

Abstract
No Special Type breast cancer (NST; commonly known as Invasive Ductal Carcinoma (IDC)) and Invasive Lobular Carcinoma (ILC) are the two major histological subtypes of breast cancer with significant differences in clinicopathological and molecular characteristics. The defining pathognomonic feature of ILC is loss of cellular adhesion protein, E-cadherin (CDH1). We have previously shown that E-cadherin functions as a negative regulator of the Insulin-like Growth Factor 1 Receptor (IGF1R) and propose that E-cadherin loss in ILC sensitizes cells to growth factor signaling which thus alters their sensitivity to growth factor signaling inhibitors and their downstream activators. To investigate this potential therapeutic vulnerability, we generated CRISPR-mediated CDH1 knockout (CDH1 KO) IDC cell lines (MCF7, T47D, ZR75.1) to uncover the mechanism by which loss of E-cadherin results in IGF pathway activation. CDH1 KO cells demonstrated enhanced invasion and migration that was further elevated in response to IGF1, serum and Collagen I. CDH1 KO cells exhibited increased sensitivity to IGF resulting in elevated downstream signaling. Despite minimal differences in membranous IGF1R levels between wildtype (WT) and CDH1 KO cells, significantly higher ligand-receptor interaction was observed in the CDH1 KO cells, potentially conferring enhanced downstream signaling activation. Critically, increased sensitivity to IGF1R, PI3K, Akt and MEK inhibitors was observed in CDH1 KO cells and ILC patient-derived organoids. Implications: Overall, this suggests that these targets require further exploration in ILC treatment and that CDH1 loss may be exploited as a biomarker of response for patient stratification.
Keywords: Invasive lobular carcinoma, E-cadherin, IGF, receptor availability, patient-derived organoids
Introduction
Despite significant diagnostic and therapeutic advancements, breast cancer remains a leading cause of malignancy associated mortality for women worldwide. Breast cancer is a heterogenous disease that can be categorized based on its histopathology and molecular features. The major histological subtypes of breast cancer are No Special Type (NST) which is often referred to as Invasive Ductal Carcinoma (IDC: ~80% of all cases) and Invasive Lobular Carcinoma (ILC: ~10–15% of all cases) (1–3). Although IDC and ILC are clinically managed as the same disease, there is an increasing appreciation of their differences in terms of molecular alterations, pathology, and prognosis (4–7). The diminished prevalence of ILC consequently has resulted in this subtype being historically understudied and therefore implores the need to better characterize the disease to establish precision medicine approaches to treatment (4, 8). ILC tumors are largely characterized by their growth in a single file, discohesive manner with presence of stromal infiltration (5–7, 9). Approximately 95% of ILC cases present with loss of CDH1, with the majority due to genomic alterations such as loss of heterozygosity and/or truncating frameshift mutations (6, 10, 11). In addition, other molecular alterations such as PIK3CA activation, PTEN loss, and mutations in TBX3 and FOXA1 are detected at higher levels in ILC tumors compared to IDC tumors (4, 12). Enhanced Akt signaling has also been observed in several ILC cell line and mouse models (13).
As one of the activators of Akt, the Insulin-like Growth Factor 1 (IGF1) pathway has long been associated with breast cancer progression (14–18). IGF1 is an essential component in cellular proliferation and survival (15, 19). Binding of IGF1 to its receptor (IGF1R) or the insulin receptor (IR) stimulates autophosphorylation and activation of the PI3K and MAPK pathways among others (15, 18, 20). Due to high levels of circulating IGF1 being associated with poor prognosis in breast cancer (21), it is imperative to understand the dysregulation of the IGF1 pathway in tumors. Numerous IGF1R inhibitors have been evaluated in preclinical and clinical settings, including humanized monoclonal antibodies that downregulate the receptor and small molecule tyrosine kinase inhibitors which inhibit pathway activation (20, 22–26). Unfortunately, these trials demonstrated little to no response in the recruited broad patient populations despite promising preclinical efficacy (13, 27–31), suggesting the necessity to better stratify patients to ensure efficient validation of therapies while maximizing response metrics.
To that end, we recently reported E-cadherin as a modulator of IGF1 signaling and a potential biomarker of inhibitor response (32). Briefly, transient E-cadherin knockdown sensitized cell lines to IGF1 signaling and to IGF1R inhibition (32, 33), and we further reported that ILC tumors have higher IGF1/2 expression and pIGF1R/IR activation compared to IDC tumors (13, 33). In this present study, we have expanded upon our earlier observations by generating CRISPR-mediated CDH1 knockout (CDH1 KO) IDC cell lines to elucidate the role of E-cadherin individually in more detail. Here we report that loss of E-cadherin renders cells sensitive to IGF1, IGF2 and insulin signaling by increasing IGF1R receptor availability for ligand binding resulting in enhanced cell migration/invasion and contextual increases in sensitivity to IGF1R, PI3K, Akt and MEK inhibitors. Given previous evidence for E-cadherin regulation of other growth factor pathways (13, 30), we also extended our analyses to additional growth factor signaling pathways. Our findings provide insights into better understanding the mechanics of E-cadherin function as well as the potential for a targeted therapeutic intervention in ILC patient populations.
Materials and Methods
Cell culture
Cell lines utilized in this study were obtained from ATCC: MCF7 (RRID: CVCL_0031), T47D (RRID: CVCL_0553), ZR75.1 (RRID: CVCL_0588), MDA-MB-134-VI (RRID: CVCL_0617), MDA-MB-231 (RRID: CVCL_0062) and Asterand for SUM44PE (RRID: CVCL_3424). Cell lines were maintained in 10% fetal bovine serum (FBS; Life Technologies) supplemented media (Thermo Fisher Scientific): MDA-MB-134 in 1:1 DMEM: L-15; MCF7 and MDA-MB-231 in DMEM; and T47D and ZR75.1 in RPMI. SUM44PE was maintained in DMEM/F12 with 2% charcoal stripped serum (CSS; Life Technologies) with additional supplements as previously described (5). Cell lines were cultured for less than 6 months at a time, routinely tested to be Mycoplasma free and authenticated by the University of Arizona Genetics Core (Tucson, Arizona) by short tandem repeat DNA profiling.
CDH1 knockout cell line generation
CRISPR mediated knockout of CDH1 in MCF7 and T47D cells was performed by utilizing the Gene Knockout Kit (V1) from Synthego (Redwood City, California) as previously described (31, 34). The parental cell line was used a comparator for MCF7 and T47D, referred as wildtype (WT). For ZR75.1, a doxycycline inducible Cas9 was utilized harboring either short guide RNA (sgRNA) for CDH1 or a non-targeting control (NTC) sequence and was introduced via an adenoviral vector. For ZR75.1, a total of 3 rounds of infection and Puromycin selection was performed before single cell sorting. 8 clones from each KO and NTC cells were isolated by single cell cloning and combined to generate a pool for subsequent experiments.
Immunoblotting
Protein for immunoblotting experiments were harvested and analyzed as described previously (33). Membrane blocking was performed with Intercept PBS blocking buffer (LiCOr #927–40000) for one hour at room temperature and probed with primary antibodies overnight at 4°C: pIGF1R/IR (Cell Signaling Technology #3024; RRID:AB_331253), IGF1R (Cell Signaling Technology #3027; RRID:AB_2122378), pAkt S473 (Cell Signaling Technology #4060; RRID:AB_2315049), Akt (Cell Signaling Technology #9272; RRID:AB_329827), InsR (Cell Signaling Technology #3025; RRID:AB_2280448), E-cadherin (BD Biosciences #610182; RRID:AB_397581), β-catenin (BD Biosciences #610154; RRID:AB_397555), p120 catenin (BD Biosciences; #610134; RRID:AB_397537), pEGFR Y1068 (Cell Signaling Technology #2234; RRID:AB_331701), EGFR (Cell Signaling Technology #4267; RRID:AB_2246311), α-Biotin (Cell Signaling Technology #5597; RRID:AB_10828011), non-phospho β-catenin (Cell Signaling Technology #1980; RRID:AB_2650576), pFGFR4 Y642 (Signalway #11836), FGFR4 (Cell Signaling Technology #8562; RRID:AB_10891199), pFRS2 Y196 (Cell Signaling Technology #3864; RRID:AB_2106222), pSTAT3 Y705 (Cell Signaling Technology #9131; RRID:AB_331586) and p-p44/42 MAPK (Cell Signaling Technology #4377; RRID:AB_331775). This was followed by 1 hour room temperature incubation with secondary antibodies (1:10,000; anti-mouse 680LT: LiCor #925–68020; anti-rabbit 800CW: LiCor #925–32211). Membranes were subsequently imaged on the LiCOr Odyssey CLx Imaging system, with band quantifications performed with built in software.
Immunofluorescence
Cells were plated at a density of 100,000–200,000 cells/well on glass coverslips (Fisher #12–545-80P) in 24-well plates, fixed on ice in ice cold methanol for 30 minutes and blocked in blocking buffer (0.3% Triton X-100, 5% BSA, 1X DPBS) for 1 hour at room temperature. Primary antibody incubation was performed overnight at 4°C: IGF1R β-subunit (Cell Signaling Technology #3027; RRID: AB_2122378; 1:100), EpCAM (Cell Signaling Technology; #2929; RRID: AB_2098657; 1:100), E-cadherin (Cell Signaling Technology; #3195; RRID: AB_2291471;1:100) and p120 catenin (BD Biosciences; #610134; RRID: AB_397537; 1:100). Secondary antibody incubation was done for 1 hour at room temperature followed by Hoechst 33342 (Thermo Scientific #62249; 1:10000) staining. Coverslips were mounted with Aqua-Poly/Mount (Polysciences #18606–20) and images were taken on a Nikon A1 confocal microscope with a 60X objective.
Immunofluorescence of organoids
Organoids were harvested with Cultrex Organoid Harvesting Solution (R&D Systems #3700–100-01) in an Eppendorf tube for 1 hour at 4°C. Centrifuged organoid pellets were fixed with 4% PFA followed by an antigen retrieval step. Pellets were blocked overnight at 4°C, incubated with E-cadherin antibody (BD 610182, RRID: AB_397581) overnight at 4°C, followed by secondary antibody incubation also overnight at 4°C. Organoids were then stained with Hoechst for 30 minutes, cleared with methanol for 1 hour and mounted with ProLong™ Gold Antifade Mountant (Invitrogen P36930). Images were taken on a Nikon A1 confocal microscope with a 60X objective.
Colony formation assay
Cells were plated at a density of 2000 cells/well in 6-well plates (Fisher #08–772-1B) in either full serum (10% FBS) or low serum (0.5% FBS) with 5nM IGF1 (GroPep Bioreagents #AQU001) supplemented media. Cells were monitored every few days and media refreshed every 4 days. Cells plated in full serum media were fixed with 100% methanol on ice and stained with 0.5% Crystal Violet (Sigma-Aldrich #C0775) in 40% methanol after 14 days while cells grown in low serum, 5nM IGF1 were stained after 21–28 days. Wells were imaged on an Olympus SZX16 dissecting microscope and de-stained with 10% acetic acid in water and quantified by spectrophotometry at 560nm. Statistical differences were tested using two-way ANOVA.
