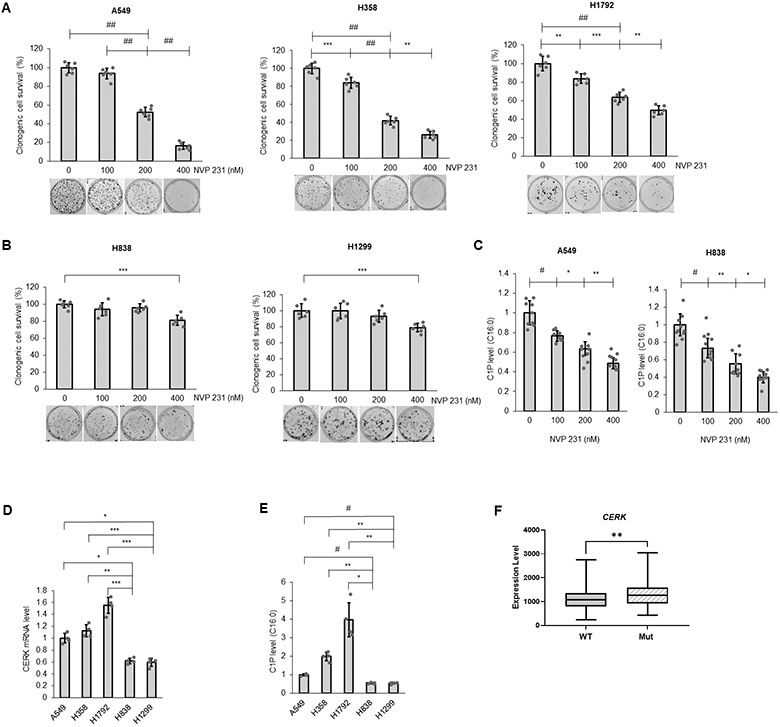
(A, B) NSCLC cells with Mut (A) or WT KRAS (B) were treated with NVP231 (0-400 nM) for 48 hours then utilized in clonogenic survival assay. (C) A549 (LUAD) and H838 (LUAD) cells were treated with NVP 231 as in (A) for 24 hours then subjected to mass spectrometry analysis for C16:0 C1P level (C16:0 C1P species represents the majority of total C1P detected in the NSCLC cell lines). (D, E) A549(LUAD)/H358(BAC)/H838(LUAD)/H1299(LCC) cells at approximately 40-50% confluency were placed in serum-free media for 16 hours, then cell lysates were subjected to RNA extraction followed by RT-qPCR assay for CERK mRNA quantification (D) or mass spectrometry analysis for C16:0 C1P level (E). (F) Comparison of CERK expression in human lung adenocarcinoma patients harboring WT versus Mut KRAS (G12 Mut); n = 305 for WT and 87 for Mut KRAS. Data in (A-E) are means ± SD; n = 6 from at least two independent occasions. *p<0.05, **p<0.005, ***p<0.0005, #p<0.00005, ##p<0.000005. Unless otherwise noted by an * or depicted p-value, data are not significant between depicted groups; p>0.05.
Inhibition of CERK induces cell death via ferroptosis
Despite the tremendous number of findings on cell death pathways reported for ceramide function, the cell survival signaling mechanisms associated with CERK and C1P are understudied and limited. Both the inhibition of apoptosis or autophagy-based cell death mechanisms have been linked to C1P (43-45); however, neither NVP-231 treatment nor CERK downregulation by siRNA affected mitochondria protein level (mitophagy indicator) or LC3 cleavage (autophagy indicator) (Sup. Fig.S3). Furthermore, the levels of apoptosis factors (cleaved PARP, cleaved caspase-9, the levels of Bcl-2 and Bcl-X, and the level of the mitochondria translocation of Bax) were unchanged by CERK inhibition (Sup. Fig.S3). Moreover, the levels of mitochondrial proteins indicative of the levels of mitochondria in the cells was also not different between Mut KRAS and WT KRAS NSCLC cell lines (Sup. Fig.4) Additionally, pre-treatment with the pan-caspase inhibitor, Z-vad-fmk (Z-vad), the necrosis inhibitor, Necrostatin-1 (Nec-1), or the autophagy inhibitor, Chloroquine (CQ), did not alter the effect of NVP-231 treatment on cell survival although these inhibitors significantly impacted the effect of cisplatin, a well-known anticancer drug to treat solid tumors, on cell survival (Fig.2A-D). NVP-231 treatment also did not affect cellular senescence (Sup. Fig.S5). In stark contrast, pre-treatment of cells with ferrostatin-1 (Fer1), a ferroptosis inhibitor, or deferoxamine (DF), an iron chelator and indirect inhibitor of ferroptosis, both significantly reversed the effect of NVP-231 treatment on cell survival (Fig. 2E), which implicated ferroptosis as the cause of cell death induced by CERK inhibition. Moreover, NVP-231 treatment increased lipid peroxidation (Fig.2F), a hallmark of ferroptosis, and the levels of the transferrin receptor 1 (TFR1), which has recently reported as a specific ferroptosis marker (46), in Mut KRAS but not in WT KRAS cells (Fig.2G, Sup. Fig.S6). These data demonstrate that NSCLC cells possessing oncogenic KRAS are addicted to CERK-derived C1P to suppress the activation of ferroptosis.
Figure 2: Ferroptosis as mediator for CERK inhibition-induced cell death.
