

The Hh gradient is translated into a phosphorylation gradient of Ci/Gli at multiple Fu/CK1 phosphorylation sites, which progressively increases CiA/GliA activity by gradually alleviating Sufu-mediated inhibition.
Abstract
Graded Hedgehog (Hh) signaling is mediated by graded Cubitus interruptus (Ci)/Gli transcriptional activity, but how the Hh gradient is converted into the Ci/Gli activity gradient remains poorly understood. Here, we show that graded Hh induces a progressive increase in Ci phosphorylation at multiple Fused (Fu)/CK1 sites including a cluster located in the C-terminal Sufu-binding domain. We demonstrated that Fu directly phosphorylated Ci on S1382, priming CK1 phosphorylation on adjacent sites, and that Fu/CK1-mediated phosphorylation of the C-terminal sites interfered with Sufu binding and facilitated Ci activation. Phosphorylation at the N-terminal, middle, and C-terminal Fu/CK1 sites occurred independently of one another and each increased progressively in response to increasing levels of Hh or increasing amounts of Hh exposure time. Increasing the number of phospho-mimetic mutations of Fu/CK1 sites resulted in progressively increased Ci activation by alleviating Sufu-mediated inhibition. We found that the C-terminal Fu/CK1 phosphorylation cluster is conserved in Gli2 and contributes to its dose-dependent activation. Our study suggests that the Hh signaling gradient is translated into a Ci/Gli phosphorylation gradient that activates Ci/Gli by gradually releasing Sufu-mediated inhibition.
Introduction
The Hedgehog (Hh) family of secreted proteins plays critical roles in embryonic development, adult tissue homeostasis and regeneration, and cancer progression (Taipale & Beachy, 2001; Jiang & Hui, 2008; Briscoe & Therond, 2013; Jiang, 2021). Initially identified as an oncogene amplified in glioma, the Gli family of transcription factors transduce the Hh signal to the nucleus to regulate Hh pathway target genes involved in the regulation of cell growth and patterning in species ranging from Drosophila to human (Hui & Angers, 2011). Gli and its Drosophila homolog Cubitus interruptus (Ci) exist in two distinct forms. In the absence of Hh, full-length Gli/Ci (GliF/CiF) is proteolytically processed into a truncated repressor form (GliR/CiR), which lacks the C-terminal coactivator–binding domain and actively represses the expression of a subset of Hh target genes (Aza-Blanc et al, 1997; Wang et al, 2000a). In the presence of Hh, Gli/Ci processing is blocked, leading to de-repression of Hh target genes. In addition, Hh stimulates the activity of full-length Gli/Ci and converts it into a transcriptionally activator form (GliA/CiA) that enters the nucleus to activate Hh target genes (Methot & Basler, 1999; Aza-Blanc et al, 2000).
A salient feature of Hh signaling is that different levels of Hh ligand trigger different developmental outcomes. It is thought that different levels of Hh signaling activity is mediated by two reversed gradients of GliR/CiR and GliA/CiA with increasing levels of Hh progressively decrease the levels of GliR/CiR activity but increase the levels of GliA/CiA activity. Much is known about how GliR/CiR is regulated. The absence of Hh allows GliF/CiF to be phosphorylated by multiple kinases including PKA, glycogen synthase kinase 3 (GSK3), and casein kinase 1 (CK1) (Jiang & Struhl, 1995; Wang et al, 1999; Wang et al, 2000a; Jia et al, 2002; Price & Kalderon, 2002), which targets it for ubiquitination by an E3 ubiquitin ligase SCFSlimb/β-TRCP, followed by proteasome-mediated partial degradation to generate GliR/CiR (Jiang & Struhl, 1998; Jia et al, 2005; Jiang, 2006; Tempe et al, 2006; Smelkinson et al, 2007). In Drosophila, Ci phosphorylation is regulated by a protein complex containing a kinesin-like protein Costal2 (Cos2) and a Ser/Thr kinase Fused (Fu), which forms a complex with Ci and its kinases including PKA, GSK3, and CK1 to facilitate Ci phosphorylation and processing (Zhang et al, 2005). Hh signaling releases Ci and its kinases from the Cos2/Fu complex, thereby inhibiting Ci phosphorylation and processing (Zhang et al, 2005; Li et al, 2014; Ranieri et al, 2014). In mammalian cells, Hh signaling is thought to regulate the local activity of PKA in the primary cilium to inhibit Gli processing (Mukhopadhyay et al, 2013; Arveseth et al, 2021; Truong et al, 2021).
Compared with the regulation of GliR/CiR, how Hh signaling converts GliF/CiF into GliA/CiA is less understood (Zhou & Jiang, 2022). In Drosophila, Fu is required for converting CiF into a labile CiA by alleviating the inhibition by Sufu, a conserved pathway regulator that binds and inhibits Ci/Gli (Ohlmeyer & Kalderon, 1998; Methot & Basler, 2000; Wang et al, 2000b; Humke et al, 2010; Tukachinsky et al, 2010; Zhang et al, 2013; Shi et al, 2014a; Han et al, 2015). Although Hh induced Sufu phosphorylation in a manner depending on Fu (Lum et al, 2003), preventing Fu-mediated phosphorylation of Sufu by mutating the relevant sites did not affect Fu-mediated activation of Ci either in vitro or in vivo (Zhou & Kalderon, 2011; Han et al, 2019). Our recent study demonstrated that Ci is a direct and physiologically relevant substrate of Fu (Han et al, 2019). We found that Fu directly phosphorylated Ci on Ser218 and Ser1230, which primed its further phosphorylation by CK1 on adjacent sties and that these phosphorylation events promoted Ci activation by attenuating Sufu/Ci interaction. Furthermore, we found that Shh stimulated phosphorylation on a conserved cluster of Ser residues in the N-terminal region of Gli proteins through the Fu-related kinases Ulk3 and Stk36 (Han et al, 2019).
In the present study, we identified a new Fu phosphorylation site located in the C-terminal Sufu-binding domain of Ci. Using in vitro kinase assay and phospho-specific antibodies, we demonstrated that Fu phosphorylated Ci on S1382 in response to Hh, which primed its further phosphorylation by CK1 on adjacent sites. We found that Fu/CK1–mediated phosphorylation at this phosphorylation cluster interfered with Sufu binding and modulated Ci activity in conjunction with other phosphorylation clusters. We showed that phosphorylation at individual Fu/CK1 sites occurs independently of one another, but each increased progressively in response to increasing levels of Hh ligand or increasing amounts of Hh exposure time. We provide evidence that increasing levels of Ci phosphorylation at Fu/CK1 sites resulted in a progressive increase in CiA activity. We also found that a similar phosphorylation cluster in the C-terminal Sufu-binding domain of Gli2 contributed to its dose-dependent activation. Taken together, our results suggest that the Hh activity gradient is translated into a Gli/Ci phosphorylation gradient that fine tunes CiA/GliA activity.
Results
Fu phosphorylates the C-terminal Sufu-binding domain of Ci
Our previous study uncovered a Sufu-binding site in the C-terminal region (SIC) of Ci from aa1371 to aa1397, which is conserved in both Gli2 and Gli3 (Han et al, 2015). Inspection of this region revealed that Ser 1382 (S1382) falls into Fu phosphorylation consensus site: S/TX5E/D (Fig 1A) (Han et al, 2019). To determine whether Fu could directly phosphorylate Ci on S1382, we carried out an in vitro kinase assay using GST fusion proteins containing the Ci C-terminal fragment (GST-CiC; Fig 1B) as substrates and Flag-tagged constitutively-active (CC-FuEE) or kinase-dead (CC-FuKR) Fu purified from insect cells as the kinase source (Shi et al, 2011; Han et al, 2019). Phosphorylation of GST fusion proteins was detected by the nonradioactive method (pIMAGO) (Han & Jiang, 2021). As shown in Fig 1C, CC-FuEE but not CC-FuKR phosphorylated the GST-CiC fusion protein containing the wild-type (WT) sequence. Substitution of S1382 (S1382A) or both S1382 and S1385 to Ala (2SA) completely abolished the phosphorylation of corresponding GST-CiC fusion proteins (Fig 1C), suggesting that S1382 is a direct Fu phosphorylation site.
Figure 1. Fu phosphorylates the C-terminal Sufu-binding domain of Ci.
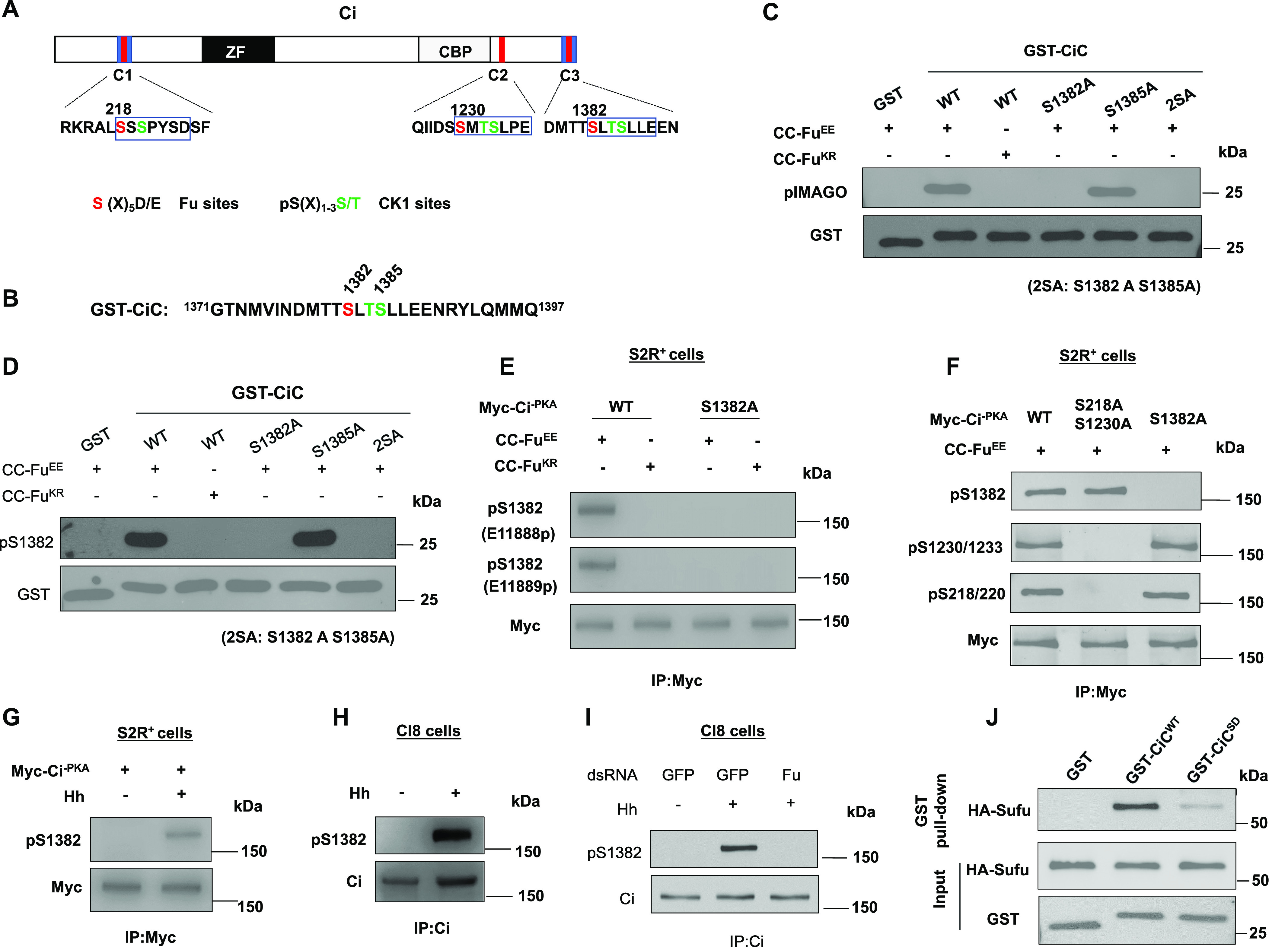
(A) Schematic diagram of Ci. Blue boxes and red bars indicate the Sufu-binding domains and Fu/CK1 phosphorylation clusters, respectively. ZF, Zinc-Finger DNA-binding domain; CBP, CBP-binding domain. The primary sequences of three phosphorylation clusters are shown with Fu and CK1 sites color-coded in red and green, respectively. (B) The primary sequence of the Ci C-terminal region in GST-CiC. (C, D) In vitro kinase assay using purified CC-FuEE or CC-FuKR as kinase and the indicated GST-CiC fusion proteins as substrates. Phosphorylation was detected by pIMAGO (C) or pS1382 antibody (D). (E) Western blot analysis of Ci phosphorylation in S2R+ cells transfected with the indicated Ci and Fu constructs. Two phospho-specific antibodies (E11888p and E11889p) were used to detect S1382 phosphorylation. (F) Western blot analysis of Ci phosphorylation on the indicated sites in S2R+ cells transfected with CC-FuEE and the indicated Myc-Ci-PKA constructs. (G) Western blot analysis of Ci phosphorylation on S1382 in S2R+ cells transfected with Myc-Ci-PKA and treated with or without Hh-conditioned medium. (H) Western blot analysis of endogenous Ci phosphorylation on S1382 in Cl8 cells treated with or without Hh-conditioned medium. (I) Western blot analysis of endogenous Ci phosphorylation on S1382 in Cl8 cells treated with the indicated dsRNA in the absence or presence of Hh. (J) Western blot analysis of HA-Sufu pulled down by the indicated GST fusion proteins. 5 μg of GST or GST-CiC fusion proteins were incubated with equal amounts of cell lysates from S2R+ cells transfected with HA-Sufu construct.
