

Abstract
Background:
An ancient family of arrestin-fold proteins, termed alpha-arrestins, may have conserved roles in regulating nutrient transporter trafficking and cellular metabolism as adaptor proteins. One alpha-arrestin, thioredoxin-interacting protein (TXNIP), is known to regulate myocardial glucose uptake. However, the in vivo role of the related alpha-arrestin, arrestin domain-containing protein 4 (ARRDC4), is unknown.
Methods:
We first tested whether interaction with glucose transporters (GLUTs) is a conserved function of the mammalian alpha-arrestins. To define the in vivo function of ARRDC4, we generated and characterized a novel Arrdc4 knockout (KO) mouse model. We then analyzed the molecular interaction between arrestin domains and the basal glucose transporter 1 (GLUT1).
Results:
ARRDC4 binds to GLUT1, induces its endocytosis, and blocks cellular glucose uptake in cardiomyocytes. Despite the closely shared protein structure, ARRDC4 and its homologue TXNIP operate by distinct molecular pathways. Unlike TXNIP, ARRDC4 does not increase oxidative stress. Instead, ARRDC4 uniquely mediates cardiomyocyte death through its effects on glucose deprivation and endoplasmic reticulum (ER) stress. At baseline, Arrdc4-KO mice have mild fasting hypoglycemia. Arrdc4-KO hearts exhibit a robust increase in myocardial glucose uptake and glycogen storage. Accordingly, deletion of Arrdc4 improves energy homeostasis during ischemia and protects cardiomyocytes against myocardial infarction (MI). Furthermore, structure-function analyses of the interaction of ARRDC4 with GLUT1 using both scanning mutagenesis and deep-learning Artificial Intelligence identify specific residues in the C-terminal arrestin-fold domain as the interaction interface that regulates GLUT1 function, revealing a new molecular target for potential therapeutic intervention against myocardial ischemia.
Conclusion:
These results uncover a new mechanism of ischemic injury in which ARRDC4 drives glucose deprivation-induced ER stress leading to cardiomyocyte death. Our findings establish ARRDC4 as a new scaffold protein for GLUT1 that regulates cardiac metabolism in response to ischemia and provide insight into the therapeutic strategy for ischemic heart disease.
Keywords: Alpha arrestin, Artificial intelligence, Glucose metabolism, Mouse model, Myocardial infarction
Graphical Abstract
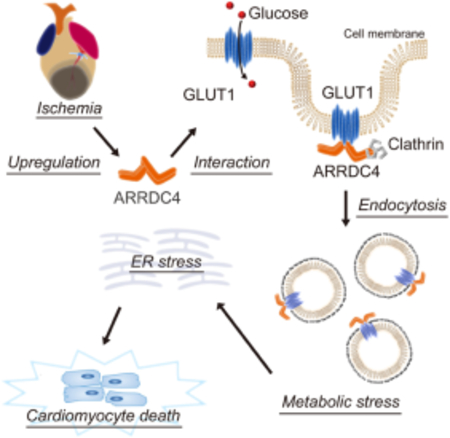
Introduction
Although acute-phase mortality after myocardial infarction (MI) has declined over the last few decades, the incidence of post-MI complications has increased1. Continuous efforts to provide new targets for therapeutic intervention remain crucial. Ischemic tissues undergo metabolic stress due to shortages in the supply of oxygen and nutrients. While fatty acids are the predominant fuel for the adult heart, the heart switches its substrate preference toward glucose during ischemia2. This confers an adaptive advantage for cardiomyocytes and prevents irreversible tissue damage, as glycolysis is the primary anaerobic source of energy. Increased glucose uptake is facilitated by translocation of glucose transporters (GLUTs), GLUT1 and GLUT4, to the plasma membrane of cardiomyocytes during ischemia3, 4. Nevertheless, the mechanisms by which acute ischemic stress regulates this process remain elusive, despite the potential to identify new strategies to reduce myocardial ischemic damage5.
The complexity of cardiomyocyte signaling requires scaffolding proteins to coordinate the cellular processes driven by receptors and transporters. Scaffold protein domains, which themselves usually lack intrinsic catalytic activity, bind with target molecules to facilitate signaling events. Beta-arrestins and visual-arrestins are prototypical intracellular scaffold proteins that bind to phosphorylated G protein-coupled receptors and arrest their activation6. More recently, a larger and more ancient family of structurally-related arrestins has been identified in yeast, which shares functions in regulation of receptor trafficking7. The function of these proteins, termed alpha-arrestins, is now emerging in mammals8.
Humans and mice have six members of alpha-arrestins, including thioredoxin-interacting protein (TXNIP) and the arrestin domain-containing (ARRDC) proteins 1–59, 10. The best-studied member of alpha-arrestins, TXNIP, has evolved a specialized and unique ability to form stable intermolecular disulfide bonds with thioredoxin and inhibit its anti-oxidative properties10. TXNIP also functions as a crucial regulator of glucose metabolism11. While TXNIP was originally thought to connect oxidative stress to metabolism, multiple lines of evidence now suggest that TXNIP inhibits glucose uptake independent of its binding to thioredoxin10, 12. TXNIP serves as an adaptor protein to facilitate endocytosis of GLUT1 and suppresses glucose influx13, 14. This mechanism may be conserved throughout evolution, since alpha-arrestins respond to glucose availability and promote endocytosis of nutrient transporters via endocytic pathways in yeast cells15.
A related mammalian alpha-arrestin, ARRDC4, has been shown to be a potent inhibitor of cellular glucose uptake in insulin-insensitive cell lines in vitro10. This evidence supports the concept that the functions of two alpha-arrestins, TXNIP and ARRDC4, are related to their conserved arrestin domains. However, unlike TXNIP16, the physiological role of ARRDC4 remains largely unknown in vivo. In this study, we hypothesized that ARRDC4 is a second example of an alpha-arrestin that plays a critical role in regulating cellular glucose uptake and the metabolic response of the myocardium to ischemia. We, therefore, generated a novel Arrdc4-knockout (KO) mouse model using CRISPR/Cas9 technology and found an essential role of ARRDC4 in glucose metabolism in the heart. The results revealed exciting findings that inhibition of ARRDC4 reduces myocardial ischemic injury and improves outcomes after acute MI. In addition, using biochemical and bioinformatics approaches, we dissected the protein-protein interaction of ARRDC4 with GLUT1. The data provide relevant insight into the therapeutic strategy that enhances cardiomyocyte survival in metabolically-challenging environments.
Methods
A detailed description of Materials and Methods can be found in the Supplemental Materials.
Results
ARRDC4 interacts with GLUT 1 and inhibits cellular glucose uptake.
Recent evidence suggests that the glucose-uptake function is intrinsic to the arrestin domains of TXNIP9. To determine which alpha-arrestins share the ability to inhibit glucose transport, we measured the uptake of a glucose analogue, 2-[3H]deoxy-glucose, in insulin-insensitive HEK293 cells transfected with different alpha-arrestins (Figure 1A). The result showed that TXNIP and ARRDC4, but not ARRDC1–3, strongly decreased glucose uptake. We tested whether, like TXNIP13, 17, ARRDC4 inhibits glucose transport due to an interaction with GLUTs. A pulldown assay using magnetic Streptactin beads confirmed the interaction of ARRDC4 with the basal glucose transporter GLUT1 but not with the insulin-regulated glucose transporter GLUT4 (Figure 1B).
Figure 1. Interaction between ARRDC4 and GLUT1.
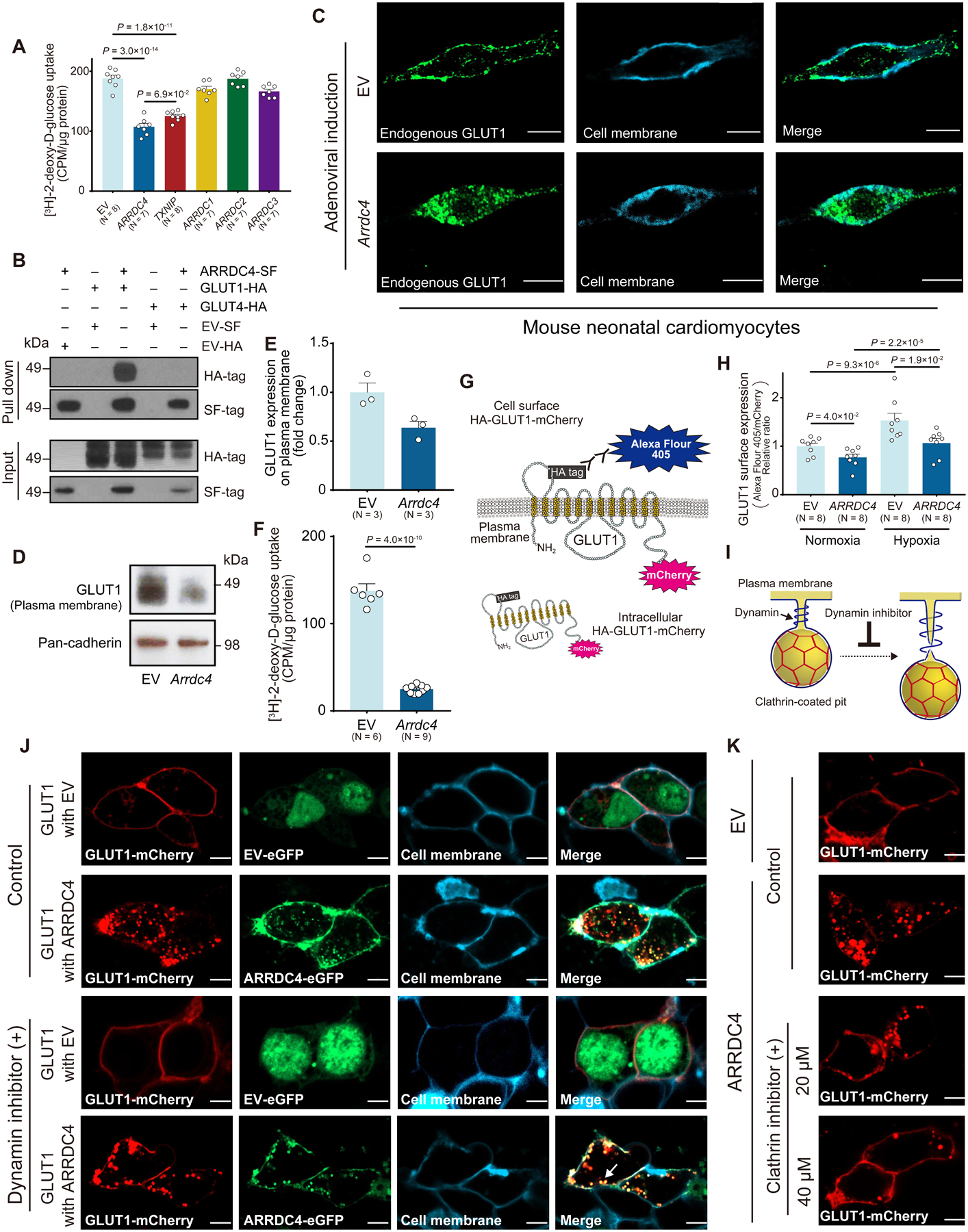
A. 2-[3H]deoxy-D-glucose uptake was measured in HEK293 cells transfected with a plasmid containing each alpha-arrestin or empty vector (EV). Uptake was normalized to the total protein content (One-way ANOVA post-hoc Bonferroni test). B. ARRDC4-Strep/FLAG (SF)-tag or EV-SF-tag (negative control) was co-expressed with GLUT1-HA-tag, GLUT4-HA-tag, or EV-HA-tag in HEK293T cells. Complexes were pulled down with Strep-Tactin resin, subjected to SDS-PAGE, and immunoblotted with anti-HA or anti-FLAG antibodies. C-F. Mouse neonatal cardiomyocytes were infected with an adenoviral vector expressing Arrdc4 or EV. Endogenous GLUT1 was detected by the anti-GLUT1 antibody conjugated with Alexa Fluor 488. Cell membrane was stained with lipophilic carbocyanine blue dye. Confocal images. Scale bars, 10 μm. Western blot analyzed GLUT1 or pan-cadherin (a membrane marker) expression in plasma membrane fractions (P=1.0 × 10−1 by Mann-Whitney U test). 2-[3H]deoxy-D-glucose uptake was measured in cardiomyocytes after adenoviral induction of Arrdc4 or EV. Uptake was normalized by total protein content (t-test). G and H. Cellular localization of GLUT1 was determined with a plasmid that encodes mCherry-conjugated GLUT1 with an HA epitope tag in the first exo-facial loop (55–64 AA). After overexpressing ARRDC4-SF or EV-SF, surface expression of GLUT1 was quantified by Alexa Fluor 405 against anti-HA antibody in non-permeabilized HEK293 cells using a microplate reader. The fluorescent levels of Alexa Fluor 405 were normalized by the total expression of GLUT1 estimated by fluorescent values of mCherry (two-way ANOVA post-hoc Bonferroni test). Cells were exposed to normoxia or hypoxia (1% O2, 3 hr). I-K. GLUT1-mCherry fusion or EV-mCherry was co-expressed in HEK293T cells with ARRDC4-eGFP fusion or EV-eGFP in the absence (control) or presence of pharmacological inhibitors. A dynamin inhibitor Dyngo (30 μM, 8 hr) inhibited dynamin to form a helical collar around the neck of a clathrin-coated pit, where it popped the vesicle from the plasma membrane. Cell membrane was detected by lipophilic carbocyanine blue dye. Arrow indicates budding clathrin-coated vesicle (J). A selective cell membrane permeable clathrin inhibitor (Pitstop1, 8 hr) blocked GLUT1-mCherry endocytosis in a dose-dependent manner. Confocal images. Scale bars, 5 μm.
