

SUMMARY
Interleukin-1 receptor-associated kinases (IRAKs) −4, −2, and −1 are involved in transducing signals from Toll-like receptors (TLRs) via the adaptor myeloid differentiation primary-response protein 88 (MYD88). How MYD88/IRAK4/2/1 complexes are formed, their redundancies, and potential non-enzymatic roles are subjects of debate. Here, we examine the hierarchical requirements for IRAK proteins in the context of TLR4 activation and confirmed that the kinase activity of IRAK4 is essential for MYD88 signaling. Surprisingly, the IRAK4 scaffold is required for activation of the E3 ubiquitin ligase TNF receptor-associated factor 6 (TRAF6) by both MYD88 and TIR domain-containing adaptor protein inducing IFN-β (TRIF), a unique adaptation in the TLR4 response. IRAK4 scaffold is, therefore, essential in integrating MYD88 and TRIF in TLR4 signaling.
Graphical abstract
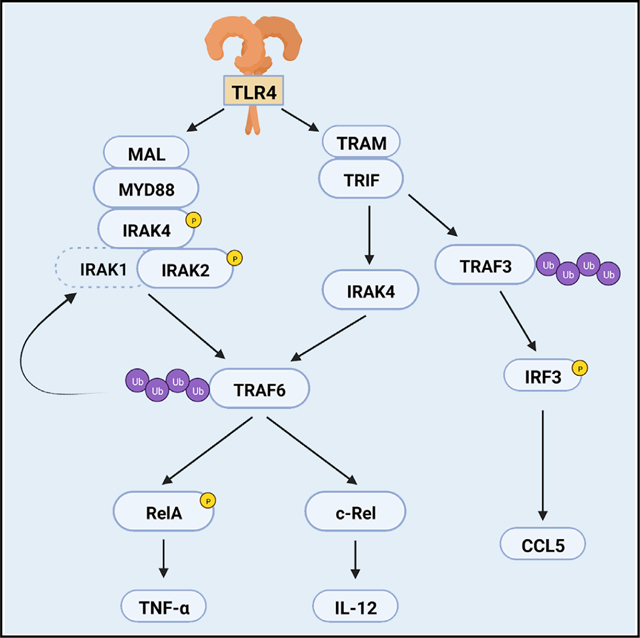
In brief
By studying the redundancies of IRAK proteins in Toll-like receptor (TLR) 4 and 7 signaling, Pereira et al. demonstrate a kinase-independent function for IRAK4 in the TRIF pathway. This provides an understanding of how TLR4 signaling triggers the production of inflammatory cytokines upon infections with Gram-negative bacteria.
INTRODUCTION
Toll-like receptors (TLRs) are pattern recognition receptors essential for protecting the host against infections (Gazzinelli and Denkers, 2006; O’Neill et al., 2013; Fitzgerald and Kagan, 2020). Upon interaction with their specific ligand, such as lipopolysaccharide (LPS) for TLR4 or single-stranded RNA for TLR7, TLRs signal via the adaptors myeloid differentiation primary-response protein 88 (MYD88) or TIR domain-containing adaptor protein inducing IFN-β (TRIF). MYD88 is the adaptor for all TLRs except TLR3, which uses TRIF (Fitzgerald and Kagan, 2020). TLR4 employs both MYD88 and TRIF in addition to bridging adaptors MYD88-adapter-like (MAL) and Trif-related adaptor molecule (TRAM) (Fitzgerald et al., 2001, 2003b; Kagan et al., 2008; Yamamoto et al., 2003b). The TLR/MYD88 interaction results in recruitment of interleukin-1 receptor-associated kinase (IRAK) 4, IRAK2, and IRAK1 (Lin et al., 2010), forming a multifunctional supramolecular organizing center termed myddosome, which regulates the activation of transcription factors such as AP-1 (via mitogen-activated protein kinases [MAPK]) and nuclear factor kappa B (NF-κB) (Fitzgerald and Kagan, 2020; Tan and Kagan, 2019; Tan et al., 2015). Similarly, TLR3 and TLR4 recruit TRIF to form the triffosome, which in addition to NF-κB, activates interferon regulatory factor (IRF) 3 via the ubiquitin ligase TNF receptor-associated factor 3 (TRAF3) (Fitzgerald and Kagan, 2020; Häcker et al., 2006; Kagan et al., 2008).
The myddosome activates the ubiquitin ligase TRAF6 (Cao et al., 1996), which initiates signaling events that result in NF-κB stimulation (Fitzgerald and Kagan, 2020; O’Neill et al., 2013). Some of these events are essential for transcription of NF-κB target genes. For instance, NF-κB is trapped in the cytosol in complex with inhibitor proteins, such as IκB-α, masking its nuclear localization sequence. Myddosome activation induces IκB-α phosphorylation and K48-ubiquitination and degradation, enabling NF-κB nuclear translocation (Silverman and Maniatis, 2001; Hayden and Ghosh, 2008). Other events have regulatory roles, such as phosphorylation at the NF-κB transactivation domain, which modulates its affinity to transcription enhancers (Chen and Greene, 2004). Different NF-κB proteins are involved in expression of a specific set of genes, such as RelA in TNF-α expression (Collart et al., 1990) and c-Rel in IL-12 (Sanjabi et al., 2000). TRIF and MYD88 signaling are integrated, so there is synergism in TLR4-mediated NF-κB activation. Both adaptors regulate cytokine expression (Adachi et al., 1998; Yamamoto et al., 2003a), but little is known about the molecular mechanisms that enables this integration. Downstream of TLR4, TRAF6 is the only known node that links MYD88 and TRIF to NF-κB (Cao et al., 1996; Sato et al., 2003), but how TRIF activates TRAF6 is not well understood.
How MYD88/IRAK-4/-2/-1 complexes are formed and the level of redundancy in their components remain subjects of debate. IRAK4 is essential for myddosome signaling, and it is assumed that Irak4−/− phenocopies MyD88−/− (Suzuki et al., 2002; Picard et al., 2003; Bernuth et al., 2008; Béla et al., 2012; von Bernuth et al., 2012; Pattabiraman et al., 2018). IRAK-1 and -2 show considerable overlap: either can be recruited to the myddosome (Lin et al., 2010), and their activities can be redundant (Kawagoe et al., 2008; Pauls et al., 2013). How IRAK4 kinase activity fits in this model is also unclear. Myddosome recruitment activates IRAK4 (Ferrao et al., 2014), which phosphorylates IRAKs 1 and 2, turning on their autophosphorylation activities (Cheng et al., 2007; Kawagoe et al., 2008). However, IRAK1 can auto-phosphorylate even when IRAK4 kinase activity is impaired (Qin et al., 2004), but it is unknown if IRAK2 has a similar mechanism. At endogenous protein levels, IRAK2 can be detected in the myddosome (Tan et al., 2015; Tan and Kagan, 2019), whereas IRAK1 recruitment is only detectable upon IRAK4 inhibition (De Nardo et al., 2018). This raises the possibility that enhanced IRAK1 recruitment compensates for the defective IRAK4 kinase activity. Macrophages deficient in IRAK4 kinase activity fail to produce pro-inflammatory cytokines in response to most TLR agonists, but TLR4-mediated cytokine production is only partially affected (Kawagoe et al., 2007; Pennini et al., 2013; Tae et al., 2007). Potentially there are IRAK1/2 compensatory mechanisms in cells expressing kinase-dead IRAK4 that require further investigation in the context of TLR4.
The current models are based on studies focused on receptors that signal through MYD88 only. The synergism between TRIF and MYD88 in TLR4 signaling is unique (Adachi et al., 1998; Gohda et al., 2004; Sasai et al., 2010; Sato et al., 2003; Verstak et al., 2014), thus generalizations based on other receptors may not apply to TLR4. Here we studied the hierarchical requirements for IRAKs 1, 2, and 4 (scaffold versus kinase) in TLR4-mediated NF-κB activation. We found that the IRAK4 scaffold integrates TRIF and MYD88 signaling at the point of TRAF6 activation. We have therefore identified a key signaling hub that coordinates MYD88 and TRIF in response to TLR4 activation.
RESULTS
IRAKs 1 and 2 are partially redundant in TLR4 but not in TLR7 signaling
Study of TLR2 signaling suggests that IRAKs 1 and 2 have redundant roles early, but late responses are IRAK2 dependent (Kawagoe et al., 2008). Here, we sought to investigate whether the same conclusions can be draw in the context of other TLRs. IRAK1 was shown dispensable for production of TNF-α and IL-12 upon stimulation with either LPS (TLR4 agonist) or R848 (TLR7 agonist). In contrast, at 6 and 24 h post-stimulation, IRAK2 deficiency resulted in complete and partial impairment of TNF-α and IL-12 responses to R848 and LPS, respectively. Importantly, up to 2 h post-stimulation, TNF-α and IL-12 produced by wild-type (WT), Irak1−/−, and Irak2−/− bone marrow-derived macrophages (BMDM) were similar (p > 0.05 one-way ANOVA) in LPS-stimulated cells. In R848-stimulated cells, however, Irak1−/− and Irak2−/− BMDMs produced similar levels of TNF-α early, but IL-12 production was deficient in Irak2−/− BMDMs. Little to no cytokine production was observed in Irak1−/−Irak2−/− cells (Figures 1A–1D). Hence, at later time points, IRAK1 and IRAK2 show some redundancy in TLR4 but not TLR7. Co-immunoprecipitation of MYD88 from cells stimulated with LPS or R848 for 60 min showed that both IRAK-1 and -2 are detectable in the myddosome, although IRAK1 is weakly detected and requires a long exposure (Figures 1E, 1F, and S1). Furthermore, in Irak1−/− BMDMs, IRAK2 is recruited to the myddosome unimpaired, whereas in LPS-, but not R848-stimulated cells, we observed increased recruitment of IRAK1 to the myddosome from Irak2−/− macrophages. Protein inputs from co-immunoprecipitation assays suggest that myddosome signaling is normal in the single knockout cells: myddosome activation leads to loss of IRAK1 signal and a shift in IRAK2 molecular weight (due its phosphorylation), events that are not observed in MyD88−/− BMDMs. Here, we observe that upon LPS and R848 stimulation, Irak1−/− macrophages have a shift in IRAK2 molecular weight, and Irak2−/− BMDMs lose the IRAK1 signal in its protein input (Figures 1E and 1F).
Figure 1. IRAK-1 and -2 have early redundancy upon TLR-4 and -7 stimulation.
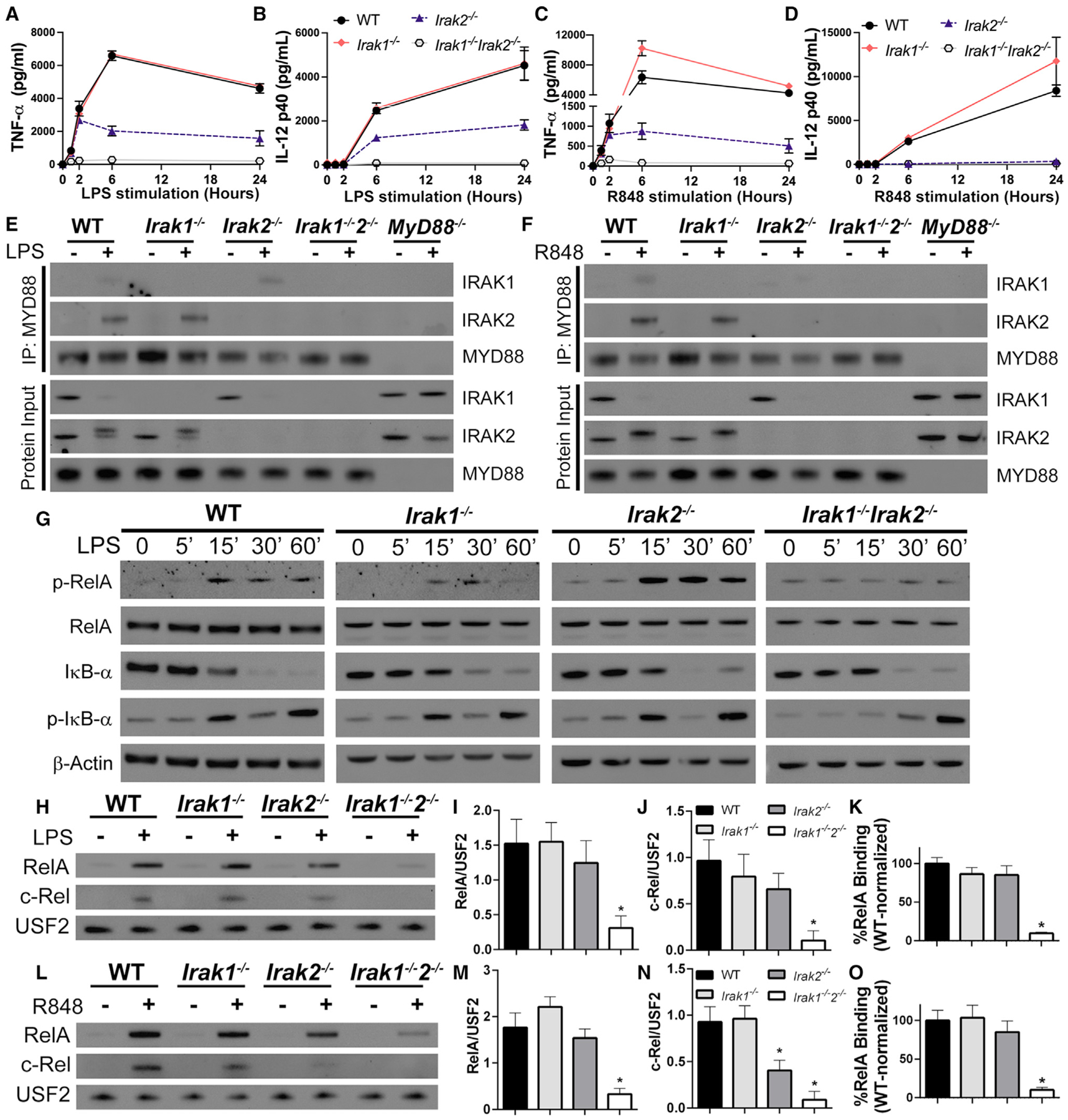
(A–D) Quantification of TNF-α and IL-12 produced by WT, Irak1−/−, Irak2−/−, and Irak1−/−Irak2−/− BMDMs treated for up to 24 h with LPS (A and B) or R848 (C and D).
