

Abstract
ProSAAS is a neuroendocrine protein that is cleaved by neuropeptide‐processing enzymes into more than a dozen products including the bigLEN and PEN peptides, which bind and activate the receptors GPR171 and GPR83, respectively. Previous studies have suggested that proSAAS‐derived peptides are involved in physiological functions that include body weight regulation, circadian rhythms and anxiety‐like behavior. In the present study, we find that proSAAS knockout mice display robust anxiety‐like behaviors in the open field, light–dark emergence and elevated zero maze tests. These mutant mice also show a reduction in cued fear and an impairment in fear‐potentiated startle, indicating an important role for proSAAS‐derived peptides in emotional behaviors. ProSAAS knockout mice exhibit reduced water consumption and urine production relative to wild‐type controls. No differences in food consumption and overall energy expenditure were observed between the genotypes. However, the respiratory exchange ratio was elevated in the mutants during the light portion of the light–dark cycle, indicating decreased fat metabolism during this period. While proSAAS knockout mice show normal circadian patterns of activity, even upon long‐term exposure to constant darkness, they were unable to shift their circadian clock upon exposure to a light pulse. Taken together, these results show that proSAAS‐derived peptides modulate a wide range of behaviors including emotion, metabolism and the regulation of the circadian clock.
Keywords: anxiety, circadian activity, fear, metabolism, mice, proSAAS knockout
Pcsk1n is located on mouse chromosome X and the proSAAS protein product is processed into more than a dozen peptides. Disruption of Pcsk1n results in phenotypes suggesting its role in a wide range of behaviors including emotion, metabolism and the regulation of the circadian clock.
1. INTRODUCTION
Peptides derived from proSAAS are among the most abundant neuropeptides in the nervous system. 1 These peptides were discovered in a search for substrates of carboxypeptidase E (CPE), an enzyme involved in the biosynthesis of the vast majority of neuropeptides. 2 , 3 This search led to the discovery of novel peptides that were named SAAS, GAV, PEN and LEN based upon amino acid sequences present within each peptide, and all four of these peptides were produced from a single precursor termed proSAAS. 4 Multiple forms of each of these peptides were found which resulted from differential proteolytic cleavage of the precursor protein; this is a hallmark of neuropeptides (e.g., dynorphin A‐17 versus dynorphin A‐8). The initial study on proSAAS showed that the protein was a potent inhibitor of the neuropeptide processing enzyme prohormone convertase 1 (PC1, also known as PC3). 4 Subsequent studies mapped the inhibitory region of proSAAS to a processing intermediate near the C‐terminal region, specifically the PEN‐LEN junction. 5 , 6 , 7 However, the inhibitory capacity of this processing intermediate was lost when PEN‐LEN was fully processed to PEN and LEN. 8 Thus, while proSAAS and its processing intermediate can function as inhibitors of PC1 within the early secretory pathway, this is not the function of the numerous secreted peptides produced from the protein and it has been proposed that these peptides function as neuropeptides. 4 Receptors for two proSAAS‐derived peptides have been identified: the full‐length form of LEN (named Big LEN) activates GPR171 and PEN activates GPR83. 9 , 10 Both are G protein‐coupled receptors that have been implicated in energy balance, anxiety, reward pathways and other behaviors. 9 , 10
The distribution of proSAAS mRNA and proSAAS‐derived peptides is consistent with multiple functions including a role in neuropeptide signaling. ProSAAS is broadly distributed throughout the neuroendocrine system, although its levels vary widely. 4 , 11 , 12 , 13 Low levels of proSAAS are present in PC1‐expressing cells in the pituitary, pancreatic islets and some brain regions, while very high levels are present in the arcuate nucleus (AN) of the hypothalamus, and other nuclei within the hypothalamus and amygdala. 14
Transgenic mice overexpressing proSAAS show elevated body weight and reduced anxiety‐like behavior. 15 Subsequently, a proSAAS knock‐out (KO) mouse was generated. 16 Preliminary characterization of these mice reported a lean phenotype and elevated anxiety, consistent with a finding opposite that for the proSAAS transgenic mice. 16 The present study describes our detailed characterization of multiple aspects of anxiety‐like and fear behaviors in proSAAS KO mice. These results greatly extend the initial report on these mice and show a number of novel functions for proSAAS‐derived peptides.
2. MATERIALS AND METHODS
2.1. Animals
Since Pcsk1n is located on mouse chromosome X, wild‐type (WT) and proSAAS knockout (KO) mice were generated as described 16 by pairing heterozygous females with wild‐type (WT) or proSAAS KO males. Heterozygous females from the same litter were assigned to breeding with respective WT and proSAAS KO males, such as to minimize genetic grift and confounding the experiment. The mice had been backcrossed onto a C57BL/6J background for more than 5 generations. Groups of 3–5 mice were housed in a temperature‐ (22°C) and humidity‐controlled (45%) room with a 14:10 h light–dark cycle (lights on at 0600 h) and given food (5001 diet; PMI Nutrition International) and water ad libitum. Parenthetically, this light cycle was used to optimize the breeding potential of some strains of mice that were difficult to breed and that were co‐housed in the colony room with the proSAAS animals.
Five cohorts of mice were examined and testing occurred during the light phase unless otherwise specified. Cohort 1 was tested in the elevated zero maze (vehicle and diazepam – one‐half of the mice in each group), a week later motor activity was evaluated in the open field where only mice that received the vehicle in the zero maze were used, and 22 days later these mice were conditioned and tested for contextual and cued fear over the next 2 days. Cohort 2 mice from Pennsylvania State University were examined only in the elevated plus maze. Cohort 3 was tested in fear‐potentiated startle (FPS). Cohort 4 was evaluated in the dark–light box, 17 days later they were tested in the elevated zero maze (vehicle and diazepam – remaining half of the mice in each group), 9 days later the vehicle‐treated were subjected to the neurophysiological screen, and 3 days later the mice entered circadian rhythm testing. Cohort 5 was tested in the comprehensive lab animal monitoring system (CLAMS). The numbers of mice in each experiment can be found in the Figure Legends (9–10 mice/group) and only the CLAMS study had both 5 males and 5 females per genotype; all other cohorts consisted of males. All behavioral testing occurred during the light phase of the circadian cycle, except the circadian rhythm experiment as noted below. Adequate measures were taken to minimize pain or discomfort of the animals. All experiments were conducted with approved protocols from the Duke University and the Pennsylvania State College of Medicine Institutional Animal Care and Use Committees in accordance with NIH guidelines for the care and use of laboratory animals.
2.2. Behavioral experiments
2.2.1. Neurophysiological screen
A rapid screen for sensory and motor function was conducted as outlined. 17 , 18 , 19 , 20 , 21 , 22 Analyses included physical appearance; orientation and reflexive behaviors; postural and righting reflexes; strength, coordination and balance; tail suspension and prepulse inhibition (PPI). In the screen for orientation and reflexive behavior, the visual placement test consisted of slowly lowering the mouse to a flat surface and noting the height at which the mouse reached for the surface. The balance test, as part of the strength, coordination and balance screen, was composed of several separate tests. The latency, duration and difficulty (e.g., incoordination of fore‐ and hind‐paws, paw‐slips, etc.) the mouse experienced climbing down and climbing up a vertical 50 cm elevated wooden pole and traversing the same 8 mm pole (wrapped with ⅛″ Ravenox Solid Braid Nylon Rope; 14.35 mm total diameter). The other tests were composed of the fore‐paw wire hang test (3 mm diameter wire) conducted over 60 s and a hind‐paw coordination test where the mouse gripped a 3 mm wire by the hind paws and the ability of the mouse to hold the wire with both hind‐paws was tested as the wire was pulled‐away. Grip‐strength was assessed with an Ugo Basile grip‐strength meter (Stoelting Company, Wood Dale, IL). Tail suspension testing for behavioral despair was conducted using a mouse apparatus (Med Associates, St. Albans, VT) with established methods where the mouse was suspended by its tail for 6 min. 20 Immobility was defined as the total time when the mouse was inactive. PPI of the acoustic startle response for sensorimotor testing was monitored in a Med Associates apparatus as described. 21 Testing comprised three different types of trials. Pulse‐only trials consisted of a 40 ms 120 dB white‐noise stimulus; pulse‐prepulse trials were composed of trials where the pulse stimulus was preceded by 100 ms with a 20 ms prepulse stimulus that was 4, 8 or 12 dB above the 64 dB white‐noise background; and null trials constituted trials where no auditory stimulus was presented above the background. Mice were acclimated to the apparatus for 5 min and testing began with 10 pulse‐only trials, followed by combinations of 36 prepulse‐pulse, 10 pulse‐only and 8 null trials presented in pseudorandom order, and ending with 10 pulse‐only trials. PPI was calculated for each prepulse intensity as the ratio of prepulse‐pulse trials to pulse‐only trials subtracted from 1 and expressed as a percentage inhibition of the startle response. Normal behaviors in all tests represented responses that did not differ from the WT controls.
2.2.2. Spontaneous activity
Spontaneous activity was conducted in an open field (21 × 21 × 30 cm; AccuScan Instruments, Columbus, OH) at lux. 18 Mice were tested for 30 min in the open field in 5‐min segments. Activity was monitored as distance traveled, vertical activity and distance traveled in the center and peripheral zones of the open field.
