

Abstract
Introduction
Genomic alterations in the juxtamembrane exon 14 splice sites in NSCLC lead to increased MET stability and oncogenesis. We present the largest cohort study of MET Exon 14 (METex14) using whole transcriptome sequencing.
Methods
A total of 21,582 NSCLC tumor samples underwent complete genomic profiling with next-generation sequencing of DNA (592 Gene Panel, NextSeq, whole exome sequencing, NovaSeq) and RNA (NovaSeq, whole transcriptome sequencing). Clinicopathologic information including programmed death-ligand 1 and tumor mutational burden were collected and RNA expression for mutation subtypes and MET amplification were quantified. Immunogenic signatures and potential pathways of invasion were characterized using single-sample gene set enrichment analysis and mRNA gene signatures.
Results
A total of 533tumors (2.47%) with METex14 were identified. The most common alterations were point mutations (49.5%) at donor splice sites. Most alterations translated to increased MET expression, with MET co-amplification resulting in synergistic increase in expression (q < 0.05). Common coalterations were amplifications of MDM2 (19.0% versus 1.8% wild-type [WT]), HMGA2 (13.2% versus 0.98% WT), and CDK4 (10.0% versus 1.5% WT) (q < 0.05). High programmed death-ligand 1 > 50% (52.5% versus 27.3% WT, q < 0.0001) and lower proportion of high tumor mutational burden (>10 mutations per megabase, 8.3% versus 36.7% WT, p < 0.0001) were associated with METex14, which were also enriched in both immunogenic signatures and immunosuppressive checkpoints. Pathways associated with METex14 included angiogenesis and apical junction pathways (q < 0.05).
Conclusions
METex14 splicing alterations and MET co-amplification translated to higher and synergistic MET expression at the transcriptomic level. High frequencies of MDM2 and CDK4 co-amplifications and association with multiple immunosuppressive checkpoints and angiogenic pathways provide insight into potential actionable targets for combination strategies in METex14 NSCLC.
Keywords: METex14, Non–small cell lung cancer, Whole transcriptome sequencing, RNA expression, MDM2, Immune signatures
Introduction
The MET proto-oncogene plays a key role in cellular proliferation, invasion, and metastases and has been reported to play an oncogenic role in NSCLC.1, 2, 3, 4 Located on chromosome 7q21–31, the MET gene encodes a receptor tyrosine kinase, which on ligand binding with its natural ligand hepatocyte growth factor or scatter factor leads to receptor dimerization, activation of the tyrosine kinase domain, and downstream signaling of RAS/RAF/MEK and PI3K/AKT/mTOR pathways.5 Tight regulation of MET expression occurs by means of the Y1003 loci located in the juxtamembrane domain encoded by exon 14. On binding of Cbl-E3 ubiquitin to Y1003, the receptor undergoes internalization and proteasomal degradation.5 Somatic mutations at or near splicing regions of exon 14 lead to skipping of transcription of exon 14, yielding the MET exon 14 (METex14) alternative splicing variant with increased receptor stability and downstream oncogenic signaling.1,6
METex14 represents approximately 2% to 4% of metastatic NSCLC and has been associated with older age, female sex, sarcomatoid histology, and generally portends a poor prognosis.2,3,7, 8, 9 Capmatinib and tepotinib have been Food and Drug Administration approved for MET-directed therapy in METex14-driven metastatic NSCLC on the basis of response rates of 68% and 43% in treatment-naive patients.10, 11, 12 Although high programmed death-ligand 1 (PD-L1) has been observed with METex14, inconsistent responses to immunotherapy have been observed.13,14 Given the aggressive nature of this key lung cancer molecular subtype and modest, variable and transient benefits with currently available agents, novel opportunities for treatment intervention through better understanding of disease biology remain a great unmet need.
Diverse genomic alterations associated with METex14 have been characterized through DNA profiling, but these assays remain an imperfect tool for identifying skipped variants as false-negative results may occur.2, 3, 4,13,15, 16, 17 RNA sequencing has improved sensitivity in identifying skipped variants,15,16 and to our knowledge, characterization of genomic alterations of METex14 using whole transcriptome sequencing (WTS) has yet to be reported. We have conducted preclinical assays on clustered, regularly interspaced, short palindromic repeat-modified cellular models of METex14-driven lung cancer and have found that METex14 NSCLC cells were associated with pathways related to cytoskeletal remodeling, cellular adhesion, epithelial to mesenchymal transition (EMT), and angiogenesis.18 The in vivo up-regulation and biological role of these pathways in METex14-driven lung cancers remain to be understood and may open novel avenues for more effective and durable therapeutic interventions.
We report the largest WTS cohort defining genomic alterations and co-alterations associated with METex14 and characterize the immune microenvironment of METex14 NSCLC using a real-world database. We also report key biological processes that may be involved in invasion and metastasis of METex14.
Materials and Methods
Subjects
A total of 21,582 NSCLC formalin-fixed, paraffin-embedded tumor samples underwent an institutional review board-approved, retrospective analysis of tumor samples submitted for molecular profiling at a CLIA-certified genomics laboratory (Caris Life Sciences, Phoenix, AZ). Analyses included next-generation sequencing (NGS) of DNA (592 Gene Panel, NextSeq, or whole exome sequencing [WES], NovaSeq), RNA (NovaSeq, WTS), and immunohistochemistry (IHC) for PD-L1. Real-world survival information was obtained from insurance claims data and calculated from tissue collection or first of treatment time to last of contact. Kaplan-Meier estimates were calculated for molecularly defined patient cohorts.