(E and F) Immunoblot analysis of MYD88 co-immunoprecipitation with IRAK-1 and -2 in WT, Irak1−/−, Irak2−/−, Irak1−/−Irak2−/−, and MyD88−/− BMDMs treated for 60 min with LPS (E) or R848 (F).
(G) Kinetic study of p-RelA, RelA, IκB-α, p-IκB-α, and β-actin by immunoblot of whole cell lysates from indicated BMDMs treated with LPS for up to 60 min.
(H–O) Immunoblot analysis of RelA and c-Rel in nuclear lysates from WT, Irak1−/−, Irak2−/−, and Irak1−/−Irak2−/− unstimulated, stimulated with LPS for 30 min (H) or R848 for 15 min (L), densitometric analysis of stimulated samples (I, J, M, and N) and RelA DNA-binding affinity in nuclear extracts from BMDMs stimulated with LPS for 30 min (K) or R848 for 15 min (O). All stimulations were done with LPS 100 ng mL−1 or R848 1 μg mL−1. *p < 0.05 in comparison to WT (one-way analysis of variance with Tukey’s multiple comparisons test). (A–D, I–K, M–O) Data from three independent experiments (mean and SEM). (E–H, L) Images are representative of three independent experiments.
Signaling events downstream of TLR/MYD88 activation further suggest IRAK-1 and -2 early redundancy. No differences were observed in RelA phosphorylation, IκB-α phosphorylation and degradation, RelA nuclear translocation, RelA DNA-binding, and MAPK activation in either Irak1−/− or Irak2−/− BMDMs stimulated with LPS or R848 (Figures 1G–1O and S2). The redundancy between these two IRAKs becomes clear when these events are studied in Irak1−/−Irak2−/− BMDMs. These cells are severely impaired in NF-κB phosphorylation, nuclear translocation, and DNA-binding induced by either LPS or R848 (Figures 1G–1O and S2). JNK activation was also reduced in LPS-stimulated Irak1−/−Irak2−/− BMDMs, while ERK and p38 activation showed a small but not statistically significant decrease in activation (Figures S2G–S2K). Consistent with early IL-12 production, Irak2−/− macrophages were deficient in c-Rel nuclear translocation upon R848 stimulation, whereas LPS-stimulated macrophages translocated c-Rel normally (Figures 1H, 1I, 1K, and 1N). Interestingly, LPS- and R848-stimulated Irak1−/−Irak2−/− BMDMs showed IκB degradation despite lack of nuclear translocation and cytokine production (Figures 1 andS2). It is at the moment unclear how this might occur, but it is possible that deficiencies in NF-κB post-translational modifications impact nuclear import.
IRAK4 kinase activity is essential for signaling via TLR7, but not TLR4
Kinase-deficient IRAK4 (Irak4 Ki) macrophages still produce significant amounts of TNF-α and IL-12 in response to LPS, but not to other TLR ligands (Kawagoe et al., 2007; Pennini et al., 2013; Tae et al., 2007). Intriguingly, IRAK4 inhibition increases IRAK1 recruitment to the myddosome in response to LPS (De Nardo et al., 2018). These data suggest that enhanced recruitment of IRAK1 to the myddosome may explain why kinase-dead IRAK4 is capable of cytokine production. To further investigate the redundancies between IRAKs 1 and 2 in LPS-stimulated cells, we generated Irak1−/−Irak4 Ki and Irak2−/−Irak4 Ki mice. Stimulation with LPS for up to 24 h resulted in partial inhibition of TNF-α and IL-12 production in Irak4 Ki BMDMs and complete impairment in Irak4−/− cells (Figures 2A–2D). Similar cytokine levels were produced by Irak4 Ki, Irak1−/−Irak4 Ki, and Irak2−/−Irak4 Ki macrophages at all time points, further indicating IRAK1 and IRAK2 redundancy in response to LPS.
Figure 2. IRAK4 kinase activity is partially required for TLR4 signaling and essential for TLR7.
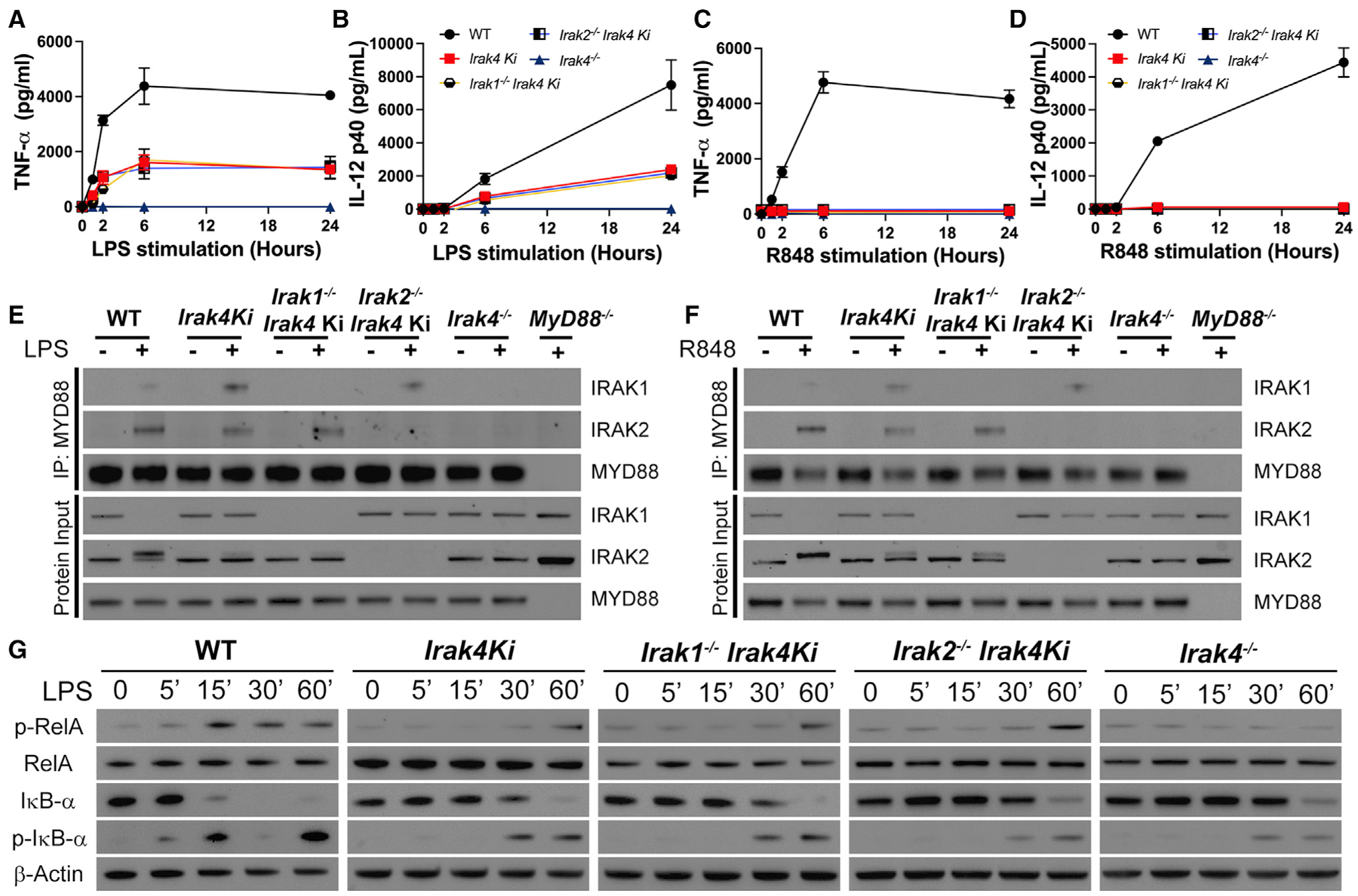
(A–D) Quantification of TNF-α (A, C) and IL-12 (B, D) produced after stimulation of WT, Irak4 Ki, Irak1−/−Irak4 Ki, Irak2−/−Irak4 Ki, and Irak4−/− BMDMs with LPS (A and B) or R848 (C and D) for up to 24 h.
(E and F) Immunoblot analysis of MYD88 co-immunoprecipitation with IRAK-1 and -2 in WT, Irak4 Ki, Irak1−/−Irak4 Ki, Irak2−/−Irak4 Ki, and Irak4−/− BMDMs treated for 60 min with LPS (E) or R848 (F).
(G) Kinetic study of p-RelA, RelA, IκB-α, p-IκB-α, and β-actin by immunoblot of whole cell lysates from indicated BMDMs treated with LPS for up to 60 min. All stimulations were done with LPS 100 ng mL−1 or R848 1 μg mL−1. *p < 0.05 in comparison with WT (one-way analysis of variance with Tukey’s multiple comparisons test). (A–D) Data from three independent experiments (mean and SEM). (E–G) Images are representative of three (E) or four (G) independent experiments.
To understand the hierarchy of IRAK recruitment to the myddosome, we immunoprecipitated MYD88 from Irak4 Ki BMDMs stimulated with either LPS or R848 for 60 min. While IRAK4 scaffold is required for IRAK1 and 2 recruitment (Kawagoe et al., 2007; Lin et al., 2010; Suzuki et al., 2002), loss of its kinase activity increased IRAK1 recruitment without affecting IRAK2, evidence that myddosome formation occurs independently of IRAK4 kinase activity. Protein inputs of Irak4 Ki BMDMs stimulated with either LPS or R848, however, revealed no IRAK1 loss of signal and deficient IRAK2 phosphorylation, suggesting that despite myddosome formation, IRAK4 kinase activity is required for signaling to occur (Figures 2E and 2F). Enhanced IRAK1 recruitment was also observed in the myddosome of Irak2−/−Irak4 Ki cells. As observed in IRAK1 single knockouts, Irak1−/−Irak4 Ki recruited IRAK2 to the myddosome similarly to WT controls. Collectively, the data confirms that the IRAK4 scaffold is sufficient for recruitment of IRAKs 1 and 2 to the myddosome, but their activation likely requires IRAK4 kinase activity. Importantly, the IRAK4 scaffold is essential for IL-12 and TNF-α production downstream of TLR4 (Figures 2A–2F).
Enhancement of RelA phosphorylation in WT cells can be observed as early as 15 min after LPS stimulation, whereas this was only evident after 60 min in either Irak4 Ki, Irak1−/−Irak4 Ki, or Irak2−/−Irak4 Ki macrophages. No RelA phosphorylation was observed in Irak4−/− BMDMs. IκB-α phosphorylation and degradation occurred slightly earlier in WT cells in comparison with IRAK4 mutant strains (Figure 2G).
IRAK4 kinase activity was, however, essential for TLR7 signaling. Irak4 Ki BMDMs stimulated with R848 for up to 24 h had severely deficient cytokine production (Figures 2C and 2D). Loss of IRAK4 kinase activity blocked RelA phosphorylation and delayed IκB-α phosphorylation and degradation, independently of IRAKs 1 or 2 (Figure S2). Irak4−/− BMDMs showed complete inhibition of RelA phosphorylation and IκB-α phosphorylation and degradation. As observed with LPS, Irak4 Ki BMDMs stimulated with R848 assemble a myddosome with increased IRAK1 and normal IRAK2 recruitment (Figure 2F), with no IRAK1 loss of signal nor IRAK2 phosphorylation in the protein input. These data suggest that IRAK1/2 cannot compensate for deficient IRAK4 kinase, and the myddosome assembled is deficient. The remaining cytokine output upon LPS stimulation, but not R848, further suggests a compensatory role of TRIF in LPS-stimulated cells.
IRAK4 scaffold is essential for NF-κB activation in TLR4-activated macrophages
Despite myddosome formation, no TNF-α and IL-12 are detected in Irak4 Ki BMDMs stimulated with R848, suggesting that without IRAK4 kinase activity the myddosome is non-functional. LPS-stimulated Irak4−/− BMDMs, however, fail to produce cytokines, but Irak4 Ki macrophages show partial impairment. This led us to speculate that the IRAK4 scaffold has a role in TLR4/TRIF signaling. To understand how this might occur, we compared LPS-stimulated WT, Irak4 Ki, Irak4−/−, MyD88−/−, Trif−/−, and MyD88−/−Trif−/− BMDMs. Both MyD88−/− and Trif−/− showed partial impairment in TNF-α and IL-12 production, whereas no production was observed in MyD88−/−Trif−/− (Figures 3A and 3B) (Adachi et al., 1998; Sato et al., 2003). No increase in RelA phosphorylation was observed in Irak4−/− or Trif−/− BMDMs, whereas MyD88−/− and Irak4 Ki showed delayed RelA phosphorylation (Figures 3C–3E). Trif−/−, MyD88−/−, Irak4−/−, and Irak4 Ki showed delayed IκB-α phosphorylation and degradation (Figure 3C). RelA and c-Rel nuclear translocation were completely impaired in Irak4−/− and MyD88−/−Trif−/− BMDMs. Irak4 Ki and MyD88−/− behaved similarly, with partial nuclear translocation. Trif−/− BMDMs translocated RelA and c-Rel similar to WT cells (Figure 3F), as TRIF deficiency impacts gene transcription without affecting RelA nuclear translocation (Sakai et al., 2017). Thus, TRIF or MYD88 deficiency severely impact cytokine production by different mechanisms, with MYD88 deficiency impacting NF-κB nuclear translocation directly, and TRIF impacting post-translational modifications, such as Ser-536 phosphorylation in RelA transactivation domain, required for interaction with transcription enhancers (Chen and Greene, 2004; Sakai et al., 2017). Interestingly, Irak4 Ki BMDMs mimicked the behavior observed in MyD88−/−. Stimulation with R848 or Pam2CSK4 (TLR2 agonist) led to no cytokine production in Irak4 Ki, Irak4−/−, MyD88−/−, and MyD88−/−Trif−/−, while Trif−/− and MyD88−/−Trif−/− BMDMs failed to respond to Poly(I:C), a TLR3 agonist (Fitzgerald et al., 2003a) (Figure S3).
Figure 3. TLR4 signaling in Irak4−/− BMDMs is more severely affected than in either Irak4 Ki or MyD88−/−.
