

Abstract
Purpose
The immunological profile of early-stage breast cancer treated with neoadjuvant PARP inhibitors has not been described. The aim of this study was to delineate the changes in the tumor immune microenvironment (TiME) induced by talazoparib.
Methods
Patients with operable germline BRCA1/2 pathogenic variant (gBRCA1/2+) breast cancer were enrolled in a feasibility study of neoadjuvant talazoparib. Thirteen patients who received 8 weeks of neoadjuvant talazoparib were available for analysis, including 11 paired pre- and post-talazoparib core biopsies. Treatment related changes in tumor infiltrating lymphocytes were examined and immune cell phenotypes and their spatial distribution in the TiME were identified and quantified by multiplex immunofluorescence using a panel of 6 biomarkers (CD3, CD8, CD68, PD-1, PD-L1, and CK).
Results
Neoadjuvant talazoparib significantly increased infiltrating intratumoral and stromal T cell and cytotoxic T cell density. There was no difference in PD-1 or PD-L1 immune cell phenotypes in the pre- and post-talazoparib specimens and PD-L1 expression in tumor cells was rare in this cohort. Spatial analysis demonstrated that pre-talazoparib interactions between macrophages and T-cells may correlate with pathologic complete response.
Conclusions
This is the first study with phenotyping to characterize the immune response to neoadjuvant talazoparib in gBRCA1/2+ breast cancer patients. These findings support an emerging role for PARP inhibitors in enhancing tumor immunogenicity. Further investigation of combinatorial strategies is warranted with agents that exploit the immunomodulatory effects of PARP inhibitors on the TiME.
Keywords: Breast cancer, BRCA1/2 mutation, Talazoparib, Tumor immune microenvironment, Multiplex immunofluorescence
Background:
Mutations in BRCA1 and BRCA2 cause defects in homologous recombination repair and lead to replication stress and genomic instability. Poly(adenosine diphosphate-ribose) polymerase (PARP) enzymes detect and repair DNA damage through the base excision repair pathway and maintain genetic stability and inhibition of PARP in BRCA1/2 deficient cancer cells results in synthetic lethality (1). Preclinical evidence suggests that PARP inhibitors may enhance the antitumor immune response by increasing tumor-infiltrating lymphocytes (TILs) and more specifically cytotoxic T cells through activation of the cGAS-STING innate immune pathway (2,3).
The importance of infiltrating immune cells and their spatial location in relation to breast cancer cells in response to PARP inhibitors remains unknown. Multiplex immunohistochemical methods allow multiparametric analysis of the dynamic immune composition and resolve spatial interactions within the tumor immune microenvironment (TiME). Comprehensive characterization of the TiME to inform strategies for harnessing the immune system for clinical benefit remains a high priority.
Talazoparib is a PARP1/2 inhibitor approved for use in advanced breast cancer patients with a germline BRCA1/2 pathogenic variant (gBRCA1/2+). A phase 3 trial showed a significant improvement in median progression-free survival (PFS) for patients receiving talazoparib compared to physician’s choice of chemotherapy (4). We previously reported a feasibility study of 8 weeks of talazoparib administered in the neoadjuvant setting (NCT02282345) to 13 patients with early-stage gBRCA1/2+ breast cancer and demonstrated a median decrease in tumor volume by 88% (range 30-98%) by ultrasound. These patients then received neoadjuvant chemotherapy prior to surgery (5). Here we report the impact of talazoparib on the TiME and describe the spatial distribution of immune and tumor cells in pre- and post-treatment specimens from that feasibility trial.
Methods:
Study design and patients
NCT02282345 was a feasibility trial of neoadjuvant talazoparib monotherapy in patients with operable breast cancer and a pathogenic gBRCA1/2+ (5). Patients received talazoparib (1 mg per day) orally for 8 weeks prior to starting a neoadjuvant anthracycline and taxane-based chemotherapy regimen of the physician’s choice. Core biopsies obtained at baseline (pre-talazoparib) and after 8 weeks of talazoparib (post-talazoparib) were analyzed for changes in the TiME. All patients included in the present analysis consented to evaluation of their archival tumor samples. This study was conducted under Institutional Review Board approved Protocol 2014-0045 and in accordance with relevant guidelines at The University of Texas MD Anderson Cancer Center (MDACC). The Institutional Review Board at The University of Texas MD Anderson Cancer Center provided approval for the use of patient samples in this study (study number 2014-0045). Samples were acquired with written informed consent from all participants included in the study and this study was performed in accordance with the ethical standards outlined in the 1964 Helsinki Declaration and its later amendments.