Fu phosphorylates Ci on S218 (N-terminal site) and S1230 (middle site), which primed Ci phosphorylation by CK1 on adjacent sites (Han et al, 2019). We found that S1382 is followed by T1384 and S1385, both of which fall into the CK1 consensus site: D/E/(p)S/T[X1-3]S/T, where S/T is the phospho-acceptor site and two amino acids (X = 2) provide an optimal spacing between the phospho-acceptor site and D/E/(p)S/T (Knippschild et al, 2005). To determine whether phosphorylation of S1382 by Fu can prime CK1 phosphorylation on adjacent sites such as S1385, we carried out sequential in vitro kinase assay in which GST-CiCWT or GST-CiCS1385A was first incubated with CC-FuEE, followed by treatment with a recombinant CK1. A phospho-Ser specific antibody, pSer, was used to detect phosphorylation on S1382/S1385. As shown in Fig S1, pSer detected equal signals from GST-CiCWT and GST-CiCS1385A when these substrates were incubated with CC-FuEE alone; however, the pSer signal of GST-CiCWT but not GST-CiCS1385A was further increased after CK1 incubation (Fig S1A). As expected, mutating both S1382 and S1385 to Ala (GST-CiC2SA) abolished CiC phosphorylation detected by the pSer antibody in the in vitro kinase assay (Fig S1B). These results suggest that CK1 can phosphorylate S1385 after primed phosphorylation of S1382 by Fu.
Figure S1. Sequential phosphorylation of CiC by Fu and CK1.
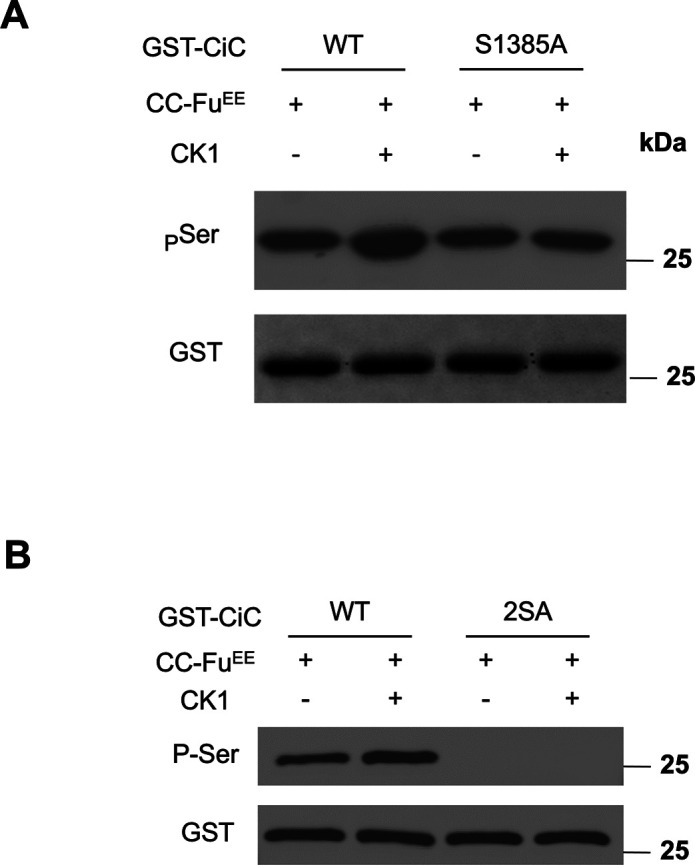
(A, B) In vitro kinase assay was carried out in which GST-CiCWT and GST-CiCS1385A (A) or GST-CiC2SA (B) were incubated with CC-FuEE purified from insect cells. After removal of the Fu kinase, the GST fusion proteins were incubated with or without a recombinant CK1, followed by Western blot analysis using the indicated antibodies.
To further investigate S1382 phosphorylation, we developed a phospho-specific antibody that recognized phosphorylation on S1382 (pS1382). The in vitro kinase assay described above showed that pS1382 specially recognized GST-CiCWT but not GST-CiCS1382A or GST-CiC2SA after incubation with purified CC-FuEE (Fig 1D). Next, CC-FuEE or CC-FuKR and a non-processed form of Ci (Myc-Ci-PKA), which has three PKA sites (S838, S856, and S892) mutated to Ala (Wang et al, 1999), were co-transfected into cultured Drosophila S2R+ cells. Cell extracts were immunoprecipitated with a Myc antibody, followed by Western blot analysis with the pS1382 antibody. We found that pS1382 only detected Myc-Ci-PKA phosphorylation in the presence of CC-FuEE but not in the presence of CC-FuKR (Fig 1E). Mutating S1382 to Ala in Myc-Ci-PKA completely abrogated the pS1382 signal (Fig 1E), suggesting that activated Fu could phosphorylate full-length Ci on S1382 in intact cells.
Because Fu can also phosphorylate Ci on S218 and S1230 (Han et al, 2019), we asked whether phosphorylation on individual Fu sites depends on the phosphorylation status of other Fu sites. We co-expressed Myc-Ci-PKA-WT, Myc-Ci-PKA-S218AS1230A, or Myc-Ci-PKA-S1382A with CC-FuEE in S2R+ cells and detected the phosphorylation of individual sites with the corresponding phospho-specific antibodies. As showed in Fig 1F, mutating S218 and S1230 to Ala did not affect phosphorylation of pS1382 by Fu. Similarly, mutating S1382 to Ala did not affect phosphorylation of S218 and S1230 by Fu (Fig 1F). Our previous study indicated that phosphorylation on S218 and S1230 was independent of each other (Han et al, 2019). Taken together, these results suggest that phosphorylation on the N-terminal, middle, and C-terminal sites by Fu occurred independently of one another.
To determine whether phosphorylation on S1382 is stimulated by Hh, S2R+ cells transfected with Myc-Ci-PKA were treated with Hh-conditioned medium. We found that Myc-Ci-PKA was phosphorylated on S1382 upon Hh treatment (Fig 1G). Moreover, Hh induced phosphorylation of endogenous Ci on S1382 in Cl8 cells (Fig 1H), which was abolished by Fu RNA interference (RNAi; Fig 1I), suggesting that Hh induces endogenous Ci phosphorylation on S1382 through Fu.
Because S1382 is located within the C-terminal Sufu-binding domain (Fig 1A) (Han et al, 2015), we asked whether phosphorylation of S1382 and adjacent CK1 sites affected Sufu binding. We generated phospho-mimetic mutations of these sites (S1382D/T1384D/S1385D) in the GST-CiC background (GST-CiCSD). GST pull-down assay showed that GST-CiCSD bound less HA-Sufu derived from S2R+ cell extracts than GST-CiCWT (Fig 1J), suggesting that Fu/CK1-mediated phosphorylation of Ci C-terminal domain attenuates Sufu/Ci interaction.
Fu-mediated phosphorylation of CiC modulates Ci activity
To determine the relative contribution of individual Fu phosphorylation sites on Ci activation, we mutated the Fu sites individually or in different combinations: SA1 (S218A), SA2 (S1230A), SA3 (S1382A), SA12 (S218A, S1230A), SA13 (S218A, S1382A), SA23 (S1230A, S1382A), and SA123 (S218A, S1230A, S1382A) (Fig 2A). These mutations were generated in the Myc-Ci-PKA background so that the effects of Fu phosphorylation site mutations on Ci activity should reflect changes in the conversion from CiF to CiA (Han et al, 2019). When transfected into S2R+ cells, these Ci variants were expressed at similar levels (Fig S2A). To determine how these mutations affect Fu-mediated Ci activation, we carried out a “de-repression” assay in which coexpression of Sufu with various Ci constructs in S2R+ cells effectively blocked their ability to activate a ptc-luciferase (ptc-luc) reporter gene, and this repression was alleviated by coexpression of CC-FuEE (Han et al, 2019). As shown in Fig 2B, mutations of individual Fu sites in Ci-PKA only slightly reduced the expression ptc-luc compared with wild-type (WT) Ci-PKA. Combined mutations of two or more Fu phosphorylation sites resulted in a more dramatic reduction in ptc-luc expression with SA123 exhibiting the strongest defect (Fig 2B). Although mutating the C-terminal site alone (SA3) had little effect on Ci activation, this mutation enhanced the defect in Ci activation when combined with other Fu site mutations (Fig 2B). Hence, Fu-mediated phosphorylation of the C-terminal region of Ci modulates Ci activity in conjunction with other phosphorylation events.
Figure 2. Dose-dependent phosphorylation and activation of Ci.