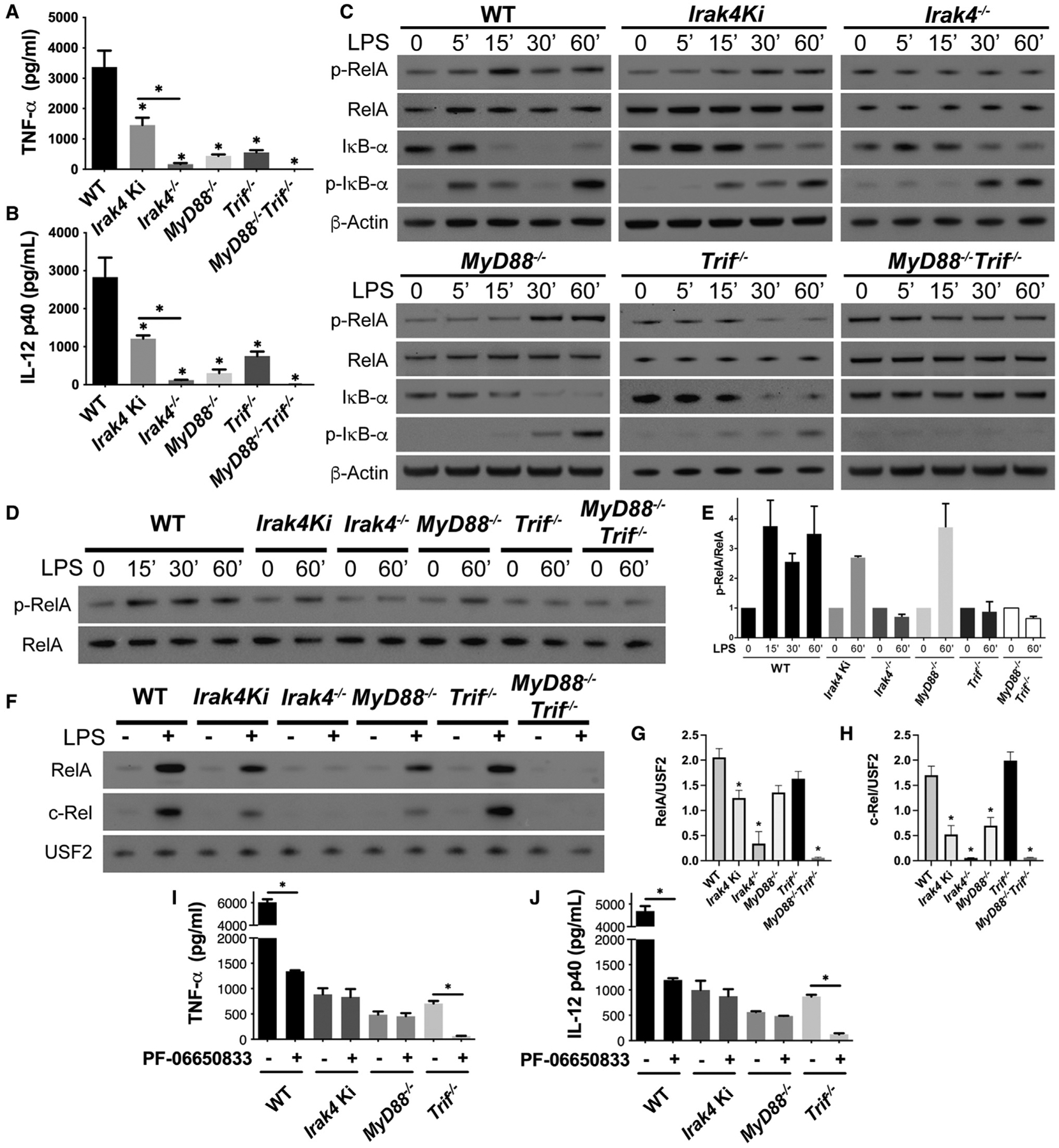
(A–H) Study of WT, Irak4 Ki, Irak4−/−, MyD88−/−, Trif−/−, and MyD88−/−Trif−/− BMDMs stimulated with LPS. (A and B) Quantification of TNF-α (A) and IL-12 (B) produced after LPS stimulation for 24 h. (C) Kinetic study of p-RelA, RelA, IκB-α, p-IκB-α, and β-actin by immunoblot of whole cell lysates from indicated BMDM strains stimulated with LPS for up to 60 min. (D and E) Immunoblot comparison of p-RelA/RelA in indicated BMDM strains stimulated with LPS (D) and densitometric quantification (E). (F) Immunoblot analysis of RelA and c-Rel in nuclear lysates of WT, Irak4 Ki, Irak4−/−, MyD88−/−, Trif−/−, and MyD88−/−Trif−/− BMDMs untreated or treated with LPS for 30 min and densitometric analysis of LPS-treated samples (G and H). (I and J) Quantification of TNF-α (G) and IL-12 (H) produced by WT, Irak4 Ki, MyD88−/−, and Trif−/− BMDMs after LPS stimulation for 24 h with or without IRAK4 inhibitor PF-06650833. All stimulations were done with LPS 100 ng mL−1 with or without PF-066503 200 nM. *p < 0.05 in comparison with WT unless otherwise indicated (one-way analysis of variance with Tukey’s multiple comparisons test). (A, B, E, and G–J) Data from three independent experiments (mean and SEM). (C, D, and F) Images are representative of three independent experiments.
To further distinguish the kinase versus scaffold function of IRAK4, we examined the effects of IRAK4 inhibition with PF-06650833 in MyD88−/− and Trif−/− BMDMs stimulated with LPS, R848, or Poly(I:C). As expected, IRAK4 inhibition suppressed cytokine production in R848-stimulated macrophages but did not affect Poly(I:C) stimulation (Figures S4A–S4E). IRAK4 inhibition had no effect on TNF-α and IL-12 produced by LPS-stimulated MyD88−/− and Irak4 Ki BMDMs. Cytokine production was abolished in Trif−/− macrophages treated with the IRAK4 inhibitor (Figures 3G and 3H). This in addition to the similarities between Irak4 Ki and MyD88−/− supports the hypothesis that IRAK4 kinase activity is essential for NF-κB activation via MYD88. Importantly, the phenotype of Irak4−/− resembles that of MyD88−/−Trif−/− macrophages, suggesting a link between TRIF and the IRAK4 scaffold that may be myddosome independent (Figures 3, S3, and S4). This IRAK4-dependent and MYD88-independent pathway may also require either IRAK1 or IRAK2, as double-deficient IRAK1 and IRAK2 macrophages fail to produce TNF-α and IL-12 (Figure 1). Importantly, these results indicate a role for IRAK4 scaffold in the TRIF pathway of LPS-stimulated BMDMs.
TRIF requires IRAK4 scaffold for TLR4-mediated NF-κB activation
We hypothesized that in LPS-stimulated Irak4 Ki BMDMs the cytokine output occurs due NF-κB activation via TRIF, while the MYD88 axis is non-functional. Based on the inhibitor experiments (Figures 3I and 3J), we expected that cytokine production in Irak4Ki/Trif−/− would be close to zero, despite myddosome formation. Indeed, TNF-α and IL-12 production in Irak4 Ki/Trif−/− BMDMs in response to LPS was undetectable (Figures 4A and 4B). Simultaneous loss of TRIF and IRAK4 kinase activity led to assembly of a myddosome containing IRAKs 1and 2 (Figure4C), but protein inputs revealed deficient IRAK2 phosphorylation and no IRAK1 loss of signal, further indicating that the myddosome assembled in Irak4 Ki BMDMs is non-functional. Importantly, TRIF itself does not impact myddosome formation or activity, as MYD88 co-immunoprecipitation in Trif−/− BMDMs revealed a pattern similar to WT cells, including IRAK1 loss of signal and IRAK2 increase in apparent molecular weight in the protein inputs. Irak4 Ki/Trif−/− failed to phosphorylate RelA and IκB-α (Figure 4D), and no RelA and c-Rel nuclear translocation was observed, as well as no RelA binding activity in the nuclear extracts (Figures 4E–4H). Irak4 Ki/Trif−/− macrophages, as expected, fail to respond to R848 and Poly(I:C) (Figures S4F–S4M).
Figure 4. Residual cytokine production in LPS-stimulated Irak4 Ki BMDMs occurs via TRIF.
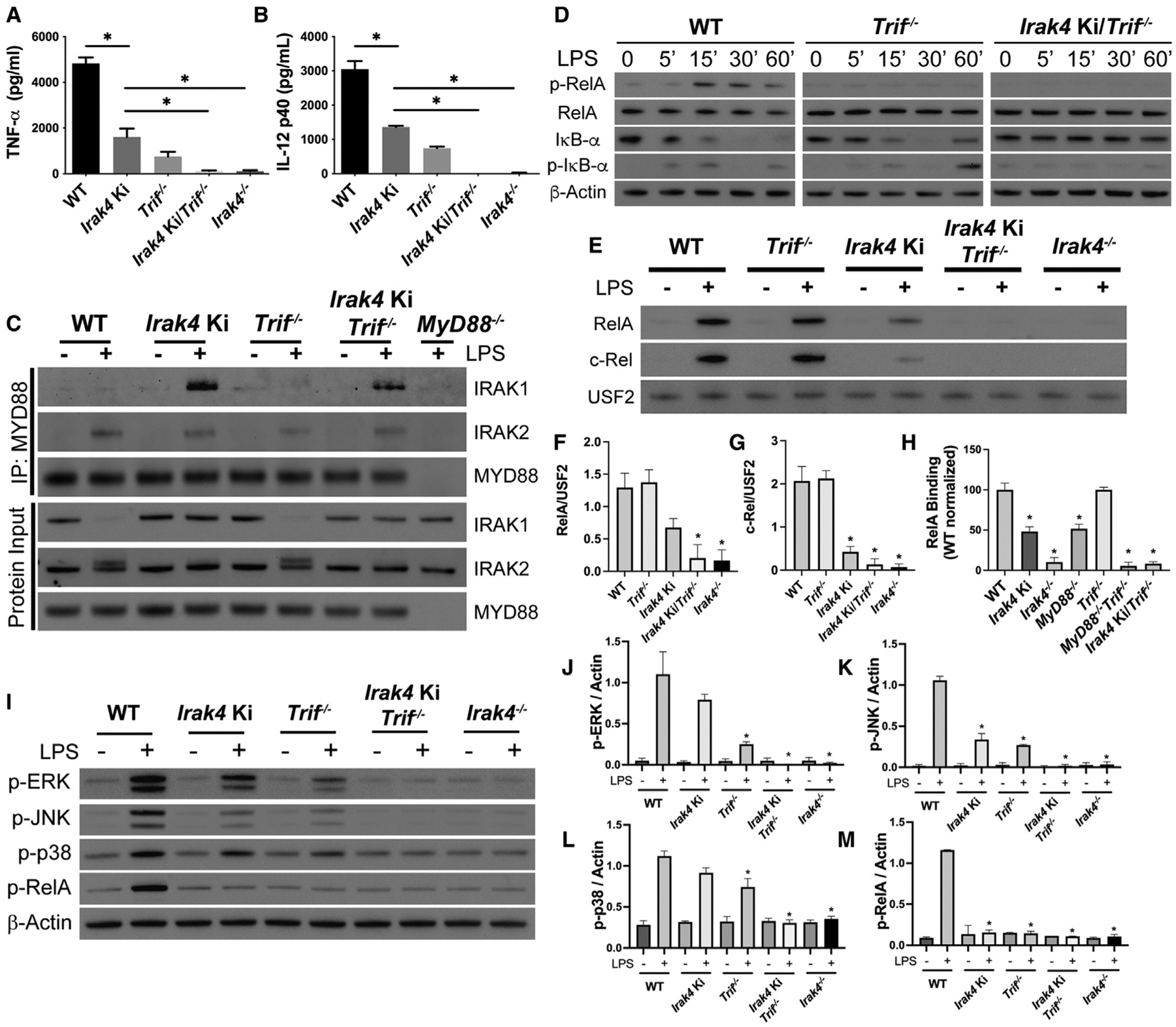
(A–M) Study of WT, Irak4 Ki, Trif−/−, Irak4 Ki/Trif−/−, and Irak4−/− BMDMs stimulated with LPS. (A and B) Quantification of TNF-α (A) and IL-12 (B) produced after LPS stimulation for 24 h. (C) Immunoblot analysis of MYD88 co-immunoprecipitations of indicated BMDMs untreated or treated with LPS for 60 min. (D) Kinetic study of p-RelA, RelA, IκB-α, p-IκB-α, and β-actin by immunoblot of whole cell lysates from indicated BMDM strains stimulated for up to 60 min with LPS. (E) Immunoblot analysis of RelA and c-Rel in nuclear lysates of WT, Irak4 Ki, Trif−/−, Irak4 Ki/Trif−/−, and Irak4−/− BMDMs untreated or treated with LPS for 30 min and their densitometric quantifications (F and G) and RelA DNA-binding affinity in nuclear extracts from BMDMs stimulated with LPS for 30 min (H). (I–M) MAPK and RelA activation profile in WT, Irak4 Ki, Trif−/−, Irak4 Ki/Trif−/−, and Irak4−/− BMDMs stimulated with LPS for 15 min. (I) Immunoblot and (J–M) densitometric analysis of LPS-treated samples. All stimulations were done with LPS 100 ng mL−1. *p < 0.05 in comparison with WT unless indicated (one-way analysis of variance with Tukey’s multiple comparisons test). (A, B, F–H, J–M) Data from three independent experiments (mean and SEM). (C–E, I) Images are representative of three independent experiments.
It was previously reported that Irak4 Ki BMDMs are impaired in JNK phosphorylation, but not p38 or ERK (Tae et al., 2007). Here, we observed that at 15 min after LPS stimulation the residual p38 and ERK activation occurs via TRIF, as Irak4 Ki/Trif−/− macrophages do not activate these MAPKs. Importantly, Irak4−/− BMDMs failed to phosphorylate p38, ERK, and JNK, further evidence that TRIF requires the IRAK4 scaffold (Figures 4I–4M). The same study saw no effects of IRAK4 kinase activity on NF-κB activation, which is in conflict with our data. This discrepancy is likely due to LPS concentration: at 1 μg mL−1 LPS (used in their study), Irak4 Ki BMDMs activate RelA similarly to WT cells. This RelA phosphorylation, however, requires TRIF and the IRAK4 scaffold, as Irak4 Ki/Trif−/− and Irak4−/− are impaired in RelA and MAPK activation (Figure S5).