Co-Immunoprecipitation
Co-IP was performed with E-cadherin (BD Biosciences #610182; RRID: AB 397581) and IGF1R (Cell Signaling Technology #3027; RRID: AB_2122378) antibodies. Cells were lysed in 20 mM Tris-HCl pH 7.4 with 1% NP-40, 137 mM NaCl and 5 mM EDTA with fresh protease and phosphatase inhibitors (1:100) and quantified using Pierce BCA Protein Assay Kit (Thermo Scientific #23225). 1mg protein from each sample was pre-cleared in 20uL of Pierce™ Protein G Agarose beads (Thermo Fisher Scientific #20398) and incubated in either 3μg of E-cadherin, IGF1R or IgG antibodies (Normal mouse IgG; Millipore #12–371; RRID: AB_145840 and Normal rabbit IgG; Millipore #12–370; RRID: AB_145841) overnight at 4°C with rotation. One 4-hour incubation was performed the next day with 45uL of Pierce™ Protein G Agarose beads at 4°C with rotation. Protein was eluted with Laemmli buffer and analyzed by immunoblotting.
qRT-PCR
RNA extraction was performed using RNeasy mini kit (Qiagen #74106) and the RNA quality and amount quantified on NanoDrop. Reverse transcription to cDNA was performed with PrimeScript™ RT Master Mix (Takara Bio #RR036B). RT-PCR was then performed with SsoAdvanced Universal SYBR (Bio-Rad #1726275) with primers as detailed in supplemental materials. Results were normalized to reads from housekeeping gene RPLPO. Statistical differences evaluated using a paired t-test.
FACS for anoikis resistance
Cells were stained with APC-Annexin V (BD Biosciences #550474) and Propidium Iodide (BD Biosciences #556463) in 1X Annexin binding buffer (BD Biosciences #556454) for 15 minutes at room temperature. Samples were analyzed on an LSR II Flow cytometer (BD Biosciences) and processed using BD FACSDiva and FlowJo software (BD Biosciences) as previously described (31). Live cell percentages in 2D conditions for each cell line was used to normalize the live cell percentages in ULA conditions. Statistical differences were tested using a two-way ANOVA.
Haptotaxis, migration and invasion assays
For haptotaxis experiments, the QCM Haptotaxis Cell Migration Assay Collagen I (EMD Millipore #ECM582) kit was used as previously described (8). For Transwell migration assays, transparent 24 well PET membranes of 8μm pore size (Fisher Scientific # 08–771-21) were used. For Collagen I invasion assays, QCM Collagen Cell Invasion Assay (EMD Millipore #ECM551) was used according to manufacturer’s protocol. For the latter two assays, cells were plated at a density of 300,000 cells/well in 300μL 0.5% FBS media in the top chamber; all bottom chambers were filled with 0.5% FBS media +/− 5nM IGF1 (GroPep Bioreagents # AQU100) or full serum (10%) media. Cells were incubated at 37°C for 72 hours. Excess cells were removed from the top chambers using cotton swabs and inserts were stained with Crystal Violet (Sigma-Aldrich #C0775) before being imaged on an Olympus SZX16 dissecting microscope and quantified with ImageJ software. Quantifications were normalized to low serum WT samples and p-values calculated with one-way ANOVA.
Receptor availability assay
Cells were seeded in 6cm plates (Fisher #08–772-E), and serum starved overnight after achieving a 70–80% confluency. Cells were then stimulated with biotinylated IGF1 (GroPep #AQU100) for 15 minutes at 4°C to reduce receptor internalization. Following PBS washes on ice, cells were treated with 2mM BS3 crosslinker (Thermo Scientific #21580) reconstituted in PBS (pH 8.0) with 6mM KCl and 10mM EGTA for 1 hour at 4°C with occasional rocking. BS3 quenching was performed with 10mM Glycine for 15 minutes and cells were harvested for subsequent immunoblotting (30).
Dose Response assays
Cells were plated in 50μL of media at 9,000 cells/well in 2D and ULA (Corning #3474) 96-well plates. Treatments were added 24 hours post seeding in an additional 50μL of respective media. IGF1R inhibitors BMS-754807 (Selleckchem #S1124) and OSI-906 (Selleckchem #S1091), PI3K inhibitor Alpelisib (Selleckchem #S2814), Akt inhibitor MK-2206 (Selleckchem #S1078), MEK inhibitor U0126 (Selleckchem #S1102) and Fulvestrant (Selleckchem #S1191) were dissolved in DMSO with a final ≤0.5% DMSO concentration in treatments. Plates were collected at day 6 and measured by CellTiter-Glo (Promega #PR-G7573) following the manufacturer’s protocol. Cell viability values were analyzed following blank cell deductions and normalization to vehicle readings. IC50 values for viability were calculated by nonlinear regression and statistical differences evaluated using sum-of-squares Global f-test (p<0.05). Synergy was assessed using SynergyFinder (35).
In silico analysis
Gene expression data from the Sweden Cancerome Analysis Network–Breast (SCAN-B) study was downloaded from Gene Expression Omnibus, accession GSE96058 (36). Differential gene expression between luminal A lobular (N = 265) and ductal (N = 1165) breast cancer samples was assessed using DESeq2 (37). Tumor purity for SCAN-B samples was estimated using the R package ESTIMATE (37). An FDR cut-off of 0.05 was used to identify significantly differentially expressed genes. Heatmap for genes of interest was created using the R package ComplexHeatmap. Gene set variation analysis (GSVA) was performed on log transformed gene FPKM matrices using the R package GSVA (38) with default parameter “Gaussian” for kernel selection. Gene sets of interest were obtained from MSigDB version 7.4. The GSVA enrichment scores were compared between luminal A lobular and ductal breast cancer samples using Mann-Whitney U test. Gene set enrichment analysis (GSEA version 4.1.0. Broad Institute) (39) was also conducted on normalized raw counts with default parameters and customized gene sets of interest obtained from MSigDB.
Organoid culture
IDC organoids (IPM-BO-56, 75, 102, 109, 126 and 134) and ILC organoids (IPM-BO-30 and 46) were established by the Institute for Precision Medicine (Pittsburgh, PA) and maintained in media as detailed in (40) with 1nM β-estradiol (Sigma-Aldrich #E8875) supplementation. For dose response assays, organoids were dissociated to single cell suspension with Trypsin and plated in 50μL of organoid media at 3,000 or 5,000 cells/well in 96-well round bottom plates (Falcon #353227) depending on the growth rate. Akt inhibitor MK2206 (Selleckchem #S1078; dissolved in DMSO) treatments were added 24 hours post seeding in an additional 50μL of organoid media. Organoids were monitored every day to ensure vehicle treated wells were growing well and remained at an appropriate density. Media was refreshed on day 6 and plates collected on day 12 with cell viability quantified by CellTiter-Glo 3D (Promega #G9681). Dose response assay analysis was performed as described above.
Data Availability Statement
All data generated in this study are available within the article and its supplementary files. SCAN-B data analyzed in this study was obtained from Gene Expression Omnibus (GEO) at GSE96058.
Results
IGF pathway activity is enhanced in ILC patient samples and cell lines in vitro
Analysis of the publicly available Sweden Cancerome Analysis Network–Breast (SCAN-B) study allowed a direct comparison of growth factor signaling differences between luminal A IDC (n=1165) and luminal A ILC (n=265) tumors. Significantly higher IGF1 and IGF2 expression were observed in ILC tumors in addition to several other growth factor signaling related genes (Fig 1A) such as IGFBPs and matricellular proteins such as CTGF, NOV and CYR61. Several genes downregulated in ILC were also observed, including IGFL1, IGF1R and WISP1. PI3K/Akt signaling was significantly higher in ILC by gene set enrichment analysis (GSEA) (Fig 1B) and gene set variation analysis (GSVA) (Fig 1C). IGF1/2 signaling activation was also higher in ILC (Fig 1D), strengthening our hypothesis regarding IGF pathway activation in ILC over IDC tumors.
Figure 1: IGF pathway activity is enhanced in ILC patient samples and cell lines in vitro.
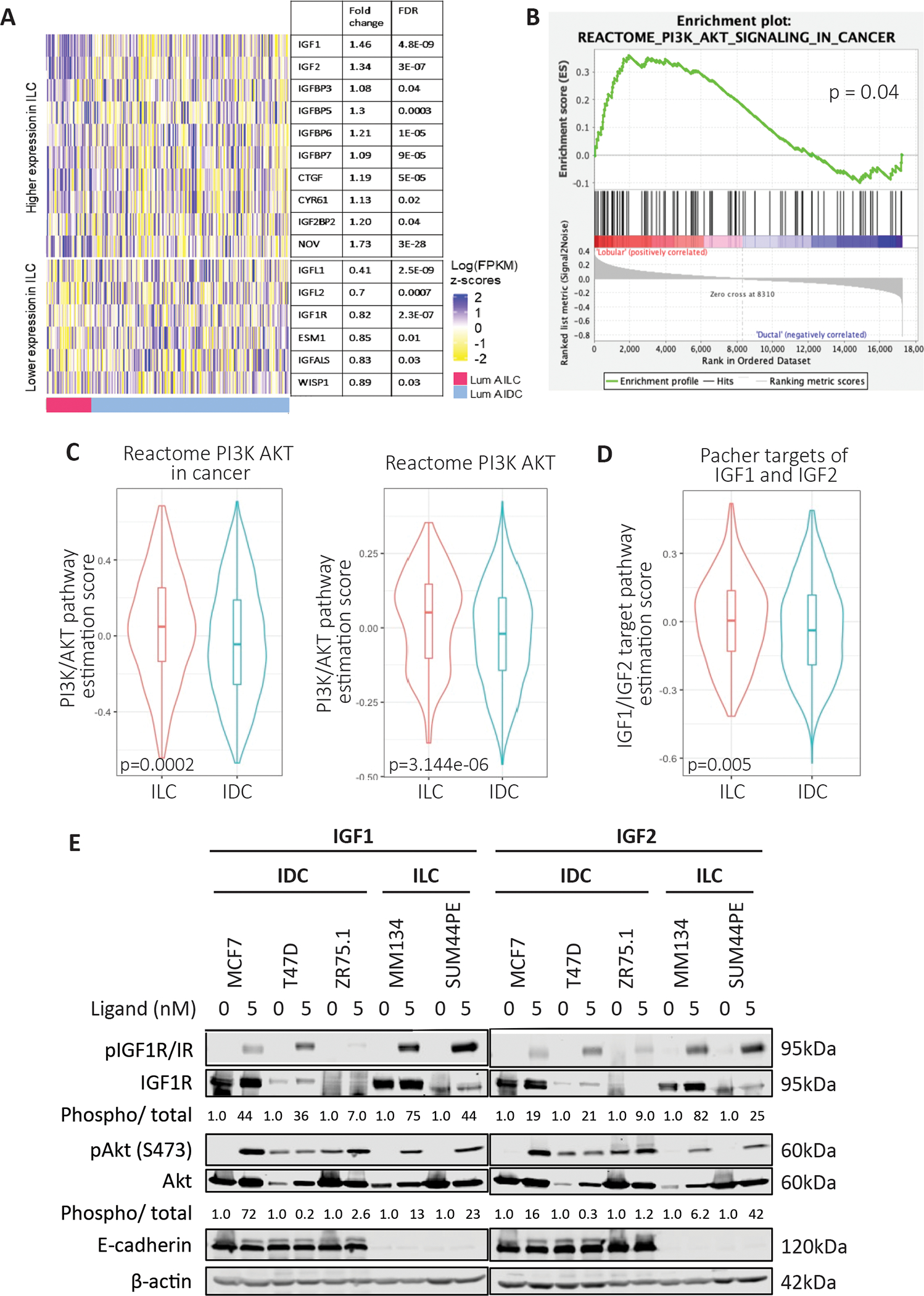
(A) Gene expression analysis of growth factor signaling related genes comparing Luminal A IDC and ILC with significantly different fold change and FDR values displayed. (B) GSEA analysis for PI3K/Akt showing significantly higher pathway activation in ILC samples obtained from the SCANB dataset. (C, D) GSVA analysis of Luminal A IDC and ILC samples using respective signature sets from MSigDB with significantly higher pathway activation in ILC. (E) IDC cells (MCF7, T47D, ZR75.1) and ILC cells (MDA-MB-134-VI, SUM44PE) were stimulated with doses of IGF1 or IGF2 (0–5nM) for 15 minutes following an overnight serum starvation. IGF1R/IR and Akt signaling was assessed by Western blotting. Phosphorylation levels of IGF1R (phospho-IGF-I Receptor β (Tyr1135/1136)/Insulin Receptor β (Tyr1150/1151)) and Akt (S473) were quantified on the LiCOr Odyssey CLx Imaging system and normalized to corresponding total protein levels and loading controls. Quantified values are indicated below the Western bands. Ligand treated sample values were further normalized to respective cell line vehicle treated samples. Representative experiment shown for all, N=2–3 for each experiment.
