

Abstract
The nuclear factor-κB (NF-κB) signaling pathway plays key roles in inflammation and the pathogenesis of many solid and hematological malignancies, including multiple myeloma (MM), a malignancy of the plasma cells. While proteasome inhibitors, such as bortezomib, employed in MM treatments may inhibit NF-κB signaling pathways, MM cells often become drug resistant in part due to non-cell autonomous mechanism(s) from the MM tumor microenvironment. We previously found that fragments of, but not full-length, hyaluronan and proteoglycan link protein 1 (HAPLN1), produced by MM bone marrow stromal cells (BMSCs), activate an atypical bortezomib-resistant NF-κB pathway in MM cells. In our present study, we found that MM cells promote HAPLN1 expression and matrix metalloproteinase (MMP)2 activity in co-cultured BMSCs and MMP2 activity is higher in BMSCs established from MM patients’ bone marrow aspirates relative to normal equivalents. Moreover, MMP2 cleaves HAPLN1 into forms similar in size to those previously observed in MM patients with progressive disease. Both HAPLN1 and MMP2 in BMSCs were required to enhance NF-κB activation and resistance to bortezomib-induced cell death in co-cultured MM cells. We propose that MMP2-processing of HAPLN1 produces a matrikine that induces NF-ĸB activation and promotes bortezomib resistance in MM cells.
Introduction
Multiple myeloma (MM) is a hematologic neoplasm characterized by the infiltration of malignant plasma cells into bone marrow (BM) and other tissues1. Currently, MM is the second most common blood cancer in the United States with a 47.8% mortality rate within 5 years due largely to patients developing therapeutic resistance.2,3 MM is a heterogenous disease with differences in pathological mechanisms, clinical manifestations and responses to therapies. For example, whole-genome and whole-exome sequencing studies have identified genetic alterations in the components of oncogenic signaling pathways including JAK2/STAT3, RAS/MEK/MAPK, and nuclear factor kappa B (NF-ĸB). In particular, the NF-ĸB family of transcription factors are well recognized to play important roles in MM pathogenesis.4,5 This family is comprised of p65(RelA), RelB, c-Rel, p100/p52, and p105/p50, which form dimeric transcription factors that are held inactive in the cytoplasm in their latent forms. NF-κB activation signaling is commonly characterized by the “canonical” and “noncanonical” pathways, in which the 26S proteasome plays an essential role in degrading the cytoplasmic inhibitors of NF-κB (IκBs) or processing p100 to p52, respectively, thus freeing corresponding NF-κB dimers to translocate to the nucleus to upregulate inflammatory, immune, and cell survival genes, among others.6–8 Over 90% of MM patients display constitutive NF-κB activity, as shown in RelA immunostaining and NF-κB-related gene expression patterns in their MM cells, further supporting the importance of NF-κB activity in MM pathogenesis.5,9,10 However, genetic alterations in the NF-ĸB pathways detected by genome/exome sequence analyses are only found in 20–30% of MM cases.5,9 Thus, it appears that many MM patients contain MM cells with NF-ĸB activities without detectable cell-intrinsic genetic aberrations within the known NF-κB signaling pathways and raise the possibility of non-cell autonomous mechanisms that arise from the BM tumor microenvironment (TME) to regulate chronic NF-κB activity in MM cells.
MM cells have been reported to respond to a plethora of MM cell-extrinsic factors in the BM TME, such as IL-6, IGF-1, TNFα, SDF-1, and BAFF, which may activate NF-ĸB in MM cells and promote their migration, survival, and/or drug resistance.11–13 The MM TME also contains cellular components, such as osteoblasts, osteoclasts, macrophages, natural killer cells, neutrophils and bone marrow stromal cells (BMSCs), which have been further implicated in MM disease progression. In particular, the functional nexus between BMSCs and MM cells has been well documented in myeloma literature.14–16 In a physiologically normal setting, BMSCs are heterogenous cells of mesenchymal origin that support hematopoietic stem cell progenitor differentiation, survival, and division.17 In MM disease, BMSCs directly adhere to malignant cells and secrete aforementioned factors, among others, which create a permissive, cancerous environment.15,17,18 While many MM therapies include proteasome inhibitors, such as bortezomib, are capable of halting canonical and noncanonical NF-ĸB activation in MM cells,19 subsequent studies have uncovered that NF-ĸB activity detected in primary MM cells is often bortezomib-resistant and sometimes paradoxically bortezomib-inducible.20,21 In an effort to identify factor(s) that can cause NF-ĸB activation in MM cells in a bortezomib-resistant manner, we previously purified and identified hyaluronan and proteoglycan link protein 1 (HAPLN1) from BMSCs conditioned media as a factor capable of inducing such an alternative NF-ĸB activity.22
HAPLN1 belongs to the link family of extracellular matrix (ECM) proteins and normally serves as structural support in cartilage and tissue formation.23 HAPLN1 contains N-terminal immunoglobulin-like (IG) domain which binds to proteoglycans, and two C-terminal proteoglycan tandem repeat (PTR1/2) domains which bind to hyaluronic acid to stabilize cartilage matrices.24,25 HAPLN1 mutant mice show developmental abnormalities, culminating in post-natal lethality.26 In contrast, its protumorigenic role is less well defined, although it has been implicated in pathogenesis of hepatocellular carcinoma, melanoma, and colorectal cancer.27–29 While investigating the potential pathological role of HAPLN1 in MM disease, we previously detected soluble HAPLN1 fragments that were immunoreactive to antibodies directed against PTR1 and PTR2 domains, but not IG domain, of HAPLN1 in the bone marrow plasma fraction obtained from MM patients.22 We also generated recombinant full-length and individual domains of HAPLN1 and found that only the smaller fragments containing PTR1 and/or PTR2 domains of HAPLN1, but not full-length HAPLN1, caused NF-ĸB activation in MM cells in a bortezomib-resistance manner.22 Recombinant PTR1 also caused bortezomib resistance in several MM cell lines analyzed. Thus, these previous findings suggested the possibility that some protease(s) might be cleaving signaling-inert full-length HAPLN1 into a novel ECM-generated pathological soluble factor, a “matrikine”, in MM TME.
In the present study, we sought to identify a protease produced by BMSCs and/or MM cells, capable of processing HAPLN1 and required for BMSC-dependent activation of NF-ĸB thereby contributing to bortezomib resistance in MM cells. We screened proteases that have been previously described to cleave HAPLN1 at the N-terminal region upstream of the IG domain30 and found that matrix metalloproteinase 2 (MMP2) mRNA was expressed at the highest level in MM BMSCs. MMP2 expression has previously been detected in MM cells from BM biopsies, and a bisphosphonate based MMP2 specific inhibitor has been reported to lower myeloma tumor burden in a U266 mouse model and reduce MM-induced bone loss in a 5TGM1 mouse model.31 We found that MM cells increased the expression of HAPLN1 and MMP2 activity in co-cultured BMSCs and MMP2 activity in conditioned media of MM patients’ BMSCs positively correlated with NF-ĸB activation observed in co-cultured MM cells. Moreover, reductions of HAPLN1 and MMP2 expression in BMSCs mitigated NF-ĸB activation observed in co-cultured MM cells and sensitized these cells to bortezomib-induced toxicity. Finally, recombinant MMP2 was capable of generating a HAPLN1 fragment that is reactive to antibodies raised against PTR1 and 2 domains, but not IG domain, similar to that we observed in bone marrow plasma fractions of progressing MM patients.22 Our study suggests that MM cells regulate expression/activity of HAPLN1 and MMP2 in BMSCs to in turn promote NF-κB signaling and bortezomib-resistance in MM cells. These results also suggest the potential for HAPLN1 and MMP2 in BMSCs, not in MM cells, as novel biomarkers and/or therapeutic targets for addressing bortezomib resistance in MM disease.
Methods
Cell Lines, Antibodies, Chemicals, and Plasmids
RPMI8226, H929, L363, MM1.S and HEK293T cell lines were purchased from American Type Culture Collection (ATCC). RPMI8226, H929, L363, and MM1.S cells were cultured in RPMI1640 media containing 10% FBS, 2% GlutaMAX (Gibco), and 1% penicillin/streptomycin, and grown in 37°C/5% CO2. HEK293T cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% FBS and 1% penicillin/streptomycin. Cell line authentication was not conducted. Early passaged cells (passage 1–4) were cryopreserved in large batches after being grown for 2 weeks to 90% confluency and subsequently thawed, expanded to 90% confluency, and tested for mycoplasma. All cell lines were found to be negative for mycoplasma in 2018 using the Universal Mycoplasma Detection Kit (ATCC). All experiments were performed within 3–10 passages after thawing from cryopreserved stocks. Antibodies against HAPLN1 (H-93, K-14, C-14) and MMP2 (2C1) were purchased from Santa Cruz Biotechnology, and that against His (A00186–100) was from GenScript. Recombinant TNFα was purchased from EMD Millipore. Antibodies were diluted to a 1:1000 concentration. Bortezomib (from Selleckchem) was dissolved in DMSO at 1000x stock solution and stored at −70°C until use. Vehicle controls were treated with DMSO alone.