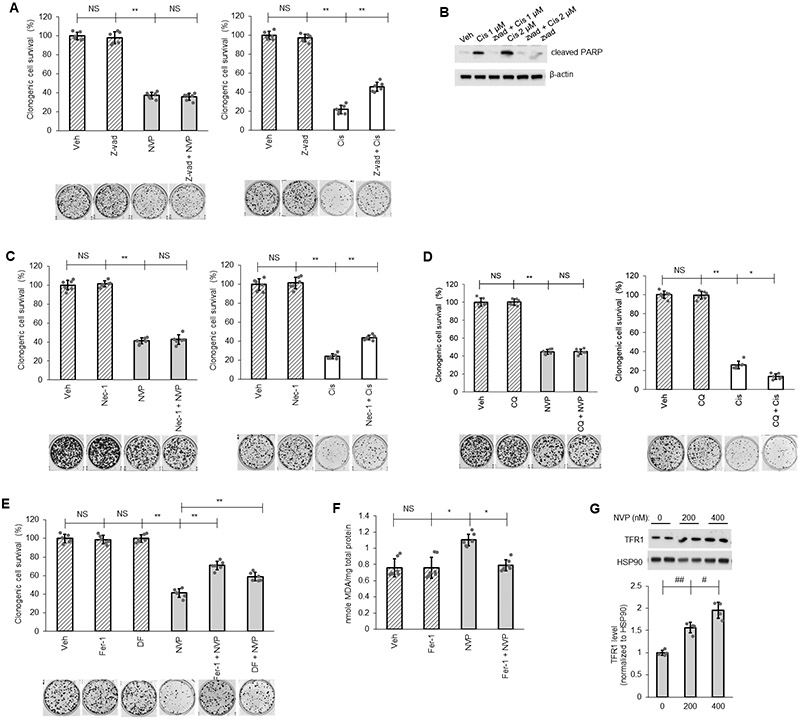
(A-E) A549 cells (LUAD) were pretreated with vehicle (Veh) or pan caspase inhibitor Z-vad-fmk (Z-vad, 25 μM) (A, B), necrosis inhibitor Necrostatin-1 (Nec-1, 10 μM) (C), autophagy inhibitor Chloroquine (CQ, 10 μM) (D), ferroptosis inhibitor Ferrostatin-1 (Fer-1, 5 μM) or iron-chelator deferoxamine (DF, 100 μM) (E) for 1 hour then treated with Veh, NVP-231 (400 nM) or Cisplatin (Cis; 1 μM except in panel B as labeled) for additional 16 hours before cells were utilized in clonogenic survival assay or Western immunoblotting. (F, G) Protein lysate from A549 cells treated with Veh or Fer-1 (5 μM) followed by 16 hours of Veh or NVP-231 (400 nM) treatment were subjected to lipid peroxidation (MDA) assay (F) or Western immunoblotting (G). Data in graphs are means ± SD; n = 6-7 in (A, C-F) or n=4 in (G) from two independent occasions. #p<0.01, ##p<0.001, *p<0.00005, **p<0.000005. Unless otherwise noted by an * or depicted p-value, data are not significant (NS) between depicted groups; p>0.05.
Inhibition of CERK reduces the survival of KRAS-mutated NSCLC cells by inducing mitochondrial dysfunction
Among organelles in mammalian cells, mitochondria are considered as a central gate controlling initiation of different cell death pathways including ferroptosis, and mitochondrial membrane potential (MMP) is an indicator for mitochondria health. Furthermore, numerous reports show that ROS generation is associated with ferroptosis (47-49). For these reasons, the effect of NVP-231 on both ROS and MMP was examined, and CERK inhibition increased MMP in A549 cells (Mut KRAS/adenocarcinoma subtype), but not in H838 cells (WT KRAS/adenocarcinoma subtype) (Fig.3A). In A549 cells, CERK inhibition also rendered a significant increase in ROS levels (Fig.3B). Importantly, pretreatment of Mut KRAS cells (A549 cells) with the antioxidant/reducing agent, N-acetylcysteine (NAC), significantly alleviated the effect of CERK inhibition on cell survival (Fig.3C), MMP (Fig.3D), and level of ferroptosis marker TFR1 (Fig.3E). These findings implicate changes in mitochondria activity and ROS levels as a key mechanism related to the ability of CERK inhibition to drive ferroptosis in NSCLC cells harboring oncogenic Mut KRAS.
Figure 3: Ceramide kinase inhibition decreases mutant KRAS NSCLC cell survival by increasing MMP and generation of ROS.

(A, B) A549 and H838 cells (both LUAD-derived) treated with NVP 231 (0-400 nM) for 24h hours in serum-free media were subjected to MMP (A) or cellular ROS (B) assay. (C-E) Cell survival assay (C), MMP assay (D) or Western immunoblotting (E) was performed from A549 cells treated with Veh or NAC [(3-9 mM) in (C) and 6 mM in (D), (E)] for 1 hour followed by NVP231 treatment [0-400 nM in (C) and 400 nM in (D)] for 24 hours. Data are means ± SD; n = 4-6 from at least two independent occasions. *p<0.05, **p<0.005, ***p<0.0005. Scale bar = 200 μm. Unless otherwise noted by an * or depicted p-value, data are not significant between depicted groups; p>0.05.