2.2.3. Dark–light emergence test
Mice were tested in the dark–light emergence test as outlined. 19 , 20 , 23 Mice were placed into the darkened side (covered with a black curtain) of a mouse shuttle box (20 × 16 × 20) separated by a solid automated door (Med Associates). The opposite chamber was the same size and was illuminated with a 40 W fluorescent bulb positioned 25.5 cm above the chamber to produce an even illumination of 760 lux. Thirty s after being placed into the darkened chamber, the door to the adjoining lighted‐chamber was opened and mice were given free access to the entire apparatus for 5 min. The latency for the mouse to enter the lighted chamber, time spent in each chamber, the number of crosses between chambers and activity in each chamber were monitored with MedPCIV software (Med Associates).
2.2.4. Elevated zero maze
The description of the maze and the test conditions have been described. 20 , 23 Naïve mice were injected (i.p.) with vehicle (Milli‐Q water with 1% Tween‐80) or diazepam (Sigma‐Aldrich, St. Louis, MO), were placed into the closed area of the maze 30 min later, and were given 5 min of free investigation. Behaviors were videotaped and scored by blinded trained observers using Observer (Noldus Information Technology, Leesburg, VA) for the percent time in the open areas, numbers of closed‐to‐open‐to‐closed area transitions, as well as the numbers of stretch‐attend postures and head‐dips.
2.2.5. Elevated plus maze
A non‐automated elevated plus maze (Med Associates), illuminated at 800 lux, was used to assess anxiety‐like behavior at the Penn State College of Medicine. The arms of the maze were 93.7 cm above the floor and consisted of 4 perpendicular arms that were 34.9 × 6 cm. The maze had two open arms located opposite to each other as well as two closed arms that were also opposite to each other with walls that were 19.1 cm high. At the intersection of all 4 arms was a 6.1 × 6.1 center area where mice were placed at the beginning of each 5 min test. During testing open arm time was scored in real time by two trained observers.
2.2.6. Fear conditioning
Fear conditioning testing was conducted in mouse fear‐conditioning chambers (Med Associates) as described. 19 , 22 Mice were tested using a 3‐day paradigm with conditioning on day 1, context testing 24 h later and cued testing on the third day. Conditioning consisted placing the mouse in the test apparatus for 2 min after which a 72 dB 12 kHz tone (conditioned stimulus or CS) was presented for 30 s and was terminated with a 2 s 0.4 mA scrambled foot‐shock (unconditioned stimulus or UCS). After 30 s the animal was returned to its home‐cage. For context testing the mouse was exposed to the conditioning chamber for 5 min in the absence of the CS and UCS. Cued testing was conducted in a novel chamber that was different from the conditioning chamber in illumination, dimensions, shape and with a different floor and walls. Mice were placed into the chamber for 2 min, after which the CS was presented for 3 min. All responses were videotaped and scored later with the Noldus Observer program for freezing behaviors by trained observers who were blinded to the genotypes of the mice. Freezing was defined by the absence of all visible movement except that required for respiration.
2.2.7. Fear potentiated startle (FPS)
Mice were examined in FPS as outlined. 17 Testing was conducted over 5 days. On day 1, startle responses to 100, 105 and 110 dB stimuli were assessed. Each stimulus was presented 6 times in a pseudo‐randomized order for 18 trials, with an average inter‐trial interval (ITI) of 30 s (range 15–60 s). On day 2, mice were presented with each startle stimulus (100, 105 and 110 dB) 3 times, followed by 9 trials where each stimulus was preceded by a 30 s 70 dB 12 kHz tone (CS), and finally where 9 additional trials were presented with the CS alone. On day 3, 10 trials were given where the CS was followed immediately with a 0.25 s 0.4 mA scrambled shock (UCS). The CS‐UCS pairings were separated by 90–180 s. Forty‐eight hours later (day 5), the mice were assessed for FPS using the same procedure as on day 2. Baseline startle responses to the 100, 105 and 100 dB startle stimuli on test day 1 are presented in arbitrary startle units (AU). Potentiation of the startle response to the CS before and after conditioning with the UCS were calculated as a ratio of the response to the CS + startle stimuli relative to startle‐only responses and was expressed as a percentage.
2.2.8. Motor activity, feeding and indirect calorimetry
Individual mice were weighed on a Mettler‐Toledo AE240 balance (Columbus, OH) and then were monitored over 3 consecutive days in a CLAMS apparatus (Columbus Instruments, Columbus, OH). Mice were provided water and ground Lab Diet rodent chow (No. 5001; PMI Nutrition, Henderson, CO) ad libitum for the duration of the study. The first 6 h of testing were discarded because of the variability typically associated with habituation to the apparatus. Feeding and drinking behaviors, motor activity, O2 intake and CO2 output were monitored with Oxymax software and a respiratory exchange ratio (RER) was calculated as described. 10 , 24 The data were recorded in 10 min intervals over the 3‐day test period. Data were collected under a 12:12 h light–dark cycle (lights on: 0800 h) where each day began with the onset of the dark cycle at 2000 h. The room was illuminated indirectly at 180 lux during the day, and with no lights (0 lux) during the dark cycle. Final output measures included the amount of food eaten (g), water consumed (ml), activity (horizontal beam‐breaks), urine output (ml), heat expenditure (kcal/kg/h) and RER.
2.2.9. Circadian activity
Circadian activity was assessed as described. 25 Twelve‐week old male mice were housed individually in cages equipped with running wheels (Coulburn Instruments, Whitehall, PA) and containing ~4 mm of bed‐o'cobs mouse bedding (Andersons, Maumee, OH); mice were provided standard mouse chow and water ad libitum. The cages and running wheels were placed into a Phenome Technologies ventilated cabinet (Lincolnshire, IL), equipped with 526 nm green wavelength LED lights to illuminate animals during the light cycle and infrared LED lights during the dark cycle. The cages were equipped with a camera so that daily assessments of the mice could be made without disturbing them. Activity data were collected using ClockLab software (Actimetrics, Wilmette, IL). Initially mice were exposed to a 12:12 h light–dark (LD) cycle (light onset: 0800 h) for 15 days to assess their wheel‐running activity before being placed into continual darkness (DD) for 54 days. Transfer from LD to DD occurred by extinguishing the lights at 2000 h on day 15. On day 70, the mice received a 6 h light‐pulse, (300 lux white light) delivered at CT18 (circadian time units). After the light‐pulse activity was monitored in DD for another 10 days, a second light pulse was given of the same duration and intensity. Following this second pulse, mice were returned to DD for 10 days. On Day 91, mice were placed back on the 12 h LD cycle for 10 additional days, to ascertain whether their circadian rhythms could be entrained to the 12:12 h LD cycle. Food and water were replenished weekly, and cages were cleaned on test days 10, 24, 38, 52, 64, 75 and 88. This cleaning schedule was selected as it was important to ensure that the cages were not disturbed for at least 4 days before or after the presentation of light pulses or during changes from the LD to DD or the DD to LD conditions. 26 For cleaning, mice were placed into a holding cage under red light illumination while the cage containing the running wheel was cleaned; this procedure took <2 min and it was performed at different times for each cleaning. Data were analyzed and actigrams were generated with Actimetrics Clock Lab analyses software and exported to SPSS 11 (SPSS Inc., Chicago, IL) for subsequent statistical analyses. Tau (τ), activity length and the average wheel running counts (revolutions/min for each h in a single circadian day) were estimated for each mouse using data from 7 consecutive days before the completion of each phase of testing. 26 , 27 These consisted of entrainment (test days 8–14), free running period 1 (test days 62–69), free running period 2 (test days 72–79), free running period 3 (test days 83–90) and the final re‐entrainment period (days 91–101).
2.3. Statistics
Statistical analyses were performed with the SPSS‐11 statistical program (SPSS Inc.) and the results were presented as means and standard errors of the mean. Independent measures t‐tests were used to analyze responses in the neurophysiological screen, in the open field (cumulative activity), light–dark emergence, elevated plus maze, and tail suspension tests, and on null and pulse‐only trials in PPI. A two‐way ANOVA was used to analyze drug responses in the elevated zero maze. Repeated‐measures ANOVA (RMANOVA) within and between subject tests were used to evaluate the activity in the open field over 30 min (5‐min intervals), the percent PPI for the 4, 8 and 12 dB prepulse stimuli, freezing behaviors during conditioning, as well as during contextual and cued fear testing, responses to the 100, 105 and 110 dB startle stimuli in FPS, and responses in the CLAMS and circadian rhythm tests. In all cases, Bonferroni corrected pair‐wise comparisons were used as the aposterori tests and a p < 0.05 was considered significant.
3. RESULTS
Both males and females were used in the CLAMS study and no main effects for sex were detected by RMANOVA. Hence, this variable was collapsed in the subsequent analyses.
3.1. Neurophysiological screen
Compared with WT animals, proSAAS KO mice displayed no differences in body posture, pelvic or tail elevation, exopthalmos, piloerection, or in basic eye, ear or whisker reflexes when lightly touched with a cotton swab (Supporting Table S1). Grip strength and grasping reflexes were also within normal limits for the mutants. Postural and righting reflexes, as estimated by horizontal and vertical placement and contact‐righting, were similar to WT controls. In addition, no genotype differences were discerned for immobility times in the tail suspension task or in tests for null, startle or PPI activities. However, in a visual placement test, mutants had to be closer to a flat surface to show vertical placement relative to WT controls (p = 0.004) and they only visually oriented to ~76% of the objects within the visual field (p < 0.001), whereas WT mice oriented 100% of the time. Although proSAAS KO mice were capable of walking across a horizontal pole or climbing up or down a vertical pole, they took slightly longer to climb down the vertical pole compared with WT mice (p = 0.046). Collectively these studies show that WT and proSAAS KO mice were similar in gross appearance, reflexes and spinocerebellar function, PPI, and that no differences in depressive‐like responses were evident. The reduced vertical placement distance and lack of orientation to a visual stimulus by the proSAAS KO mice indicate that the mutants may be near‐sighted relative to WT controls.