DNA NGS, WES, and RNA WTS
DNA sequencing was performed using the NextSeq or NovaSeq platform (Illumina, Inc., San Diego, CA) after microdissection. Identified genetic variants were further annotated as “pathogenic,” “likely pathogenic,” “variant of unknown significance,” “likely benign,” or “benign,” according to the 2015 American College of Medical Genetics and Genomics standards,19 a core foundation for a recently published guideline for classifying oncogenicity of somatic variants.20 Only “pathogenic” and “likely pathogenic” mutations were counted toward mutation frequency calculation in our study. A hybrid-capture method using Agilent SureSelect Human All Exon V7 bait panel (Agilent Technologies, Santa Clara, CA) and the Illumina NovaSeq platform (Illumina, Inc., San Diego, CA) were used for RNA sequencing. A minimum of 10% tumor content in the area for microdissection was required for enrichment and extraction of tumor-specific RNA. If no correlative DNA alteration was identified, METex14 required a read depth of at least 10. FASTQ files were aligned with STAR aligner (Alex Dobin, release 2.7.4a GitHub). Data were then produced by Salmon, which provides fast and bias-aware quantification of transcript expression.21 BAM files from STAR aligner were processed for RNA variants using a proprietary custom detection pipeline. The reference genome was GRCh37/hg19.
Tumor Mutational Burden, PD-L1 Status
All nonsynonymous missense, nonsense, in-frame insertion/deletion and frameshift mutations that had not been previously described as germline alterations in dbSNP151, Genome Aggregation Database or benign variants were counted toward tumor mutational burden (TMB) measurement.22 A cutoff point of more than or equal to 10 mutations per megabase (mut/Mb) was used to define TMB-high tumors. PD-L1 status was assessed by means of 22c3 anti–PD-L1 antibody (Dako) on formalin-fixed, paraffin-embedded sessions and evaluated for percentage positively stained tumor cells to derive a tumor proportion score.
Single-Sample Gene Enrichment Analysis
Single-sample gene enrichment analysis (ssGSEA) was used to calculate pathway enrichment scores of 50 hallmark gene pathways per tumor using WTS data set without normalization.23,24 Interferon (IFN)-γ signature and T-cell inflammation signature scores were defined by 18-gene25 and 160-gene mRNA signatures,26 respectively, and were used to estimate the likelihood of a tumor’s response to anti–programmed cell death protein-1 (PD-1) therapy. QuanTISeq was performed to quantify fractions of 10 different types of infiltrated immune cells.27
Statistical Methods
Chi-square and Fisher’s exact tests were used to assess statistical differences between categorical variables and Wilcoxon or Mann-Whitney U tests for comparisons between numerical variables. Benjamin-Hochberg correction was applied to minimize the false discovery rate when multiple tests were performed, where the reported p or q value was the statistic without or with correction, respectively. R (version 4.1.2) was used for ssGSEA analysis and QuanTISeq. Python (version 3.9.12) was used for other analyses.
Results
Clinicopathologic Features
Of the 21,582 cases, 533 (2.47%) were identified with METex14 and 21,049 without METex14 (wild type [WT]) (Table 1). METex14-positive tumors occurred more frequently in females (302, 56.7% versus 231, 43.3% males, p < 0.05) and in older patients (median 77 y versus 69 y, p < 0.0001). The most frequently represented histology was adenocarcinoma (324, 60.8%), followed by squamous cell (57, 10.7%), adenosquamous (15, 2.8%), sarcomatoid (21, 3.9%), and large cell (1, 0.2%). Of all histologies, METex14 was enriched in patients with sarcomatoid histology (21 of 202, 10.4%) followed by adenosquamous (15 of 196, 7.65%). A total of 115 cases (21.6%) were classified as “other non-small cell carcinoma” histology in which accurate histologic information was not available or was of mixed histology. Staging information was unavailable; however, METex14 was more often identified through primary site of biopsy. Of the 104 patients with METex14 with smoking data, there was a higher proportion of light or never smokers compared with patients with WT (p < 0.0001).
Table 1.
Clinicopathologic Characteristics of Patients With NSCLC With and Without METex14
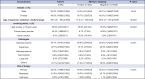 |
Subtype of Genomic Alterations of METex14
To better understand the heterogeneity of MET mutations which yield METex14, we characterized the subtypes of MET mutations by their spatial chromosomal locations (Fig. 1A). The most common genomic alterations were base substitutions in the splice donor site (49.5%), located in 5′ splice site of intron 14, followed by deletions in the polypyrimidine tract (17.6%), located in intron 13, which play a role in spliceosome assembly, and deletions in the splice donor site (16.5%) (Fig. 1B). Less common alterations were deletions in splice acceptor site (8.8%), located in 3′ splice site of intron 13, insertions and deletions (indels) at polypyrimidine tract (2.4%), base substitutions at splice acceptor site (2.6%), indels at splice donor site (1.7%), and indels at splice acceptor site (1.1%). The most common alteration overall was c.3082G > C (Fig. 1C). Y1003X, a rare alteration directly affecting the Y1003 Cbl-binding sites, was found to be mutually exclusive to METex14 skipping variants (p < 0.01).
Figure 1.

(A) Spatial chromosomal representation of METex14 mutation subtypes. (B) Distribution of METex14 alteration on the basis of mutation subtype. (C) Frequency of often identified DNA alterations. (D) Frequency of co-alterations (left) or co-amplifications (right) of METex14 and MET WT. (E) Oncoprint of METex14 and co-alterations in TP53, MDM2, CDK4, and HMGA2 (green: mutation detected; red: amplification detected; gray: wild type; white: data not available). CNA > 6. CNA, copy number alteration; METex14, MET exon 14; NGS, next-generation sequencing; WT, wild type.
Of the 533 fusion variants identified by WTS, 11.3% did not have a corresponding genetic alteration, regardless of sequencing platform (Fig. 1E and Supplementary Fig. 1). Although speculative, this lack of corresponding fusion variant to its genomic alternation may be explained by possible alterations occurring within introns 13 and 14 that were distant from the skipped event, which would not be captured by the WES platform. In addition, oncogenic events with low allele frequency at the DNA level that may not be captured in the WES platform may also gain transcription advantage and overexpress above the detection level in WTS.