Primary BMSCs
Primary human normal and MM BMSCs (Table 1) were obtained from fresh whole BM aspirates with written informed consent from the patients, and the studies were conducted in accordance with recognized ethical guidelines (U.S. Common Rule). Studies were approved by the University of Wisconsin-Madison Institutional Review Board (HO07403) at the University of Wisconsin Hospital and Clinics. Clinical information for MM samples included patient age (44–78), sex (60% male, 40% female), and disease stage (newly diagnosed or relapse/refractory). Normal BMSCs and MM BMSCs were isolated as previously described.22,32 Briefly, bone marrow was diluted in OptiMEM media containing 2% GlutaMAX, 1% NEAA (Gibco), and 1% penicillin/streptomycin supplemented with heparin sulfate and DNase I and centrifuged with lymphocyte separation medium (Cellgro) to isolate the mononuclear population. This fraction was cultured with OptiMEM media including 10% FBS at 37°C in a 5% CO2 incubator, and nonadherent cells were removed 24 hours after plating to allow BMSCs to grow out. For MM BMSCs, the mononuclear fraction was first positively sorted for myeloma plasma cells using CD138+ magnetic MACS beads (Miltenyi Biotec) to 90% purity, and CD138− fraction was cultured as described for normal BMSCs.22 Adherent BMSCs, derived from normal and MM barrow, were expanded by passaging 3–4 times and cryopreserved as previously described.22,32 To maintain consistency, only BMSCs passaged 3–4 times ex vivo were used in the current study. Number of normal and MM BM patient samples utilized in the present study can be found in Table 1.
Table 1:
Number of normal and MM BM patient samples utilized in the present study
| # Normal BM Samples | # MM BM Samples |
|---|---|
| 9 | 37 |
Co-culture Assays
6 × 104 BMSCs were plated per well in 6-well dishes and allowed to grow for 48 hours. BMSCs were washed once with 1X PBS and 1 × 106 RPMI8226 cells were plated on top of the BMSC monolayer for an additional 24 hours in serum-free OptiMEM media containing 2% GlutaMAX, 1% NEAA (Gibco), and 1% penicillin/streptomycin. MM cells were then removed from the stromal layer by gentle mechanical tapping while the BMSC monolayer was undisturbed.
qRT-PCR Analysis
Total RNA from 1 × 106 CD138− cells and CD138+ cells, and 6 × 104 BMSCs was isolated using Trizol reagent (Invitrogen). RNA was used to make cDNA and to perform qRT-PCR on a BioRad iQ5 with primers found in Supplementary Table 1. Relative expression was determined by ΔΔCt calculation and normalized to GAPDH mRNA levels of the same sample.
Conditioned Media Immunoblot Analysis
Serum-free OptiMEM media was harvested from co-culture wells and protein was precipitated using trichloroacetic acid (TCA). Briefly, CM was mixed with 10% volume of 2% deoxycholate, followed by 10% volume of 100% TCA. CM was spun at 1.6K RPM with tabletop centrifuge for 5 min and removed of DOC/TCA. The protein pellet was resuspended in 50 uL of 1.5M Tris-HCl (pH 8.8) and an equal volume of 2X Laemmli SDS sample buffer was added, which was followed by incubation at ~95°C for 10 min. Protein concentration was measured by RCDC assay. 100 ug of protein for each sample were run on denaturing 10% SDS-polyacrylamide gel and transferred onto a polyvinylidene fluoride or nitrocellulose membrane (GE Healthcare). Ponceau S Red (Thermo Fisher Scientific) was used to stain the proteins on the membrane to confirm equal loading of total proteins in gel lanes. Membranes were incubated with appropriate antibodies, analyzed via enhanced chemiluminescence (GE Healthcare), and quantified via ImageJ analysis (National Institutes of Health) to calculate the signal intensity of protein(s) of interest normalized to total Ponceau S Red stain protein quantified per lane.
Gelatin Zymography
Conditioned medium harvested from 6 × 104 BMSCs in mono- or co-culture with 1 × 106 RPMI8226 cells was spun at 1.6K RPM using tabletop centrifuge for 5 min to remove debris. An equal volume of 2X loading buffer (containing 0.5 M HCL at pH 6.8, 75% glycerol, 10% SDS, and bromophenol blue) was added to each sample. 30 uL of each sample was run on 10% SDS-polyacrylamide gel containing 10% gelatin (resuspended in deionized water). Gels were washed twice for 30 min each time with 2.5% Triton X-100 solution (containing deionized water and Triton X-100), equilibrated once in MMP2 Tris buffer (containing 50 mM Tris, 200 mM NaCl, and 10 mM CaCl2 at pH 7.4) for 30 minutes, and incubated in Tris buffer for 18 hours at 37°C. Zymograms were stained with 4 mL 0.1% Coomassie blue for 10 min and destained in destaining solution (deionized water, 40% methanol, and 10% acetic acid) for one hour.
MMP2 and HAPLN1 Recombinant Protein Cleavage Assays
Recombinant C-terminal His-tagged full-length HAPLN1 (R&D Systems) was treated with APMA pre-activated recombinant MMP2 (Millipore Sigma) for 30 minutes at 37⁰C at a 1:5 ratio in 50 uL of MMP2 Tris buffer.
Electrophoretic Mobility Shift Assays (EMSAs)
NF-ĸB activation in MM cell lines was measured by EMSA using 32P-labeled double-stranded DNA probes for NF-ĸB (5’- TCAACAGAGGGACTCCGAGAGGCC −3’) and Oct-1 loading control (5’- TGTCGAATGCAAATCACTAGAA −3’) and total cell extracts as previously described.22 The EMSA gels were dried and exposed to X-ray films. The dried gels were also exposed to phosphor-image screens and NF-ĸB and Oct-1 DNA binding activities were quantified through ImageQuant software (GE Healthcare). NF-ĸB activation in each sample was normalized to Oct-1 binding value from the same sample, and the fold change in NF-ĸB activation in experimental groups (NF-ĸB/Oct-1) were normalized to vehicle-treated control values (NF-ĸB/Oct-1) from the same sample.
Lentiviral shHAPLN1 Construct Generation and BMSC HAPLN1 and MMP2 Knockdowns
HAPLN1 and MMP-2 was knocked down from BMSCs using shHAPLN1 and shMMP-2 lentiviral constructs purchased from Millipore Sigma: (SHCLNG-NM_001884) 5’-CCGGGGAGTCAGGAACTACGGATTTCTCGAGAAATCCGTAGTTCCTGACTCCTTTTTTG-3’ and (SHCLNG-NM_004530) 5’-GCTGAAGGACACACTAAAGAA-3’, respectively. These constructs also harbor a GFP expression cassette to monitor infection efficiency. HEK293T cells in DMEM containing 10% FBS were transfected with shHAPLN1 (4 ug/10 cm dish), envelope pVSV-G (2 ug/10 cm dish), and packaging pCMVΔ8.91 (4 ug/10 cm dish) constructs via Lipofectamine 2000. Supernatants containing packaged lentiviral particles were collected after 48 hours and pre-incubated with protamine sulfate at 100 ng/mL for 15 min at 37°C/5% CO2 before 6 × 104 BMSC cells in DMEM containing 10% FBS, 2% Glutamax, and 1% NEAA were added, drop-wise, on top of the supernatant. BMSCs were then plated in 6-well dish and GFP and HAPLN1 expression in BMSCs was measured 48–72 hours after knockdown.