3.2. Spontaneous activity
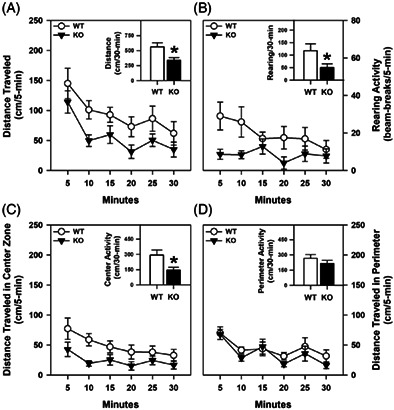
Spontaneous motor activity was examined in the open field. Locomotor and rearing activities were significantly reduced in the proSAAS KO relative to the WT controls (p‐values ≤0.044) (Figure 1A–B). When activities in the center and peripheral zones were examined separately, locomotor activities were higher in the center zone for WT than the mutant mice (p = 0.025) (Figure 1C), whereas both genotypes covered similar distances in the peripheral zone (Figure 1D). Collectively, these data show that spontaneous activities of the proSAAS KO mice are decreased relative to those of the WT controls and this is due primarily to reduced activity in the center zone.
FIGURE 1.
Spontaneous motor activity in a novel open field with proSAAS KO mice. All motor activities were monitored over 30 min in 5‐min blocks or as cumulative activities. (A) Horizontal activity as distance traveled for WT and proSAAS KO mice. A RMANOVA found significant effects of time [F (5,90) = 10.277, p < 0.001] and genotype [F (1,18) = 5.569, p = 0.030]. Inset, cumulative locomotion over 30 min [t (18) = 2.360, p = 0.030]. (B) Vertical activity as beam‐breaks. A RMANOVA detected significant effects of genotype [F (1,18) = 3.523, p = 0.044]. Inset, cumulative rearing over 30 min [t (18) = 1.804, p = 0.044]. (C) Horizontal activity as distance traveled in the center zone. A RMANOVA detected significant time [F (5,90) = 7.195, p < 0.001] and genotype effects [F (1,18) = 5.957, p = 0.025]. Inset, cumulative center distance traveled over 30 min [t (18) = 2.441, p = 0.025]. (D) Horizontal activity as distance traveled in the perimeter of the open field. A RMANOVA detected a significant time effect [F (5,90) = 7.968, p < 0.001]. Inset, cumulative perimeter distance traveled over 30 min. N = 10 mice/genotype; *p < 0.05, KO versus WT controls
3.3. Anxiety‐like behaviors
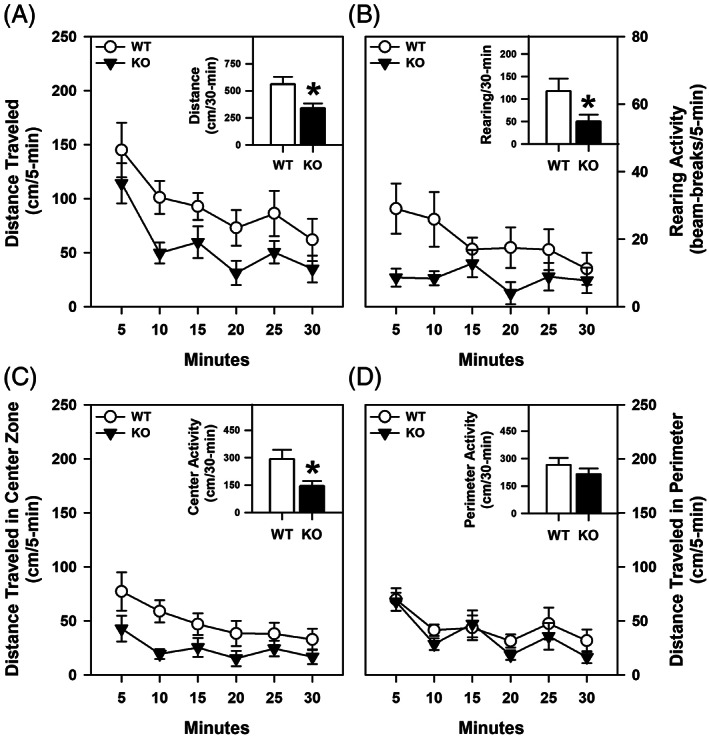
Decreased activity in the center of the open field is often interpreted as evidence for an anxiety‐like phenotype. 28 To examine this possible phenotype in greater detail, mice were tested in the dark–light emergence test and in the zero maze. In the former test, naïve proSAAS KO mice took longer to leave the darkened chamber than the WT controls (p < 0.001) (Supporting Table S2). All other behavioral indices did not differ between the genotypes, including the percent time and motor activity in the lighted chamber, total activities in the lighted and darkened chambers and the total number of crossings between the chambers. In the zero maze, vehicle‐treated proSAAS KO mice spent less time in the open areas (p < 0.001), they failed to engage in closed‐to‐open‐to‐closed area transitions (p = 0.045) and they showed fewer stretch‐attend postures (p = 0.002) and head‐dips (p < 0.001) than the vehicle‐treated WT animals (Figure 2A–D). Anxiety‐like behavior was tested independently in the elevated plus maze at Pennsylvania State University College of Medicine. On a brightly illuminated (800 lux) maze, proSAAS KO mice (3.9 ± 2.5 s) spent less time in the open arms than WT littermates (17.9 ± 5.13 s) [t (1,19) = 2.503, p < 0.05] showing the robustness of this phenotype. Together the open field, dark–light emergence, elevated zero maze and elevated plus maze results indicate that the proSAAS KO mice display robust and highly‐reproducible anxiety‐like responses.
FIGURE 2.
Behavioral responses in the elevated zero maze to diazepam for WT and proSAAS KO mice. (A) Percent time spent in open areas of the maze for mice given the vehicle, or 0.5 or 1 mg/kg diazepam. A two‐way ANOVA identified significant genotype [F (1,54) = 32.615, p < 0.001] and treatment effects [F (2,54) = 17.025, p < 0.001]; the genotype by treatment interaction was significant [F (2,54) = 7.025, p = 0.001]. (B) Open area transitions for mice administered the same regimen. A two‐way ANOVA found the genotype [F (1,54)=31.770, p < 0.001] and treatment effects [F (2,54) = 7.484, p = 0.001], and the genotype by treatment interaction to be significant [F (2,54) = 7.868, p = 0.001]. (C) Stretch‐attend postures into the open areas. A two‐way ANOVA showed the main effects of treatment [F (2,54) = 4.812, p = 0.012] and the genotype by treatment interaction [F (2,54) = 9.925, p < 0.001] were significant. (D) Head‐dip behaviors. The two‐way ANOVA observed significant genotype [F (1,54) = 11.647, p < 0.001] and treatment effects [F (2,54) = 5.099, p = 0.009], and a significant genotype by treatment interaction [F (2,54) = 4.879, p = 0.011]. N = 10 mice/genotype/treatment; *p < 0.05, KO versus WT controls; + p < 0.05, compared with vehicle‐treated mice within genotype; # p < 0.05, 0.05 versus 1 mg/kg diazepam within genotype
To test whether proSAAS KO mice respond to anxiolytic drugs, mice were administered diazepam and tested in the zero maze. When the percent time in the open areas was examined, the mutants given 1 mg/kg diazepam were found to spend less time in the open areas than the WT mice given the same dose (p < 0.001) (Figure 2A). For WT animals, 1 mg/kg diazepam increased open area time compared with their vehicle‐control (p < 0.001). By comparison, 0.5 and 1 mg/kg diazepam were equipotent in increasing open area time in the proSAAS KO mice relative to their vehicle‐controls (p‐values ≤0.007) and the enhancement with 0.5 mg/kg diazepam was not significantly different from that of WT animals given the same dose. For transitions, 1 mg/kg diazepam significantly increased these responses in WT mice relative to the proSAAS KO animals (p < 0.001) (Figure 2B). Within WT mice, 1 mg/kg diazepam significantly stimulated transitions relative to vehicle and the 0.5 mg/kg dose (p‐values<0.001), whereas in mutants both diazepam doses were without effect where transitions remained low. An examination of stretch‐attend postures showed 0.5 mg/kg diazepam increased the presentation of this behavior in proSAAS KO compared with WT mice (p = 0.005) (Figure 2C). Diazepam was without effect in WT animals, whereas both diazepam doses increased these responses in the proSAAS KOs compared with their vehicle‐control (p‐values ≤0.009) and to levels that were similar to those of the WTs given the vehicle. When head‐dips over the side of the elevated maze were examined, WT mice treated with 1 mg/kg diazepam engaged in more of these responses than similarly‐treated proSAAS KO animals (p = 0.009) (Figure 2D). The numbers of head dips were unaffected by diazepam in WT mice, while both doses enhanced these behaviors in proSAAS KOs relative to the vehicle control (p‐values ≤0.042). Collectively, these data show that some of the anxiety‐like behaviors in proSAAS KO mice can be alleviated with diazepam.