Glucose uptake is normally associated with GLUT expression on the cell surface18. To determine the effect of ARRDC4 on GLUT1 localization, mouse neonatal cardiomyocytes were infected with an adenoviral gene vector of Arrdc4 or an empty vector. Following confirmation of the successful gene transduction (Figure S1A), confocal microscopic analysis visualized endogenous GLUT1 (Figure 1C). Immunofluorescence for GLUT1 showed rich and relatively uniform expression at the plasma membrane in empty-vector transfected cardiomyocytes. By contrast, GLUT1 localized in intracellular puncta in cells overexpressing ARRDC4, indicating that GLUT1 had been internalized. Consistently, Western blot showed a weaker band of GLUT1 protein in the plasma membrane fraction from ARRDC4-overexpressed cardiomyocytes than from empty-vector transfected control cells (Figure 1D–E). As a result, 2-[3H]deoxy-glucose uptake was repressed by overexpression of Arrdc4 in cardiomyocytes (Figure 1F). To more rigorously quantify GLUT1 surface expression, we generated a plasmid that encodes mCherry-conjugated GLUT1 with a HA epitope tag in the first exofacial loop (55–64 amino acid). After overexpressing ARRDC4, surface expression of GLUT1 was quantified by fluorescence levels of Alexa Fluor 405-conjugated secondary antibody against anti-HA antibody in non-permeabilized HEK293 cells (Figure 1G). In agreement with confocal images and Western blot analyses, ARRDC4 significantly decreased HA-GLUT1 surface expression compared with empty vector control (Figure 1H). We also found that hypoxia increased the relative amount of HA-GLUT1 on the cell surface of control cells; however, overexpression of ARRDC4 reduced translocation of GLUT1 to the plasma membrane in response to hypoxia.
To further scrutinize the effect of ARRDC4 on GLUT1 trafficking, co-localization was analyzed by confocal imaging of HEK293T cells expressing GLUT1-mCherry fusion and ARRDC4-GFP fusion. Cells transfected with an empty vector showed higher expression of surface GLUT1-mCherry on the plasma membrane (Figure 1J). In contrast, in cells overexpressing ARRDC4, GLUT1-mCherry was predominantly found inside cytoplasmic vesicles co-localized with ARRDC4-GFP. Then, we tested whether ARRDC4 promotes GLUT1 endocytosis through a clathrin-dependent pathway. Immunostaining showed that upon internalization, both clathrin and its adaptor protein AP-2 were co-localized with GLUT1 in intracellular puncta (Figure S1B–C). During clathrin-mediated endocytosis, dynamin, a GTPase, drives vesicle scission from the plasma membrane (Figure 1I)19. When cells were treated with a dynamin inhibitor Dyngo, which blocks membrane scission at the neck of a clathrin-coated pit, the puncta of GLUT1-mCherry remained at the edge of the plasma membrane with intracytoplasmic granules of ARRDC4-GFP (Figure 1J). Further, when cells were preincubated with a selective cell membrane permeable clathrin inhibitor Pitstop120, ARRDC4-mediated GLUT1 endocytosis was abolished in a dose-dependent manner (Figure 1K). These data suggest that ARRDC4 inhibits cellular glucose uptake by promoting GLUT1 endocytosis through a clathrin-dependent pathway.
ARRDC4 overexpression promotes cellular stress.
We previously showed that TXNIP mediates cell death by inhibiting glucose metabolism in a thioredoxin-independent manner10, 12. We, therefore, hypothesized that cytotoxicity is also a function conserved between TXNIP and ARRDC4. We overexpressed ARRDC4 in mouse neonatal cardiomyocytes or in efficiently-transfectable HT1080 cells and found that ARRDC4 was cytotoxic to both cell types. The cytotoxicity was demonstrated by multiple techniques, including increased lactate dehydrogenase (LDH) release (Figure 2A), cellular trypan blue uptake (Figure 2B), activation of caspase-3/7 (Figure 2C), increased Annexin V-positive apoptosis (Figure 2D) and DNA Nuclear Green DCS1-positive necrosis (Figure 2E and Figure S2A). These results are consistent with the depletion of intracellular ATP (Figure 2F), which is a part of the common machinery of apoptosis and necrosis21.
Figure 2. ARRDC4 as a metabolic stress inducer.
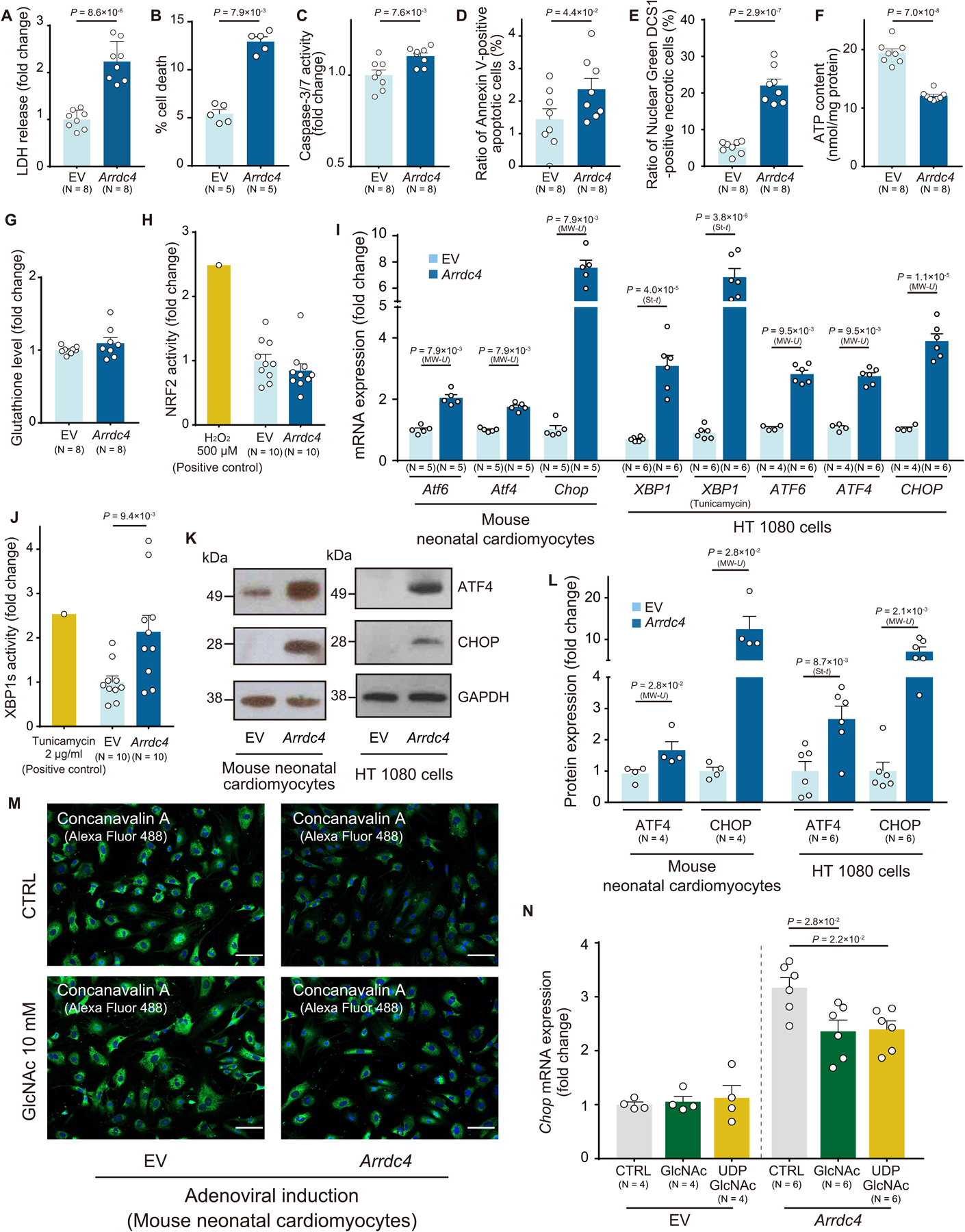
A. Adenoviral overexpression of Arrdc4 increased lactate dehydrogenase (LDH) release into culture media compared to empty vector (EV) control in mouse neonatal cardiomyocytes (t-test). B-F. After adenoviral induction of ARRDC4 in HT1080 cells, cellular damage was detected by trypan blue staining (B), activation of caspase-3/7 (C), FITC-Annexin V-positive apoptosis (D), nuclear green DCS1-positive necrosis (E) and cellular ATP levels (F) (A and C-F, t-test; B, Mann-Whitney U test). G and H. The amount of glutathione was estimated in live cells. NRF2 activation was quantified as a cellular response to oxidative stress. HT1080 cells were transfected with a plasmid containing a reporter gene in which luciferase was fused with the NRF2 ubiquitination domain; thus, oxidative stressor would stabilize the luciferase-fusion protein and increase luciferase activities. H2O2 (500 μM for 2 hr) as a positive control. P=N.S. (t-test). I. The induction of major endoplasmic reticulum (ER) stress markers was quantified by quantitative PCR in Arrdc4-overexpressing mouse neonatal cardiomyocytes and HT1080 cells. Tunicamycin (1 μg/ml, 2 hr) was added to aggravate ER stress (St-t, t-test; MW-U, Mann-Whitney U test). J. XBP1s activation was quantified by luciferase assay in HT 1080 cells (t-test). K and L. Protein expression of ATF4 or CHOP was analyzed by Western blot analysis and quantified by densitometry (St-t, t-test; MW-U, Mann-Whitney U test). M and N. N-linked protein glycosylation was detected by fluorescently-labeled concanavalin A. The addition of N-acetylglucosamine (GlcNAc, 10 mM, 8 hr) or uridine diphosphate-GlcNAc (UDP-GlcNAc, 1 mM, 8 hr) partially restored ARRDC4-mediated induction of CHOP (one-way ANOVA post-hoc Bonferroni test) in Arrdc4-overexpressing mouse neonatal cardiomyocytes.