Time-lapse Microscopy Analysis
BMSC, RPMI8226 cell, and collagen layer preparation
1×104 BMSCs were plated in a 96-well plate to adhere overnight. 1.5×104 RPMI8226 cells were embedded in a thin layer (~400 μm) of 3D collagen layer over BMSCs for 24 hours. Collagen layer was made by dispensing 100 uL 10X PBS, 85.5 uL tissue culture grade water, 14.5 uL 1 N NaOH, and 800 uL PureCol collagen (cat# 5005, Advanced BioMatrix) in a 1.5 mL Eppendorf tube without mixing. The mixture was vortexed for 7 seconds, spun down in tabletop centrifuge for 3 seconds, and pH was tested (between 7.6–8) with Litmus paper. RPMI8226 cells (1.5×104 cells per well) were mixed with the collagen mixture. BMSC media in 96-well was removed and RPMI8226 collagen mixture was pipetted directly into center bottom of the well. 96-well plate was spun at 250xg for 45 seconds at room temperature using Centrifuge 5804 R to disperse collagen mixture evenly. Plate was incubated for 45 minutes in 37°C incubator to solidify collagen gels before adding BMSC media on top (100 uL total). Outer wells not used for experiment were filled with 1X PBS to combat evaporation during timelapse studies. Cells were rested overnight in 37°C incubator. 5 nM of bortezomib treatment and 400 ng Hoechst 33258 dye were added on top of BMSC and RPMI collagen-containing wells at a final volume of 200 uL per well. Timelapse was started after addition of drug and Hoechst. The details of this timelapse imaging and analysis methods are published elsewhere.33
Statistical Analysis
qRT-PCR, immunoblot, and substrate zymography data were processed using Prism v.6 (GraphPad Software, San Diego, CA, USA). Student’s t-test was used to derive statistical significance between two groups. Pearson correlation was used to derive significant correlation across multiple groups. Time-lapse data was processed using an open-source software, JEX, to perform quantitative phase imaging, cell identification and tracking from phase images, and quantification of both phase and fluorescence signals.33 Statistical analysis of drug sensitivity was performed in R (‘survminer’ package), utilizing Kaplan Meier survival curve analysis for individual single cell survival times. Log ratio of normalized phase and fluorescence signals were used to represent viability. The details of this analysis method are published elsewhere.33
Results
HAPLN1 expression in BMSCs is increased by co-cultured MM cells
We previously reported that BMSCs established from MM patient bone marrow (BM) aspirates express variable levels of HAPLN1 mRNA22. However, it is not known whether primary MM cells also express HAPLN1. To determine this, qRT-PCR was used to compare HAPLN1 mRNA expression between CD138+ MM cell fraction and the CD138− non-MM cell fraction obtained from MM BM aspirates (Figure 1A). HAPLN1 mRNA expression was minimally detectable in the CD138+ fraction while it was 35 to 65-fold higher in the CD138− fraction (Figure 1B), indicating that HAPLN1 is expressed primarily in CD138- fraction, not CD138+ MM cell fraction. Western blot analysis of conditioned media (CM) obtained from BMSCs grown from a CD138− fraction showed secreted HAPLN1 (Supplementary Figure 1). To determine whether MM cells can influence HAPLN1 expression in BMSCs, we co-cultured BMSCs with RPMI8226 MM cell line for 48 hours and measured HAPLN1 protein expression in the resulting CM (Figure 1C). The presence of RPMI8226 cells increased the level of HAPLN1 detected in BMSC CM (Figure 1D, anti-IG antibody immunoblot). Moreover, upon probing with a HAPLN1-PTR1 reactive antibody, smaller HAPLN1 species were detectable in the CM (Figure 1D, anti-PTR1 antibody immunoblot). Incubation of BMSCs obtained from different MM patients with RPMI8226 cells caused 25 to 150-fold increases in HAPLN1 mRNA expression (Figure 1E). Several other MM cell lines, in addition to RPMI8226, also caused increases in HAPLN1 mRNA expression in co-cultured BMSCs (Figure 1F). These results revealed that MM cells cause BMSCs to increase HAPLN1 expression.
Figure 1: HAPLN1 expression in BMSCs is augmented by MM cells.
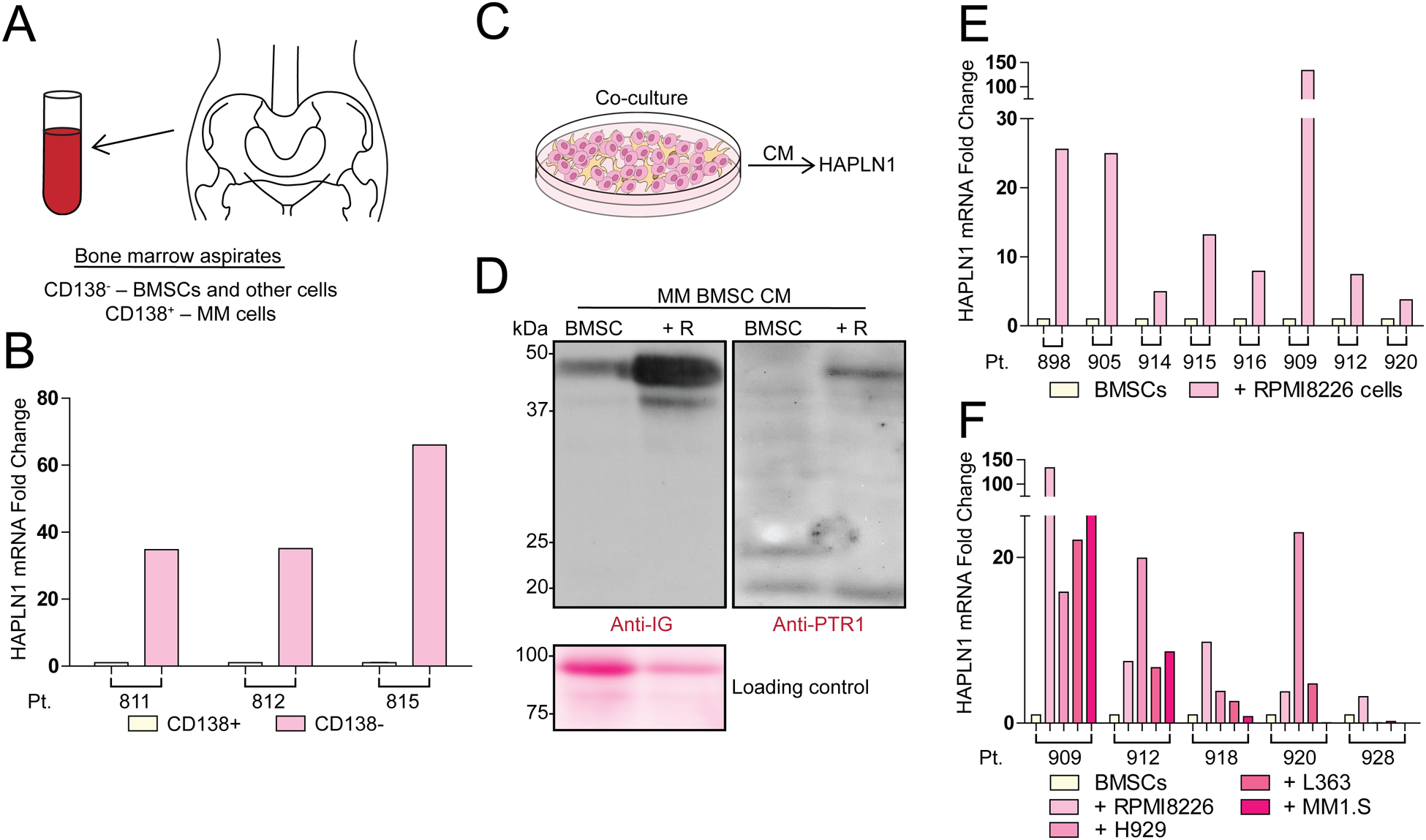
(A) An illustration of MM bone marrow aspirates and CD138+ (MM cells) and CD138− fraction containing BMSCs and other cell types. (B) A graph showing relative HAPLN1 mRNA expression in CD138− and CD138+ cell fractions from 3 MM patient-derived bone marrow aspirates. (C) Experimental design of BMSC and MM cell co-culture conditioned media (CM) collection to measure HAPLN1 secretion. (D) Representative immunoblot of HAPLN1 protein detected in MM BMSC alone (BMSC) and those co-cultured with RPMI8226 MM cells (+R). CM were collected 48 hours after co-culture and probed with Anti-IG (Immunoglobulin-like domain) and Anti-PTR1 (PTR1 domain) antibodies. Ponceau S Red stain used as protein loading control is also shown. Immunoblot results are representative of two technical replicates. (E) A graph showing HAPLN1 mRNA expression in eight MM BMSCs samples with or without RPMI8226 MM cell co-culture. (F) A graph showing HAPLN1 mRNA expression in five MM BMSCs samples, with or without co-culture with RPMI8226, H929, L363, or MM1.S MM cell lines. The mRNA analysis was done in duplicate and normalized to GAPDH mRNA expression. Pt. refers to patient ID.
MMP2 activity is also increased by the presence of MM cells
The causes of smaller HAPLN1 species detected in BMSC CM could be due to secretion of alternative splice forms of HAPLN1 or its proteolytic processing. Several splice variants of HAPLN1 mRNAs are known but none of them would be expected to produce 20–25kDa products detectable by anti-PTR1 domain antibody.23 Thus, we next considered the possibility of the proteolytic HAPLN1 processing as HAPLN1 has been shown to be cleaved by matrix metalloproteinases (MMPs) at sites N-terminal of the IG domain30. To determine if any MMPs were expressed by MM BMSCs, a protease mRNA expression screen was performed by qRT-PCR analysis of MMPs associated with MM in the literature.34 Among these, MMP2 mRNA was found to be expressed at the highest level (Supplementary Figure 2). Next, a gelatin zymography activity assay was employed to measure MMP2 enzymatic activity in CMs obtained from cultured normal and MM-derived BMSCs (Figure 2A). Its activity was significantly higher in CMs of BMSCs established from MM patients compared to those from normal donors (Figure 2B).
Figure 2: MMP2 activity is higher in BMSCs from MM patients than normal donors and is increased by co-cultured MM cells.