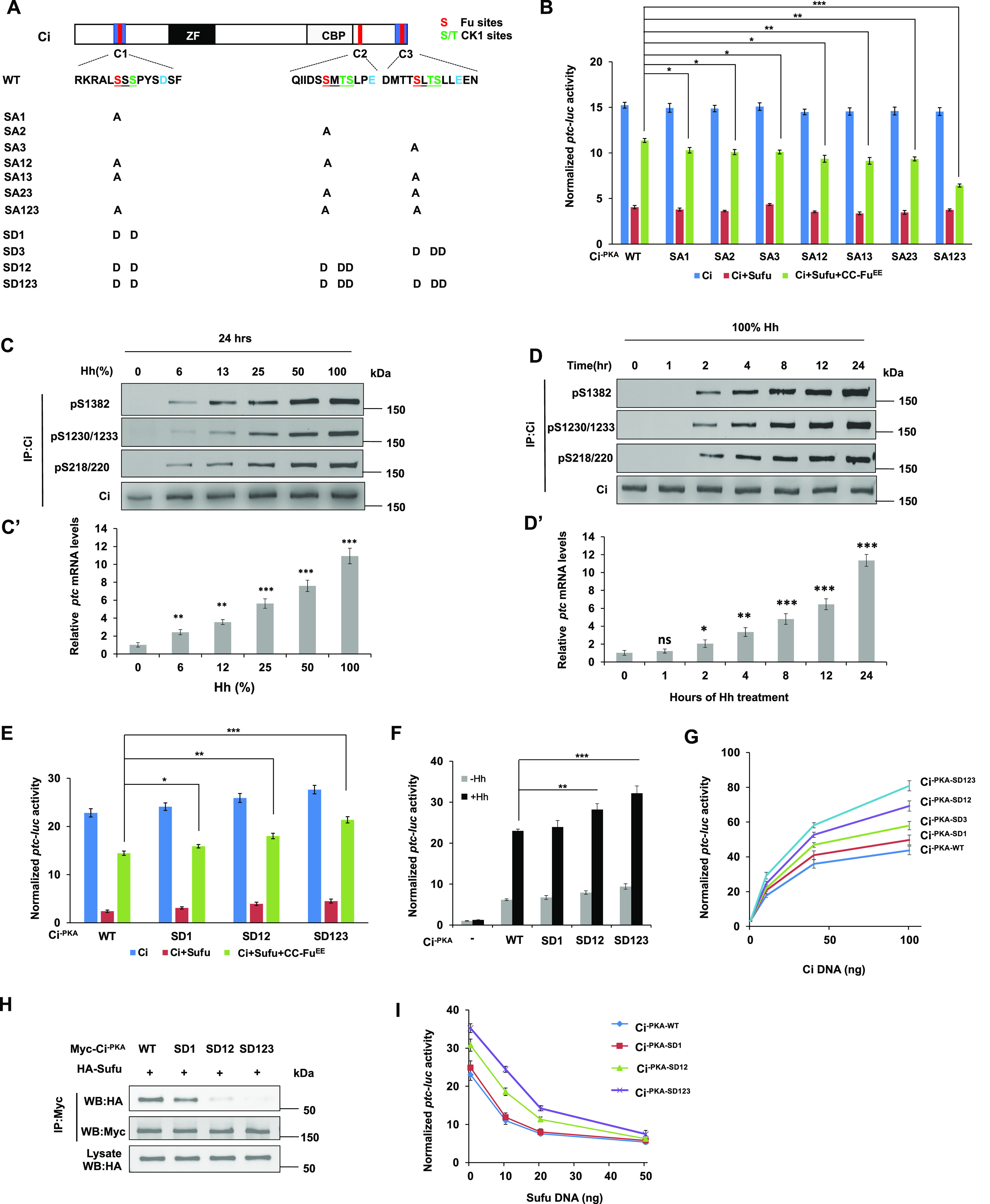
(A) Schematic diagram of Ci with primary sequences of the three phosphorylation clusters shown underneath. Fu and CK1 sites are color coded in “red” and “green,” respectively. Amino acid substitutions for individual Ci constructs are indicated. (B) ptc-luc reporter assay in S2R+ cells transfected with the indicated Myc-Ci-PKA constructs in the presence or absence of Sufu/CC-FuEE. (C, C’, D, D’) Western blot analyses of endogenous Ci phosphorylation on the indicated sites in Cl8 cells exposed to Hh at increasing concentrations for 24 h (C) or with 100% Hh for increasing amounts of time (D). Hh-conditioned medium was used at a 6:4 dilution ratio with fresh medium (defined as 100%). (C’, D’) Quantification of ptc mRNA expression by RT-qPCR in Cl8 cells exposed to Hh at increasing concentrations (C’) or duration (D’). (E) ptc-luc reporter assay in S2R+ cells transfected with the indicated Myc-Ci-PKA constructs in the presence or absence of Sufu/CC-FuEE. (F) ptc-luc reporter assay in S2R+ cells transfected with the indicated Ci constructs at low concentrations (5 ng/well) and treated with (+Hh) or without (−Hh) Hh-conditioned medium. (G) ptc-luc reporter assay in S2R+ cells transfected with increasing amounts of the indicated Ci constructs. (H) Co-immunoprecipitation assay in S2R+ cells transfected with the indicated Myc-Ci-PKA variants and HA-Sufu. (I) ptc-luc reporter assay in S2R+ cells transfected with a fixed amount of the indicated Ci constructs (50 ng/well) and increasing amounts of Sufu. Data are mean ± SD from three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 (t test).
Figure S2. Expression levels of Ci variants in S2R+ cells.

(A, B) Western blot analysis of protein expression levels of the indicated phosphorylation deficient (A) or phosphorylation-mimetic (B) Ci variants transfected into S2R+ cells indicated that these Ci constructs were expressed at similar levels.
Dose-dependent phosphorylation of Ci in response to the Hh gradient
We next determined how phosphorylation of individual Fu sites are regulated in response to graded Hh signaling. We used the phospho-specific antibodies pS218/220, pS1230/1233, and pS1382 to monitor phosphorylation of endogenous Ci at the N-terminal, middle, and C-terminal Fu sites, respectively, in Cl8 cells treated with conditioned medium containing increasing levels of HhN. We found that phosphorylation detected by pS218/220, pS1230/1233, and pS1382 increased progressively in response to increasing levels of Hh ligand (Fig 2C), which correlated with increasing levels of pathway activity reflected by the levels of ptc expression (Fig 2C’).
Hh signaling outcomes not only depend on the concentration of Hh ligand but also depend on the duration of Hh ligand exposure (Stamataki et al, 2005). Therefore, we examined how phosphorylation at each Fu site changes over the course of Hh stimulation. Cl8 cells were treated with HhN-conditioned medium for different periods of time and Ci phosphorylation levels at individual sites were monitored by Western blot analyses of immunoprecipitated Ci with the corresponding phospho-specific antibodies. As shown in Fig 2D, phosphorylation at individual Fu phosphorylation sites occurred at about 2 h after Hh exposure, gradually increased over time, and plateaued at 24 h of Hh exposure. The gradual increase in Ci phosphorylation correlated with the progressive increase in ptc mRNA expression (Fig 2D’). These results suggest the Ci phosphorylation gradient is determined not only by Hh signal strength but also by signal duration.
Increasing Fu-mediated phosphorylation progressively increases Ci activity in vitro
To examine whether different levels of Ci phosphorylation determine different levels of Ci activity, we generated a set of phospho-mimetic Ci variants with one, two, or three Fu/CK1 phosphorylation clusters converted to acidic residues (D) to mimic different levels of phosphorylation (Fig 2A): SD1 has cluster 1 mutated to D; SD12 has clusters 1 and 2 mutated to D; and SD123 has all three clusters mutated to D in the Ci-PKA background. When transfected into S2R+ cells, these phospho-mimetic Ci variants were expressed at similar levels (Fig S2B). In the “de-repression” assay, increasing the number of phospho-mimetic mutations progressively increased the ptc-luc activity (Fig 2E). When individual Ci variants were expressed at low levels, increasing the number of phospho-mimetic mutations progressively increased both the basal and Hh-induced Ci activity (Fig 2F). In another assay, individual Ci variants were expressed at different levels by transfecting different amounts of DNA constructs and their relative activities were compared at a given expression level. Again, we found that increasing the number of phospho-mimetic mutations resulted in a progressive increase in Ci activity (Fig 2G).
Fu induces the formation of CiA by antagonizing Sufu (Ohlmeyer & Kalderon, 1998). In addition, Fu/CK1-mediated phosphorylation of Ci attenuates the binding of Sufu to Ci (Han et al, 2019). Consistent with a gradual change in pathway activity, increasing the number of phospho-mimetic mutations in Ci progressively decreased its binding affinity to Sufu as measured by CoIP experiments (Fig 2H). When fixed amount of WT Myc-Ci-PKA or SD variants were coexpressed with increasing amounts of Sufu, Ci variants with more phospho-mimetic mutations (SD123 and SD12) consistently exhibited higher pathway activity than those with less or no phospho-mimetic mutations (SD1 and WT) before reaching a saturated amount of Sufu which inhibited all the Ci constructs (Fig 2I). These results suggest that increasing levels of Ci phosphorylation by Fu/CK1 render Ci more resistant to Sufu-mediated inhibition.
Increasing Fu-mediated phosphorylation progressively activates Ci in vivo
To determine how increasing levels of Ci phosphorylation modulate pathway activity in vivo, we generated transgenic flies expressing various Ci constructs under the control of the UAS promoter. The phiC31 integrase system was used to introduce Ci constructs into the same genetic locus to ensure similar expression of different Ci transgenes (Bischof et al, 2007). These Ci transgenes were expressed under the control of a weak Gal4 driver, C765, which drives the expression of Ci transgenes at a level close to that of endogenous Ci (Han et al, 2019).
During the development of Drosophila wing, cells in the posterior (P) compartment of wing imaginal discs express and secrete Hh, whereas anterior (A) compartment cells express Ci, and consequently, only A-compartment cells near the anterior-posterior (A/P) compartment boundary receive and transduce the Hh signal to activate the Hh pathway target genes such as ptc (Fig 3A–A”). When expressed under the control of the C765 Gal4 driver (C765>) that drives the expression of UAS transgenes at low levels throughout wing discs (Chen et al, 2010; Ma et al, 2016), both Ci-PKA_WT and Ci-PKA_SD1 induced ectopic expression of ptc only in P-compartment cells but not in A-compartment cells distant from the A/P boundary (Fig 3B’–C”), suggesting that neither Ci-PKA_WT nor Ci-PKA_SD1 exhibited Hh-independent pathway activity when expressed at levels close to that of endogenous Ci. Under the same condition, Ci-PKA_SD12 induced weak ectopic expression of ptc, whereas Ci-PKA_SD123 induced relatively strong ectopic ptc expression in A-compartment cells distant from the A/P boundary (Fig 3D’–E”), suggesting that Ci-PKA_SD123 exhibited higher Hh-independent activity than Ci-PKA_SD12. Of note, Ci-PKA_SD123 activated ptc expression more robustly in P-compartment cells than in A-compartment cells distant from the A/P boundary, suggesting that the activity of Ci-PKA_SD123 is still up-regulated by Hh.
Figure 3. The effect of increasing phospho-mimetic mutations on Ci activity in vivo.