Co-alterations and Co-amplifications
METex14 skipping variants were largely mutually exclusive with other known driver mutations, including KRAS, EGFR, BRAF, ALK fusion, and ROS-1 fusion (Fig. 1D). Co-alterations with TP53 were represented in 43.1% of METex14 (67.5% WT) and 5.4% of METex14 were associated with POT1 mutation (1.4% WT). Furthermore, 2.7% of patients with METex14 had MET co-amplifications (copy number alteration [CNA] ≥ 6), of which 0.19% also had EGFR alterations compared with 0.12% in MET WT, which may represent mechanisms of EGFR resistance. The most common co-alteration that distinguished METex14 from WT were co-amplifications (CNA ≥6) in three genes mapping to chromosome 12q14–15–MDM2 (12q15) (19.0% versus 1.8% WT), HMGA2 (12q14.3) (13.2% versus 0.98% WT), and CDK4 (12q14.1) (10.0% versus 1.5% WT) (Fig. 1D). Frequent co-amplification of MDM2 in METex14 compared with MET WT suggests MDM2 as an important event in MET skipping variants. As expected, MDM2 amplification was mutually exclusive to TP53 alterations (Fig. 1E). No considerable differences in co-alterations between WES and 592 gene panel platforms were observed in our cohort (Supplementary Fig. 1).
The functional relevance of co-amplification of MDM2, CDK4, and HMGA2 were further assessed by correlation with RNA expression. As anticipated, MDM2 amplification correlated with higher MDM2 expression levels than in nonamplified cases and also higher CDK4 and HMGA2 expression (Fig. 2A). Amplification of CDK4 was also associated with higher CDK4, MDM2, and HMGA2 expression and HMGA2 amplification correlated with increased expression of HMGA2, MDM2, and CDK4 (Supplementary Fig. 2). In contrast to MDM2 and CDK4 in which amplification revealed positive correlation with expression, HMGA2 amplification was not associated with expression (Fig. 2B). The correlation between gene amplification and expression in MDM2 and CDK4 and lack of correlation in HMGA2 may point toward MDM2 and CDK4 as common and functionally relevant co-oncogenic events in METex14 tumors. CDK4 co-amplification resulted in relatively high MDM2 expression regardless of MDM2 co-amplification in METex14, which may suggest CDK4 to be involved in regulation of MDM2 expression (Supplementary Fig. 3).
Figure 2.

(A) mRNA expression of MDM2, CDK4, and HMGA2 on the basis of MDM2 co-amplification in METex14 and MET WT (y axis, log2-transformed TPM; error bars represented interquartile range and median presented). (B) Oncoprint of MDM2, CDK4, and HMGA2 represented with mRNA expression levels. Red indicates higher expression and blue lower expression. (C) MET expression on the basis of METex14 mutation subtype (left) and MET co-amplification (right). (D) Ratios of METex14 mutation junction reads to WT junction reads in METex14/Amp+ versus METex14/Amp- (left) and Spearman correlation (right) of MET expression and ratio of METex14 junction reads to WT junction reads (gray dots: METex14/Amp-; red dots: METex14/Amp+). (E) Oncoprint of METex14 and MET co-amplification represented against MET expression and METex14 junction reads. Amp, amplification; METex14, MET exon 14; TPM, Trusted Platform Module; WT, wild type.
MET mRNA Expression on the Basis of Alteration Type
A 3-fold increase in MET RNA expression in METex14 was observed compared with MET WT, with most splice site alteration subtypes (donor splice site base substitutions, donor splice site deletion, polypyrimidine site deletion, and acceptor splice site deletion) translating to significantly increased MET expression in (all q < 0.01) (Fig. 2C, left). A trend toward increased mRNA expression was observed in indels at splice donor site, polypyrimidine tract, and splice acceptor site (p < 0.01). There was no difference in MET expression between splice acceptor site base substitution and WT, which may suggest differential expression of MET on the basis of mutation subtype. Strikingly, co-amplification of MET with METex14 was observed to have a 24-fold increase in MET expression compared with double WT (METex14 WT and MET nonamplified tumor), with 13-fold increase in expression with MET amplification alone and a threefold increase in expression with METex14 (Fig. 2C, right). Within the MET co-amplified tumors, the METex14 allelic variant was preferentially expressed (p < 0.01, Fig. 2D and E), on the basis of increased number of METex14 junction reads compared with WT junction reads. In patients with available prognostic data, MET amplification was observed to be associated with worse overall survival compared with METex14 (Fig. 3).
Figure 3.

(A) Kaplan-Meier plot of overall survival (date of tissue collection to last contact) of METex14 patients with and without amplification. (B) Kaplan-Meier plot of overall survival (date of tissue collection to last contact) of MET WT patients with and without amplification. (C) Kaplan-Meier plot of overall survival (date of tissue collection to last contact) of MET-amplified patients with and without METex14. (D) Kaplan-Meier plot of overall survival (date of tissue collection to last contact) of non–MET-amplified patients with and without METex14. HR, hazard ratio; CI, confidence interval; METex14, MET exon 14; WT, wild type.
Prognosis of Patients Treated With Crizotinib Based on Mutation Subtype
Among 25 patients whose treatment data with crizotinib were available, patients with donor splice site alterations were observed to have improved prognosis compared with patients with acceptor site alterations using start of treatment to last of contact (hazard ratio = 0.24, p = 0.006), suggesting possible differential response to treatment on the basis of mutation subtype (Supplementary Fig. 4). Co-alterations in copy number alteration of WIF1, CDKN2A, and RB1 were more frequent in acceptor site alterations treated with crizotinib and no difference in immunogenic signatures was observed, though evaluation is limited by a small sample size (Supplementary Fig. 5). No difference in prognosis was observed between all patients, either treated or untreated, with donor splice site and acceptor splice site mutations, which may suggest mutation subtype as a potential predictive, but not a prognostic biomarker.