3.4. Fear conditioning
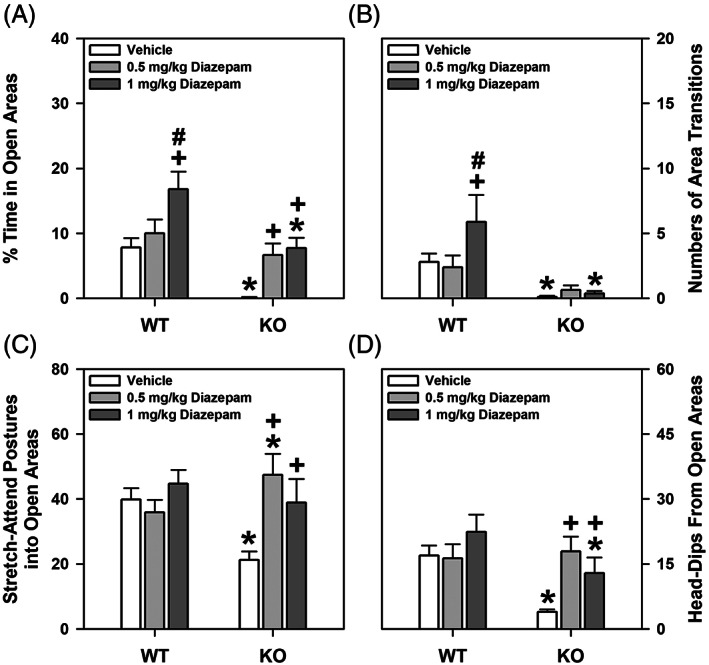
Since human anxiety disorders can share features of excessive anxiety and fear, 29 we examined fear memories in the proSAAS mice. During the first 2 min of conditioning and during the CS‐UCS presentation, no significant genotype differences in freezing behavior were observed. However, following the CS‐UCS pairing, WT mice engaged in more freezing than the mutant animals (p = 0.003) (Figure 3A). When tested for retention of contextual fear memory, no genotype differences were discerned (Figure 3B). Similarly, during the first 2 min of testing for cued fear in the absence of the CS, no genotype differences were observed (Figure 3C). However, during CS presentation in the final 3 min of testing, freezing was enhanced in WT relative to mutant animals (p‐values = 0.053).
FIGURE 3.
Freezing responses of WT and proSAAS KO mice in fear conditioning. (A) Percent freezing in the 2 min prior to CS presentation, during the CS interval, and following the CS‐UCS pairing. A RAMONA found a significant main effect of time [F (3,51) = 10.132, p < 0.001] and genotype [F (1,17) = 12.611, p = 0.002], as well as a significant time by genotype interaction [F (3,51) = 6.132, p = 0.001]. (B) Percent freezing during context testing shown in 1 min blocks across the 5 min test. An ANOVA failed to detect any significant effects. (C) Percent time spent freezing during cued testing depicted in 1 min blocks across the 5 min test. During the first 2 min no CS or UCS were presented, while in the final 3 min the CS alone was present. A RAMONA noted a significant effect of time [F (4,68) = 85.041, p < 0.001] and genotype [F (1,17) = 29.666, p < 0.001]; the time by genotype interaction was also significant [F (4,68) = 3.048, p = 0.023]. N = 9–10 mice/genotype; *p < 0.05, KO versus WT controls
To test whether the genotype effects in fear conditioning were because of differential sensitivities to foot‐shock, mice were exposed to different levels of shock. Since both WT and proSAAS KO mice responded similarly to the different intensities of shock (Supporting Figure S1), the genotype effects for fear conditioning cannot be attributed to differential responses to foot‐shock. Conjointly, these data show that proSAAS KO mice are not deficient in contextual fear but they are impaired in cued recall.
3.5. Fear‐potentiated startle (FPS)
Since the proSAAS KO mice were impaired in cued fear conditioning and because deficiencies on this task have been related to amygdala‐associated dysfunctions, 30 , 31 , 32 fear responses were assessed further using FPS. 33 On day 1, baseline startle responses were similar for WT and KO mice where overall startle responses were enhanced with increasing dBs (p‐values ≤0.003) (Figure 4A). On the second day, potentiation to the startle stimulus by the CS was similar between the genotypes to stimuli at 100 dB (WT = 21.2 ± 4.7; KO = 15.5 ± 8.4 mA), 105 dB (WT = 22.3 ± 3.8; KO = 16.4 ± 7.2 mA) and 110 dB (WT = 19.9 ± 5.1; KO = 17.7 ± 6.8 mA).
FIGURE 4.
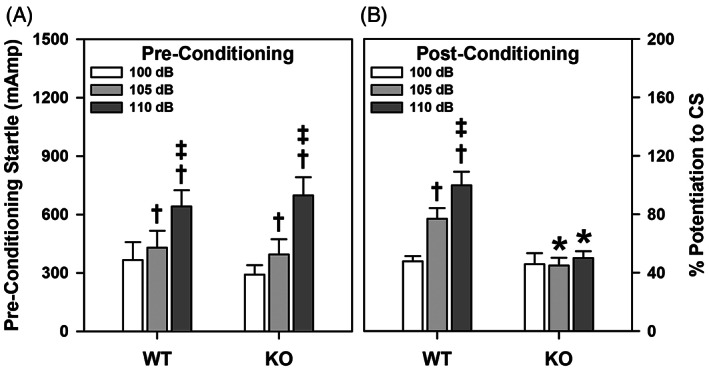
Fear‐potentiated startle in WT and proSAAS KO mice. (A) Startle responses to the 100, 105 and 110 dB white noise stimuli during pre‐conditioning. A RMANOVA found only the main effects of dB on startle intensity [F (2,34) = 38.246, p < 0.001] to be significant. (B) FPS to the CS preceding the 100, 105, and 110 dB white noise stimuli examined 48 h after CS‐UCS pairings. For post‐conditioning, a RMANOVA detected significant effects of dB on startle intensity [F (2,34) = 30.033, p < 0.001] and genotype [F (1,17) = 5.586, p = 0.030], as well as a significant effect of dB on the startle intensity by genotype interaction [F (2,34) = 19.364, p < 0.001]. N = 9–10 mice/genotype; *p < 0.05, KO versus WT controls; † p < 0.05, compared with the 100 dB response; ‡ p < 0.05, compared with the 105 dB response
Forty‐eight h following conditioning, the mice were tested. At post‐conditioning, FPS was significantly lower at the 105 and 110 dB intensities in the proSAAS KO animals than WT controls (p‐values ≤0.022) (Figure 4B). Although the WT mice showed intensity‐dependent potentiation of responses (p‐values ≤0.004), proSAAS KO mice exhibited no differences in potentiation among the 3 intensities. These data show that the proSAAS KO mice are deficient in FPS.
3.6. Motor activity, food and water intake and indirect calorimetry
ProSAAS‐derived peptides are highly expressed in the hypothalamus, 4 , 34 , 35 , 36 and are highly enriched within NPY‐expressing cells. 14 Transgenic overexpression of proSAAS leads to increases in body weight, 15 whereas Pcsk1n deletion promotes decreased body weight. 16 To further examine the physiological basis for the change in body weight in proSAAS KO mice, food and water intake were monitored and indirect calorimetry was used to access energy expenditure and metabolism in the WT and proSAAS KO mice. The body weights did not differ between the WT (30.51 ± 1.32 g) and proSAAS KO mice (30.9 ± 2.08 g). Data analyses over 20‐min blocks across the 3 days showed that the onset of motor activity, feeding and drinking behaviors in the dark cycle did not differ between genotypes or across test days. Hence, the data were collapsed into blocks of time that corresponded to the light (0800–2000 h) and dark (2000–0800 h) cycles and were averaged across the test days for each animal. In addition, the data were evaluated within each light and dark cycle for times when the mice were active or inactive (i.e., <60 beam‐breaks of horizontal activity). No genotype differences were observed for motor activity or feeding (Figure 5A–D). As anticipated overall motor activities were higher during times of activity than inactivity for both phases of the light–dark cycle, with activities highest during the night (p‐values<0.001) (Figure 5A–B). Times of inactivity were highest during the light cycle (p < 0.001). An examination of feeding behavior showed it was higher overall during times of activity than inactivity over both phases of the light–dark cycle (p‐values<0.001) (Figure 5C–D). Feeding was enhanced at times of inactivity during the light cycle (p = 0.018).
FIGURE 5.
