

Abstract
Background
Enoyl-CoA hydratase short-chain 1 (ECHS1) is a key mitochondrial enzyme that is involved in valine catabolism and fatty acid beta-oxidation. Mutations in the ECHS1 gene lead to enzymatic deficiency, resulting in the accumulation of certain intermediates from the valine catabolism pathway. This disrupts the pyruvate dehydrogenase complex and the mitochondrial respiratory chain, with consequent cellular damage. Patients present with a variable age of onset and a wide spectrum of clinical features. The Leigh syndrome phenotype is the most frequently reported form of the disease. Herein, we report a case of a male with ECHS1 deficiency who was diagnosed at 8 years of age. He presented severe dystonia, hyperlordosis, moderate to severe kyphoscoliosis, great difficulty in walking, and severe dysarthria. A valine-restricted and total fat-restricted diet was considered as a therapeutic option after the genetic diagnosis. An available formula that restricted branched-chain amino acids and especially restricted valine was used. We also restricted animal protein intake and provided a low-fat diet that was particularly low in dairy fat.
Results
This protein- and fat-restricted diet was initiated with adequate tolerance and adherence. After three years, the patient noticed an improvement in dystonia, especially in walking. He currently requires minimal support to walk or stand. Therefore, he has enhanced his autonomy to go to school or establish a career for himself. His quality of life and motivation for treatment have greatly increased.
Conclusions
There is still a substantial lack of knowledge about this rare disorder, especially knowledge about future effective treatments. However, early diagnosis and treatment with a valine- and fat-restricted diet, particularly dairy fat-restricted diet, appeared to limit disease progression in this patient with ECHS1 deficiency.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13023-022-02468-6.
Keywords: Children, Enoyl-CoA hydratase, Leigh syndrome, Diet, Valine
Background
Enoyl-CoA hydratase, short-chain 1 (ECHS1), EC 4.2.1.17, is also known as SCEH. It is a protein encoded by gene ID 1892, which is located on chromosome 10. The gene product is a member of the hydratase/isomerase superfamily that localizes to the mitochondrial matrix. It is broadly expressed in the liver, kidney, and adipose tissue, and its expression in 22 other tissues, including the whole gastrointestinal tract, brain, and numerous secretory glands, has been reported. Transcript variants utilizing alternative transcription initiation sites have been described [1].
ECHS1 catalyzes the hydration of the double bond between the second and third carbons of 2-trans-enoyl-coenzyme A (CoA) intermediates to L-3-hydroxy-acyl-CoAs [2, 3]. It catalyzes several metabolic pathways. In particular, this crotonase catalyzes the second step of beta-oxidation of fatty acids [4] and plays a key role in valine catabolism. Furthermore, it takes part in the fourth step of this metabolic pathway by catalyzing the hydration of methacrylyl-CoA to 3-hydroxyisobutyryl-CoA and the conversion of acryloyl-CoA to 3-hydroxypropyl-CoA [5, 6]. ECHS1 is also involved in the metabolism of isoleucine in the transformation of tiglyl-CoA to 2-methyl-3-hydroxybutyryl-CoA (Fig. 1).
Fig. 1.

Illustration of the short-chain fatty acid, odd-chain fatty acid and valine catabolic pathways, showing the formation of certain metabolites as a result of short-chain enoyl-CoA hydratase 1 (ECHS1) deficiency. The concept that 2,3-dihydroxy-2-methylbutyric acid originates from acryloyl-CoA was indirectly inferred based on Peters et al. [6]
Mutations in the ECHS1 gene (OMIM* 602292) result in ECHS1 deficiency, which is a rare autosomal recessive disorder that was first reported by Peters et al. [7] as a cause of Leigh syndrome (OMIM* 256000). Both the age at onset and clinical manifestations vary greatly from patient to patient. Masnada et al. [8] distinguished four phenotypes of ECHS1 deficiency based on clinical and neurological features. The first is of severe neonatal onset with a rapidly fatal course and white matter changes. The second is a severe infantile presentation with slower neurological deterioration and deep gray nuclei degeneration, developmental delay, pyramidal and extrapyramidal signs, feeding difficulties, and optic atrophy. The third phenotype is another slowly progressive juvenile form that is similar to the previous one but with less severity and mainly basal ganglia involvement. The fourth phenotype is a less common form of presentation that is based on paroxysmal exercise-induced dystonia with a normal neurological examination between these attacks and isolated degeneration of the pallidum on magnetic resonance imaging [8]. A characteristic magnetic resonance imaging (MRI) pattern of basal ganglia lesions has been reported, with small cysts in the putamen and pallidum [9]. Cardiac involvement [4] and cutis laxa are also clinical features of this disease [10].