Tissue samples
Formalin-fixed, paraffin-embedded (FFPE) core biopsy specimens were obtained from patients at baseline and after 8 weeks of talazoparib.
Tumor-infiltrating lymphocyte assessment
TILs were quantified manually by hematoxylin-eosin (H&E) stained FFPE sections and scored as a percentage of tumor area (tumor cells and stroma) by a breast pathologist (E.R.P.C.) according to the International Immuno-Oncology Working Group method for assessing TILs (6).
Immune profiling by multiplex immunofluorescence staining
Immune profiling was performed on pre- and post-talazoparib biopsies with multiplex immunofluorescence (mIF) to simultaneously evaluate 6 biomarkers (pancytokeratin AE1/AE3, CD8, CD3, CD68, PD-1, and PD-L1). Using Vectra multispectral imaging system v3.0 (Akoya/PerkinElmer), FFPE sections stained with the mIF panel and Opal 7-color Kit were scanned as previously described (7,8) and detailed in Supplemental Methods. Cell phenotypes were quantified as cell density (cells/mm2) and co-localization of the biomarkers was performed on invasive tumor and associated stromal regions by two pathologists (F.Y. and E.R.P.C.) using Inform 2.3 image analysis software (Akoya/PerkinElmer). Lineages and corresponding identification biomarkers are as follows: total T cells, CD3+; CD8− T cells, CD3+CD8−; cytotoxic T cells, CD3+CD8+; epithelial breast cancer cells, AE1/AE3+ (CK+); and macrophages, CD68+. Co-expression of PD-1 and PD-L1 on T cell subsets, and PD-L1 on tumor cells and macrophages were also examined.
Spatial interaction analysis
Scanned images were analyzed by Inform 2.3 (Akoya/PerkinElmer) to determine the spatial location of each cell phenotype. Pairwise combinations from spatial interactions based on the nearest neighbor distribution were computed with spatial G-function to quantify spatial interactions of cells of interest as previously described and detailed in Supplemental Methods (9). The probability of pairwise cell phenotype interactions were compared before and after 8 weeks of talazoparib and in correlation to pathologic complete response (pCR) after completion of neoadjuvant chemotherapy.
Gene expression analysis
RNA-seq data was generated by the Core Genomics Lab at MD Anderson. cDNA was generated from 150 ng of RNA using the NuGEN Ovation system and amplified using both 3’ poly(A) selection and random priming. Amplified products were shared using a Covaris E220 ultrasonicator and fragment sizes confirmed with an Agilent Bioanalyzer. Libraries were prepped using the NuGEN Ovation Ultralow library prep protocol, and sequenced on an Illumina HiSeq 2000 system.
We verified the quality of the sequencing data using FASTQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Then, we mapped reads to the hg19 human genome assembly using a GTF gene model from ENSEMBL (PMID 33137190) using the STAR aligner (PMID 23104886). Gene-level counts were quantified with HTSeq-count (PMID 25260700), and transcripts per million (TPM) were calculated using RSEM (PMID 20022975). To deconvolute the immune cell phenotypes from the bulk transcriptional profiles, we applied CIBERSORT in absolute mode (10).
Statistical analysis
The Wilcoxon signed-rank test was used to evaluate associations between pre- and post-TALA biomarker changes. Unpaired patient specimens were excluded from pre- versus post-talazoparib analyses. Correlations were evaluated using Spearman rank correlation. Statistical comparison of gene expression analysis was done using a two-tailed Student’s T-test. A nominal p < .05 was considered statistically significant All data outputs are provided as median values. Statistical calculations were performed by R v3.6.1 software (http://www.r-project.org) and GraphPad Prism v8.
Data Availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Results:
Of the 13 patients, twenty-four tissue biopsies were available for analysis and included 11 paired, one pre-, and one post-talazoparib patient specimens (Fig. 1A). Clinicopathologic characteristics of paired specimens are summarized in Supplemental Table 1 (5).
Figure 1:

Tumor-infiltrating lymphocyte variation before and after neoadjuvant talazoparib. A) Schema of study analysis. B) Representative TILs by H&E in pre- and post-talazoparib specimens. Arrowhead depicts TILs surrounded by invasive breast ductal carcinoma. Scale bar, 100 μM. C) Percentage of TILs in paired pre- and post-talazoparib specimens (n=6). Bars, boxes, and whiskers represent median, interquartile range, and range, respectively. Individual data points are shown and color encodes underlying biology with endocrine receptor (ER+) (red) or triple negative breast cancer (TNBC) (black). Data points with a white asterix in the center represents two paired specimens with overlapping values.