(A, B, C, D, E, F, G, H, I, J) Control (A, A”) or wing discs expressing UAS-Myc-Ci-PKA_WT (B, B”), UAS-Myc-Ci-PKA_SD1 (C, C”), UAS-Myc-Ci-PKA_SD12 (D, D”), or UAS-Myc-Ci-PKA_SD123 (E, E”) under the control of C765 Gal4 driven from larvae grown at 25°C were immunostained to the expression of Ci (white in A, B, C, E; red in A”, B”, C”, D”, E”) and Ptc (white in A’, B’, C’, D’, E’; green in A”, B”, C”, D”, E”). Insets in D” and E” are large magnification views of the regions outlined in D’ and E’, respectively, which demarcate A-compartment cells distant from the A/P boundary. (F, G, H, I, J) Adult wings of the indicated genotypes are shown. Arrows in (G) and (H) indicate the gaps on wing vein 4. Scale bars are 50 μM for wing discs and 500 μM for adult wings.
Compared with control wings (Fig 3F), wings expressing Ci-PKA_WT or Ci-PKA_SD1 appear normal in overall morphology with only minor defects such as gaps in wing vein 4 (arrows; Fig 3G and H). By contrast, wings expressing Ci-PKA_SD12 showed enlarged size and abnormal wing vein patterns (Fig 3I). The overgrowth phenotype and vein patterning defect were exacerbated in wings expressing Ci-PKA_SD123 (Fig 3J), consistent with Ci-PKA_SD123 being more potent than Ci-PKA_SD12 in the transduction of Hh signaling. Hence, the severity of wing patterning defects caused by various Ci mutants correlated with their relative pathway activities, which were revealed by their abilities to induce ectopic ptc expression.
Gal4 activity is temperature-sensitive and drives UAS transgene expression at higher levels at 30°C than at room temperature (Brand et al, 1994). To differentiate the activity between Ci-PKA_WT and Ci-PKA_SD in vivo, larvae expressing C765>Ci-PKA_WT or C765>Ci-PKA_SD1 were grown at 30°C 3 d before dissection and immunostaining. Under this condition, both Ci-PKA_WT and Ci-PKA_SD1 induced ectopic expression of ptc in A-compartment cells distant from the A/P boundary and wing overgrowth, with Ci-PKA_SD1 causing more severe phenotypes (Fig S3), suggesting that Ci-PKA_SD1 is more active than Ci-PKA_WT. Taken together, these observations suggest that increasing Fu/CK1-mediated phosphorylation progressively activates Ci in vivo.
Figure S3. Phosphorylation of Ci N-terminal Fu/CK1 sites increased Ci activity in vivo.

(A, B) Adult wings expressing UAS-Myc-Ci-PKA_WT (A) or UAS-Myc-Ci-PKA_SD1 (B) under the control of the C765 Gal4 driver at 30°C. (C, C’, C”, D, D”) Wing discs expressing UAS-Myc-Ci-PKA_WT (C, C’, C”) or UAS-Myc-Ci-PKA_SD1 (D, D’, D”) under the control of the C765 Gal4 driver at 30°C were immunostained for Ci (red) and Ptc (blue).
C-terminal phosphorylation modulates Gli2 activity
Both Gli2 and Gli3 contain a conserved Sufu-binding domain in their C-terminal region (Han et al, 2015). Sequence alignment between Ci and mouse Gli2/3 identified a similar phosphorylation cluster in the Gli2/3 C-terminal region (Figs 4A and S4). S1528 is a putative Fu/Ulk3 site because there is an acidic residue (E) at +7 position although the previously identified Fu/Ulk3 sites all have an acidic residue at +6 position (Han et al, 2019). We speculated that the spacing between the phospho-acceptor site and the acidic residue could be flexible. Indeed, in vitro kinase assay showed that immunopurified wild-type Ulk3 (HA-Ulk3WT) but not a kinase dead form (HA-Ulk3KR) phosphorylated a GST fusion protein carrying the Gli2 C-terminal region (GST-Gli2C; Fig 4B). Substitution of S1528 to Ala or combined mutation of S1528 and two downstream CK1 consensus sites (3SA: S1528A/S1532A/T1531A) abolished the phosphorylation (Fig 4B), suggesting that Ulk3 can directly phosphorylate Gli2 on S1528.
Figure 4. Multisite phosphorylation of Gli2 promotes its activation.
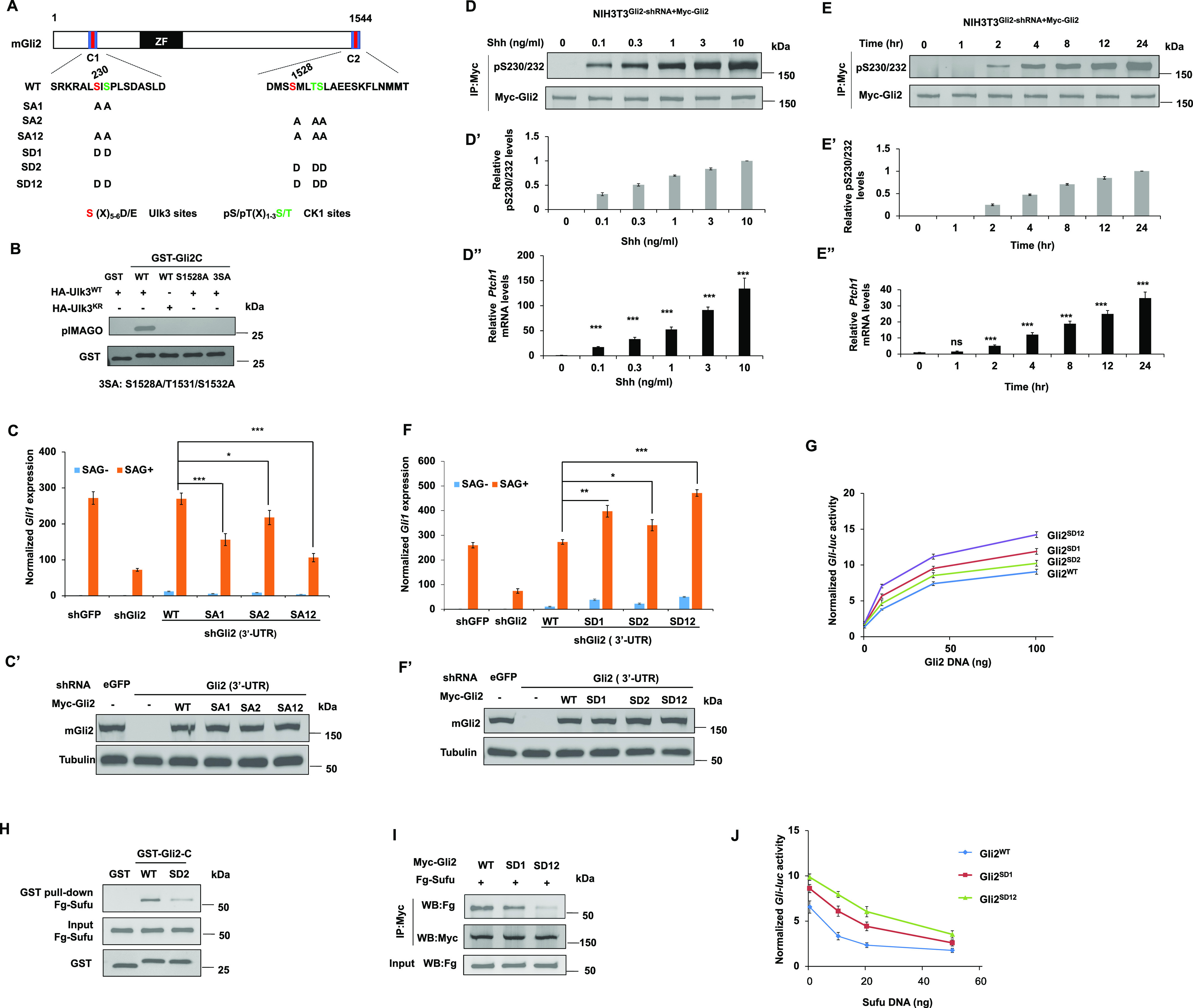
(A) Schematic diagram of mouse Gli2 with primary sequences of the three phosphorylation clusters shown underneath. Ulk3 and CK1 sites are color-coded in “red” and “green,” respectively. Amino acid substitutions for individual Gli2 constructs are indicated. (B) In vitro kinase assay using immunopurified HA-Ulk3WT or HA-Ulk3KR as kinase and the indicated GST-fusion proteins as substrates. Phosphorylation was detected by pIMAGO. (C, C’, F, F’) Relative Gli1 mRNA expression (C, F) or Gli2 protein levels (C’ and F’) in NIH3T3 cells expressing the indicated shRNA and Myc-Gli2WT or its variants with or without SAG. (D, D’, D”, E, E’, E”) Western blot analysis of Myc-Gli2 phosphorylation on S230/232 in NIH3T3 cells infected with lentivirus expressing shRNA targeting the 3′ UTR of Gli2 and lentivirus expressing Myc-Gli2WT (NIH3T3Gli2-shRNA/Myc-Gli2) in the presence of increasing amounts of Shh-N for 24 h (D) or exposed to a fixed amount of Shh-N (1 ng/ml) for increasing amounts of time (E). Quantification of pS230/232 signals (D’, E’) or Ptch1 mRNA (D”, E”) under these conditions. (G, J) Gli-luc reporter activity in NIH3T3 cells transfected with increasing amounts of the indicated Gli2 constructs (G) or fixed amount of the indicated Gli2 constructs and increasing amounts of Sufu constructs (J). (H) Western blot analysis of Fg-Sufu pulled down by the indicated GST fusion proteins from HEK293T cells expressing a Fg-Sufu construct. (I) Western blot analysis of Flag-Sufu coimmunoprecipitated with Myc-Gli2WT, Myc-Gli2SD1, and Myc-Gli2SD12. Data are mean ± SD from three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 (t test).
Figure S4. The C-terminal phosphorylation cluster is conserved in Gli2 and Gli3.

Alignment of the C-terminal sequence from Ci (aa1371-1397), mouse Gli2 (aa1517-1544), and mouse Gli3 (aa1556-1583). Ulk3 and CK1 sites are color-coded in “red” and “green,” respectively. The acidic residues at +6/7 position of Ulk3 site is indicated in magenta.
Gli proteins contain a conserved Ulk3/CK1 phosphorylation cluster near their N-terminal Sufu-binding domain and mutating this cluster to Ala in Gli2 (SA1:S230A/S232A) attenuated its activation by Shh in NIH3T3 cells (Han et al, 2019). To determine the contribution of the C-terminal phosphorylation sites to Gli2 activation, we mutated them to Ala either alone (SA2: S1528A/S1532A/T1531A) or in combination with the N-terminal sites (SA12: SA230A/S232A/S1528A/S1532A/T1531A) (Fig 4A). Lentiviral infection was used to stably express wild type Gli2 (WT) and Gli2 variants (SA1, SA2, and SA12) in NIH3T3 cells with endogenous Gli2 knocked down via an shRNA targeting the 3′ UTR of Gli2 (Han et al, 2019). Western blot analysis confirmed that the exogenously expressed WT Gli2 and SA variants were expressed at levels close to the endogenous Gli2 level in these cell lines (Fig 4C’). As shown in Fig 4C, the C-terminal phosphorylation cluster mutation (SA2) reduced Gli2 activation mediated by the Smo agonist SAG, albeit less dramatically compared with the N-terminal phosphorylation cluster mutation (SA1). Combined mutation of the two clusters (SA12) further reduced SAG-induced Gli2 activation compared with the single-cluster mutations (Fig 4C), suggesting that phosphorylation of Gli2 at both its N- and C-terminal sites contributes to its activation.