TLR4 signals from the plasma membrane and from endosomes, and LPS stimulation increases TLR4 endocytosis in a CD14-dependent mechanism. This endocytic TLR4 is required for LPS-induced TRIF signaling (Kagan et al., 2008; Tan et al., 2015; Tanimura et al., 2008; Zanoni et al., 2011). Study of cell surface TLR4 revealed similar endocytosis in LPS-stimulated WT and Irak4 Ki, whereas Irak4−/− BMDMs showed a small decrease in endocytosis (Figures 5A and 5B). This decrease, however, was not sufficient to impact TRAF3 activation, as suggested by pull-down assay using beads containing tandem ubiquitin binding entities (TUBEs) (Figure 5C). LPS stimulation of IRAK-deficient and WT macrophages also resulted in similar levels of IRF3 phosphorylation (a readout of its activation) and C-C motif chemokine ligand 5 (CCL5) production (Figures 5D–5F). Similarly, deficiencies in IRAKs 1 and 2 did not impact TLR4 endocytosis, IRF3 activation, CCL5 production, and TLR3-mediated cytokine production (Figure S6). Accordingly, we found no changes in CD14 endocytosis in IRAK-deficient BMDMs (Figures S6–S6J). Thus, the small decrease in TLR4 endocytosis observed in Irak4−/− BMDMs is insufficient to impact TRIF signaling, and the IRAK4 scaffold likely plays a role downstream of TLR4 endocytosis. Additionally, these data suggests that the IRAK4 scaffold is not required for the TRIF/TRAF3/IRF3 pathway but is essential for NF-κB activation via TLR4/TRIF.
Figure 5. IRAK4 is not involved in TLR4 endocytosis or TRAF3 and IRF3 activation.
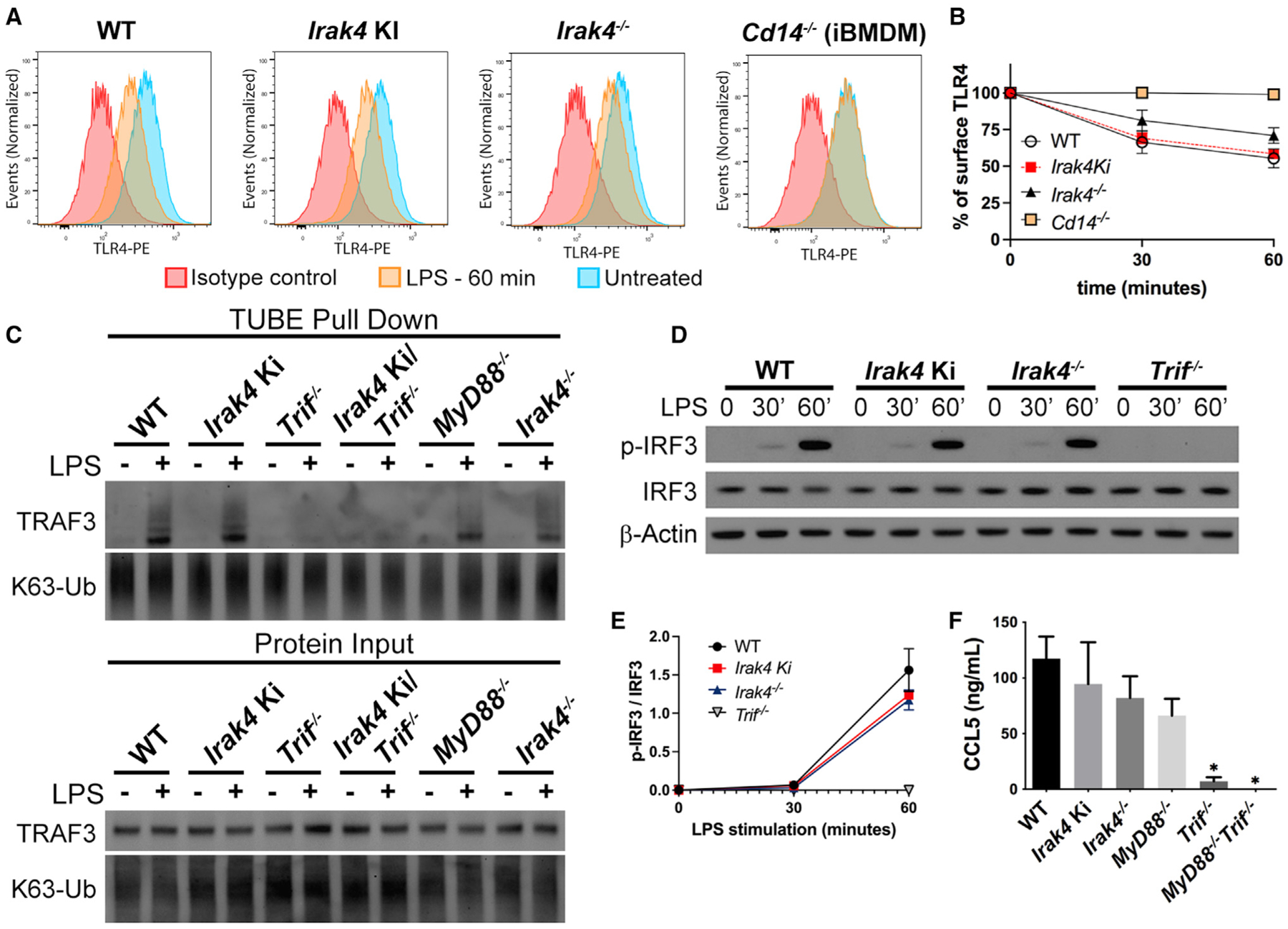
(A and B) Study of TLR4 endocytosis in WT, Irak4 Ki, and Irak4−/− BMDMs and control Cd14−/− immortalized BMDMs, stimulated with LPS for up to 60 min. (A) Histogram of surface TLR4 in the indicated strains unstimulated, stimulated with LPS for 60 min, or unstimulated cells stained with isotype control antibody. (B) Quantification of surface TLR4 after LPS stimulation for up to 60 min. (C) Immunoblot of ubiquitinated TRAF3 pulled down from indicated BMDMs treated with LPS for 30 min.
(D and E) Kinetic study of IRF3 phosphorylation by immunoblot analysis of whole cell lysates from WT, Irak4 Ki, Irak4−/−, and Trif−/− BMDMs (D) and densitometric analysis (E).
(F) Quantification of CCL5 produced by WT, Irak4 Ki, and Irak4−/−, MyD88−/−, Trif−/−, and MyD88−/−Trif−/− BMDMs after LPS stimulation for 24 h. All stimulations were done with LPS 100 ng mL−1. *p < 0.05 in comparison with WT (one-way analysis of variance with Tukey’s multiple comparisons test). (A) Data are representative of three independent experiments. Data from two (E) or three (B, F) independent experiments (mean and SEM). (C and D) Images are representative of two independent experiments.
IRAK4 scaffold is required for MYD88 and TRIF activation of TRAF6
TRIF and MYD88 activate NF-κB via the E3 ubiquitin ligase TRAF6 (Cao et al., 1996; Sasai et al., 2010; Sato et al., 2003). Interestingly, TRIF-mediated TRAF6 activation is myddosome independent and occurs only downstream of TLR4 as TRAF6 is not required by TLR3 (Gohda et al., 2004). TRAF6 is, therefore, a unique node between TRIF and MYD88. IRAK2 and IRAK4 are important for TRAF6 activation via MYD88 (Suzuki et al., 2002; Keating et al., 2007; Pauls et al., 2013), and we hypothesized that downstream of TLR4/TRIF, TRAF6 activation also requires the IRAK4 scaffold and either IRAK1 or IRAK2.
TRAF6 is activated by auto-ubiquitination and is involved in NF-κB signaling via K63 ubiquitination of specific targets, including IRAK1 (Cao et al., 1996; Conze et al., 2008). To investigate endogenous TRAF6 signaling, we performed pull-down assays using TUBE-containing beads. Stimulation of WT BMDMs with LPS for up to 1 hr followed by TUBE pull-down revealed enrichment of ubiquitinated TRAF6 and IRAK1 between 15 and 60 min post-stimulation, with no clear differences in the pool of K63-ubiquitinated proteins. R848 stimulation showed faster kinetics, with ubiquitinated TRAF6 and IRAK1 detectable between 5 and 30 min post-stimulation. The ubiquitination was followed by “degradation (or lack of antibody reactivity with)” of IRAK1 in input material (Figure 6A). We sought to evaluate the contribution of IRAK1 and IRAK2 to TRAF6 activation. Knockout of either IRAKs 1 or 2 revealed no impairment in TRAF6 activation by either agonist, whereas a marked deficiency was observed in the double knockout cells (Figure 6B), further evidence of early IRAK-1 and -2 redundancy (Figure 1).
Figure 6. IRAK4 scaffold is required for TRAF6 activation, while IRAK1 and IRAK2 show redundancy.
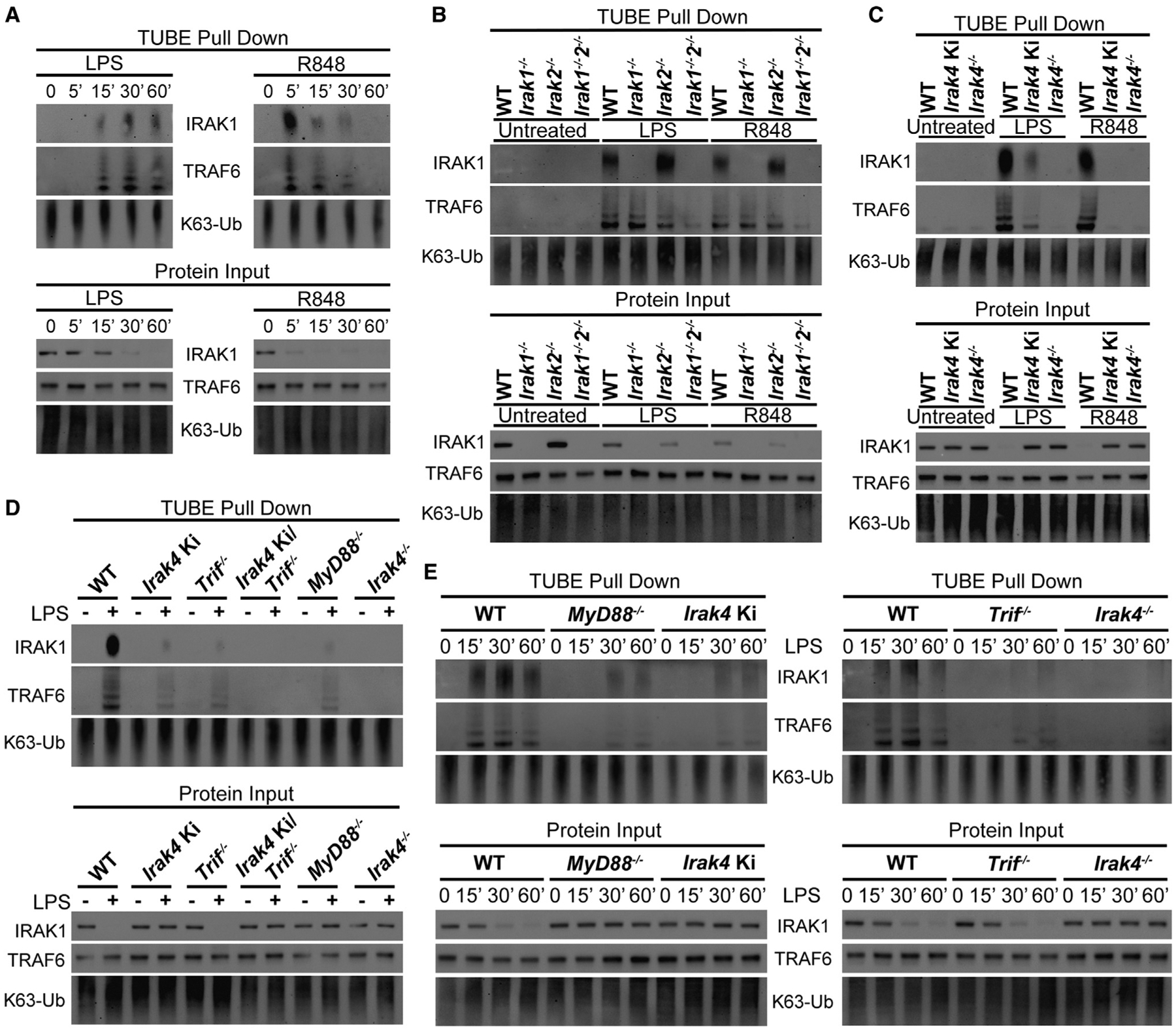
(A) Immunoblot of ubiquitinated TRAF6 and IRAK1 pulled down from WT BMDMs treated with LPS or R848 for up to 60 min.
(B–D) Immunoblot of ubiquitinated TRAF6 and IRAK1 pulled down from indicated BMDMs treated with LPS for 30 min or R848 for 15 min.
(E) Immunoblot of ubiquitinated TRAF6 and IRAK1 pulled down from indicated BMDMs treated with LPS for up to 60 min. All stimulations were done with LPS 100 ng mL−1 or R848 1 μg mL−1. Images are representative of two (E) or three (A–D) independent experiments.
Next, we investigated how the IRAK4 scaffold and kinase activities affect TRAF6 activity. LPS stimulation of Irak4 Ki BMDMs showed partial impairment in TRAF6 and IRAK1 ubiquitination, whereas these events were below the detection limit in Irak4−/−. Upon R848 stimulation, however, loss of IRAK4 kinase activity was sufficient to block TRAF6 activation (Figure 6C). These results are consistent with the hypothesis that the IRAK4 scaffold is required for TRIF-mediated TRAF6 activation. To further test this hypothesis, we stimulated WT, Irak4 Ki, Irak4−/−, Trif−/−, MyD88−/−, and Irak4 Ki/Trif−/− BMDMs with LPS. Trif−/−, MyD88−/−, and Irak4 Ki cells showed partial impairment in TRAF6 and IRAK1 ubiquitination. TRAF6 activation in Irak4−/− and Irak4 Ki/Trif−/− macrophages was not detected (Figure 6D). To further elaborate on this, we evaluated the kinetics of TRAF6 activation in WT, Irak4 Ki, Irak4−/−, Trif−/−, and MyD88−/− BMDMs. TRAF6 activation was observed at 15, 30, and 60 min after LPS stimulation in WT cells. Irak4 Ki, Trif−/−, and MyD88−/− macrophages displayed no TRAF6 activation at 15 min and diminished activation at 30 and 60 min, whereas Irak4−/− BMDMs showed only very weak detection of ubiquitinated TRAF6 and IRAK1 at 60 min (Figure 6E).