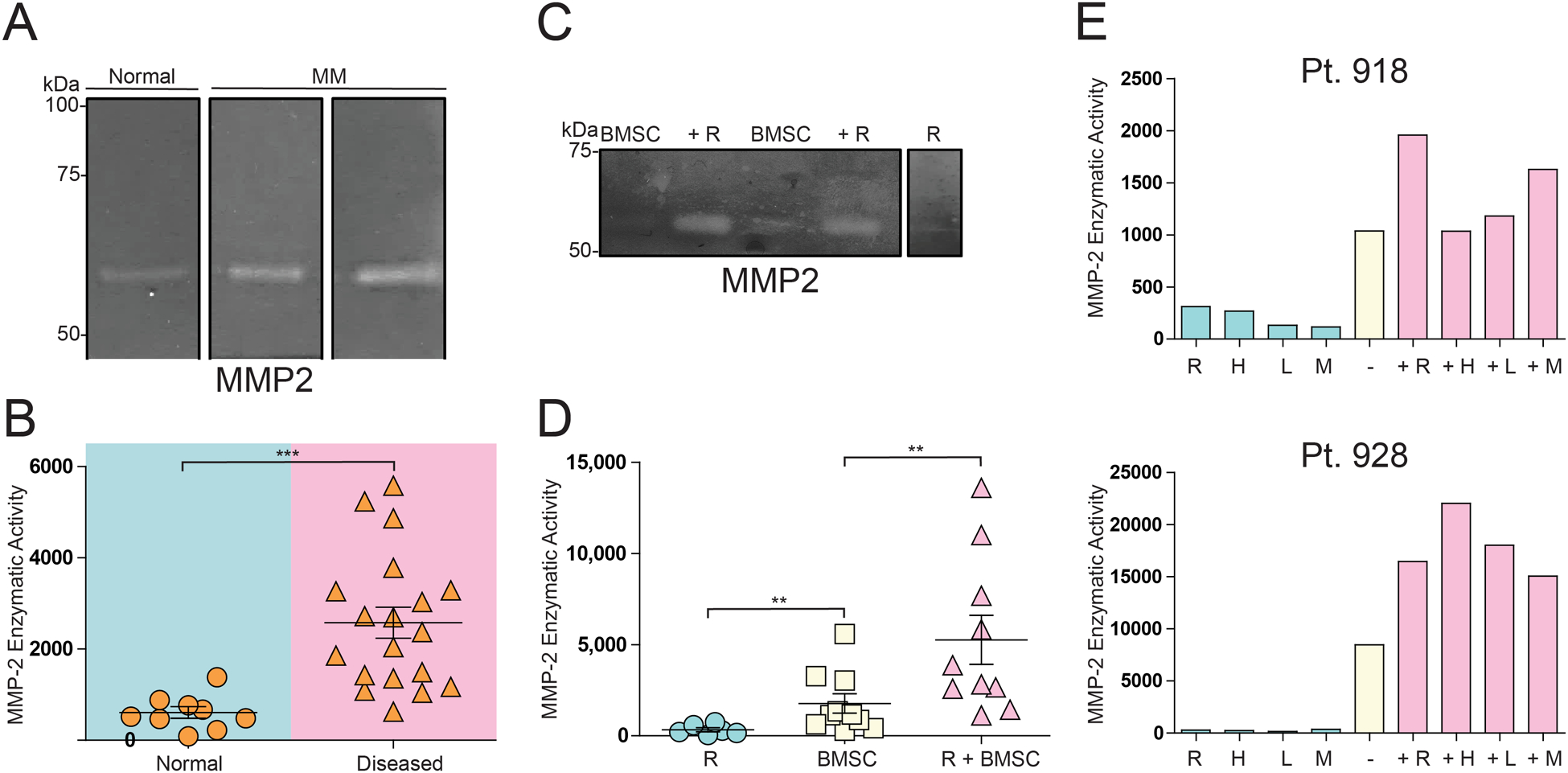
(A) A representative zymogram gel detecting MMP2 activity in 48 hour-CM from BMSCs from normal and MM patient donors. (B) Dot plot of MMP2 activity from zymogram of multiple independent BMSCs samples as in (A). (C) A representative zymogram gel detecting MMP2 activity in CM from BMSCs alone (BMSC), those co-cultured for 48 hours with RPMI8226 cells (+R), or RPMI8226 cells alone (R). (D) Dot plot of MMP2 activity from zymograms from multiple independent samples as in (C). (E) Bar graph of MMP2 enzymatic activity from different MM cell lines CM alone or CM from MM patient 918 or 928 BMSCs co-cultured with different MM cell lines. −, BMSC CM alone; +, with cell lines: R, RPMI8226; H, H929; L, L363; M, MM1.S. Quantification of zymogram activity of MMP2 bands performed by Image-J analysis. In B, ***p<0.001 T-test. In D, **p<0.005 T-test and **p<0.005 one-way ANOVA test.
To test whether MMP2 activity could be regulated by the presence of MM cells, as was the case with HAPLN1 expression (Figure 1D), BMSCs were co-cultured with RPMI8226 cells and MMP2 activity from the co-culture CMs was measured by zymogram (Figure 2C). MMP2 activity was significantly higher in CMs of the co-culture than that in BMSCs alone and was minimally detectable in CM of RPMI8226 cells alone (Figure 2D). To assess whether the increase in MMP2 activity in BMSCs seen in the presence of RPMI8226 cells was limited to the RPMI8226 MM cell line, two patient BMSCs were co-cultured with other MM cell lines and the resulting MMP2 activity was measured as above. MMP2 activity increased in virtually all BMSCs co-culture CMs relative to BMSCs alone CM (Figure 2E). As before, MMP2 activity in CMs from MM cell lines alone only had minor activities. Combined, these results demonstrated that MM cells cause both increased HAPLN1 expression and MMP2 activity in BMSCs.
MMP2 cleaves HAPLN1 into a C-terminal fragment immunoreactive to antibodies against PTR1 and PTR2 domains
We have previously reported a HAPLN1 fragment around 20 kDa that was immunoreactive to antibodies against PTR1 and PTR2, but not IG, domains in the bone marrow plasma fraction of MM patients whose disease was deemed progressive at the time of BM aspirate collection.22 While MMP2 recombinant protein has been reported to cleave HAPLN1 between the signal peptide and the IG domain resulting in a 37kDa product30, the generation of ~20kDa products containing PTR1 and PTR2 domains has yet to be described. To test whether MMP2 is capable of generating ~20kDa HAPLN1 C-terminal fragment containing PTR1 and PTR2 domains, we incubated recombinant full-length HAPLN1 protein with a C-terminal His-tag with recombinant MMP2 protein. Significantly, the full-length protein was cleaved into a ~20 kDa fragment that was detected by antibodies reactive to PTR1, PTR2 and His, but not IG, along with a few intermediate products (Figure 3A). Time course analysis of the reaction showed that the reaction is nearly complete within the first 30 minutes and the ~20kDa product was mostly resistant to further proteolysis (Figure 3B). These results indicated that MMP2 is capable of cleaving HAPLN1 between IG and PTR1 domains and PTR1-PTR2 fragment is mostly resistant to further proteolysis by MMP2 (Figure 3C).
Figure 3: MMP2 generates a C-terminal fragment containing PTR1 and PTR2 domains from full-length HAPLN1.
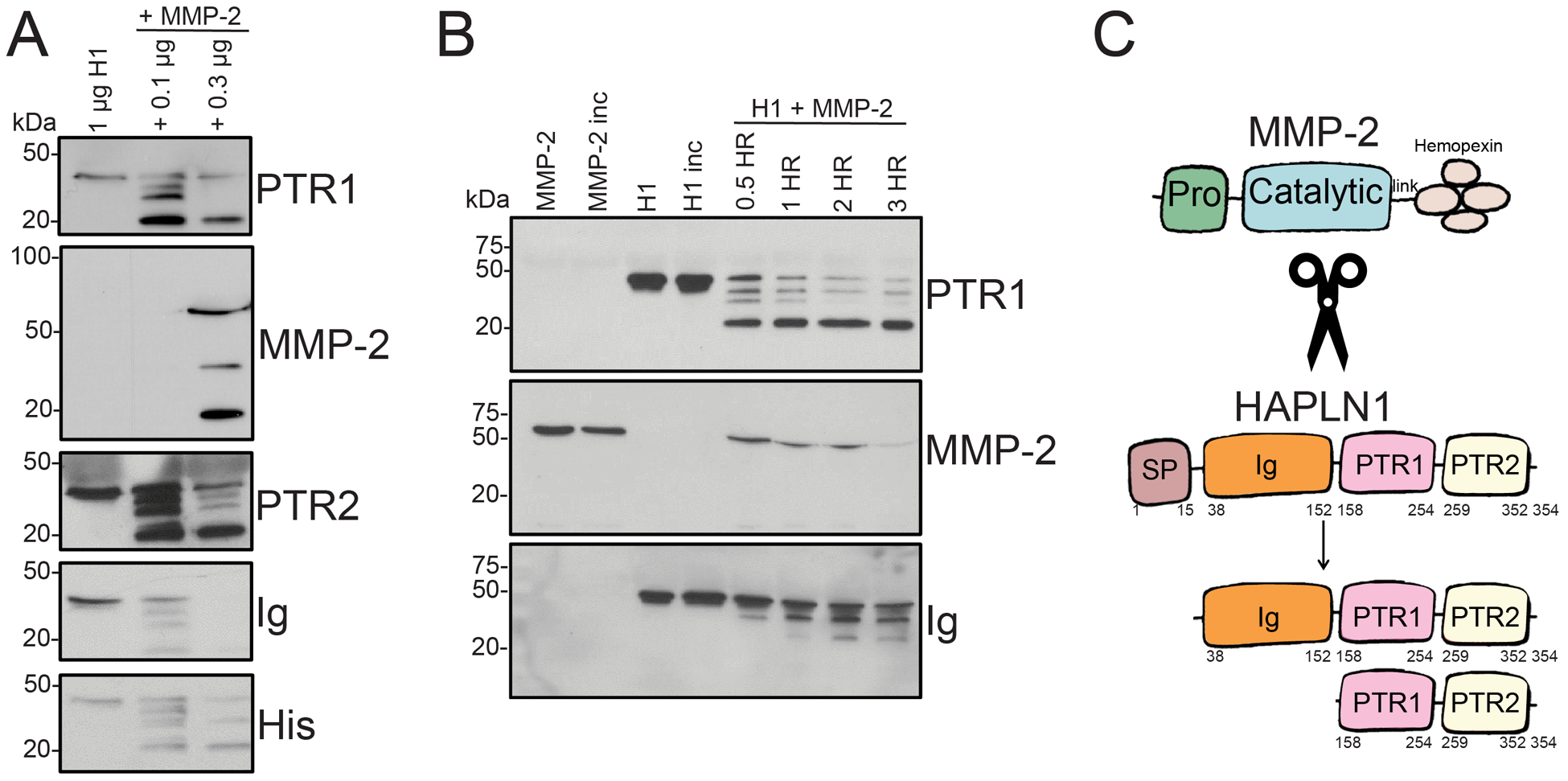
(A) Representative immunoblot of recombinant HAPLN1 protein alone or treated with 0.1 or 0.3 ug recombinant and activate MMP2, collected after 24 hours of reaction. HAPLN1 immunoreactivities were detected via anti-HAPLN1 antibodies, H-93, K-14, and C-14 (anti-IG, anti-PTR1, and anti-PTR2, respectively). MMP2 immunoreactivity was detected via anti-MMP2 antibody, 2C1. C-terminal His tag immunoreactivity was detected via anti-His antibody. (B) Representative immunoblot of recombinant HAPLN1 protein alone or treated with 0.3 ug MMP2, collected after 0.5, 1, 2, and 3 hours of reaction and detected as in (A). (C) Illustration of putative MMP2 cleavage products of HAPLN1 at region between the signal peptide, SP, and immunoglobulin-like, IG, domains, and between the IG and proteoglycan tandem repeat 1, PTR1, domains.