PD-L1 and TMB
Of 20,694 patients with available PD-L1 IHC data, nearly twice as many patients in the METex14 cohort had a high PD-L1 more than or equal to 50% (52.5% versus 27.3% WT, q < 0.0001) with similar proportions observed in the PD-L1 1% to 49% (30.0% versus 29.7% WT) (Fig. 4A, left). Of 21,152 patients with TMB status, lower proportion of METex14 patients had a high TMB (≥10 mut/Mb), (8.3% versus 36.7% WT) with median TMB, 4 mut/Mb METex14 and 7 mut/Mb WT (q < 0.0001) (Fig. 4A, right). A similar pattern of lower proportion of patients having median TMB greater than or equal to 15 mut/Mb and greater than or equal to 20 mut/Mb were observed in METex14 compared with WT (Supplementary Fig. 6). In addition, of seven METex14 patients with high TMB who had smoking status available, none were non-smokers (Fig. 4B) and of 2402 WT patients with high TMB, only 13 (0.5%) were non-smokers, highlighting high TMB correlating with smoking status. Every METex14 tumor with high TMB fell into the high PD-L1 greater than or equal to 50% strata.
Figure 4.

(A) METex14 and MET WT patients stratified by PD-L1 less than 1%, 1% to 49%, and greater than or equal to 50% (left). Percentage of patients with high TMB (>10 mut/Mb) in METex14 and MET WT cohorts (right). (B) Oncoprint of METex14 with smoking history against TMB, PD-L1, IFN-γ signature, and T-cell inflammation signature. (C) IFN-γ signature (left) and T-cell inflammation signature (right) in METex14 and MET WT. (D) mRNA expression of immune checkpoints in METex14, MET WT, KRAS mutant, and EGFR mutant cohorts. IFN-γ, interferon-γ; METex14, MET exon 14; mut/Mb, mutations per megabase; PD-L1, programmed death-ligand 1; WT, wild type.
Immune Infiltrate Signature
A previous proof-of-principle study revealed that an 18-gene mRNA signature associated with IFN-γ and T-cell inflammation predicted response to immunotherapy across multiple tumor types, and larger immunogenic gene signatures (160-gene) have also been established.25,26 Variable outcomes with immunotherapy in METex14 as reported in the literature despite association with high PD-L1 prompted evaluation of tumor microenvironment (TME) in METex14 and WT subsets using the IFN-γ (18-gene) and T-cell inflammation signatures (160-gene) at the transcriptomic level. We found significant enrichment of IFN-γ and T-cell inflammation signatures in METex14 compared with WT (Fig. 4C). There was no significant difference in IFN-γ or T-cell inflammation signatures on the basis of smoking or high TMB (≥10 mut/Mb) status within METex14 which suggests smoking or TMB to not be confounding factors in explaining differences in inflammatory signatures (Supplementary Fig. 7A and B). Significantly higher IFN-γ signatures were observed with increasing PD-L1 expression in METex14 and MET WT (Supplementary Fig. 7C), with the difference in IFN-signatures more significant in the PD-L1 greater than or equal to 50% subgroups (Supplementary Fig. 7D). Moreover, METex14 also displayed significantly higher immune cell infiltrates of macrophage M1, macrophage M2, CD4+ T-cells, CD8+ T-cells, regulatory T-cells, and dendritic cells compared with WT (Supplementary Fig. 8).
Higher expression of IFN-γ previously revealed inhibition of immune activation through a negative feedback loop,28 and multiple genes involved in immune checkpoints in addition to CD274 (PD-L1), including CD80, CD86, CTLA4, HAVCR2 (TIM-3), LAG3, IDO1, PDCD1, and PDCD1LG2 were also more up-regulated in METex14 compared with WT (Fig. 4D). This increase in immune checkpoints was observed when compared against KRAS and EGFR-mutant cohorts suggesting a more immunosuppressive TME with METex14 compared with alternate driver mutations. A similar trend of higher PD-L1, lower TMB, higher IFN-γ/T-cell inflammation signatures, and higher immunosuppressive checkpoints was also observed in METex14 when compared with a cohort of pan-WT patients, who did not harbor co-alterations in EGFR, KRAS, ALK, and ROS-1, which could potentially affect immune TME (Supplementary Figs. 6 and 9). Strong association of higher IFN-γ and T-cell inflammation signatures in METex14 irrespective of smoking status or TMB and increased expression of multiple immune checkpoints suggest both an inflammatory and an immunosuppressive TME in METex14.
ssGSEA Pathway Analysis
To further explore differentially regulated pathways in METex14, we performed ssGSEA analysis and observed a consistent up-regulation of pathways involved in inflammatory response. In addition to up-regulation of IFN-γ, pathways involved in EMT, angiogenesis, and apical junction pathways were found to be enriched in METex14 on univariate analysis (Fig. 5A). When stratified by histology, EMT signature had a trend toward higher association with non-adenocarcinoma histologies (squamous, adenosquamous, and sarcomatoid) (Supplementary Fig. 10). mRNA expression of individual markers involved in invasion and metastases of MET, including PLAU, SERPINE1, CSF2, MMP3, EFNB2, PLAUR, IL1A, CXCL2, and VEGFC, which were up-regulated in our previously established preclinical cell-based models of METex1418 was also found to be significantly overexpressed in METex14 patients (q < 0.00001 for all) (Fig. 5B). An up-regulation in tumor necrosis factor in METex14 was observed, which was not observed in our previous preclinical model.18 On 70 cases with data available for logistic regression on multivariate analysis, apical junction pathway was observed to be associated with METex14 (p < 0.05). These up-regulated pathways and genes highlight several potential vulnerabilities that can be explored for potential therapeutic benefit.
Figure 5.

(A) ssGSEA pathway analysis of METex14 patients. (B) mRNA expression of genes previously found to be up-regulated in METex14 in in vitro models in METex14 and MET WT cohorts. METex14, MET exon 14; mut/Mb, mutations per megabase; FC, fold change; ssGSEA, single-sample gene enrichment analysis; WT, wild type.