Since ARRDC4 greatly decreased cell viability associated with inhibition of glucose uptake, we next sought to explore a regulatory relationship between ARRDC4-mediated metabolic inhibition and cellular stress. Based on the established role of TXNIP in regulating redox homeostasis, we estimated levels of reactive oxygen species (ROS) in ARRDC4-overexpressed cells. Unlike TXNIP12, ARRDC4 did not change the level of the anti-oxidant glutathione (Figure 2G) or the activation of nuclear factor erythroid 2-related factor 2 (NRF2), a sensitive marker for ROS (Figure 2H). As an alternative mechanism, glucose depletion triggers the accumulation of misfolded proteins in the endoplasmic reticulum (ER) lumen to activate the unfolded protein response (UPR)22. The canonical UPR transcriptional factors include activating transcription factor 6 (ATF6), ATF4, C/EBP homologous protein (CHOP), and X-box binding protein 1 (XBP1), which function downstream of major ER-localized stress sensors23. Interestingly, ARRDC4 increased transcriptional expression of multiple UPR genes, including ATF6, ATF4, CHOP, and XBP1 (Figure 2I). ARRDC4 rendered cells more sensitive to the UPR under tunicamycin treatment than in control groups. Then, we employed a more quantitative method to detect a spliced form of XBP1 to measure ER stress. Cells were transfected with a plasmid containing a reporter gene in which luciferase was fused with the 3’ exon side of the intron region of XBP1. Overexpression of ARRDC4 caused ER stress, which spliced the XBP1 intron sequence, leading to a formation of the luciferase-fused XBP1s (activated XBP1) with an increased luciferase activity (Figure 2J). These results indicate that ARRDC4 led to a pronounced increase in splicing of XBP1, revealing ARRDC4 as an ER stress inducer. Of the UPR gene regulatory network, the ATF4/CHOP arm mediates the expression of genes that promote glucose deprivation-induced cell death24. Western blot analysis confirmed that overexpression of ARRDC4 induced protein expression of ATF4 and CHOP (Figure 2K–L).
Given that glucose provides glycosylation precursors, we determined whether inadequate intracellular levels of glucose disturb the ER apparatus by interfering with N-linked protein glycosylation25. Overexpression of Arrdc4 decreased the level of N-linked protein glycosylation in cardiomyocytes detected by concanavalin A26 (Figure 2M). Because N-acetylglucosamine (GlcNAc) and uridine diphosphate-GlcNAc (UDP-GlcNAc) are donor molecules for post-translational modification of N-glycosylation25, we tested the ability of supplemental GlcNAc or UDP-GlcNAc to restore N-glycosylation (Figure 2M) and suppress ER stress (Figure 2N). ARRDC4-induced CHOP expression was partially but significantly inhibited by these substrates. Thus, the results support the concept that inhibition of GLUT1 trafficking and intracellular glucose starvation by ARRDC4 is involved in the UPR signaling network that determines cell fate.
Generation of the Arrdc4-knockout mouse model.
To investigate the physiological function of ARRDC4 in vivo, we created a novel Arrdc4-KO mouse model by CRISPR/Cas9 genome editing. A pair of gRNA was designed to target the Arrdc4 gene locus and microinjected with Cas9 mRNA into C57BL/6 embryos, resulting in deletion of exon 1–8 in the Arrdc4 gene (Figure 3A). PCR of genomic DNA demonstrated targeted deletion of Arrdc4 with primers F, R1, and R2, allowing amplification of the deleted region; a 964-bp fragment for the wild-type allele and a 768-bp fragment for the Arrdc4-KO (Figure 3B). DNA sequencing verified the loss of 13630 base pairs of genomic DNA within the Arrdc4 gene locus in a homozygous Arrdc4-KO mouse (Figure 3C). Quantitative PCR of multiple tissues detected no significant expression of Arrdc4 mRNA in Arrdc4-KO animals (Figure 3D). These results confirmed the successful generation of an Arrdc4-KO animal. To profile the transcriptional response to Arrdc4 deficiency, we performed unbiased high-throughput RNA-seq in heart tissues. Among 14411 genes identified (the Gene Expression Omnibus accession number GSE186336), Arrdc4 was the only gene whose expression was greatly changed in Arrdc4-KO hearts (log2 fold change: −7.0) compared with wild-type hearts (Figure 3E and Online Dataset 1).
Figure 3. The novel Arrdc4-knockout mouse model.
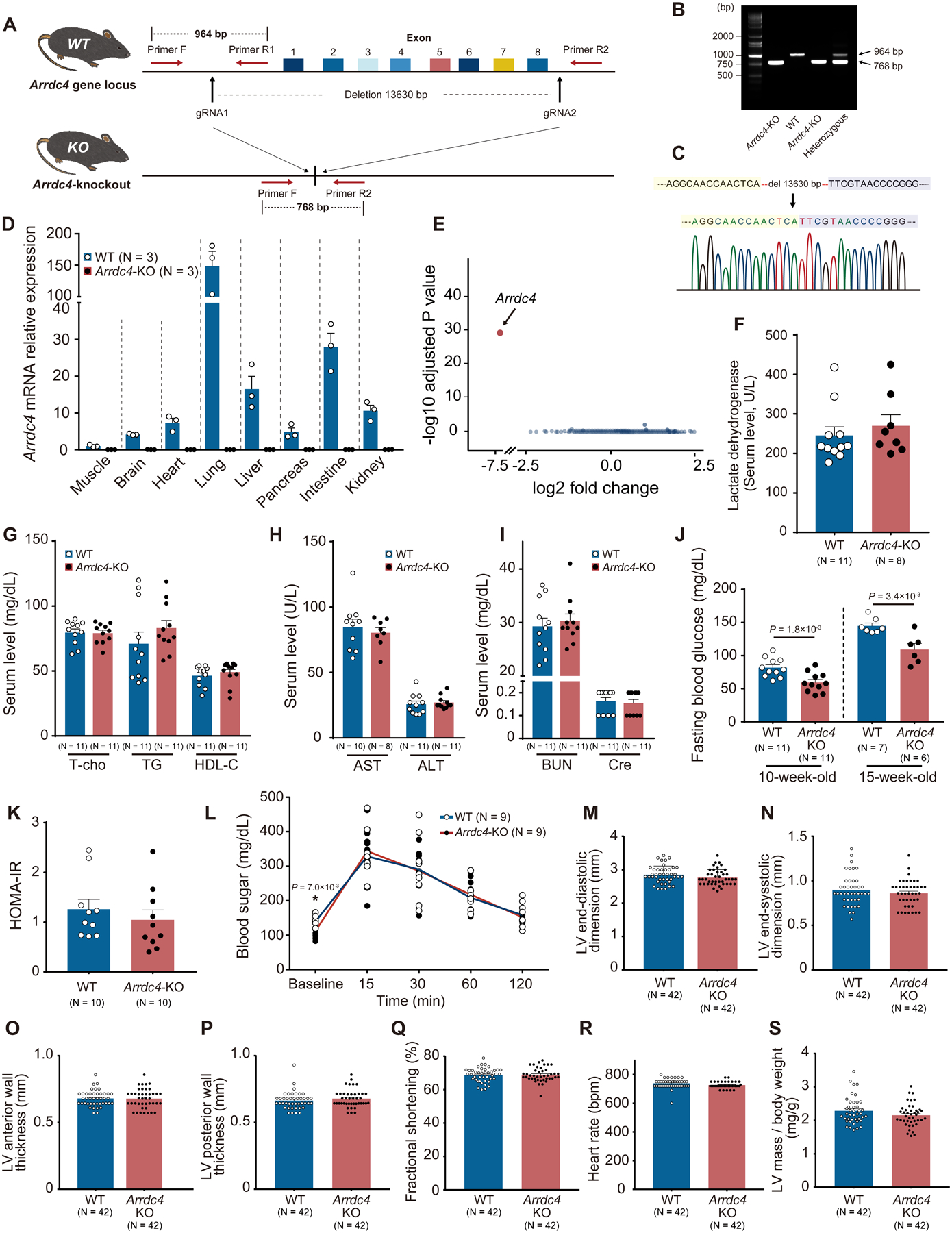
A. A pair of gRNA was designed to target the Arrdc4 locus. The gRNAs and Cas9 mRNA were microinjected into C57BL/6 embryos. B. Homozygous and heterozygous Arrdc4-knockout (KO) and wild-type (WT) mice were characterized by PCR with primers F, R1, and R2. C. Successful deletion of Arrdc4 exon1–8 was confirmed by DNA sequencing in the mouse. D. No significant Arrdc4 mRNA was expressed in multiple tissues from Arrdc4-KO mice quantified by quantitative PCR. E. Volcano plot of differentially expressed transcripts with spots representing up- or down-regulated genes in Arrdc4-KO hearts with fold change on X-axis and P-value on Y-axis. F-J. Detailed clinical chemistry was analyzed. After a fasting period of 6 hr, blood was obtained for biochemical analysis in 13-week-old mice (F-I, Mann-Whitney U test; J, t-test). K. Homeostatic model assessment of insulin resistance (HOMA-IR) was calculated from blood sugar and responsive serum insulin levels. P=N.S. (Mann-Whitney U test). L. Glucose tolerance test was performed by an intraperitoneal injection of 2 g glucose/kg body mass in mice (two-way ANOVA with repeated measures followed by Bonferroni test or *t-test for baseline comparison). M-S. Echocardiographic left ventricular (LV) parameters were measured in WT and Arrdc4-KO mice at baseline. P=N.S. between the genotypes (Mann-Whitney U test).
Since we discovered that ARRDC4 is a crucial regulator of GLUT1 function in vitro, the systemic metabolic phenotype was examined. Arrdc4-KO and wild-type littermates aged 13 weeks old did not significantly differ in body weight (Figure S2B). The serum levels of lactate dehydrogenase, lipid profile (triglycerides, total and HDL cholesterol), hepatic enzymes (alanine transaminase and aspartate transaminase), and renal function (blood urea nitrogen and creatinine) were all within the normal range in Arrdc4-KO mice, and comparable to wild-type controls in a 6-hr fasted state (Figure 3F–I). Interestingly, fasting blood glucose levels were significantly lower in Arrdc4-KO mice than in wild-type mice at two different ages (Figure 3J), even though no difference was found in random blood glucose levels between wild-type (112±5 mg/dL) and Arrdc4-KO mice (110±5 mg/dL, N=10, P=N.S.). The serum insulin levels were comparable between the genotypes in the fasted state (wild-type 3.1±0.4 vs. Arrdc4-KO 3.7±0.8 μIU/mL, N=10, P=N.S.). To determine whether loss of Arrdc4 changes insulin-mediated glucose uptake, a homeostatic model assessment of insulin resistance (HOMA-IR) was calculated. There was no difference in HOMA-IR between the genotypes (Figure 3K). We examined the kinetics of serum glucose clearance after an intraperitoneal glucose bolus in fasted mice (Figure 3L). The glucose tolerance test showed a similar rise in serum glucose levels between Arrdc4-KO and wild-type mice, suggesting a normal rate of glucose clearance by peripheral tissues under hyperglycemia in the Arrdc4-KO animals. Thus, the hypoglycemic phenotype was only evident under a fasted state in the Arrdc4-KO animals.
To assess cardiac structure and function, echocardiographic parameters were measured. There were no differences in left ventricular (LV) parameters between wild-type and Arrdc4-KO mice at baseline (Figure 3M–S).
Arrdc4-KO mice survive better after acute myocardial infarction.
The heart responds to oxygen deprivation by increasing glucose uptake and glycolytic metabolism to meet the demands under anaerobic metabolism27, 28. This shift prevents tissue damage in the ischemic heart. To address whether GLUT1 inhibition by ARRDC4 mediates cardiac ischemic injury, 12-week-old Arrdc4-KO and their age- and gender-matched wild-type mice were subjected to sham or experimental MI surgeries. Following coronary artery ligation, there was no difference between the genotypes in the degree of ST-segment elevation on electrocardiograms or in the area at risk evaluated by phthalocyanine blue staining (Figure 4A–C). However, the infarct size was significantly smaller at day 7 post-MI in Arrdc4-KO mice than in wild-type mice (Figure 4D).
Figure 4. Arrdc4-knockout mice with myocardial infarction.
