

Abstract
Basal-like breast cancers (BLBC) are the most common triple-negative subtype (hormone receptor and HER2 negative) with poor short-term disease outcome and are commonly identified by expression of basal cytokeratins (CKs) 5 and 17. The goal of this study was to investigate whether CK5 and CK17 play a role in adverse behavior of BLBC cells. BLBC cell lines contain heterogeneous populations of cells expressing CK5, CK17 and the mesenchymal filament protein vimentin. Stable shRNA knockdown of either CK5 or CK17 compared to non-targeting control in BLBC cells was sufficient to promote an epithelial-mesenchymal transition (EMT) gene signature with loss of E-cadherin and an increase in vimentin expression. Relative to control cells, CK5 and CK17 knockdown cells acquired a more spindle-like morphology with increased cell scattering and were more invasive in vitro. However, CK5 or CK17 knockdown compared to control cells generated decreased lymph node and lung metastases in vivo. Loss of CK5 or CK17 moderately reduced the IC50 dose of doxorubicin in vitro and led to increased doxorubicin efficacy in vivo. Single-cell RNA-sequencing of BLBC patient-derived xenografts identified heterogeneous populations of CK5/CK17, vimentin, and dual basal CK/vimentin positive cells that fell on an EMT spectrum of epithelial, mesenchymal, and intermediate, respectively, while knockdown of CK5 transitioned cells towards a more mesenchymal score.
Keywords: basal-like breast cancer, cytokeratin 5, cytokeratin 17, vimentin, epithelial-mesenchymal transition
Introduction
Triple negative breast cancers (TNBC) are a diverse group of tumors primarily defined by lack of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2). TNBCs have relatively poor disease-free survival (DFS) and overall survival (OS) within the first 5 years post diagnosis compared to ER+ and HER2+ subtypes (1). Basal-like breast cancers (BLBC) constitute around half of TNBC, and are typically identified by high expression of basal cytokeratins (CKs), primarily CK5, CK14, and CK17, and other markers of basal/myoepithelial cells such as p63 and epidermal growth factor receptor (EGFR) (2–4). BLBC are often associated with BRCA1 mutations and have worse short term DFS compared to non-basal TNBCs (5,6). Numerous studies have reported that immunohistochemical detection of basal CKs alone, particularly CK5/6, is an independent indicator of reduced metastasis free and OS (Reviewed in 7). The poor short-term prognosis of BLBC, relative to other subtypes, has been attributed to higher proliferation, lack of effective targeted therapies, higher mutational burden, and incomplete pathological response to anthracycline and taxane chemotherapies (8–10). Whether basal CKs actively contribute to the aggressive properties of BLBC cells has not been extensively studied.
CKs are the major intermediate filament proteins of epithelial cells that form a dynamic cytoskeleton that provides mechanical stability and protects the cell from stress (11). CK filaments are composed of one high molecular weight (i.e. CK1–8) and one low molecular weight (i.e. CK9–28) protein that assemble into heterodimers that then polymerize to form tetramers and are expressed in a tissue-specific manner (11). CKs are frequently used in the histopathological diagnosis of adenocarcinomas. All breast cancer subtypes express abundant simple or “luminal” CKs 8, 18, and usually 19. While the stratified or “basal” CKs 5, 14, and 17 are mostly restricted to BLBC, they are sporadically expressed in luminal and HER2+ breast cancers (12,13). The term “basal-like” is controversial as basal CKs, especially CK5, are found in sporadic luminal layer cells in normal breast tissue (14). CKs impact multiple cellular processes including growth, migration, adhesion, energy metabolism, inflammation, and immunity though protein-protein interactions (15). Basal CKs 5 and 17 have been reported to regulate cell size and proliferation in oral squamous carcinoma, and cervical, pancreatic and breast cancer through interaction with proteins such as p27KIP1 and 14-3-3σ (16–20). CK14 and CK5 are also associated with collective invasion of breast cancer cells (21–23). The mechanisms by which basal CKs mediate these activities in breast cancer cells and are not well defined.
CK expression is integrally tied to the process of epithelial-mesenchymal transition (EMT), where epithelial cells lose apical-basal polarity, weaken cell-cell adhesions, and shift to a spindle-like morphology, leading to increased motility and invasion (24). EMT is accompanied by loss of epithelial markers including CKs and E-cadherin and gain of mesenchymal markers such as N-cadherin, fibronectin, and the mesenchymal intermediate filament protein vimentin (24,25). These molecular changes are orchestrated by several EMT transcription factors (EMT-TFs) (25). In cancer, this process has long been proposed as a primary mechanism for tumor cells to invade the basement membrane during the initial step of metastasis (26). However, more recent studies have divulged that EMT alone is not sufficient for metastasis and requires cooperation between tumor cells in both epithelial and mesenchymal states (27,28). Hybrid breast cancer cells that co-express E-cadherin, basal CKs, and vimentin were found to possess the most metastatic and tumor initiating potential (27,29). Therefore, the partial retention of epithelial traits is emerging as essential for tumor progression.
Our previous work identified that BLBC cells rely on CK5 to maintain E-cadherin expression and an epithelial morphology (19). In the present study, we sought to investigate how CK5 and its frequent dimeric partner CK17 impact the behavior of BLBC cells. Surprisingly, knockdown of either CK5 or CK17 was sufficient to transition cells to a predominantly mesenchymal phenotype characterized by loss of basal CKs and E-cadherin, and increased expression of vimentin and EMT-TFs, a change in cell morphology, and increased invasion in vitro. However, CK5 and CK17 knockdown increased sensitivity to anthracycline therapy and decreased the efficiency of metastatic colonization in vivo. Our findings highlight that CK5 and CK17 are important guardians of an aggressive basal epithelial phenotype and insinuates that their protein interactome could be a unique weakness of BLBC cells.
Materials and Methods
Cell culture and reagents
Breast cancer cell lines were obtained from the University of Colorado Cancer Center Cell Technologies Shared Resource. EWD8 and MDA-MB-468 cells were maintained as previously described (19). BT20 cells were maintained in DMEM/F12 1:1 medium with L-glutamine supplemented with 10% fetal bovine serum. The generation and propagation of breast cancer PDX-derived cell line UCD46 was previously described (30). Cells were maintained in Dulbecco’s Modified Eagle Media supplemented with 10% FBS, 100 ng/mL cholera toxin, and 10−9 M insulin. Cell lines were authenticated using short tandem repeat analysis and tested negative for mycoplasma using the MycoAlert mycoplasma detection kit (Lonza, Basel, Switzerland). Sigma Mission shRNAs targeting KRT5 (shCK5-22 [TRCN0000425222], shCK5-88 [TRCN0000426388]), KRT17 (shCK17-73 [TRCN0000082873], shCK17-77 [TRCN0000082877]), and non-targeting clone (SHC0002) were obtained from the University of Colorado Cancer Center Functional Genomics Shared Resource. Cells were transduced with virus containing the shRNAs and stable pools selected with puromycin as previously described (31).
Proliferation and invasion assays
Cell proliferation was measured with the IncuCyte S3 Live Cell Analysis System (Sartorius, Göttingen, Germany) using the Cell-by-Cell module essentially as described (30). A collagen-based spheroid invasion assay was used to measure invasion. 1000 cells were seeded into 96-well ultra-low attachment round bottom plates in 100 uL of culture media. The plate was centrifuged at 125 g for 10 min and returned to a 37C incubator for three days to allow spheroids to form. Rat tail collagen I (Corning, Corning, NY, USA) was diluted to 1.5 mg/mL using 10X PBS, 1N NaOH, and water. 100 uL of the collagen I mix was gently added to each well and plates centrifuged at 300 g for 10 min to center spheroids in each well. The plate was returned to the incubator for 30 min to allow collagen to polymerize then 50 uL of media was added per well and spheroids incubated for 5 days. Spheroids were imaged using the 4X objective with a Nikon Eclipse Ti-S microscope (Nikon, Tokyo, Japan). The area measurement tool on the NIS-Elements AR Analysis software (Nikon) was used to measure the core spheroid area and the outer invasion area. The invasion area was normalized to the spheroid area. Fold change in invasion area normalized to control cells was plotted. ANOVA with Tukey post-tests were used to determine statistical significance.
Chemotherapy treatment and IC50 values
MDA-MB-468 and BT20 shCont, shCK5-22, and shCK17-73 cells were plated at 5000 cells/well in sexplicate in 96 well plates. Doxorubicin-HCl (15007) or Paclitaxel (10461, Cayman Chemical, Ann Arbor, MI, USA) were serially diluted and added at the indicated concentrations the following day. Wells were imaged using the IncuCyte S3 (Sartorius) using the 10X objective every 4 hours for 96 hours. Two images were acquired per well. Percent confluency was measured using IncuCyte S3 (Sartorius). GraphPad Prism 9 (GraphPad, San Diego, CA, US) was used to determine IC50 after 72 h treatment by plotting drug concentration vs. fold change, Log10 transforming concentration, normalizing, and performing nonlinear regression. Experiment was independently performed 3–4 times and 95% confidence intervals were calculated with GraphPad Prism 9 to determine statistical significance.
Cell morphology and scatter assays
Cells were plated at 2 × 105 cells/well in 6 well plates in complete media. After 24 h, 5 fields/well were imaged using the 10X objective of a Nikon Ti-S microscope. ImageJ was used to measure aspect ratios by using the length tool to measure the length and the width of each cell. Aspect ratio was calculated by dividing the longer length by the shorter length. Aspect ratios were plotted, and statistical significance was determined using one-way ANOVA/Tukey. To measure cell scatter, single cells, loose clusters (2–10 cells), and tight clusters (>10 cells) were counted in each field according to published methods (32). The proportion of cluster type was plotted for each cell line. Chi-square tests were used to determine the statistical significance.