These results are consistent with the hypothesis that the IRAK4 scaffold coordinates TRIF-mediated TRAF6 activation, although a compensatory mechanism involving other molecules might exist. Taken together, these data suggest that in TLR4 signaling, activation of TRAF6 by MYD88 and TRIF requires the IRAK4 scaffold and may also require either IRAK1 or IRAK2.
IRAK4 scaffold is essential for the response to Gram-negative bacteria
Mutations in IRAK proteins are commonly associated with deficient innate immune response and susceptibility to pyogenic bacterial infections, which can lead to sepsis (Picard et al., 2003; von Bernuth et al., 2012). To investigate whether the responses observed here with ultrapure LPS correlate with responses to a live pathogen, we conducted infection assays in BMDMs using Escherichia coli “Bort,” a strain with enhanced murine toxicity (Bortolussi et al., 1978). In this model, the production of cytokines is TLR4 dependent, with TNF-α and IL-12 production occurring via MYD88 and TRIF, and CCL5 predominantly via TRIF (Figures S7A–S7D). E. coli infection of WT, Irak1−/−, Irak2−/−, and Irak1−/−Irak2−/− BMDMs confirmed the IRAK1/2 redundancies. Loss of IRAK1 did not impact E. coli-induced TNF-α and IL-12 production, whereas loss of IRAK2 led to decreased production. Simultaneous loss of IRAK-1 and -2 further decreased cytokine production. None of the mutations affected CCL5, evidence that IRF3 responses are unaffected (Figures 7A–7C).
Figure 7. IRAK4 scaffold is essential to the response against Gram-negative bacteria, while IRAK-1 and -2 show redundancy.
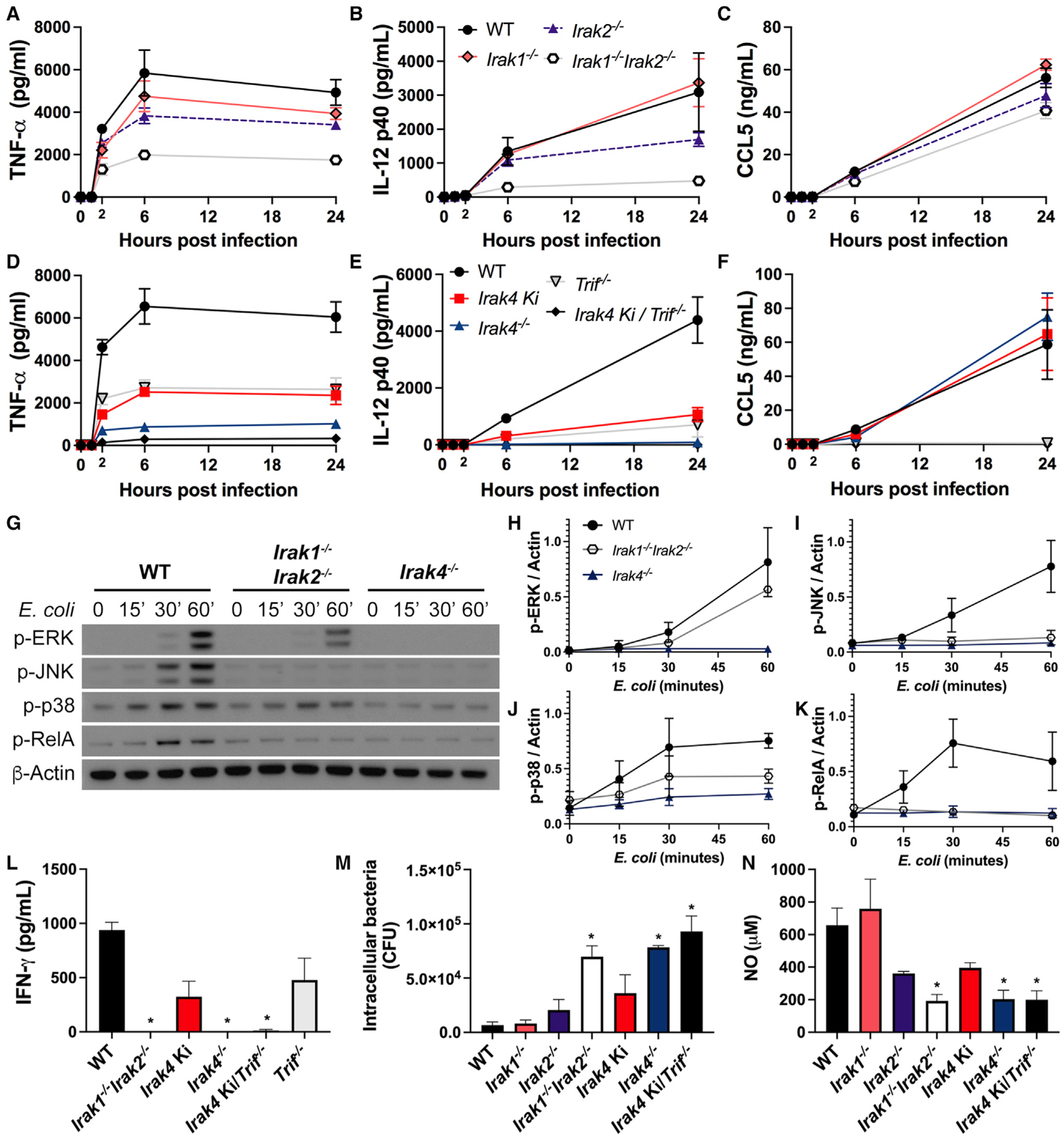
(A–F) Production of TNF-α (A, D), IL-12 (B, E), and CCL5 (C, F) in BMDMs infected with E. coli Bort for up to 24 h at MOI 1. (A–C) WT, Irak1−/−, Irak2−/−, and Irak1−/−Irak2−/− BMDMs. (D–F) WT, Irak4 Ki, Trif−/−, Irak4 Ki/Trif−/−, and Irak4−/− BMDMs.
(G–K) Immunoblot of p-ERK, p-JNK, p-p38, p-RelA, and β-actin from WT, Irak1−/−Irak2−/−, and Irak4−/− BMDMs infected with E. coli Bort at MOI 1 for up to 60 min (G) and densitometric analysis (H–K).
(L) Production of IFN-γ in splenocytes infected with E. coli Bort for 24 h at MOI 1.
(M and N) Viable intracellular bacteria counts (M) and NO production (N) in IFN-γ-primed BMDMs (100 ng mL−1, 20 h) infected with E. coli Bort for 6 h at MOI 10. (G) Image is representative of three independent experiments. Data from three (A–F, H–K, M, and N) or two (L) independent experiments (mean and SEM). *p < 0.05 in comparison with WT (one-way analysis of variance with Tukey’s multiple comparisons test).
Next, we focused on macrophages from IRAK4-deficient strains. In previous reports, pro-inflammatory cytokine production induced by R848 or Gram-positive bacteria required IRAK4 kinase activity (Kawagoe et al., 2007; Tae et al., 2007; Pattabiraman et al., 2018). Our LPS data, however, highlighted a substantial difference between loss of the IRAK4 scaffold and kinase activity (Figures 2–4). E. coli infection experiments further corroborate this interpretation where Irak4 Ki macrophages produced lower amounts of TNF-α and IL-12 than WT cells but were absent in Irak4−/−cells. BMDMs from Irak4 Ki/Trif−/− secreted neither TNF-α nor IL-12. In contrast, similar quantities of CCL5 were produced in all mutants except Irak4 Ki/Trif−/− and Trif−/−, which were deficient (Figures 7D–7F). The differences in cytokine output in these experiments are not due to cell viability or bacterial load, as they remained similar in all cell types (Figures S7E–S7H).
Interestingly, cytokine output in E. coli-infected Irak1−/−Irak2−/− BMDMs was higher than that observed in Irak4−/− BMDMs or in LPS-stimulated Irak1−/−Irak2−/− BMDMs (Figures 1 and 7), despite absence of RelA activation (Figures 7G and 7K). Although the reason for this discrepancy is unknown, it possibly involves other pathways that are active in Irak1−/−Irak2−/− BMDMs but not in in Irak4−/−. For instance, activation of MAPKs ERK and p-38 is absent in Irak4−/− but not in Irak1−/−Irak2−/− macrophages stimulated with LPS or infected with E. coli (Figure S2, Figures 4I–M, Figures 7G–7K).
Cytokines such as IL-12 stimulate the production of interferon-γ (IFN-γ) by lymphocytes (Trinchieri, 1995), which enhances the response of macrophages to Gram-negative bacteria (Schroder et al., 2004). Stimulation of total splenocytes with E. coli led to severe impairment in IFN-γ production in Irak1−/−Irak2−/−, Irak4−/−, and Irak4 Ki/Trif−/−, with an intermediate production in Irak4 Ki splenocytes (Figure 7L). IFN-γ-primed BMDMs from Irak1−/−Irak2−/−, Irak4−/−, and Irak4 Ki/Trif−/− mice showed deficient bacteria clearance, which correlated with lower nitric oxide (NO) production (Figures 7M and 7N). Collectively, the data suggest that deficiency in IRAK4 scaffold or combined deficiency of TRIF and IRAK4 kinase activity impairs the host response to Gram-negative bacteria.
DISCUSSION
Our current assumptions about how IRAKs work are based on overexpression studies and/or research focused on TLRs that signal through a single adaptor. These approaches are extremely valuable but need further validation when applied to TLR4, which synergistically uses MYD88 and TRIF and their respective bridging adaptors MAL and TRAM. Removal of any single adaptor severely impacts TRAF6-mediated NF-κB activation (Adachi et al., 1998; Fitzgerald et al., 2001, 2003b; Gohda et al., 2004; Sasai et al., 2010; Sato et al., 2003; Verstak et al., 2014; Yamamoto et al., 2003b).
First, we investigated the redundancies between IRAK1 and IRAK2. Previous work suggested that in the context of TLR2, loss of IRAK2 does not impact early signaling events due compensatory mechanisms involving IRAK1 (Kawagoe et al., 2008). IRAK2 was, however, necessary for a “late response,” shown by cytokine measurements. Similarly, stimulation of BMDMs carrying IRAK2 E525A (deficient in TRAF6 activation) with R848 or Pam3CSK4 does not impact early signaling, likely due to IRAK1 redundancy (Pauls et al., 2013). Here we explored the same questions in the context of TLR4 and TLR7 and reached similar conclusions. Early signaling events are largely unaffected by either IRAK-1 or -2, and only loss of both proteins inhibited TRAF6 and NF-κB activation. We did, however, observed divergences regarding TLR-4 and -7 in “late phase” signaling and showed cytokine production is not completely absent in LPS-stimulated Irak2−/− macrophages, but it is in Irak1−/−Irak2−/−. This contrasts to TLR7, where loss of IRAK2 is enough to completely block cytokine production. These observations suggests that IRAK2 is essential for MYD88 signaling, whereas NF-κB activation via TLR4/TRIF can utilize either IRAK-1 or -2 to bypass this dependency.
Myddosome formation at the cell membrane occurs within minutes of stimulation. These early membrane-bound complexes are short-lived and act as a nucleating step that presumably results in formation of stable signaling complexes that sustain NF-κB activation (Tan and Kagan, 2019; Tan et al., 2015; Latty et al., 2018; Moncrieffe et al., 2020; Deliz-Aguirre et al., 2021). Upon LPS stimulation, IRAK1 is recruited to the myddosome of WT and Irak2−/− macrophages, while R848-stimulated Irak2−/− failed to recruit IRAK1. This enhanced IRAK1 co-immunoprecipitation could occur due increased myddosome stability and failure to propagate signal (De Nardo et al., 2018). This suggests that despite differences in IRAK1 recruitment, the LPS-induced myddosome assembled in Irak2−/− BMDMs cannot sustain NF-κB activation, and the residual cytokine production occurs via TRIF, which is not involved in TLR7 signaling (Yamamoto et al., 2003a). Since we observe a near complete inhibition in cytokine production in LPS-stimulated Irak1−/−Irak2−/− BMDMs, we speculate that sustained LPS-induced NF-κB activation via MYD88 requires IRAK2, while TRIF may require either IRAK1 or IRAK2.
Next, we examined how IRAK-1 and -2 redundancies work in the context of IRAK4 kinase activity. IRAK4 inhibition increases myddosome stability and recruitment of IRAK-1 and -4, while partially impacting cytokine output (De Nardo et al., 2018; Kawagoe et al., 2007; Pennini et al., 2013; Tae et al., 2007). This led us to speculate that IRAK1/2 recruitment to the myddosome could partially compensate for loss of IRAK4 enzymatic activity. Despite enhanced myddosome stability in Irak4 Ki BMDMs, IRAK-1 or -2 do not individually affect cytokine responses. Importantly, loss of IRAK4 kinase activity greatly decreases IRAK1 and IRAK2 post-translational modifications, suggesting that the phosphorylation events triggered by IRAK4 cannot be compensated by downstream IRAKs. Since we and others observed that Irak4 Ki does not produce cytokines via MYD88-only TLRs, phenocopying MyD88−/− (Kawagoe et al., 2007; Pennini et al., 2013; Tae et al., 2007), we tested whether the same is true for TLR4. IRAK4 inhibition and stimulation of Irak4 ki/Trif−/− cells demonstrates that residual cytokine production occurs via TRIF, despite myddosome formation. These observations suggest that additional loss of IRAK-1 or -2 in a Irak4 Ki background has no impact because this myddosome is hypofunctional. We cannot completely rule out the possibility that IRAK4 Ki-containing myddosomes have vestigial activity in other contexts: human IRAK4 D329A has no kinase activity, but IRAK4D329A fibroblasts stimulated with IL-1β show partial cytokine production and lower IRAK4/1 interactions (De et al., 2018). It is possible that in this context IRAK1 and/or IRAK 2 partially compensate for loss of IRAK4 kinase activity.