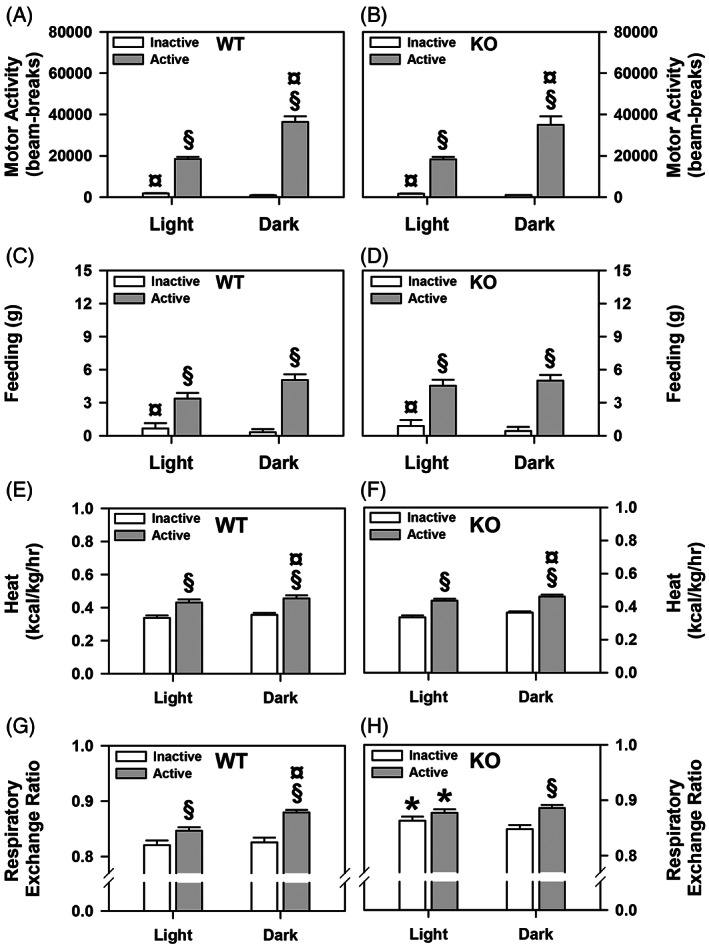
Cumulative motor activity and feeding behavior, heat production, and indirect calorimetry during the light and dark cycles for WT and proSAAS KO mice. (A–B) Cumulative motor activities of WT and proSAAS KO mice over 3 days of testing. A RMANOVA detected significant main effects of diurnal rhythm [F (1,18) = 30.765, p < 0.001] and activity periods [F (1,18) = 345.728, p < 0.001], and a significant diurnal rhythm by activity period interaction [F (1,18) = 49.920, p < 0.001]. (C–D) Cumulative feeding behaviors of WT and proSAAS KO animals. A RMANOVA for feeding behavior found a significant main effect of activity period [F (1,18) = 49.138, p < 0.001] and a significant diurnal rhythm by time interaction [F (1,18) = 6.494, p < 0.020]. (E–F) Cumulative energy expenditure by WT and proSAAS KO mice. The RMANOVA revealed significant main effects of diurnal rhythm [F (1,18) = 46.397, p < 0.001] and activity periods [F (1,18) = 287.736, p < 0.001]. (G–H) Cumulative calorimetry in WT and proSAAS KO animals. A RMANOVA for the respiratory exchange ratio noted significant effects of diurnal rhythm [F (1,18) = 12.519, p < 0.001] and activity period [F (1,18) = 109.829, p < 0.001), with the diurnal rhythm by activity period [F (1,18) = 10.691, p < 0.001] and the diurnal rhythm by activity period by genotype interactions being significant [F (1,18) = 4.040, p < 0.051]. N = 10 (5 males/genotype, 5 females/genotype) mice/genotype/cycle; *p < 0.05, versus the WT controls; § p < 0.05, active versus the inactive period within genotype; ¤ p < 0.05, light versus dark cycle within genotype and activity phase
In contrast to motor activities and feeding, genotype differences were discerned for drinking (Supporting Figure S2A–B). WT mice drank more water at times of inactivity during the light and dark cycle‐phases than the proSAAS KO animals (p‐values ≤0.002). In addition, the WTs also drank more during times of activity at night (p = 0.023). As expected, drinking was higher for both genotypes at times of activity than inactivity during the day and night (p‐values ≤0.005). Interestingly, WT mice consumed more water at times of activity during the night than the day (p < 0.001), whereby this distinction was not evident for the proSAAS KO animals. Urine output largely reflected fluid intake (Supporting Figure S2C–D). WT mice urinated more than the mutants when they were active at night (p = 0.026). Urination was higher in both genotypes during active than inactive times during both phases of the light cycle (p‐values ≤0.022). WT animals urinated more at times of activity during the dark than light phase (p = 0.001).
Energy expenditure and indirect calorimetry were examined also. Energy expenditure, as heat production, did not differ between the genotypes (Figure 5E–F). As expected heat production was higher during periods of activity than inactivity (p < 0.001) and during the dark than light cycle (p < 0.001). Indirect calorimetry was evaluated through analysis of the respiratory exchange ratio or RER (Figure 5G–H). The RER was significantly higher in the proSAAS KO mice at times of activity and inactivity during the light cycle than in the WT controls (p‐values ≤0.011). The RER was increased also in mutants relative to WT animals when they were inactive during the night, but this effect did not reach significance. In WT animals the RER was enhanced during times of activity relative to inactivity over the light and dark cycles (p‐values<0.007) and the RER at times of activity during the dark was higher than during the light cycle (p = 0.033). In proSAAS KO mice the RER was increased at times of activity relative to inactivity during the dark cycle (p = 0.006). By contrast, no differences in RER in mutants were seen between active or inactive periods during the day and these values were not different from those at active times during the night or from values for active WT mice at night. Collectively, these analyses indicate that motor activities, food intake and heat expenditure are similar between the genotypes. Nevertheless, RER was selectively elevated in the mutant mice during the inactive and active periods of the light cycle relative to the WT controls.
3.7. Circadian activity
The light–dark cycle differences in the food and water intake, and calorimetry experiments suggest there may be some differences in circadian rhythms of the WT and proSAAS KO mice. Indeed, little SAAS is found within the suprachiasmatic nucleus of the hypothalamus and exogenous application of this peptide produces a phase delay in circadian timing in vitro. 37 , 38 To examine a possible role for proSAAS in circadian rhythms in vivo, wheel running activities were examined in WT and proSAAS KO mice during 12:12 h LD cycles (lights on 0800 h), during constant DD cycles, following pulses of light during the DD cycle, and at re‐introduction to a 12:12 h LD cycle (Figure 6A–B). When exposed to a 12:12 h LD cycle for the first 15 days, wheel‐running cycles of all mice were readily entrained and no genotypic differences were found for the onset, length of the activity period or τ (Supporting Table S3, Entrainment). When placed on a DD cycle for 54 days, all mice displayed no change in τ or activity periods (Supporting Table S3, Free‐Running Period 1). Nevertheless, the onset of activity for proSAAS KO mice had a larger negative phase shift compared with WT controls (p = 0.041). Following a 6 h light pulse on day 70, WT mice exhibited a marked phase shift in wheel‐running activity (Supporting Table S3, Free‐Running Period 1), whereas mutants displayed little or no phase shift to this light pulse (p < 0.001). Following this first light pulse and the return to DD conditions, τ and the length of the activity period were similar between genotypes (Supporting Table S3, Free‐Running Period 2). Upon presentation of a second light pulse on day 80, WT mice again showed a marked phase shift in wheel‐running activity (p = 0.010), while this shift was less prominent in the mutants and was significantly different from the WT controls (Supporting Table S3, Free‐Running Period 2). Upon the return to DD for 10 days following the second light pulse, no significant genotype differences were observed for τ or the activity period (Supporting Table S3, Free‐Running Period 3). Moreover, all mice exhibited robust re‐entrainment to the 12:12 h LD cycle over the final 10 days of testing (Supporting Table S3, Re‐Entrainment).
FIGURE 6.