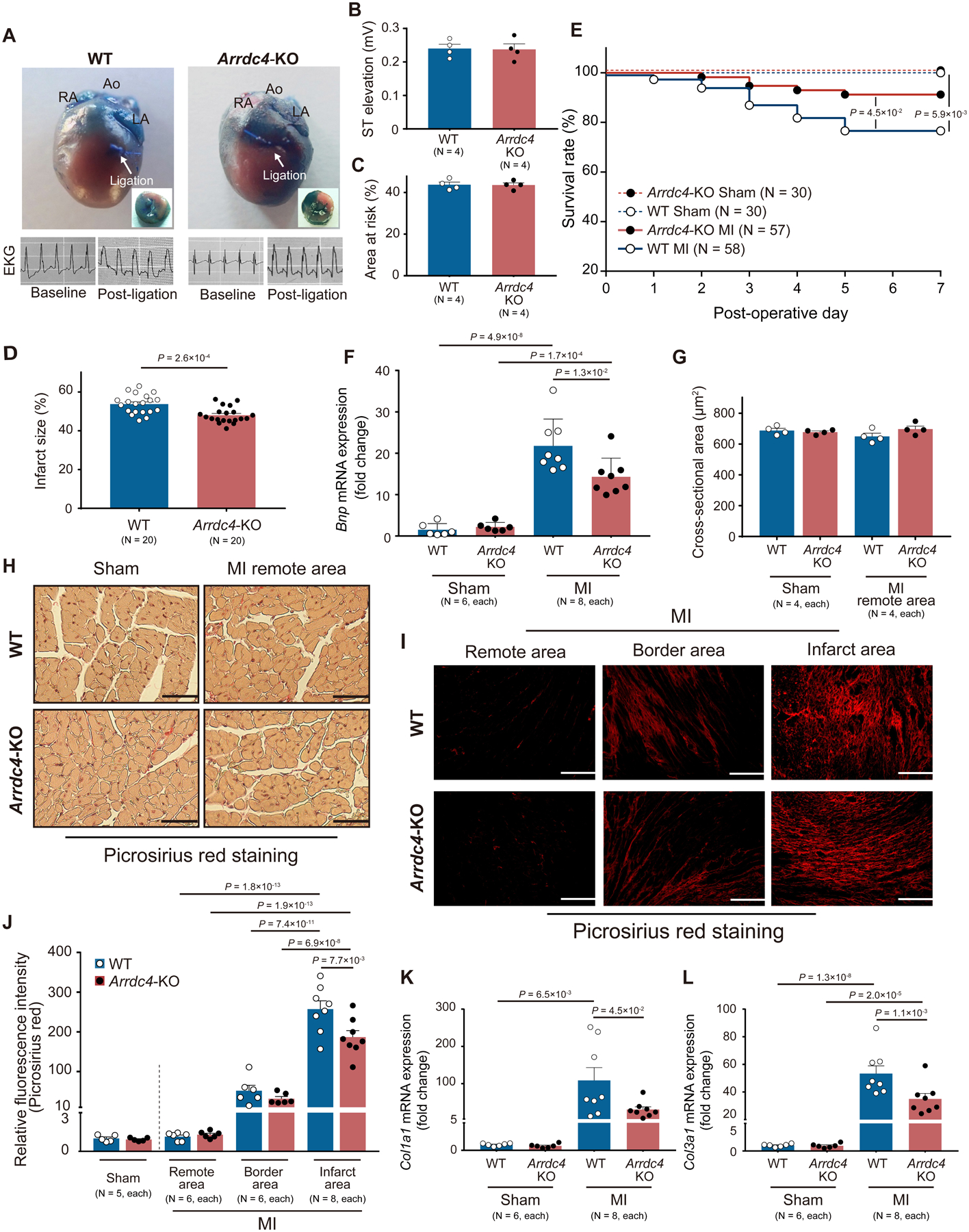
A-D. Ligation of the left anterior descending coronary artery was performed. The heart was excised to mount on an isolated heart perfusion apparatus and perfused with 0.3% phthalocyanine blue. Non-jeopardized or ischemic left ventricular (LV) tissues were identified by blue or non-blue coloration, respectively. The ST-segment elevation was confirmed in the limb lead of EKG during and after the myocardial infarction (MI) surgery. Planimetry image analysis was performed to estimate the size of the area at risk (P=N.S., Mann-Whitney U test) and infarction (t-test). E. Kaplan-Meier curves show a better survival rate at day 7 following experimental MI in Arrdc4-knockout (KO) mice than in wild-type (WT) mice (Log-rank test). F. The mRNA expression level of Bnp was measured by quantitative PCR in the apical areas of the heart tissue (two-way ANOVA post-hoc Bonferroni test). Values are expressed as fold change of the parameter with respect to WT Sham. G and H. Histological assessments with picrosirius red staining revealed no significant change in myocyte cross-sectional area between the genotypes in sham hearts and the remote regions of MI hearts (P=N.S., Kruskal-Wallis post-hoc Dunn’s test). Scale bars, 50 μm. I and J. Myocardial collagen (red) content were assessed by picrosirius red fluorescence intensity. Values are expressed as fold change of the parameter with respect to WT Sham. Scale bars, 100 μm (two-way ANOVA post-hoc Bonferroni test in MI hearts). K and L. The mRNA expression levels of Col1a1 and Col3a1 were measured by quantitative PCR in whole heart homogenates (two-way ANOVA post-hoc Bonferroni test). Values are expressed as fold change of the parameter with respect to WT Sham.
Of 30 Arrdc4-KO (15 males and 15 females) and 30 wild-type (16 males and 14 females) mice that underwent sham surgeries, no mouse died from the sham procedure during the entire experimental study period. Of 57 Arrdc4-KO (28 males and 29 females) and 58 wild-type (28 males and 30 females) mice that underwent MI surgeries, 5 Arrdc4-KO (2 males and 3 females) and 13 wild-type (5 males and 8 females) mice died throughout the 7-day protocol. No mouse died during the perioperative phase (within 12 hr after surgery). The survival rates were statistically different between the genotypes, as determined by Kaplan-Meier analysis (Figure 4E). Acute MI increased heart weight normalized by the tibial length in both Arrdc4-KO (sham 48±1 vs. MI 54±1 mg/cm, P<0.05) and wild-type (sham 50±1 vs. MI 56±1 mg/cm, P<0.05) mice (P=N.S. between the genotypes).
To assess cardiac structure and function, 2-dimensional echocardiographic parameters were measured at 7 days after MI or sham surgeries (Table 1). While LV function was significantly reduced by MI in both genotypes, Arrdc4-KO mice subjected to MI had smaller LV dimensions/volumes and higher fractional shortening than wild-type mice. Consistent with better cardiac function, Arrdc4-KO post-MI hearts exhibited a decreased mRNA expression of B-type natriuretic peptide (Bnp), a biomarker for heart failure, compared with wild-type post-MI hearts (Figure 4F).
Table 1.
Echocardiographic parameters in wild-type (WT) and Arrdc4 knock-out (KO) mice 7 days after sham or MI surgeries.
| Sham | MI | P-value | |||||
|---|---|---|---|---|---|---|---|
| WT | Arrdc4-KO | WT | Arrdc4-KO | Sham WT vs. MI WT | Sham Arrdc4-KO vs. MI Arrdc4-KO | MI WT vs. MI Arrdc4-KO | |
| Echocardiography, n | 12 | 12 | 30 | 30 | |||
| Heart rate, beat/min | 750 (720–750) | 720 (720–750) | 720 (720–750) | 720 (720–750) | N.S. | N.S. | N.S. |
| End-diastolic dimension, mm | 2.9±0.1 | 2.8±0.1 | 4.5±0.1 | 4.0±0.1 | 1.5×10−11 | 9.0×10−7 | 4.1×10−3 |
| End-systolic dimension, mm | 0.9±0.1 | 0.9±0.1 | 3.5±0.2 | 2.8±0.1 | 1.0×10−15 | 1.2×10−13 | 1.9×10−4 |
| End-diastolic volume, μL | 31.6 ±2.0 | 30.6±2.3 | 90.8 (66.4–118.2) | 65.6 (50.3–89.0) | 1.3×10−7 | 1.3×10−4 | *5.0×10−3 |
| End-systolic volume, μL | 1.3 (0.8–2.7) | 1.5±0.7 | 50.9 (31.1–64.9) | 28.3 (19.3–41.1) | 1.5×10−8 | 2.1×10−4 | *4.4×10−3 |
| Anterior wall thickness, mm | 0.65±0.02 | 0.69±0.02 | 0.37 (0.36–0.39) | 0.39 (0.36–0.43) | 8.7×10−7 | 1.6×10−5 | N.S. |
| Posterior wall thickness, mm | 0.64±0.02 | 0.68 (0.64–0.71) | 0.64 (0.57–0.71) | 0.64 (0.60–0.68) | N.S. | N.S. | N.S. |
| Fractional shortening, % | 69±2 | 69±1 | 23±1 | 30±1 | 1.0×10−15 | 1.0×10−15 | 1.0×10−4 |
| LV mass / body weight, mg/g | 2.2±0.1 | 2.3±0.1 | 3.6±0.2 | 2.8 (2.3–3.2) | 1.9×10−5 | *4.4×10−3 | 4.7×10−2 |
LV mass, Left ventricular mass. The normally distributed data are expressed as mean ± SEM (two-way ANOVA post-hoc Bonferroni test), while not normally distributed data are expressed as median and quartiles (Kruskal-Wallis test followed by post-hoc Dunn’s test or *Mann-Whitney U test).
The echocardiographic LV mass over body weight was significantly increased by MI in both genotypes, but the increase was smaller in Arrdc4-KO mice than in wild-type mice (Table 1). Histologically, MI did not induce cardiomyocyte hypertrophy in the remote area measured by the myocyte cross-sectional area (Figure 4G–H). Since posterior wall thickness was minimally changed in the non-infarcted myocardium (Table 1), the discrepancy between LV mass/body weight and myocyte cross-sectional area might be due to the type of LV remodeling post-MI which is primarily a state of volume overload29, leading to eccentric hypertrophy.
MI caused significant fibrosis within the infarct and borderline myocardium (Figure 4I–J) in both wild-type and Arrdc4-KO animals. In accordance with smaller infarct size, collagen deposition was less prominent in Arrdc4-KO hearts than in wild-type hearts. The transcript levels of collagen type I (Col1a1) and III (Col3a1) alpha 1 chains supported the histological evidence, showing less induction of the fibroblast markers in MI hearts from Arrdc4-KO mice than in wild-type animals (Figure 4K–L). These results demonstrate that deletion of Arrdc4 allowed animals to have improved survival with smaller infarct size and better cardiac function following acute MI.
Deletion of Arrdc4 protects cells from ischemic stress.
Hypoxia or lactic acidosis (Figure S2C) has been shown to trigger ARRDC4 induction in cells30. Since glucose deprivation is a pivotal contributor to cell death during hypoxic stress, we tested the hypothesis that ARRDC4 is a key determinant of cell survival under hypoxia. Hypoxic stimuli (1% O2, 24 hr) increased cellular LDH release in HT1080 cells (Figure 5A). Adenoviral overexpression of ARRDC4 further enhanced the hypoxia-induced cytotoxicity, suggesting that cellular survival is dependent on ARRDC4 expression under hypoxia. We then determined whether hypoxia-induced cytotoxicity is rescued by gene-silencing ARRDC4 with siRNA. Our approach produced a knockdown efficiency of 62±1% (Figure S2D), sufficient to decrease cellular LDH release under hypoxia relative to the non-targeting siRNA control (Figure 5B). Downregulation of ARRDC4 preserved higher levels of cellular ATP content (Figure 5C) and suppressed activation of XBP1 splicing (Figure 5D) during hypoxia. We validated the role of ARRDC4 in isolated adult mouse cardiomyocytes (Figure S2E). Hypoxic stimuli (1% O2) increased cellular LDH release (Figure 5E) and expression of Chop (Figure 5F) in a time-dependent manner. Interestingly, cardiomyocytes derived from adult Arrdc4-KO mice showed greater resistance to hypoxia than cardiomyocytes from wild-type animals. These results indicate that ARRDC4 plays a causal role in the induction of ER stress and cytotoxicity in hypoxia.
Figure 5. ARRDC4 mediates stress response to ischemia.