We assessed the differences in IGF pathway activity in IDC (MCF7, T47D, ZR75.1) and ILC (MDA-MB-134-VI, SUM44PE) cell lines. Both ILC cell lines showed higher pIGF1R/IR expression after IGF1 stimulation compared to the IDC cell lines, though the expression of total IGF1R/IR varied across cell models (Fig 1E). pAkt (S473) levels, as a measure of downstream pathway activation, did not always correlate with pIGF1R/IR levels. We observed Akt activation in the absence of ligands in T47D and ZR75.1 cells, while MCF7 cells demonstrated robust pAkt induction despite modest pIGF1R/IR levels. These results are not entirely surprising given the existing PI3K activating mutations in MCF7 and T47D (41–43), and PTEN loss in ZR75.1 cells (43) which may contribute to the Akt activation. We further assessed the effects of insulin and again observed that ILC cell lines demonstrated enhanced activation of pIGF1R/IR, although with this ligand ZR75.1 also showed robust activation (Supp Fig 1A). To test whether there was a difference between ILC and IDC cell lines in response to ligands targeting other cell surface receptors that may be regulated by E-cadherin (30, 44–46), we assessed the effects of EGF stimulation in our panel of cell lines (Supp Fig 1B). Little EGFR activation could be detected in ILC cell lines suggesting cellular context specific changes in growth factor signaling activation. Taken together these data support increased IGF pathway activity in ILC compared to IDC in both representative patient tumors and in a cell line models.
CDH1 knockout cells as a model to study the role of E-cadherin in regulating the IGF pathway
As previously reported (32, 33), and expanded herein (Fig. 1), E-cadherin negatively regulates IGF pathway activity. To better understand this regulation, we generated CRISPR-mediated CDH1 knockout (CDH1 KO) IDC cell lines of MCF7, T47D and ZR75.1. Both MCF7 and T47D demonstrate successful and complete knockout of CDH1 while ZR75.1 CDH1 KO cells retained some expression of E-cadherin though markedly reduced compared to WT cells (Fig 2A, B). ZR75.1 cells harbor copy number amplification of CDH1 which we believe made complete knockout of CDH1 in this model challenging (42, 47).
Figure 2: CDH1 knockout cells as a model to study the role of E-cadherin in regulating the IGF pathway.
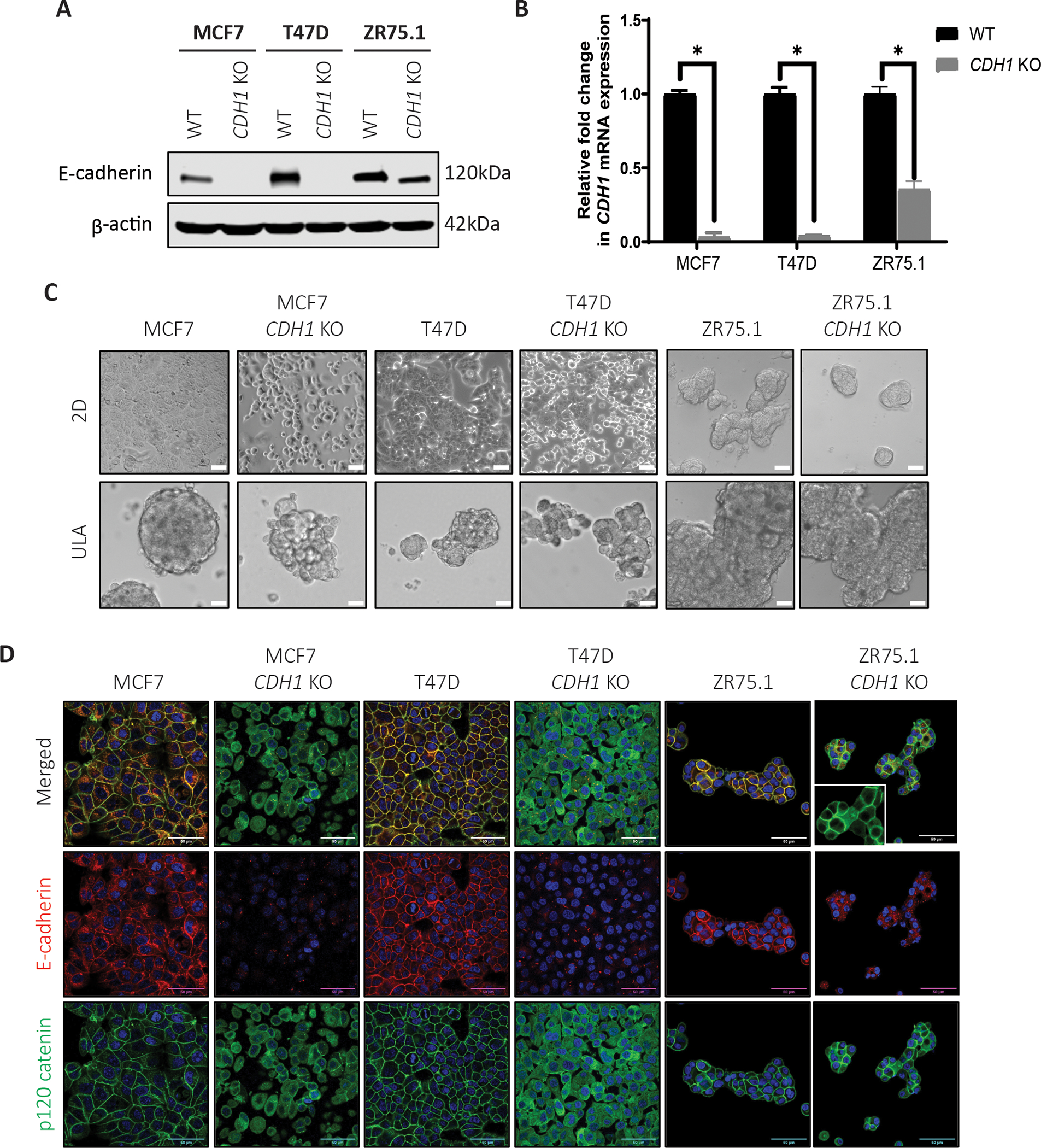
(A) Western blotting and (B) qRT-PCR confirms reduction of E-cadherin in CDH1 KO MCF7, T47D and ZR75.1 CRISPR cell lines compared to wildtype (WT) parental cells. Statistical differences evaluated using paired t-test (*p < 0.05). Representative experiment shown, N=2 (each with two biological and 3 technical replicates). (C) Representative brightfield images (10X magnification) of WT and CDH1 KO cell line models plated in 2D and Ultra-low attachment (ULA) plates. (D) E-cadherin (red) and p120 catenin (green) staining of WT and CDH1 KO cells confirms CDH1 loss and p120 re-localization in CDH1 KO cell models by confocal microscopy (60X objective). Inset in the ZR75.1 CDH1 KO panel shows a zoomed in image. Scale bar: 50μm. Representative experiment shown for all, N=2–3 for each experiment.
We first assessed cell morphology given the known role of E-cadherin in formation of cell-cell junctions. As shown in Fig 2C, MCF7 and T47D CDH1 KO cells have a rounded morphology compared to their parental (WT) cells in 2D culture. In addition, they also demonstrated decreased cell-cell attachment as well as decreased cell-plate attachment. In ultra-low attachment (ULA) conditions, MCF7 and T47D CDH1 KO cells appeared to form less tight, more grape-like cell clusters synonymous with ILC cell aggregates (8, 48). We did not note any obvious difference in either 2D or ULA conditions between ZR75.1 WT and CDH1 KO cells. Examination of E-cadherin by immunofluorescence confirmed loss in all three cell lines models (Fig 2D). Furthermore, similar to ILC cell lines (8, 49), p120 catenin was re-localized to the cytoplasm in the E-cadherin null CDH1 KO cells. Consistent with our earlier observations, ZR75.1 CDH1 KO cells showed residual E-cadherin expression, and p120 expression was visualized in both the cytoplasm and at the cell membrane. Due to the incomplete knockout of CDH1 in our ZR75.1 cells, we did not perform any additional experiments with this model.
As a function of the cadherin complex, E-cadherin stabilizes β-catenin on the cell membrane and prevents its release into the cytoplasm. The release of β-catenin results in free β-catenin being targeted for proteasomal degradation and/or its accumulation resulting in subsequent activation of Wnt signaling. We thus examined expression of β-catenin following treatment with a GSK-3α and GSK-3β inhibitor (CHIR-99021; Selleckchem #S2924) which functions as a Wnt/β-catenin activator. With vehicle treatment, both MCF7 and T47D CDH1 KO cells exhibited little appreciable β-catenin expression, which was only elevated following 24-hour GSK inhibitor treatment (Supp Fig 1C), suggesting that CDH1 KO alone does not alter β-catenin accumulation. Together, these data support the successful generation of CDH1 KO IDC cell line models which display phenotypes consistent with loss of E-cadherin functionality including the characteristic re-localization of p120 as seen in human ILC tumors.
Loss of E-cadherin sensitizes cells to IGF signaling pathway activation
To understand the signaling effects of E-cadherin loss, we subjected WT and CDH1 KO cells to IGF1 stimulation. We observed enhanced sensitivity in both CDH1 KO cell lines by proxy of higher pIGF1R/IR and pAkt activation (Fig 3A). This enhanced signaling response of CDH1 KO cells to WT cells was evident to the greatest extend upon 5nM IGF1 stimulation: 2.72-fold and 1.3-fold higher signaling in MCF7 and T47D CDH1 KO cells respectively. We next assessed the effects of E-cadherin loss on IGF2, and insulin signaling and observed enhanced IGF1R/IR activation in MCF7 CDH1 KO cells following ligand stimulation (Fig 3B, C) which did not translate into enhanced Akt activation. T47D CDH1 KO cells showed similar sensitivity to both IGF2 and insulin when compared to WT cells, suggesting an IGF1 specific differential signaling response.