Inhibition or downregulation of CERK decreases cell survival and sensitizes NSCLC cells with oncogenic KRAS to cisplatin via increased MMP and VDAC modulation
VDAC (voltage-dependent anion-selective channel) is one of the key proteins that regulates mitochondrial activity and maintains MMP. Importantly, VDAC has also been strongly implicated in ferroptosis activation (50-52). Due to the link between VDAC and MMP as well as ferroptosis, we examined whether VDAC1 was a critical component of the susceptibility of Mut KRAS NSCLC cells to CERK inhibition. In Mut KRAS NSCLC cells, downregulation of VDAC1, the most abundant VDAC isoform, resulted in higher cell survival upon treatment with ferroptosis inducer, erastin (Sup Fig. S7). Importantly, downregulation of VDAC1 significantly alleviated the effect of NVP-231 on the survival and MMP of NSCLC cells with Mut KRAS (Fig.4A-C). Also, reduction of VDAC1 level significantly “rescued” the effect of CERK downregulation (siCE) on MMP, cell viability and level of ferroptosis marker TFR1 observed in Mut KRAS NSCLC cells (Fig.4D-G). The effect NVP-231 on the clonogenic survival of Mut KRAS cells was specific for VDAC1 as downregulation of VDAC2 showed insignificant effects on the ability of CERK inhibition or downregulation to inhibit cell survival (Sup. Fig.8). These data demonstrate that through VDAC1, CERK regulates ferroptosis to augment cell survival in NSCLC cells possessing Mut KRAS.
Figure 4: Downregulation of VDAC reduces the effect of CERK inhibition/downregulation on Mut KRAS NSCLC cell survival and MMP.

(A-C) A549/H838 cells (both LUAD-derived) transfected with control/VDAC1 siRNA (siCon or siVD1) for 48h were subjected to SDS-PAGE/immunoblotting (A) or treated with NVP 231 [(0-400 nM) in (B), 400 nM on (C)]; 24 hour-post-NVP treatment, cells were utilized in clonogenic cell survival assay (B) or MMP assay (C). (D-F) Clonogenic cell survival assay (D), MMP assay (E) or Western immunoblotting (F) was performed from cells transfected with siCon or siVD1 for 24 hours then with control (siCon) or CERK (siCE) siRNA for additional 48 hours. (G) RT-qPCR was utilized from RNA samples prepared from cells transfected with siCon or siCE for 72 hours. Data in graphs are means ± SD; n = 4-6 from at least two independent occasions. *p<0.01, **p<0.0001, ***p<0.00001. Scale bar = 200 μm. Unless otherwise noted by an * or depicted p-value, data are not significant between depicted groups; p>0.05.
Cisplatin, a platinum-based chemotherapeutic drug, is a current first-line treatment of NSCLC including those with a Mut KRAS oncogenotype, but with varying effectiveness (53-55). In this study, we investigated the inhibitory efficacy of combining the CERK inhibitor, NVP-231, with cisplatin on the survival of NSCLC cells as we previously showed the cell death mechanisms to be different. NVP-231 pre-treatment amplified the survival inhibitory effect of cisplatin treatment, but only in NSCLC cells harboring a KRAS mutation (Sup. Fig.S9A,B, Sup. Fig.S1B,C). Additionally, downregulation of CERK sensitized Mut KRAS, but not WT KRAS cells to cisplatin treatment in respect of suppressing cell viability (Sup. Fig.S9C,D, Sup. Fig.S10A,D, Sup. Fig.S11A,C) as well as tumor growth (Fig.5A-C, Sup. Fig.S10E, Sup. Fig.S11D, Sup. Fig.S12). CERK reduction also rendered a boost in ferroptosis activation ± cisplatin in tumors (Fig.5D). Importantly, the NVP-231/cisplatin combination treatment reduced cell survival in a synergistic manner (Fig.6A,B), and the sensitizing effect of NVP-231 on cisplatin treatment regarding cell survival was diminished by downregulation of VDAC1 (Fig.6C). Taken together, these findings show that CERK inhibition/downregulation synergistically sensitizes Mut KRAS NSCLC cells to cisplatin treatment via action on VDAC1.
Figure 5: Ceramide kinase inhibition/downregulation sensitizes Mut KRAS NSCLC cells to cisplatin treatment.

(A-D) A549 cells (LUAD histology) with CERK WT (+/+) or Het (+/−) were injected into the flanks of nude mice (106 cells/mice). Mice developing tumors with approximate 100 mm3 volume were treated with cisplatin (3 mg/kg body weight) twice/week for 2 weeks (4 treatments). Tumor size was measured 2-3 times/week (A, B) then mice were euthanized, and tumors were excised for weight measurement (C). Tumor sections were subjected to IHC analysis for ferroptosis marker TFR1 (D). Scale bar = 200 μm. Data in (B,C) are means ± SD; data in (A) are mean; n=9-10 in (A-D). *p<0.05, **p<0.005, ***p<0.0005. Unless otherwise noted by an * or depicted p-value, data are not significant between depicted groups; p>0.05.
Figure 6: NVP 231 synergizes with cisplatin on Mut KRAS NSCLC cells in a VDAC-dependent manner.

(A) Clonogenic cell survival assay was performed from A549 cells (LUAD-derived) treated with varying concentrations of NVP 231 (0-350 nM) for 8 hours, then with varying concentration of cisplatin (Cis) for additional 16 hours. (B) Synergistic curve was generated from the half-maximal effective concentration (EC50) of NVP/cisplatin on cell survival. (C) Cell survival assay was performed from A549 cells transfected with control or VDAC1 siRNA (siCon or siVD1) for 48 hours followed by 8-hour-vehicle (Veh) or NVP (300 nM) then 16-hour-Veh or Cis- (0.5 μM) treatment. Data in (A) and (C) are means ± SD; n = 4 from two independent occasions. Data in (B) are EC50 values calculated from mean values in (A). *p<0.0005. Unless otherwise noted by an * or depicted p-value, data are not significant between depicted groups; p>0.05.