Discussion
We present the most comprehensive characterization of METex14 alterations to date encompassing 533 METex14 tumors using WTS data. Similar to previous cohorts identified through DNA genomic profiling, patients with METex14 skipping were predominantly female, older, and enriched in sarcomatoid and adenosquamous histologies.3,4,9,13,29,30 This is in contrast to other driver alterations such as EGFR and ALK, which are associated with younger age and adenocarcinoma, and suggests the importance of unbiased NGS testing in patients irrespective of age or adenocarcinoma histology.31 Association of smoking status with METex14 has previously widely varied,3,4,9,13,29,32 and in our limited cohort of patients whose smoking status was available, METex14 patients were predominantly of light smokers (<15 pack years) or nonsmokers, though data were lacking on previous heavy (>15 pack years) smokers.
The frequency and subtype of genomic alterations of METex14 were also comparable to previous DNA profiling data, with METex14 identified in 2.47% by WTS and the donor splice site base substitutions as the most common alteration leading to a skipped variant.2,3,9,11,17,32 Approximately 11% of patients with a skipped variant had no associated DNA alteration, which may be secondary to underlying long deletions not captured owing to limitations in detection, and support RNA-based testing as a more sensitive assay for METex14.15,16 Near mutual exclusivity was observed with KRAS and EGFR mutations supporting METex14 as an independent oncogenic driver,2,9 and co-mutations were common in TP53 and POT1. POT1 (protection of telomeres 1), responsible for telomere maintenance, is enriched in pulmonary sarcomatoid subtypes, and its co-alteration with METex14 may represent histologic association.33
A novel observation of differential MET expression on the basis of mutation subtype was reported in our study, with at least three-fold increase in expression in nearly all mutation subtypes, except for point mutations in splice acceptor site. Although crizotinib,11 capmatinib,10 and tepotinib12 have not revealed disparate treatment response on the basis of mutation subtypes, molecular characterization among splicing mutations in these studies was more broadly categorized and not uniformly defined across studies. Thus, whether observed differences in mRNA expression on the basis of splicing mutation subtypes may have treatment implications requires further understanding. A more striking finding in our study was that in cases with MET co-amplification (CNA > 6), a near 24-fold increase in MET expression was found, higher than 14-fold increase with MET amplification alone and three-fold increase with METex14 alone, suggesting synergistic MET expression with MET co-amplification. The clinical significance of this increased MET expression with MET co-amplification may be both prognostic and therapeutic. MET amplification was previously associated with strong c-MET expression and poor prognosis, also revealed in our study on the basis of worse survival curves in patients with MET amplification.9 Both capmatinib10 and crizotinib34 were found to have an increased response rate with high levels of MET amplification and support high-level MET amplification as a possible prognostic and predictive biomarker. Interestingly, co-amplification of MET with METex14 did not have an increased response to capmatinib,10 but it did have high response rates of more than 60% to tepotinib.12 Nevertheless, levels of co-amplification were not reported in these studies. Whether high-level co-amplification, which correlates with higher mRNA expression and presumably higher protein expression, that in a previous study revealed improved response to MET tyrosine kinase inhibitor (TKI),35 can serve as a predictive biomarker, warrants further investigation.
Dysregulation of p53 has well been associated in lung cancer.36 Inactivation of p53 can occur in multiple ways, including mutation of TP53 or inactivation of WT p53, such as through MDM2 amplification, a negative regulator of p53. Our study revealed a significantly higher MDM2 amplification in METex14 which was also observed in previous DNA profiling studies.2,4,13,17 As anticipated, MDM2 co-amplification was mutually exclusive to TP53 mutation and despite lower TP53 mutations found in METex14 compared with MET WT, the high combined proportion of patients having either mutant TP53 (43.08%) or MDM2 amplification (19.03%) in METex14 highlights impaired TP53 pathway as a crucial actionable target and driver of oncogenesis in METex14. Co-amplifications in HMGA2 was not previously reported and its frequent co-amplification with METex14 highlights the role of HMGA2, which promotes EMT through transforming growth factor-β/Smads, PI3K/AKT, and Wnt/B-catenin pathways,37 as a functionally relevant gene important in the invasive properties of METex14.
Interestingly, the most frequent co-amplified genes, MDM2, CDK4, and HMGA2, all co-localized to chromosome 12q14–15. MDM2 and CDK4 seemed co-dependent of each other, on the basis of increased expression of CDK4 with MDM2 amplification and increased expression of MDM2 with CDK4 amplification. CDK4 amplification resulted in high MDM2 expression regardless of MDM2 amplification in METex14, and this finding could suggest CDK4 as an important regulator of MDM2. Both MDM2 inhibitors and CDK4/6 inhibitors have antitumor activity in alternate tumor types when combined with chemotherapy,38 targeted therapy,39,40 and hormonal therapy,41 and reveal potential novel combination treatment strategies with MET TKI in METex14 patients with MDM2 or CDK4 co-amplification.
Despite high PD-L1 expression, patients with METex14 were found to have in published series low to modest responses to immunotherapy.13,14,42,43 Low TMB in METex14 compared with WT was observed in our cohort, in which only 8% of METex14 had high TMB (≥10 mut/Mb) and may possibly explain lower than expected responses to immunotherapy in METex14 despite high PD-L1 expression. Furthermore, higher expression of immunosuppressive checkpoints was observed in METex14 compared with WT, contributing to potential bypass pathways conferring resistance to anti–PD-1/PD-L1. Interestingly, METex14 was also observed to have higher IFN-γ/T-cell inflammatory gene signatures compared with WT, and both an up-regulation of inflammatory and immunosuppressive TME in patients previously known to only have modest responses to immunotherapy could suggest immune suppression as a dominating mechanism in METex14. On the basis of promising data of combination checkpoint inhibitors in NSCLC,44 combination immunotherapy with MET TKI may be potential therapeutic strategies in overcoming anti–PD-L1/PD-1 resistance.