We first used H&E to examine the impact of talazoparib on TIL expressions. Six paired samples did not have residual tumor cells after treatment with talazoparib were excluded from the analysis. For those with residual tumor, there was a trend towards an increase in TILs after treatment. Pre-talazoparib, the median TIL level was 3.0% (range 1-40%); and post-talazoparib, the median TIL level was 17.5% (range 1-90%) (p=0.058) (Fig. 1B&C).
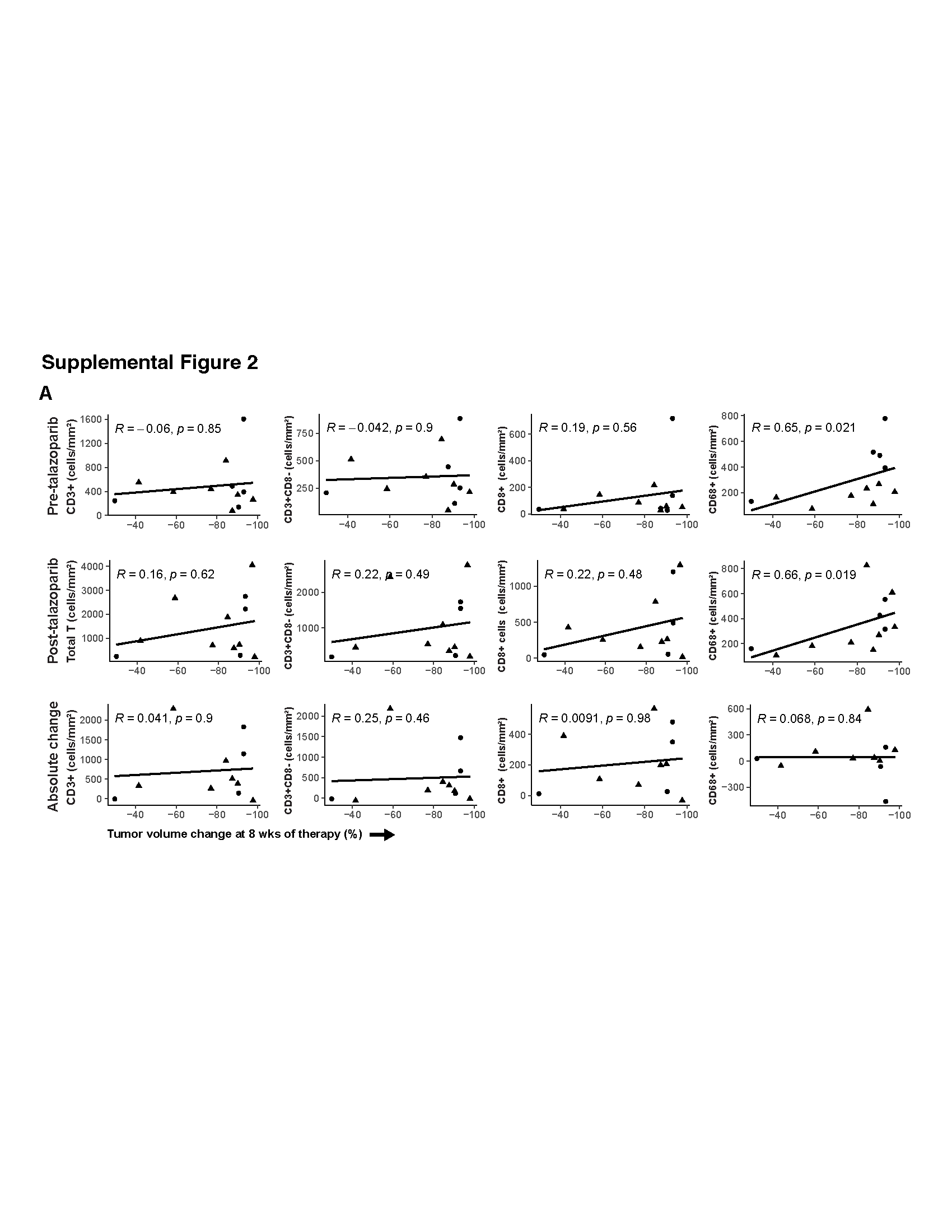
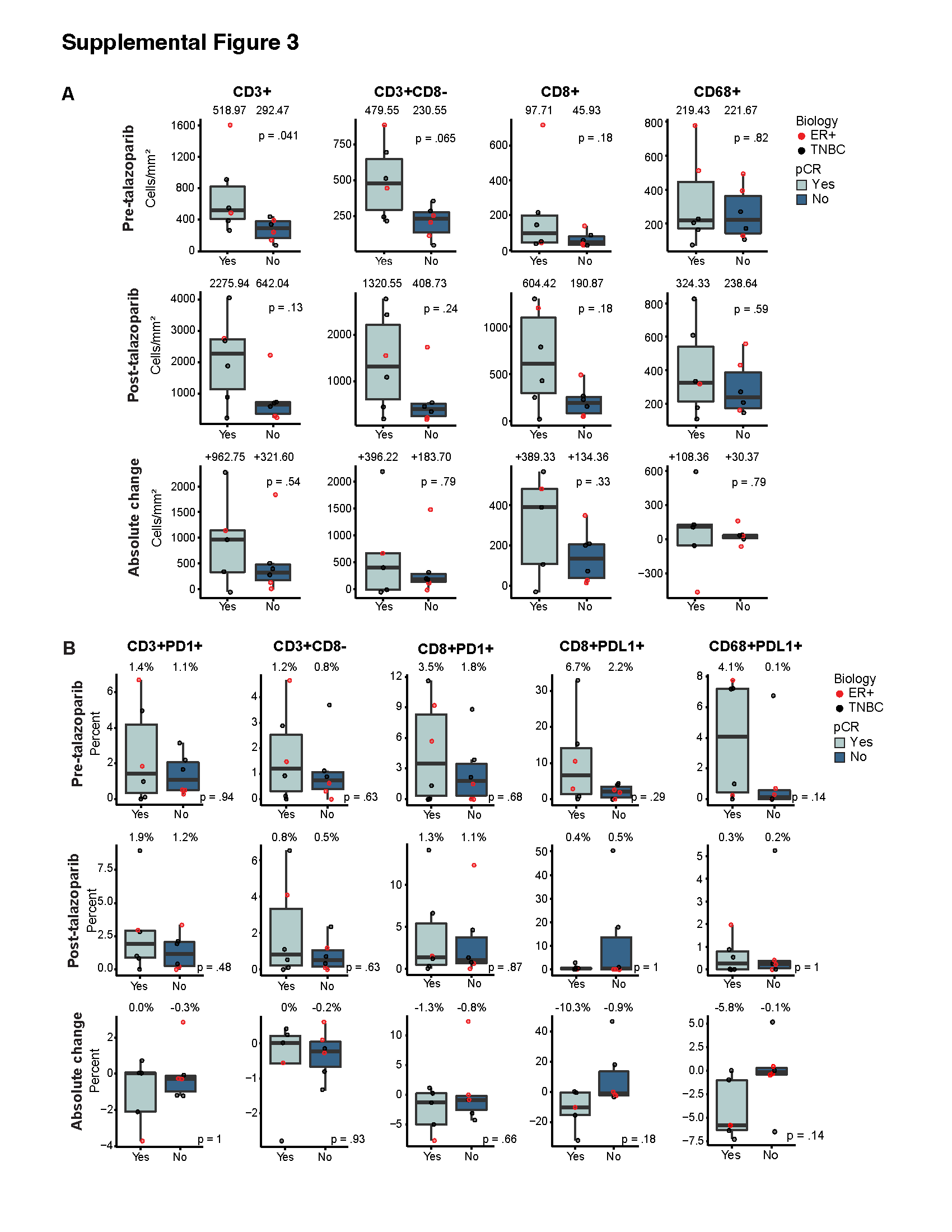
Major constituents of the TiME were determined using mIF (Fig. 2A). The complexity of the immune cell subpopulations in the intratumoral and intrastromal compartment for each patient is shown as a percentage of the total number of immune cells (CD3+ and CD63+ cells) (Fig. 2B). Some samples were dominated by CD8− T cells (patient 10, pre-talazoparib), whereas others were dominated by macrophages (patient 2, pre-talazoparib). PD-1+ and PD-L1+ immune cell subsets represented a minority of the total immune cells across specimens. Total T cells, CD8− T cells, and cytotoxic T cells significantly increased after treatment in the tumor and the surrounding stroma; there was no change in macrophages (Fig. 2C). The median percentage of PD-1+ T cells and PD-1+ cytotoxic T cells out of the total number of T cells and cytotoxic T cells respectively, was approximately 1% in pre- and post-talazoparib specimens and the percentage of PD-L1+ T cells and macrophages was similarly low (Fig. S1). The percentage of PD-L1+ tumor cells was absent/low and detected in 1/12 pre-talazoparib specimens (0.02% PD-L1+ tumor cells) and in 2/7 post-talazoparib specimens (3.14% and 6.35% PD-L1+ tumor cells for each patient). Four post-talazoparib specimens had treatment related effects, but no tumor cells were detected for analysis. There was no difference in the pre- and post-talazoparib immune cell phenotypes by clinical stage, subtype, or BRCA mutational status (Table S1). Pre- and post-talazoparib macrophages correlated with a decrease in tumor volume by ultrasound after treatment (Fig. S2). Pre-talazoparib total T cell densities predicted pCR on surgical specimens after completion of 8 weeks of talazoparib and neoadjuvant chemotherapy (Fig. S3). Other immune cell phenotype densities pre- or post-talazoparib did not predict response by ultrasound or pCR (Fig. 3 & Fig. S3).