Figure 3: Loss of E-cadherin sensitizes cells to IGF signaling pathway activation.
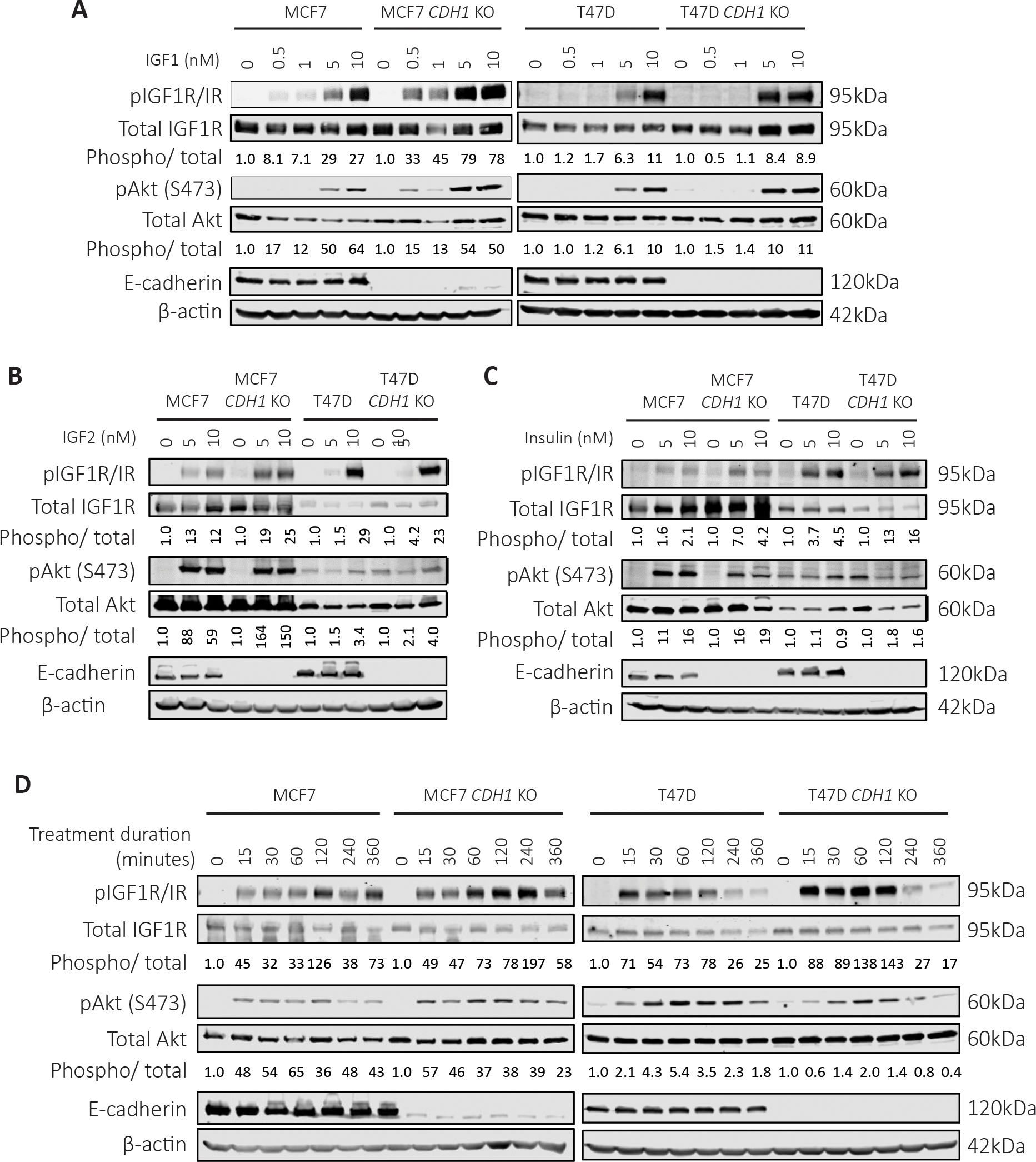
Cells were serum starved overnight and stimulated with (A) IGF1, (B) IGF2 or (C) Insulin (0–10nM) for 15 minutes. Cells were harvested for Western blot to assess IGF1R/IR and Akt signaling. For quantification, phosphorylated protein levels were normalized to corresponding total protein levels and loading controls. Quantified values are indicated below the Western bands. Ligand treated sample values were further normalized to respective cell line vehicle treated samples. (D) Cells were treated with 10nM IGF1 for a time course from 0–6 hours to assess the duration of signaling activity between WT and CDH1 KO cells. Representative experiment shown for all, N=2–3 for each experiment.
To explore if our findings were related to a general growth factor sensitivity following loss of E-cadherin, we stimulated cells with EGF but did not observe any consistent differences in pEGFR Tyr1068 activation, or pAkt levels between CDH1 KO and WT cells (Supp Fig 2A). We specifically examined the effects of E-cadherin deletion on FGFR activity given recent studies on the role of FGFR4 as a driver of endocrine resistance (50), particularly in ILC (51). Cells were stimulated with a cocktail of FGF ligands (FGF 1, 2, 4, 6, 8, 17, 19, 21 and 23) resulting in strong enhancement in both raw and normalized pFGFR4 levels in both MCF7 and T47D CDH1 KO cells, despite decreased total FGFR4 levels in the CDH1 KO cells (Supp Fig 2B). Surprisingly, while pFGFR4 was elevated in CDH1 KO cells, downstream activators did not display a difference between WT and CDH1 KO cells. Additionally, in both MCF7 and T47D CDH1 KO cells, we observed elevated pathway activity that was sustained for up to 4 hours in the MCF7 cells and up to 2 hours in the T47D cells (Fig 3D). These results were replicated for pAkt activation in the MCF7 CDH1 KO cells but not in the T47D CDH1 KO, warranting additional studies on the sustained receptor activity and its effects on downstream signaling response. Collectively, these results suggest that there is an enhancement of IGF1 response in IDC cell lines harboring E-cadherin loss under the conditions tested, but that this increased signaling sensitivity may not be generalizable to other cell surface receptors and is mostly applicable to the IGF1 pathway.
Loss of E-cadherin increases IGF1R availability on the membrane to allow ligand binding
To understand why and how loss of E-cadherin leads to sensitized IGF1R signaling, we first compared the expression of IGF1R in the WT and CDH1 KO cells. Using cell fractionation, minimal differences between the MCF7 WT and CDH1 KO cells were noted (Fig 4A). Higher IGF1R expression was observed in both whole cell and membrane fractions of the T47D CDH1 KO cells compared to WT, which was consistent with increased IGF1R mRNA (Supp Fig 3A). We assessed the availability of the receptor for ligand binding using a biotinylated IGF1 ligand, which was crosslinked to its receptor for subsequent complex quantification via α-biotin immunoblotting. MCF7 and T47D CDH1 KO cells showed a 2.1-fold and 4-fold higher ligand-receptor complex respectively when compared to their corresponding WT cell line controls (Fig 4B). This finding was stronger in the T47D CDH1 KO cells, likely due to the higher baseline IGF1R expression in these cells. As E-cadherin has previously been reported to create a repressive EGFR complex (30), we examined for the presence of a physical interaction between IGF1R and E-cadherin on the cell membrane which potentially could physically and spatially prevent ligands from binding IGF1R. An immunoprecipitation performed for both IGF1R and E-cadherin did not reveal co-IP of either protein despite multiple attempts to optimize conditions (Fig 4C). Imaging of IGF1R to assess localization on the membrane showed no significant differences between the WT and CDH1 KO cells (Fig 4D, E). Our results suggest that increased receptor availability to bind ligand in CDH1 KO cells likely leads to the observed elevated signaling induction upon ligand binding.
Figure 4: Loss of E-cadherin increases IGF1R availability on the membrane to allow ligand binding.
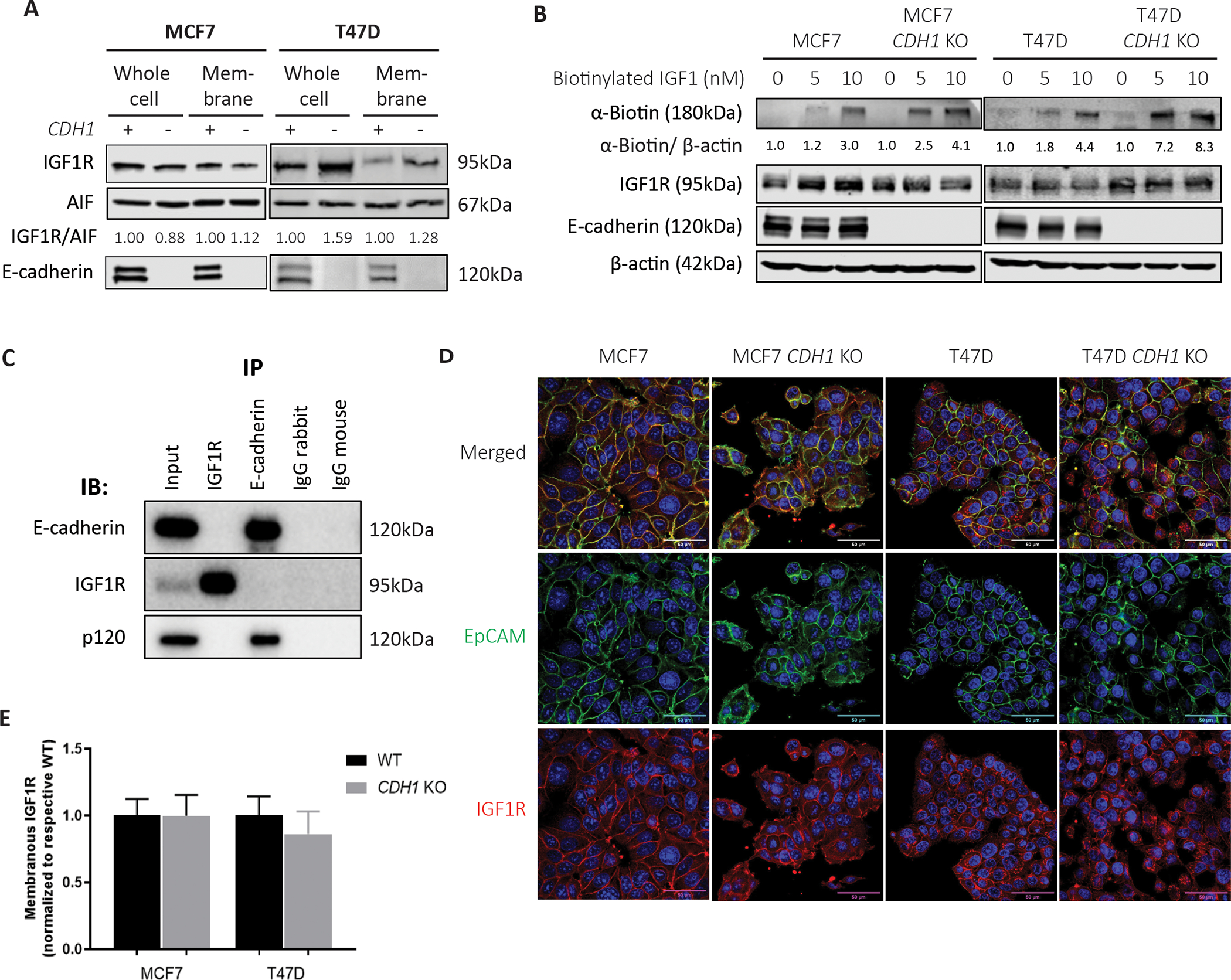
(A) Cell fractionation assay performed on MCF7 and T47D WT and CDH1 KO cells to assess whole cell and membrane IGF1R expression levels. IGF1R bands were quantified and normalized to membrane control, AIF1. Quantified values are indicated below the Western bands. (B) Cells were stimulated with biotinylated IGF1 (0–10nM) for 10 minutes and crosslinked to assess ligand-receptor complex levels between WT and CDH1 KO cells. (C) Immunoprecipitation of IGF1R and E-cadherin in T47D cells was assessed for a co-IP of other proteins, with p-120 catenin assessed as a known interactor of E-cadherin. (D, E) Cell lines were dual stained for EpCAM (green) and IGF1R (red) and imaged by confocal microscopy at a 60X objective. Scale bar: 50μm. 12 images for each cell line were quantified and graphed (E). Representative experiment shown for all, N=2–3 for each experiment.