The role of ceramide kinase in regulation of MMP and cell survival involves the AKT-regulated VDAC-tubulin interaction
Previously in this study, we demonstrated that VDAC1 was required for CERK inhibition to induce MMP, ROS generation, and ferroptosis. Accumulating evidence has shown the importance of VDAC-tubulin interaction on the opening/closure state of VDAC related to ROS and MMP (56,57). To examine this mechanism in regard to CERK-derived C1P in Mut KRAS NSCLC, we again treated our NSCLC cell lines with NVP-231, which dramatically decreased tubulin binding to VDAC in NSCLC cells with Mut KRAS (Fig.7A,B). Moreover, this effect on the VDAC-tubulin interaction was reversed by pre-treatment of Mut KRAS cells with a common microtubule destabilizer, colchicine, (Fig.7C) to induce a surge in available free tubulin for VDAC binding. Cells pre-treated with colchicine were also less impacted by NVP-231 treatment in respect of MMP (Fig.7D). These findings indicate that CERK inhibition dysregulates MMP in Mut KRAS cells via blocking free tubulin binding to VDAC.
Figure 7: CERK inhibition increased MMP in Mut KRAS NSCLC cells by limiting VDAC-tubulin interaction and AKT activation.

(A, B) CERK inhibition decreased VDAC-tubulin binding and increased MMP in Mut KRAS NSCLC cells, which was reverted by tubulin-destabilizer, colchicine (Col), treatment. (A) A549/H838 cells (both LUAD-derived) were treated with NVP 231 (0-400 nM) for 24h hours, then mitochondria fractions prepared from the cell samples were subjected to co-IP assay for VDAC1 followed by SDS-PAGE/immunoblotting. (B) A549 cells were treated with vehicle (Veh) or NVP-231 (400 nM) for 24 hours then cells were subjected to VDAC-tubulin colocalization assay. (C, D) A549 cells were treated with Veh or NVP 231 (400 nM) for 24h hours followed by 2-hour treatment with Col (1 μM), then mitochondria fractions prepared from the cell samples were subjected to co-IP assay for VDAC followed by SDS-PAGE/immunoblotting (C) or post-treatment cells were subjected to MMP assay (D). (E) A549 cells treated with Veh, NVP-231 (400 nM) or AKT inhibitor, BEZ235 (AKTi; 50 nM) were subjected to Western immunoblotting. (F, G) AKT inhibition decreased VDAC-tubulin binding and increased MMP in Mut KRAS cells, which was reverted by Col treatment. Cells treated with Veh or AKTi as in (E) were additionally treated with Col (1 μM) for 2 hours before utilized in co-IP assay for VDAC followed by SDS-PAGE/immunoblotting (F) or MMP assay (G). (H-J) A549 cells were transfected with adenovirus control (Ad-Null) or expressing constitutively active AKT2 (Ad-caAKT) for 24 hours followed by 24-hour treatment of Veh or NVP 231 (400 nM); cells were then subjected to SDS-PAGE/immunoblotting (H), MMP assay (I) or clonogenic cell survival assay (J). Data in graphs are means ± SD; n = 12 in (B); n=6-7 in (D), (G), (I), (J) from two independent occasions. *p<0.01, **p<0.005, ***p<0.0005. Unless otherwise noted by an * or depicted p-value, data are not significant (NS) between depicted groups; p>0.05.
The cellular mechanism as to how CERK-derived C1P regulates the VDAC-tubulin interaction to maintain MMP was further explored by examining the PI3K/AKT pathway for several reasons: 1) RAS mutations have been widely reported to enable AKT pathway activation (58,59); 2) CERK has been implicated in AKT activation (18); 3) VDAC gating and the VDAC-tubulin interaction has been reported to be affected by phosphorylation state of VDAC (60); and 4) the phosphorylation state of VDAC1, which modulates cell survival, is linked to AKT signaling (61-64). In this regard, decreased AKT phosphorylation was observed following NVP-231 treatment and stable downregulation of CERK in Mut, but not WT KRAS NSCLC cells (Fig. 7E; Sup. Figs. S5;S10A-C;S11A,B;S13A-C). Indeed, AKT inhibition mimicked the effect of NVP-231 treatment on cell survival, MMP and VDAC-tubulin binding in Mut KRAS NSCLC cells. Specifically, treatment of Mut KRAS NSCLC cells with PI3K/AKT axis inhibitor, BEZ235 (AKTi), significantly decreased the level of AKT phosphorylation (Fig.7E), tubulin binding to VDAC (Fig.7F), increased MMP (Fig.7G), and induced ferroptotic cell death (Sup. Fig.S14). Additionally, the effect of AKT inhibitor on cell survival and tubulin-VDAC interaction was reversed by pre-treating the cells with colchicine (Fig.7F,G). The contribution of AKT activation for the roles of CERK in regulating MMP and cell survival was further validated by either expressing constitutively active (ca) AKT2 in Mut KRAS cells or dominant negative (DN) AKT2 prior NVP-231 treatment. Expressing of caAKT2 (Ad-caAKT) in Mut KRAS cells ablated the effect of NVP-231 treatment on AKT signaling, cell survival and MMP (Fig.7H-J); whereas expressing DN AKT2 did not show an additive effect with CERK inhibition on cell survival in Mut KRAS cells (Sup. Fig.S15). Interestingly, mitochondrial AKT directly bound to VDAC, and treatment with NVP-231 not only decreased AKT level in the mitochondria, but reduced AKT-VDAC binding (Fig.8A). Treating Mut KRAS cells with NVP-231 also lowered AKT-mediated phosphorylation of VDAC (Fig.8B) and a significant decrease in total phospho-serine VDAC was also observed in A549 cells with one allele of CERK ablated by CRISPR/cas9 (Sup. Fig.S16), which was linked to decreased AKT phosphorylation, clonogenic survival, and tumor growth along with increased cellular ROS generation (Sup. Fig.13). This effect was specific as re-expression of CERK “rescued” these effects in the heterozygous CERK ablation model regarding AKT activation and cell survival (Sup. Fig.S17). Intriguingly, NSCLC cells with Mut KRAS were shown to have higher levels of AKT activation and mitochondria AKT (Fig.8C, Sup. Fig.1D) as well as more VDAC-tubulin binding and AKT-mediated phosphorylation of VDAC (Fig.8D) despite a lack of differences in the levels of mitochondria proteins among those cell lines (Sup. Fig.S4). As VDAC phosphorylation has been reported to have significant impact on tubulin-VDAC association (60), our findings suggest an attractive model for mechanistic action of CERK in Mut KRAS NSCLC cells (Fig.8E): 1) In the presence of constitutively-activated KRAS, CERK is upregulated/activated, and either directly or indirectly, activates AKT to promote its translocation to mitochondria for enhancement of VDAC phosphorylation and tubulin binding in order to maintain MMP and prevents overproduction of ROS, which can lead to cell death; and 2) When CERK activity or C1P levels are low, inactive AKT is likely dominant, which renders less mitochondria AKT-mediated phosphorylation of VDAC and tubulin-VDAC association, causing out-of-control VDAC activity, a surge in ROS-mediated lipid peroxidation and ferroptotic cell death in Mut KRAS NSCLC.