Figure 2:

Neoadjuvant talazoparib increases infiltration of T cell lymphocyte populations by multiplex immunofluorescence. A) Representative 7-color multiplex immunofluorescence microscopy. Staining of pre- and post-talazoparib biopsies with lymphoid biomarker panel by pseudo-coloring (orange, PD-L1; green, PD-1; red, CD3; lavender, CD8; yellow, CD68; cyan, CK; blue, DAPI). Scale bar, 100 μM. Enlarged subsection of core to the right shows individual markers for CD3 and CD8 single channel and composite images, with DAPI nuclear marker (pseudocolored blue). B) Immune cell phenotypes by intratumor (T) and intrastromal (S) compartments grouped by patient and by pre- (upper panel) and post-talazoparib (lower panel) specimens. Patient characteristics are described on the bottom. Stacked bar graphs are shown as proportion of total immune cells evaluated (CD3+ and CD63+). Immune cell subsets are indicated by different colors. C) Pre- and post-talazoparib immune cell densities (cells/mm2) in paired samples (n=11). Bars, boxes, and whiskers represent median, interquartile range, and range, respectively.
Figure 3.

Multiplex immunofluorescence spatial analysis. A) Representative spatial analysis in a paired pre-talazoparib specimens with low T cell infiltration (upper panel, low AUC) and a post-talazoparib specimen with high T cell infiltration (lower panel, high AUC). Spatial analysis of mIF data report of all pseudocolored cell types (left); tumor cells and total T cell X- and Y-spatial coordinate plots (middle); and G(r) function versus r (μm) plots with Kaplan Meier (km) estimate of AUC with radius 0-20μm (shaded blue) of tumor cell and cytotoxic T cell interaction (right). B) Pre-talazoparib G-function analysis probability of interaction between macrophages onto CD8− T cells (upper panel) and macrophages onto cytotoxic T cells (lower panel) stratified by pCR (pCR Yes, n=6, pCR No, n=6). C) Heatmap of probability of pre-talazoparib spatial interactions between each cell phenotype, grouped by pCR. Km: Kaplan Meier estimate of AUC with darker shading representing higher probability of interaction between two cell phenotypes.
Spatial analysis revealed significant heterogeneity in the probability of cell phenotype interactions. Some pre-talazoparib specimens with a low probability of tumor cells interacting with CD8− T cells, converted to a high probability after treatment with talazoparib with a representative paired specimen (Fig. 3A). The probability of spatial interactions of cell phenotypes on pre-talazoparib specimens was correlated with pCR. Of these pairwise interactions, macrophages were in closer proximity to CD8− T cells in patients who achieved pCR versus not (probability of interaction: 0.22 vs 0.09, p = 0.017) (Fig. 3B). The probability of spatial interaction of other cell phenotypes pre-talazoparib and pCR was not significant (Fig. S4). A spatial matrix analyses of cell phenotype interactions in pre-talazoparib biopsies of individual patients is depicted by heatmap clustered by pCR response (Fig. 3C).
To further characterize the TiME changes by mIF we analyzed bulk gene expression data. In agreement with the findings of mIF, talazoparib significantly increased in gene expression levels of T cell markers, CD3 and CD8 subunits, and CD4 (Fig. S5); there was no significant difference in granzyme B or FOXP3 (Fig. S5). Post-talazoparib, there was modest enrichment in immune checkpoints ICOS, CTLA4, LAG3, PDCD1 (PD-1), and CD274 (PD-L1) (Fig 4A). To assess the differential immune cell infiltration, we applied CIBERSORT (Fig. 4B). The two main immune subpopulations were memory resting CD4+ T cells and M2 macrophages (Fig 4C). Post-talazoparib specimens were enriched in CD4+ T memory resting, M2 macrophages, CD8+ T cell, memory B cell, T cell gamma delta and activated mast cell subsets clustered in post-talazoparib specimens. Expression of T regulatory and other immune cell subsets were less frequent.