Silencing HAPLN1 and MMP2 expression in BMSCs diminishes BMSC-induced NF-κB activity in MM cells
The results thus far indicate that (i) MM cells increase HAPLN1 mRNA expression and secretion while also increasing the activity of MMP2 in BMSCs and (ii) MMP2 is capable of processing HAPLN1 into a C-terminal ~20kDa fragment that can cause NF-ĸB activation in MM cells.22 Thus, these results suggest that MM cells cause BMSCs to produce increased levels of both HAPLN1 and MMP2 that can act in concert to in turn cause NF-ĸB signaling in MM cells via a MMP2-processed HAPLN1 C-terminal fragment. To test this concept, we looked for a correlation between the level of MMP2 activity in BMSC CMs and that of NF-ĸB signaling capacity in MM cells. As before32, RPMI8226 NF-ĸB activation was increased to variable levels when co-cultured with BMSCs established from MM patients (Figure 4A). When such activities were compared to MMP2 activity in each CMs measured by gelatin zymography, a statistically significant positive correlation was observed (Figure 4B). To delineate if these activities were causally related, we next employed RNA interference to functionally knock down HAPLN1 and MMP2 expression from BMSCs and measured their impacts on NF-ĸB activation in MM cells co-cultured with these knockdown cells. Given that efficient knockdown of genes proved to be challenging in primary BMSCs, we first screened several gene transduction methods and found that lentiviral infection of suspended BMSCs in the presence of protamine yielded most robust and reproducible knockdowns (Supplementary Figure 3A–B; see methods). Flow cytometry analysis confirmed the purity of BMSCs and the lack of alterations in several BMSCs cell surface markers after knockdowns (Supplementary Figure 3C). While HAPLN1 and MMP2 single knockdown BMSCs partially reduced NF-ĸB activation in co-cultured RPMI8226 cells compared to control BMSCs, the double knockdown BMSCs nearly completely lost the activity (Figure 4C). When a similar knockdown experiment was performed on multiple BMSC samples, similarly significant reductions in NF-ĸB activity were observed (Figure 4D, left plot). We noted that some BMSCs caused NF-ĸB activation in MM cells while others did not. Significantly, those BMSCs with higher NF-ĸB inducing activity expressed higher HAPLN1 mRNA when cultured with RPMI8226 cells than those without such activities (Supplementary Figure 4A and 4B). When these patient BMSC samples were separately analyzed based on HAPLN1 expression, BMSCs with higher HAPLN1 expression induced NF-ĸB activity in co-cultured RPMI8226 cells to a significantly greater degree than BMSCs with lower HAPLN1 expression. Moreover, knockdown of HAPLN1 and MMP2 in those with higher HAPLN1 expression reduced NF-ĸB activity in co-cultured RPMI8226 cells to a significantly greater degree than BMSCs with lower HAPLN1 expression (Figure 4D, right plots). The reductions in NF-kB activity in MM cells co-cultured with BMSCs with reduced HAPLN1 and MMP2 expression also correlated with a reduction of two known NF-ĸB target genes, IL8 and Bcl2a1 (Supplementary Figure 5). Thus, expression of both HAPLN1 and MMP2 in BMSCs is necessary for BMSCs to induce NF-ĸB activation in MM cells, particularly in those BMSCs that express relatively high HAPLN1 levels following co-culture with MM cells.
Figure 4: Reductions of HAPLN1 and MMP2 expression in BMSCs diminish BMSC’s capacity to increase NF-κB activity in MM cells.
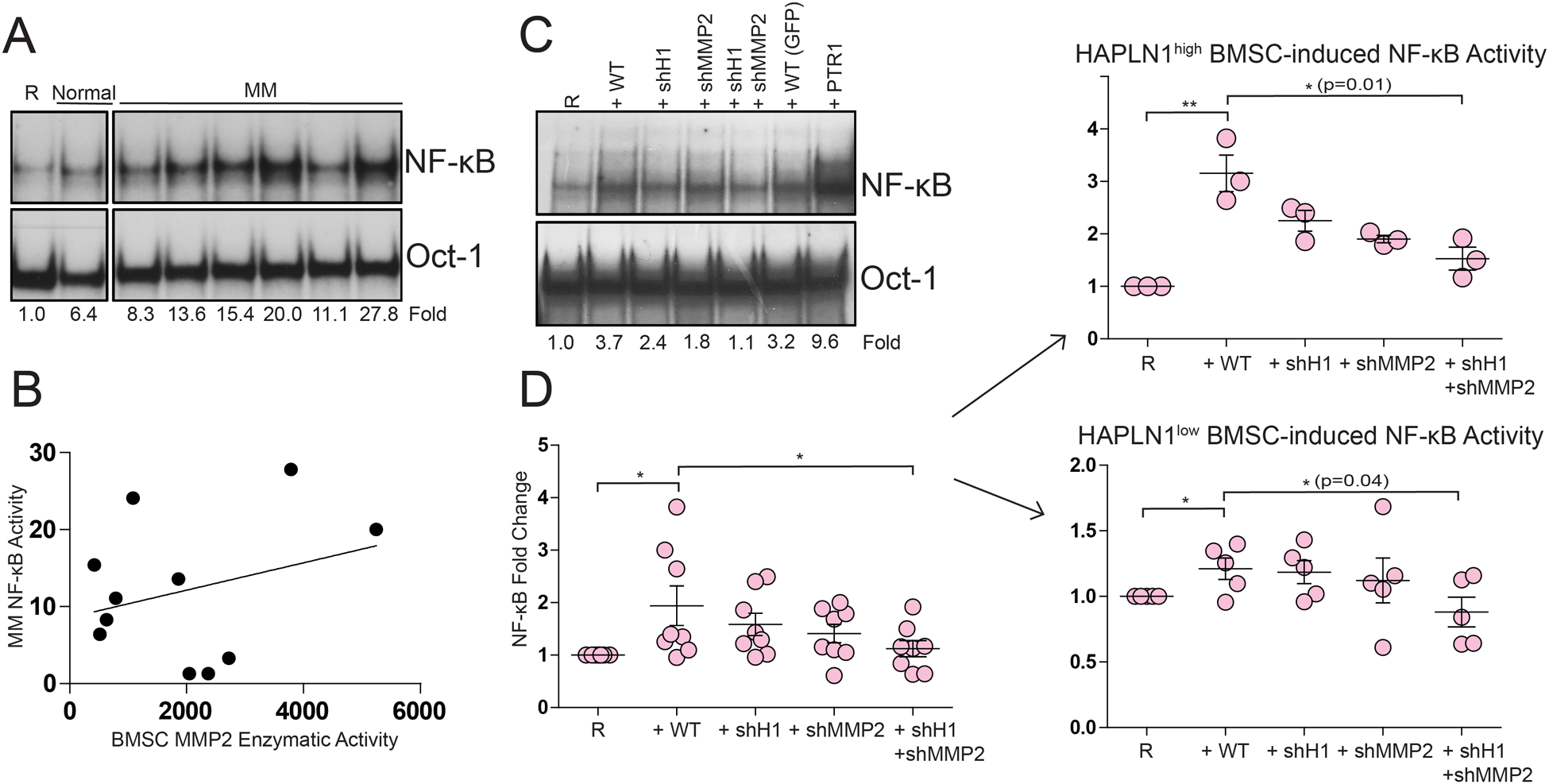
(A) Representative EMSA gels of NF-ĸB and Oct-1 DNA-binding in RPMI8226 cells co-cultured for 24 hours without (R) or with BMSCs from normal donors (Normal) or MM patients (MM). NF-ĸB and Oct-1 DNA-binding activities were quantified via Phosphoimager and the fold-change in NF-ĸB activity normalized to the Oct-1 loading control are shown below the gels. (B) NF-ĸB activity detected in RPMI8226 cells co-cultured with MM BMSCs as in (A) was plotted against MMP2 activity in the CMs detected by zymography as in Figure 2A. *p=0.05, logistic regression test; *p=0.04, Spearman rank-order correlation; r2=0.4. (C) Representative EMSA gels of NF-ĸB and Oct-1 DNA-binding in RPMI8226 cells co-cultured for 24 hours without (R) or with control BMSCs transduced with irrelevant GFP lentiviral particles (WT) or those with HAPLN1 and/or MMP2 knockdowns (+shH1, HAPLN1 knockdown; +shMMP2, MMP2 knockdown; +shH1+shMMP2, HAPLN1 and MMP2 double knockdown; +PTR1, 100 nM MBP-PTR1 protein (positive control). (D) Dot plots depicting NF-ĸB DNA-binding fold-change of RPMI8226 cells alone (R) or those treated with indicated control or knockdown BMSCs from eight MM patients and analyzed as in (C). *p<0.05 T-test. Plot stratified to HAPLN1high (upper right panel) and HAPLN1low BMSCs (lower right panel). *p<0.05, **p<0.005 T-test (upper right); *p<0.05 T-test (lower right).