Figure 8: Ceramide kinase inhibition decreased mitochondrial AKT level, AKT-VDAC binding and AKT-mediated VDAC phosphorylation.

(A, B) A549 cells (LUAD-derived) were treated with vehicle (Veh) or NVP 231 (400 nM) for 24 hours, mitochondria fraction prepared from the post-treatment cells were subjected to co-IP (A) or IP (B) assay for VDAC1 followed by SDS-PAGE/immunoblotting; cytosol and mitochondria fraction was also included in the Western immunoblotting. (C, D) Compared to WT KRAS NSCLC cells, Mut KRAS cells had more AKT activation, less VDAC-tubulin binding and AKT-mediated phosphorylation level of VDAC1. A549/H358/H1792/H838/H1299 cells at approximately 40-50% confluency were placed in serum-free media for 16 hours, then cell lysates were prepared and utilized in SDS-PAGE/immunoblotting (C) or mitochondria fractions prepared from the cell samples were subjected to co-IP or IP assay for VDAC1 followed by SDS-PAGE/immunoblotting (D). (E) Mechanistic model of how CERK inhibition attenuates Mut KRAS NSCLC cell survival. Left panel: in NSCLC cells with constitutively active KRAS by mutation, high CERK activity/C1P level elevates PI3K signaling and activates AKT by phosphorylation; active AKT then transports to mitochondria where it interacts with and phosphorylates VDAC; phosphorylation of VDAC by AKT results in more tubulin recruitment to VDAC, which reduces VDAC function and ROS generation to limit ROS-induced ferroptotic cell death. Right panel: low level or the absence of CERK/C1P prevents AKT activation, VDAC phosphorylation by AKT and subsequent VDAC-tubulin interaction; with less tubulin binding, VDAC activity were enhanced, causing more ROS production, ROS-mediated ferroptosis and cell death.
DISCUSSION
CERK inhibition, partial genetic ablation, and downregulation in Mut KRAS, but not WT KRAS NSCLC cells and tumors was observed to increase MMP, ferroptosis, and cisplatin sensitivity as well as significantly lower AKT activation, cell survival, tumor growth capacity. Hence, the study strongly suggests the involvement of AKT pathway and mitochondria function in how CERK controls ferroptosis activation to maintain the homeostasis of Mut KRAS NSCLC cells. Indeed, pretreatment of Mut KRAS cells with ROS scavenger or ferroptosis inhibitor Ferrostatin-1 alleviated the effect of NVP-231 treatment on cell survival, but interestingly, did not completely revert the survival back to control levels. This lack of a complete “rescue” may be due to the limited capability of these agents to completely remove ROS or block ferroptosis. On the other hand, it does not rule out the possibility that CERK activates other pro-survival pathway(s) such as those linked to AKT. In support, caAKT2 expression completely abrogated the effect of CERK inhibition on cell survival, and future investigations will be needed to determine these additional survival mechanisms. Regardless, this study strongly suggests that one mode by which CERK aids the survival and cellular homeostasis of Mut KRAS NSCLC cells is via AKT regulation of VDAC to suppress ROS production and ferroptosis.
This study also demonstrates that function of CERK to suppress ferroptosis is specific to KRAS versus other RAS family members as a NSCLC cell line with an activating NRAS mutation (H1299) was not susceptible to NVP-231. This specificity for KRAS signaling as to CERK-derived C1P suggested that common regulatory factors or cellular locations specific to CERK and KRAS may be involved. On plausible example of a regulatory factor is calmodulin, a modulator of CERK activity that will associate specifically with KRAS versus other RAS family members (65-67). Another plausible link are bioactive phosphoinositides as CERK possesses a PH-domain (68) and KRAS specifically is linked to enzymes that modulate the levels of this lipid class (61). The link of calmodulin and specific phosphoinositides (e.g., PI5P, PI(3,5)P2, PI(4,5)P2, PI(3,4,5)P3) to only KRAS signaling may explain the specificity of the increased levels of C1P in these NSCLC cells, and one supposition is the existence of a stable complex between these factors driving AKT activation as this enzyme possesses a PH-domain and also associates with specific phosphoinositides (e.g., PIP3) (69). As to a specific cellular location, KRAS is the only RAS isoform that localizes to the mitochondria where CERK is also localized (14,70-72). Interestingly, CERK requires cardiolipin for full activation (73,74), and KRAS signaling is linked to increased levels of a cardiolipin regulatory enzyme, PLSCR3 (61,75). Alternatively, CERK expression is higher in NSCLC cells with Mut KRAS, and thus, activation of KRAS may simply increase CERK levels via a transcription mechanism, although this would not explain the specificity for Mut KRAS versus NRAS. Future studies will interrogate these possibilities as to the specific link of CERK to KRAS as a direct versus indirect mechanism.