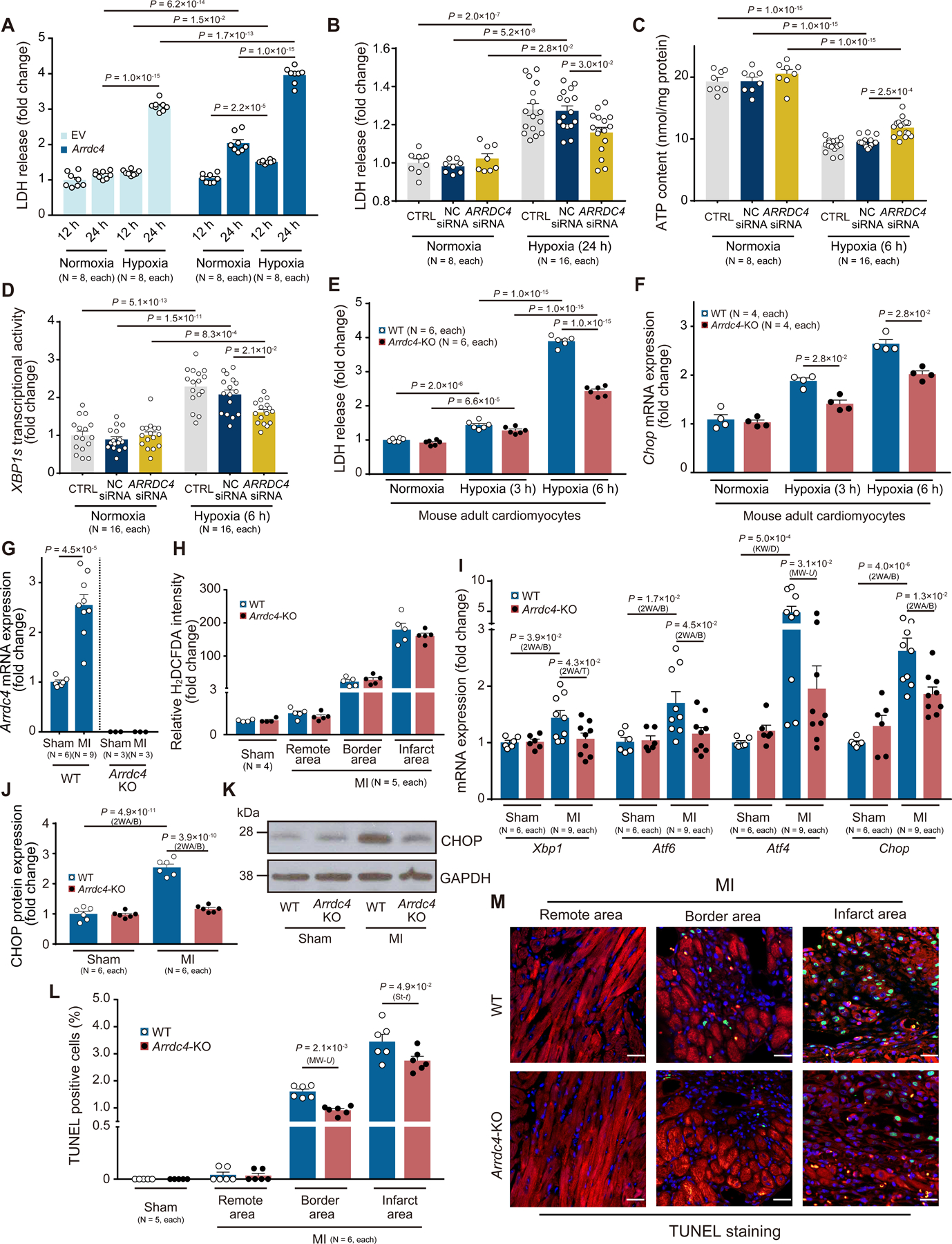
A. Adenoviral overexpression of Arrdc4 enhanced lactate dehydrogenase (LDH) release into culture media in HT1080 cells under hypoxia (1% O2, 24 hr) compared with empty vector (EV) control (two-way ANOVA post-hoc Bonferroni test). B. Mild ARRDC4 knockdown decreased LDH release under hypoxia (1% O2, 24 hr) relative to the non-targeting (NC) siRNA control. CTRL indicates cells without siRNA (two-way ANOVA post-hoc Bonferroni test). C and D. Hypoxia (1% O2, 6 hr)-induced ATP deprivation and XBP1s activation were diminished by ARRDC4 knockdown (two-way ANOVA post-hoc Bonferroni test). E and F. Adult mouse cardiomyocytes were isolated from wild-type (WT) or Arrdc4-knockout (KO) animals. LDH release (two-way ANOVA post-hoc Bonferroni test) and gene expression of Chop (Mann-Whitney U test) were analyzed under hypoxia (1% O2). G. Arrdc4 mRNA expression was upregulated at day 7 in the myocardial infarction (MI) heart from WT mice. No significant mRNA expression was detected by quantitative PCR in whole heart homogenates from Arrdc4-KO mice (t-test). H. Frozen sections of the mouse heart were stained with a fluorogenic dye H2DCFDA that measures ROS within the myocardium. The fluorescence intensity was expressed in the remote, border, or infarct area as a relative change over the WT Sham group. P=N.S. between genotypes in all areas (Kruskal-Wallis test followed by post-hoc Dunn’s test). I-K. Relative transcript levels of Xbp1, Atf6, Atf4, and Chop were measured in heart homogenates by quantitative PCR and normalized to the level of Gapdh. Protein levels of CHOP were analyzed by Western blot analysis in heart homogenates and normalized to the level of GAPDH (2WA/B, two-way ANOVA post-hoc Bonferroni test; 2WA/T, two-way ANOVA post-hoc Tukey test; KW/D, Kruskal-Wallis post-hoc Dunn’s test; MW-U, Mann-Whitney U test). Values are expressed as fold change of the parameter with respect to WT Sham. L-M. Apoptotic cells were analyzed by triple staining with TUNEL (green), an anti-sarcomeric alpha-actinin antibody (red), and DAPI (MW-U, Mann-Whitney U test; St-t, t-test). Values are expressed as fold change of the parameter with respect to WT Sham.
Consistent with the in vitro evidence, we found that the heart responded to ischemia and upregulated Arrdc4 in wild-type mice but not in Arrdc4-KO animals in vivo (Figure 5G). To determine whether MI-induced Arrdc4 expression involves oxidative stress, tissue levels of H2O2 were probed by H2DCFDA on the myocardium (Figure 5H). Although the ROS level surged by MI in border and infarct areas in wild-type hearts, Arrdc4-KO hearts had equivalent levels of ROS in MI. On the other hand, upregulation of Arrdc4 by MI was accompanied by parallel elevations in the UPR as measured by multiple transcriptional markers of Xbp1, Atf6, Atf4, and Chop in wild-type hearts (Figure 5I). Upregulation of CHOP was also confirmed by Western blotting analysis (Figure 5J–K). The induction of ER stress was significantly blocked by deletion of Arrdc4 in Arrdc4-KO MI hearts. CHOP favors ER-stress-mediated apoptosis31. MI significantly increased the number of TUNEL-positive apoptotic cardiomyocytes in border and infarct areas in wild-type hearts. However, Arrdc4-KO hearts exhibited fewer apoptotic cells than wild-type hearts (Figure 5L–M). These data demonstrate that deletion of Arrdc4 protects the heart during severe ischemia and that the beneficial effect was correlated with decreased levels of ER stress and cardiomyocyte death in Arrdc4-KO hearts.
Arrdc4-KO hearts are tolerant of ischemia with greater myocardial glucose uptake.
We found that GLUT1 was more abundantly expressed on the plasma membrane in Arrdc4-KO hearts than in wild-type hearts (Figure 6A–C). Normally, the rate of glucose transport in cardiomyocytes is GLUT1 dependent under insulin-deficient conditions28. Thus, we tested whether Arrdc4 deficiency increases myocardial glucose utilization and modulates cardiac mechanical function in isolated perfused hearts from adult mice. Arrdc4-KO hearts exhibited a two-fold increase in myocardial 2-[3H]deoxy-glucose uptake over wild-type hearts during insulin-free aerobic perfusion (Figure 6D). An increase in glucose uptake in isolated cardiac fibroblasts or inflammatory monocytes was relatively mild in Arrdc4-KO mice (a 1.3-fold or 1.2-fold increase over wild-type cells, respectively), suggesting that a major contributor to the cardiac metabolic shift is cardiomyocytes (Figure S2F). Myocardial glycogen storage was greater in Arrdc4-KO hearts than in wild-type hearts at baseline (Figure 6E). During anaerobic perfusion (with buffer equilibrated with 95% N2-5% CO2), deletion of Arrdc4 even more robustly enhanced myocardial glucose uptake (Figure 6D). In contrast, the rate of palmitate oxidation was equivalent between wild-type and Arrdc4-KO hearts during 20 min of hypoxic perfusion (Figure 6F). No significant difference in tissue ATP content was detected under normoxic perfusion between the genotypes, but following 60 min of hypoxia, ATP content was better preserved in Arrdc4-KO hearts than in wild-type hearts (Figure 6G).
Figure 6. Cardiac metabolism and ischemic tolerance in Arrdc4-knockout hearts.
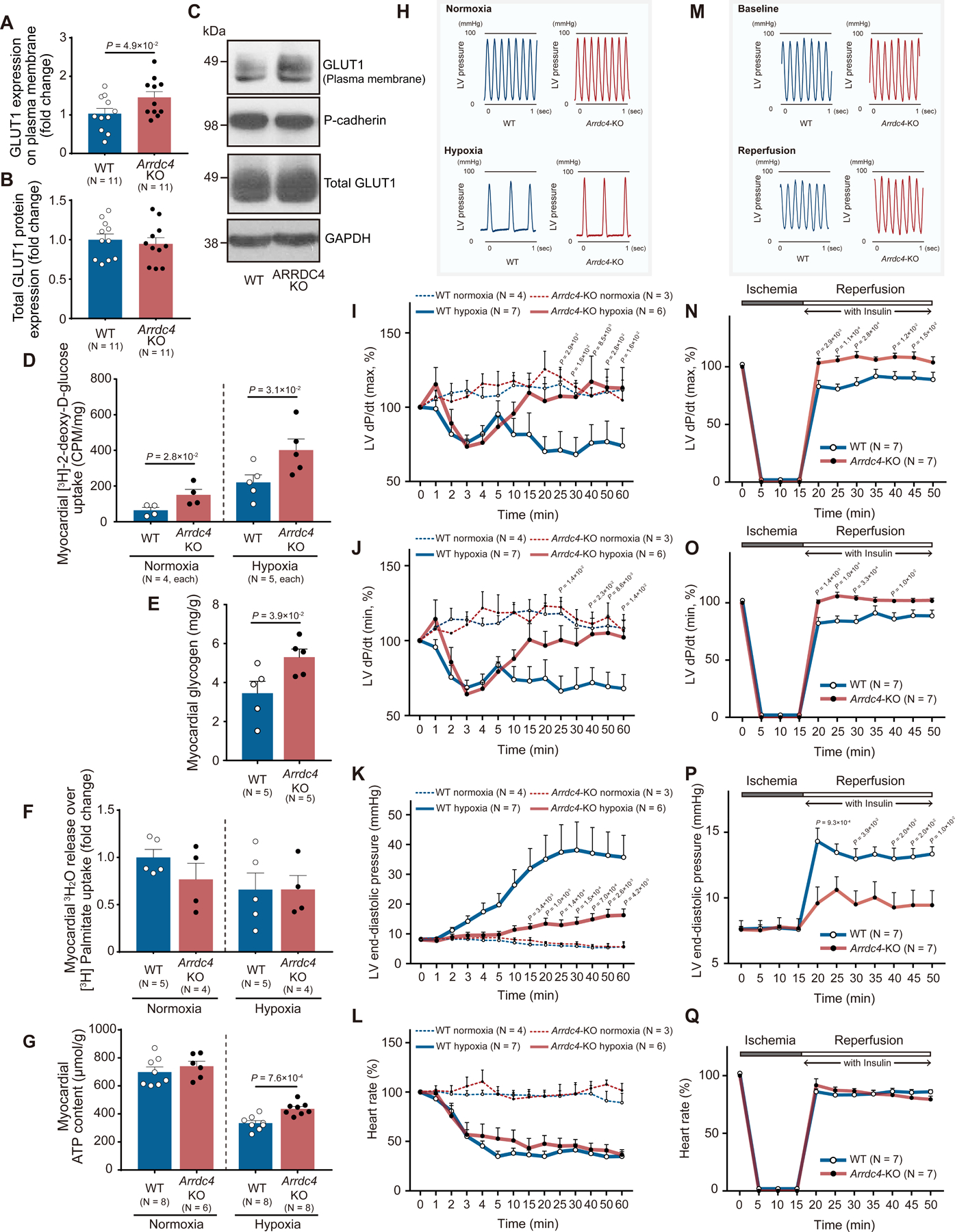
A-C. Protein expression of GLUT1 was analyzed by Western blot analysis in the membrane fraction and total tissue homogenates from the mouse heart. Pan-cadherin and GAPDH served as loading control for membrane and cytosolic proteins (t-test). D. The Arrdc4-knockout (KO) or wild-type (WT) heart was subjected to global hypoxia (with buffer equilibrated with 95% N2, 5% CO2) or normoxia (95% O2, 5% CO2) in the Langendorff perfusion system. The perfusate was switched from Krebs-Henseleit (KH) buffer to a solution containing [3H] 2-deoxy-D-glucose. The heart was perfused with the radiolabeled glucose for 20 min and washed with the standard KH buffer for 5 min. The radioactivity was measured in whole heart homogenates by liquid scintillation counting (Mann-Whitney U test). E. Myocardial glycogen storage was quantified in heart homogenates at baseline and normalized by heart weight (Mann-Whitney U test). F. Exogenous oxidation rates of [3H]-palmitate were determined by measuring the release of 3H2O into the perfusion buffer and normalized by myocardial uptake of [3H]-palmitate. P=N.S. between the genotypes (Mann-Whitney U test). G. Cellular ATP content per heart weight was measured in isolated hearts following 60 min of normoxic or hypoxic perfusion (t-test). H and M. Representative tracing of left ventricular (LV) pressure in the WT or Arrdc4-KO heart under hypoxic perfusion or following global ischemia-reperfusion. I-L and N-Q. Arrdc4-KO hearts maintained better cardiac mechanical function during hypoxic perfusion (1 hr) or after ischemia (15 min) and reperfusion, as shown by the higher LV dP/dt maximum (I or N) and minimum (J or O) and lower LV end-diastolic pressure (K or P) than those in WT hearts. Heart rate slowed down during hypoxic perfusion (L) or after reperfusion (Q) in both genotypes (the P-value indicates the comparison between Arrdc4-KO hypoxia or ischemia vs. WT hypoxia or ischemia by two-way ANOVA with repeated measures followed by Bonferroni test).