Deficiency of ECHS1 usually leads to the accumulation of methacrylyl-CoA and acryloyl-CoA. These harmful substrates are highly reactive with sulfhydryl groups and tend to form adducts with cysteine or cysteamine [6, 7, 11]. High concentrations of these intermediate metabolites seem to cause dysfunction in the pyruvate dehydrogenase complex (PDC) and the mitochondrial respiratory chain, resulting in cellular damage [12]. Additionally, increased excretion of 2-methyl-2,3-dihydroxybutyric acid in urine is frequently observed [13]. S-(2-carboxypropyl)-cysteine, S-(2-carboxypropyl)-cysteamine and N-acetyl-S-(2-carboxypropyl)-cysteine have also been proposed as useful diagnostic markers [2, 14]. Other patients present with increased plasma levels of lactate and pyruvate in a conserved ratio [15] (Fig. 1).
Over 50 patients from more than 35 families have been reported to date [9, 16]. Whole-exome sequencing (WES) has increased the recognition of pathogenic variants in ECHS1. Most of these variants are missense variants [3], suggesting that entire loss-of-function mutations may be incompatible with life [17].
The majority of patients with ECHS1 deficiency are initially diagnosed by their clinical features and do not undergo molecular diagnosis until later in life. They usually receive standard treatments that are used for other mitochondrial diseases. Anecdotal reports of treatment with N-acetylcysteine to detoxify the reactive metabolites of methacrylyl-CoA and acryloyl-CoA have been published [18]. A valine-restricted diet is a rational biochemical approach. Recently, a valine-restricted diet appeared to be beneficial for five patients with ECHS1 defects, improving the biochemical and clinical manifestations of these disorders [19]. The administration of cofactors, i.e., coenzyme Q10, vitamins C and E, riboflavin, creatine monohydrate and other antioxidants, that are used in mitochondrial disorders with limited evidence of benefit has also been considered [18, 20, 21]. Furthermore, restricting the consumption of specific fatty acids has been tested in several FAO disorders [22]; this approach aims to reduce the synthesis of toxic intermediates [23]. In addition, although high-fat, reduced-carbohydrate ketogenic diets have been used in some cases of moderate ECHS1 deficiency, they have been ineffective in improving the symptoms in severe cases of the disease [24, 25].
Based on the previous background, here, we report the case of an ECHS1-deficient child and how the use of a valine- and fat-restricted diet partially improved his condition.
Materials and methods
Participant
The patient was a boy with ECHS1 deficiency who was diagnosed in 2017 in the Pediatric Neurology Unit of a third-level hospital in Spain. He was the first born from a set of twins and was delivered by scheduled cesarean section in 2006. He was conceived via the in vitro fertilization of two of the mother's eggs and the pregnancy was uneventful. His twin sister is healthy. His parents are also healthy, and their marriage is not consanguineous.
He had normal neurological development up to 15 months of age. Ten days after receiving a vaccination, he suffered from neurological regression, with acute ataxia onset, gait loss, and pyramidal signs. In addition, severe dystonia affected his daily activities, including walking, handling, and speech.
Generally, he was smiling, happy, and engaged in good social interactions. Feeding and sleep were both adequate, and the height-to-weight ratio was normal. He received speech therapy, physical therapy, and psycho-pedagogical support.
Molecular genetic analysis
This patient was diagnosed from a multicenter project of patients with basal ganglia diseases of unknown cause [9]. The ECHS1 variants were validated and segregated using Sanger sequencing. The genetic analysis of a whole-blood sample identified two heterozygous variants, c.123_124del p.(Gly42Glufs*3) and c.830C > T p.(Thr277Ile), in the ECHS1 gene (NM_004092.3, sequencing). Therefore, the result was reported to be compatible with the diagnosis of Leigh syndrome.