Figure 4.

Immune checkpoint expression and composition of immune cells in TiME by CIBERSORT. A) Heatmap analysis of immune checkpoints. Red denotes highly expressed genes, and blue denotes lower expression levels by bulk RNA sequencing. B) Heatmap showing absolute abundance of each immune cell type predicted by CIBERSORT. Red denotes higher cell abundance and blue denotes lower cell abundance. Heatmaps grouped by pre- and post-talazoparib specimens. C) Scatterplots showing enrichment of memory resting CD4+ T cells and M2 macrophages post-talazoparib treatment.
Discussion:
This study is the first description of the impact of talazoparib monotherapy, administered in the neoadjuvant setting to gBRCA1/2+ breast cancer patients, on the TiME. Our findings show that 8 weeks of talazoparib results in increased T cell and cytotoxic T cell density in the tumor and the adjacent stroma.
Application of CIBERSORT to enumerate the immune cell composition within the TiME suggests that the increase in T cells by mIF are predominantly CD4+ memory resting cells, CD8+ T cells, and gamma delta T cells. Additionally, CIBERSORT revealed that talazoparib may also lead to infiltration of plasma and memory B cells, and mast cells. Post-talazoparib there was an increase in targetable immune checkpoints, ICOS, CTLA4, LAG3, PD-1 and PD-L1, possibly suggesting future combinatorial strategies after additional validation.
Correlating TiME changes and outcomes in this study cohort is limited; all patients had a decrease in tumor volume by ultrasound of at least 30% and went on to receive interval chemotherapy prior to surgery and determination of pCR. Nonetheless, baseline total T cells by mIF predicted pCR, consistent with prior reports (11). The effect of neoadjuvant chemotherapy on the TiME and early and late outcomes is inconsistent and limited to a few smaller patient studies. In SWOG S0800, a decrease in TILs correlated with pCR, however in another smaller study an increase in post-treatment TILs were associated with longer 5-year recurrence free survival. This is potentially an important observation as in early-stage gBRCA1/2+ breast cancer, baseline TILs are associated with a greater response to neoadjuvant chemotherapy and long-term survival (Yam C, 2021) (12) and clinical response, defined by clinical and radiographic evaluation, to short-course neoadjuvant olaparib, in unselected early-stage TNBC (13).
Interestingly, increased densities of macrophages in both pre- and post-talazoparib treated specimens correlated with radiographic response, but not the absolute change. Application of CIBERSORT to enumerate the immune cell composition within the TiME suggests the predominant macrophage detected by mIF is of the immunosuppressive M2 phenotype.
In our cohort of patients, PD-1 and PD-L1 expression was low in immune cell populations and tumor cells, respectively, and unchanged after treatment with talazoparib. This is in contrast to other studies of early-stage breast cancer with staining of PD-1+ mononuclear cells (40-50%) and PD-L1+ tumor cells (20-50%), with variations by molecular subtype, grade and stage (14,15). Low expression of PD-1 and PD-L1 in our analysis may be due to variation in staining intensity by different antibody clones and/or the heterogeneous staining distribution of PD-L1 in core biopsies as previously described (16). Additionally, in preclinical models of gBRCA1/2+ breast cancer, there is conflicting evidence that PARP inhibitors induce PD-L1 expression on tumor cells, which may be due to transient PD-L1 induction (2,17). Interestingly, a recent study found olaparib increased PD-L1+ macrophages in murine BRCA-deficient xenografts, possibly counteracting PARP inhibitor T cell mediated immunity (18). In our study, although not statistically significant, PD-L1+ macrophage cell density either remained low (<1%) or decreased after treatment, with the exception of one outlier. The mIF biomarker panel used for our analysis did not include additional macrophage markers such as CD163 or CSFR1 to differentiate macrophages from CD63+ tumor-associated fibroblasts of monocyte-derived fibrocytes that have been described in the breast cancer TiME (19). In future studies, an expanded mIF biomarker panel should be considered in a larger cohort.
It has previously been reported that in early-stage breast cancer, cytotoxic T cells within cancer islands compared to the overall tumor tissue or stroma are associated with relapse-free survival by mIF (20). By spatial analysis we found heterogeneity in samples exhibiting immune infiltrated and non-infiltrated phenotype in relation to tumor cells. Additionally, spatial analysis suggested that pretreatment interaction of macrophages and T cells may predict pCR. Additional investigation is required to confirm this observation and better characterize macrophage phenotypes, with additional biomarkers as above (19).