Last, consistent with our previous preclinical studies,18 an up-regulation of genes related to cellular adhesion, extracellular matrix disassembly, and angiogenesis, mechanisms of invasion and metastases in METex14 were also up-regulated in our METex14 cohort by pathway analysis. Tumor angiogenesis is hypothesized to promote an immunosuppressive environment45 and may serve as an additional factor in immunotherapy resistance in METex14. Efficacy of dual driver-alteration and vascular endothelial growth factor (VEGF)-inhibition in NSCLC have been found with first-line EGFR inhibitor, erlotinib,46,47 and in preclinical studies with c-MET and VEGF/vascular epithelial growth factor receptor (VEGFR) in epithelial cancers.48 Combination anti-VEGF therapy and MET-directed TKI with or without immunotherapy may thus be potential alternate treatment strategies in improving treatment outcomes in METex14 which rely on angiogenesis for invasion and metastases.
Limitations of our study include the inclusion of alternate driver mutations in METex14 and MET WT, which may confound results; however, most cohort designated as WT did not have an alternate driver mutation and the focus of the study was to characterize METex14 against samples without METex14. In addition, although MET mRNA expression was differentially up-regulated with distinct METex14 mutation subtypes and synergistically increased with MET co-amplification, whether this translates into increased c-MET expression and improved response to novel MET TKI remains to be further characterized with IHC and detailed treatment data. The variability of time in tissue collection, pretreatment versus post-treatment, also may confound characterization of co-alterations or results on immune TME because co-alterations may represent bypass resistance mechanisms rather than de novo co-mutations and up-regulation of checkpoint inhibition may be a response to immunotherapy, respectively, in those treated with immunotherapy. Overall, an improved understanding of METex14 subsets is needed to select patients who may have improved responses to MET TKI, and for those patients with suboptimal response, further research on newer combination treatment strategies such as MET TKI with anti-VEGF, selective small molecule inhibitors of MDM-2 and CDK4/6, or with combination checkpoint inhibitors are needed.
CRediT Authorship Contribution Statement
So Yeon Kim: Conceptualization, Methodology, Analysis, Investigation, Original draft preparation, Writing—review and editing.
Jun Yin: Data curation, Methodology, Analysis, Software, Investigation, Visualization, Writing—review and editing.
Stephen Bohlman: Analysis.
Phillip Walker, Sanja Dacic, Chul Kim, Hina Khan, Stephen V. Liu, Patrick C. Ma, Misako Nagasaka, Karen L. Reckamp, Jim Abraham, Dipesh Uprety, Joanne Xiu: Writing—review and editing.
Feng Wang: Conceptualization.
Jian Zhang: Data curation, Investigation.
Haying Cheng: Writing—review and editing, Supervision.
Balazs Halmos: Conceptualization, Methodology, Analysis, Investigation, Writing—review and editing, Supervision.
Footnotes
Drs. Kim and Yin contributed equally as co-first authors.
Disclosures: Dr. Dacic has received consulting fees from AstraZeneca, has received honoraria from Takeda and Merck, and participates in the Pulmonary Pathology Society. Dr. Kim has received institutional grants from AstraZeneca, Bristol-Myers Squibb, Novartis, Regeneron, Karyopharm, Debiopharm, Janssen, Genentech, Spectrum, and Merck and has received consulting fees from AstraZeneca, Novartis, Janssen, PierianDx, Sanofi, Diffusion Pharmaceuticals, Mirati, and Jazz Pharmaceuticals. Dr. Khan has received a grant from Bristol-Myers Squibb/Winn CDA and consulting fees from Sanofi-Genzyme. Dr. Liu has received grants from Alkermes, Bayer, Blueprint, Bristol-Myers Squibb, Elevation Oncology, Genentech, Gilead, Merck, Merus, Nuvalent, Pfizer, Rain Therapeutics, RAPT, and Turning Point Therapeutics; has received consulting fees from Amgen, AstraZeneca, Bayer, Beigene, Blueprint, Boehringer Ingelheim, Bristol-Myers Squibb, Catalyst, Daiichi Sankyo, Eisai, Elevation Oncology, Genentech/Roche, Gilead, Guardant Health, Janssen, Jazz Pharmaceuticals, Lilly, Merck/Merck Sharp & Dohme, Novartis, Regeneron, Sanofi, Takeda, and Turning Point Therapeutics; and took part in data safety monitoring for Candel Therapeutics. Dr. Ma has received institutional grants from Merck, Genentech-Roche, AbbVie, Apollomics, OncoC4, Genmab, BeiGene, Mirati, and Elevation Oncology; has received consulting fees and honoraria from AstraZeneca; and is a Steering Committee Member for Big Ten Cancer Research Consortium. Dr. Nagasaka has received consulting fees from AstraZeneca, Caris Life Sciences, Daiichi Sankyo, Novartis, EMD Serono, Janssen, Pfizer, Lilly, Genentech, and Mirati; honoraria from Takeda and Blueprint; and support for attending meetings from AnHeart. Dr. Reckamp has received institutional grants from Genentech, Blueprint, Calithera, Daiichi Sankyo, Elevation Oncology, and Janssen and consulting fees from Amgen, AstraZeneca, Blueprint, Daiichi Sankyo, EMD Serono, Genentech, GlaxoSmithKline, Janssen, Lilly, Merck KGA, Mirati, Seattle Genetics, Takeda, and Tesaro. Dr. Uprety has received consultant fees from AstraZeneca, Daiichi Sankyo, and Sanofi. Dr. Halmos has received grants from Boehringer Ingelheim, AstraZeneca, Merck, Bristol-Myers Squibb, Advaxis, Amgen, AbbVie, Daiichi, Pfizer, GlaxoSmithKline, Beigene, and Janssen; has received consultant fees from Veracyte; and is part of the advisory board for AstraZeneca, Boehringer Ingelheim, Apollomics, Janssen, Takeda, Merck, Bristol-Myers Squibb, Genentech, Pfizer, Eli-Lilly, and TPT. The remaining authors declare no conflict of interest.