Loss of HAPLN1 and MMP2 also diminishes BMSC-mediated protection of MM cells against bortezomib toxicity
As reductions of HAPLN1 and MMP2 in BMSCs reduced NF-ĸB activation in co-cultured MM cells, we next asked whether BMSC’s ability to protect MM cells from bortezomib-induced toxicity is also reduced by their combined deficiencies. To this end, we previously developed a time-lapse cell viability assay to track single cells undergoing apoptosis up to a period of 72 hours and to statistically measure the rate at which the cells die when introduced to bortezomib.33 RPMI8226 cells were cultured in collagen in the presence or absence of BMSCs with or without bortezomib. Over 48 hours, the fates of individual MM cells were tracked by measuring cell phase intensity and cell nuclear intensity based on Hoechst 33258 staining. Survival curves were then generated from these individual cell fate measurements. This analysis showed that >80% of RPMI8226 cells survived without any drug treatment during the 48-hour period and the presence of BMSCs did not improve this basal survival probability (Figure 5A). In contrast, RPMI8226 cells treated with bortezomib alone had the lowest survival probability, and the presence of BMSCs significantly increased the survival probability in the presence of bortezomib (Figure 5A and B). BMSCs with combined HAPLN1 and MMP2 knockdown were then incorporated into the time-lapse and compared to wildtype BMSCs. While wildtype BMSCs and HAPLN1 and MMP2 knockdown BMSCs did not show significant differences in their abilities to improve basal survival probability in RPMI8226 cells, MM cells treated with HAPLN1 and MMP2 knockdown BMSCs and bortezomib had a significantly less survival probability than ones treated with wildtype BMSCs (Figure 5C and D). Collectively, these results revealed that MM cells can cause increased HAPLN1 expression and MMP2 activity in BMSCs, which in turn promote increased NF-ĸB activity and bortezomib resistance in MM cells.
Figure 5: Loss of HAPLN1 and MMP2 diminishes BMSC protection against MM bortezomib toxicity.
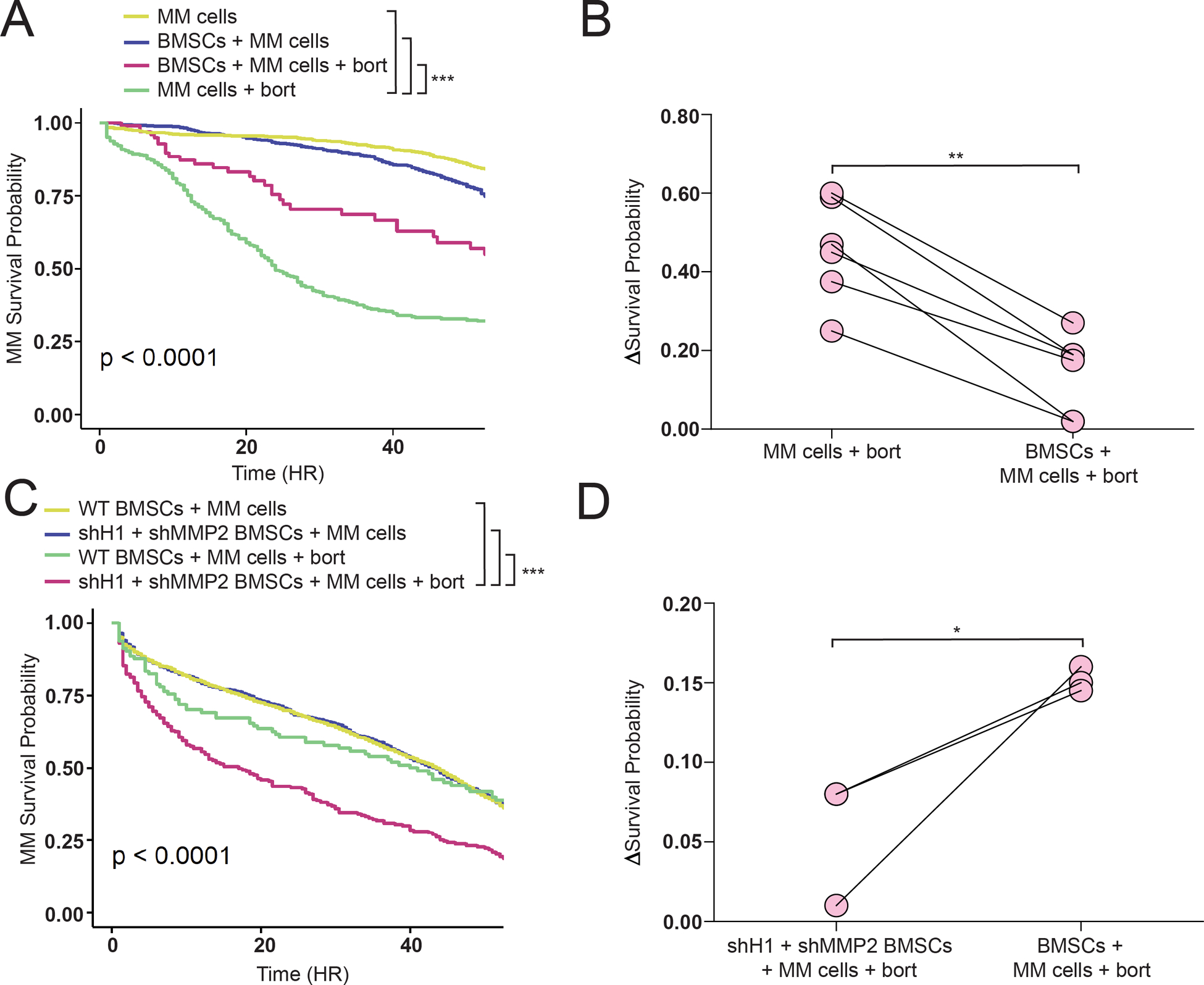
(A) Representative survival graph of RPMI8226 cells alone or co-cultured with MM BMSCs, treated or not with 4 nM bortezomib (+bort), and analyzed by time lapse imaging as in the materials and methods section and in Mark et al.33 (B) Statistical analysis of experiments replicated in (A) representing the change in survival probability between RPMI8226 cells co-cultured with or without BMSCs derived from 5 patient samples. (C) Representative survival graph of RPMI8226 cells as in (A), except for BMSCs with or without HAPLN1 and MMP2 knockdowns (shH1+shMMP2). ***p<0.0005, Kaplan Meier analysis in (A) and (B). (D) Statistical analysis of experiments replicated in (C) representing the change in survival probability between RPMI8226 cells co-cultured with or without wildtype or shH1+shMMP2 BMSCs derived from 3 patient samples. R2=0.89, **p<0.005 in (B); R2=0.45, *p<0.05 in (D), both T-test.
Discussion
In our previous study, we identified HAPLN1 as a BMSC-derived ECM factor whose C-terminal PTR1 and PTR2 domains, but not full-length form, robustly activated NF-ĸB in MM cells.22 In the present study, we found: (i) MM cells increase HAPLN1 expression and MMP2 activity in co-cultured BMSCs; (ii) BMSCs from MM patients display higher MMP2 activity than those generated from normal BM samples; (iii) MMP2 activity in BMSC CM positively correlates with the ability of BMSCs to increase NF-ĸB activity in co-cultured MM cells; (iv) MMP2 is capable of generating C-terminal HAPLN1 fragments containing PTR1 and PTR2 domains; and (v) reductions in expression of HAPLN1 and MMP2 in BMSCs reduces NF-ĸB activation and bortezomib-induced toxicity in co-cultured MM cells. Together, these results support the model in which MMP2 proteolytically cleaves HAPLN1 into a novel matrikine capable of increasing NF-ĸB activity and bortezomib resistance in MM cells (Figure 6). In addition, they also reveal a novel bi-directional regulatory relationship between MM cells and BMSCs wherein MM cells instruct BMSCs to produce a matrikine that in turn benefits MM cell drug resistance.