Because Arrdc4 deficiency reprogrammed glucose metabolism to maintain higher anaerobic ATP production within the myocardium under hypoxia, we investigated whether ARRDC4 is a causal determinant of myocardial mechanical function. In these experiments, the perfusion buffer contained glucose as the sole oxidative energy substrate; thus, the mechanical function was primarily supported by glucose uptake and glycolysis. Exposure to hypoxic perfusion fluid caused bradycardia and impaired LV mechanical performance in wild-type and Arrdc4-KO mice (Figure 6H). LV developed pressure fell (Figure 6I–J), and LV end-diastolic pressure rose (Figure 6K) progressively throughout 60 min of hypoxia in wild-type hearts. In contrast, Arrdc4-KO hearts maintained better mechanical performance after 10 min of hypoxia. We also tested this hypothesis in isolated perfused hearts subjected to a simulated period of global ischemia followed by reperfusion in the presence of insulin (6.0 μIU/mL) (Figure 6M). A 15-minute period of ischemia substantially impaired cardiac function in both genotypes. Upon reperfusion, LV developed pressure fell (Figure 6N–O) and LV end-diastolic pressure rose (Figure 6P) compared with pre-ischemic baseline in wild-type hearts. However, Arrdc4-KO hearts had improved post-ischemic recovery of LV mechanical function. There was no difference in heart rate between the genotypes (Figure 6L and Q). These results support our hypothesis that Arrdc4 deletion provides a functional advantage to the hypoxic or ischemic myocardium through enhanced myocardial glucose utilization. The data also suggest that the beneficial effect of Arrdc4 deficiency is cardiac autonomous as the cardioprotection was demonstrated in the isolated heart.
Loss of the ARRDC4-GLUT1 interaction restores glucose metabolism.
Given that the ARRDC4-GLUT1 interaction plays a role in ischemic injury, the question arises as to whether disruption of the interaction may confer an adaptive advantage against metabolic stress. To address this issue, we determined the details of the molecular interaction between ARRDC4 and GLUT1.
First, to identify a specific domain that contributes to the metabolic function of ARRDC4, we tested N-terminal or C-terminal truncations of ARRDC4 protein (Figure 7A). Strep/FLAG (SF)-tagged full-length ARRDC4 (1–419) and five truncations were successfully subcloned into an expression plasmid (Figure 7B) and transfected into HEK293T cells. Similar to previous findings on TXNIP32, the yield of protein expression of N-terminal truncated ARRDC4 was significantly less than that of full-length or C-terminal truncations. Thus, we only used full-length or C-terminal truncations for the pulldown assay. Western blot analysis for elution of protein complexes revealed that the anti-HA antibody detected a strong band with a molecular weight of 45 kDa and a weaker band of 55 kDa (Figure 7C), which corresponded to the known molecular weights of the GLUT1 protein33. The pulldown assay showed that ARRDC4 truncated at the tail domain (1–318) retained the ability to interact with GLUT1. Further truncation into the C-terminal domain (1–222) fully abolished the interaction with GLUT1. The results indicate that the C-terminal domain containing the region 222–318 is vital in the interaction with GLUT1.
Figure 7. Identification of the ARRDC4-GLUT1 docking sites.
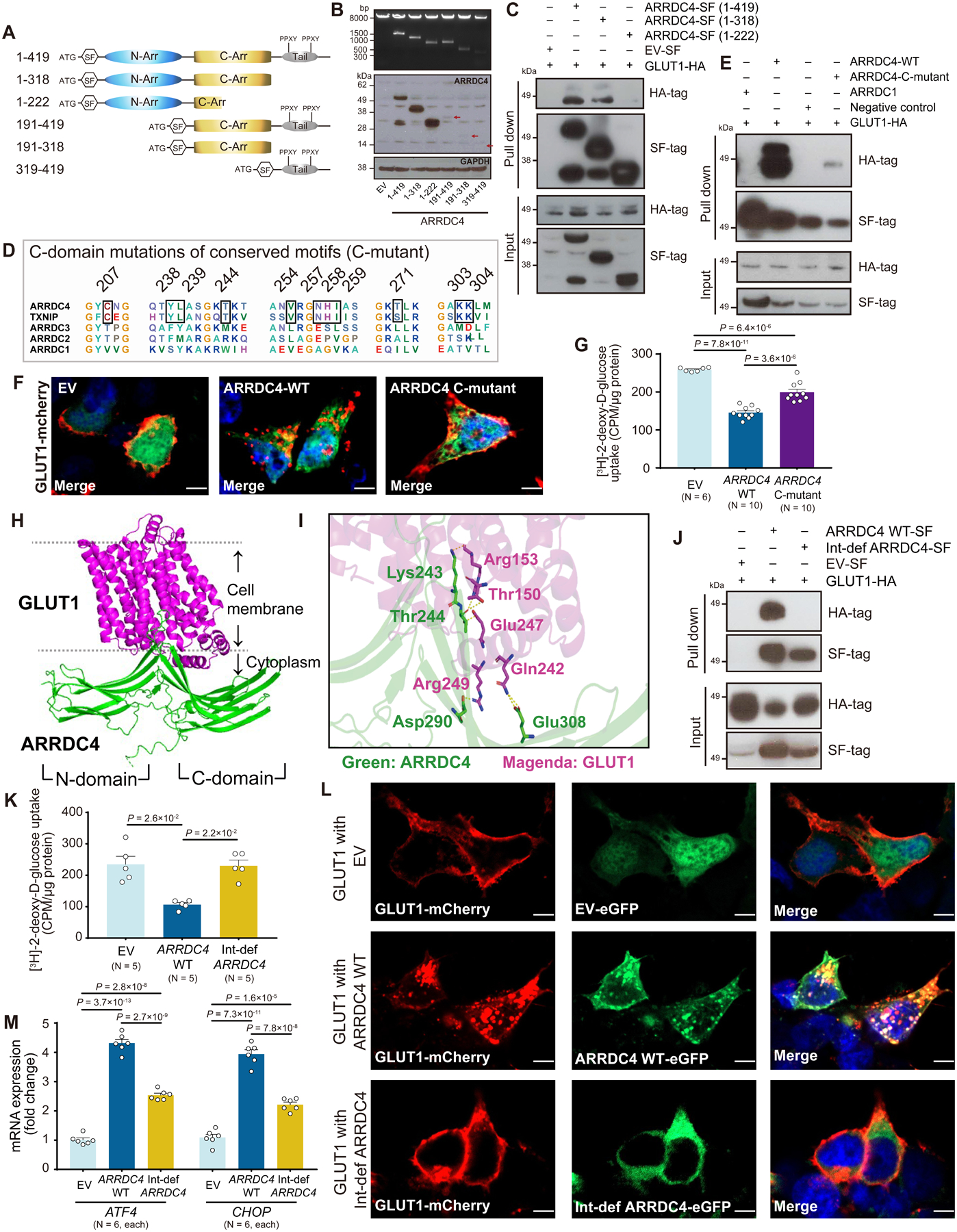
A. Schematic of Strep/FLAG (SF)-tagged constructs for full-length human ARRDC4 (residues 1–419) and five truncations. B. Expression of all ARRDC4 constructs was verified by Western analysis of cell lysates with anti-FLAG and GAPDH antibodies. Arrows indicate protein expression of N-terminal domain truncations. C. Full length or truncation of ARRDC4-SF-tag or empty vector (EV)-SF-tag was co-expressed with GLUT1-HA-tag in HEK293T cells. Complexes were pulled down with Strep-Tactin resin, subjected to SDS-PAGE, and immunoblotted with anti-HA or anti-FLAG antibodies. D. Alignment of C-terminal domain of human alpha arrestins identified candidate motifs conserved between ARRDC4 and TXNIP. E. ARRDC4-wild-type (WT)-SF-tag, ARRDC4-C-terminal domain motifs mutant (C-mutant)-SF-tag or ARRDC1-SF-tag was co-expressed with GLUT1-HA-tag in HEK293 cells. Complexes were pulled down as described above. F. Confocal microscopic images showed GLUT1-mCherry fusion (red) on the plasma membrane in HEK293T cells transfected with EV containing tandem GFP marker gene (green). GLUT1 localized in intracellular puncta in cells overexpressing ARRDC4 WT or C-mutant. Blue, DAPI. Scale bars, 5 μm. G. 2-[3H]deoxy-D-glucose uptake was measured in HEK293 cells overexpressing EV, ARRDC4 WT, or C-mutant. Uptake was normalized to total protein content (one-way ANOVA post-hoc Bonferroni test). H and I. Artificial Intelligence predicted Lys243, Thr244, Asp290, and Glu 308 in the C-terminal domain as possible docking sites with GLUT1. J. Interaction-defective (Int-def) ARRDC4 was created by mutating all four amino acids to alanine. Pull-down assay was performed for ARRDC4 WT-SF-tag, Int-def ARRDC4-SF-tag, or EV-SF-tag in HEK293 cells co-expressed with GLUT1-HA-tag. K and L. Int-def ARRDC4 restored the inhibitory effect of ARRDC4 WT on 2-[3H]deoxy-D-glucose uptake in HEK293 cells (Kruskal-Wallis post-hoc Dunn’s test). Confocal images showed intracellular internalization of GLUT-mCherry by overexpression of ARRDC4 WT-eGFP but not by Int-def ARRDC4-eGFP. Blue, DAPI. Scale bars, 5 μm. M. The mRNA expression of ATF4 and CHOP in cellular lysates was measured by quantitative PCR and normalized to the level of GAPDH (one-way ANOVA post-hoc Bonferroni test).
Then, because ARRDC4 and TXNIP had similar metabolic regulatory effects while ARRDC1–3 did not (Figure 1A), we hypothesized that motifs conserved in ARRDC4 and TXNIP but not ARRDC1–3 might identify molecular mechanisms to control GLUT1 function. Alignments of alpha arrestins revealed seven-candidate motifs in the C-terminal domains (Figure 7D). We constructed point mutations in all these conserved motifs in the C-terminal domain by replacing the ARRDC4 residue with alanine by alanine-scanning mutagenesis34. The mutation (C-mutant) only partially blocked the interaction with GLUT1 (Figure 7E) and did not fully recover the cellular glucose uptake (Figure 7G). GLUT1 expression remained in intracellular puncta in cells expressing ARRDC4 C-mutant, indicating that GLUT1 had been still endocytosed (Figure 7F).