Using the phospho-specific antibody that recognizes the N-terminal sites (pS230/S232) (Han et al, 2019), we found that Gli2 phosphorylation at these sites in NIH3T3 cells increased progressively in response to increasing levels of Shh or increasing duration of Shh exposure, which correlated with a progressive increase in Shh pathway activity determined by Ptch1 mRNA expression (Fig 4D–E”), suggesting that both Shh signal strength and duration determine the levels of Gli2 phosphorylation by Ulk3/CK1.
To determine whether increasing levels of Gli2 phosphorylation by Ulk3/CK1 could progressively increase Gli2 activity, we generated phospho-mimetic mutations in N- (SD1) or C-terminal sites (SD2) either alone or in combination (SD12) (Fig 4A). These Gli2 variants were introduced via lentivirus into NIH3T3 cells with endogenous Gli2 depleted by shRNA targeting the Gli2 3′ UTR. Stable Cell lines expressing Gli2 variants at levels close to that of the endogenous Gli2 were selected for further analysis (Fig 4F’). In agreement with our previous finding (Han et al, 2019), phospho-mimetic mutation of the N-terminal sites (SD1) enhanced SAG-induced Gli2 activation (Fig 4F). We found that phospho-mimetic mutation of the C-terminal sites (SD2) also increased SAG-induced Gli2 activity albeit as lower magnitude compared with SD1 and that combined mutation (SD12) further increased Gli2 activity compared with single-site mutation (Fig 4F). In another assay, Gli2WT, Gli2SD1, Gli2SD2, and Gli2SD12 were expressed at increasing levels by transfecting increasing amounts of DNA constructs and their relative activities (measured by Gli-luc activity) were compared at a given expression level. As shown in Fig 4G, increasing the number of phospho-mimetic mutations resulted in a progressive increase in Gli2 activity.
By GST pull-down assay, we found that SD2 mutation reduced the binding of Sufu to the C-terminal region of Gli2 (Fig 4H), suggesting that C-terminal phosphorylation of Gli2 interfered with its binding to Sufu. Co-IP experiments showed that increasing the number of phospho-mimetic mutations in Gli2 gradually diminished Sufu binding to full-length Gli2 (Fig 4I), suggesting that increasing levels of Ulk3/CK1–mediated phosphorylation activate Gli2 by gradually alleviating Sufu-mediated repression. In support of this notion, we found that Gli2SD1 and Gli2SD12 were more resistant to Sufu-mediated inhibition than Gli2WT (Fig 4J).
To determine the effect of Gli2 phosphorylation on its activation in a more physiological context, we turned to an in vitro differentiation assay where mouse embryonic stem cells (mESCs) were induced to differentiate into spinal cord neural progenitor cells that can respond to different levels of Shh and express different Shh target genes that specify progenitor subtypes (Fig 5A and B; Gouti et al, 2014; Pusapati et al, 2018). We depleted endogenous Gli2 from mESCs by shRNA targeting the 3′ UTR of Gli2 and introduced Myc-tagged Gli2WT, Gli2SD1 and Gli2SD12 via lentiviral infection (Fig 5C). mESCs that expressed Gli2WT, Gli2SD1, or Gli2SD12 at similar levels were cultured in N2B27 medium supplemented with bFGF for 2 d and then treated with retinoic acid (RA) alone or RA plus ShhN for 24 h, followed by RT-qPCR to determine the expression of Shh responsive genes (Fig 5A and B). Foxa2 and Nkx6.1 are two Hh target genes that respond to high levels of Shh. In response to Shh stimulation, Gli2SD1 variants induced higher expression of Nkx6.1 and Foxa2 than Gli2WT, and Gli2SD12 exhibited the highest activity toward activating these high threshold Shh target genes (Fig 5D and E). On the other hand, Gli2WT, Gli2SD1, and Gli2SD12 activated the expression of a low threshold Shh target gene (Olig2) equally well (Fig 5F), consistent with Gli2 being required for high but not low threshold response to Shh (Ding et al, 1998; Matise et al, 1998). These results suggest that the Ulk3/CK1–mediated phosphorylation modulates Gli2 activation during spinal cord neural progenitor cell differentiation.
Figure 5. The effect of increasing phospho-mimetic mutations on Gli2 activity in neural progenitors derived in vitro.
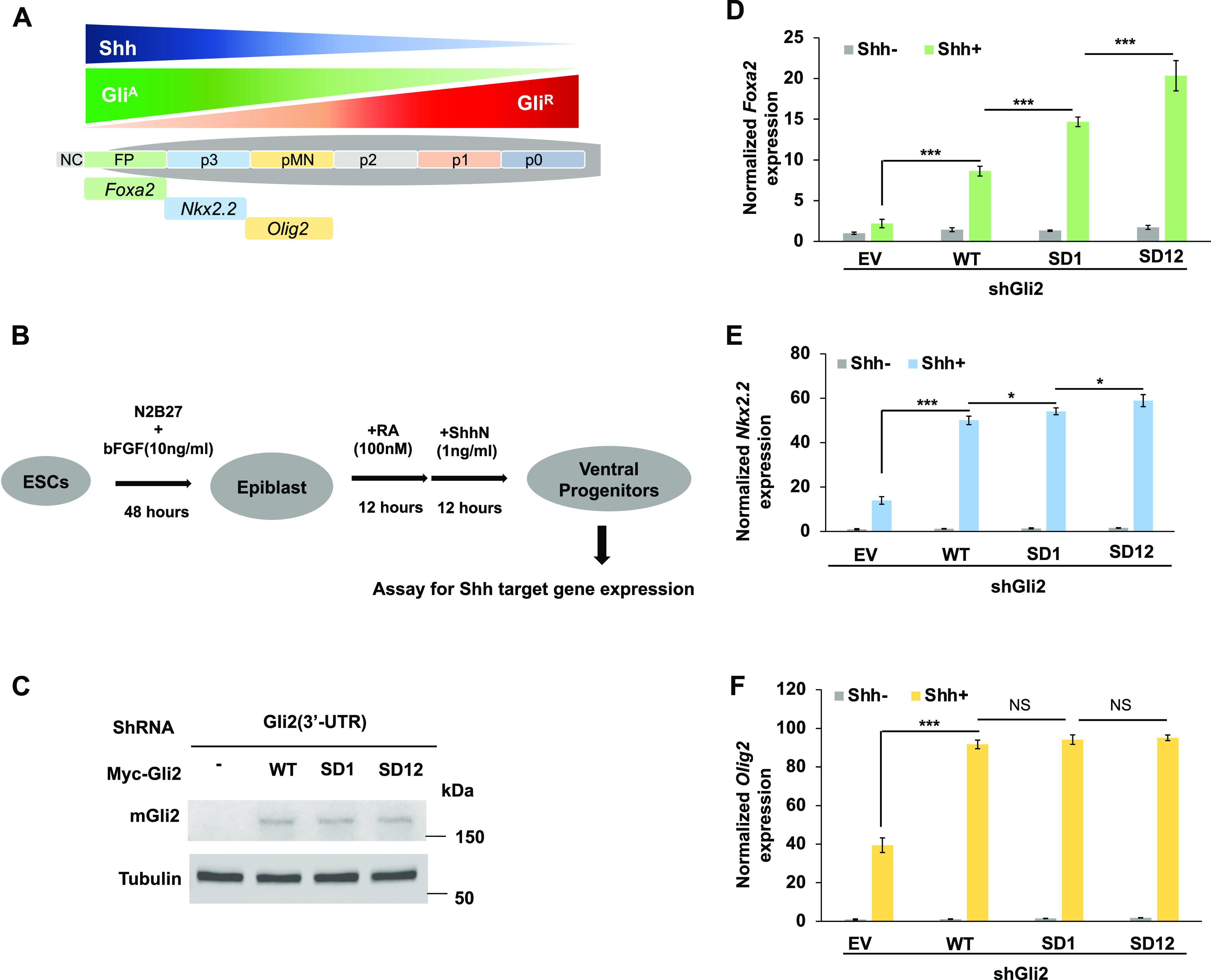
(A) A schematic drawing of ventral patterning of vertebrate neural tube by Shh and GliA/GliR gradients. NC, notochord; FP, floor plate; P3-P0, progenitors for V3-V0 interneurons; pMN, progenitors for motoneurons. (B) A flowchart of in vitro differentiation scheme from mouse ES cells to neural progenitor cells. (C) Western blot analysis of Gli2 protein from mouse ES cells expressing the indicated shRNA and Myc-Gli2 lentiviral constructs. (D, E, F) Relative mRNA levels of Foxa2 (D), Nkx2.2 (E), and Olig2 (F) measured by RT-qPCR in neural progenitor cells derived from mouse ES cells expressing the indicated shRNA and Myc-Gli2 lentiviral constructs with or without Shh-N. Data are mean ± SD from three independent experiments. *P < 0.05 and ***P < 0.001 (t test). NS, not significant.
Discussion
The Hh morphogen gradient governs cell growth and patterning by generating opposing gradients of CiR/GliR and CiA/GliA. It has been well-established that PKA/GSK3/CK1–mediated multisite phosphorylation regulates the formation of CiR/GliR. Here, we provided evidence that Fu/Ulk3/CK1–mediated multisite phosphorylation plays a conserved role in the regulation of the CiA/GliA activity gradient. We showed that Hh induces Ci/Gli phosphorylation at conserved Fu/Ulk3/CK1 sites in a dose- and time-dependent manner. Furthermore, we found that increasing the number of phospho-mimetic mutations of the physiologically relevant Fu/Ulk3/CK1 sites progressively increased CiA/GliA activity by gradually alleviating Sufu-mediated inhibition (Fig 6). We propose that the Hh morphogen gradient is translated into a Ci/Gli phosphorylation gradient that contributes to the formation of a CiA/GliA activity gradient.
Figure 6. Hh activates Ci/Gli through differential phosphorylation.

Graded Hh signals gradually increase the overall levels of Ci/Gli phosphorylation on multiple Fu/Ulk3/CK1 sites, leading to gradual release of Sufu repression and progressive increase in CiA/GliA activity. See text for details.