Figure 6: A model depicting bidirectional signaling relationship between MM cells and BMSCs involving HAPLN1 and MMP2.
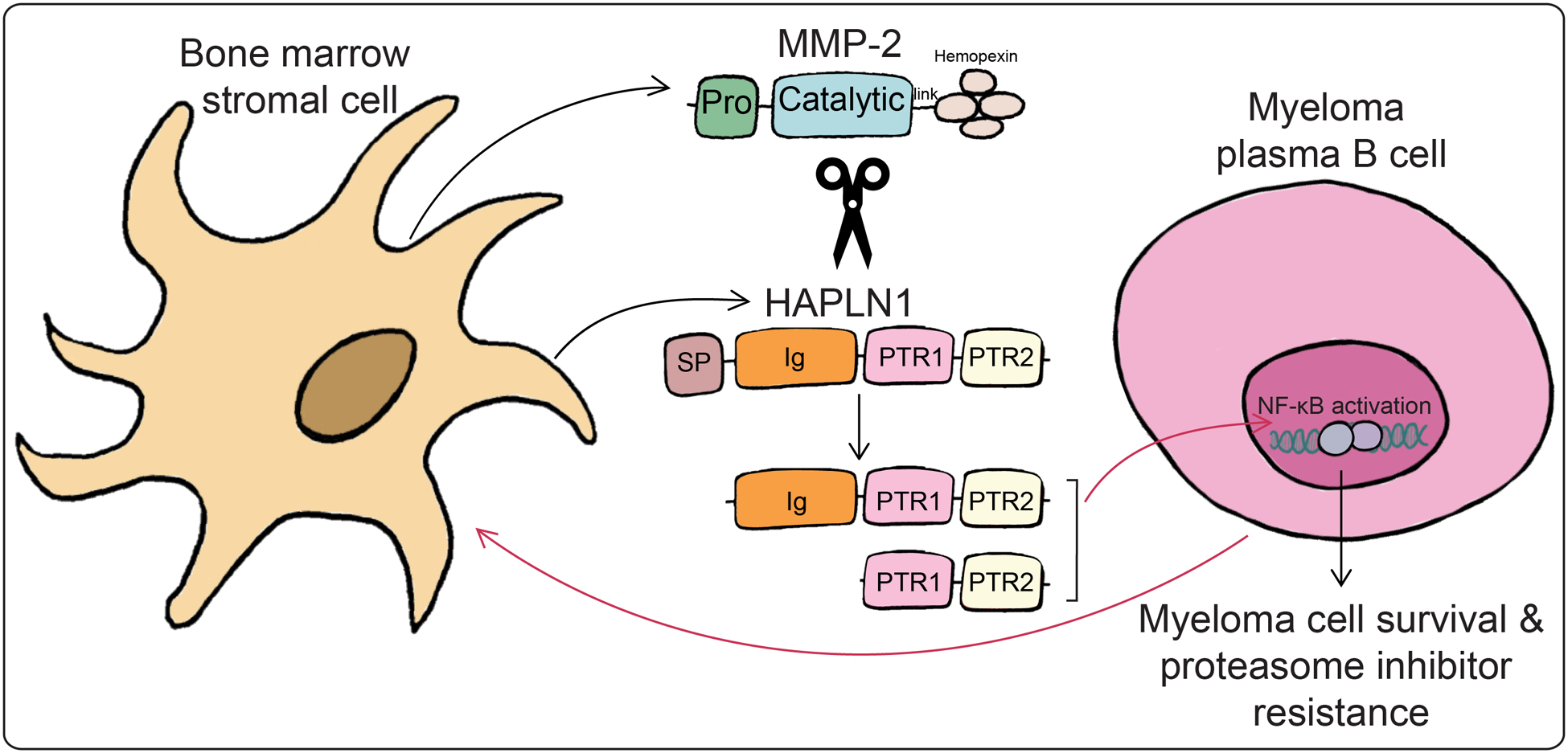
MM cells influence BMSCs to produce HAPLN1 and MMP2, leading to cleaved C-terminal HAPLN1 products, which are capable of inducing NF-ĸB activation and bortezomib resistance in MM cells.
A pathological role of HAPLN1 is not unique to MM disease but has also been implicated in various other diseases. For example, in aging lymphatic ECM, loss of HAPLN1 permitted melanoma cells to escape from lymphatic sites to distal metastatic ones.27 In hepatocellular carcinoma, expression of HAPLN1 in cancer cells themselves is linked to cancer cell stemness via β-catenin-related mechanism.28 HAPLN1 polymorphism is a marker for susceptibility to ankylosing spondylitis, an inflammatory disease that can result in vertebrate fusion.35 Moreover, proteolytic generation of short HAPLN1 fragments N-terminal to the IG domain have been reported to yield signaling peptides which can bind to bone morphogenic protein (BMP) type II receptor or stimulate synthesis of cartilage proteoglycans.36 The HAPLN1 N-terminal fragments are also suggested to participate in asbestos-induced mesothelioma as mesothelioma cells transfected with HAPLN1 plasmids lacking PTR1 and PTR2 domains increased in proliferation compared to vehicle-transfected cells.37 In contrast, we previously reported that the C-terminal HAPLN1 products containing PTR1 and/or PTR2 domains can induce NF-kB signaling and bortezomib resistance in MM cells.22 However, how an active C-terminal HAPLN1 fragment was generated from the inactive full-length HAPLN1 remained obscure. Thus, to our knowledge, the MMP2-processed HAPLN1 product we report here is the first example of a matrikine capable of inducing NF-ĸB signaling and drug resistance in MM cells. Our study also suggests the possibility that some of HAPLN1’s role in pro-tumorigenic and pro-inflammatory actions reported above could be related to its ability to induce NF-ĸB signaling following proteolytic processing.
In addition to HAPLN1, a new pathological role of MMP2 that we uncovered here in MM disease also adds to growing evidence for MMP2’s role in MM and other malignancies. In nasopharyngeal carcinoma and breast cancer, for instance, MMP2 is responsible for promoting tumor invasion and metastasis through degradation of collagen IV, a major constituent of basement membrane.38 In MM, osteoblasts are reported to increase MMP2 expression in MM cells, which then leads to subsequent bone destruction.39 Inhibition of MMP2 by bone-seeking bisphosphonate based MMP-2 specific inhibitors decreased myeloma tumor burden in a U266 mouse model and reduced MM induced bone loss in the 5TGM1 model.31 Moreover, endothelial cells are reported to secrete MMP2 which aids in MM cell dissemination and invasion.40 Other groups have linked the increase of MMP1 production in BMSCs after being stimulated by MM cells to increase collagen I degradation in the BM through induction of myeloma cell-derived MMP7, which can then activate MMP2 through specific cleavage.41 BMSCs exposed to the chemotherapeutic drug VP16 increase in MMP2 expression followed by diminished expression of CXCL12, an MMP2 receptor, and reduced chemotactic support. This reduction of BMSC support of hematopoietic cell migration can be recovered by feeding BMSCs with physiologically relevant concentrations of recombinant MMP2 protein.42 Despite these prior studies, the role of MMP2 in generating a matrikine in MM disease is unprecedented. Therefore, processing of HAPLN1 to generate an NF-ĸB signaling and drug resistance factor appears to represent a new function for MMP2 in any malignancy.
The findings reported here contribute to our overall understanding of how BMSCs and MM cells behave in a coordinated manner by utilizing a BMSC-processed ECM protein to facilitate MM disease progression. Our study suggests that the levels of HAPLN1 and MMP2 expression have a causal relationship with the degree of NF-ĸB activation and BMSC-induced protection from bortezomib. Although we do not know the nature of factor(s) produced by MM cells that causes HAPLN1 and MMP2 increases in BMSCs, a preliminary study suggests that BMSC-MM cell contact is not an absolute requirement for MM cells to increase the MMP2 activity in BMSCs (Supplementary Figure 6). In future studies, it would be of great interest to identify the nature of the MM cell produced factor(s) that increases HAPLN1 expression and MMP2 activity in BMSCs to generate such a matrikine. In addition to HAPLN1, versican, a proteoglycan EMC protein related to HAPLN1, was also found to be processed by ADAM-TS proteases into a matrikine, versikine, which facilitates immune-sensing of MM cells.43 Together, these findings provide new insights into how ECM-derived matrikines may contribute to MM disease progression via acting directly on MM cells or the MM immune environment.
Despite current MM therapies, patients have a median survival of 5–7 years after diagnosis due to intrinsic or acquired drug resistance.22 Therefore, the findings in this study highlight the novel roles of HAPLN1 and MMP2 as MM pathologic drivers, and suggest the possibility to utilize them as two measurable markers to distinguish patients who may present as bortezomib resistant. Moreover, elucidation of the regulatory relationship between HAPLN1 and MMP2 may allow for the development of new therapeutic targets in MM.2,44 Thus, HAPLN1 and MMP2, along with a novel HAPLN1 matrikine, may serve as new biomarkers or therapeutic targets for addressing bortezomib resistance problems in MM patients.