Genomic DNA was extracted from peripheral blood samples using standard methods. The sequencing of all exons and in the flanking intronic regions ± 20 bp was performed by Centogene (Rostock, Germany) using the CentoXome Gold protocol and guaranteeing a depth of reads of 100X. Typically, 97–98% of the target bases are > 10 × covered. Next, a proprietary bioinformatics method was applied that includes annotation of bases, alignment to the hg19 reference human genome (Genome Reference Consortium GRCh37), filtering of low-quality reads and possible artifacts, and annotation of variants (Centogene, Rostock, Germany). For the medical evaluation, we considered all the pathogenic variants reported in The Human Gene Database (HGMD®) and ClinVar and all variants with minimal allelic frequency (MAF) below 1% in Genome Aggregation Database (gnomAD). All possible inheritance patterns were considered based on the clinical information contributed. Only variants related to the phenotype of the patient were reported. The patient’s clinical information and family history were used to evaluate the identified variants. All identified variants were evaluated based on their pathogenicity and causality, and those associated with the phenotype of the patient that were not benign or probably benign were reported.
Consistent with the genetic analysis, respiratory chain activity was decreased in the CII and CI + III complexes and PDC activity was decreased [9]. The results of the muscle biopsy study showed a deficit of mitochondrial respiratory chain complex I (level 8.03, pathological value < 10). Additionally, signs of mitochondrial proliferation, such as a citrate synthase value of 1353 (reference value 200–900), were observed. We only have qualitative data for PDC activity.
Physical examination and additional tests
After the confirmation of the diagnosis and just before starting treatment, the physical examination showed the progressive development of scanning speech, severe dystonia, ataxia, hypotonia with hyperlordosis and moderate to severe kyphoscoliosis, and the extremities were in internal rotation with great difficulty walking. No abnormal eye movements were observed. The Burke-Fahn-Marsden Dystonia Rating Scale (BFMDRS) showed an 18/30 disability score.
The Gross Motor Function Classification System (GMFCS) for functional motor impairment, psychomotor delay, and intellectual disability was determined to be level IV by principal clinical judgments.