Previous studies showed that graded Hh signaling in Drosophila is translated into Smo phosphorylation and activity gradients, whereby increasing Smo phosphorylation levels progressively increase Smo cell surface accumulation and switch Smo C-terminal intracellular tail (C-tail) from a closed and inactive formation to an open and active formation, leading to dimerization/oligomerization of Smo C-tails (Jia et al, 2004; Zhao et al, 2007; Chen et al, 2010; Li et al, 2016). Smo transduces the Hh signal by directly interacting with Cos2/Fu and inducing Fu dimerization, autophosphorylation, and activation (Jia et al, 2003; Lum et al, 2003; Ogden et al, 2003; Ruel et al, 2003; Shi et al, 2011; Zhang et al, 2011; Zhou & Kalderon, 2011). The Smo gradient is thought to be translated into a Fu activity gradient by inducing increasing levels of its kinase activation loop phosphorylation (Shi et al, 2011). In addition, phosphorylation of Fu regulatory domain may also contribute to its activation (Zhou & Kalderon, 2011). Here, we provided evidence that the Fu activity gradient is further translated into Ci phosphorylation and activity gradients. We found that Ci phosphorylation at multiple Fu sites increased progressively overtime in response to increasing concentrations of Hh, which may reflect the progressively increased Smo and Fu activities (Lum et al, 2003; Fan et al, 2012; Ranieri et al, 2012). Phosphorylation at individual Fu sites on Ci exhibited similar dynamics (Fig 2D), suggesting that phosphorylation of Ci in different regions did not occur in a sequential fashion. Consistent with this notion, mutating one phosphorylation cluster does not appear to affect the phosphorylation on other clusters (Fig 1F) (Han et al, 2019). It is possible that low Hh or short exposure time only resulted in hypophosphorylation of Ci with only one or two Fu/CK1 clusters phosphorylated whereas high Hh or long exposure time resulted in hyperphosphorylation of Ci with all three Fu/CK1 clusters phosphorylated (Fig 6). Alternatively, phosphorylation of the three Fu/CK1 clusters on individual Ci protein could occur simultaneously and that the number of fully phosphorylated Ci proteins might increase overtime when cells were exposed with a fixed concentration of Hh or in cells treated with increasing concentrations of Hh. At the cellular level, the observed increase in Ci phosphorylation in response to increasing Hh concentration and exposure time could be because of increased responsiveness of individual cells or/and increased number of cells responding to Hh in the population. To address these questions, tools for quantifying Ci phosphorylation at single molecule or cell level need to be developed.
Our previous study identified a phosphorylation consensus sequence for the Fu family of kinases: S/TX5D/E (Han et al, 2019). However, we speculate that the spacing between the phosphorylation acceptor site and the downstream acidic residual is likely to be flexible. Indeed, the C-terminal site in Gli2 (S1528) has a spacing of six amino acids instead of five (Fig S4). This flexibility may increase the number of target sites for this family of kinases and can serve as guidance for identifying additional phosphorylation events and physiological relevant substrates regulated by the Fu family of kinases. Of note, the sequence surrounding Gli2 S1528 confers to the CK1 consensus site (Fig S4), raising the possibility that S1528 phosphorylation could also be catalyzed by CK1. Consistent with this, CK1 has been implicated as a positive regulator of both Smo and Ci/Gli2 (Chen et al, 2011; Shi et al, 2014b). We found that phosphorylation at the N-terminal Fu/Ulk3/CK1 cluster in Gli2 increased progressively in response to increasing amounts of Shh or ligand exposure time, but whether phosphorylation at the C-terminal cluster follows the same dynamics remains to be determined.
How does Fu/Ulk3/CK1–mediated phosphorylation of Ci/Gli contribute to Ci/Gli activation? Our previous studies identified multiple Sufu-binding sites in Ci and Gli proteins including Gli2 and Gli3 (Han et al, 2015). Binding of Sufu to Ci/Gli inhibits Ci/Gli by masking its nuclear localization signal and coactivator-binding domain (Shi et al, 2014a; Han et al, 2015). The N- and C-terminal Fu/Ulk3/CK1 phosphorylation clusters are located near the N- and C-terminal Sufu-binding sites, respectively (Figs 1A and 4A). Phospho-mimetic mutations at Fu/Ulk3/CK1 phosphorylation clusters decreased Sufu binding to Ci/Gli and increased the binding of Transportin (Trn) and Drosophila CBP (dCBP) to Ci (Figs 1J, 2H, and 4H and I) (Han et al, 2019), suggesting that Fu/Ulk3/CK1–mediated phosphorylation activates Ci/Gli by altering the binding of Sufu and thereby increasing Ci/Gli nuclear translocation and coactivator recruitment. However, it remains possible that Sufu may not completely dissociate from Ci/Gli, and the decreased binding affinity between Sufu and Ci/Gli may reflect a change in the conformation of Ci/Gli-Sufu complex (Zhang et al, 2013). Indeed, Sufu has been shown to accompany activated Ci/Gli to enter the nucleus in response to Hh (Zhang et al, 2017; Roberto et al, 2022). Further study is needed to determine the precise mechanism by which Fu/Ulk3/Stk36 releases Sufu-mediated inhibition of Ci/Gli.
We noticed that Ci-PKA_SD123 was still activated by CC-FuEE and Hh (Figs 2E and F and 3E’), suggesting that additional mechanisms regulated by Hh/Fu may contribute to Ci/Gli activation. For example, Hh signaling could activate Ci by releasing the inhibition of full-length Ci by PKA and Cos2 independent of Ci processing (Price & Kalderon, 1999; Wang et al, 1999; Little et al, 2020). Hh signaling could induce Ci phosphorylation by CK1 on multiple HIB/SPOP degrons to protect CiA from premature degradation by Cul3HIB/SPOP (Zhang et al, 2006; Shi et al, 2014b). Fu could activate Ci by phosphorylating additional sites on Ci or other substrates. Finally, Fu could allosterically regulate Ci/Sufu interaction independent of its kinase activity. Future work is needed to explore these possibilities.
Materials and Methods
DNA constructs
Myc-Ci-PKA was described previously (Han et al, 2019). Myc-Ci-PKA variants with point mutations in different phosphorylation sites shown in Fig 2A were generated using PCR-based site directed mutagenesis. Myc-Ci-PKA and its variants were subclones into the pUAST vector (Han et al, 2019). All GST-fusion proteins were subcloned into the pGEX 4T-1 vector using EcoRI and XhoI sites. N-terminally 6XMyc-tagged mouse Gli2WT, Gli2SA1, Gli2SA2, Gli2SA12, Gli2SD1, Gli2SD2, and Gli2SD12 and N-terminally 3XHA-tagged mouse Ulk3WT (HA-Ulk3WT) and Ulk3KR (HA-Ulk3KR) were subcloned into the pcDNA3.1(+) vector using EcoRI and XbaI sites.
Drosophila strains and husbandry
Drosophila culture and crosses were carried out according to standard procedure. Transgenic flies expressing Ci constructs (UAS-Myc-Ci-PKA_WT, UAS-Myc-Ci-PKA_SD1, UAS-Myc-Ci-PKA_SD12, and UAS-Myc-Ci-PKA_SD123) were generated using the phiC31 integration system (Bischof et al, 2007; Jia et al, 2009). Gal4 driver C765 (BL#36523) was used to drive the expression of UAS transgenes.
Cell culture, transfection, and lentiviral production
Drosophila S2R+ cells were cultured in Drosophila serum-free medium (Gibco) containing 10% FBS (Gibco) at 24°C. Clone-8 cells were cultured in Shields and Sang M3 Insect Medium (Sigma-Aldrich) with 2.5% FBS (Gibco), 2.5% fly extract (1645670, DGRC), insulin (0.125 IU/ml; Sigma-Aldrich) at 24°C. The Calcium Phosphate Transfection Kit (Specialty Media) was used to perform the transfection experiments according to the manufacturer’s instruction. The NIH3T3 cells were cultured in DMEM (Sigma-Aldrich) containing 10% bovine calf serum (Gibco) and 1% penicillin/streptomycin (Sigma-Aldrich). The transfection was carried out by using the GenJet Plus in vitro DNA transfection kit (SignaGen). HEK293T cells were cultured in DMEM (Sigma-Aldrich) supplemented with 10% FBS (Gibco) and 1% penicillin/streptomycin (Sigma-Aldrich). The PolyJet in vitro DNA transfection kit (SignaGen) was used to do the transfection according to the manufacturer’s instruction. For lentivirus production, the N-terminal 6× Myc-tagged mouse WT and Gli2SD12 (Myc-Gli2SD12) and Gli2SA12 (Myc-Gli2SA12) were subcloned into the lentiviral vector FUGW. The expression vectors and package vectors (PSPAX2 and PMDG2) were co-transfected into HEK293T cells using PolyJet (SignaGen). After 48 h, the recombinant viruses were harvested for cell line infection. The recombinant viruses were infected into the NIH3T3 cells using the standard method.
HhN-conditioned medium treatment was performed as previously described (Han et al, 2019). Briefly, the hygromycin (200 μg/ml) was used to select the S2R+ cells stably expressing the HhN (the N-terminal fragment of Hh) which is responsible for the hedgehog signaling. The cells were cultured in the medium without hygromycin but with 0.7 mM CuSO4 for 1 d. Then the medium was harvested and sterilized by filtration. Hh-conditioned medium was used at a 6:4 dilution ratio with fresh medium (defined as 100% in Fig 2C and D). For Shh or SAG treatment, cells were starved for 12 h (0.5% BCS), recombinant human Shh N-terminal fragment (1 ng/ml, #8908-SH-005; R&D Systems) or SAG (200 nm, Cat. no. 566660; Sigma-Aldrich) was added to the medium for 12 h or overnight.
Immunostaining, immunoprecipitation, and Western blotting analysis
Immunostaining of imaginal discs was carried out as previously described (Jiang & Struhl, 1995). Immunofluorescence imaging was performed using a Zeiss 710 laser scanning microscope. For immunoprecipitation assay, after transfection for 48 h, cells were washed twice with PBS and then lysed on ice for 10 min with lysis buffer containing 1M Tris, pH 8.0, 5M NaCl, 1MNaF, 0.1M Na3VO4, 1% CA630, 10% glycerol, and 0.5M EDTA (pH 8.0). Cell lysates were incubated with protein A-Sepharose beads (Thermo Fisher Scientific) for 1 h at 4°C to eliminate nonspecific binding proteins. After removal of the protein-A beads by centrifugation, the cleared lysates were incubated with Myc (HA or Flag) antibody for 2 h or overnight. Protein complexes were collected by incubation with protein A-Sepharose beads for 1 h at 4°C, followed by centrifugation. Immunoprecipitates were washed three times for 5 min each with lysis buffer and were separated on SDS–PAGE. Western blot was carried out using standard protocol. Antibodies used in this study were as follows: mouse anti-Myc (Santa Cruz Biotechnology), mouse anti-Flag (Sigma-Aldrich), mouse anti-HA (Santa Cruz Biotechnology), rat anti-Ci 2A1, and mouse anti-Ptc (Developmental Studies Hybridoma Bank).
Phospho antibodies generation
pS218/220 and pS1230/1233 phospho-specific antibodies were previously described (Han et al, 2019). The pS1382 antibody was made by Abmart using the following phospho-peptides as antigens: TTS(p)LTSLLEE. The phospho-specific antibodies were purified by positive and negative selection using the affinity column conjugated with the corresponding phosphorylated and non-phosphorylated peptides sequentially.