Loss of E-cadherin enhances anoikis resistance and increases cell survival
We previously showed increased anchorage independent growth in ILC cells compared to IDC (8). In IDC cells with CDH1 KO, we similarly found an increase in ULA growth compared to WT cells in the MCF7 and T47D models (31) as has also been seen by others (52). ZR75.1 WT and CDH1 KO cells did not show any significant differences in either growth condition (Supp Fig 4A), potentially due to the incomplete CDH1 deletion and a copy number amplification (42). To explore the previously observed ability for MCF7 and T47D CDH1 KO cells to thrive in ULA conditions, we examined anoikis resistance using an Annexin V and Propidium Iodide staining technique followed by flow cytometry after a 3-day culture in 2D or ULA plates. While the percentage of live cells in 2D were similar between WT and CDH1 KO cells, the fraction of live cells in ULA were significantly higher in the T47D CDH1 KO cells (Fig 5A), consistent with anoikis resistance. No major differences were observed between the MCF7 WT and CDH1 KO cells in either condition.
Figure 5: Loss of E-cadherin enhances anoikis resistance and increases cell survival.
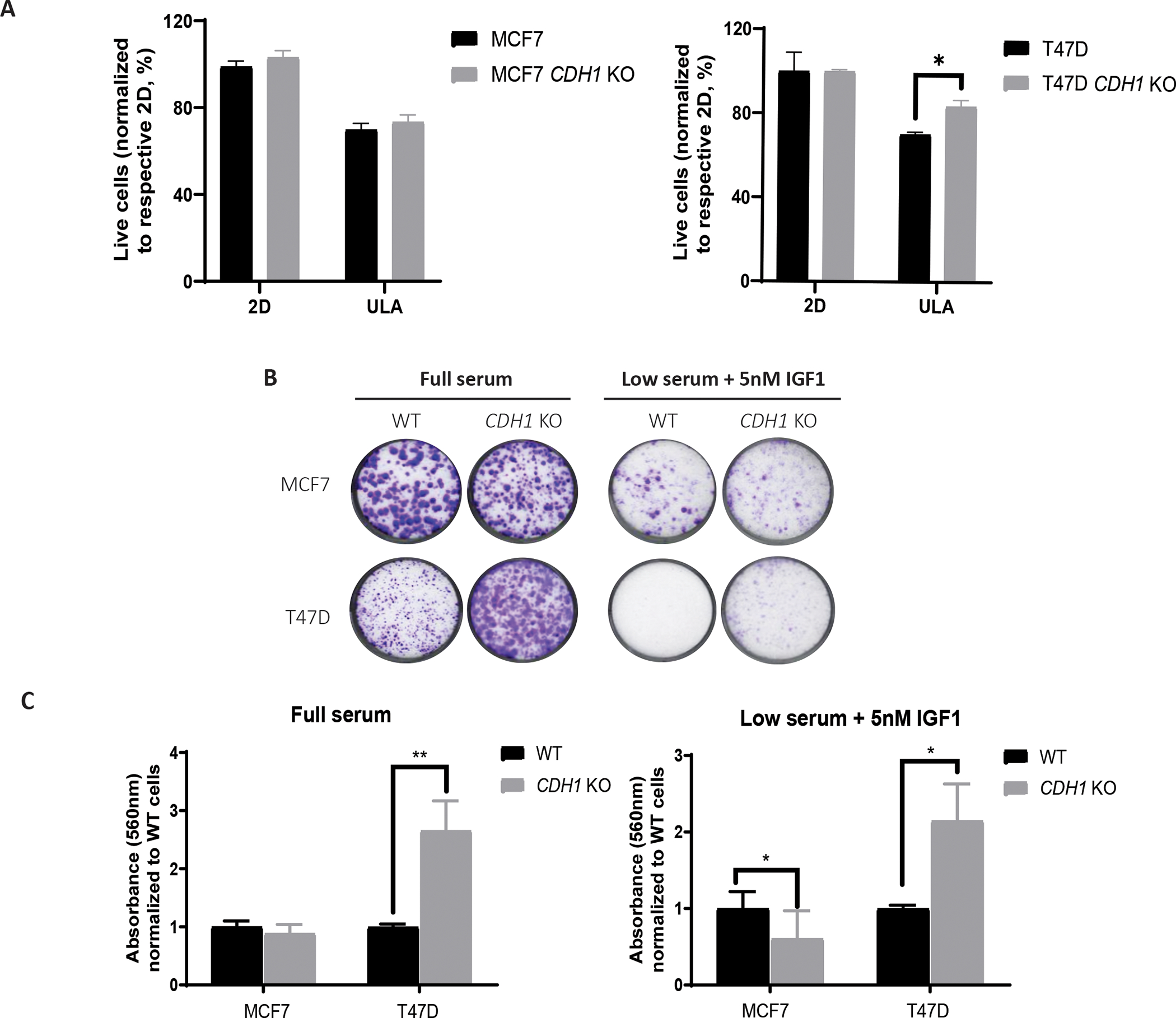
(A) MCF7 and T47D cells were grown in 2D and ULA plates for 3–4 days and stained with Annexin V and propidium iodide to measure live and apoptotic cells, respectively. Live cell percentage in 2D for each cell line was used to normalize the live cell percentages in ULA plates. Representative experiment shown; N=3 (each with three biological replicates). (B, C) Cells were plated at 2,000 cells/well in full serum and low serum supplemented with 5nM IGF1 media and stained with 0.5% Crystal Violet after two to three weeks. Representative images are shown. Plates were de-stained with 10% acetic acid, absorbance quantified and graphed after normalization to WT cells of corresponding conditions. Statistical differences were evaluated using two-way ANOVA (*p < 0.05, **p<0.01, ***p<0.001, representative experiment shown, N=3 (each with three biological replicates)).
To further explore cell survival, we performed colony formation assays by plating cells at a low density in either full serum (10% FBS) or low serum (0.5% FBS) containing 5nM IGF1 in 2D plates. T47D CDH1 KO cells showed increased clonogenic survival in both conditions (Fig 5B–C). CDH1 KO cells also demonstrated increased colonies in full serum compared to WT, but with no colonies observed in low serum conditions. MCF7 CDH1 KO cells showed no clear differences in full serum quantifications, however, they formed a greater number of smaller colonies than the WT cells, with similar observations in low serum conditions. The presence of enhanced cell survival phenotype in the low serum plus IGF1 condition further highlights the IGF-specific phenotype in our CDH1 KO cells. Our data support that loss of E-cadherin may play a context-dependent role in anoikis resistance and cell survival.
Loss of E-cadherin increases collagen I haptotaxis, IGF1-driven migration and serum-driven collagen invasion
We investigated if deletion of CDH1 in IDC cell lines confers haptotactic migration towards the extracellular matrix (ECM), a phenotype that is pronounced in ILC cell lines (8). We observed a significantly higher migration towards Collagen I by MCF7 and T47D CDH1 KO cells (Fig 6A) compared to their respective WT cells. Multiple studies have reported on E-cadherin loss permitting cell migration and invasion (53–55); conflicting reports on the requirement of E-cadherin for metastasis have also been reported (56). To explore this inconsistency while assessing the importance of IGF1, CDH1 KO cells were subjected to Transwell migration assays with chemotactic low serum (0.5% FBS) media +/− 5nM IGF1 or full serum (10% FBS) media. Both CDH1 KO cell models showed increased migration towards serum compared to WT cells (Fig 6B, C). Consistent with the IGF1 sensitive nature of these CDH1 KO cell lines, MCF7 and T47D CDH1 KO cells also showed significant migration towards IGF1, a phenotype which was successfully halted upon IGF1R/IR inhibitor, BMS-754807 addition (Supp Fig 4B–D). Higher migration of T47D CDH1 KO cells without the presence of any chemotactic gradient highlights the migratory phenotype driven by the independent loss of E-cadherin in this model. Given the haptotactic migration towards Collagen I and migration towards IGF1 and serum, we hypothesized that CDH1 KO cells may also be able to invade through Collagen I (8) towards a gradient of IGF1 and serum. While there was no Collagen I invasion observed with the MCF7 cells (Supp Fig 4E), T47D CDH1 KO cells showed a significant Collagen I invasion towards serum (Fig 6D). It is important to note that neither of these three cell lines are invasive by nature and the invasive phenotypes observed in T47D CDH1 KO cells is promising and requires greater investigation in the context of tumor invasion.
Figure 6: Loss of E-cadherin increases collagen I haptotaxis, IGF1-driven migration and serum-driven collagen invasion.