Magnetic resonance imaging (MRI) performed in 2016 showed T2 hyperintensity in both the caudate and putamen nuclei, with more marked hyperintensity in the posterior and medial left putamen, suggesting cavitation. No brain atrophy or white matter involvement was observed (Fig. 2, Panels A and B).
Fig. 2.
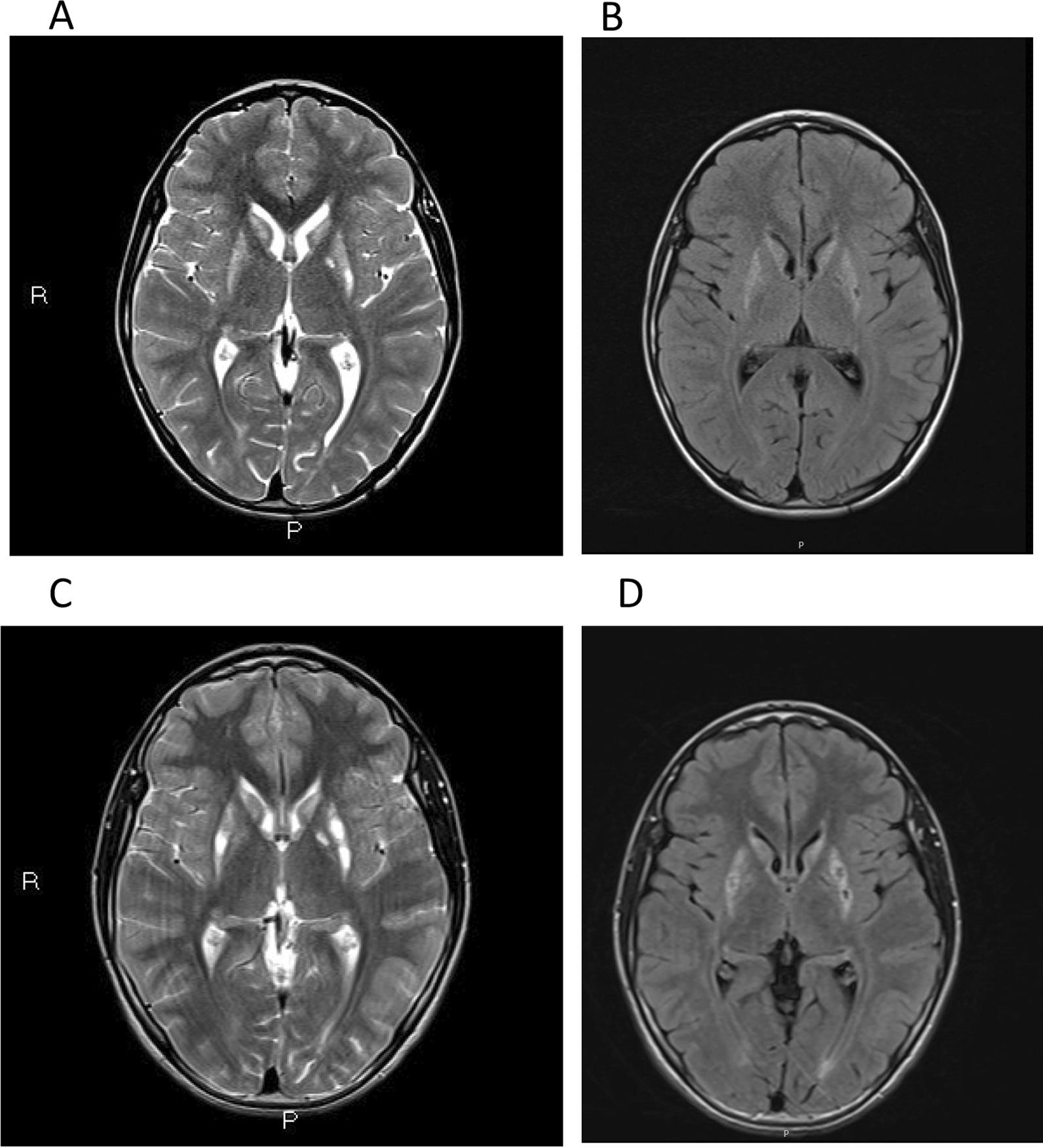
The axial T2 sequence in cerebral magnetic resonance at diagnosis (Panel A) and after 3 years of dietary intervention (Panel C) and the corresponding fluid-attenuated inversion recovery (FLAIR) images (Panel B, D)
Bilateral optic atrophy was detected after ophthalmic evaluation. Concerning the biochemical features, the venous acid–base analysis showed a base excess of 2.80 mmol/L, HCO3 27.20 mmol/L, pH 7.44 and lactate 1.10 mmol/L. Additionally, the cerebrospinal fluid (CSF) study, including amino acid and thiamine determination, was normal, and the amino acid and acylcarnitine profiles measured in the plasma and organic acids in urine were also normal. In particular, normal urinary excretion of 2-methyl-2,3-hydroxybutyric acid was found.
Treatment
After genetic diagnosis and based on the potentially affected metabolic pathways, the diet of this patient was modified. Valine and fat, particularly dairy fat, were restricted. This protein- and fat-restricted diet was initiated with adequate tolerance and adherence.
The nutritional plan included the consumption of a convenient, reduced volume, ready-to-drink protein substitute twice a day from 3 years of age. This substitute is also used for other congenital inborn errors of metabolism, i.e., methylmalonic and propionic acidemias. The product (MMA/PA PA cooler™) was supplied by Vitaflo®, Spain, a Nestlé Health Science company, in a 130 ml pouch (https://www.nestlehealthscience.es/vitaflo).
Table 1 shows the nutritional composition of the supplement. Indeed, the protein substitute provided all of the amino acids found in dietary protein except methionine, threonine, and valine and contained only a small amount of isoleucine. This protein substitute was used in combination with the amount of protein tolerated from the individual’s usual diet. The foods in this diet varied. However, the dairy products were from skimmed milk, and the diet was low in fatty foods. This diet also contained relatively high amounts of complex carbohydrates at each feeding, especially through the intake of bread and pasta. The natural proteins of this diet were provided by small quantities of dairy products, certain cereals, legumes (in a controlled way), gelatin, and a minimum quantity of fish. The maximum daily amount of these dietary proteins (in grams) were given to the family. In addition, free fruit and vegetables were given.
Table 1.