Immunoblotting
Immunoblots of whole cell lysates were essentially as previously described (19). Primary antibodies were as follows: CK5 (mouse NCL-L-CK5, Leica Biosystems, Buffalo Grove, IL, USA, 1:1500), E-cadherin (mouse, 14472, Cell Signaling Technologies, 1:1000), CK17 (mouse, NBP2–29421, Novus Biologicals, Littleton, CO, 1:1000), CK8/18 (mouse, NCL-L-5D3, Leica Biosystems, 1:2000), Vimentin (rabbit, 5741, Cell Signaling Technologies, 1:1000), ZEB1 (rabbit, NBP1–05987, Novus Biologicals, 1:1000) or β-actin (mouse, A5441, Sigma, 1:1000). Secondary antibodies were IRDye800CW Goat-Anti-Mouse IgG (926-32210, Li-Cor Biosciences, Lincoln, NE, USA) and IRDye 680LT Goat-Anti-Rabbit IgG (926-68021, Li-Cor Biosciences) both at 1:10,000. Immunoblots were imaged and analyzed with the Odyssey Infrared Imaging System and Image Studio Lite (Li-Cor Biosciences).
Immunocytochemistry and immunohistochemistry
Immunocytochemistry (ICC) was essentially as described (19). Primary antibodies as noted above CK5 (1:100), CK17 (1:200); vimentin (1:200), or CK8/18 (1:2000) for 2 h, secondary antibodies (A11029, A11037, A11008, A11032, Invitrogen, 1:200) for 1 h, and counterstained with DAPI. Cells were imaged using the Olympus BX40 fluorescent microscope and channels merged in Adobe Photoshop 2021. Immunohistochemistry (IHC) was performed as previously described (33) using CK5 (rabbit, ab75869, Abcam, 1:200), CK17 (rabbit, ab109725, Abcam, 1:100), vimentin (mouse, 5G3F10, Cell Signaling Technologies, 1:100), or pan-CK (mouse ab86734, 1:400, abcam, Waltham, MA) antibodies and either fluorescent secondary antibodies as described and imaged using an Olympus BX40 fluorescent microscope, or ImmPRESS Peroxidase detection kit (Vector Laboratories).
RNA-sequencing (RNA-seq), single-cell (sc) RNA-seq, and EMT scoring
For RNA-seq, MDA-MB-468 and BT20 cells with stable shCont or shCK5-22 were plated in 6 well plates at 2×105 cells/well. RNA isolation, library preparation, RNA-seq, and data analysis were essentially as previously described (30). Pathway analysis was performed using the GSEA platform.
For sc RNA-seq, derivation of breast cancer PDX was previously described (33,34). For this study, three PDX that are characterized as basal-like using the PAM50 qualifier (UCD46, UCD115, UCD18) were used (30,33). Clinical descriptions are as follows: UCD46 (primary tumor, IDC multifocal, Grade 3, ER(0%)/PR(20%)/Her2(0%)), UCD115 (primary tumor, IDC, no grade information, ER−PR−HER2−), UCD18 (primary tumor, metaplastic carcinoma, Grade 3, no ER/PR/HER2 information). PDX grown in female NOD-scid IL2Rgammanull (NSG) mice were processed into single cell suspensions and enriched for human cells as previously described (35). Sc RNA seq was performed using the 10X Genomics system, libraries were aligned to the human (GRCh38) and unique molecular identifiers (UMIs) de‐duplicated using Cell Ranger 2.1.1. Data quality control, processing, and dimensionality reduction were as previously described (36).
For EMT scoring, the published Core EMT gene signature from Taube et al. was used (37). This signature contains 159 genes that are downregulated during EMT, which we designated as “epithelial” and 87 genes that are upregulated during EMT, which we designated as “mesenchymal”. The average expression of the epithelial genes and mesenchymal genes was extracted for each cell. The ratio of epithelial:mesenchymal gene expression (EMT) followed by log2(EMT) was calculated for each cell to generate the EMT score.
In vivo experiments
Animal experiments were performed under an approved University of Colorado Institutional Animal Care and Use Committee protocol. MDA-MB-468 shCont, shCK5-22, and shCK17-73 cells were labeled with GFP using eGFP-Lv151 Lentifect purified lentiviral particles (GeneCopoeia, Rockville, MD, USA) and cells stably expressing GFP selected with neomycin. Cells were harvested, diluted in Cultrex (Trevigen, Gaithersburg, MD, USA), and 1 × 106 cells were injected bilaterally into opposing fourth mammary fat pads of 8-week-old female NSG mice. Five-six mice were used per group. Tumors were measured weekly and volumes estimated by the formula l(w2)/2. Mice were euthanized when the average tumor volume reached 500 mm3 (7 weeks for shCK17, 9 weeks for shCont, 10 weeks for shCK5). A dissecting microscope and Illumatool (Lightools Research, Encinitas, CA) was used to visualize and photograph gross lymph node metastases. The number of lymph nodes containing GFP+ tumor cells between the #4 and #2 mammary fat pads was counted and plotted. Lungs were perfused with PBS prior to paraffin embedding. Gross lung metastases were photographed using an Olympus MVX10 Microscope. Two different lung sections (per animal per group) were stained by IHC with pan CK antibodies as described. Lung metastases were quantified using a trained algorithm for CKs with the Aperio eSlide manager software (Leica) and calculating the number or strong CK+ cells per μm2 of lung tissue.
For treatment experiments, MDA-MB-468 shCont or shCK5 cells were injected into female NGS mice as described and grown to an average volume of 450–500 mm3, then stratified into treatment groups of 8 mice each. Animals were treated with vehicle (PBS) or 5 mg/kg doxorubicin (in 100 μL PBS) by intraperitoneal injection once/week for two weeks. Tumors were harvested one week following dose number two and processed as described. One animal in each control group was removed from the study prior to endpoint.
Statistical Methods
Data are represented as mean ± SEM unless otherwise noted and analyzed by two-tailed Student’s t-test, one-way ANOVA followed by a Tukey or Dunnett’s post hoc tests as indicated, two-way ANOVA, or Chi-squared analyses as noted in each figure. Graphpad Prism 9 was used for analyses when samples met variance and normality tests. P<0.05 were considered significant.
Data Availability
The data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE136260.
Results
BLBC are unique in their co-expression of epithelial and mesenchymal intermediate filament proteins
To initially assess the relationship between CK5 and CK17 and several EMT markers and hormone receptors, we determined correlation coefficients in 307 TNBCs from TCGA (38). Transcripts for CK5 and CK17 are positively associated with each other and CK14, modestly associated with mesenchymal markers vimentin and N-cadherin, and negatively associated with E-cadherin, ER, PR, and luminal CKs 8/18 (Fig. 1A). To refine these associations in BLBC we compared RNA-seq data from two hormone receptor positive breast cancer cell lines (T47D and ZR75–1) and two BLBC cell lines (MDA-MB-468 and BT20) (Fig. 1B). Each of the four cell lines had abundant transcripts for luminal CKs 8 and 18 and E-cadherin, while transcripts for basal CKs 5, 14, and 17 and vimentin were only abundant in the two BLBC cell lines. Notably, CK5 and CK17 transcripts were more abundant than CK14 in BLBC. We next assessed protein expression of several of these genes by immunoblot in a panel of breast cancer cell lines that represent hormone receptor positive (MCF7, T47D, ZR75–1), an intermediate luminal-basal cell line derived from T47D cells by long-term estrogen withdrawal (EWD8) (39), BLBC (MDA-MB-468, BT20, and UCD46 (30)), and mesenchymal-like TNBC (SUM159) (Fig. 1C). Protein levels were normalized to β-actin by densitometry and individually plotted as arbitrary units (AU) times 100 (Fig. 1D). ER+ cell lines have abundant CK8/18 and E-cadherin, lack CK5 and CK17 except for low CK17 expression in MCF7, and are absent for vimentin. The luminal-basal cell line EWD8 expresses CK5 and CK17 with lower CK18 and E-cadherin but lacks vimentin. BLBC UCD46, MDA-MB-468, and BT20 express CK5 and 17, lower E-cadherin and CK8/18 relative to ER+ cell lines, and vimentin, except for MDA-MB-468 cells where vimentin protein is not detectable by immunoblot. The mesenchymal cell line SUM159 lacked expression of luminal and basal CKs and E-cadherin and expressed vimentin. These results confirm that BLBC are unique among breast cancer cell lines in containing dual expression of epithelial/mesenchymal markers including luminal and basal CKs, vimentin, and E-cadherin.
Figure 1. BLBC cell lines contain heterogeneous expression of basal CKs and vimentin.