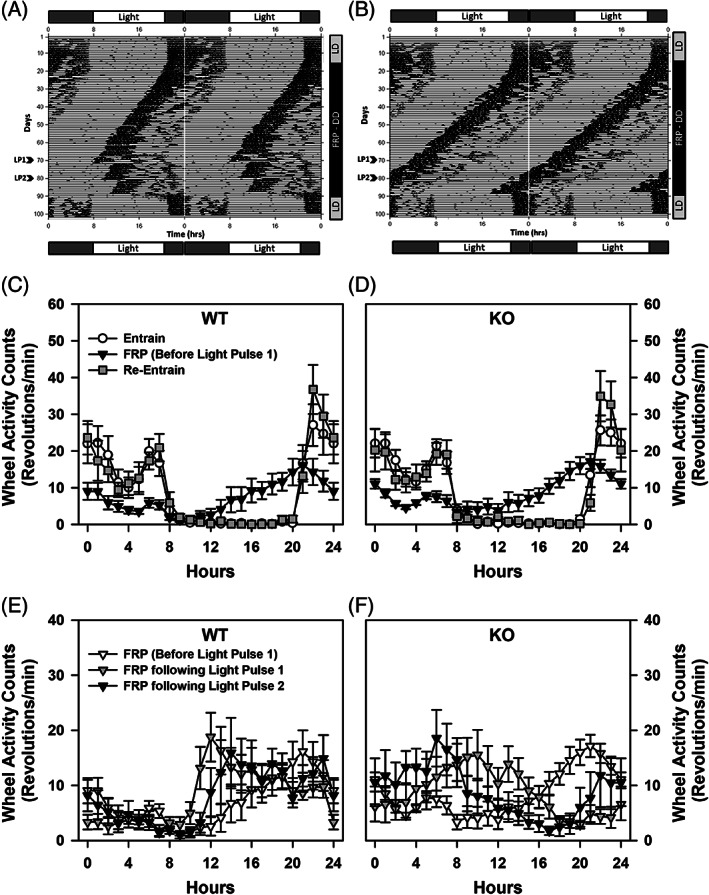
Wheel running circadian activity for WT and proSAAS KO mice. (A) Representative Actigram showing circadian wheel running activity for a WT mouse across 101 days of testing. Mice were introduced to a 12:12 h LD cycle for 15 days, followed by DD for a 75 day free‐run period during which a 6 h light pulse was given at days 70 (LP1) and 80 (LP2); mice were re‐entrained to a 12:12 h LD cycle for the final 10 days of testing. Note the shift in onset of circadian activity during DD phase and the subsequent shifts in activity to each light pulse. (B) Representative Actigram showing wheel running activity of a proSAAS KO mouse across 101 days of testing as described for the WT animal. (C–D) Mean running wheel activity counts (rpm) for WT and proSAAS KO mice during entrainment (black circle), the DD free run period (FRP) before light pulse 1 (open triangle), and during re‐entrainment (filled square), showing the onset and offset of circadian activity. A RMANOVA reported a significant effect of time [F (23,1173) = 61.234, p < 0.001] and a significant time by test‐period interaction [F (46,1173) = 12.736, p < 0.001]. (E–F) Mean running wheel activity counts (rpm) for WT mice for the 3 phases of the 75‐day DD FRP [before light pulse 1 is given (clear triangle), following the light pulse 1 on day 70 (filled triangle), and following light pulse 2 on day 80 (black triangle)]. Note that the light pulses increased wheel activity counts at 11–13 h, with very low activity prior to 10 h. (F) Mean running wheel activity counts (rpm) for all proSAAS KO mice for the 3 phases of the 75‐day DD FRP as described for WT animals. Note that the light pulses do not prevent the leftward shift in increased running wheel activity before 10 h, as seen with the WT controls. A RMANOVA found the effects of time [F (23,1173) = 2.685, p = 0.027] and the time and genotype [F (23,1173) = 6.849, p < 0.001], time by test‐period [F (23,1173) = 2.533, p = 0.008], and time by test‐period by genotype interactions [F (23,1173) = 1.655, p = 0.040] to be significant. n = 9–10 mice/genotype/condition
The circadian rhythms of the proSAAS mice were examined in greater detail in 24 h segments. These times included the end of entrainment in the LD cycle (lights on 0800 h), during the free running period (FRP) in the DD phase just before the first light pulse, just after the first and before the second light pulses, following the second light pulse and at re‐entrainment to the LD cycle (lights on 0800 h). Wheel running activity during entrainment and re‐entrainment were similar between the genotypes under a 12:12 h light–dark cycle; however, wheel‐running activities during the FRP during the DD phase (before light pulse 1) were lower for both groups compared with entrainment and re‐entrainment from 0–7, 14–20 and 22–24 h (p‐values = 0.046) (Figure 6C–D). In short, during the entrainment and re‐entrainment periods WT and proSAAS KO mice showed similar levels of wheel running during the light cycle (0–8 and 20–24 h for a total of 12 h), with rapid reductions in this behavior at the onset of the dark cycle at 8 h and marked increases at the onset of the light cycle at 20 h. Additionally, WT and mutant animals displayed similar reductions in wheel running activities during the first FRP in the DD phase prior to presentation of light‐pulse 1.
Using the activity levels at the first FRP during the DD phase as a comparison, the effects of light pulses on wheel running activity counts were examined following the first and second light pulses. No significant genotype differences were found for wheel running activity count prior to light pulse 1 (Figure 6E–F). By comparison, following the first light pulse activity counts were lower for WT mice at 3–10 h (p‐values ≤0.049), but higher at 15–19 h (p‐values ≤0.040) than for proSAAS KO animals. Essentially an identical relationship was observed after light pulse 2 where wheel running counts were lower for WT mice at 4–10 h (p‐values ≤0.049) and higher at 14–19 h (p‐values ≤0.040) than for the mutants. Within the WT group, activities following light pulse 1 were higher at 11–13 h compared with counts before the light pulse (p‐values ≤0.055) (Figure 6E). Additionally, activity counts were enhanced following light pulse 2 at 11 h (p = 0.055) relative to the first light pulse. In proSAAS KO mice, wheel running activity following the light pulse 1 was enhanced at 8 and 10 h (p‐values ≤0.013), but reduced at 17 and 19–21 h (p‐values 0.052) compared with activity before this pulse (Figure 6F). Wheel‐running counts following light pulse 2 were significantly higher at 6–8 h (p‐values ≤0.052) and they were lower at 17–20 h (p‐values ≤0.029) than those following the first light pulse. These data show that WT responses to both light pulses produced leftward shifts in the onset of wheel running activity during the FRP where counts were increased relative to those observed before light pulses 1 and 2 at approximately 11–13 h. By contrast, although the two light pulses produced changes in wheel running counts in proSAAS KO mice, their patterns of activity changes were different from those of the WT animals. Collectively, these data show that entrainment of running‐wheel activity, the onset of activity and the length of the activity periods during a LD cycle were similar between genotypes. The alterations in circadian rhythmicity during the DD FRP could be restored in WT mice with the 6‐h light pulses. However, the proSAAS mutants were refractory to these light pulses.
4. DISCUSSION
ProSAAS was first identified in 2000. 4 Although proSAAS‐derived peptides are among the most abundant neuropeptides in brain, relatively little is known about their function. Originally, proSAAS was reported to be an endogenous inhibitor of PC1/3, 4 , 7 but neither overexpression nor knockout of Pcsk1n were found to alter the levels this convertase. 15 , 16 An abundance of recent evidence shows that peptides produced from proSAAS are secreted by neurons and act as neuropeptide signals. Specifically, bigLEN activates GPR171, 10 PEN activates GPR83, 9 and other peptides derived from proSAAS produce biological effects through unidentified receptors. 38 , 39 Expression of proSAAS‐derived peptides in the amygdala, as well as the AN and SCN of the hypothalamus suggest a potential role for these peptides in reward signaling, fear and anxiety, feeding behavior and metabolism, and circadian rhythms. 4 , 12 , 40 The subsequent production of mutant mice lacking proSAAS and transgenic mice that overexpressed proSAAS confirmed many of these predictions, with mice presenting with phenotypes associated with obesity and metabolic dysregulation. 15 , 16 In addition, we have shown recently that proSAAS KO mice fail to sensitize to the locomotor‐activating effects of cocaine. 40 In the present study, we have extended these previous results by conducting detailed behavioral studies on proSAAS KO mice.
4.1. Anxiety and fear behaviors
Human anxiety disorders can involve conditions that can include fear and excessive anxiety. 29 However, while anxiety and fear share some of the same neural circuits, there are differences between them. 33 , 41 , 42 With respect to conditioned fear, the traditional view has been that deficiencies in contextual fear are due to abnormalities in hippocampal processes, whereas impairments in contextual and cued fear may be attributed to amygdala dysfunction. 30 , 31 More recently, this view has become more nuanced where contextual fear appears to be encoded by a circuit from the ventral hippocampus to the amygdala and within the amygdala itself. 43 , 44 In our experiment, we find contextual fear to be intact, while cued fear was deficient in the proSAAS KO mice. This distinction between contextual and cued fear in the proSAAS KO animals suggests at least amygdala‐associated functions may be aberrant in these mutants. 30 , 31 Nevertheless, there is some evidence that contextual fear learning also requires the amygdala. 33 , 43 , 44 The results from the FPS experiments also implicate the amygdala in the control of fear responses in the proSAAS KO mice 31 , 33 ; however, other upstream brain regions to the amygdala may be involved in this response. 31 , 32 , 33 , 41 , 42 , 43 , 45
In a separate series of studies, anxiety‐like behaviors in the proSAAS mice were examined. Previous work has demonstrated that open field activity is decreased in proSAAS KO mice compared with WT controls. 16 This reduction in spontaneous motor activity may be because of increased anxiety‐like behavior. Similar to previous work, 16 we found that open field locomotion and rearing activities in proSAAS KO mice were decreased compared with WT littermates. In support of an anxiety‐like phenotype, these mutants traveled over shorter distances in the center of the open field than WT controls, while covering similar distances in the periphery. Additionally, results from the dark–light emergence test, and the elevated zero and plus mazes were consistent with an anxiety‐like phenotype. While these findings confirmed face validity for anxiety, in a test for predictive validity the proSAAS mice were administered diazepam since this drug is known to reduce anxiety in human patients. 46 Diazepam was efficacious in alleviating some of the mutant's anxiety‐like responses in the elevated zero maze. Future experiments will examine effects of additional anxiolytics in the proSAAS KO mice and analyze what neural systems may be responsible for their anxiety‐like behaviors.
Recent work has demonstrated that direct administration of MS 21570, a GPR171 antagonist, into the basolateral amygdala (BLA) reduces anxiety‐like behavior in the elevated plus maze and in fear conditioning. 47 In addition, shRNA‐induced knockdown of GPR171 in the BLA produces a decrease in anxiety‐like behavior in the elevated plus maze. 47 While these results appear contrary to our current findings of the increased anxiety‐like behavior in proSAAS KO mice, they may be attributed to several conditions. First, the transcript and the peptides derived from proSAAS are detected widely throughout the brain, including the amygdala, cortex and hippocampus. 4 , 11 , 35 , 36 , 48 , 49 , 50 In our global proSAAS KO mice, all proSAAS‐derived peptides are absent throughout brain. Hence, the loss of these or any of the proSAAS‐derived peptides (except bigLEN, the ligand for GPR171) may be responsible for the anxiety‐like behavior. Second, anxiety may be modulated not only through proSAAS and other peptides, 51 but also through additional neural systems. 52 , 53 In this case, loss of proSAAS‐derived peptides may reduce the tone or activation of these other systems to control anxiety. Finally, deletion of Pcsk1n leads to the loss of bigLEN and its signaling at GPR171. 10 While pharmacological antagonism of bigLEN's actions at GPR171 or knockdown of this receptor in the BLA reduce anxiety‐like behavior, GPR171 is expressed in other brain regions that can exert effects on anxiety and modulate stress. 39 In this expression profile, GPR171 signaling may mediate multiple responses, some of which may counter activities in neural circuits in other brain areas that modulate anxiety. Additionally, there is evidence that GPR171 can interact with GPR83 which binds to PEN, a proSAAS‐derived peptide, and this interaction can modify the signaling through both receptors. 9 Because proSAAS KO mice lack both bigLEN and PEN, signaling through both receptors may be modified and transduction related to controlling anxiety can be compromised. Regardless, anxiety is mediated through many different mechanisms and the loss of proSAAS‐derived peptides leads to angiogenic actions.