Composition of the ready-to-drink protein substitute (1 pouch of 130 ml) used for the dietary management of the ECHS1 case
| Energy (kcal) | 103 | Minerals | |
| Total amino acids (g) | 18.2 | Sodium (mg) | 127 |
| Protein equivalent (g) | 15.0 | Potassium (mg) | 235 |
| Carbohydrates (g)1 | 7.0 | Chlore (mg) | 182 |
| Sugars (g) | 5.9 | Calcium (mg) | 299 |
| Lipids (g) | 1.7 | Phosphorus (mg) | 267 |
| Saturated fat (g) | 0.33 | Magnesium v | 94 |
| DHA (mg) | 100 | Iron (mg) | 5.4 |
| EPA (mg) | 23 | Copper (mg) | 0.55 |
| Amino acids (g) | Zinc (mg) | 5.4 | |
| Alanine | 1.68 | Manganese (mg) | 0.8 |
| Arginine | 1.73 | Iodine (μg) | 63 |
| Aspartic acid | 3.0 | Molybdenum (μg) | 36 |
| Cystine | 0.73 | Selenium (μg) | 22 |
| Glutamine | 0.00 | Chromium (μg) | 22 |
| Glycine | 0.79 | Vitamins | |
| Histidine | 1.13 | A (μg) | 208 |
| Isoleucine | 0.05 | D (μg) | 7.5 |
| Leucine | 2.37 | E (mg) | 3.9 |
| Lysine | 1.85 | K (μg) | 25 |
| Methionine | 0.00 | C (mg) | 27 |
| Phenylalanine | 1.13 | Thiamine (B1) (mg) | 0.50 |
| Proline | 0.83 | Riboflavin (B2) (mg) | 0.57 |
| Serine | 1.25 | Niacin (mg) (B5) | 6.2 |
| Threonine | 0.00 | Pyridoxamine (B6) (mg) | 0.70 |
| Tryptophan | 0.47 | Folic acid (B9) (μg) | 100 |
| Tyrosine | 1.13 | B12 (μg) | 1.2 |
| Valine | 0.00 | Biotin (B7) | 47 |
| Carnitine (mg) | 17 | Pantothenic acid (B3) (mg) | 2.0 |
| Taurine (mg) | 33 | Choline (mg) | 150 |
EPA, eicosapentaenoic acid; DHA, docosahexaenoic acid
aSucrose, glucose syrup, and modified corn starch
A quantitative 24 h dietary record was evaluated during every visit. On average, the patient had a daily energy intake of 1312 kcal, of which 16.7% was derived from dietary protein (54.8 g/d total protein, of which 29.9 g of protein equivalent was obtained from the amino acid supplement), 70.4% (231 g/d) was derived from carbohydrates, and 12.9% (18.7 g/d) was derived from lipids.
Results
After two years of this treatment, the child exhibited better stability. He required minimal support to walk or stand, which enhanced his autonomy at school and improved his hygiene and personal care routines. He had no adverse effects and had a good nutritional profile and growth.
After three years of treatment, his current weight is 29.5 kg (percentile 4), his height is 144 cm (percentile 10), his body mass index (BMI) is 14.04 with a prepubertal status. The family continues to perform dietary calculations as expected, complying with what was prescribed. Clinical evaluation and neurological tests are carried out every 6 months. Gross motor function has improved, and his GMFCS classification was reduced from level IV to level III. The child now shows greater stability. He requires less support when walking or standing, his language speed has increased, and he has more autonomy (i.e., he is now able to get dressed and shower alone). Minor hyperlordosis and kyphoscoliosis and fewer dystonic positions were observed, improving the ability of the patient to walk. He is even able to walk upright for 1 h a day. He turns with less difficulty, and he now has better fine motor skills and minor tremors. For example, he is now able to eat a yogurt. He has a better understanding and is conscientious of the concepts of his interest. His family seeks social integration. He is adapting to his high-school class, where he usually moves autonomously with a tricycle. On the BFMDRS, the disability score after 3 years improved from 18/30 to 9/30. His quality of life and motivation to continue this treatment have greatly increased. After treatment, there was no significant progression as evaluated by neuroimaging. In fact, MRI showed more marked hyperintensities in both the caudate and putamen nuclei, suggesting increased cavitation (Fig. 2, Panels C and D).
A video with images of the patient immediately after diagnosis and 3 years after treatment is included in the Additional file 1: Video.
Regarding the biochemical features, Table 2 shows the plasma concentrations of valine, leucine, and isoleucine at the time of diagnosis and after 1.5 and 3 years of follow-up. Valine was decreased during the clinical course compared with the baseline level, while isoleucine and leucine remained fairly constant. In the last analysis, the excretion of 2-methyl-2,3-hydroxybutyric acid was 1 nmol/mol creatinine, remaining within the normal values (< 2 nmol/mol creatinine). Additionally, no urinary methacrylyl-glycine was detected. In addition, the S-(2-carboxypropyl) cysteine concentration was 0.18 mmol/mol creatinine (normal value < 0.39) and that of S-(2-carboxyethyl)-cysteine was 0.23 mmol/mol creatinine (normal value < 0.15). However, N-acetyl-S-(2-carboxyethyl)-cysteine, N-acetyl-S-(2-carboxypropyl)-cysteine and S-(2-carboxypropyl)-cysteine carnitine were undetectable. In addition, the plasma levels of acylcarnitines were within the range of normality, and the levels of C4-OH [3-OH-butyryl/3-OH-isobutyryl-carnitine] (0.03 µmol/L) were also within the range of normality (0.01–0.06 µmol/L).