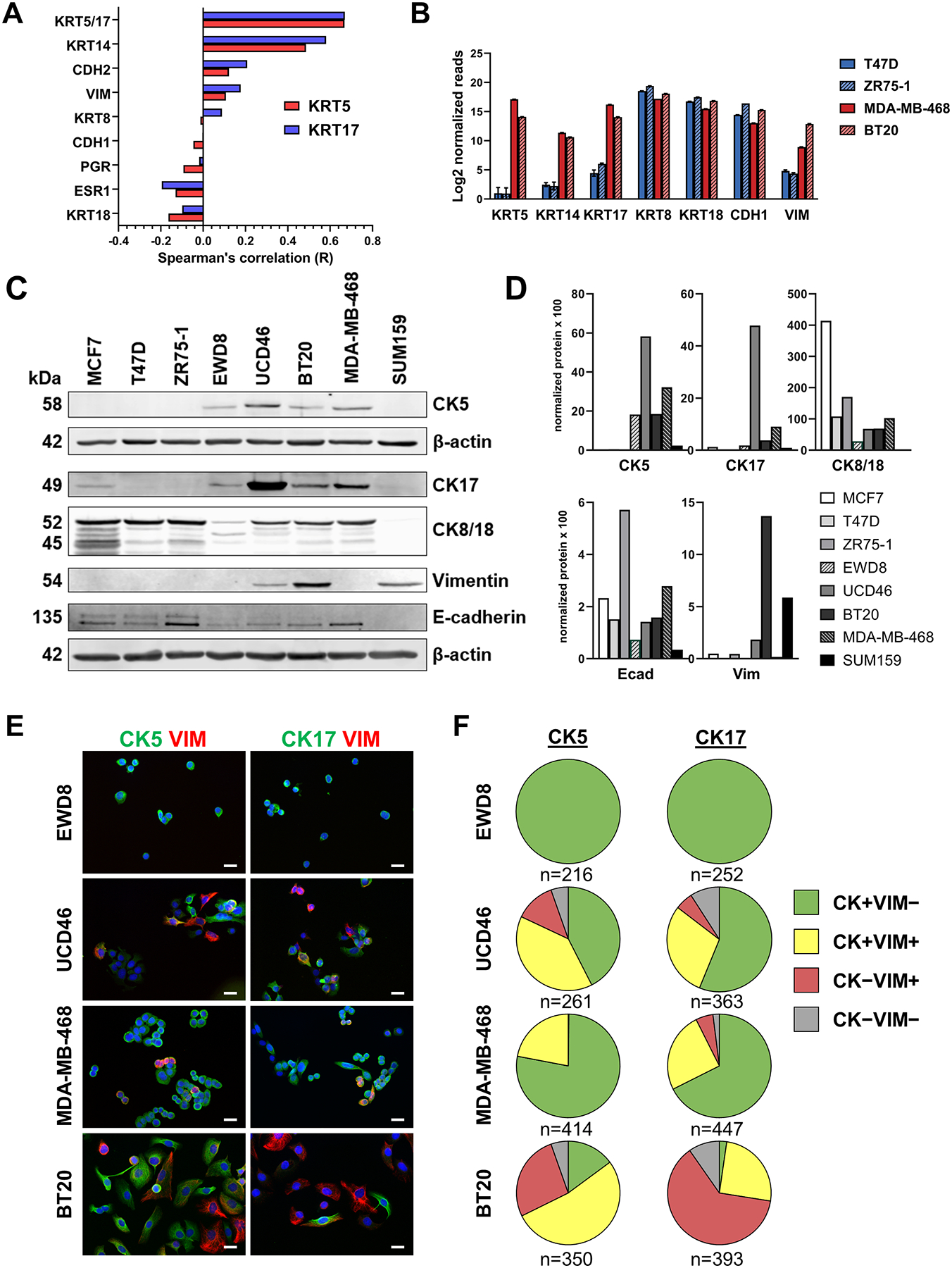
A. Spearman correlation coefficients for KRT5 (red bars) and KRT17 (blue bars) for the indicated genes derived from mRNA expression data for The Cancer Genome Atlas (TCGA, cbioportal) for 307 TNBCs. B. Log2 normalized mRNA expression of the indicated 7 genes from RNA-seq data of two ER+ (T47D, ZR75–1) and two BLBC (MDA-MB-468, BT20) cell lines. C. Cell lysates were collected from the indicated 8 cell lines and analyzed by immunoblot for the indicated proteins. β-actin was used as a loading control. CK5 was run on a separate blot. D. Protein levels were normalized to β-actin by densitometry and individually plotted as arbitrary units (AU) times 100. E. Dual-fluorescent ICC for CK5 (green) or CK17 (green) and vimentin (VIM, red), plus DAPI (blue) counterstain in luminal-basal cell line EWD8 and BLBC cell lines UCD46, MDA-MB-468, and BT20. Scale bars, 20 μm. F. Pie charts indicating the proportion of cells that are positive for CK5 (left) or CK17 (right) only (green), VIM only (red), CK5/17 plus VIM (yellow), or none (gray) for the four cell lines. Number of cells counted is indicated.
To determine the degree of co-expression of CK5, CK17, and vimentin in BLBC cells we performed dual ICC in four cell lines that are CK5/CK17+ (EWD8, MDA-MB-468, BT20, and UCD46). We observed significant co-expression of CK5 and CK17 within the same cells (60–87% of cells), with <5% of cells negative for both CKs in each of these cell lines (Supplementary Fig. S1). We next looked at co-expression of CK5 or CK17 with vimentin by ICC (Fig. 1E). Pie charts indicate the proportion of cells positive for CK5 or CK17 only, VIM only, CK5/17 plus VIM, or none in the four cell lines (Fig. 1F). Consistent with immunoblot data, no vimentin was detected in luminal-basal EWD8 cells. UCD46, MDA-MB-468, and BT-20 BLBC cells showed various degrees of basal CK, vimentin, and dual basal CK/vimentin positive cells. MDA-MB-468 are predominantly basal CK+, with a moderate dual positive population and few vimentin only cells (consistent with immunoblots). Vimentin positive and dual basal CK/vimentin positive populations were more abundant in BT20 vs MDA-MB-468 and UCD46 cells. All cells were ubiquitous for luminal CK8/18 expression (Supplementary Fig. S2), suggesting basal CKs are more heterogeneous with vimentin expression and could be pivotal in determining cell phenotype and behavior.
Genetic knockdown of basal CKs enhances an EMT molecular signature in BLBC
In our previous study, we observed that shRNA knockdown of CK5 in MDA-MB-468 cells led to complete loss of E-cadherin, partial loss of membrane β-catenin, and a more spindle-like morphology (19). To study this further, we generated additional stable CK5 and CK17 knockdown BT20 and MDA-MB-468 cells using two independent shRNAs each (shCK5-22 & shCK5-88; shCK17-73 & shCK17-77) and a control construct (shCont). To assess global changes in gene expression, we performed RNA-seq on control (shCont) vs CK5 knockdown (shCK5-22) MDA-MB-468 and BT20 cells. Gene Set Enrichment Analysis (GSEA) identified several common pathways affected by loss of CK5 including an EMT gene signature (Fig. 2A). Additional overlap and pathways are shown in Fig. S3. Heatmaps of core EMT genes in shCont vs shCK5-22 cells illustrates a striking shift in EMT marker expression (Fig. 2B). Knockdown of CK5 produced a dramatic decrease in basal CK5, 14, and 17 as well as E-cadherin transcripts in both cell lines. Luminal CK8/18 transcripts were also reduced in MDA-MB-468 cells. The canonical mesenchymal marker vimentin was increased in both cell lines, while N-cadherin (CDH2) and fibronectin (FN1) increased specifically in MDA-MB-468 cells. EMT-TF transcripts were also upregulated with CK5 knockdown, most notably ZEB1 and ZEB2 in both cell lines. By immunoblot (Fig. 2C), CK5 knockdown led to decreased CK5 and CK17 expression and vice versa in both cell lines. shCK5-88 was less efficient at reducing CK5 and CK17 in MDA-MB-468 cells (Fig. 2C). Vimentin levels increased substantially after CK5 or CK17 knockdown in both cell lines while E-cadherin levels decreased. The EMT-TF ZEB1 generally increased with CK5 and CK17 knockdown, except for shCK17-73 BT20 cells. Thus, decrease of a single basal CK is sufficient to decrease the expression of epithelial and increase the expression of mesenchymal genes in BLBC cells.
Figure 2. Knockdown of CK5 or 17 in BLBC cells produces an EMT molecular signature.
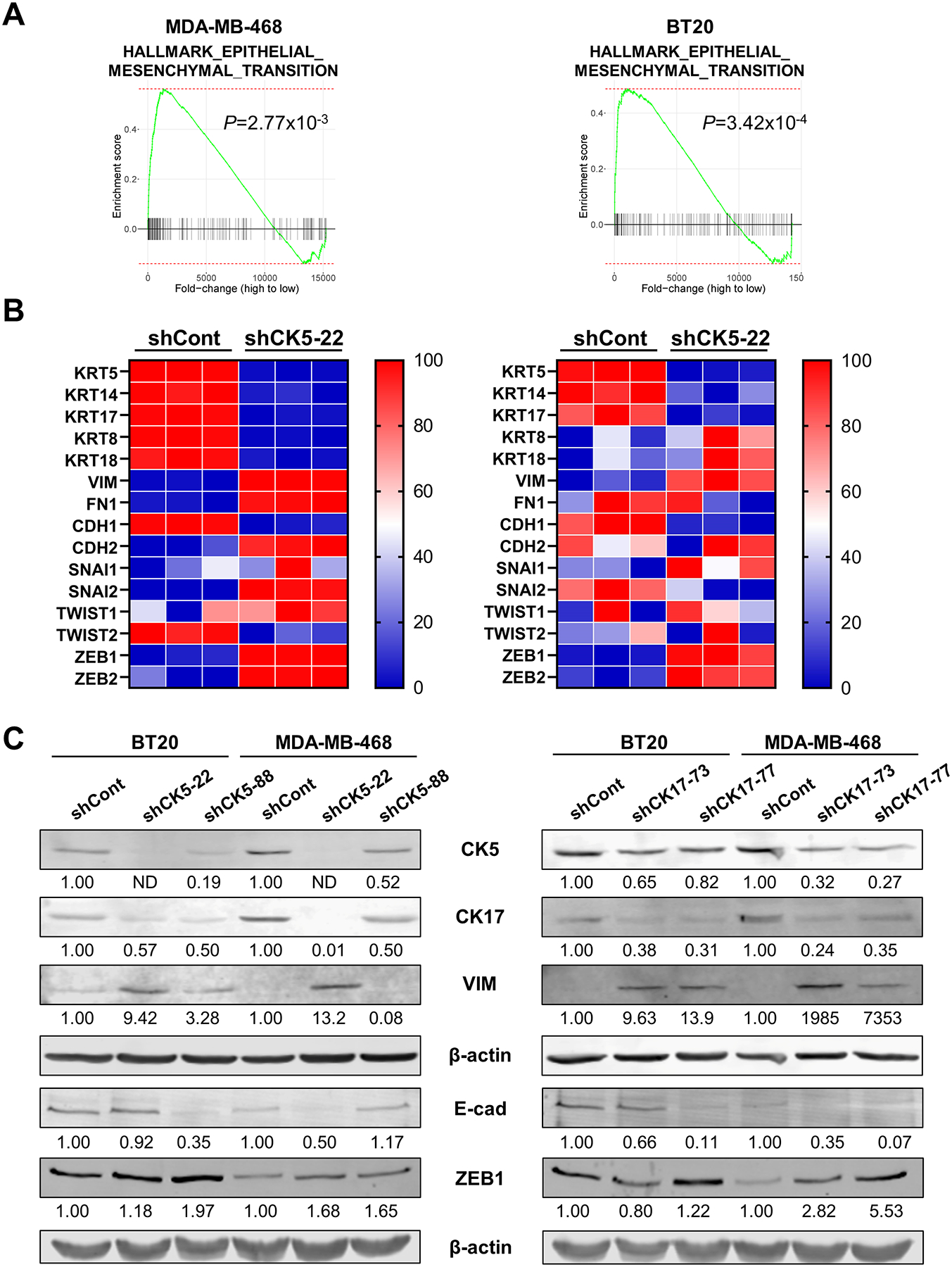
A. Gene set enrichment analysis of RNA-seq data from shCont and shCK5-22 MDA-MB-468 (left) and BT20 (right) cells identified Hallmark EMT as a significantly enriched pathway in both cell lines. B. Heatmaps of select EMT genes were generated from triplicate RNA-seq samples of shCont and shCK5-22 in MDA-MB-468 (left) and BT20 (right) cells, normalized to each gene.