Preclinical evidence has demonstrated overwhelming synergy with the combination of PARP inhibitors and PD-1/PD-L1 immune checkpoint blockade (2,3). However, in the initial phase 1/2 single-arm trials, MEDIOLA and TAPACIO/Keynote-162, this combination demonstrated response rates similar to historical trials with PARP inhibitor monotherapy in BRCA1/2-mt metastatic breast cancer. Further studies and follow-up are ongoing to determine if this combination leads to a longer duration of response (4,21-23). Thus, there is an urgent need for in depth prospective investigations of the TiME. Our analysis highlights the potential of mIF to characterize the complexities of PARP inhibitor mediated changes on immune cell phenotypes within the TiME and to inform combinations of immune modulating agents in future studies.
In summary, our descriptive analysis of the changes in the TiME before and after PARP inhibitor treatment provides a provocative preliminary signal that talazoparib enhances tumor immunity. The small sample size precluded more rigorous statistical analysis and correlation with histologic grade, subtype, BRCA mutation status, and response. Investigation continues into the relative importance of these findings in a phase II multicenter trial (NCT03499353) underway with single agent talazoparib for 6 months prior to surgery.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Statement of translational relevance:
Growing preclinical evidence clearly suggests that PARP inhibitors modulate the TiME. This study further evaluates these findings in gBRCA1/2-mt breast cancer patients treated with neoadjuvant talazoparib. Using multiplex immunofluorescence to quantitatively monitor and spatially resolve immune cell phenotypes, we demonstrated PARP inhibition increased T-cell trafficking into the TiME. This study sets forth a rationale to further characterize PARP inhibitors novel immunomodulatory function in future translational studies and for the development of combination strategies with immunotherapy.
Funding:
The Toomim Family Fund. JTC was supported by the Cancer Prevention and Research Institute of Texas (RP160710, RP170668). TK is funded by the NCI T32 Translational Genomics Fellowship and Rosalie B. Hite fellowship.
Footnotes
Trial registration: ClinicalTrials.gov, NCT02282345. Registered November 4, 2014.
Conflicts of interest:
E. A. Mittendorf reports personal financial interests: sponsored research agreement from Glaxo SmithKline; honoraria from Physician Education Resource; compensated service on Scientific Advisory Boards for Exact Sciences (formerly Genomic Health), Merck, and Roche/Genentech; and uncompensated service on Steering Committees for BMS, Lilly, Roche/Genentech. E. A. Mittendorf reports institutional financial interests: Clinical trial funding from Roche/Genentech (via SU2C grant). G. J. Whitman is a shareholder of Pfizer and editor for UpToDate. JLitton reports personal financial interests: sponsored research agreements from Pfizer/Medivation, Genentech, BMS, Novartis, GSK, Zenith, Merck, EMD-Serono and Astra-Zenica. And has had uncompensated service on advisory Boards and Steering Committees for Pfizer, Astra-Zeneca and Ayala Pharmaceuticals. The other authors declare no potential conflicts of interest.