Table 2.
The plasma concentrations of branched-chain amino acids in a case of ECHS1 deficiency treated with a valine- and fat-restricted diet
| Amino acid (µmol/L) | Diagnosis | 1.5 y after diagnosis | 3 y after diagnosis | Range of normality |
|---|---|---|---|---|
| Valine | 158 | 95 | 104 | 219 ± 47 |
| Isoleucine | 39 | 40 | 36 | 58 ± 15 |
| Leucine | 88 | 92 | 93 | 115 ± 26 |
Discussion
The main finding of the present study was that treatment of male child with an ECHS1 deficiency with a valine- and fat-restricted diet resulted in an improvement in his quality of life and the amelioration of clinical features. In fact, after three years of treatment, the patient showed greater stability, required less support when walking or standing, had improved language skills, and was more autonomous.
In our patient, at the time of diagnosis, MRI detected T2 hyperintensity in the caudate and putamen nuclei. However, no brain atrophy or white matter involvement was observed. In addition, our patient exhibited bilateral optic atrophy soon after the diagnosis. In ECHS1 deficiency patients, microcephaly and impaired vision are usually detected by physical examination [9]. These patients also have difficulty fixing or following objects, nystagmus, and optic nerve atrophy, which could explain vision problems [26]. In addition, they show difficulties in speech. However, they are aware of their surroundings and communicate in other ways, such as by sounds or smiling. These patients have poor head control, so they are unable to sit up or ambulate. This symptom occurred in our patient.
Although clinical and biochemical features could suggest ECHS1 deficiency, the diagnosis of this condition is mostly established by the identification of biallelic pathogenic variants in the ECHS1 gene by molecular genetic testing. In the present case report, the first variant in the ECHS1 gene, c.123_124del p. (Gly42Glufs*3), alters the reading frame at codon 42. The new reading frame ends 2 positions later in a stop codon. It is classified as likely pathogenic (class 2) according to the American College of Medical Genetics and Genomics (ACMG) recommendations. The second variant in the ECHS1 gene, c.830C > T p.(Thr277Ile), causes an amino acid change from threonine to isoleucine at position 277. In ClinVar, this variant is listed as likely pathogenic but does not provide evidence supporting its pathogenicity (clinical studies, ID: 397,546). Therefore, it is classified as a variant of uncertain significance (class 3) according to ACMG recommendations.
Urinary excretion of 2-methyl-2,3-dihydroxybutyric acid and other metabolites, such as S-(2-carboxypropyl)-cysteine, S-(2-carboxypropyl)-cysteamine, and N-acetyl-S-(2-carboxypropyl)-cysteine, is useful to diagnose ECHS1 deficiency. These compounds have been proposed as helpful biomarkers for initial screening processes [6]. Despite these observations, neither 2-methyl-2,3-dihydroxybutyric acid nor abnormal fatty acylcarnitines and organic acids in the urine were detected in our patient. Moreover, lactate levels in the plasma were not significantly elevated (1.10 mmol/L), adding evidence to the relatively mild activity of the PDC.
A definitive cure for the ECHS1 disorder has not yet been discovered. Despite the encouraging results that some studies have reported about clinical improvement in adapting patients’ diets, there is scarce literature about dietary treatment. Loupatty et al. [27] first suggested that treating patients with frequent carbohydrate-rich meals, in which the main source of energy is carbohydrates and protein intake is restricted, could be a good therapeutic option for 3-hydroxyisobutyryl-CoA hydrolase (HIBCH) deficiency. HIBCH deficiency is a mitochondrial disease that is similar to ECHS1 deficiency. Later, Soler-Alfonso et al. [28] showed that a HIBCH-deficient child clinically responded to a valine-restricted diet using a targeted formula for maple syrup urine disease. Given the clinical and pathophysiological similarities between HIBCH and ECHS1 deficiencies, it was thought that the same restricted dietary treatment could be potentially beneficial in ECHS1 subjects. According to what had been assumed, some studies demonstrated improvement in clinical outcomes after a valine-restricted diet with N-acetylcysteine supplementation [29, 30]. Recently, Abdenur et al. [19] showed enhancement in both clinical and biochemical manifestations, with a decrease in S-(2-carboxypropyl)-cysteine in patients with either HIBCH or ECHS1 deficiencies.