C. Cell lysates were collected from shCont and shCK5 (left) or shCK17 (right) BT20 and MDA-MB-468 cells (2 shRNA constructs each) and analyzed by immunoblot for CK5, CK17, vimentin (VIM), E-cadherin (E-cad), and ZEB1. β-actin was used as a loading control; E-cad and ZEB1 were run on a separate blot. Protein levels were normalized to β-actin and indicated as fold change over shCont. ND = not detected.
Loss of basal CKs increases mesenchymal morphology and behavior in BLBC cells in vitro
Since CK5 and 17 knockdown altered expression of key EMT genes, we next investigated whether their inhibition conferred phenotypic changes associated with EMT. Brightfield microscopy of control vs CK5 or CK17 knockdown cells shows notable changes in cell morphology (Fig. 3A). MDA-MB-468 cells transitioned from a more rounded to a more spindle-like morphology with less cell aggregates. While BT20 cells have a more innate spindle cell appearance, loss of CK5 or CK17 still notably altered cell morphology with fewer aggregates. To quantify these observations, we first measured the aspect ratio (cell length/width) and found that compared to shCont cells, shCK5 and shCK17 MDA-MB-468 cells had significantly higher aspect ratios (Fig. S4), while the aspect ratio did not significantly differ in BT20 cells, possibly due to their more naturally elongated shape. Second, we quantified cell scatter according to a published method that is frequently used to evaluate intercell cohesion as a measure of EMT (32). The number of single cells and loose clusters increased while the number of tight clusters decreased in both cell lines with shCK5-22 and both shCK17 constructs (Fig. 3B). shCK5-88 cells with less efficient knockdown (Fig. 2C) were not significantly different. To test whether morphological changes with basal CK knockdown were associated with more mesenchymal behavior we assessed invasion using a collagen I matrix and spheroid-based invasion assay. Representative images of spheroids derived from control vs CK5 or CK17 knockdowns are shown in Fig. 3C. Loss of either CK5 or CK17 significantly increased invasion of MDA-MB-468 and BT20 cells compared to shCont cells (Fig. 3D). These data support long held observations that cells that undergo EMT are more invasive in vitro.
Figure 3. Knockdown of CK5 or 17 increases mesenchymal morphology and behavior of BLBC cells.
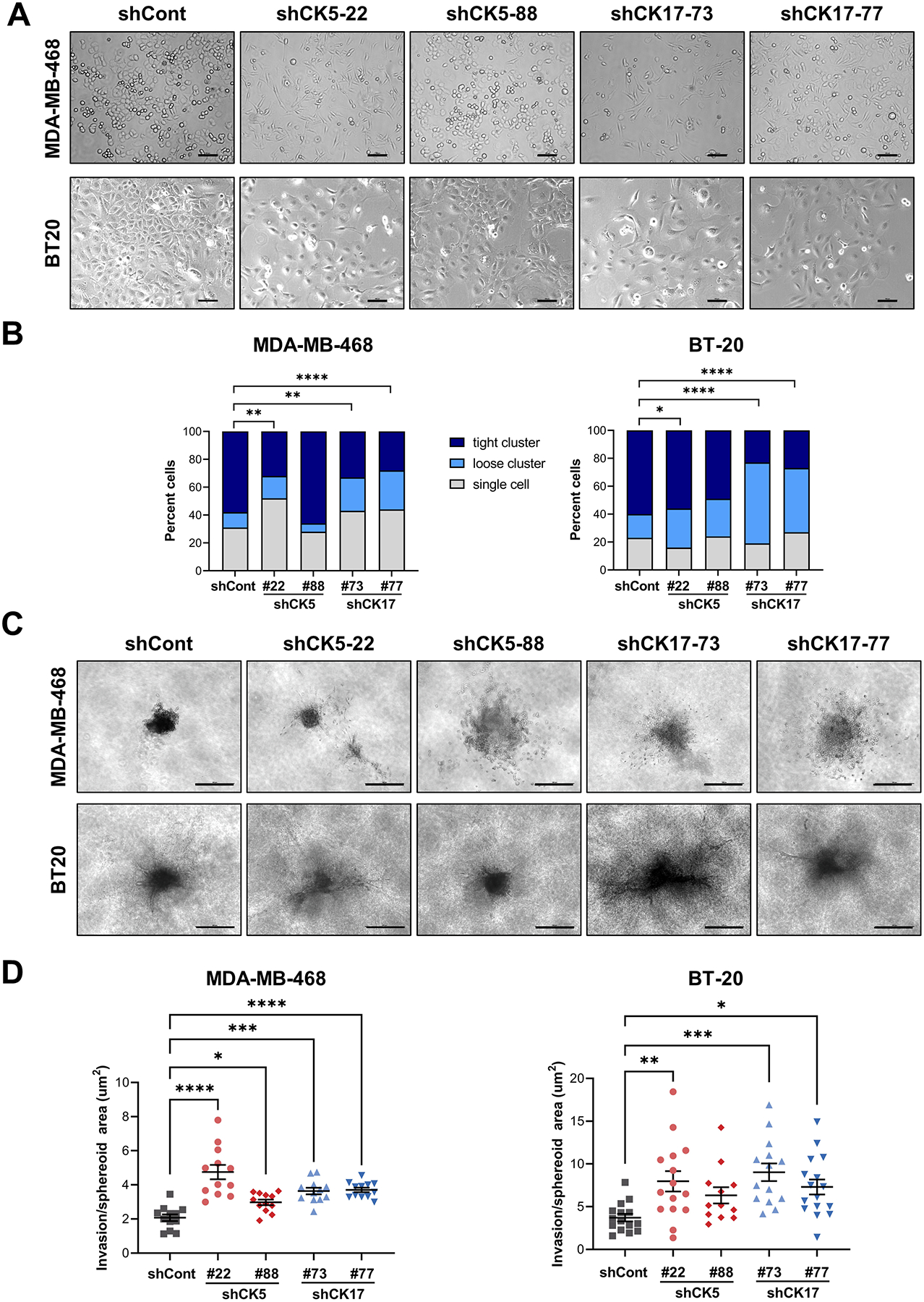
A. Bright-field images of MDA-MB-468 (top) and BT20 (bottom) cells with stable shCont or two constructs of shCK5 and shCK17. Scale bars, 100 μm. B. Cell scatter was measured by counting the number of single cells, loose clusters (2–10 cells), and tight clusters (>10 cells) in five bright-field images. The number of counted events ranged from 81–126 in MDA-MB-468 and 60–100 in BT20. The percent of cells in each category is graphed. Chi-square tests of contingency tables comparing each shRNA construct to shCont are indicated. C. Invasion was measured using a spheroid invasion assay. Spheroids were generated from the indicated cell lines on a Collagen I matrix for five days. Representative images are shown. Scale bars, 500 μm. D. The spheroid core and invasion area were measured using NIS-Elements AR analysis software and plotted as invasion/spheroid area. Bars represent mean ± SEM. One-way ANOVA/Dunnett’s test compared to shCont was used to determine statistical significance. N=12–15/group. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Knockdown of CK5 or CK17 decreases metastasis efficiency in vivo
To test the impact of knockdown of CK5 or CK17 on metastatic potential, we implanted GFP-labeled MDA-MB-468 shCont, shCK5-22, and shCK17-73 cells orthotopically and bilaterally into the fourth mammary fat pads of female NSG mice and measured tumor growth, and lymph node and lung metastasis. We controlled for tumor size by collecting tumors at the same average volume of 500 mm3 (Fig. 4A). While shCK5-22 tumors took 1–2 weeks longer to achieve similar tumor volume vs shCont and shCK17-73 tumors, final tumor burden was not significantly different among the groups (Fig. 4B). We assessed lymph node metastasis by measuring the number of lymph nodes that were GFP+ between the #4 and #2 mammary glands at necropsy (Fig. 4C). The number of positive lymph nodes was significantly higher in mice bearing shCont vs shCK5-22 or shCK17-73 tumors (Fig. 4D). To assess spontaneous hematogenous metastasis, lungs were dissected, perfused, and imaged (Fig. 4E). Lungs from mice with shCont tumors had observably more and larger foci than mice bearing shCK5 and shCK17 tumors. Paraffin sections of lungs were stained by IHC for pan-CK and the number of strong CK+ cells/area quantified using digital analysis. Lungs from mice with shCont tumors yielded significantly more lung disseminated tumor cells compared to lungs from mice with shCK5 or shCK17 tumors (Fig. 4F). We repeated this experiment and harvested tumors at the same time point with similar results (Supplementary Fig. S5). Dual IHC for CK5/vimentin or CK17/vimentin on spontaneous lung metastases for each cohort reflect loss of basal CKs in knockdown lesions (Supplementary Fig. S6). These results support that BLBC shCont tumors with heterogeneous expression of CKs 5 and 17 and vimentin are more efficient at spontaneous metastasis compared to isogenic cells that are predominantly vimentin positive through CK5 or CK17 knockdown.