Our findings also indicate the role of CERK in AKT activation to regulate VDAC function and implies CERK-VDAC axis as contributor for cisplatin resistance in Mut KRAS cells. Indeed, AKT was implicated to directly phosphorylate VDAC1, which was linked to enhanced tubulin association. This is in opposition to the current indirect model via AKT inhibition of GSK-3β, which leads to dephosphorylation of VDAC1 on ser13 and thr107 with subsequent suppression of intrinsic apoptosis (63,76). Our data suggest that an additional direct phosphorylation mechanism also exists, and previous studies have identified additional phosphorylation sites linked to AKT, which have not been investigated (ser57, thr65, thr98)) (77,78). Thus, context (ferroptosis vs apoptosis suppression) as to a particular cell signaling cascade may be important as to how VDAC1 activity is modulated. A similar mechanism of direct VDAC1 phosphorylation blocking cell death, in this case apoptosis, was shown for NEK1 phosphorylation of VDAC1 on Ser193, but the sequence does not correspond to an AKT consensus site (79). Regardless, this study suggests a distinct mechanism where CERK inhibition sensitizes Mut KRAS cells to cisplatin by suppressing the AKT pathway to directly impact VDAC activity. Therefore, therapeutic drugs targeting CERK would be a promising component in combinational approach for targeting NSCLC in a very precise manner (e.g., the Mut KRAS subtype).
In conclusion, this study demonstrated intriguing findings regarding a new mechanism of how the inhibition of CERK reduces the survival and increases the sensitivity to cisplatin of NSCLC cells harboring KRAS mutations. Our findings suggest an attractive model for mechanistic action of CERK in Mut KRAS NSCLC cells (Fig.8E): 1) In the presence of constitutively-activated KRAS, CERK is upregulated/activated, and either directly or indirectly, activates AKT to promote its translocation to mitochondria for enhancement of VDAC phosphorylation and tubulin binding in order to maintain MMP and prevents overproduction of ROS, which can lead to cell death; and 2) When CERK activity or C1P levels are low, inactive AKT is likely dominant, which renders less mitochondria AKT-mediated phosphorylation of VDAC and tubulin-VDAC association, causing out-of-control VDAC activity, a surge in ROS-mediated lipid peroxidation and ferroptotic cell death in Mut KRAS NSCLC. The study also provides a basis for the development of highly effective anti-cancer combinations for the treatment of NSCLC with specific oncogenic mutation (i.e., Mut KRAS).
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health – National Institute of Allergy and Infectious Diseases grant R01 AI139072 (to CEC), and National Institute of General Medical Sciences grants R01 GM137578 and R01 GM137394 (to CEC). This work was also supported by research grants from the Veteran’s Administration (VA Merit Review, I BX001792 (CEC) and a Senior Research Career Scientist Award, IK6BX004603 (CEC)). The contents of this manuscript do not represent the views of the Department of Veterans Affairs or the United States Government.
Footnotes
The authors have declared that no conflict of interest exists.
COMPETING INTERESTS
There are not any competing financial interests in relation to the work described.
LITERATURE CITED
- 1.Baran Y et al. Alterations of ceramide/sphingosine 1-phosphate rheostat involved in the regulation of resistance to imatinib-induced apoptosis in K562 human chronic myeloid leukemia cells. J Biol Chem. 2007; 282:10922–10934. [DOI] [PubMed] [Google Scholar]
- 2.Bose R et al. Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals. Cell. 1995; 82:405–414. [DOI] [PubMed] [Google Scholar]
- 3.Bruno A et al. Lack of ceramide generation in TF-1 human myeloid leukemic cells resistant to ionizing radiation. Cell Death Differ. 1998; 5:172–182. [DOI] [PubMed] [Google Scholar]
- 4.Camgoz A et al. Roles of ceramide synthase and ceramide clearence genes in nilotinib-induced cell death in chronic myeloid leukemia cells. Leuk Lymphoma. 2011; 52:1574–1584. [DOI] [PubMed] [Google Scholar]
- 5.Hernández-Tiedra S et al. Dihydroceramide accumulation mediates cytotoxic autophagy of cancer cells via autolysosome destabilization. Autophagy. 2016; 12:2213–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maurer B Increase of ceramide and induction of mixed apoptosis/necrosis by N-(4-hydroxyphenyl)-retinamide in neuroblastoma cell lines. J Natl Cancer Inst. 1999; 91:1138–1146. [DOI] [PubMed] [Google Scholar]
- 7.Zhu Q et al. C6-ceramide synergistically potentiates the anti-tumor effects of histone deacetylase inhibitors via AKT dephosphorylation and α-tubulin hyperacetylation both in vitro and in vivo. Cell Death Dis. 2011; 2:e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wijesinghe D et al. Ceramide kinase is required for a normal eicosanoid response and the subsequent orderly migration of fibroblasts. J Lipid Res. 2014; 55:1298–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacKnight H et al. The interaction of ceramide 1-phosphate with group IVA cytosolic phospholipase A2 coordinates acute wound healing and repair. Sci Signal. 2019; 12:eaav5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salas A Sphingosine kinase-1 and sphingosine 1-phosphate receptor 2 mediate Bcr-Abl1 stability and drug resistance by modulation of protein phosphatase 2A. Blood. 2011; 117:5941–5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu S et al. Ceramide kinase mediates intrinsic resistance and inferior response to chemotherapy in triple-negative breast cancer by upregulating Ras/ERK and PI3K/Akt pathways. Cancer Cell International. 2021; 21:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rovina P et al. Subcellular localization of ceramide kinase and ceramide kinase-like protein requires interplay of their pleckstrin homology domain-containing N-terminal regions together with C-terminal domains. Biochim. Biophys. Acta. 2009; 1791:1023–1030. [DOI] [PubMed] [Google Scholar]