In the present case, dietary treatment was initiated after several tolerance tests using an available formula that was designed for patients with methylmalonic and propionic acidemia (MMA/PA). This MMA/PA cooler 15 is a ready-to-drink methionine-free, threonine-free, valine-free and low isoleucine medical food, and it was given in different amounts during the day. In addition, the patient’s diet was restricted to animal proteins, legumes, and defatted or skimmed products. However, the intake of natural proteins and carbohydrates had to be ensured. We set the consumption limit for high-biological value proteins at 1 g/kg/day. Plasma amino acids were monitored during treatment, and the family to was asked to adjust the diet when low levels were detected.
In our case, the aim of specifically restricting valine was focused on reducing neurotoxicity due to enzymatic blockade and the accumulation of metabolites in the catabolic pathway.
Mitochondria play a key role in the consumption of energy in eukaryotic cells by oxidizing fatty acids and sugars to generate ATP. Mitochondrial fatty acid β-oxidation (FAO) and oxidative phosphorylation (OXPHOS) are two key pathways involved in this process, and ECHS1 is a key enzyme involved in mitochondrial FAO. For the very-long-chain and long-chain fatty acid (C14–24) fatty acyl-CoA esters, the second, third and fourth steps of β-FAO are performed by the multidomain mitochondrial trifunctional protein (MTP). However, medium-(C8–C12) and short-chain (C4–C6) fatty acyl-CoA esters are metabolized via a different set of enzymes that perform hydration (step 2; ECHS1).
Disruption of FAO can cause human disease. However, patients with deficiencies in the FAO enzyme ECHS1 are typically diagnosed with Leigh syndrome, which is normally associated with OXPHOS dysfunction [31]. In our patient, the result of the muscle biopsy study showed a deficit of the mitochondrial respiratory chain complex I (level 8.03, pathological value < 10) together with signs of mitochondrial proliferation, such as a citrate synthase value of 1352.98 (reference value 200–900). For PDC activity, we only have qualitative data that supports mild activity. Interestingly, there may be a correlation between the ECHS1 phenotypic severity and PDC activity [31]. Patients with low PDC activity also have high lactate levels. Several of these patients present with a more severe prognosis [4, 7, 32], including death within 48 h of birth [2, 33]. In contrast, milder cases of ECHS1 deficiency do not show reduced PDC activity or lactic acidosis [6, 12, 34, 35].
Since ECHS1 deficiency is associated with severe acidosis and low ATP from impaired aerobic oxidation usually occurs, we hypothesized that a diet with a relatively low content of fat and a particularly low content of medium- and short-chain fatty acids might be beneficial for patients with impaired OXPHOS.
Restricting the consumption of specific fatty acids has been tested in several FAO disorders [22], aiming to reduce the synthesis of toxic intermediates [23]. In addition, although high-fat, reduced-carbohydrate ketogenic diets have been used in some cases of moderate ECHS1 deficiency, they have been ineffective in improving the symptoms of the disease in severe cases [24, 33].
Bovine milk lipids contain approximately 12 fatty acids in amounts greater than 1%. Butyric acid (4:0) and caproic acid (6:0) are the most important substrates for ECHS1, and they represent 11.8% and 4.6%, respectively, of the total triacylglycerol fatty acids. In addition, all of the 4:0, 83% of 6:0 and 63% of 8:0 are esterified at the sn-3 position and are easily digested by gastric and pancreatic lipases [36, 37]. These short and medium-chain fatty acids reach the systemic circulation and freely diffuse across the plasma membrane. They enter directly into mitochondria without the assistance of the carnitine shuttle system [38]. They are rapidly oxidized in the liver, muscle, and other peripheral tissues. Hence, reducing the amount of total fat, particularly dairy fat, in the diet could reduce the impact of FAO on the symptoms of mitochondrial ECHS1 deficiency.
A limitation of this study is that important biochemical markers, such as S-(2-carboxypropyl)-cysteine, S-(2-carboxyethyl)-cysteine, N-acetyl-S-(2-carboxyethyl)-cysteine, N-acetyl-S-(2-carboxypropil)-cysteine, and S-(carboxypropyl)-cysteine carnitine, were not measured at the beginning of dietary treatment, so we cannot compare them with current levels. Another limitation of our study is that the patient has a mild phenotype, as evidenced by low levels of altered metabolites as well as an improved clinical course. In only two years of treatment, the patient has progressed from great difficulty walking, severe dystonia and hypotonia with severe hyperlordosis and kyphoscoliosis to showing better stability, less dystonic positions, and much more autonomy in walking and daily routine. He has also demonstrated great social integration in school due to more fluent language and better understanding.