4.2. Metabolism
Previous work has demonstrated that both PEN and bigLEN are involved in feeding behavior and body weight regulation. 14 Early studies have found that body weight is decreased in proSAAS deficient mice, whereas proSAAS overexpressing transgenic mice are obese. 15 , 16 Peptides derived from proSAAS are robustly expressed in regions of the brain involved in feeding behavior that include the AN and paraventricular nuclei (PVN) of the hypothalamus. 4 , 11 , 48 , 49 , 50 The receptor for bigLEN, GPR171, is highly expressed in these nuclei, as well as in the dorsomedial hypothalamus. Systemic administration of a GPR171 agonist stimulates feeding behavior and increases body weight gain in mice. 54 Although a GPR171 antagonist has been identified, its possible impact on feeding behavior or body weight regulation remain to be examined.
Besides GPR171, the receptor for PEN, GPR83, is expressed also in the AN and PVN of the hypothalamus, and GPR83 has been shown to co‐localize and functionally interact with GPR171 in the PVN. 9 GPR83 has additional receptor interactions. For instance, GPR83 forms heterodimers with the ghrelin receptor in the AN and these interactions negatively modulate ghrelin receptor activation and ghrelin‐induced feeding behaviors. 55 GPR83 KO mice have a ~40% decrease in fat body mass with no change in lean mass and they consume similar amounts of regular chow as WT controls. When given a high fat diet GPR83 KO animals are resistant to diet‐induced obesity and increased energy expenditure, despite having slightly elevated food consumption. 55 These results suggest that resistance to diet‐induced obesity is due to increased energy expenditure and lipid metabolism rather than reduced food intake. Collectively, these reports indicate that the proSAAS‐derived peptides, bigLEN and PEN, can influence feeding and metabolism not only through their respective cognate receptors (GPR171 and GPR83), but also through interactions with themselves or other receptors.
In the present study, we have examined the effect of removing all proSAAS‐derived peptides on feeding and metabolism. Since body weights are reduced in proSAAS deficient mice, 16 whereas proSAAS overexpressing transgenic mice are obese, 15 we used WT and proSAAS KO mice with virtually identical weights in the present studies. Although no genotype differences were observed for feeding or heat production, the RER was increased in the proSAAS KO mice during the light portion of the light–dark cycle relative to WT controls. Additionally, there were non‐significant increases in this ratio at times of inactivity during the dark cycle. An increase in the RER suggests that the proSAAS KO mice are preferentially burning carbohydrates for fuel. Thus, over time there may be a reduced tendency for carbohydrate to be stored as lipids with the consequence of a phenotype leaner than that of the WT animals. Future studies will examine whether the proSAAS KO mice are resistant to high‐fat diet‐induced obesity and hyperglycemia.
4.3. Circadian rhythms
In a previous report the proSAAS‐derived neuropeptide, little‐SAAS, is found to be released by the SCN in response to stimulation of the retino‐hypothalamic tract. 38 , 56 Addition of little‐SAAS to SCN brain slices produces a phase delay in circadian‐mediated peak firing and an activation of SCN neurons. 38 Subsequent investigations have shown that little‐SAAS co‐localizes with light‐activated c‐FOS in a subset of neurons in the central SCN, indicating that neurons expressing little‐SAAS can be activated by light signals. 37 The present study represents the first to assess whether circadian rhythms may be disrupted in vivo in mice lacking proSAAS. We find that basal circadian patterns of wheel running activity are not significantly different between WT and proSAAS KO mice. Nevertheless, during free‐run periods in continual darkness (DD) the mutants show a phase shift that is longer than in the WT animals. Moreover, when both phenotypes are exposed to light pulses in this DD phase, circadian rhythmicity is restored during this phase in WT mice, whereas the proSAAS mutants are refractory to light pulses. These findings are consistent with the previous report showing that little‐SAAS can shift the firing pattern in the SCN. 38 Together, these results indicate that proSAAS or peptides derived from this precursor play an important role in resetting the SCN in response to light cues. However, to date the receptor for little‐SAAS has not been identified.
5. CONCLUSIONS
ProSAAS protein is the precursor for over a dozen distinct peptides (including big and little forms of some of its peptides) and these peptides are among the most abundant and are broadly expressed throughout the brain. 4 , 11 , 12 , 13 , 35 , 36 Hence, it should not be surprising that deletion of proSAAS expression leads to many different phenotypes. Results from the present studies show these phenotypes fall into discrete categories that include at least anxiety‐like and fear behaviors, metabolic regulation and entrainment of circadian rhythms. Currently, only two receptors have been identified for the proSAAS‐derived peptides. BigLEN is the ligand for GPR171, whereas PEN is the ligand for GPR83. 9 , 10 While the responses to bigLEN and PEN produce actions at their own receptors, intracellular responses can become complicated since GPR171 and GPR83 can transduce not only their own signals, but they can bind also to themselves as well as other receptors to affect signaling. 9 , 55 This nuanced arrangement may become even more complex as receptors for the other proSAAS‐derived peptides are identified. In this context of receptor specificity and interactions, a study of the actions of proSAAS‐derived peptides and their receptors may reveal new paradigms for understanding a basis for associations between or among different neuropsychiatric conditions.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
Supporting information
Appendix S1. Supporting Information.
ACKNOWLEDGMENTS
This work was supported in part by National Institutes of Health grants R21 DA036385 (D.J.M.), K01 DA037355 (D.J.M.) and DA008622 (to J.P.), and discretionary funds (W.C.W.). Work was funded also, in part, under a grant (SAP # 4100057673) from the Pennsylvania Department of Health using Tobacco CURE Funds (D.J.M.). Some of the behavioral experiments were conducted with equipment and software purchased with a North Carolina Biotechnology Center grant.
Aryal DK, Rodriguiz RM, Nguyen NL, et al. Mice lacking proSAAS display alterations in emotion, consummatory behavior and circadian entrainment. Genes, Brain and Behavior. 2022;21(7):e12827. doi: 10.1111/gbb.12827
Funding information National Institute on Drug Abuse, Grant/Award Numbers: DA008622, R21 DA036385, K01 DA037355; Pennsylvania Department of Health: SAP # 4100057673
Contributor Information
Lloyd D. Fricker, Email: lloyd.fricker@einsteinmed.edu
William C. Wetsel, Email: william.wetsel@duke.edu, Email: wetse001@mc.duke.edu.
DATA AVAILABILITY STATEMENT
Data available upon request from the authors.