Figure 4. Loss of CK5 and CK17 reduces lymph node and lung metastasis.
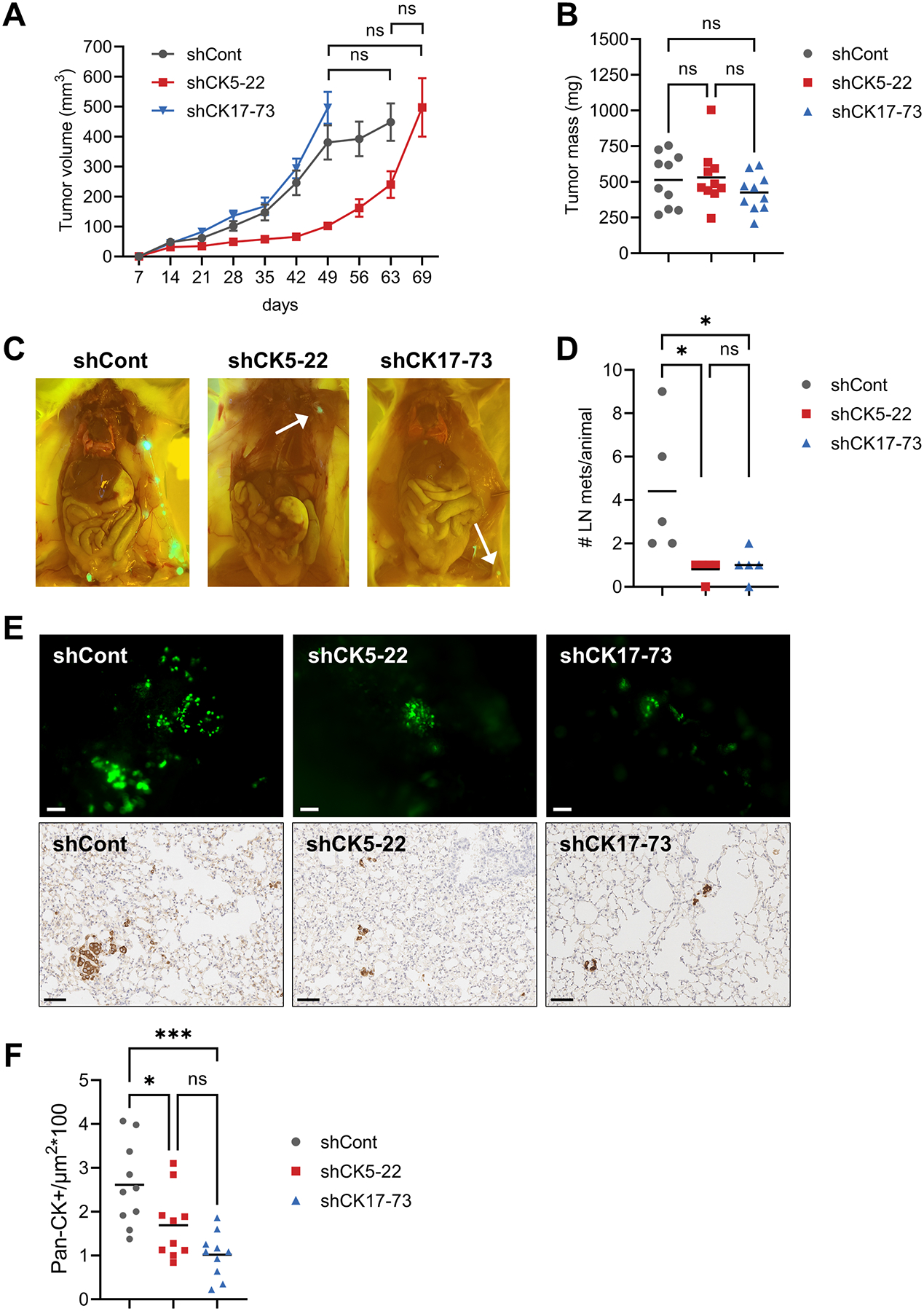
A. Growth of GFP-labeled MDA-MB-468 shCont, shCK5-22, and shCK17-73 as xenografts in female NSG mice. Tumors were grown until each group reached an average volume of 500 mm3. Tumor volume over time is depicted. N=10 tumors/group. One-way ANOVA/Tukey test of tumor volumes at the final timepoints is indicated. ns, not significant. B. Final tumor mass for each group. One-way ANOVA/Tukey test. C. Representative images of lymph node metastases at necropsy for each group. D. Number of positive lymph nodes (LN)/animal. One-way ANOVA/Tukey test is indicated. *P<0.05. E. Representative gross images of GFP+ lung metastases in each group (top), scale bars, 100 μm, and representative images of IHC for pan-CK in lung sections for each group (bottom), scale bars, 60 μm. F. Quantitation of lung metastases for each group. Two different sections of lung per animal were stained by IHC with pan-CK antibody and counted as number of strong CK+ cells/μm2 using a trained algorithm on the Aperio digital e-slide analyzer. One-way ANOVA/Tukey was used to determine significance.
Basal CK deficiency enhances doxorubicin therapy
Chemotherapy is the current standard of care for TNBC tumors, typically an anthracycline-taxane sequence (40). CK17 has been associated with reduced sensitivity to chemotherapy in cervical cancer (16). To test how loss of CK5 and CK17 impact chemotherapeutic response in BLBC cells, we performed dose response curves for doxorubicin and paclitaxel in MDA-MB-468 and BT20 shCont, shCK5-22, and shCK17-73 cells. Basal growth rates were lower in shCK5 vs shCont cells in both cells lines (Fig. 5A). Representative dose response curves for doxorubicin are depicted in Fig. 5B. Knockdown of CK5 or CK17 reduced the half-maximal inhibitory (IC50) dose of doxorubicin in half for each cell line (Fig. 5C); replicates are shown in Fig. S7. Knockdown of CK5 or CK17 did not significantly impact paclitaxel sensitivity (Supplementary Fig. S7).
Figure 5. Loss of basal CKs increases chemosensitivity to doxorubicin.
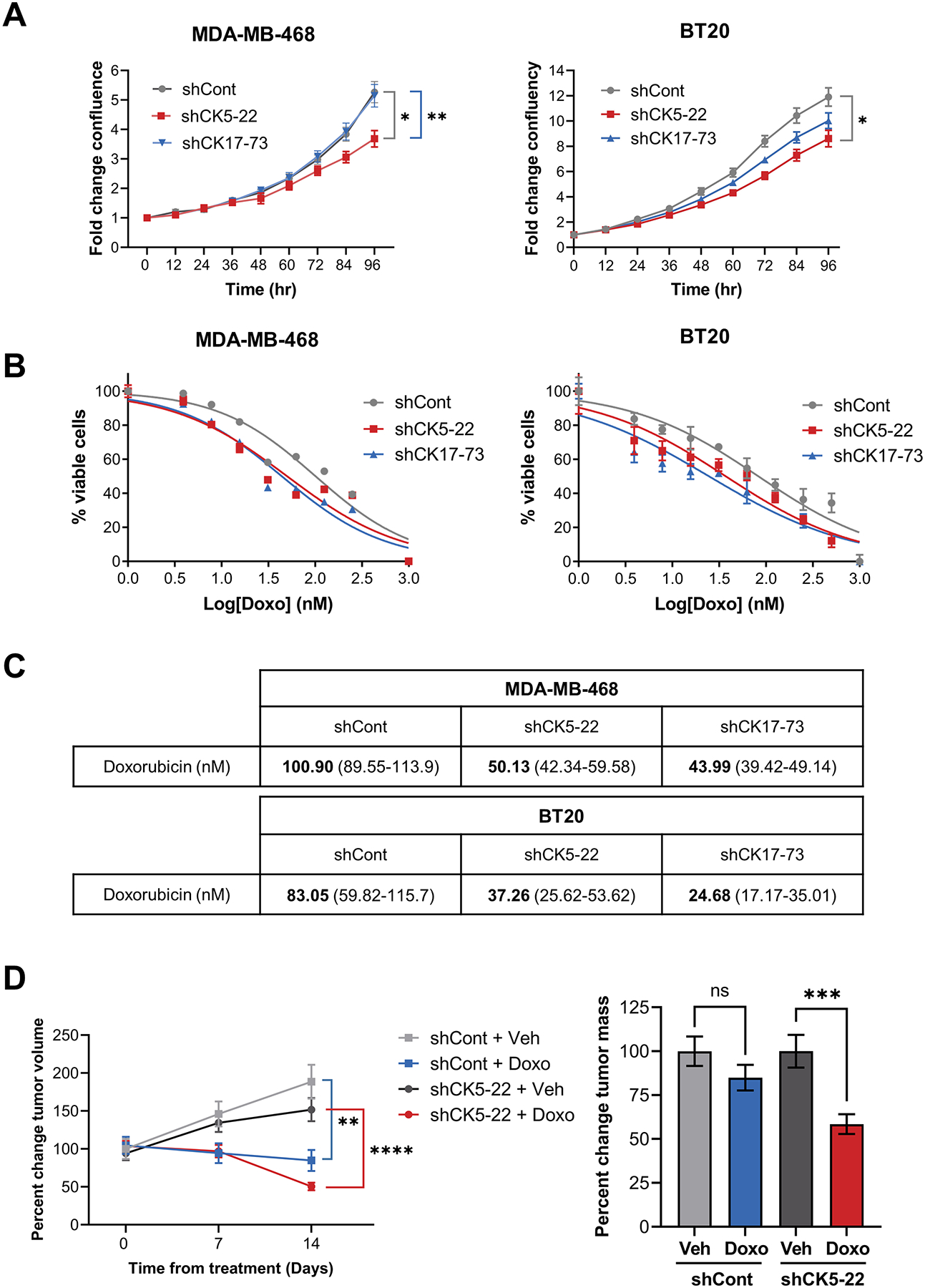
A. Growth of shCont, sh-CK5-22, and shCK17-73 MDA-MB-468 (left) and BT20 (right) cells was measured over time using the IncuCyte. Significant differences are indicated for the final timepoint, two-way repeated measures ANOVA, N=6/group. B. The indicated cell lines were treated with varying concentrations of doxorubicin (Doxo) (B) in sextuplicate and growth monitored using the IncuCyte. Normalized transformed graphs are plotted for percent cell viability vs Log(conc) drug at 72 h. Error bars represent mean ± SEM. Experiments was repeated twice, representative graphs are shown. C. Table shows IC50 values for doxorubicin in the indicated cell lines. 95% confidence intervals are shown in parentheses. D. MDA-MB-468 shCont and shCK5 tumors were grown until an average volume of 450–500 mm3, stratified into treatment groups of vehicle (Veh) or Doxo for two weeks. Mean changes in tumor volume from time zero (left) and percent change in final tumor mass normalizing the Doxo groups to their corresponding vehicle group post-treatment (right) are plotted. T-tests at the final timepoint are indicated comparing Veh vs Doxo. N=14 for shCont tumors and N=16 for shCK5-22 tumors.