Supplementary Material
Implications:
HAPLN1 and MMP2 produced by BMSCs obtained from MM patients promote NF-ĸB activity and resistance to bortezomib toxicity in MM cells, uncovering their potential as biomarkers or therapeutic targets to address bortezomib resistance in MM patients.
Acknowledgments
This work was funded by Cancer Biology Training Grant T32 CA009135 NRSA Award (to CM), Collaborative Health Sciences Program Award #AAB7473 under Wisconsin Partnership Program (to SM and JW), NIH/NCI R01 CA155192 and R01 CA251595 (to SM), A RIDE Scholar Award (to SM), and a pilot fund from the UW Carbone Cancer Center Grant P30 CA014520 (to SM). We thank the members of the Miyamoto laboratory for helpful comments on the project and the manuscript. We also thank Dr. Caroline Alexander for helpful discussions and suggestions regarding MMPs and the donors of normal and MM bone marrow samples.
Financial Support:
This work was funded by Cancer Biology Training Grant T32 CA009135 NRSA Award (to Christina Mark), Collaborative Health Sciences Program Award #AAB7473 under Wisconsin Partnership Program (to Shigeki Miyamoto and Jay Warrick), NIH/NCI R01 CA155192 and R01 CA251595 (to Shigeki Miyamoto), A RIDE Scholar Award (to Shigeki Miyamoto), Trillium Fund (to Shigeki Miyamoto, Natalie Callander, and Peiman Hematti), and a pilot fund from the UW Carbone Cancer Center Grant P30 CA014520 (to Shigeki Miyamoto).
Footnotes
Conflict of Interest: The authors have no conflicts of interest
References
- 1.Hideshima T, Mitsiades C, Tonon G, Richardson PG & Anderson KC Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nature Reviews Cancer 7, 585–598 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Kumar SK & Rajkumar SV The multiple myelomas — current concepts in cytogenetic classification and therapy. Nature Reviews Clinical Oncology 15, 409–421 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Myeloma - Cancer Stat Facts. SEER; https://seer.cancer.gov/statfacts/html/mulmy.html. [Google Scholar]
- 4.Chapman MA et al. Initial genome sequencing and analysis of multiple myeloma. Nature 471, 467–472 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keats JJ et al. Promiscuous Mutations Activate the Non-Canonical NF-kB Pathway in Multiple Myeloma. Cancer Cell 12, 131–144 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Staudt LM Oncogenic Activation of NF-κB. Cold Spring Harb Perspect Biol 2, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gilmore TD Multiple Myeloma: Lusting for NF-κB. Cancer Cell 12, 95–97 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Perkins ND The diverse and complex roles of NF-κB subunits in cancer. Nat Rev Cancer 12, 121–132 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Annunziata CM et al. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 12, 115–130 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bharti AC et al. Nuclear factor–κB and STAT3 are constitutively active in CD138+ cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood 103, 3175–3184 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Moser-Katz T, Joseph NS, Dhodapkar MV, Lee KP & Boise LH Game of Bones: How Myeloma Manipulates Its Microenvironment. Frontiers in Oncology 10, 3386 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gupta VA et al. Bone marrow microenvironment–derived signals induce Mcl-1 dependence in multiple myeloma. Blood 129, 1969–1979 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rellick SL et al. Melphalan exposure induces an interleukin-6 deficit in bone marrow stromal cells and osteoblasts. Cytokine 58, 245–252 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furukawa M et al. Autocrine and Paracrine Interactions between Multiple Myeloma Cells and Bone Marrow Stromal Cells by Growth Arrest-specific Gene 6 Cross-talk with Interleukin-6. J. Biol. Chem 292, 4280–4292 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boise LH & Shanmugam M Stromal Support of Metabolic Function through Mitochondrial Transfer in Multiple Myeloma. Cancer Res 79, 2102–2103 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Mekhloufi A et al. Bone Marrow Stromal Cell-Derived IL-8 Upregulates PVR Expression on Multiple Myeloma Cells via NF-kB Transcription Factor. Cancers (Basel) 12, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garayoa M et al. Mesenchymal stem cells from multiple myeloma patients display distinct genomic profile as compared with those from normal donors. Leukemia 23, 1515–1527 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Hideshima T & Anderson KC Signaling Pathway Mediating Myeloma Cell Growth and Survival. Cancers 13, 216 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson KC Proteasome Inhibitors in Multiple Myeloma. Seminars in Oncology 36, S20–S26 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Markovina S et al. Bortezomib-Resistant Nuclear Factor-κB Activity in Multiple Myeloma Cells. Mol Cancer Res 6, 1356–1364 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hideshima T et al. Bortezomib induces canonical nuclear factor-κB activation in multiple myeloma cells. Blood 114, 1046–1052 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huynh M et al. Hyaluronan and proteoglycan link protein 1 (HAPLN1) activates bortezomib-resistant NF-κB activity and increases drug resistance in multiple myeloma. J. Biol. Chem 293, 2452–2465 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spicer AP, Joo A & Bowling RA A Hyaluronan Binding Link Protein Gene Family Whose Members Are Physically Linked Adjacent to Chrondroitin Sulfate Proteoglycan Core Protein Genes THE MISSING LINKS. J. Biol. Chem 278, 21083–21091 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Doege K, Hassell JR, Caterson B & Yamada Y Link protein cDNA sequence reveals a tandemly repeated protein structure. Proc Natl Acad Sci U S A 83, 3761–3765 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perkins SJ, Nealis AS, Dudhia J & Hardingham TE Immunoglobulin fold and tandem repeat structures in proteoglycan N-terminal domains and link protein. Journal of Molecular Biology 206, 737–748 (1989). [DOI] [PubMed] [Google Scholar]
- 26.Watanabe H & Yamada Y Mice lacking link protein develop dwarfism and craniofacial abnormalities. Nat Genet 21, 225–229 (1999). [DOI] [PubMed] [Google Scholar]
- 27.Ecker BL et al. Age-Related Changes in HAPLN1 Increase Lymphatic Permeability and Affect Routes of Melanoma Metastasis. Cancer Discov 9, 82–95 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mebarki S et al. De novo HAPLN1 expression hallmarks Wnt-induced stem cell and fibrogenic networks leading to aggressive human hepatocellular carcinomas. Oncotarget 7, 39026–39043 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ashktorab H et al. Distinct Genetic Alterations in Colorectal Cancer. PLoS ONE 5, e8879 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen Q, Murphy G, Hughes CE, Mort JS & Roughley PJ Matrix metalloproteinases cleave at two distinct sites on human cartilage link protein. Biochem J 295, 595–598 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shay G et al. Selective inhibition of matrix metalloproteinase-2 in the multiple myeloma-bone microenvironment. Oncotarget 8, 41827–41840 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Markovina S et al. Bone marrow stromal cells from multiple myeloma patients uniquely induce bortezomib resistant NF-κB activity in myeloma cells. Molecular Cancer 9, 176 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mark C, Callander N, Chng K, & Warrick J & Miyamoto S Timelapse viability assay to detect division and death of primary multiple myeloma cells in response to drug treatments with single cell resolution. Integ. Biol (2022) in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tauro M, McGuire J & Lynch CC New approaches to selectively target cancer-associated matrix metalloproteinase activity. Cancer Metastasis Rev 33, 1043–1057 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Lin Z et al. A genome-wide association study in Han Chinese identifies new susceptibility loci for ankylosing spondylitis. Nature Genetics 44, 73–77 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Wang Z, Weitzmann MN, Sangadala S, Hutton WC & Yoon ST Link Protein N-terminal Peptide Binds to Bone Morphogenetic Protein (BMP) Type II Receptor and Drives Matrix Protein Expression in Rabbit Intervertebral Disc Cells. J Biol Chem 288, 28243–28253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ivanova AV et al. Protumorigenic Role of HAPLN1 and Its IgV Domain in Malignant Pleural Mesothelioma. Clin Cancer Res 15, 2602–2611 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu X et al. Matrix Metalloproteinase-2 Contributes to Cancer Cell Migration on Collagen. Cancer Res 65, 130–136 (2005). [PubMed] [Google Scholar]
- 39.Hecht M et al. Osteoblasts promote migration and invasion of myeloma cells through upregulation of matrix metalloproteinases, urokinase plasminogen activator, hepatocyte growth factor and activation of p38 MAPK. British Journal of Haematology 138, 446–458 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Vacca A et al. Endothelial cells in the bone marrow of patients with multiple myeloma. Blood 102, 3340–3348 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Barille S & Harousseau J-L Metalloproteinases in Multiple Myeloma: Production of Matrix Metalloproteinase-9 (MMP-9), Activation of proMMP-2, and Induction of MMP-1 by Myeloma Cells. 8. [PubMed] [Google Scholar]
- 42.Clutter SD, Fortney J & Gibson LF MMP-2 Is Required for Bone Marrow Stromal Cell Support of Chemotaxis. Exp Hematol 33, 1192–1200 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hope C et al. Immunoregulatory roles of versican proteolysis in the myeloma microenvironment. Blood 128, 680–685 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chauhan D, Hideshima T, Mitsiades C, Richardson P & Anderson KC Proteasome inhibitor therapy in multiple myeloma. Mol. Cancer Ther 4, 686–692 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.