To map the specific binding sites between ARRDC4 and GLUT1, we next employed a deep-learning method based on the AlphaFold database35 to build the structural model of the ARRDC4-GLUT1 complex (Figure 7H). Using the HDOCK algorithm36, our computational analyses constructed the model (Table S1) that predicted the contact possibility between the C-terminal domain of ARRDC4 (the side chain of Lys243, thr244, Asp290, and Glu308) and the intracellular loops of GLUT1 (Figure 7I). Surprisingly, when these residues were all mutagenized, it caused a total loss of interaction with GLUT1 (Figure 7J). Wild-type ARRDC4 inhibited cellular glucose transport robustly, but the interaction-defective ARRDC4 restored the level of glucose transport to that of empty-vector control (Figure 7K). Consistently, confocal imaging verified that, while cells expressing wild-type ARRDC4 had a granular GLUT1-mCherry expression in cytoplasmic vesicles, the mutation of the docking site preserved the level of GLUT1-mCherry on the plasma membrane (Figure 7L). The interaction-defective ARRDC4 failed to fully induce the ATF4/CHOP mRNA expression compared to wild-type ARRDC4 (Figure 7M). These data suggest that the C-terminal domain element is a prerequisite for the ability of ARRDC4 to control GLUT1 function and that disruption of the ARRDC4-GLUT1 interaction rescues cells from the ARRDC4-mediated metabolic stress.
Discussion
Nutrient limitation is a feature of the cardiomyocyte environment under severe ischemia. The characteristics of cell injury involve rapid disruption of cell membrane integrity and an accumulation of unfolded or misfolded proteins in the ER, rendering cardiomyocytes more susceptible to cell death. The mediators that regulate crosstalk between energy deprivation and the maladaptive response to ischemia are still elusive. In this study, using a combination of molecular and physiologic approaches, we demonstrate a unique role of an alpha-arrestin ARRDC4 in glucose metabolism and cardiomyocyte survival. Building on the unexpected finding that MI upregulates Arrdc4, our model integrates the observations that (a) ARRDC4 serves as an adaptor protein for GLUT1 to promote its endocytosis, (b) upregulation of ARRDC4 inhibits glucose transport, (c) inhibition of glucose transport is linked to ER stress, and (d) this mediates deleterious effects on cardiomyocyte survival in MI. Importantly, we discovered that deletion of Arrdc4 mitigates the energy crisis during ischemia by enhancing glucose transport, rescuing cardiomyocytes from enhanced vulnerability. The data also suggest that this mechanism of action of ARRDC4 gives new and relevant insight into the therapeutic strategy through the direct interaction with GLUT1.
The results presented here, along with previous studies13, suggest that alpha-arrestins are part of a feedback loop in glucose homeostasis. Ischemia recruits GLUTs from intracellular stores to the plasma membrane, and if oxygen deprivation is prolonged, the transcription is modified27, 28. The net result of these changes by ischemia is an increase in the maximal velocity of glucose transport. Upregulation of ARRDC4, in turn, promotes the internalization of GLUT1 to brake further glucose uptake. However, under the condition where ischemic stress is severe and persistent, this feedback may lead to a maladaptive response by blocking a preferential anaerobic bioenergetics pathway. The remarkable shared phenotypes between Arrdc4-KO and Txnip-KO hearts strongly support their common regulatory mechanisms in cardiac metabolism, including enhanced myocardial glucose uptake, increased myocardial glycogen storage, and less cardiomyocyte death during ischemia16. The concept that shifting the energy substrate preference toward glucose metabolism effectively treats acute MI has been long proposed37. Cardiac-specific overexpression of GLUT1 protects the heart from ischemic injury in mice38. Hence, it is reasonable to conclude that cellular homeostasis is better preserved through the interplay with GLUT1 in the Arrdc4-KO heart under myocardial ischemia.
GLUT1 internalization proceeds through clathrin-dependent or -independent endocytosis pathways13. Early studies demonstrated that alpha-arrestins interact with subunits of the endosomal sorting complexes required for transport machinery7, 39. Two distant relatives of alpha-arrestins (Art4 and Art8) cause endocytosis of high-affinity glucose transporter (Hxt6) in budding yeast40. TXNIP functions as an adaptor protein to recruit GLUT1 into clathrin-coated pits and promotes endocytosis of GLUT1 in human cells13. Given that ARRDC4 is most closely related to TXNIP in homology10, it is intriguing that ARRDC4 and TXNIP share the same ability to regulate GLUT1 trafficking. Whereas TXNIP usually is localized in the nucleus, ARRDC4 is already present at the plasma membrane10 showing a greater inhibition of glucose uptake than does TXNIP. Because two members of the alpha-arrestin family are now shown to interact with GLUT1, these results emphasize the importance of the evolutionally-conserved arrestin fold in transporter trafficking and suggest the possibility that multiple arrestins may work together to control basal glucose metabolism.
Although TXNIP has been shown to interact with GLUT in a 1:1 molecular ratio14, the interaction interface of TXNIP that controls GLUT1 has been uncharacterized. Our truncation and targeted mutagenesis studies focused on 7 conserved motifs between TXNIP and ARRDC4 in the C-terminal arrestin domain. Then, we took the investigation one step further using Artificial Intelligence to map the specific binding sites within the domain. Based on its high accuracy in structure determination35, this approach was successful in detailed predictions such that those motifs, when mutated, showed significant effects. Among the set of 4 amino acid residues identified, one was conserved (Thr244) in ARRDC4 and TXNIP, while the others (Lys243, Asp290, and Glu308) were not. We speculate that Thr244 is a crucial site responsible for alpha-arrestin to bind to GLUT1, and the others may support rigid docking. Despite the ability of the interaction-deficient mutant to completely restore cellular glucose uptake (Figure 7K), the induction of ER stress was only partially inhibited by the mutant (Figure 7M). This raises the possibility that there are alternative mechanisms involved. For example, TXNIP has pleiotropic functions by interacting with multiple binding partners, including protein disulfide isomerase41 and nucleotide-binding domain nod-like receptor protein 3 (NLRP3) inflammasome42. While no other signaling molecule has been identified so far as a binding partner for ARRDC4, ARRDC4 may also be involved in various biological processes other than GLUT1 regulation.
GLUT1 is the most ubiquitously distributed of the transporter isoforms and is constitutively present on peripheral tissue surfaces. Transgenic mice that overexpress GLUT1 in skeletal muscle exhibit reduced plasma glucose levels43. The present study showed that deletion of Arrdc4 exerts mild but significant effects on systemic glucose metabolism. The beneficial impact of Arrdc4 deficiency was demonstrated in the isolated heart (Figure 6); thus, the cardioprotection likely accounts for the mechanisms within the heart. Further investigation will be required to address the cardiac-specific function of ARRDC4 fully. Nevertheless, we believe that the current study with the systemic-KO model provides valuable information. For instance, protein-protein interaction (PPI) inhibitors emerge as a new therapeutic approach44. We envision a possible therapeutic strategy that disrupts the ARRDC4-GLUT1 interaction with a commonly applicable approach (e.g., oral drugs). Therefore, the cardioprotection by systemic inhibition of ARRDC4 highlights the translational potential.
In conclusion, our work forges a new mechanistic link between alpha-arrestin domains and glucose transport regulation. These results expand our understanding of structure-function relationships for mammalian alpha-arrestin recruitment to GLUT1. Notably, the data shed light on the ARRDC4-GLUT1 axis and emphasize its importance to energy homeostasis under metabolic stress, during which GLUT1 is a crucial player. The molecular pathways defined herein provide a new target for therapeutic intervention aimed at improving cardioprotection under myocardial ischemia and infarction.
Supplementary Material
Novelty and Significance.
What is known?
ARRDC4 (arrestin domain-containing protein 4) is closely related to TXNIP (thioredoxin-interacting protein) in homology among the alpha arrestin superfamily.
TXNIP acts as an adaptor protein to facilitate endocytosis of GLUT (glucose transporter) 1 and suppresses glucose influx, although the direct interface between TXNIP and GLUT1 remains uncharacterized.
Unlike TXNIP, the physiological role of ARRDC4 in the heart is largely unknown.
What new information does the article contribute?
The present study is the first to demonstrate that ARRDC4 is a second example of an arrestin that acts as an adaptor protein for GLUT1, suggesting that the control of basal glucose metabolism is an evolutionarily conserved function of the alpha-arrestin family.
Furthermore, we identify completely novel molecular structure-function characterization of the interaction between alpha-arrestins and GLUT1 and uncover that interaction-defective ARRDC4 restores GLUT1 function.
Finally, we provide direct in vivo evidence that ARRDC4 inhibits myocardial glucose uptake and accelerates myocardial ischemic injury.
The heart responds to oxygen deprivation by increasing glucose uptake and glycolytic metabolism for the anaerobic energy source. In cardiomyocytes, glucose uptake is facilitated by glucose transporters GLUT1 and GLUT4. Here, we discovered that ARRDC4, a member of a previously obscure protein family termed alpha-arrestin, serves as a regulatory scaffold protein for GLUT1 and facilitates GLUT1 endocytosis in cardiomyocytes. ARRDC4 inhibits cellular glucose utilization, induces endoplasmic reticulum (ER) stress, and sensitizes cardiomyocytes to cellular damage under hypoxia. By creating a novel Arrdc4-knockout (KO) mouse model, we found that inhibition of ARRDC4 augments myocardial glucose uptake and mitigates the energy crisis, thereby improving cardiac outcomes following acute myocardial infarction (MI). Using biochemical and bioinformatics approaches, we dissected the structure-function relationship between ARRDC4 and GLUT1. We found that the C-terminal domain element in ARRDC4 is a prerequisite for the ability of ARRDC4 to control the GLUT1 function. Further analysis revealed that disruption of the ARRDC4-GLUT1 interaction rescues cells against metabolic stress. These results emphasize the regulatory mechanism of glucose homeostasis by ARRDC4 as an adaptor protein for GLUT1 and provide insight into the new therapeutic strategy to protect the heart against MI.
Sources of Funding
This work was supported by awards from the E.G. Watkins Family Foundation, the Philip V. & Anna S. Brown Foundation, the LaRue S. Fisher and Walter F. Fisher Memorial Trust, and an NIH grant 1R01HL130861.