To assess doxorubicin sensitivity in vivo, MDA-MB-468 shCont and shCK5-22 tumors were grown to an average volume of 450–500 mm3 then treated with vehicle or doxorubicin for two weeks (Fig. 5D). By tumor volume measurement, doxorubicin treatment decreased tumor volume in both shCont and shCK5-22 tumors. However, final tumor mass was only significantly reduced in shCK5 tumors (by >40%). Raw tumor growth curves and tumor mass, and IHC for CK5/vimentin in shCont and shCK5 vehicle and doxorubicin treated tumors are shown in Supplementary Fig. S8. These data support the BLBC cells that retain basal CKs are slightly less sensitive to doxorubicin treatment.
BLBC PDX contain heterogeneous populations of basal CK and vimentin positive cells that exist across an EMT spectrum
To investigate intermediate filament expression in solid tumor models of BLBC, we analyzed several of our PDX that classify as basal-like on the PAM50 classifier (UCD46, UCD115, and UCD18) (30,33). Dual IHC for CK5/CK17, CK5/vimentin or CK17/vimentin in each tumor uncovered a high degree of CK5/CK17 co-expression with populations of dual CK5/vimentin or CK17/vimentin varying between tumors from rare (UCD46) to common (UCD18) (Fig. 6A). Single-cell RNA-seq (sc RNA-seq) was performed on the three PDX. A binary epithelial or mesenchymal classification in addition to UMAP plots for the genes for CK5 (KRT5), CK17 (KRT17), and vimentin (VIM) are shown in Supplementary Fig. S9. We applied a published EMT gene signature consisting of 246 genes (37) to generate an EMT score for individual cells expressing CK5 or CK17, vimentin, or all three markers (Fig. 6B). A positive, higher score indicates a more epithelial state and a negative, lower score indicates a more mesenchymal state. Not surprisingly, within each tumor model, CK5/CK17+ cells are the most epithelial, vimentin+ cells the most mesenchymal, and CK5/CK17/vimentin+ cells intermediate. There were also intertumoral differences in EMT scores. For example, all three populations in UCD46 were more epithelial vs the same groups in UCD115 and UCD18, underscoring general differentiation state differences between tumors. Application of the same EMT score to RNA-seq data from MDA-MB-468 and BT20 shCont vs shCK5 cells confirm that CK5 knockdown induces a shift to a more mesenchymal state – this was more pronounced in MDA-MB-468 cells (Fig. 6C). These experiments confirm the innate epithelial-mesenchymal heterogeneity of BLBC cells and coupled with our genetic knockdown data, implicate that retention of basal CK expression in epithelial or intermediate populations is central to the rapid progression of this disease.
Figure 6. CK5/17 denote more epithelial cells on the EMT spectrum in TNBC.
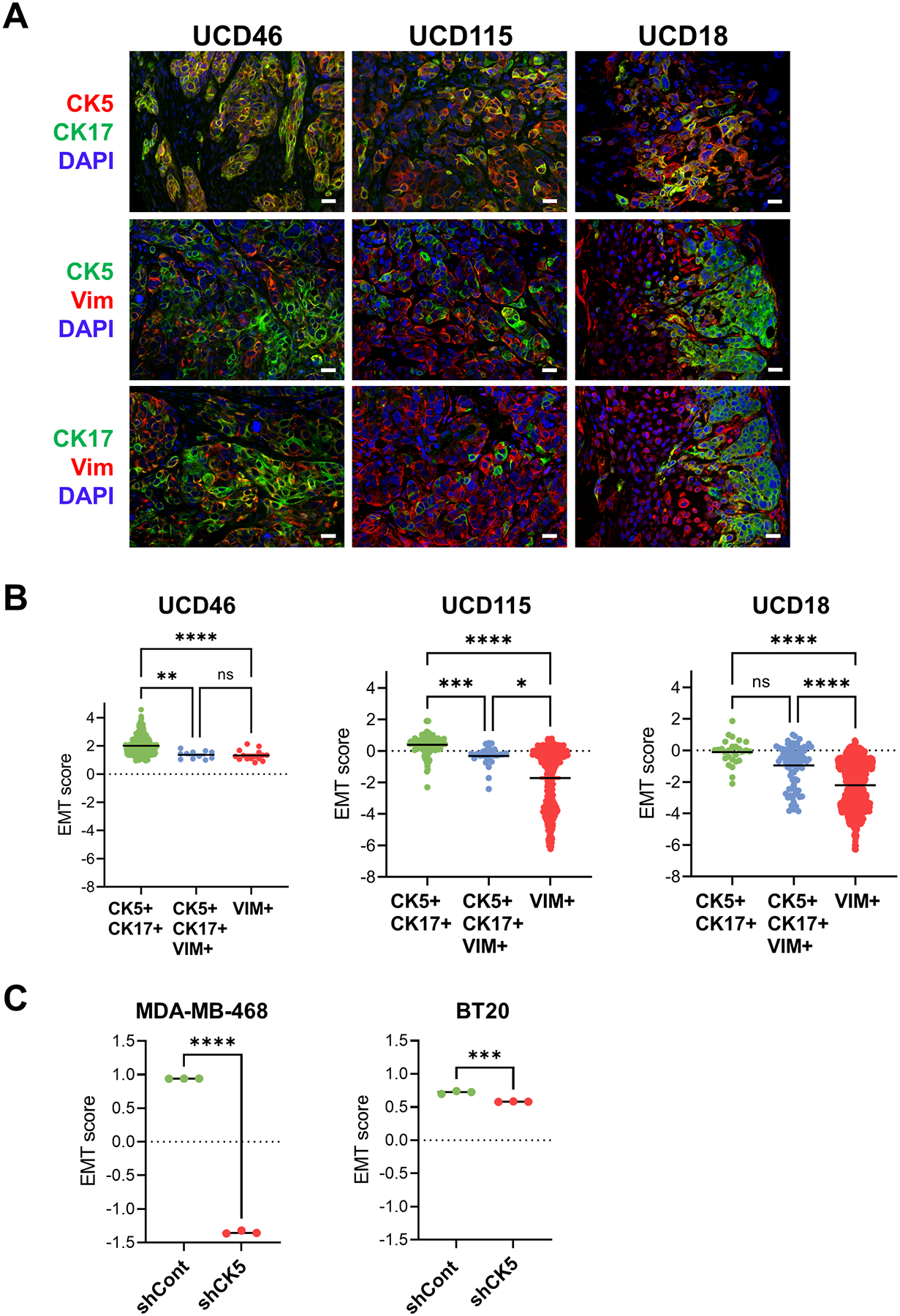
A. Dual fluorescent IHC of paraffin sections of BLBC PDX UCD46, UCD115, and UCD18 for CK5 (red)/CK17(green), CK5(green)/vimentin(VIM, red) or CK17(green)/vimentin(VIM, red) plus DAPI (blue) counterstain. Scale bars, 20 μm. B. sc RNA-seq of BLBC PDX was used to generate EMT scores based on a published 156 gene signature (37) in individual cells that were CK5/CK17+, CK5/CK17/vimentin(VIM)+, or vimentin(VIM)+. y-axes (positive, higher scores are more epithelial, negative, lower scores are more mesenchymal). Scatter plot plus means. One-way ANOVA/Kruskal-Wallis test was performed to assess statistical significance. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, NS=not significant. C. EMT scores were generated as in (B) for RNA-seq data from MDA-MB-468 and BT-20 cells, comparing each of the three replicates of shCont and shCK5-22. Unpaired T-tests were used to determine statistical significance. ***P<0.001, ****P<0.0001.