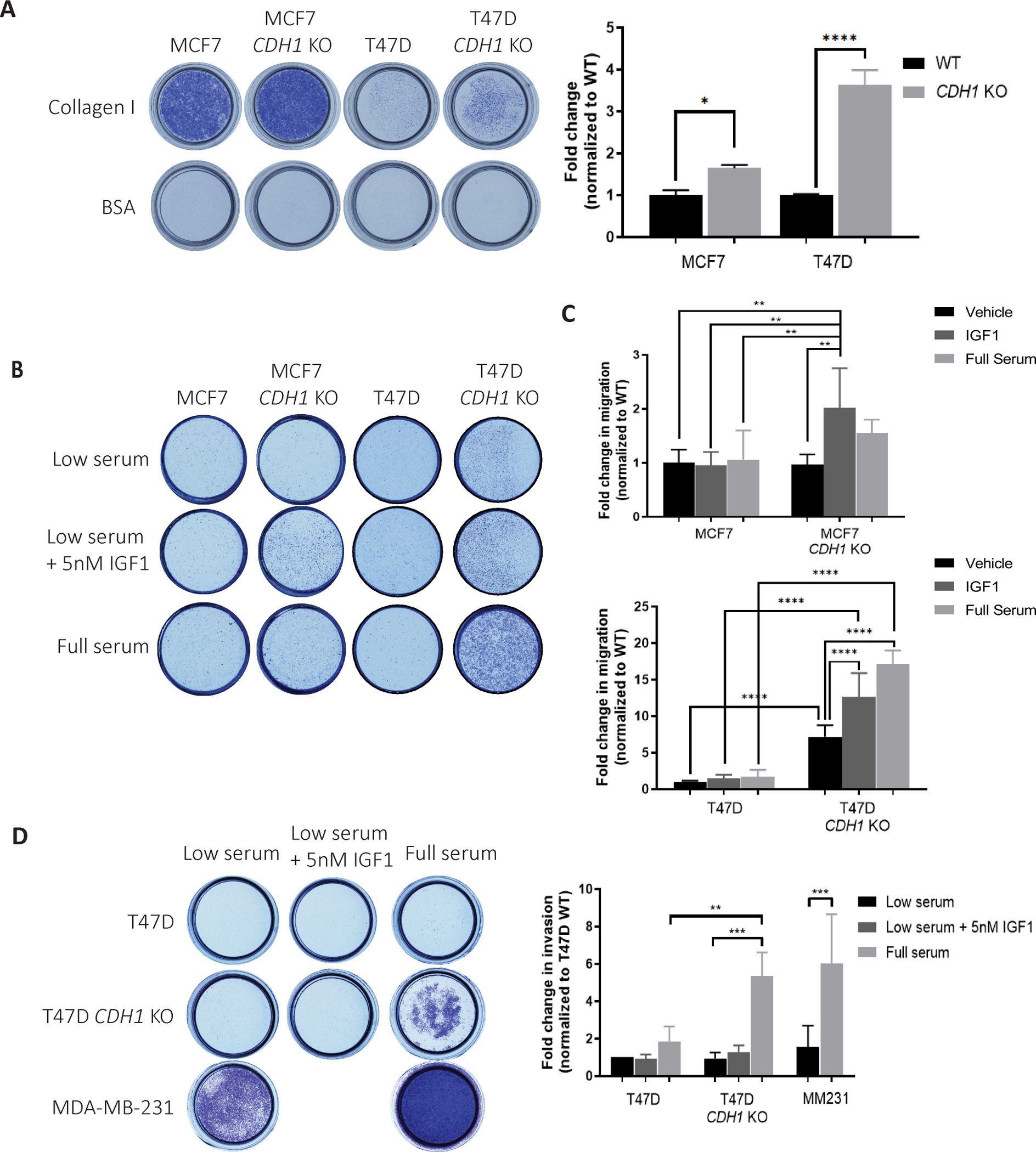
(A) Images and quantification of Crystal Violet-stained Collagen I coated inserts from haptotaxis assays after 72 hours. Migrated cell colonies were quantified with ImageJ and plotted. (B) Images and (C) quantification of Crystal Violet-stained Transwell inserts from migration assays towards the indicated attractants after 72 hours. (D) Images and quantification of Crystal Violet-stained Collagen I inserts from invasion assays towards the indicated attractants after 72 hours. Graphs show representative data normalized to low serum WT cell samples from two to three independent experiments (N=2 biological replicates). p-values from one-way ANOVA statistical testing. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001.
Loss of E-cadherin sensitizes cells to Fulvestrant and IGF1R/PI3K/Akt/MEK inhibitors in a context dependent manner
Motivated by the increased sensitivity of CDH1 KO cells to IGF ligands and the potential for clinical translation, we sought to determine if these cells are also sensitive to IGF1R inhibitors. Only T47D CDH1 KO cells were more sensitive to the tested IGF1R inhibitors compared to WT cells (BMS-754807 and OSI-906) (Fig 7A, B, E). MCF7 CDH1 KO cells did not show any significant differences when compared to WT cells (Supp Fig 5A) although BMS754807 treatment did inhibit IGF1R and Akt signaling in these cell lines, albeit at a lower efficacy in CDH1 KO cells, potentially owing to their increased ligand sensitivity (Supp Fig 5B). Given the higher incidence of PIK3CA hotspot mutations and occurrence of PTEN loss in ILC (4, 12, 57), we examined if the loss of E-cadherin in these IDC cell lines also sensitizes them to PI3K and Akt inhibitors, Alpelisib and MK-2206, respectively. As with the IGF1R inhibitors, only T47D CDH1 KO cells showed increased sensitivity to MK2206 and an overall increased sensitivity trend to Alpelisib upon averaging statistical analysis of repeated experiments (Fig 7C–E, Supp Fig 5A, C). Next, we assessed if the absence of susceptibility to IGF1R and Akt inhibitors in the MCF7 CDH1 KO cells might be explained by alternative pathways being activated. We performed combination treatments of BMS-754807 with MEK inhibitor, U0126, and observed a strong additive effect where both MCF7 and T47D CDH1 KO cells were more sensitive than their corresponding WT cells (Fig 7F, G). While this was an expected result in the T47D cells, it is interesting to note the additive effect in the MCF7 CDH1 KO cells as this suggests the activation of alternative pathways following IGF1R, PI3K and Akt independent inhibitions. We further assessed the synergy effect utilizing the SynergyFinder platform (35), with the ZIP method as output (Supp Fig. 6A, B). In MCF7 and T47D WT and CDH1 KO cells, synergy scores were between −9.717 to 10.995, which validates an additive but not synergistic drug combination effect. As over 90% of ILC tumors are estrogen receptor (ER) positive, we also investigated the combination effects of Akt inhibition with an ER degrader, Fulvestrant, and observed a higher susceptibility in the T47D CDH1 KO cells (Fig 7H, Supp Fig 6C), supporting the notion of targeting the IGF pathway concurrently to targeting ER. These results support the study of combinatory therapies in pre-clinical studies to better target the activation of the IGF pathway following E-cadherin loss. Finally, to analyze pathway inhibition in a more physiologically relevant model, we utilized patient derived ILC and IDC breast organoids (PDO) to compare Akt inhibition sensitivity. Loss of E-cadherin in ILC PDOs (PDO 30 and 46) is depicted by immunofluorescence (Fig 7I), while membranous E-cadherin expression is still present in IDC PDOs (PDO 56, 75, 102, 109, 126 and 134). Comparing the IDC and ILC PDOs, ILC organoids did demonstrate a stronger trend in sensitivity to MK2206 treatment (Fig 7J, Supp Fig 6D). This was further shown by a significantly lower area under dose response curves comparing the ILC to the IDC organoids (Fig 7K), strengthening potential translational implications. Although our sample sizes are small given the limitations of patient-derived organoids, we are able to confidently conclude on the higher sensitivity to Akt inhibitor in ILC PDOs.
Figure 7: Loss of E-cadherin sensitizes cells to Fulvestrant and IGF1R/PI3K/Akt/MEK inhibitors in a context dependent manner.
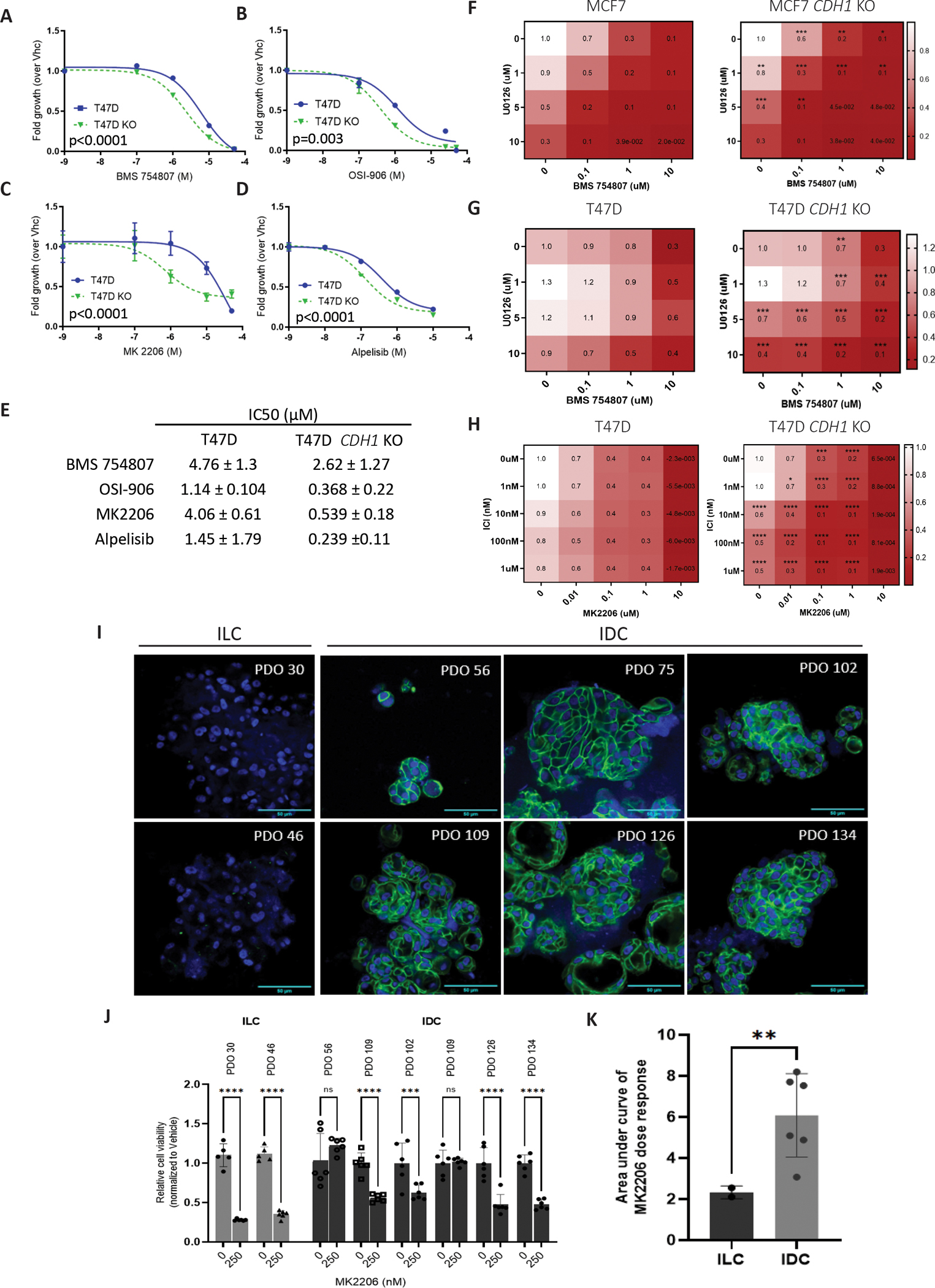
T47D parental and CDH1 KO cells were seeded in 96-well 2D plates and treated with IGF1R inhibitors (OSI-906 or BMS-754807), PI3K inhibitor (Alpelisib) or Akt inhibitor (MK2206) for 6 days. Conditions in the panels as follows: (A) BMS-754807; (B) OSI-906; (C) MK2206, (D) Alpelisib and (E) shows a summary of IC50 values. CellTiter Glo assay was used to assess cell viability (relative luminescence) and data was normalized to vehicle treated control. (F, G) Cells were treated a combination of MEK inhibitor (U0126) and BMS-754807 for 6 days and viability assessed as above. (H) Cells were treated a combination of Akt inhibitor (MK2206) and Fulvestrant for 6 days and viability assessed as above. IC50 values for viability were calculated by nonlinear regression and statistical differences evaluated using sum-of-squares Global f-test (p < 0.05; representative experiment shown; N=3 (each with six biological replicates)). (I) E-cadherin immunofluorescence was performed on patient derived IDC and ILC organoids. Green: E-cadherin and blue: Hoechst. Scale bar: 50um. (J) ILC and IDC organoids were treated with Akt inhibitor (MK2206) for 12 days and viability assessed with CellTiter Glo 3D (relative luminescence), and data was normalized to vehicle treated cells. Statistical differences were evaluated using 2way-ANOVA (**p<0.005, ***p<, 0.0005***p<0.0001; representative experiment shown; n=1 (each with six biological replicates)). (K) Area under curve of IDC and ILC organoids dose response with MK2206 was calculated from (J), with statistical differences evaluated using unpaired t-tests (p=0.0056).