In vitro kinase assay
In vitro kinase assay was performed by incubating 25 μl of reaction mixtures containing 150 mM Tris–HCl (pH 7.5), 0.2 mM Mg2+/ATP, 1 μg of purified GST-Ci/Gli2 fusion proteins, together with appropriate amount of kinase for 30 min at 30°C. The reaction was terminated by adding 2× SDS loading buffer. The resultant samples were load on SDS–PAGE and subjected to the pIMAGO phospho-protein detection kit with the fluor-680 (Sigma-Aldrich) detection kit and LI-COR Odyssey platform for Western blot analyses of phosphorylated proteins (Millipore Sigma). Flag-CC-FuEE and Flag-CC-FuKR were purified from insect cells as described previously (Han & Jiang, 2021). HA-Ulk3WT and HA-Ulk3KR were immunopurified from HEK293 cells transfected with the corresponding constructs. Recombinant CK1 was purchased from Abcam (ab102102).
Luciferase reporter assay and RNAi
The Dual-Luciferase Reporter Assay System (Promega) was applied to detect the dual–luciferase activity according to the manufacturer’s instructions. Briefly, S2R+ cells were cultured in 12-well plates, and on the second day, each well was transfected with 0.5 μg ptc-luc reporter construct, 25 ng RL-PolIII Renilla construct, and 0.5 μg Ci construct. Samples were collected after 48-h incubation. For Hh treatment, 1 d after the transfection, two-thirds of the medium was replaced with HhN-conditioned medium, and cells were incubated for 24 h before harvest. Each sample was measured in triplicate by using a FLUOstar OPTIMA plate reader (BMG Labtech). The Gli-luc reporter assay was carried out by transfecting the NIH3T3 cells in 12-well plate with 0.4 mg 8XGli-Luc reporter, 0.1 mg PRL-SV40 Renilla vector, and 0.5 mg Gli2 construct. The luciferase activity of each sample was measured in triplicate by using a FLUOstar OPTIMA plate reader (BMG Labtech). dsRNAs were generated using the MEGAscript High Yield Transcription Kit (Ambion). The dsRNA targeting the coding sequence of GFP was used as a negative control. Fu dsRNA targets the coding sequence between amino acids 324–434. For RNAi experiments, cells were treated with corresponding dsRNA in serum-free culture medium for 8 h at 24°C before switching to regular growth medium. The dsRNA-treated cells were cultured for 2 d before analysis. shRNA plasmids against eGFP (control, SHC005) and mGli2 (TRCN0000219066) were purchased from Sigma-Aldrich.
Quantitative RT-PCR
Total RNA was extracted from 1 × 106 cells using the RNeasy Plus Mini Kit (QIAGEN), and cDNA was synthesized with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems), and qPCR was performed using Fast SYBR Green Master Mix (Applied Biosystems) and a Bio-Rad CFX96 real-time PCR system. qRT-PCR was performed in triplicate for each of three independent biological replicates. Quantification of mRNA levels was calculated using the comparative CT method. Primers: Ptc, 5′-ATGGACCGCGACAGCCTCCCA-3′ and 5′-CGACGCAGAAGGTGCTCAGCA-3′; Gli1, 5′-GTGCACGTTTGAAGGCTGTC-3′ and 5′-GAGTGGGTCCGATTCTGGTG-3′; Ptch1, 5′-GAAGCCACAGAAAACCCTGTC-3′ and 5′-GCCGCAAGCCTTCTCTACG-3′; Foxa2, 5′-GGAGTGTACTCCAGGCCTATTA-3′ and 5′-CTCCACTCAGCCTCTCATTTC-3′; Nkx2.2, 5′-CAGCCTCATCCGTCTCAC-3′ and 5′-TCACCTCCATACCTTTCTCC-3′; GAPDH, 5′-GTGGTGAAGCAGGCATCTGA-3′ and 5′-GCCATGTAGGCCATGAGGTC-3′.
Neuron progenitor differentiation
A stable cell line derived from HM1 mESCs harboring 8XGBS-Venus reporter at hypoxanthine-guanine phosphoribosyl transferase (HPRT) locus was first transduced by shRNA lentivirus targeting 3′ UTR of Gli2. The Knock down efficiency was validated by qRT-PCR (>90%). Myc-tagged WT and mutant mGli2 were introduced by lentivirus and further G418 selection. The resultant mESCs were cultured with feeder MEFs onto dishes pre-coated with 0.1% gelatin in medium (DMEM, 15% Optima FBS, 1× MEM nonessential amino acids, 2 mM L-glutamine, 1% Embryo Max nucleosides, 55 μM 2-mercaptoethanol, and 1,000 U/ml ESGRO LIF). For differentiation, feeder cells were eliminated by trypsinization followed by 2× 10 min incubation for settlement. mESC were then cultured in CellBIND plates at the density of 60,000 cells per six well in N2B27 medium (DMEM F12 and neurobasal medium at 1:1 ratio, N2 and B27 supplement, 2 mM L-glutamine 40 μg/ml BSA, 55 μM 2-mercaptoethanol). For the first 2 d (day 0 and 1) after the seeding, cells were cultured in N2B27 medium supplemented with 10 ng/ml bFGF. On day 2, the differentiation was induced by adding RA (100 nM; Sigma-Aldrich) for 12 h. After that, the cells were treated with Shh N (1 ng/ml, R&D System) for another 12 h. Then samples were collected for qRT-PCR.
Supplementary Material
Acknowledgements
We thank Dr. Xiaochun Li and Dr. James Briscoe for reagents, the Developmental Studies Hybridoma Bank for antibodies, and the Bloomington Drosophila Stock Center for fly stocks. This work was supported by grants from NIGMS (R35GM118063) and Welch Foundation (I-1603) to J Jiang. J Jiang is a Eugene McDermott Endowed Scholar in Biomedical Science at UTSW.
Author Contributions
M Zhou: data curation, formal analysis, investigation, and writing—review and editing.
Y Han: conceptualization, data curation, formal analysis, investigation, and methodology.
B Wang: resources, investigation, and project administration.
YS Cho: data curation and formal analysis.
J Jiang: conceptualization, formal analysis, supervision, funding acquisition, project administration, and writing—original draft, review, and editing.
Conflict of Interest Statement
The authors declare that they have no conflict of interest.
References
- Arveseth CD, Happ JT, Hedeen DS, Zhu JF, Capener JL, Klatt Shaw D, Deshpande I, Liang J, Xu J, Stubben SL, et al. (2021) Smoothened transduces Hedgehog signals via activity-dependent sequestration of PKA catalytic subunits. PLoS Biol 19: e3001191. 10.1371/journal.pbio.3001191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aza-Blanc P, Lin HY, Ruiz i Altaba A, Kornberg TB (2000) Expression of the vertebrate Gli proteins in Drosophila reveals a distribution of activator and repressor activities. Development 127: 4293–4301. 10.1242/dev.127.19.4293 [DOI] [PubMed] [Google Scholar]
- Aza-Blanc P, Ramírez-Weber FA, Laget MP, Schwartz C, Kornberg TB (1997) Proteolysis that is inhibited by Hedgehog targets Cubitus interruptus protein to the nucleus and converts it to a repressor. Cell 89: 1043–1053. 10.1016/s0092-8674(00)80292-5 [DOI] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K (2007) An optimized transgenesis system for Drosophila using germ-line-specific φC31 integrases. Proc Natl Acad Sci U S A 104: 3312–3317. 10.1073/pnas.0611511104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Manoukian AS, Perrimon N (1994) Ectopic expression in Drosophila. Methods Cell Biol 44: 635–654. 10.1016/s0091-679x(08)60936-x [DOI] [PubMed] [Google Scholar]
- Briscoe J, Therond PP (2013) The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol 14: 416–429. 10.1038/nrm3598 [DOI] [PubMed] [Google Scholar]
- Chen Y, Li S, Tong C, Zhao Y, Wang B, Liu Y, Jia J, Jiang J (2010) G protein-coupled receptor kinase 2 promotes high-level Hedgehog signaling by regulating the active state of Smo through kinase-dependent and kinase-independent mechanisms in Drosophila. Genes Dev 24: 2054–2067. 10.1101/gad.1948710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Sasai N, Ma G, Yue T, Jia J, Briscoe J, Jiang J (2011) Sonic hedgehog dependent phosphorylation by CK1α and GRK2 is required for ciliary accumulation and activation of smoothened. PLoS Biol 9: e1001083. 10.1371/journal.pbio.1001083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Motoyama J, Gasca S, Mo R, Sasaki H, Rossant J, Hui C (1998) Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development 125: 2533–2543. 10.1242/dev.125.14.2533 [DOI] [PubMed] [Google Scholar]
- Fan J, Liu Y, Jia J (2012) Hh-induced smoothened conformational switch is mediated by differential phosphorylation at its C-terminal tail in a dose- and position-dependent manner. Dev Biol 366: 172–184. 10.1016/j.ydbio.2012.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouti M, Tsakiridis A, Wymeersch FJ, Huang Y, Kleinjung J, Wilson V, Briscoe J (2014) In vitro generation of neuromesodermal progenitors reveals distinct roles for wnt signalling in the specification of spinal cord and paraxial mesoderm identity. PLoS Biol 12: e1001937. 10.1371/journal.pbio.1001937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Jiang J (2021) Expression and purification of fused kinase from insect cells for in vitro kinase assay. STAR Protoc 2: 100376. 10.1016/j.xpro.2021.100376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Shi Q, Jiang J (2015) Multisite interaction with Sufu regulates Ci/Gli activity through distinct mechanisms in Hh signal transduction. Proc Natl Acad Sci U S A 112: 6383–6388. 10.1073/pnas.1421628112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Wang B, Cho YS, Zhu J, Wu J, Chen Y, Jiang J (2019) Phosphorylation of Ci/Gli by fused family kinases promotes hedgehog signaling. Dev Cell 50: 610–626.e4. 10.1016/j.devcel.2019.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui CC, Angers S (2011) Gli proteins in development and disease. Annu Rev Cell Dev Biol 27: 513–537. 10.1146/annurev-cellbio-092910-154048 [DOI] [PubMed] [Google Scholar]
- Humke EW, Dorn KV, Milenkovic L, Scott MP, Rohatgi R (2010) The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev 24: 670–682. 10.1101/gad.1902910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H, Liu Y, Yan W, Jia J (2009) PP4 and PP2A regulate Hedgehog signaling by controlling Smo and Ci phosphorylation. Development 136: 307–316. 10.1242/dev.030015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Amanai K, Wang G, Tang J, Wang B, Jiang J (2002) Shaggy/GSK3 antagonizes Hedgehog signalling by regulating Cubitus interruptus. Nature 416: 548–552. 10.1038/nature733 [DOI] [PubMed] [Google Scholar]
- Jia J, Tong C, Jiang J (2003) Smoothened transduces Hedgehog signal by physically interacting with Costal2/Fused complex through its C-terminal tail. Genes Dev 17: 2709–2720. 10.1101/gad.1136603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Tong C, Wang B, Luo L, Jiang J (2004) Hedgehog signalling activity of smoothened requires phosphorylation by protein kinase A and casein kinase I. Nature 432: 1045–1050. 10.1038/nature03179 [DOI] [PubMed] [Google Scholar]
- Jia J, Zhang L, Zhang Q, Tong C, Wang B, Hou F, Amanai K, Jiang J (2005) Phosphorylation by double-time/CKIε and CKIα targets cubitus interruptus for slimb/β-TRCP-mediated proteolytic processing. Dev Cell 9: 819–830. 10.1016/j.devcel.2005.10.006 [DOI] [PubMed] [Google Scholar]