Discussion
This study provides evidence that basal CKs are important contributors to the pathology of BLBC. While typically utilized as biomarkers in molecular and immunophenotyping of breast and other cancers, emerging data support that specific CKs impart regulatory functions important for cancer cell pathobiology. The basal-like subclass of TNBC became more widely recognized with the application of molecular classifiers in the early 2000s (3). While these tumors have gene signatures reflective of basal/myoepithelial cells in the normal breast including regular expression of basal CKs, evidence suggests BLBC, particularly in women with BRCA1 germline mutations, originate from CK5+ luminal progenitor cells (41). The role of CKs beyond structural integrity in BLBC has remained an enigma. In the current study, we find that basal CKs 5 and 17 protect an epithelial state that is precariously plastic and subject to EMT upon loss of basal CK stoichiometry. BLBC cells with genetic loss of CKs are modestly more sensitive to doxorubicin therapy and are less efficient at metastatic colonization in vivo (Figs. 4 & 5), providing evidence that the basal epithelial state is important for tumor progression. Further analysis of basal CK function may discover unconventional vulnerabilities for this aggressive disease.
TNBC in general has suffered from lack of long-term effective targeted therapies that have transformed ER+ and HER2+ cancers into a long-term survivable disease for many patients. The poor short-term (5-year) prognosis of BLBC is due to multiple factors that include a high mitotic index and increased genomic instability due to frequent deficiency in p53 and BRCA1. Although BLBC have relative (vs other subtypes) increased expression of several druggable targets including EGFR, c-kit, and MEK/ERK, numerous clinical trials testing drugs targeting these molecules failed to reach study endpoints. Currently, approved targeted therapies for TNBC include poly(ADP-ribose) polymerase (PARP) inhibitors (i.e. olaparib, talazoparib) for BRCA1/2 mutant tumors and immune checkpoint inhibitors targeting programmed death ligand 1 (i.e. pembrolizumab, atezolizumab) in early high risk and metastatic TNBC, and antibody-drug conjugates for refractory disease (i.e. sacituzumab) (42). These treatments provide improved DFS in a subset of patients but rarely provide long-term disease stabilization (42). Thus, there remains an unmet need to understand BLBC biology and novel disease targets. Our observations that loss of CK5 and CK17 improves sensitivity to doxorubicin implicates that basal CKs provide some protection against cytotoxic drugs, although we did not observe the same trend with paclitaxel. This is presumably due to different mechanisms of action with doxorubicin targeting topisomerase II and DNA damage repair, critical for BLBC, while paclitaxel stabilizes microtubules. Interestingly, Escobar-Hoyos et al observed that CK17 knockdown was associated with improved response to cisplatin in cervical cancer cells (16). Their studies found that CK17 can solubilize and enter the nucleus where it promotes cyclin-dependent kinase inhibitor 1B (p27kip) export and cell cycle progression (16). We previously reported that CK5 mobilizes β-catenin to the nucleus and is necessary for Wnt signaling activity in MDA-MB-468 cells (19). Collectively, these studies insinuate that basal CKs may provide some protection from cytotoxic stress, in part, by regulating the subcellular location of cell cycle inhibitory and pro-survival signaling molecules.
Our studies utilizing BLBC cell lines and PDX confirm that this subset of breast cancers, compared to others, is unique in its frequent co-expression of epithelial and mesenchymal intermediate filament proteins. Populations of basal CK+ only, vimentin+ only, and dual positive cells co-exist with intertumoral differences in the extent of heterogeneity (Figs. 1 & 6). Interestingly, luminal CKs 8/18 were ubiquitous in BLBC cell lines and PDX, suggesting that the innate heterogeneity between basal CKs and vimentin is central to BLBC. Genetic knockdown of either CK5 or CK17 was sufficient to transition cells towards a more ubiquitous vimentin+ mesenchymal phenotype, accompanied by loss of E-cadherin, an EMT gene signature, a spindle-like morphology, and increased cell scatter (Figs. 2 & 3). CKs essentially act as cytosolic scaffolding proteins that move or sequester molecules (as noted above) and can control cell adhesion, motility and signaling (11). Phospho-modifications control CK filament assembly and key protein-protein interactions and influence cell plasticity and metastasis (43). While several key basal CK phosphoresidues have been mapped in skin blistering diseases (44), the key sites that regulate CK assembly and disassembly and protein-protein interactions in breast cancer are unknown. Kinases such as MAPK, ERK1, and p38 that are upregulated in BLBC cells are all predicted to phosphorylate CK5 at key head domain residues involved in filament assembly (44). Recently, Bollong et al discovered a small molecule inhibitor of vimentin phosphorylation that leads to its disassembly and blocks the growth of mesenchymal breast cancer cells (45). Identifying the key sites and kinases involved in basal CK phosphorylation critical for their function in BLBC is a first step towards discovering novel mechanisms for their disruption.
BLBC are prone to early relapse accompanied by distant organ metastasis predominantly to lung and central nervous system, leading to a high 5-year mortality rate vs other breast cancer subtypes and even non-basal TNBC (46). In our studies, while MDA-MB-468 tumors with shCont, shCK5 and shCK17 all formed spontaneous lung metastases, we found a significant reduction in metastatic tumor cells with CK5 and CK17 deficiency (Fig. 4). This is interesting as shCK5 and shCK17 vs shCont BLBC cells were relatively more invasive using standard in vitro matrix invasion assays (Fig. 3) which could be partially due to increased expression of matrix metalloprotein genes such as MMP14 observed by RNA-seq upon CK5 knockdown. Furthermore, the role of EMT in metastasis has undergone dogmatic changes; while it is recognized that EMT is an integral part of the metastatic process, it is not alone sufficient for overt metastatic colonization of distant organs (27). Several studies found that a cell intrinsic “reversible EMT” was required for breast cancer metastases where overexpression of EMT-transcription factors TWIST1, SNAIL1, and Pfrrx1 promote dissemination, but their silencing and EMT reversion is required for metastatic outgrowth (47–49). Furthermore, circulating tumor cells (CTCs) with the highest metastatic colonization efficiency travel in clusters containing both epithelial and mesenchymal populations (36,50,51). Recent studies have found that cells intermediate on the EMT spectrum cells may have higher intrinsic metastatic potential. For example, Kroger et al found that cells with a hybrid epithelial-mesenchymal (E/M) phenotype that includes co-expression of CK5/8 and vimentin were the most tumorigenic and metastatic in vivo (29). Using genetic mouse models with conditional loss of E-cadherin, Padmanaban et al demonstrated that while loss of E-cadherin increased invasion, it decreased CTCs and metastatic outgrowth (52). Lawson et al. reported that early micrometastases in mice bearing TNBC PDX were enriched for a basal/stem-like gene signature including CK5 (53). Thus, robust metastasis involves the basal epithelial state – which could prospectively cooperate and synergize with co-existent mesenchymal cells or even involve collaboration between basal CKs and vimentin within the same cells.
In summary, this study highlights that basal CKs 5 and 17 have a unique role in regulating the plasticity and behavior of BLBC cells. Their tandem regulation in breast cancer cells, previously recognized in keratinocytes, is poorly understood. Furthermore, understanding the function of these molecules, their role in cell plasticity, their key protein-protein interactions and signaling mechanisms has potential for novel approaches to tackle the rapid and poor clinical course of BLBC. Furthermore, expression of basal CKs, cell plasticity, and invasion are influenced by the tumor microenvironment (23) - which could play a unique role in the progression of this disease. While several new targeted therapies have been approved for TNBCs; the disease lags behind other subtypes in sustainable therapies and BLBC in particular disproportionately affects younger women (<50) and African-American women (8). Investigating the intermediate filament networks that distinguish BLBC cells has potential to advance our understanding and treatment of this disease.
Supplementary Material
Implications:
This study supports that basal CKs 5 and 17 contribute to the adverse behavior of BLBC cells and could be an untapped source of therapeutic vulnerability for this aggressive disease.
Acknowledgements
We thank the University of Colorado Cancer Center Genomics, Cell Technologies, and Pathology shared resources supported by P30CA046934. This work was supported by grants from the National Institutes of Health NIH F31 CA232456 (OM), NIH 2R01 CA140985 (CAS), NIH R01CA205044 (PK), and the Breast Cancer Research Foundation 21-144 (CAS).
Support:
This work was supported by grants from the National Institutes of Health grants NIH F31 CA232456 (OM), NIH 2R01 CA140985 (CAS), NIH R01CA205044 (PK), and the Breast Cancer Research Foundation 21-144 (CAS).