Irak4 Ki BMDMs showed partial inhibition in TLR4-dependent TNF-α and IL-12 induction, whereas these cytokines were nearly completely absent in LPS-stimulated Irak4−/− BMDMs (Tae et al., 2007). Irak4 Ki closely resembles MyD88−/−, with partial NF-κB nuclear translocation via TLR4, so we hypothesized that IRAK4 scaffold is required for NF-κB activation via TLR4/TRIF. LPS-stimulated Irak4−/− and MyD88−/−Trif−/− BMDMs showed no NF-κB nuclear translocation, in support of our hypothesis. This raises the possibility that in some contexts IRAK4 may signal independently of MYD88. Since we were able to rule out IRAK4 involvement in TLR4 endocytosis and TRIF/TRAF3/IRF3 activation, we focused our attention on TRAF6, a known node between MYD88 and TRIF in TLR4-mediated NF-κB activation (Cao et al., 1996; Gohda et al., 2004; Sasai et al., 2010; Sato et al., 2003; Verstak et al., 2014). We found that TRAF6 activation is impaired in Irak4−/− and Irak4 ki/Trif−/−, while this level of inhibition is not observed in MYD88 or TRIF knockouts. These data put IRAK4 between MYD88/TRAF6 (kinase activity required) and TRIF/TRAF6 (scaffold required). Though conflicting reports exist regarding the involvement of TRAF6 in TLR3 signaling (Gohda et al., 2004; Jiang et al., 2003; Sato et al., 2003), potential links between TRAF6 and TLR3 are unlikely to involve IRAK4 (Jiang et al., 2003; Kawagoe et al., 2007; Pennini et al., 2013; Tae et al., 2007).
These findings beg the question: how does IRAK4 mediate TRAF6 activation via TLR4/TRIF? One possibility involves TRAM. Upon LPS stimulation, TRAM and TRAF6 are found in the same molecular complex (Verstak et al., 2014), and biochemical evidence suggests that TRAM can physically interact with IRAK4 and IRAK1 (Bin et al., 2003). The nature of these interactions and whether they occur in physiological conditions remain unexplored. We also speculate that, in addition to IRAK4 scaffold, NF-κB activation via TLR4/TRIF may require either IRAK1 or IRAK2. We raise this hypothesis based on the observations that LPS-treated Irak1−/−Irak2−/− BMDMs are impaired in TRAF6 and NF-κB activation, similar to Irak4−/− and MyD88−/−Trif−/− macrophages. TRAF6 binding motifs are absent in IRAK4 but present in IRAK1 and IRAK2 (Flannery and Bowie, 2010). Although IRAK2 is of major importance to TRAF6 activation (Keating et al., 2007), its deficiency can be compensated by IRAK1 in the first 2 hr after stimulation (Pauls et al., 2013). We hypothesize that IRAK1 is more efficient at compensating IRAK2 deficiencies in the TRIF/TRAF6 than in the MYD88/TRAF6 pathway, since MYD88 activation leads to IRAK1 degradation (Ferrao et al., 2014; Kubo-Murai et al., 2008). Interestingly, MAPK activation was not completely deficient in Irak1−/−Irak2−/− BMDMs, suggesting that TRAF6 activation and/or signaling downstream of TRAF6 might involve additional components. Generation of additional mouse strains such as Irak1−/−Irak2−/−Trif−/− are required to further study these previously overlooked interactions.
Signaling through TLR4 is more complex than other TLRs. Presumably this is because TLR4 is so critical to the host that it has developed multiple fail-safe signaling mechanisms. The reasons for this are unclear, but our findings suggest that the IRAK4 scaffold is central to protecting TLR4 responsiveness by integrating MYD88 and TRIF signaling, one of the key features in the TLR4 pathway.
Limitations of the study
This study focused on signaling events involving NF-κB and used the cytokines TNF-α and IL-12 as readout, as they are commonly used as surrogates for NF-κB activation. It is possible that other transcription factors can contribute to the expression of these and other cytokines.
The data presented in this article were obtained in vitro using mouse cells. Generalizations to other species, such as humans, and in vivo infections should be done carefully.
STAR⋆METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and resource requests can be directed to and will be fulfilled by the lead contact, Ricardo Gazzinelli (ricardo.gazzinelli@umassmed.edu).
Materials availability
Mouse lines generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
The quantitative data and uncropped immunoblots generated in this study are available at Mendeley Data: https://doi.org/10.17632/tt3grr9zj7.1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice
WT C57BL/6, Irak1−/− (Thomas et al., 1999) and Irak2−/− (Wan et al., 2009) mice were obtained from The Jackson Laboratory. Tlr4−/−, Trif−/−, and MyD88−/− mice were provided by Dr. Shizuo Akira (Department of Host Defense, Osaka University, Osaka, Japan) (Adachi et al., 1998; Hoshino et al., 1999; Yamamoto et al., 2003a). Irak4−/− were provided by Dr. Tak Mak (Princess Margaret Cancer Centre, University of Toronto, Toronto, Canada) (Suzuki et al., 2002). Irak4 Ki mice were provided by Dr. Xiaoxia Li (Department of Inflammation and Immunity, Lerner Research Institute, Cleveland Clinic, Cleveland, USA) (Tae et al., 2007). MyD88−/−Trif−/−, Irak1−/−Irak2−/−, Irak1−/−Irak4 Ki, Irak2−/−Irak4 Ki, Irak4 Ki/Trif−/− were generated by in-house crossing. All mice were bred and maintained in pathogen-free conditions in accordance with the Institutional Animal Care and Use Committee (IACUC) at University of Massachusetts Medical School (UMMS). 8- to -12-week-old mice, male and female in similar proportions, were used in this work.
METHOD DETAILS
Chemicals
The following chemicals were routinely used in this work: NaF (Sigma-Aldrich), NaVO4 (Sigma-Aldrich), β-Glycerophosphate disodium (Santa Cruz), Nonidet P-40 Substitute (NP-40) (Boston Bioproducts), iodoacetamide (Sigma-Aldrich), dithiothreitol (DTT) (Boston Bioproducts), Dulbecco’s Modified Eagle Medium (DMEM) (Corning), Dulbecco’s Phosphate Buffered Saline (PBS) (Corning), Tris-HCl (Roche), NaCl (Sigma-Aldrich), glycerol (Fisher Scientific), Ethylenediamine tetraacetic acid (EDTA) (Boston Bioproducts) and Tween-20 (Boston bioproducts).
Cell culture and stimulations
For isolation of bone marrow cells and generation of BMDM, mice were sacrificed by CO2 exposure followed by cervical dislocation in accordance with UMMS IACUC guidelines. The skin was then sterilized with 70% isopropanol, the legs removed, the tibia and femur were collected and cleaned of muscle. The proximal and distal epiphysis were cut away and the bone marrow was flushed out of the bone using BMDM complete media (DMEM supplemented with 10% fetal bovine serum (R&D Systems), 5 mM L-Glutamine (GIBCO), 25 mM HEPES (Thermo-Fisher) and 20% L929-conditioned media). For each animal, the bone marrow cells were placed in 3 untreated 150 mm × 15 mm petri dishes (Corning) cultured at 37°C and 5% CO2 in BMDM complete media for 6 to 9 days, with the media replenished on day 5. For isolation of splenocytes, mice were sacrified as described above, the spleens were collected, macerated in a 100 μm nylon cell strainer, and the red blood cells lysed in ACK Lysing Buffer (Gibco) for 5 min at room temperatute. The splenocytes were then washed twice and resuspended in DMEM containing 10% FBS.
Stimulations were done with 100 ng mL−1 ultrapure LPS from E. coli O 111:B4 (Invivogen), 1 μg mL−1 R848 (Invivogen), 10 μg mL−1 Poly(I:C) (Invivogen) or 200 ng mL−1 PAM3CSK4 (Invivogen). For IFN-γ priming, BMDMs were incubated with 100 ng mL−1 IFN-γ (BioLegend) for 20 h. Experiments using the IRAK4 inhibitor PF-06650833 (Tocris) included a pre-incubation step (30 min, 37°C and 5% CO2) with 200 nM of the inhibitor, followed by stimulation with the specific agonist in the presence of 200 nM PF-06650833.
Preparation of whole cell lysates and nuclear extracts and NF-κB binding assay
For preparation of whole cell lysates, 4 × 106 cells were washed three times in cold PBS containing 25 mM β-Glycerophosphate disodium, 10 mM NaF and 1 mM NaVO4, and lysed for 10 min on ice with Tris-HCl 50 mM, NaCl 150 mM, EDTA 5 mM, 1% NP-40, Halt Protease inhibitor (Thermo-Fisher) and Halt Phosphatase inhibitor (Thermo-Fisher). Alternatively, after stimulation and washes, nuclear extracts were prepared using NE-Per Nuclear and Cytoplasmic Extraction Reagents (Thermo-Fisher) as described by the manufacturer. Nuclear extracts were then used for immunoblot as described below. Alternatively, nuclear extracts were used in NF-kB (p65) Transcription Factor binding assay (Cayman Chemical). Briefly, 1 μg per well of nuclear extracts were added to the plate and the assay was performed according to the manufacturer’s instructions. RelA binding in LPS- or R848-treated samples was calculated by subtracting the OD of respective untreated control and normalized to WT.
Immunoblot
Whole cell lysates or nuclear extracts were incubated with Pierce Lane Reducing Sample Buffer (Thermo-Fisher) at 100°C for 10 min, cooled on ice for 5 min, and loaded on polyacrylamide gels. Following electrophoresis, the proteins were transferred to 0.45 μm nitrocellulose membrane (Amersham Protran 0.45 NC) in wet conditions (25 mM tris-base, 192 mM glycine, 20% methanol, pH 8.5). The membranes were blocked for 2 h at room temperature in blotting-grade blocker (Bio-Rad), incubated with primary antibody in TBS containing BSA 1% for 16 h at 4°C, washed three times for five minutes each with TBS containing 0.1% Tween 20 (TBS-T), incubated with secondary antibody in blotting-grade blocker for one hour and washed three times in TBS-T. Proteins were detected using Clarity Max ECL Substrate (Bio-Rad) and imaged in ChemiDoc MP Imaging System (Bio-Rad). Densitometric analysis were performed using ImageJ (NIH) (Schneider et al., 2012).
The following antibodies and dilutions were used for immunoblots: IRAK1 (Cell Signaling, 4504S, 1:1000), IRAK2 (Abcam, ab62419, 1:500), TRAF6 (Abcam, ab33915, 1 in 500), TRAF3 (Abcam, ab239357, 1:400), MyD88 (R&D Systems, AF3109, 1:1000), phospho-IRF3 (Cell Signaling, 4947, 1:1000), IRF3 (Cell Signaling, 4302, 1:1000), USF2 (Novus Biologicals, NBP1–92649, 1:750), c-Rel (Cell Signaling, 67489, 1:1000), phospho-IκB-α (Ser32) (Cell Signaling, 2859, 1:750), IκB-α (Cell Signaling, 4812S, 1:1000), phospho-RelA/NF-κB (Ser 536) (Cell Signaling, 3033, 1:1000), RelA/NF-κB (Novus Biologicals, NB100–2176, 1:1000), Actin (Sigma-Aldrich, A2066, 1:3000), K63-linkage specific polyubiquitin (Cell Signaling, 5621S, 1:1000), phospho-SAPK/JNK (Thr 183/Tyr 185) (Cell Signaling, 4668T, 1:1000), phospho-p38 MAPK (Thr 180/Tyr 182) (Cell Signaling, 4511T, 1:1000), phospho-p44/42 MAPK (Erk 1/2) (Thr 202/Tyr 204) (Cell Signaling, 4370T, 1:2000), anti-Goat-IgG HRP-conjugated (R&D Systems, HAF017, 1:10000), and anti-Rabbit-IgG HRP-conjugated (Sigma-Aldrich, A0545, 1:10000).
MYD88 co-Immunoprecipitation
MYD88 co-immunoprecipitations were performed as described previously (Tan and Kagan, 2018). Briefly, 4 × 106 cells were stimulated with LPS (100 ng mL−1) or R848 (1 μg mL−1), washed three times in cold PBS containing 25 mM β-Glycerophosphate disodium, 10 mM NaF and 1 mM NaVO4, and lysed for 10 min on ice with 0.6 mL of IP lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 5% glycerol, 2 mM dithiothreitol (DTT), 1% NP-40, Halt Protease inhibitor (Thermo-Fisher) and Halt Phosphatase inhibitor (Thermo-Fisher)). The lysates were then centrifuged at 14,000 × G for 10 min at 4°C, and the supernatants collected. A small fraction of the supernatants was kept for analysis of the protein input, whilst the remainder (550 μL) were used for co-immunoprecipitation. 1 μL of anti-MyD88 (R&D Systems, AF3109) and 30 μL of Protein G Sepharose 4 Fast Flow (Millipore Sigma) pre-equilibrated with IP lysis buffer was added to each sample. The mix was then incubated with gentle agitation for 4 h at 4°C, washed three times with IP lysis buffer, and eluted with 50 μL of sample buffer (Pierce Lane Reducing Sample Buffer (Thermo-Fisher)). Immunoblots of eluted samples and protein inputs were then carried out as described under “Immunoblot”.
TUBE pull down
For enrichment of ubiquitinated proteins, 4 × 106 cells were stimulated with LPS (100 ng mL−1) or R848 (1 μg mL−1), washed three times in cold PBS containing 25 mM β-Glycerophosphate disodium, 10 mM NaF and 1 mM NaVO4, and lysed for 10 min on ice in 0.6 mL of TUBE lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 10% glycerol, 10 mM iodoacetamide, Halt Protease inhibitor (Thermo-Fisher) and Halt Phosphatase inhibitor (Thermo-Fisher)). A small fraction of the supernatants was kept for analysis of the protein input, whilst the remainder were used for protein pull down. For this, 550 μL of protein lysate was added to 30 μL of TUBE2-Agarose (Life Sensors) pre-equilibrated in TUBE lysis buffer, and the mix was then incubated with gentle agitation for 3 h at 4°C. After three washes with TBS-T, the proteins were eluted with 50 μL of sample buffer (Pierce Lane Reducing Sample Buffer (Thermo-Fisher)). Immunoblots of samples eluted from the beads and their protein inputs were then carried out as described under “Immunoblot”.
Flow cytometry
TLR4 and CD14 endocytosis was quantified by flow cytometry as described previously (Perkins et al., 2018). Briefly, 1 × 106 viable BMDMs were incubated with LPS for the indicated time points (37°C, 5% CO2) at a volume of 2 mL in sterile flow cytometry polypropylene round-bottom tubes (Corning). After incubation, 2 mL of ice-cold FACS Buffer (PBS containing 0.5% FBS and 2 mM EDTA) was added, the cells were centrifuged (400 × G, 5 min, 4°C), and washed twice in FACS Buffer. The cells were then resuspended in 100 μL of FACS Buffer containing 20 μg mL−1 anti-CD16/32 (Fcγ RII-Blocking antibody) (BioLegend, 101302), transferred to a 96-wells U-bottom plate (Corning) and incubated for 20 min on ice. The plates were then centrifuged (500 × G, 5 min, 4°C), the supernatant discarded, and the cells resuspended in 100 μL of FACS Buffer containing 4 μg mL−1 PE-conjugated anti-TLR-4 (BioLegend, 145403), 1 μg mL−1 APC-conjugated anti-CD14 (BioLegend, 123311) or 100 μL of FACS Buffer containing 4 μg mL−1 PE-conjugated IgG2a κ-chain isotype-matched control (BioLegend, 400507) and incubated on ice for 30 min in the dark. After three washes, the samples were then resuspended in FACS Buffer and read on a LSR II flow cytometer (BD). Analysis were done on FlowJo v10 (BD), and percentage of surface TLR4 was calculated as follows: (MFIt=x – MFIisotype control)/(MFIt=0 – MFIisotype control).