Discussion
Recent advancements have led to personalized cancer treatments; however, more progress needs to be made to effectively target tumors. Our study highlights the role of E-cadherin in regulating IGF signaling and tumorigenic properties in vitro. Genetic deletion of E-cadherin in IDC cells caused reduced cell-cell attachment and morphology transformation into looser cell clusters in suspension. Furthermore, E-cadherin loss was associated with enhanced anchorage independence and anoikis resistance, all of which are features observed in ILC tumors. Additionally, CDH1 KO cells were more sensitive to IGF1, IGF2 and insulin signaling attributable to an increased receptor availability for ligand binding. This was an IGF-specific phenotype and was not observed with other growth factor signaling pathways tested albeit FGFR4 activations were higher in the CDH1 KO cells but without significant downstream signaling. Further characterization of CDH1 KO cells also showed an increased cell survival in clonogenic assays, and enhanced cell migration/invasion in the majority of models, suggesting a role for E-cadherin in controlling metastatic phenotypes. Importantly, a context dependent increase in sensitivity to IGF1R, PI3K, Akt and MEK inhibitors was also observed in our models. This study contributes to an increasing body of work delineating ILC as a unique breast cancer subtype and suggests a potential for targeted therapeutic approaches towards the IGF1R/PI3K/Akt axis owing to the diagnostic loss of E-cadherin in this histological subtype.
Studies have attributed spatial accessibility of IGF1R for ligand binding as a limiting factor in signaling activation (30, 58), and consistent with this notion, we found that loss of E-cadherin confers increased IGF ligand binding by IGF1R, and subsequent pathway activation. Despite previous reports on E-cadherin repressing growth factor receptors (30, 33, 58) and co-localization (58), we were unable to find an association by co-IP despite numerous attempts, suggesting that this is not the mechanism affecting the availability of IGF1R for ligand binding when E-cadherin is present. The formation of a ternary complex involving E-cadherin, IGF1R and integrins is an ongoing topic of investigation thought to affect cell mobility (59), which could additionally complicate immunoprecipitation. Similar interactions have been reported for E-cadherin and EGFR, although we did not observe an increased sensitivity to the EGF ligand in our CDH1 KO cell lines (30, 60). We, and others, have shown that downregulation of E-cadherin by growth factors can promote EMT (58, 61); others have additionally demonstrated growth factor signaling regulation by E-cadherin, suggesting that a bidirectional feedback loop may exist. These studies were initially almost exclusively performed in the context of EGFR where multiple groups had shown that E-cadherin inhibits ligand-dependent activation of EGFR signaling (30, 62, 63). However, a recent study by Teo and colleagues (13) supported our findings by reporting on ILC cell lines generated from a p53-deficient metastatic mouse model exhibiting had PI3K/AKT pathway activation and enhanced sensitivity to pathway inhibition.
Due to the function of E-cadherin in maintaining cell-cell adhesion, its loss through EMT or genetic deletion is often thought to correlate with increased tumor invasion and metastasis (62, 64). Other studies, however, have suggested a requirement for E-cadherin in metastasis (56). Here we show that CDH1 KO cells demonstrate increased migration towards Collagen I (8). The CDH1 KO cells also showed an enhanced migration towards serum and IGF1, with T47D CDH1 KO cells showing increased migration even in the absence of a chemoattractant. T47D CDH1 KO also showed invasion through Collagen I, providing additional in vitro evidence supporting that the loss of E-cadherin may enhance metastatic phenotypes. The in vitro nature of these findings, however, is an interpretation limitation and in vivo experimentation is needed to better understand the role of E-cadherin in metastasis and validate its effect on IGF signaling.
Inhibitors of downstream activators such as PI3K, Akt and MEK have successfully been developed and approved for the clinic. This is paramount in malignancy with a high percentage of mutations leading to PIK3CA activation and PTEN loss such as in ILC (4), and maybe more so important given the high IGF pathway activation found in the SCAN-B dataset. Other studies with CRISPR MCF7 CDH1 KO cells lines have shown that loss of E-cadherin leads to dependency upon ROS1 and enhanced sensitivity to crizotinib (65) for which a clinical trial is currently underway. We report increased sensitivity to IGF1R/PI3K/Akt and MEK inhibitors in T47D CDH1 KO cells and an increased trend in sensitivity to an Akt inhibitor in ILC PDOs compared to IDC PDOs. Given the genetic landscape of MCF7 and T47D cells, these models are primed for hyperactive Akt signaling, which has been shown to be further enhanced by loss of E-cadherin. The helical domain mutation in MCF7 cells (E545K) is reported to have a more aggressive phenotype over the kinase domain mutation in T47D (H1047R) (66), which may explain the absence of additional sensitivity in MCF7 CDH1 KO cells to compounds tested since the PIK3CA mutation may effectively mask any potential effect from E-cadherin loss. With the possibility of IGF1R inhibitor efficacy being reduced by hyperactive downstream pathways such as MEK as seen in colon carcinoma (67) and the possibilities of downstream activation inhibition being overhauled by hyperactive receptor tyrosine kinases (68), we tested and observed an additive effect of treating cells with an IGF1R inhibitor in combination with a MEK inhibitor. Further, additive effects of combining Akt inhibitors with Fulvestrant was also found to be beneficial in E-cadherin deficient cells, proving novel therapeutical benefits which require further exploration in vivo. Our studies with ILC PDOs showing higher sensitivity to Akt inhibitor is very encouraging and requires additional investigation. Future studies will need to test the efficacy of inhibitors targeting the IGF pathway comparing cells +/− CDH1 in vivo and investigate the potential of efficiently exploiting E-cadherin as a biomarker of therapeutic response.
In conclusion, this study delineates the role of E-cadherin in regulating enhanced IGF signaling, controlling metastatic phenotypes, and identifying use of specific tyrosine kinase inhibitors that can effectively target elevated IGF1 signaling activation. We reveal novel findings of increased receptor availability for ligand binding following the loss of E-cadherin and increased susceptibility to IGF1R/PI3K/Akt and MEK inhibitors upon CDH1 deletion. Our study adds to a growing body of evidence suggesting that CDH1 may represent a biomarker of response to kinase inhibitors and further exemplifies the need for investigation into the clinical translation of these findings. The in vitro nature of our studies in a small panel of cell lines is an important, notable limitation, but notwithstanding, we have robustly revealed important cellular- and contextual-dependent functions of E-cadherin in IGF driven tumor phenotypes. Our findings require translation into in vivo models with the goal of validating E-cadherin loss as a biomarker of growth factor receptor inhibitor response in breast cancer as well as the utilization of E-cadherin expression as a vital patient stratification tool in clinical trials.
Supplementary Material
Acknowledgements
The authors would like to thank Osama Shah for data acquisition and bioinformatic input and the Institute for Precision Medicine, a partnership of the University of Pittsburgh and UPMC, for providing the breast cancer patient derived organoids used in these studies. This research was supported in part by the University of Pittsburgh Center for Research Computing through the resources provided.
Financial support:
Research funding for this project was provided in part by Susan G. Komen Scholar awards (SAC110021 to AVL and SAC160073 to SO), the Breast Cancer Research Foundation (to AVL and SO) and the National Cancer Institute (R01CA224909).
Footnotes
Conflict of Interest Disclosure: The authors declare no potential conflicts of interest.
References
- 1.Pestalozzi BC, Zahrieh D, Mallon E, Gusterson BA, Price KN, Gelber RD, et al. Distinct Clinical and Prognostic Features of Infiltrating Lobular Carcinoma of the Breast: Combined Results of 15 International Breast Cancer Study Group Clinical Trials. Journal of Clinical Oncology. 2008;26(18):3006–14. [DOI] [PubMed] [Google Scholar]
- 2.Corben AD. Pathology of invasive breast disease. Surg Clin North Am. 2013;93(2):363–92. [DOI] [PubMed] [Google Scholar]
- 3.Mamtani A, King TA. Lobular Breast Cancer: Different Disease, Different Algorithms? Surg Oncol Clin N Am. 2018;27(1):81–94. [DOI] [PubMed] [Google Scholar]
- 4.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell. 2015;163(2):506–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Du T, Zhu L, Levine KM, Tasdemir N, Lee AV, Vignali DAA, et al. Invasive lobular and ductal breast carcinoma differ in immune response, protein translation efficiency and metabolism. Sci Rep. 2018;8(1):7205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rakha EA, Ellis IO. Lobular breast carcinoma and its variants. Semin Diagn Pathol. 2010;27(1):49–61. [DOI] [PubMed] [Google Scholar]
- 7.Chen Z, Yang J, Li S, Lv M, Shen Y, Wang B, et al. Invasive lobular carcinoma of the breast: A special histological type compared with invasive ductal carcinoma. PLoS One. 2017;12(9):e0182397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tasdemir N, Bossart EA, Li Z, Zhu L, Sikora MJ, Levine KM, et al. Comprehensive Phenotypic Characterization of Human Invasive Lobular Carcinoma Cell Lines in 2D and 3D Cultures. Cancer Res. 2018;78(21):6209–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barroso-Sousa R, Metzger-Filho O. Differences between invasive lobular and invasive ductal carcinoma of the breast: results and therapeutic implications. Therapeutic Advances in Medical Oncology. 2016;8(4):261–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vos CB, Cleton-Jansen AM, Berx G, de Leeuw WJ, ter Haar NT, van Roy F, et al. E-cadherin inactivation in lobular carcinoma in situ of the breast: an early event in tumorigenesis. Br J Cancer. 1997;76(9):1131–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoshiura K, Kanai Y, Ochiai A, Shimoyama Y, Sugimura T, Hirohashi S. Silencing of the E-cadherin invasion-suppressor gene by CpG methylation in human carcinomas. Proc Natl Acad Sci U S A. 1995;92(16):7416–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Desmedt C, Zoppoli G, Gundem G, Pruneri G, Larsimont D, Fornili M, et al. Genomic Characterization of Primary Invasive Lobular Breast Cancer. J Clin Oncol. 2016;34(16):1872–81. [DOI] [PubMed] [Google Scholar]
- 13.Teo K, Gomez-Cuadrado L, Tenhagen M, Byron A, Ratze M, van Amersfoort M, et al. E-cadherin loss induces targetable autocrine activation of growth factor signalling in lobular breast cancer. Sci Rep. 2018;8(1):15454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Becker MA, Ibrahim YH, Cui X, Lee AV, Yee D. The IGF Pathway Regulates ERα through a S6K1-Dependent Mechanism in Breast Cancer Cells. Molecular Endocrinology. 2011;25(3):516–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim H-J, Litzenburger BC, Cui X, Delgado DA, Grabiner BC, Lin X, et al. Constitutively Active Type I Insulin-Like Growth Factor Receptor Causes Transformation and Xenograft Growth of Immortalized Mammary Epithelial Cells and Is Accompanied by an Epithelial-to-Mesenchymal Transition Mediated by NF-κB and Snail. Molecular and Cellular Biology. 2007;27(8):3165–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yee D, Lee AV. Journal of Mammary Gland Biology and Neoplasia. 2000;5(1):107–15. [DOI] [PubMed] [Google Scholar]