- Jiang J (2021) Hedgehog signaling mechanism and role in cancer. Semin Cancer Biol 10.1016/j.semcancer.2021.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Hui CC (2008) Hedgehog signaling in development and cancer. Dev Cell 15: 801–812. 10.1016/j.devcel.2008.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J (2006) Regulation of Hh/Gli signaling by dual ubiquitin pathways. Cell Cycle 5: 2457–2463. 10.4161/cc.5.21.3406 [DOI] [PubMed] [Google Scholar]
- Jiang J, Struhl G (1995) Protein kinase A and hedgehog signaling in Drosophila limb development. Cell 80: 563–572. 10.1016/0092-8674(95)90510-3 [DOI] [PubMed] [Google Scholar]
- Jiang J, Struhl G (1998) Regulation of the Hedgehog and Wingless signalling pathways by the F- box/WD40-repeat protein Slimb. Nature 391: 493–496. 10.1038/35154 [DOI] [PubMed] [Google Scholar]
- Knippschild U, Gocht A, Wolff S, Huber N, Lohler J, Stoter M (2005) The casein kinase 1 family: Participation in multiple cellular processes in eukaryotes. Cell Signal 17: 675–689. 10.1016/j.cellsig.2004.12.011 [DOI] [PubMed] [Google Scholar]
- Li S, Li S, Han Y, Tong C, Wang B, Chen Y, Jiang J (2016) Regulation of smoothened phosphorylation and high-level hedgehog signaling activity by a plasma membrane associated kinase. PLoS Biol 14: e1002481. 10.1371/journal.pbio.1002481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Ma G, Wang B, Jiang J (2014) Hedgehog induces formation of PKA-Smoothened complexes to promote Smoothened phosphorylation and pathway activation. Sci Signal 7: ra62. 10.1126/scisignal.2005414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little JC, Garcia-Garcia E, Sul A, Kalderon D (2020) Drosophila hedgehog can act as a morphogen in the absence of regulated Ci processing. Elife 9: e61083. 10.7554/elife.61083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum L, Zhang C, Oh S, Mann RK, von Kessler DP, Taipale J, Weis-Garcia F, Gong R, Wang B, Beachy PA (2003) Hedgehog signal transduction via smoothened association with a cytoplasmic complex scaffolded by the atypical kinesin, Costal-2. Mol Cell 12: 1261–1274. 10.1016/s1097-2765(03)00426-x [DOI] [PubMed] [Google Scholar]
- Ma G, Li S, Han Y, Li S, Yue T, Wang B, Jiang J (2016) Regulation of smoothened trafficking and hedgehog signaling by the SUMO pathway. Dev Cell 39: 438–451. 10.1016/j.devcel.2016.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matise MP, Epstein DJ, Park HL, Platt KA, Joyner AL (1998) Gli2 is required for induction of floor plate and adjacent cells, but not most ventral neurons in the mouse central nervous system. Development 125: 2759–2770. 10.1242/dev.125.15.2759 [DOI] [PubMed] [Google Scholar]
- Methot N, Basler K (1999) Hedgehog controls limb development by regulating the activities of distinct transcriptional activator and repressor forms of Cubitus interruptus. Cell 96: 819–831. 10.1016/s0092-8674(00)80592-9 [DOI] [PubMed] [Google Scholar]
- Methot N, Basler K (2000) Suppressor of fused opposes hedgehog signal transduction by impeding nuclear accumulation of the activator form of Cubitus interruptus. Development 127: 4001–4010. 10.1242/dev.127.18.4001 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Wen X, Ratti N, Loktev A, Rangell L, Scales SJ, Jackson PK (2013) The ciliary G-protein-coupled receptor Gpr161 negatively regulates the sonic hedgehog pathway via cAMP signaling. Cell 152: 210–223. 10.1016/j.cell.2012.12.026 [DOI] [PubMed] [Google Scholar]
- Ogden SK, Ascano M Jr., Stegman MA, Suber LM, Hooper JE, Robbins DJ (2003) Identification of a functional interaction between the transmembrane protein Smoothened and the kinesin-related protein Costal2. Curr Biol 13: 1998–2003. 10.1016/j.cub.2003.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlmeyer JT, Kalderon D (1998) Hedgehog stimulates maturation of Cubitus interruptus into a labile transcriptional activator. Nature 396: 749–753. 10.1038/25533 [DOI] [PubMed] [Google Scholar]
- Price MA, Kalderon D (1999) Proteolysis of cubitus interruptus in Drosophila requires phosphorylation by protein kinase A. Development 126: 4331–4339. 10.1242/dev.126.19.4331 [DOI] [PubMed] [Google Scholar]
- Price MA, Kalderon D (2002) Proteolysis of the hedgehog signaling effector cubitus interruptus requires phosphorylation by glycogen synthase kinase 3 and casein kinase 1. Cell 108: 823–835. 10.1016/s0092-8674(02)00664-5 [DOI] [PubMed] [Google Scholar]
- Pusapati GV, Kong JH, Patel BB, Gouti M, Sagner A, Sircar R, Luchetti G, Ingham PW, Briscoe J, Rohatgi R (2018) G protein-coupled receptors control the sensitivity of cells to the morphogen Sonic Hedgehog. Sci Signal 11: eaao5749. 10.1126/scisignal.aao5749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranieri N, Ruel L, Gallet A, Raisin S, Therond PP (2012) Distinct phosphorylations on kinesin costal-2 mediate differential hedgehog signaling strength. Dev Cell 22: 279–294. 10.1016/j.devcel.2011.12.002 [DOI] [PubMed] [Google Scholar]
- Ranieri N, Therond PP, Ruel L (2014) Switch of PKA substrates from Cubitus interruptus to Smoothened in the Hedgehog signalosome complex. Nat Commun 5: 5034. 10.1038/ncomms6034 [DOI] [PubMed] [Google Scholar]
- Roberto N, Becam I, Plessis A, Holmgren RA (2022) Engrailed, suppressor of fused and Roadkill modulate the Drosophila GLI transcription factor Cubitus interruptus at multiple levels. Development 149: dev200159. 10.1242/dev.200159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruel L, Rodriguez R, Gallet A, Lavenant-Staccini L, Therond PP (2003) Stability and association of smoothened, Costal2 and fused with cubitus interruptus are regulated by hedgehog. Nat Cell Biol 5: 907–913. 10.1038/ncb1052 [DOI] [PubMed] [Google Scholar]
- Shi Q, Han Y, Jiang J (2014. a) Suppressor of fused impedes Ci/Gli nuclear import by opposing Trn/Kapβ2 in Hedgehog signaling. J Cell Sci 127: 1092–1103. 10.1242/jcs.142828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Li S, Jia J, Jiang J (2011) The Hedgehog-induced Smoothened conformational switch assembles a signaling complex that activates fused by promoting its dimerization and phosphorylation. Development 138: 4219–4231. 10.1242/dev.067959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Li S, Li S, Jiang A, Chen Y, Jiang J (2014. b) Hedgehog-induced phosphorylation by CK1 sustains the activity of Ci/Gli activator. Proc Natl Acad Sci U S A 111: E5651–E5660. 10.1073/pnas.1416652111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smelkinson MG, Zhou Q, Kalderon D (2007) Regulation of Ci-SCFSlimb binding, Ci proteolysis, and hedgehog pathway activity by Ci phosphorylation. Dev Cell 13: 481–495. 10.1016/j.devcel.2007.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamataki D, Ulloa F, Tsoni SV, Mynett A, Briscoe J (2005) A gradient of Gli activity mediates graded Sonic Hedgehog signaling in the neural tube. Genes Dev 19: 626–641. 10.1101/gad.325905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale J, Beachy PA (2001) The Hedgehog and Wnt signalling pathways in cancer. Nature 411: 349–354. 10.1038/35077219 [DOI] [PubMed] [Google Scholar]
- Tempe D, Casas M, Karaz S, Blanchet-Tournier MF, Concordet JP (2006) Multisite protein kinase A and glycogen synthase kinase 3β phosphorylation leads to Gli3 ubiquitination by SCFβTrCP. Mol Cell Biol 26: 4316–4326. 10.1128/mcb.02183-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong ME, Bilekova S, Choksi SP, Li W, Bugaj LJ, Xu K, Reiter JF (2021) Vertebrate cells differentially interpret ciliary and extraciliary cAMP. Cell 184: 2911–2926.e18. 10.1016/j.cell.2021.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukachinsky H, Lopez LV, Salic A (2010) A mechanism for vertebrate hedgehog signaling: Recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol 191: 415–428. 10.1083/jcb.201004108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Fallon JF, Beachy PA (2000. a) Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100: 423–434. 10.1016/s0092-8674(00)80678-9 [DOI] [PubMed] [Google Scholar]
- Wang G, Amanai K, Wang B, Jiang J (2000. b) Interactions with Costal2 and suppressor of fused regulate nuclear translocation and activity of cubitus interruptus. Genes Dev 14: 2893–2905. 10.1101/gad.843900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Wang B, Jiang J (1999) Protein kinase A antagonizes Hedgehog signaling by regulating both the activator and repressor forms of Cubitus interruptus. Genes Dev 13: 2828–2837. 10.1101/gad.13.21.2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Zhang L, Wang B, Ou CY, Chien CT, Jiang J (2006) A hedgehog-induced BTB protein modulates hedgehog signaling by degrading Ci/Gli transcription factor. Dev Cell 10: 719–729. 10.1016/j.devcel.2006.05.004 [DOI] [PubMed] [Google Scholar]
- Zhang W, Zhao Y, Tong C, Wang G, Wang B, Jia J, Jiang J (2005) Hedgehog-regulated costal2-kinase complexes control phosphorylation and proteolytic processing of cubitus interruptus. Dev Cell 8: 267–278. 10.1016/j.devcel.2005.01.001 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Fu L, Qi X, Zhang Z, Xia Y, Jia J, Jiang J, Zhao Y, Wu G (2013) Structural insight into the mutual recognition and regulation between suppressor of Fused and Gli/Ci. Nat Commun 4: 2608. 10.1038/ncomms3608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Mao F, Lu Y, Wu W, Zhang L, Zhao Y (2011) Transduction of the Hedgehog signal through the dimerization of Fused and the nuclear translocation of Cubitus interruptus. Cell Res 21: 1436–1451. 10.1038/cr.2011.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Shen L, Law K, Zhang Z, Liu X, Hua H, Li S, Huang H, Yue S, Hui CC, et al. (2017) Suppressor of fused chaperones Gli proteins to generate transcriptional responses to sonic hedgehog signaling. Mol Cell Biol 37: e00421-16. 10.1128/mcb.00421-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Tong C, Jiang J (2007) Hedgehog regulates smoothened activity by inducing a conformational switch. Nature 450: 252–258. 10.1038/nature06225 [DOI] [PubMed] [Google Scholar]
- Zhou M, Jiang J (2022) Gli phosphorylation code in hedgehog signal transduction. Front Cell Dev Biol 10: 846927. 10.3389/fcell.2022.846927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Kalderon D (2011) Hedgehog activates fused through phosphorylation to elicit a full spectrum of pathway responses. Dev Cell 20: 802–814. 10.1016/j.devcel.2011.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.