Infection assay, cellular viability, and intracellular bacteria quantification
2 × 105 BMDMs per well were plated in sterile flat-bottom 96 well-plates and kept in BMDM complete media without antibiotics at 37°C and 5% CO2 for 16 h 1 × 106 splenocytes were plated in sterile flat-bottom 96 well-plates in DMEM containing 10% FBS without antibiotics.
E. coli strain “Bort” (ATCC 700973) was grown to stationary phase in LB broth (Sigma-Aldrich) for 18 h at 37°C and 250 rpm and diluted in DMEM containing 10% FBS. The MOIs were confirmed retroactively by plating the bacteria in LB agar (Sigma-Aldrich).
The plates had their supernatants removed, washed once in DMEM containing 10% FBS, and infected with 200 μL of bacteria or left with media alone (uninfected controls), followed by plate centrifugation to facilitate contact between bacteria and cells (400 × G, 5 min, room temperature), and incubation (37°C, 5% CO2) for one hour. The supernatant was collected (1 h post infection, hpi), the plates washed three times in media and the extracellular bacteria was killed by adding media containing 50 μg mL−1 gentamicin (Millipore Sigma) for one hour. Then, the supernatant was collected (2 hpi), the cells washed three times, media replenished with DMEM 10% FBS containing 10 μg mL−1 gentamicin and incubated for additional 4 h (6 hpi) and 22 h (24 hpi).
At 2, 6 and 24 hpi, viable intracellular bacteria were counted by lysing the cells in cold-PBS containing 0.5% Triton X-100 for 30 min and plating them in LB agar.
LDH activity was quantified as a surrogate for cellular death using the 2, 6 and 24 hpi supernatants, according to the manufacturer’s instructions (Cytotox 96 Non-Radioactive Cytotoxicity Assay, Promega). Controls with 100% LDH activity were obtained by lysing uninfected cells in cold-PBS containing 0.5% Triton X-100 for 30 min.
Cytokine and NO quantifications
For cytokine, chemokines and NO quantifications, 2 × 106 cells were plated in flat-bottom 96-well plates (Corning) and stimulated with agonists for 24 h, unless stated otherwise. The supernatants were collected and kept at −80°C until analyzed. All cytokines and chemokines were measured by ELISA according to the manufacturer’s instructions: IL-12 p40−Mouse uncoated ELISA kit (Invitrogen, 88-7120-88), Mouse CCL5/Rantes DuoSet ELISA (R&D Systems, DY478), Mouse TNF-alpha DuoSet ELISA (R&D Systems, DY410), Mouse Interferon-gamma DuoSet ELISA (R&D Systems, DY485). NO was measured in the supernatants according to the manufacturer’s instructions (Abcam, Nitric Oxide Assay Kit, colorimetric, ab65328).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical tests were performed using GraphPad Prism version 9 for macOS (GraphPad Software). Unless otherwise stated, experiments in this work employed one-way analysis of variance (ANOVA) with Tukey post-comparison tests using a confidence interval of 95%. Asterisk denotates statistical significance (p < 0.05). Error bars represent standard error of the mean (s.e.m.). Statistical details of experiments can be found in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit mAb anti-IRAK1 (D51G7) | Cell Signaling | 4504S; RRID AB_1904032 |
| Rabbit pAb anti-IRAK2 | Abcam | ab62419; RRID AB_956084 |
| Rabbit Recombinant mAb anti-TRAF6 (EP591Y) | Abcam | ab33915; RRID AB_778572 |
| Rabbit Recombinant mAb anti-TRAF3 (EPR22992-93) | Abcam | ab239357; RRID AB_2915900 |
| Goat pAb anti-MyD88 | R&D Systems | AF3109; RRID AB_2146703 |
| Rat mAb PE anti-mouse CD284 (TLR4) | BioLegend | 145404; RRID AB_2561874 |
| Rat mAb anti-mouse CD16/32 | BioLegend | 101301; RRID AB_312800 |
| Rat mAb anti-mouse CD14 | BioLegend | 123311; RRID AB_940574 |
| Rat PE IgG2a k-chain isotype-matched control | BioLegend | 400507; RRID AB_326530 |
| Rabbit mAb anti-phospho-IRF-3 (Ser396) (4D4G) | Cell Signaling | 4947; RRID AB_823547 |
| Rabbit mAb anti-IRF-3 (D83B9) | Cell Signaling | 4302; RRID AB_1904036 |
| Rabbit pAb anti-USF2 | Novus Biologicals | NBP1-92649; RRID AB_11007053 |
| Rabbit mAb anti-c-Rel (D3B8S) | Cell Signaling | 67489; RRID AB_2799726 |
| Rabbit mAb anti-phospho-IkBa (Ser32) (14D4) | Cell Signaling | 2859; RRID AB_561111 |
| Rabbit mAb anti-phospho-NF-kB p65 (Ser 536) (93H1) | Cell Signaling | 3033; RRID AB_331284 |
| Rabbit pAb anti-Actin | Sigma-Aldrich | A2066; RRID AB_476693 |
| Rabbit mAb anti-IkB-a (44D4) | Cell Signaling | 4812S; RRID AB_10694416 |
| Rabbit mAb anti-K63-linkage Specific Polyubiquitin (D7A11) | Cell Signaling | 5621S; RRID AB_10827985 |
| Rabbit pAb anti-RelA/NF-kB p65 | Novus Biologicals | NB100-2176; RRID AB_535932 |
| Rabbit pAb anti-Goat-IgG HRP-conjugated | R&D Systems | HAF017; RRID AB_562588 |
| Goat pAb anti-Rabbit IgG (whole molecule) Peroxidase | Sigma-Aldrich | A0545; RRID AB_257896 |
| Rabbit mAb anti-phospho-SAPK/JNK MAPK (81E11) | Cell Signaling | 4668T; RRID AB_823588 |
| Rabbit mAb anti-phospho-p38 MAPK (D3F9) | Cell Signaling | 4511T; RRID AB_2139682 |
| Rabbit mAb anti-phospho-p44/42 (Erk1/2) (D13.14.4E) | Cell Signaling | 4370T; RRID AB_2315112 |
| Bacterial and virus strains | ||
| Escherichia coli (Migula) Castellani and Chalmers, C5 (Bort) | ATCC | ATCC 700973 |
| Chemicals, peptides, and recombinant proteins | ||
| ACK Lysing Buffer | Gibco | A1049201 |
| Blotting-Grade Blocker | Bio-Rad | 1706404 |
| di-ABZI | Invivogen | tlrl-diabzi |
| Dithiothreitol (DTT, Cleland’s Reagent) | Boston Bioproducts | #P-765-10G |
| Dulbecco’s Modification of Eagle’s Medium | Corning | 10-013-CV |
| Dulbecco’s Phosphate-Buffered Saline | Corning | 21-031-CV |
| Ethylenediamine tetraacetic acid (EDTA) | Boston Bioproducts | #BM-150 |
| Fetal Bovine Serum - Premium | R&D Systems | S11150 |
| Gentamicin | Millipore Sigma | G1397 |
| Glycerol | Fisher Scientific | BP229 |
| Halt Phosphatase Inhibitor Cocktail (100x) | Thermo-Fisher | 1862495 |
| Halt Protease Inhibitor Cocktail (100x) | Thermo-Fisher | 87786 |
| HEPES | Thermo-Fisher | 15630106 |
| Interferon-Gamma, Recombinant | BioLegend | 575308 |
| Iodoacetamide (IAA) | Sigma-Aldrich | I6125 |
| L-Glutamine | Gibco | 25030-081 |
| LB Broth (Miller) | Sigma-Aldrich | L3522 |
| LB Broth with Agar (Miller) | Sigma-Aldrich | L3147 |
| NaF | Sigma-Aldrich | 201154 |
| NaVO4 | Sigma-Aldrich | 567540 |
| NP-40 (Nonidet P-40 Substitute) | Boston Bioproducts | #P-877 |
| Pam3CSK4 | invivogen | tlrl-pms |
| Penicilin Streptomycin Solution, 100x | Corning | 30-002-CI |
| PF-06650833 | Tocris | 6373 |
| Pierce™ Lane Marker Reducing Sample Buffer | Thermo-Fisher | 39000 |
| Poly(I:C) HMW | Invivogen | tlr-pic |
| Protein G Sepharose 4 Fast Flow | Millipore Sigma | GE17-0618-01 |
| R848 (Resiquimod) | Invivogen | tlrl-r848 |
| Sodium Chloride | Sigma-Aldrich | S5886 |
| TMB Substrate Reagent Set | BD | 555214 |
| Tris hydrochloride | Roche | 10812846001 |
| TUBE2 Agarose | LifeSensors | UM-0402-1000 |
| Tween-20 | Boston Bioproducts | #P-934 |
| Ultrapure LPS, E. coli O111:B4 | Invivogen | tlrl-3pelps |
| β-Glycerophosphate disodium | Santa Cruz | sc-203323 |
| Critical commercial assays | ||
| CytoTox 96(R) Non-Radioactive Cytotoxicity Assay | Promega | G1780 |
| Clarity Max Western ECL Substrate | Bio-Rad | 1705062 |
| NE-Per™ Nuclear and Cytoplasmic Extraction Reagents | Thermo-Fisher | 78833 |
| IL-12/IL-23 p40 (Total) Mouse Uncoated ELISA Kit | Invitrogen | 88-7120-88 |
| Mouse CCL5/RANTES DuoSet ELISA | R&D Systems | DY478 |
| Mouse IFN-gamma DuoSet ELISA | R&D Systems | DY485 |
| Mouse TNF-alpha DuoSet ELISA | R&D Systems | DY410 |
| NF-κB (p65) Transcription Factor Assay Kit | Cayman Chemical | 10007889 |
| Nitric Oxide Assay Kit (Colorimetric) | Abcam | ab65328 |
| Deposited data | ||
| Raw data | This paper | https://doi.org/10.17632/tt3grr9zj7.1 |
| Software and algorithms | ||
| GraphPad Prism | Graphpad Software | www.graphpad.com |
| FlowJo | BD | www.flowjo.com |
| ImageJ | NIH | imagej.nih.gov/ij/ |
Highlights.
IRAK1 and IRAK2 are partially redundant in TLR4 signaling, but not in TLR7 signaling
IRAK4 kinase activity is required for TLR4/MYD88 signaling
TRIF and MYD88 require the IRAK4 scaffold for TRAF6 activation
TRIF-mediated TRAF3 activation occurs independently of IRAKs
ACKNOWLEDGMENTS
We are grateful to Larissa Pereira, Dr. Theresa Ramalho, and Dr. Rosane DeOliveira for the technical support. This work was partially supported by the National Institutes of Health (R01NS098747, R01AI079293, R01AI060025, 2U19 AI089681); Biotechnology and Biological Sciences Research Council (BB/V000276/1); Fundação de Amparo a Pesquisa do Estado de São Paulo (Fapesp, 2016/23618-8), Brazilian National Institute of Science and Technology for Vaccines granted by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq)/Fundação de Amparo à Pesquisa do Estado de Minas Gerais (Fapemig)/Coordenação de Aperfeiçoamento de Pessoal de Ensino Superior (CAPES) (465293/2014-0).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111225.