REFERENCES
- 1. Fricker LD. Neuropeptides and other bioactive peptides: from discovery to function. Colloquium Series on Neuropeptides. Morgan & Claypool Life Sciences; 2012:122‐133. [Google Scholar]
- 2. Fricker LD. Carboxypeptidase E. Annu Rev Physiol. 1988;50:309‐321. [DOI] [PubMed] [Google Scholar]
- 3. Fricker LD, Snyder SH. Enkephalin convertase: purification and characterization of a specific enkephalin‐synthesizing carboxypeptidase localized to adrenal chromaffin granules. Proc Natl Acad Sci U S A. 1982;79:3886‐3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fricker LD, McKinzie AA, Sun J, et al. Identification and characterization of proSAAS, a granin‐like neuroendocrine peptide precursor that inhibits prohormone processing. J Neurosci. 2000;20:639‐648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cameron A, Fortenberry Y, Lindberg I. The SAAS granin exhibits structural and functional homology to 7B2 and contains a highly potent hexapeptide inhibitor of PC1. FEBS Lett. 2000;473:135‐138. [DOI] [PubMed] [Google Scholar]
- 6. Basak A, Koch P, Dupelle M, et al. Inhibitory specificity and potency of proSAAS‐derived peptides toward proprotein convertase 1. J Biol Chem. 2001;276:32720‐32728. [DOI] [PubMed] [Google Scholar]
- 7. Qian Y, Devi LA, Mzhavia N, Munzer S, Seidah NG, Fricker LD. The C‐terminal region of proSAAS is a potent inhibitor of prohormone convertase 1. J Biol Chem. 2000;275:23596‐23601. [DOI] [PubMed] [Google Scholar]
- 8. Sayah M, Fortenberry Y, Cameron A, Lindberg I. Tissue distribution and processing of proSAAS by proprotein convertases. J Neurochem. 2001;76:1833‐1841. [DOI] [PubMed] [Google Scholar]
- 9. Gomes I, Bobeck EN, Margolis EB, et al. Identification of GPR83 as the receptor for the neuroendocrine peptide PEN. Sci Signal. 2016;9(ra43):ra43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gomes I, Aryal DK, Wardman JH, et al. GPR171 is a hypothalamic G protein‐coupled receptor for BigLEN, a neuropeptide involved in feeding. Proc Natl Acad Sci U S A. 2013;110:16211‐16216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Feng Y, Reznik SE, Fricker LD. ProSAAS and prohormone convertase 1 are broadly expressed during mouse development. Brain Res Gene Expr Patterns. 2002;1:135‐140. [DOI] [PubMed] [Google Scholar]
- 12. Fricker LD. Carboxypeptidase E and the identification of novel neuropeptides as potential therapeutic targets. Adv Pharmacol. 2018;82:85‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Morgan DJ, Mzhavia N, Peng B, Pan H, Devi LA, Pintar JE. Embryonic gene expression and pro‐protein processing of proSAAS during rodent development. J Neurochem. 2005;93:1454‐1462. [DOI] [PubMed] [Google Scholar]
- 14. Wardman JH, Berezniuk I, Di S, Tasker JG, Fricker LD. ProSAAS‐derived peptides are colocalized with neuropeptide Y and function as neuropeptides in the regulation of food intake. PLoS One. 2011;6:e28152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wei S, Feng Y, Che FY, et al. Obesity and diabetes in transgenic mice expressing proSAAS. J Endocrinol. 2004;180:357‐368. [DOI] [PubMed] [Google Scholar]
- 16. Morgan DJ, Wei S, Gomes I, et al. The propeptide precursor proSAAS is involved in fetal neuropeptide processing and body weight regulation. J Neurochem. 2010;113:1275‐1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Irwin S. Comprehansive observational assessment, 1A: a systematic, quantitative procedure for assessing the behavioral and physiological stste of the mouse. Psychopharmacologia (Berl). 1968;13:222‐257. [DOI] [PubMed] [Google Scholar]
- 18. Pogorelov VM, Rodriguiz RM, Insco ML, Caron MG, Wetsel WC. Novelty seeking and stereotypic activation of behavior in mice with disruption of the Dat1 gene. Neuropsychopharmacology. 2005;30:1818‐1831. [DOI] [PubMed] [Google Scholar]
- 19. Rodriguiz RM, Wetsel WC. Assessments of cognitive deficits in mutant mice. In: Levin ED, Buccafusco JJ, eds. Animal Models of Cognitive Impairment. CRC Press; 2006:223‐282. [PubMed] [Google Scholar]
- 20. Fukui M, Rodriguiz RM, Zhou J, et al. Vmat2 heterozygous mutant mice display a depressive‐like phenotype. J Neurosci. 2007;27:10520‐10529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodriguiz RM, Gadnidze K, Ragnauth A, et al. Animals lacking endothelin‐converting enzyme‐2 are deficient in learning and memory. Genes Brain Behav. 2008;7:418‐426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Taylor GA, Rodriguiz RM, Greene RI, et al. Behavioral characterization of P311 knockout mice. Genes Brain Behav. 2008;7:786‐795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rodriguiz RM, Wilkins JJ, Creson TK, et al. Emergence of anxiety‐like behaviours in depressive‐like Cpe fat/fat mice. Int J Neuropsychopharmacol. 2013;16:1623‐1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thupari JN, Landree LE, Ronnett GV, Kuhajda FP. C75 increases peripheral energy utilization and fatty acid oxidation in diet‐induced obesity. Proc Natl Acad Sci U S A. 2002;99:9498‐9502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pappas AL, Bey AL, Wang X, et al. Deficiency of Shank2 causes mania‐like behavior that responds to mood stabilizers. JCI Insight. 2017;2(20):e92052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu Y, Toh KL, Jones CR, Shin JY, Fu YH, Ptacek LJ. Modeling of a human circadian mutation yields insights into clock regulation by PER2. Cell. 2007;128:59‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jud C, Schmutz I, Hampp G, Oster H, Albrecht U. A guideline for analyzing circadian wheel‐running behavior in rodents under different lighting conditions. Biol Proced Online. 2005;7:101‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Treit D, Fundytus M. Thigmotaxis as a test for anxiolytic activity in rats. Pharmacol Biochem Behav. 1988;31:959‐962. [DOI] [PubMed] [Google Scholar]
- 29. American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders. 5th ed. American Psychiatric Publishing; 2013:189‐233. [Google Scholar]
- 30. Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274‐285. [DOI] [PubMed] [Google Scholar]
- 31. Fendt M, Fanslow MS. The neuroanatomical and neurochemical basis of conditioned fear. Neurosci Biobehav Rev. 1999;23:743‐760. [DOI] [PubMed] [Google Scholar]
- 32. Bergstrom HC. The neurocircuitry of remote cued fear memory. Neurosci Biobehav Rev. 2016;71:409‐417. [DOI] [PubMed] [Google Scholar]
- 33. Davis M. Neural systems involved in fear and anxiety measured with fear‐potentiated startle. Am Psychol. 2006;213:29‐42. [DOI] [PubMed] [Google Scholar]
- 34. Mzhavia N, Berman Y, Che FY, Fricker LD, Devi LA. ProSAAS processing in mouse brain and pituitary. J Biol Chem. 2001;276:6207‐6213. [DOI] [PubMed] [Google Scholar]
- 35. Heintz N. Gene expression nervous system atlas (GENSAT). Nat Neurosci. 2004;7:483. [DOI] [PubMed] [Google Scholar]
- 36. Lein ES, Hawrylycz MJ, Ao N, et al. Genome‐wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168‐176. [DOI] [PubMed] [Google Scholar]
- 37. Atkins N Jr, Mitchell JW, Romanova EV, et al. Circadian integration of glutamatergic signals by little SAAS in novel suprachiasmatic circuits. PLoS One. 2010;5:e12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hatcher NG, Atkins N Jr, Annangudi SP, et al. Mass spectrometry‐based discovery of circadian peptides. Proc Natl Acad Sci U S A. 2008;105:12527‐12532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fricker LD, Devi LA. Orphan neuropeptides and receptors: novel therapeutic targets. Pharmacol Ther. 2018;185:26‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Berezniuk I, Rodriguiz RM, Zee ML, et al. ProSAAS‐derived peptides are regulated by cocaine and are required for sensitization to the locomotor effects of cocaine. J Neurochem. 2017;143:268‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tovote P, Fadok JP, Luthi A. Neuronal circuits for fear and anxiety. Nat Rev Neurosci. 2015;16:317‐331. [DOI] [PubMed] [Google Scholar]
- 42. Apps R, Strata P. Neuronal circuits for fear and anxiety ‐ the missing link. Nat Rev Neurosci. 2015;16:642. [DOI] [PubMed] [Google Scholar]
- 43. Kim W, Cho J‐H. Encoding of contextual fear memory in hippocampus‐amygdala circuit. Nat Commun. 2020;11:1‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Goosens KA, Maren S. Contextual and auditory fear conditioning are mediated by the lateral, basal, and cantral amygdaloid nuclei in rats. Learn Mem. 2001;8:148‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shi C, Davis M. Pain pathways involved in fear conditioning measured with fear‐potentiated startle: lesion studies. J Neurosci. 1999;19:420‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Uhlenhuth EH, Turner DA, Purchatzke G, Gift T, Chassan J. Intensive design in evaluating anxiolytic agents. Psychopharmacology (Berl). 1977;52:79‐85. [DOI] [PubMed] [Google Scholar]
- 47. Bobeck EN, Gomes I, Pena D, et al. The BigLEN‐GPR171 peptide receptor system within the basolateral amygdala regulates anxiety‐like behavior and contextual fear conditioning. Neuropsychopharmacology. 2017;42:2527‐2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Feng Y, Reznik SE, Fricker LD. Distribution of proSAAS‐derived peptides in rat neuroendocrine tissues. Neuroscience. 2001;105:469‐478. [DOI] [PubMed] [Google Scholar]
- 49. Mzhavia N, Qian Y, Feng Y, Che FY, Devi LA, Fricker LD. Processing of proSAAS in neuroendocrine cell lines. Biochem J. 2002;361:67‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lanoue E, Day R. Coexpression of proprotein convertase SPC3 and the neuroendocrine precursor proSAAS. Endocrinology. 2001;142:4141‐4149. [DOI] [PubMed] [Google Scholar]
- 51. Kormos V, Gaszner B. Role of neuropeptides in anxiety, stress, and depression: from animals to humans. Neuropeptides. 2013;47:401‐419. [DOI] [PubMed] [Google Scholar]
- 52. Millan MJ. The neurobiology and control of anxious states. Prog Neurobiol. 2003;70:83‐244. [DOI] [PubMed] [Google Scholar]
- 53. Viveros MP, Marco EM, File SE. Endocannabinoid system and stress and anxiety responses. Pharmacol Biochem Behav. 2005;81:331‐342. [DOI] [PubMed] [Google Scholar]
- 54. Wardman JH, Gomes I, Bobeck EN, et al. Identification of a small‐molecule ligand that activates the neuropeptide receptor GPR171 and increases food intake. Sci Signal. 2016;9(ra55):ra55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Muller TD, Muller A, Yi CX, et al. The orphan receptor Gpr83 regulates systemic energy metabolism via ghrelin‐dependent and ghrelin‐independent mechanisms. Nat Commun. 2013;4:1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bora A, Annangudi SP, Millet LJ, et al. Neuropeptidomics of the supraoptic rat nucleus. J Proteome Res. 2008;7:4992‐5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information.
Data Availability Statement
Data available upon request from the authors.