Cite this article as: Kim SY, Yin J, Bohlman S, et al. Characterization of MET exon 14 skipping alterations (in NSCLC) and identification of potential therapeutic targets using whole transcriptome sequencing. JTO Clin Res Rep. 2022;3:100381.
Note: To access the supplementary material accompanying this article, visit the online version of the JTO Clinical and Research Reports at www.jtocrr.org and at https://doi.org/10.1016/j.jtocrr.2022.100381.
Supplementary Data
References
- 1.Ma P.C., Jagadeeswaran R., Jagadeesh S., et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005;65:1479–1488. doi: 10.1158/0008-5472.CAN-04-2650. [DOI] [PubMed] [Google Scholar]
- 2.Frampton G.M., Ali S.M., Rosenzweig M., et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5:850–859. doi: 10.1158/2159-8290.CD-15-0285. [DOI] [PubMed] [Google Scholar]
- 3.Schrock A.B., Frampton G.M., Suh J., et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thorac Oncol. 2016;11:1493–1502. doi: 10.1016/j.jtho.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Awad M.M., Oxnard G.R., Jackman D.M., et al. MET exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-met overexpression. J Clin Oncol. 2016;34:721–730. doi: 10.1200/JCO.2015.63.4600. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y., Xia M., Jin K., et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol Cancer. 2018;17:45. doi: 10.1186/s12943-018-0796-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kong-Beltran M., Seshagiri S., Zha J., et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006;66:283–289. doi: 10.1158/0008-5472.CAN-05-2749. [DOI] [PubMed] [Google Scholar]
- 7.Vuong H.G., Ho A.T.N., Altibi A.M.A., Nakazawa T., Katoh R., Kondo T. Clinicopathological implications of MET exon 14 mutations in non-small cell lung cancer—a systematic review and meta-analysis. Lung Cancer. 2018;123:76–82. doi: 10.1016/j.lungcan.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 8.Yeung S.F., Tong J.H.M., Law P.P.W., et al. Profiling of oncogenic driver events in lung adenocarcinoma revealed MET mutation as independent prognostic factor. J Thorac Oncol. 2015;10:1292–1300. doi: 10.1097/JTO.0000000000000620. [DOI] [PubMed] [Google Scholar]
- 9.Tong J.H., Yeung S.F., Chan A.W., et al. MET amplification and exon 14 splice site mutation define unique molecular subgroups of non-small cell lung carcinoma with poor prognosis. Clin Cancer Res. 2016;22:3048–3056. doi: 10.1158/1078-0432.CCR-15-2061. [DOI] [PubMed] [Google Scholar]
- 10.Wolf J., Seto T., Han J.Y., et al. Capmatinib in MET exon 14–mutated or MET-amplified non–small-cell lung cancer. N Engl J Med. 2020;383:944–957. doi: 10.1056/NEJMoa2002787. [DOI] [PubMed] [Google Scholar]
- 11.Drilon A., Clark J.W., Weiss J., et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat Med. 2020;26:47–51. doi: 10.1038/s41591-019-0716-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paik P.K., Felip E., Veillon R., et al. Tepotinib in non–small-cell lung cancer with MET exon 14 skipping mutations. N Engl J Med. 2020;383:931–943. doi: 10.1056/NEJMoa2004407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee J.K., Madison R., Classon A., et al. Characterization of non–small-cell lung cancers with MET exon 14 skipping alterations detected in tissue or liquid: clinicogenomics and real-world treatment patterns. JCO Precis Oncol. 2021;5 doi: 10.1200/PO.21.00122. PO.21.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sabari J.K., Leonardi G.C., Shu C.A., et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann Oncol. 2018;29:2085–2091. doi: 10.1093/annonc/mdy334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies K.D., Lomboy A., Lawrence C.A., et al. DNA-based versus RNA-based detection of MET exon 14 skipping events in lung cancer. J Thorac Oncol. 2019;14:737–741. doi: 10.1016/j.jtho.2018.12.020. [DOI] [PubMed] [Google Scholar]
- 16.Benayed R., Offin M., Mullaney K., et al. High yield of RNA sequencing for targetable kinase fusions in lung adenocarcinomas with no mitogenic driver alteration detected by DNA sequencing and low tumor mutation burden. Clin Cancer Res. 2019;25:4712–4722. doi: 10.1158/1078-0432.CCR-19-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Awad M.M., Lee J.K., Madison R., et al. Characterization of 1,387 NSCLCs with MET exon 14 (METex14) skipping alterations (SA) and potential acquired resistance (AR) mechanisms. J Clin Oncol. 2020;38(suppl 15) 9511–9511. [Google Scholar]
- 18.Wang F., Liu Y., Qiu W., et al. Functional analysis of MET exon 14 skipping alteration in cancer invasion and metastatic dissemination. Cancer Res. 2022;82:1365–1379. doi: 10.1158/0008-5472.CAN-21-1327. [DOI] [PubMed] [Google Scholar]
- 19.Richards S., Aziz N., Bale S., et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horak P., Griffith M., Danos A.M., et al. Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): joint recommendations of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC) Genet Med. 2022;24:986–998. doi: 10.1016/j.gim.2022.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Merino D.M., McShane L.M., Fabrizio D., et al. Establishing guidelines to harmonize tumor mutational burden (TMB): in silico assessment of variation in TMB quantification across diagnostic platforms: phase I of the Friends of Cancer Research TMB Harmonization Project. J Immunother Cancer. 