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
REFERENCES
- Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, and Akira S (1998). Targeted Disruption of the MyD88 Gene Results in Loss of IL-1- and IL-18-Mediated Function. Immunity 9, 143–150. 10.1016/S1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- Béla SR, Dutra MS, Mui E, Montpetit A, Oliveira FS, Oliveira SC, Arantes RME, Antonelli LR, Mcleod R, and Gazzinelli RT (2012). Impaired innate immunity in mice deficient in interleukin-1 receptor-associated kinase 4 leads to defective type 1 t cell responses, b cell expansion, and enhanced susceptibility to infection with toxoplasma gondii. Infect. Immun 80, 4298–4308. 10.1128/IAI.00328-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bin LH, Xu LG, and Shu HB (2003). TIRP, a Novel Toll/Interleukin-1 receptor (TIR) Domain-containing Adapter Protein Involved in TIR Signaling. J. Biol. Chem 278, 24526–24532. 10.1074/jbc.M303451200. [DOI] [PubMed] [Google Scholar]
- Bortolussi R, Ferrieri P, and Wannamaker LW (1978). Dynamics of Escherichia coli infection and meningitis in infant rats. Infect. Immun 22, 480–485. 10.1128/iai.22.2.480-485.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, Xiong J, Takeuchi M, Kurama T, and Goeddel DV (1996). TRAF6 is a signal transducer for interleukin-1. Nature 383, 443–446. 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- Chen L-F, and Greene WC (2004). Shaping the nuclear action of NF-κB. Nat. Rev. Mol. Cell Biol 5, 392–401. 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- Cheng H, Addona T, Keshishian H, Dahlstrand E, Lu C, Dorsch M, Li Z, Wang A, Ocain TD, Li P, Parsons TF, Jaffee B, and Xu Y (2007). Regulation of IRAK-4 kinase activity via autophosphorylation within its activation loop. Biochem. Biophys. Res. Commun 352, 609–616. 10.1016/j.bbrc.2006.11.068. [DOI] [PubMed] [Google Scholar]
- Collart MA, Baeuerle P, and Vassalli P (1990). Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol. Cell. Biol 10, 1498–1506. 10.1128/mcb.10.4.1498-1506.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conze DB, Wu C-J, Thomas JA, Landstrom A, and Ashwell JD (2008). Lys63-Linked Polyubiquitination of IRAK-1 Is Required for Interleukin-1 Receptor- and Toll-Like Receptor-Mediated NF-κB Activation. Mol. Cell. Biol 28, 3538–3547. 10.1128/mcb.02098-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nardo D, Balka KR, Cardona Gloria Y, Rao VR, Latz E, and Masters SL (2018). Interleukin-1 receptor-associated kinase 4 (IRAK4) plays a dual role in myddosome formation and Toll-like receptor signaling. J. Biol. Chem 293, 15195–15207. 10.1074/jbc.RA118.003314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S, Karim F, Kiessu E, Cushing L, Lin L-L, Ghandil P, Hoarau C, Casanova J-L, Puel A, and Rao VR (2018). Mechanism of dysfunction of human variants of the IRAK4 kinase and a role for its kinase activity in interleukin-1 receptor signaling. J. Biol. Chem 293, 15208–15220. 10.1074/jbc.RA118.003831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deliz-Aguirre R, Cao F, Gerpott FHU, Auevechanichkul N, Chupanova M, Mun Y, Ziska E, and Taylor MJ (2021). MyD88 oligomer size functions as a physical threshold to trigger IL1R Myddosome signaling. J. Cell Biol 220, e202012071. 10.1083/jcb.202012071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrao R, Zhou H, Shan Y, Liu Q, Li Q, Shaw DE, Li X, and Wu H (2014). IRAK4 Dimerization and trans-Autophosphorylation Are Induced by Myddosome Assembly. Mol. Cell 55, 891–903. 10.1016/j.mol-cel.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KA, and Kagan JC (2020). Toll-like Receptors and the Control of Immunity. Cell 180, 1044–1066. 10.1016/j.cell.2020.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao S-M, and Maniatis T (2003a). IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol 4, 491–496. 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, Brint E, Dunne A, Gray P, Harte MT, McMurray D, Smith DE, Sims JE, Bird TA, and O’Neill LA (2001). Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature 413, 78–83. 10.1038/35092578. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, Monks B, Pitha PM, and Golenbock DT (2003b). LPS-TLR4 Signaling to IRF-3/7 and NF-κB Involves the Toll Adapters TRAM and TRIF. J. Exp. Med 198, 1043–1055. 10.1084/jem.20031023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery S, and Bowie AG (2010). The interleukin-1 receptor-associated kinases: Critical regulators of innate immune signalling. Biochem. Pharmacol 80, 1981–1991. 10.1016/j.bcp.2010.06.020. [DOI] [PubMed] [Google Scholar]
- Gazzinelli RT, and Denkers EY (2006). Protozoan encounters with Toll-like receptor signalling pathways: implications for host parasitism. Nat. Rev. Immunol 6, 895–906. 10.1038/nri1978. [DOI] [PubMed] [Google Scholar]
- Gohda J, Matsumura T, and Inoue J.i. (2004). Cutting Edge: TNFR-Associated Factor (TRAF) 6 Is Essential for MyD88-Dependent Pathway but Not Toll/IL-1 Receptor Domain-Containing Adaptor-Inducing IFN-β (TRIF)-Dependent Pathway in TLR Signaling. J. Immunol 173, 2913–2917. 10.4049/jimmunol.173.5.2913. [DOI] [PubMed] [Google Scholar]
- Häcker H, Redecke V, Blagoev B, Kratchmarova I, Hsu L-C, Wang GG, Kamps MP, Raz E, Wagner H, Häcker G, Mann M, and Karin M (2006). Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439, 204–207. 10.1038/nature04369. [DOI] [PubMed] [Google Scholar]
- Hayden MS, and Ghosh S (2008). Shared Principles in NF-κB Signaling. Cell 132, 344–362. 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, and Akira S (1999). Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol 162, 3749–3752. [PubMed] [Google Scholar]
- Jiang Z, Zamanian-Daryoush M, Nie H, Silva AM, Williams BRG, and Li X (2003). Poly(dI$dC)-induced Toll-like receptor 3 (TLR3)-mediated activation of NFκB and MAP kinase is through an interleukin-1 receptor-associated kinase (IRAK)-independent pathway employing the signaling components TLR3-TRAF6-TAK1-TAB2-PKR. J. Biol. Chem 278, 16713–16719. 10.1074/jbc.M300562200. [DOI] [PubMed] [Google Scholar]
- Kagan JC, Su T, Horng T, Chow A, Akira S, and Medzhitov R (2008). TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat. Immunol 9, 361–368. 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawagoe T, Sato S, Jung A, Yamamoto M, Matsui K, Kato H, Uematsu S, Takeuchi O, and Akira S (2007). Essential role of IRAK-4 protein and its kinase activity in Toll-like receptor-mediated immune responses but not in TCR signaling. J. Exp. Med 204, 1013–1024. 10.1084/jem.20061523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawagoe T, Sato S, Matsushita K, Kato H, Matsui K, Kumagai Y, Saitoh T, Kawai T, Takeuchi O, and Akira S (2008). Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat. Immunol 9, 684–691. 10.1038/ni.1606. [DOI] [PubMed] [Google Scholar]
- Keating SE, Maloney GM, Moran EM, and Bowie AG (2007). IRAK-2 participates in multiple Toll-like receptor signaling pathways to NFκB via activation of TRAF6 ubiquitination. J. Biol. Chem 282, 33435–33443. 10.1074/jbc.M705266200. [DOI] [PubMed] [Google Scholar]
- Kim TW, Staschke K, Bulek K, Yao J, Peters K, Oh KH, Vandenburg Y, Xiao H, Qian W, Hamilton T, Min B, Sen G, Gilmour R, and Li X (2007). A critical role for IRAK4 kinase activity in Toll-like receptor-mediated innate immunity. J. Exp. Med 204, 1025–1036. 10.1084/jem.20061825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenerman D, Gay NJ, Li B, Penczek PA, Hopkins L, Bryant CE, Moncrieffe MC, Bollschweiler D, Li B, Penczek PA, Hopkins L, and Bryant CE (2020). MyD88 Death-Domain Oligomerization Determines Myddosome Architecture : Implications for Toll-like Receptor Signaling. Struct. Des 28, 281–289.e3. 10.1016/j.str.2020.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo-Murai M, Hazeki K, Nigorikawa K, Omoto T, Inoue N, and Hazeki O (2008). IRAK-4-dependent Degradation of IRAK-1 is a Negative Feedback Signal for TLR-mediated NF-κB Activation. J. Biochem. (Tokyo) 143, 295–302. 10.1093/jb/mvm234. [DOI] [PubMed] [Google Scholar]
- Latty SL, Sakai J, Hopkins L, Verstak B, Paramo T, Berglund NA, Cammarota E, Cicuta P, Gay NJ, Bond PJ, et al. (2018). Activation of toll-like receptors nucleates assembly of the MyDDosome signaling hub. eLife 7, e31377–15. 10.7554/eLife.31377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SC, Lo YC, and Wu H (2010). Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 465, 885–890. 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LAJ, Golenbock D, and Bowie AG (2013). The history of Toll-like receptors — redefining innate immunity. Nat. Rev. Immunol 13, 453–460. 10.1038/nri3446. [DOI] [PubMed] [Google Scholar]
- Pattabiraman G, Murphy M, Agliano F, Karlinsey K, and Medvedev AE (2018). IRAK4 activity controls immune responses to intracellular bacteria Listeria monocytogenes and Mycobacterium smegmatis. J. Leukoc. Biol 104, 811–820. 10.1002/JLB.2A1117-449R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauls E, Nanda SK, Smith H, Toth R, Arthur JSC, and Cohen P (2013). Two Phases of Inflammatory Mediator Production Defined by the Study of IRAK2 and IRAK1 Knock-in Mice. J. Immunol 191, 2717–2730. 10.4049/jimmunol.1203268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennini ME, Perkins DJ, Salazar AM, Lipsky M, and Vogel SN (2013). Complete Dependence on IRAK4 Kinase Activity in TLR2, but Not TLR4, Signaling Pathways Underlies Decreased Cytokine Production and Increased Susceptibility to Streptococcus pneumoniae Infection in IRAK4 Kinase–Inactive Mice. J. Immunol 190, 307–316. 10.4049/jimmunol.1201644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins DJ, Richard K, Hansen A-M, Lai W, Nallar S, Koller B, and Vogel SN (2018). Autocrine–paracrine prostaglandin E2 signaling restricts TLR4 internalization and TRIF signaling. Nat. Immunol 19, 1309–1318. 10.1038/s41590-018-0243-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard C, Puel A, Bonnet M, Ku C-L, Bustamante J, Yang K, Soudais C, Dupuis S, Feinberg J, Fieschi C, Elbim C, et al. (2003). Pyogenic Bacterial Infections in Humans with IRAK-4 Deficiency. Science 299, 2076–2079. 10.1126/science.1081902. [DOI] [PubMed] [Google Scholar]
- Qin J, Jiang Z, Qian Y, Casanova J-L, and Li X (2004). IRAK4 Kinase Activity Is Redundant for Interleukin-1 (IL-1) Receptor-associated Kinase Phosphorylation and IL-1 Responsiveness. J. Biol. Chem 279, 26748–26753. 10.1074/jbc.M400785200. [DOI] [PubMed] [Google Scholar]
- Sakai J, Cammarota E, Wright JA, Cicuta P, Gottschalk RA, Li N, Fraser IDC, and Bryant CE (2017). Lipopolysaccharide-induced NF-κB nuclear translocation is primarily dependent on MyD88, but TNFα expression requires TRIF and MyD88. Sci. Rep 7, 1428. 10.1038/s41598-017-01600-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjabi S, Hoffmann A, Liou HC, Baltimore D, and Smale ST (2000). Selective requirement for c-Rel during IL-12 P40 gene induction macrophages. Proc. Natl. Acad. Sci. USA 97, 12705–12710. 10.1073/pnas.230436397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai M, Tatematsu M, Oshiumi H, Funami K, Matsumoto M, Hatakeyama S, and Seya T (2010). Direct binding of TRAF2 and TRAF6 to TICAM-1/TRIF adaptor participates in activation of the Toll-like receptor 3/4 pathway. Mol. Immunol 47, 1283–1291. 10.1016/j.molimm.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Sato S, Sugiyama M, Yamamoto M, Watanabe Y, Kawai T, Takeda K, and Akira S (2003). Toll/IL-1 Receptor Domain-Containing Adaptor Inducing IFN-β (TRIF) Associates with TNF Receptor-Associated Factor 6 and TANK-Binding Kinase 1, and Activates Two Distinct Transcription Factors, NF-κB and IFN-Regulatory Factor-3, in the Toll-Like Receptor S. J. Immunol 171, 4304–4310. 10.4049/jimmunol.171.8.4304. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Hertzog PJ, Ravasi T, and Hume DA (2004). Interferon-γ: an overview of signals, mechanisms and functions. J. Leukoc. Biol 75, 163–189. 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- Silverman N, and Maniatis T (2001). NF-κB signaling pathways in mammalian and insect innate immunity. Genes Dev. 15, 2321–2342. 10.1101/gad.909001. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Suzuki S, Duncan GS, Millar DG, Wada T, Mirtsos C, Takada H, Wakeham A, Itie A, Li S, Penninger JM, Wesche H, Ohashi PS, Mak TW, and Yeh WC (2002). Severe impairment of interleukin-1 and toll-like receptor signalling in mice lacking IRAK-4. Nature 416, 750–756. 10.1038/nature736. [DOI] [PubMed] [Google Scholar]
- Tan Y, and Kagan JC (2019). Innate Immune Signaling Organelles Display Natural and Programmable Signaling Flexibility. Cell 177, 384–398.e11, e11. 10.1016/j.cell.2019.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, and Kagan JC (2018). Biochemical Isolation of the Myddosome from Murine Macrophages. Methods Mol. Biol 1714, 79–95. 10.1007/978-1-4939-7519-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Zanoni I, Cullen TW, Goodman AL, and Kagan JC (2015). Mechanisms of Toll-like Receptor 4 Endocytosis Reveal a Common Immune-Evasion Strategy Used by Pathogenic and Commensal Bacteria. Immunity 43, 909–922. 10.1016/j.immuni.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura N, Saitoh S, Matsumoto F, Akashi-Takamura S, and Miyake K (2008). Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem. Biophys. Res. Commun 368, 94–99. 10.1016/j.bbrc.2008.01.061. [DOI] [PubMed] [Google Scholar]
- Thomas JA, Allen JL, Tsen M, Dubnicoff T, Danao J, Liao XC, Cao Z, and Wasserman SA (1999). Impaired Cytokine Signaling in Mice Lacking the IL-1 Receptor-Associated Kinase. J. Immunol 163, 978–984. [PubMed] [Google Scholar]
- Trinchieri G (1995). Interleukin-12: A Proinflammatory Cytokine with Immuno-regulatory Functions that Bridge Innate Resistance and Antigen-Specific Adaptive Immunity. Annu. Rev. Immunol 13, 251–276. 10.1146/annurev.iy.13.040195.001343. [DOI] [PubMed] [Google Scholar]
- Verstak B, Stack J, Ve T, Mangan M, Hjerrild K, Jeon J, Stahl R, Latz E, Gay N, Kobe B, Bowie AG, and Mansell A (2014). The TLR signaling adaptor TRAM interacts with TRAF6 to mediate activation of the inflammatory response by TLR4. J. Leukoc. Biol 96, 427–436. 10.1189/jlb.2a0913-487r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku C-L, Chrabieh M, Mustapha IB, Ghandil P, Camcioglu Y, et al. (2008). Pyogenic Bacterial Infections in Humans with MyD88 Deficiency. Science 321, 691–696. 10.1126/science.1158298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bernuth H, Picard C, Puel A, and Casanova J-L (2012). Experimental and natural infections in MyD88- and IRAK-4-deficient mice and humans. Eur. J. Immunol 42, 3126–3135. 10.1002/eji.201242683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Xiao H, Affolter J, Kim TW, Bulek K, Chaudhuri S, Carlson D, Hamilton T, Mazumder B, Stark GR, Thomas J, and Li X (2009). Interleukin-1 Receptor-associated Kinase 2 Is Critical for Lipopolysaccharide-mediated Post-transcriptional Control. J. Biol. Chem 284, 10367–10375. 10.1074/jbc.M807822200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, and Akira S (2003a). Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301, 640–643. 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, Takeuchi O, Takeda K, and Akira S (2003b). TRAM is specifically involved in the Toll-like receptor 4–mediated MyD88-independent signaling pathway. Nat. Immunol 4, 1144–1150. 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
- Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, Granucci F, and Kagan JC (2011). CD14 controls the LPS-induced endocytosis of toll-like receptor 4. Cell 147, 868–880. 10.1016/j.cell.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The quantitative data and uncropped immunoblots generated in this study are available at Mendeley Data: https://doi.org/10.17632/tt3grr9zj7.1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.