Footnotes
The authors declare that they have no conflict of interest
References
- 1.Caan BJ, Sweeney C, Habel LA, Kwan ML, Kroenke CH, Weltzien EK, et al. Intrinsic subtypes from the PAM50 gene expression assay in a population-based breast cancer survivor cohort: prognostication of short- and long-term outcomes. Cancer Epidemiol Biomarkers Prev 2014;23(5):725–34 doi 10.1158/1055-9965.EPI-13-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheang MC, Voduc D, Bajdik C, Leung S, McKinney S, Chia SK, et al. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res 2008;14(5):1368–76 doi 14/5/1368 [pii] 10.1158/1078-0432.CCR-07-1658. [DOI] [PubMed] [Google Scholar]
- 3.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A 2001;98(19):10869–74 doi 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res 2004;10(16):5367–74 doi 10.1158/1078-0432.CCR-04-0220. [DOI] [PubMed] [Google Scholar]
- 5.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer 2004;4(10):814–9 doi 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 6.Turner NC, Reis-Filho JS. Basal-like breast cancer and the BRCA1 phenotype. Oncogene 2006;25(43):5846–53 doi 10.1038/sj.onc.1209876. [DOI] [PubMed] [Google Scholar]
- 7.Rakha EA, Reis-Filho JS, Ellis IO. Basal-like breast cancer: a critical review. J Clin Oncol 2008;26(15):2568–81 doi 26/15/2568 [pii] 10.1200/JCO.2007.13.1748. [DOI] [PubMed] [Google Scholar]
- 8.Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F, et al. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res 2007;13(8):2329–34. [DOI] [PubMed] [Google Scholar]
- 9.Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016;534(7605):47–54 doi 10.1038/nature17676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun 2016;7:11479 doi 10.1038/ncomms11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moll R, Divo M, Langbein L. The human keratins: biology and pathology. Histochem Cell Biol 2008;129(6):705–33 doi 10.1007/s00418-008-0435-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horwitz KB, Dye WW, Harrell JC, Kabos P, Sartorius CA. Rare steroid receptor-negative basal-like tumorigenic cells in luminal subtype human breast cancer xenografts. Proc Natl Acad Sci U S A 2008;105(15):5774–9 doi 10.1073/pnas.0706216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato T, Tran TH, Peck AR, Girondo MA, Liu C, Goodman CR, et al. Prolactin suppresses a progestin-induced CK5-positive cell population in luminal breast cancer through inhibition of progestin-driven BCL6 expression. Oncogene 2014;33(17):2215–24 doi 10.1038/onc.2013.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gusterson B Do ‘basal-like’ breast cancers really exist? Nat Rev Cancer 2009;9(2):128–34 doi nrc2571 [pii] 10.1038/nrc2571. [DOI] [PubMed] [Google Scholar]
- 15.Pan X, Hobbs RP, Coulombe PA. The expanding significance of keratin intermediate filaments in normal and diseased epithelia. Curr Opin Cell Biol 2013;25(1):47–56 doi 10.1016/j.ceb.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Escobar-Hoyos LF, Shah R, Roa-Pena L, Vanner EA, Najafian N, Banach A, et al. Keratin-17 Promotes p27KIP1 Nuclear Export and Degradation and Offers Potential Prognostic Utility. Cancer Res 2015;75(17):3650–62 doi 10.1158/0008-5472.CAN-15-0293. [DOI] [PubMed] [Google Scholar]
- 17.Kim S, Wong P, Coulombe PA. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature 2006;441(7091):362–5 doi 10.1038/nature04659. [DOI] [PubMed] [Google Scholar]
- 18.Ku NO, Michie S, Resurreccion EZ, Broome RL, Omary MB. Keratin binding to 14-3-3 proteins modulates keratin filaments and hepatocyte mitotic progression. Proc Natl Acad Sci U S A 2002;99(7):4373–8 doi 10.1073/pnas.072624299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McGinn O, Ward AV, Fettig LM, Riley D, Ivie J, Paul KV, et al. Cytokeratin 5 alters beta-catenin dynamics in breast cancer cells. Oncogene 2020;39(12):2478–92 doi 10.1038/s41388-020-1164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mikami T, Maruyama S, Abe T, Kobayashi T, Yamazaki M, Funayama A, et al. Keratin 17 is co-expressed with 14-3-3 sigma in oral carcinoma in situ and squamous cell carcinoma and modulates cell proliferation and size but not cell migration. Virchows Arch 2015;466(5):559–69 doi 10.1007/s00428-015-1735-6. [DOI] [PubMed] [Google Scholar]
- 21.Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013;155(7):1639–51 doi 10.1016/j.cell.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheung KJ, Padmanaban V, Silvestri V, Schipper K, Cohen JD, Fairchild AN, et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc Natl Acad Sci U S A 2016;113(7):E854–63 doi 10.1073/pnas.1508541113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanley CJ, Henriet E, Sirka OK, Thomas GJ, Ewald AJ. Tumor-Resident Stromal Cells Promote Breast Cancer Invasion through Regulation of the Basal Phenotype. Mol Cancer Res 2020;18(11):1615–22 doi 10.1158/1541-7786.MCR-20-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2020;21(6):341–52 doi 10.1038/s41580-020-0237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gooding AJ, Schiemann WP. Epithelial-Mesenchymal Transition Programs and Cancer Stem Cell Phenotypes: Mediators of Breast Cancer Therapy Resistance. Mol Cancer Res 2020;18(9):1257–70 doi 10.1158/1541-7786.MCR-20-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yeung KT, Yang J. Epithelial-mesenchymal transition in tumor metastasis. Mol Oncol 2017;11(1):28–39 doi 10.1002/1878-0261.12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bill R, Christofori G. The relevance of EMT in breast cancer metastasis: Correlation or causality? FEBS Lett 2015;589(14):1577–87 doi 10.1016/j.febslet.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 28.Lourenco AR, Ban Y, Crowley MJ, Lee SB, Ramchandani D, Du W, et al. Differential Contributions of Pre- and Post-EMT Tumor Cells in Breast Cancer Metastasis. Cancer Res 2020;80(2):163–9 doi 10.1158/0008-5472.CAN-19-1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kroger C, Afeyan A, Mraz J, Eaton EN, Reinhardt F, Khodor YL, et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc Natl Acad Sci U S A 2019;116(15):7353–62 doi 10.1073/pnas.1812876116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finlay-Schultz J, Jacobsen BM, Riley D, Paul KV, Turner S, Ferreira-Gonzalez A, et al. New generation breast cancer cell lines developed from patient-derived xenografts. Breast Cancer Res 2020;22(1):68 doi 10.1186/s13058-020-01300-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fettig LM, McGinn O, Finlay-Schultz J, LaBarbera DV, Nordeen SK, Sartorius CA. Cross talk between progesterone receptors and retinoic acid receptors in regulation of cytokeratin 5-positive breast cancer cells. Oncogene 2017;36(44):6074–84 doi 10.1038/onc.2017.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xue B, Krishnamurthy K, Allred DC, Muthuswamy SK. Loss of Par3 promotes breast cancer metastasis by compromising cell-cell cohesion. Nat Cell Biol 2013;15(2):189–200 doi 10.1038/ncb2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kabos P, Finlay-Schultz J, Li C, Kline E, Finlayson C, Wisell J, et al. Patient-derived luminal breast cancer xenografts retain hormone receptor heterogeneity and help define unique estrogen-dependent gene signatures. Breast Cancer Res Treat 2012;135(2):415–32 doi 10.1007/s10549-012-2164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobrolecki LE, Airhart SD, Alferez DG, Aparicio S, Behbod F, Bentires-Alj M, et al. Patient-derived xenograft (PDX) models in basic and translational breast cancer research. Cancer Metastasis Rev 2016;35(4):547–73 doi 10.1007/s10555-016-9653-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hanna C, Kwok L, Finlay-Schultz J, Sartorius CA, Cittelly DM. Labeling of Breast Cancer Patient-derived Xenografts with Traceable Reporters for Tumor Growth and Metastasis Studies. J Vis Exp 2016(117) doi 10.3791/54944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brechbuhl HM, Vinod-Paul K, Gillen AE, Kopin EG, Gibney K, Elias AD, et al. Analysis of circulating breast cancer cell heterogeneity and interactions with peripheral blood mononuclear cells. Mol Carcinog 2020;59(10):1129–39 doi 10.1002/mc.23242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A 2010;107(35):15449–54 doi 10.1073/pnas.1004900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cancer Genome Atlas N Comprehensive molecular portraits of human breast tumours. Nature 2012;490(7418):61–70 doi 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haughian JM, Pinto MP, Harrell JC, Bliesner BS, Joensuu KM, Dye WW, et al. Maintenance of hormone responsiveness in luminal breast cancers by suppression of Notch. Proc Natl Acad Sci U S A 2012;109(8):2742–7 doi 10.1073/pnas.1106509108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, et al. Breast cancer. Nat Rev Dis Primers 2019;5(1):66 doi 10.1038/s41572-019-0111-2. [DOI] [PubMed] [Google Scholar]
- 41.Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med 2009;15(8):907–13 doi 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 42.Malhotra MK, Emens LA. The evolving management of metastatic triple negative breast cancer. Semin Oncol 2020;47(4):229–37 doi 10.1053/j.seminoncol.2020.05.005. [DOI] [PubMed] [Google Scholar]
- 43.Omary MB, Ku NO, Tao GZ, Toivola DM, Liao J. “Heads and tails” of intermediate filament phosphorylation: multiple sites and functional insights. Trends Biochem Sci 2006;31(7):383–94 doi 10.1016/j.tibs.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 44.Sawant M, Schwarz N, Windoffer R, Magin TM, Krieger J, Mucke N, et al. Threonine 150 Phosphorylation of Keratin 5 Is Linked to Epidermolysis Bullosa Simplex and Regulates Filament Assembly and Cell Viability. J Invest Dermatol 2018;138(3):627–36 doi 10.1016/j.jid.2017.10.011. [DOI] [PubMed] [Google Scholar]
- 45.Bollong MJ, Pietila M, Pearson AD, Sarkar TR, Ahmad I, Soundararajan R, et al. A vimentin binding small molecule leads to mitotic disruption in mesenchymal cancers. Proc Natl Acad Sci U S A 2017;114(46):E9903–E12 doi 10.1073/pnas.1716009114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kennecke H, Yerushalmi R, Woods R, Cheang MC, Voduc D, Speers CH, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol 2010;28(20):3271–7 doi 10.1200/JCO.2009.25.9820. [DOI] [PubMed] [Google Scholar]
- 47.Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 2012;22(6):709–24 doi 10.1016/j.ccr.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 48.Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012;22(6):725–36 doi 10.1016/j.ccr.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tran HD, Luitel K, Kim M, Zhang K, Longmore GD, Tran DD. Transient SNAIL1 expression is necessary for metastatic competence in breast cancer. Cancer Res 2014;74(21):6330–40 doi 10.1158/0008-5472.CAN-14-0923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Micalizzi DS, Maheswaran S, Haber DA. A conduit to metastasis: circulating tumor cell biology. Genes Dev 2017;31(18):1827–40 doi 10.1101/gad.305805.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013;339(6119):580–4 doi 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Padmanaban V, Krol I, Suhail Y, Szczerba BM, Aceto N, Bader JS, et al. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019;573(7774):439–44 doi 10.1038/s41586-019-1526-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015;526(7571):131–5 doi 10.1038/nature15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE136260.