- 13.Kim T et al. The interaction between the pleckstrin homology domain of ceramide kinase and phosphatidylinositol 4,5-bisphosphate regulates the plasma membrane targeting and ceramide-1-phosphate levels. Biochem. Biophys. Res. Commun. 2006; 342:611–617. [DOI] [PubMed] [Google Scholar]
- 14.Lamour N et al. Ceramide kinase uses ceramide provided by ceramide transport protein: localization to organelles of eicosanoid synthesis. J Lipid Res. 2007; 48:1293–1304. [DOI] [PubMed] [Google Scholar]
- 15.Mishra S et al. Cptp: A sphingolipid transfer protein that regulates autophagy and inflammasome activation. Autophagy. 2018; 14:862–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simanshu D et al. Non-vesicular trafficking by a ceramide-1-phosphate transfer protein regulates eicosanoids. Nature. 2013; 500:463–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rivera I et al. Ceramide 1-phosphate regulates cell migration and invasion of human pancreatic cancer cells. Biochem. Pharmacol. 2016; 102:107–119. [DOI] [PubMed] [Google Scholar]
- 18.Schwalm S et al. Ceramide Kinase Is Upregulated in Metastatic Breast Cancer Cells and Contributes to Migration and Invasion by Activation of PI 3-Kinase and Akt. J Mol Sci. 2020; 21: 1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goehe R et al. hnRNP L regulates the tumorigenic capacity of lung cancer xenografts in mice via caspase-9 pre-mRNA processing. J Clin Invest. 2010; 120:3923–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shultz J et al. SRSF1 regulates the alternative splicing of caspase 9 via a novel intronic splicing enhancer affecting the chemotherapeutic sensitivity of non-small cell lung cancer cells. Mol Cancer Res. 2011; 9:889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shultz J et al. Alternative splicing of caspase 9 is modulated by the phosphoinositide 3-kinase/Akt pathway via phosphorylation of SRp30a. Cancer Res. 2010; 70:9185–9196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson R et al. The Alternative Splicing of Cytoplasmic Polyadenylation Element Binding Protein 2 Drives Anoikis Resistance and the Metastasis of Triple Negative Breast Cancer. J Biol Chem. 2015; 290:25717–25727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shapiro B et al. Melanoma Differentiation-associated Gene 7/IL-24 Exerts Cytotoxic Effects by Altering the Alternative Splicing of Bcl-x Pre-mRNA via the SRC/PKCδ Signaling Axis. J Biol Chem. 2016; 291:21669–21681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shultz J et al. The Proto-oncogene PKCι regulates the alternative splicing of Bcl-x pre-mRNA. MCR. 2012; 5:660–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vu N et al. Caspase-9b Interacts Directly with cIAP1 to Drive Agonist-Independent Activation of NF-κB and Lung Tumorigenesis. Cancer Res. 2016; 76:2977–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vu N et al. hnRNP U enhances caspase-9 splicing and is modulated by AKT-dependent phosphorylation of hnRNP L. J Biol Chem. 2013; 12:8575–8584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nogueira C et al. Cooperative interactions of PTEN deficiency and RAS activation in melanoma metastasis. Oncogene. 2010; 29:6222–6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lamour N et al. Ceramide kinase regulates the production of tumor necrosis factor α (TNFα) via inhibition of TNFα-converting enzyme. J Biol Chem. 2011; 286:42808–42817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mietla J et al. Characterization of eicosanoid synthesis in a genetic ablation model of ceramide kinase. J Lipid Res. 2013; 54:1834–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shaner R et al. Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J Lipid Res. 2009; 50:1692–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wijesinghe D et al. Use of high performance liquid chromatography-electrospray ionization-tandem mass spectrometry for the analysis of ceramide-1-phosphate levels. J. Lipid Res. 2010; 51:641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wijesinghe D et al. Systems-Level Lipid Analysis Methodologies for Qualitative and Quantitative Investigation of Lipid Signaling Events During Wound Healing. Adv Wound Care (New Rochelle). 2013; 2:538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoadley K et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell. 2018; 173:291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu J et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell. 2018; 173:400–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez-Vega F et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell. 2018; 173:321–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor A et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell. 2018; 33:676–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cerami et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring ultidimensional Cancer Genomics Data. Cancer Discovery. 2012; 2:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wickham et al. Welcome to the tidyverse. Journal of Open Source Software. 2019; 4:1686. [Google Scholar]
- 40.Motulsky H, Brown R Detecting outliers when fitting data with nonlinear regression – a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics. 2006; 7:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Payne A et al. Ceramide kinase promotes tumor cell survival and mammary tumor recurrence. Cancer Res. 2014; 74:6352–6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Graf C et al. Targeting ceramide metabolism with a potent and specific ceramide kinase inhibitor. Mol Pharmacol. 2008; 74:925–932. [DOI] [PubMed] [Google Scholar]
- 43.Granado M et al. Ceramide 1-phosphate inhibits serine palmitoyltransferase and blocks apoptosis in alveolar macrophages. Biochim Biophys Acta. 2009; 1791:263–272. [DOI] [PubMed] [Google Scholar]