References:
- 1.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005;434(7035):913–7 doi 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 2.Wang Z, Sun K, Xiao Y, Feng B, Mikule K, Ma X, et al. Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Scientific reports 2019;9(1):1853 doi 10.1038/s41598-019-38534-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, et al. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer research 2019;79(2):311–9 doi 10.1158/0008-5472.can-18-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Litton JK, Rugo HS, Ettl J, Hurvitz SA, Goncalves A, Lee KH, et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. The New England journal of medicine 2018;379(8):753–63 doi 10.1056/NEJMoa1802905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Litton JK, Scoggins M, Ramirez DL, Murthy RK, Whitman GJ, Hess KR, et al. A feasibility study of neoadjuvant talazoparib for operable breast cancer patients with a germline BRCA mutation demonstrates marked activity. NPJ breast cancer 2017;3:49 doi 10.1038/s41523-017-0052-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hendry S, Salgado R, Gevaert T, Russell PA, John T, Thapa B, et al. Assessing tumor-infiltrating lymphocytes in solid tumors: a practical review for pathologists and proposal for a standardized method from the international immunooncology biomarkers working group: part 1: assessing the host immune response, TILs in invasive breast carcinoma and ductal carcinoma in situ, metastatic tumor deposits and areas for further research. Advances in anatomic pathology 2017;24(5):235–51 doi 10.1097/pap.0000000000000162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parra ER, Uraoka N, Jiang M, Cook P, Gibbons D, Forget MA, et al. Validation of multiplex immunofluorescence panels using multispectral microscopy for immune-profiling of formalin-fixed and paraffin-embedded human tumor tissues. Scientific reports 2017;7(1):13380 doi 10.1038/s41598-017-13942-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stack EC, Wang C, Roman KA, Hoyt CC. Multiplexed immunohistochemistry, imaging, and quantitation: a review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods (San Diego, Calif) 2014;70(1):46–58 doi 10.1016/j.ymeth.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 9.Barua S, Solis L, Parra ER, Uraoka N, Jiang M, Wang H, et al. A functional spatial analysis platform for discovery of immunological interactions predictive of low-grade to high-grade transition of pancreatic intraductal papillary mucinous neoplasms. Cancer informatics 2018;17:1176935118782880 doi 10.1177/1176935118782880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12(5):453–7 doi 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sonderstrup IMH, Jensen MB, Ejlertsen B, Eriksen JO, Gerdes AM, Kruse TA, et al. Evaluation of tumor-infiltrating lymphocytes and association with prognosis in BRCA-mutated breast cancer. Acta oncologica (Stockholm, Sweden) 2019;58(3):363–70 doi 10.1080/0284186x.2018.1539239. [DOI] [PubMed] [Google Scholar]
- 12.Yam C, Y. E. (2021). Immune Phenotype and Response to Neoadjuvant Therapy in Triple-Negative Breast Cancer. Clin Cancer Res, Oct 1;27(19):5365–5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eikesdal HP, Yndestad S, Elzawahry A, Llop-Guevara A, Gilje B, Blix ES, et al. Olaparib monotherapy as primary treatment in unselected triple negative breast cancer. Ann Oncol 2021;32(2):240–9 doi 10.1016/j.annonc.2020.11.009. [DOI] [PubMed] [Google Scholar]
- 14.Yuan C, Liu Z, Yu Q, Wang X, Bian M, Yu Z, et al. Expression of PD-1/PD-L1 in primary breast tumours and metastatic axillary lymph nodes and its correlation with clinicopathological parameters. Scientific reports 2019;9(1):14356 doi 10.1038/s41598-019-50898-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dill EA, Gru AA, Atkins KA, Friedman LA, Moore ME, Bullock TN, et al. PD-L1 expression and intratumoral heterogeneity across breast cancer subtypes and stages: an assessment of 245 primary and 40 metastatic tumors. The American journal of surgical pathology 2017;41(3):334–42 doi 10.1097/pas.0000000000000780. [DOI] [PubMed] [Google Scholar]
- 16.Sun WY, Lee YK, Koo JS. Expression of PD-L1 in triple-negative breast cancer based on different immunohistochemical antibodies. Journal of translational medicine 2016;14(1):173 doi 10.1186/s12967-016-0925-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clinical cancer research : an official journal of the American Association for Cancer Research 2017;23(14):3711–20 doi 10.1158/1078-0432.ccr-16-3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mehta AK, Cheney EM, Hartl CA, Pantelidou C, Oliwa M, Castrillon JA, et al. Targeting immunosuppressive macrophages overcomes PARP inhibitor resistance in BRCA1-associated triple-negative breast cancer. 2021;2(1):66–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruffell B, Au A, Rugo HS, Esserman LJ, Hwang ES, Coussens LM. Leukocyte composition of human breast cancer. Proc Natl Acad Sci USA 2012;109(8):2796–801 doi 10.1073/pnas.1104303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Egelston CA, Avalos C, Tu TY, Rosario A, Wang R, Solomon S, et al. Resident memory CD8+ T cells within cancer islands mediate survival in breast cancer patients. JCI insight 2019. doi 10.1172/jci.insight.130000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vinayak S, Tolaney SM, Schwartzberg L, Mita M, McCann G, Tan AR, et al. Open-label clinical trial of niraparib combined with pembrolizumab for treatment of advanced or metastatic triple-negative breast cancer. JAMA oncology 2019. doi 10.1001/jamaoncol.2019.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Domchek SM, Postel-Vinay S, Im SA, Park YH, Delord JP, Italiano A, et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol 2020;21(9):1155–64 doi 10.1016/s1470-2045(20)30324-7. [DOI] [PubMed] [Google Scholar]
- 23.Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. The New England journal of medicine 2017;377(6):523–33 doi 10.1056/NEJMoa1706450. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.