Footnotes
Disclosures
None
References
- 1.Liang CS and Delehanty JD. Increasing post-myocardial infarction heart failure incidence in elderly patients a call for action. J Am Coll Cardiol. 2009;53:21–3. [DOI] [PubMed] [Google Scholar]
- 2.Shao D and Tian R. Glucose Transporters in Cardiac Metabolism and Hypertrophy. Compr Physiol. 2015;6:331–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brosius FC 3rd, Liu Y, Nguyen N, Sun D, Bartlett J and Schwaiger M. Persistent myocardial ischemia increases GLUT1 glucose transporter expression in both ischemic and non-ischemic heart regions. J Mol Cell Cardiol. 1997;29:1675–85. [DOI] [PubMed] [Google Scholar]
- 4.Tian R and Abel ED. Responses of GLUT4-deficient hearts to ischemia underscore the importance of glycolysis. Circulation. 2001;103:2961–6. [DOI] [PubMed] [Google Scholar]
- 5.Chen Z, Dudek J, Maack C and Hofmann U. Pharmacological inhibition of GLUT1 as a new immunotherapeutic approach after myocardial infarction. Biochem Pharmacol. 2021;190:114597. [DOI] [PubMed] [Google Scholar]
- 6.Lefkowitz RJ, Rajagopal K and Whalen EJ. New roles for beta-arrestins in cell signaling: not just for seven-transmembrane receptors. Mol Cell. 2006;24:643–652. [DOI] [PubMed] [Google Scholar]
- 7.Kahlhofer J, Leon S, Teis D and Schmidt O. The alpha-arrestin family of ubiquitin ligase adaptors links metabolism with selective endocytosis. Biol Cell. 2021;113:183–219. [DOI] [PubMed] [Google Scholar]
- 8.Puca L and Brou C. Alpha-arrestins - new players in Notch and GPCR signaling pathways in mammals. J Cell Sci. 2014;127:1359–67. [DOI] [PubMed] [Google Scholar]
- 9.Patwari P and Lee RT. An expanded family of arrestins regulate metabolism. Trends Endocrinol Metab. 2012;23:216–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patwari P, Chutkow WA, Cummings K, Verstraeten VL, Lammerding J, Schreiter ER and Lee RT. Thioredoxin-independent regulation of metabolism by the alpha-arrestin proteins. J Biol Chem. 2009;284:24996–5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chutkow WA, Patwari P, Yoshioka J and Lee RT. Thioredoxin-interacting protein (Txnip) is a critical regulator of hepatic glucose production. J Biol Chem. 2008;283:2397–406. [DOI] [PubMed] [Google Scholar]
- 12.Nakayama Y, Mukai N, Wang BF, Yang K, Patwari P, Kitsis RN and Yoshioka J. Txnip C247S mutation protects the heart against acute myocardial infarction. J Mol Cell Cardiol. 2021;155:36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu N, Zheng B, Shaywitz A, Dagon Y, Tower C, Bellinger G, Shen CH, Wen J, Asara J, McGraw TE, Kahn BB and Cantley LC. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Molecular cell. 2013;49:1167–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dykstra H, LaRose C, Fisk C, Waldhart A, Meng X, Zhao G and Wu N. TXNIP interaction with GLUT1 depends on PI(4,5)P2. Biochim Biophys Acta Biomembr. 2021;1863:183757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hovsepian J, Albanese V, Becuwe M, Ivashov V, Teis D and Leon S. The yeast arrestin-related protein Bul1 is a novel actor of glucose-induced endocytosis. Mol Biol Cell. 2018;29:1012–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshioka J, Chutkow WA, Lee S, Kim JB, Yan J, Tian R, Lindsey ML, Feener EP, Seidman CE, Seidman JG and Lee RT. Deletion of thioredoxin-interacting protein in mice impairs mitochondrial function but protects the myocardium from ischemia-reperfusion injury. J Clin Invest. 2012;122:267–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waldhart AN, Dykstra H, Peck AS, Boguslawski EA, Madaj ZB, Wen J, Veldkamp K, Hollowell M, Zheng B, Cantley LC, McGraw TE and Wu N. Phosphorylation of TXNIP by AKT Mediates Acute Influx of Glucose in Response to Insulin. Cell Rep. 2017;19:2005–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carruthers A, DeZutter J, Ganguly A and Devaskar SU. Will the original glucose transporter isoform please stand up! American journal of physiology Endocrinology and metabolism. 2009;297:E836–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cocucci E, Gaudin R and Kirchhausen T. Dynamin recruitment and membrane scission at the neck of a clathrin-coated pit. Mol Biol Cell. 2014;25:3595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ, MacGregor KA, Tomilin N, Pechstein A, Chau N, Chircop M, Sakoff J, von Kries JP, Saenger W, Krausslich HG, Shupliakov O, Robinson PJ, McCluskey A and Haucke V. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell. 2011;146:471–84. [DOI] [PubMed] [Google Scholar]
- 21.Tsujimoto Y Apoptosis and necrosis: intracellular ATP level as a determinant for cell death modes. Cell Death Differ. 1997;4:429–34. [DOI] [PubMed] [Google Scholar]
- 22.Iurlaro R, Puschel F, Leon-Annicchiarico CL, O’Connor H, Martin SJ, Palou-Gramon D, Lucendo E and Munoz-Pinedo C. Glucose Deprivation Induces ATF4-Mediated Apoptosis through TRAIL Death Receptors. Mol Cell Biol. 2017;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hetz C, Zhang K and Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21:421–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leon-Annicchiarico CL, Ramirez-Peinado S, Dominguez-Villanueva D, Gonsberg A, Lampidis TJ and Munoz-Pinedo C. ATF4 mediates necrosis induced by glucose deprivation and apoptosis induced by 2-deoxyglucose in the same cells. FEBS J. 2015;282:3647–58. [DOI] [PubMed] [Google Scholar]
- 25.Palorini R, Cammarata FP, Balestrieri C, Monestiroli A, Vasso M, Gelfi C, Alberghina L and Chiaradonna F. Glucose starvation induces cell death in K-ras-transformed cells by interfering with the hexosamine biosynthesis pathway and activating the unfolded protein response. Cell Death Dis. 2013;4:e732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishino K, Kudo M, Peng WX, Kure S, Kawahara K, Teduka K, Kawamoto Y, Kitamura T, Fujii T, Yamamoto T, Wada R and Naito Z. 2-Deoxy-d-glucose increases GFAT1 phosphorylation resulting in endoplasmic reticulum-related apoptosis via disruption of protein N-glycosylation in pancreatic cancer cells. Biochem Biophys Res Commun. 2018;501:668–673. [DOI] [PubMed] [Google Scholar]
- 27.Young LH, Russell RR 3rd, Yin R, Caplan MJ, Ren J, Bergeron R, Shulman GI and Sinusas AJ. Regulation of myocardial glucose uptake and transport during ischemia and energetic stress. Am J Cardiol. 1999;83:25H–30H. [DOI] [PubMed] [Google Scholar]
- 28.Depre C, Vanoverschelde JL and Taegtmeyer H. Glucose for the heart. Circulation. 1999;99:578–88. [DOI] [PubMed] [Google Scholar]
- 29.French BA and Kramer CM. Mechanisms of Post-Infarct Left Ventricular Remodeling. Drug Discov Today Dis Mech. 2007;4:185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen JL, Merl D, Peterson CW, Wu J, Liu PY, Yin H, Muoio DM, Ayer DE, West M and Chi JT. Lactic acidosis triggers starvation response with paradoxical induction of TXNIP through MondoA. PLoS Genet. 2010;6:e1001093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szegezdi E, Duffy A, O’Mahoney ME, Logue SE, Mylotte LA, O’Brien T and Samali A. ER stress contributes to ischemia-induced cardiomyocyte apoptosis. Biochem Biophys Res Commun. 2006;349:1406–11. [DOI] [PubMed] [Google Scholar]
- 32.Xiao X, Xu Y and Chen H. Sodium butyrate-activated TRAF6-TXNIP pathway affects A549 cells proliferation and migration. Cancer Med. 2020;9:3477–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu S and Ding WG. The 45 kDa form of glucose transporter 1 (GLUT1) is localized in oligodendrocyte and astrocyte but not in microglia in the rat brain. Brain Res. 1998;797:65–72. [DOI] [PubMed] [Google Scholar]
- 34.Cunningham BC and Wells JA. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244:1081–5. [DOI] [PubMed] [Google Scholar]
- 35.Humphreys IR, Pei J, Baek M, Krishnakumar A, Anishchenko I, Ovchinnikov S, Zhang J, Ness TJ, Banjade S, Bagde SR, Stancheva VG, Li XH, Liu K, Zheng Z, Barrero DJ, Roy U, Kuper J, Fernandez IS, Szakal B, Branzei D, Rizo J, Kisker C, Greene EC, Biggins S, Keeney S, Miller EA, Fromme JC, Hendrickson TL, Cong Q and Baker D. Computed structures of core eukaryotic protein complexes. Science. 2021;374:eabm4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yan Y, Tao H, He J and Huang SY. The HDOCK server for integrated protein-protein docking. Nat Protoc. 2020;15:1829–1852. [DOI] [PubMed] [Google Scholar]
- 37.Lopaschuk GD and Stanley WC. Glucose metabolism in the ischemic heart. Circulation. 1997;95:313–5. [DOI] [PubMed] [Google Scholar]
- 38.Luptak I, Yan J, Cui L, Jain M, Liao R and Tian R. Long-term effects of increased glucose entry on mouse hearts during normal aging and ischemic stress. Circulation. 2007;116:901–9. [DOI] [PubMed] [Google Scholar]
- 39.Peterson YK and Luttrell LM. The Diverse Roles of Arrestin Scaffolds in G Protein-Coupled Receptor Signaling. Pharmacol Rev. 2017;69:256–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nikko E and Pelham HR. Arrestin-mediated endocytosis of yeast plasma membrane transporters. Traffic. 2009;10:1856–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee S, Min Kim S, Dotimas J, Li L, Feener EP, Baldus S, Myers RB, Chutkow WA, Patwari P, Yoshioka J and Lee RT. Thioredoxin-interacting protein regulates protein disulfide isomerases and endoplasmic reticulum stress. EMBO Mol Med. 2014;6:732–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou R, Tardivel A, Thorens B, Choi I and Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–40. [DOI] [PubMed] [Google Scholar]
- 43.Marshall BA, Ren JM, Johnson DW, Gibbs EM, Lillquist JS, Soeller WC, Holloszy JO and Mueckler M. Germline manipulation of glucose homeostasis via alteration of glucose transporter levels in skeletal muscle. J Biol Chem. 1993;268:18442–5. [PubMed] [Google Scholar]
- 44.Maculins T, Garcia-Pardo J, Skenderovic A, Gebel J, Putyrski M, Vorobyov A, Busse P, Varga G, Kuzikov M, Zaliani A, Rahighi S, Schaeffer V, Parnham MJ, Sidhu SS, Ernst A, Dotsch V, Akutsu M and Dikic I. Discovery of Protein-Protein Interaction Inhibitors by Integrating Protein Engineering and Chemical Screening Platforms. Cell Chem Biol. 2020;27:1441–1451 e7. [DOI] [PubMed] [Google Scholar]
- 45.Yoshioka J, Schulze PC, Cupesi M, Sylvan JD, MacGillivray C, Gannon J, Huang H and Lee RT. Thioredoxin-interacting protein controls cardiac hypertrophy through regulation of thioredoxin activity. Circulation. 2004;109:2581–6. [DOI] [PubMed] [Google Scholar]
- 46.Zhou YY, Wang SQ, Zhu WZ, Chruscinski A, Kobilka BK, Ziman B, Wang S, Lakatta EG, Cheng H and Xiao RP. Culture and adenoviral infection of adult mouse cardiac myocytes: methods for cellular genetic physiology. Am J Physiol Heart Circ Physiol. 2000;279:H429–36. [DOI] [PubMed] [Google Scholar]
- 47.Alam P, Maliken BD, Ivey MJ, Jones SM and Kanisicak O. Isolation, Transfection, and Long-Term Culture of Adult Mouse and Rat Cardiomyocytes. J Vis Exp. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Melzer M, Beier D, Young PP and Saraswati S. Isolation and Characterization of Adult Cardiac Fibroblasts and Myofibroblasts. J Vis Exp. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Myers RB, Fomovsky GM, Lee S, Tan M, Wang BF, Patwari P and Yoshioka J. Deletion of thioredoxin-interacting protein improves cardiac inotropic reserve in the streptozotocin-induced diabetic heart. Am J Physiol Heart Circ Physiol. 2016;310:H1748–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crowley LC, Marfell BJ, Christensen ME and Waterhouse NJ. Measuring Cell Death by Trypan Blue Uptake and Light Microscopy. Cold Spring Harb Protoc. 2016;2016. [DOI] [PubMed] [Google Scholar]
- 51.Stypmann J, Engelen MA, Troatz C, Rothenburger M, Eckardt L and Tiemann K. Echocardiographic assessment of global left ventricular function in mice. Lab Anim. 2009;43:127–37. [DOI] [PubMed] [Google Scholar]
- 52.Mukai N, Nakayama Y, Abdali SA and Yoshioka J. Cardiomyocyte-specific Txnip C247S mutation improves left ventricular functional reserve in streptozotocin-induced diabetic mice. Am J Physiol Heart Circ Physiol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chabowski A, Gorski J, Calles-Escandon J, Tandon NN and Bonen A. Hypoxia-induced fatty acid transporter translocation increases fatty acid transport and contributes to lipid accumulation in the heart. FEBS Lett. 2006;580:3617–23. [DOI] [PubMed] [Google Scholar]
- 54.Lindsey ML, Yoshioka J, MacGillivray C, Muangman S, Gannon J, Verghese A, Aikawa M, Libby P, Krane SM and Lee RT. Effect of a cleavage-resistant collagen mutation on left ventricular remodeling. Circ Res. 2003;93:238–45. [DOI] [PubMed] [Google Scholar]
- 55.Yan J, Young ME, Cui L, Lopaschuk GD, Liao R and Tian R. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation. 2009;119:2818–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.