- 44.Mitra P et al. Ceramide kinase regulates growth and survival of A549 human lung adenocarcinoma cells. FEBS Letters. 2007; 581:735–740. [DOI] [PubMed] [Google Scholar]
- 45.Li Y et al. The pleiotropic roles of sphingolipid signaling in autophagy. Cell Death & Disease. 2014;5:e1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feng H et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020; 30:3411–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen X et al. Ferroptosis: machinery and regulation. Autophagy. 2021;17:2054–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li J et al. Ferroptosis: past, present and future. Cell Death Dis. 2020; 11:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park E et al. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019;10:822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lemasters J et al. Evolution of Voltage-Dependent Anion Channel Function: From Molecular Sieve to Governator to Actuator of Ferroptosis. Front Oncol. 2017; 7:303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lipper C et al. Redox-dependent gating of VDAC by mitoNEET. Proc Natl Acad Sci U S A. 2019; 116:19924–19929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang Y et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat Commun. 2020; 11:433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fournel L et al. Cisplatin increases PD-L1 expression and optimizes immune check-point blockade in non-small cell lung cancer. Cancer Lett. 2019; 464:5–14. [DOI] [PubMed] [Google Scholar]
- 54.He L et al. FEN1 promotes tumor progression and confers cisplatin resistance in non-small-cell lung cancer. Mol Oncol. 2017; 11:640–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xiao L et al. Cytoplasmic RAP1 mediates cisplatin resistance of non-small cell lung cancer. Cell Death Dis. 2017; 8:e2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Puurand M et al. Tubulin betaII and betaIII Isoforms as the Regulators of VDAC Channel Permeability in Health and Disease. Cells. 2019; 8:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rostovtseva T et al. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc Natl Acad Sci U S A. 2008; 105:18746–18751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kennedy A et al. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Cell. 2011; 42:36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shaw. Nature. R et al. Ras, PI(3)K and mTOR signalling controls tumour cell growth 2006; 441:424–430. [DOI] [PubMed] [Google Scholar]
- 60.Sheldon K et al. Phosphorylation of Voltage-Dependent Anion Channel by Serine/ Threonine Kinases Governs Its Interaction with Tubulin. PLoS One. 2011; 6:e25539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Frame S et al. (2001) GSK3 takes centre stage more than 20 years after discovery. Biochem. J 359, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cross DA et al. (1995) Inhibition of GSK-3 by insulin mediated by protein kinase B. Nature 378, 785–789 [DOI] [PubMed] [Google Scholar]
- 63.Li L et al. Plasminogen kringle 5 induces endothelial cell apoptosis by triggering a voltage-dependent anion channel 1 (VDAC1) positive feedback loop. J Biol Chem. 2014. Nov 21;289(47):32628–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yuan S et al. Voltage-dependent anion channel 1 is involved in endostatin-induced endothelial cell apoptosis. FASEB J. 2008. Aug;22(8):2809–20. [DOI] [PubMed] [Google Scholar]
- 65.Adhikari H et al. Interrogating the protein interactomes of RAS isoforms identifies PIP5K1A as a KRAS-specific vulnerability. Nat Commun. 2018; 9:3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nussinov R et al. Calmodulin and PI3K Signaling in KRAS Cancers. Trends Cancer. 2017; 3:214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Villalonga P et al. Calmodulin binds to K-Ras, but not to H- or N-Ras, and modulates its downstream signaling. Mol Cell Biol. 2001; 21:7345–7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sugiura M et al. Ceramide kinase, a novel lipid kinase. Molecular cloning and functional characterization. J Biol Chem. 2002; 277:23294–23300. [DOI] [PubMed] [Google Scholar]
- 69.Park W et al. Comprehensive identification of PIP3-regulated PH domains from C. elegans to H. sapiens by model prediction and live imaging. Mol Cell. 2008; 30:381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ashery U et al. Nonconventional trafficking of Ras associated with Ras signal organization. Traffic. 2006; 7:119–126. [DOI] [PubMed] [Google Scholar]
- 71.Miller T et al. Depletion of phosphatidylinositol 4-phosphate at the Golgi translocates K-Ras to mitochondria. J Cell Sci. 2019; 132:jcs231886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Philips M Compartmentalized signalling of Ras. Biochem Soc Trans. 2005; 33:657–661. [DOI] [PubMed] [Google Scholar]
- 73.Bajjalieh S et al. Ceramide kinase. Methods Enzymol. 2000;311:207–215. [DOI] [PubMed] [Google Scholar]
- 74.Wijesinghe D et al. Ceramide kinase and ceramide-1-phosphate. Methods Enzymol. 2007;434:265–292. [DOI] [PubMed] [Google Scholar]
- 75.Kovalski J et al. The Functional Proximal Proteome of Oncogenic Ras Includes mTORC2. Mol Cell. 2019; 73:830–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sharma K et al. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014;8(5):1583–94. [DOI] [PubMed] [Google Scholar]
- 77.Humphrey SJ et al. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 2013;17(6):1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gnad F et al. Systems-wide analysis of K-Ras, Cdc42, and PAK4 signaling by quantitative phosphoproteomics. Mol Cell Proteomics. 2013;12(8):2070–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen Y, Craigen WJ, Riley DJ. Nek1 regulates cell death and mitochondrial membrane permeability through phosphorylation of VDAC1. Cell Cycle. 2009. Jan 15;8(2):257–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.