- 17.Hawsawi Y, El-Gendy R, Twelves C, Speirs V, Beattie J. Insulin-like growth factor — Oestradiol crosstalk and mammary gland tumourigenesis. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 2013;1836(2):345–53. [DOI] [PubMed] [Google Scholar]
- 18.Baserga R, Peruzzi F, Reiss K. The IGF-1 receptor in cancer biology. Int J Cancer. 2003;107(6):873–7. [DOI] [PubMed] [Google Scholar]
- 19.Litzenburger BC, Kim HJ, Kuiatse I, Carboni JM, Attar RM, Gottardis MM, et al. BMS-536924 reverses IGF-IR-induced transformation of mammary epithelial cells and causes growth inhibition and polarization of MCF7 cells. Clin Cancer Res. 2009;15(1):226–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iams WT, Lovly CM. Molecular Pathways: Clinical Applications and Future Direction of Insulin-like Growth Factor-1 Receptor Pathway Blockade. Clinical Cancer Research. 2015;21(19):4270–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgillo F, De Vita F, Antoniol G, Orditura M, Auriemma PP, Diadema MR, et al. Serum insulin-like growth factor 1 correlates with the risk of nodal metastasis in endocrine-positive breast cancer. Curr Oncol. 2013;20(4):e283–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crudden C, Girnita A, Girnita L. Targeting the IGF-1R: The Tale of the Tortoise and the Hare. Front Endocrinol (Lausanne). 2015;6:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boone DN, Lee AV. Targeting the Insulin-like Growth Factor Receptor: Developing Biomarkers from Gene Expression Profiling. Critical Reviews™ in Oncogenesis. 2012;17(2):161–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh P, Alex JM, Bast F. Insulin receptor (IR) and insulin-like growth factor receptor 1 (IGF-1R) signaling systems: novel treatment strategies for cancer. Med Oncol. 2014;31(1):805. [DOI] [PubMed] [Google Scholar]
- 25.Ekyalongo RC, Yee D. Revisiting the IGF-1R as a breast cancer target. npj Precision Oncology. 2017;1(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Y, Yee D. Targeting insulin and insulin-like growth factor signaling in breast cancer. J Mammary Gland Biol Neoplasia. 2012;17(3–4):251–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pappo AS, Patel SR, Crowley J, Reinke DK, Kuenkele K-P, Chawla SP, et al. R1507, a Monoclonal Antibody to the Insulin-Like Growth Factor 1 Receptor, in Patients With Recurrent or Refractory Ewing Sarcoma Family of Tumors: Results of a Phase II Sarcoma Alliance for Research Through Collaboration Study. Journal of Clinical Oncology. 2011;29(34):4541–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pappo AS, Vassal G, Crowley JJ, Bolejack V, Hogendoorn PCW, Chugh R, et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: Results of a Sarcoma Alliance. Cancer. 2014;120(16):2448–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langer CJ, Novello S, Park K, Krzakowski M, Karp DD, Mok T, et al. Randomized, phase III trial of first-line figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin alone in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2014;32(19):2059–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J. 2004;23(8):1739–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tasdemir N, Ding K, Savariau L, Levine KM, Du T, Elangovan A, et al. Proteomic and transcriptomic profiling identifies mediators of anchorage-independent growth and roles of inhibitor of differentiation proteins in invasive lobular carcinoma. Sci Rep. 2020;10(1):11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Erdem C, Nagle AM, Casa AJ, Litzenburger BC, Wang YF, Taylor DL, et al. Proteomic Screening and Lasso Regression Reveal Differential Signaling in Insulin and Insulin-like Growth Factor I (IGF1) Pathways. Mol Cell Proteomics. 2016;15(9):3045–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagle AM, Levine KM, Tasdemir N, Scott JA, Burlbaugh K, Kehm J, et al. Loss of E-cadherin Enhances IGF1–IGF1R Pathway Activation and Sensitizes Breast Cancers to Anti-IGF1R/InsR Inhibitors. Clinical Cancer Research. 2018;24(20):5165–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen F, Ding K, Priedigkeit N, Elangovan A, Levine KM, Carleton N, et al. Single-Cell Transcriptomic Heterogeneity in Invasive Ductal and Lobular Breast Cancer Cells. Cancer Res. 2021;81(2):268–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ianevski A, He L, Aittokallio T, Tang J. SynergyFinder: a web application for analyzing drug combination dose-response matrix data. Bioinformatics. 2020;36(8):2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brueffer C, Gladchuk S, Winter C, Vallon-Christersson J, Hegardt C, Hakkinen J, et al. The mutational landscape of the SCAN-B real-world primary breast cancer transcriptome. EMBO Mol Med. 2020;12(10):e12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell. 2018;172(1–2):373–86 e10. [DOI] [PubMed] [Google Scholar]
- 41.Ikediobi ON, Davies H, Bignell G, Edkins S, Stevens C, O’Meara S, et al. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol Cancer Ther. 2006;5(11):2606–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marcotte R, Sayad A, Brown KR, Sanchez-Garcia F, Reimand J, Haider M, et al. Functional Genomic Landscape of Human Breast Cancer Drivers, Vulnerabilities, and Resistance. Cell. 2016;164(1–2):293–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hollestelle A, Nagel JHA, Smid M, Lam S, Elstrodt F, Wasielewski M, et al. Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Research and Treatment. 2010;121(1):53–64. [DOI] [PubMed] [Google Scholar]
- 44.Andl CD, Rustgi AK. No one-way street: Cross-talk between E-cadherin and receptor tyrosine kinase (RTK) signaling—A mechanism to regulate RTK activity. Cancer Biology & Therapy. 2005;4(1):35–8. [DOI] [PubMed] [Google Scholar]
- 45.De-Freitas-Junior JCM, Carvalho S, Dias AM, Oliveira P, Cabral J, Seruca R, et al. Insulin/IGF-I Signaling Pathways Enhances Tumor Cell Invasion through Bisecting GlcNAc N-glycans Modulation. An Interplay with E-Cadherin. PLoS ONE. 2013;8(11):e81579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Curto M, Cole BK, Lallemand D, Liu CH, McClatchey AI. Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J Cell Biol. 2007;177(5):893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lombaerts M, Van Wezel T, Philippo K, Dierssen JWF, Zimmerman RME, Oosting J, et al. E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. British Journal of Cancer. 2006;94(5):661–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kenny PA, Lee GY, Myers CA, Neve RM, Semeiks JR, Spellman PT, et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Molecular Oncology. 2007;1(1):84–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sarrió D, Pérez-Mies B, Hardisson D, Moreno-Bueno G, Suárez A, Cano A, et al. Cytoplasmic localization of p120ctn and E-cadherin loss characterize lobular breast carcinoma from preinvasive to metastatic lesions. Oncogene. 2004;23(19):3272–83. [DOI] [PubMed] [Google Scholar]
- 50.Garcia-Recio S, Thennavan A, East MP, Parker JS, Cejalvo JM, Garay JP, et al. FGFR4 regulates tumor subtype differentiation in luminal breast cancer and metastatic disease. J Clin Invest. 2020;130(9):4871–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Levine KM, Priedigkeit N, Basudan A, Tasdemir N, Sikora MJ, Sokol ES, et al. FGFR4 overexpression and hotspot mutations in metastatic ER+ breast cancer are enriched in the lobular subtype. NPJ Breast Cancer. 2019;5:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schackmann RCJ, Van Amersfoort M, Haarhuis JHI, Vlug EJ, Halim VA, Roodhart JML, et al. Cytosolic p120-catenin regulates growth of metastatic lobular carcinoma through Rock1-mediated anoikis resistance. Journal of Clinical Investigation. 2011;121(8):3176–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-Cadherin Promotes Metastasis via Multiple Downstream Transcriptional Pathways. Cancer Research. 2008;68(10):3645–54. [DOI] [PubMed] [Google Scholar]
- 54.Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147(2):275–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Petrova YI, Schecterson L, Gumbiner BM. Roles for E-cadherin cell surface regulation in cancer. Molecular Biology of the Cell. 2016;27(21):3233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Padmanaban V, Krol I, Suhail Y, Szczerba BM, Aceto N, Bader JS, et al. E-cadherin is required for metastasis in multiple models of breast cancer. Nature. 2019;573(7774):439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Michaut M, Chin SF, Majewski I, Severson TM, Bismeijer T, de Koning L, et al. Integration of genomic, transcriptomic and proteomic data identifies two biologically distinct subtypes of invasive lobular breast cancer. Sci Rep. 2016;6:18517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guvakova MA, Surmacz E. Overexpressed IGF-I receptors reduce estrogen growth requirements, enhance survival, and promote E-cadherin-mediated cell-cell adhesion in human breast cancer cells. Exp Cell Res. 1997;231(1):149–62. [DOI] [PubMed] [Google Scholar]
- 59.Canonici A, Steelant W, Rigot V, Khomitch-Baud A, Boutaghou-Cherid H, Bruyneel E, et al. Insulin-like growth factor-I receptor, E-cadherin and alpha v integrin form a dynamic complex under the control of alpha-catenin. Int J Cancer. 2008;122(3):572–82. [DOI] [PubMed] [Google Scholar]
- 60.Bae G-Y, Choi S-J, Lee J-S, Jo J, Lee J, Kim J, et al. Loss of E-cadherin activates EGFR-MEK/ERK signaling, which promotes invasion via the ZEB1/MMP2 axis in non-small cell lung cancer. Oncotarget. 2013;4(12):2512–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.St John MA. Inflammatory mediators drive metastasis and drug resistance in head and neck squamous cell carcinoma. Laryngoscope. 2015;125 Suppl 3:S1–11. [DOI] [PubMed] [Google Scholar]
- 62.Jeanes A, Gottardi CJ, Yap AS. Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene. 2008;27(55):6920–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fedor-Chaiken M, Hein PW, Stewart JC, Brackenbury R, Kinch MS. E-Cadherin Binding Modulates EGF Receptor Activation. Cell Communication & Adhesion. 2003;10(2):105–18. [PubMed] [Google Scholar]
- 64.Petrova YI, Schecterson L, Gumbiner BM. Roles for E-cadherin cell surface regulation in cancer. Mol Biol Cell. 2016;27(21):3233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bajrami I, Marlow R, van de Ven M, Brough R, Pemberton HN, Frankum J, et al. E-Cadherin/ROS1 Inhibitor Synthetic Lethality in Breast Cancer. Cancer Discov. 2018;8(4):498–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zardavas D, Phillips WA, Loi S. PIK3CA mutations in breast cancer: reconciling findings from preclinical and clinical data. Breast Cancer Research. 2014;16(1):201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Q, Zhang Y, Zhu J, Zheng H, Chen S, Chen L, et al. IGF-1R inhibition induces MEK phosphorylation to promote survival in colon carcinomas. Signal Transduction and Targeted Therapy. 2020;5(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, et al. AKT Inhibition Relieves Feedback Suppression of Receptor Tyrosine Kinase Expression and Activity. Cancer Cell. 2011;19(1):58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated in this study are available within the article and its supplementary files. SCAN-B data analyzed in this study was obtained from Gene Expression Omnibus (GEO) at GSE96058.