2020;8 doi: 10.1136/jitc-2019-000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subramanian A., Tamayo P., Mootha V.K., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mootha V.K., Lindgren C.M., Eriksson K.F., et al. PGC-1 α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 25.Ayers M., Lunceford J., Nebozhyn M., et al. IFN-γ–related Mrna profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127:2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spranger S., Luke J.J., Bao R., et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci U S A. 2016;113:E7759–E7768. doi: 10.1073/pnas.1609376113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finotello F., Mayer C., Plattner C., et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019;11:34. doi: 10.1186/s13073-019-0638-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bellucci R., Martin A., Bommarito D., et al. Interferon-γ-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology. 2015;4 doi: 10.1080/2162402X.2015.1008824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim E.K., Kim K.A., Lee C.Y., et al. Molecular diagnostic assays and clinicopathologic implications of MET exon 14 skipping mutation in non-small-cell lung cancer. Clin Lung Cancer. 2019;20:e123–e132. doi: 10.1016/j.cllc.2018.10.004. [DOI] [PubMed] [Google Scholar]
- 30.Rahouma M., Kamel M., Narula N., et al. Pulmonary sarcomatoid carcinoma: an analysis of a rare cancer from the Surveillance, Epidemiology, and End Results database. Eur J Cardiothorac Surg. 2017;53:828–834. doi: 10.1093/ejcts/ezx417. [DOI] [PubMed] [Google Scholar]
- 31.Sacher A.G., Dahlberg S.E., Heng J., Mach S., Jänne P.A., Oxnard G.R. Association between younger age and targetable genomic alterations and prognosis in non–small-cell lung cancer. JAMA Oncol. 2016;2:313–320. doi: 10.1001/jamaoncol.2015.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Le X., Hong L., Hensel C., et al. Landscape and clonal dominance of co-occurring genomic alterations in non–small-cell lung cancer harboring MET exon 14 skipping. JCO Precis Oncol. 2021;5 doi: 10.1200/PO.21.00135. PO.21.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen E., Xiu J., Bentley R., López G.Y., Walsh K.M. Frequent mutations of POT1 distinguish pulmonary sarcomatoid carcinoma from other lung cancer histologies. Clin Lung Cancer. 2020;21:e523–e527. doi: 10.1016/j.cllc.2020.04.002. [DOI] [PubMed] [Google Scholar]
- 34.Camidge D.R., Otterson G.A., Clark J.W., et al. Crizotinib in patients with MET amplified NSCLC. J Thorac Oncol. 2021;16:1017–1029. doi: 10.1016/j.jtho.2021.02.010. [DOI] [PubMed] [Google Scholar]
- 35.Guo R., Offin M., Brannon A.R., et al. MET Exon 14-altered lung cancers and MET inhibitor resistance. Clin Cancer Res. 2021;27:799–806. doi: 10.1158/1078-0432.CCR-20-2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Momand J., Jung D., Wilczynski S., Niland J. The MDM2 gene amplification database. Nucleic Acids Res. 1998;26:3453–3459. doi: 10.1093/nar/26.15.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang S., Mo Q., Wang X. Oncological role of HMGA2 (review) Int J Oncol. 2019;55:775–788. doi: 10.3892/ijo.2019.4856. [DOI] [PubMed] [Google Scholar]
- 38.Konopleva M.Y., Röllig C., Cavenagh J., et al. Idasanutlin plus cytarabine in relapsed or refractory acute myeloid leukemia: results of the MIRROS trial. Blood Adv. 2022;6:4147–4156. doi: 10.1182/bloodadvances.2021006303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu M., Xu S., Wang Y., et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, sensitizes lung cancer cells to treatment with epidermal growth factor receptor tyrosine kinase inhibitors. Oncotarget. 2016;7:84951–84964. doi: 10.18632/oncotarget.13069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y., Li X., Liu X., et al. Simultaneous inhibition of PI3Kα and CDK4/6 synergistically suppresses KRAS-mutated non-small cell lung cancer. Cancer Biol Med. 2019;16:66–83. doi: 10.20892/j.issn.2095-3941.2018.0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hortobagyi G.N., Stemmer S.M., Burris H.A., et al. Overall survival with ribociclib plus letrozole in advanced breast cancer. N Engl J Med. 2022;386:942–950. doi: 10.1056/NEJMoa2114663. [DOI] [PubMed] [Google Scholar]
- 42.Mazieres J., Drilon A., Lusque A., et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol. 2019;30:1321–1328. doi: 10.1093/annonc/mdz167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guisier F., Dubos-Arvis C., Viñas F., et al. Efficacy and safety of anti-PD-1 immunotherapy in patients with advanced NSCLC with BRAF, HER2, or MET mutations or RET translocation: GFPC 01-2018. J Thorac Oncol. 2020;15:628–636. doi: 10.1016/j.jtho.2019.12.129. [DOI] [PubMed] [Google Scholar]
- 44.Paz-Ares L., Ciuleanu T.E., Cobo M., et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): an international, randomized, open-label, phase 3 trial. Lancet Oncol. 2021;22:198–211. doi: 10.1016/S1470-2045(20)30641-0. [DOI] [PubMed] [Google Scholar]
- 45.Huang Y., Goel S., Duda D.G., Fukumura D., Jain R.K. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013;73:2943–2948. doi: 10.1158/0008-5472.CAN-12-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakagawa K., Garon E.B., Seto T., et al. Ramucirumab plus erlotinib in patients with untreated, EGFR-mutated, advanced non-small-cell lung cancer (RELAY): a randomized, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20:1655–1669. doi: 10.1016/S1470-2045(19)30634-5. [DOI] [PubMed] [Google Scholar]
- 47.Kawashima Y., Fukuhara T., Saito H., et al. Bevacizumab plus erlotinib versus erlotinib alone in Japanese patients with advanced, metastatic, EGFR-mutant non-small-cell lung cancer (NEJ026): overall survival analysis of an open-label, randomized, multicentre, phase 3 trial. Lancet Respir Med. 2022;10:72–82. doi: 10.1016/S2213-2600(21)00166-1. [DOI] [PubMed] [Google Scholar]
- 48.Awazu Y., Nakamura K., Mizutani A., et al. A novel inhibitor of c-Met and VEGF receptor tyrosine kinases with a broad spectrum of in vivo antitumor activities. Mol Cancer Ther. 2013;12:913–924. doi: 10.1158/1535-7163.MCT-12-1011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.