In this rare disease, clinical results should be considered with caution because there is no literature about how to perform a clinical follow-up and what tests could be performed to best carry out a specific evaluation to control its progression or improvement. The validated international tests used, BFMDRS and GMFCS, were applied to perform a neurological evaluation based on motor function and disability. The BFMDRS gives a disability score, and it is a universally applied instrument for the quantitative assessment of dystonia in both children and adults. It should be used longitudinally, as it was used for this patient, every 6 months, and the age of the patient should be considered. The GMFCS measures the changes in gross motor function in children with cerebral palsy or similar symptoms. Therefore, it is used both to describe a child’s current abilities and to quantify changes in function over time as a result of development, therapy, or training.
In addition, extensive biochemical evaluation, particularly those that are related to the metabolites associated with ECHS1 deficiency, was not performed by clinicians before treatment. This is because of difficulties in the hospital in carrying out specific biochemical analyses but also because of scarce knowledge about the biochemical pathways related to this ultrarare disease. Moreover, the direct effect of a low-valine and low-fat diet on those metabolites in the brain cannot be directly known, and perhaps these metabolites are found in different concentrations in the brain than in blood. Therefore, whether the diet resulted in biochemical amelioration, as it seems to have done to an extent, cannot be proven in the present case.
Indeed, the clinical improvements after 3 years on this low-valine, low-fat diet could be a fortuitous result, as we did not obtain clear biochemical evidence of improvement. However, the progression of the patient, prior to the genetic diagnosis, was evident, and clinical stabilization with some improvements would seem to be associated with our reasonable assumption of a direct effect of the diet. In the future, we hope to be able to provide new evidence related to the continuity of the diet in the long term since it does not cause adverse effects. Through these future studies, its effect can be confirmed.
Conclusion
There is still a substantial lack of knowledge about this rare disorder. especially knowledge of future effective treatments. The dietary approach of ECHS1 has shown benefits in multiple patients. However, the correct formulation between a valine-restricted diet or a combined valine-fat restriction will need additional patient information. Indeed, early diagnosis and the therapeutic use of a valine- and fat-restricted diet supported by a specific and longitudinal clinical evaluation and metabolite measurements could help to confirm its effects on clinical disease progression in patients with ECHS1 deficiency.
Supplementary Information
Additional file 1. This video shows the clinical evolution of the patient with ECHS1 deficiency before and 3 years after treatment with a low-valine and low-fat diet. One can observe how after treatment he shows better stability, less walking and turning difficulties, an improvement in fine motor skills and tremors.
Acknowledgements
The researchers thank the patient and his family and Raquel Muñoz Sánchez, a radiologist at the Reina Sofía University Hospital, for their participation in the present study.
Abbreviations
- ACMG
American College of Medical Genetics and Genomics
- BFMDRS
Burke-Fahn-Marsden Dystonia Rating Scale
- BMI
Body Mass Index
- CFS
Cerebrospinal fluid
- CoA
Coenzyme A
- C4OH
3-OH-isobutyryl carnitine
- ECHS1
Enoyl-CoA hydratase, short-chain 1
- FAO
Fatty acid oxidation
- GMFCS
Gross motor function classification system
- GnomAD
Genome aggregation database
- HGMD®
The human gene database
- HIBCH
3-Hydroxyisobutyryl-CoA hydrolase
- MAF
Minimal allelic frequency
- MMA/PA
Methylmalonic and propionic acidemia
- MRI
Magnetic resonance imaging
- MS
Mass spectroscopy
- MTP
Mitochondrial trifunctional protein
- OXPHOS
Oxidative phosphorylation
- WES
Whole exome sequencing
Author contributions
AG designed the manuscript and contributed to the biochemical explanation of the relationship between biochemical events and dietary responses. S.P. and MG-C obtained the clinical results and wrote the manuscript. KF-R included the information related to the diet. EL-L incorporated neurologic evaluations and data. LM-S, HB-M, and BP-D were responsible for the biochemical and molecular analyses. All the authors have reviewed and approved the final version of this manuscript.
Funding
The biochemical and genetic analyses were funded by Instituto de Salud Carlos III, Grant/Award Number: PI18/01319, CERCA Programme/Generalitat de Catalunya, Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR), Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), and Instituto de Salud Carlos III (ISCIII), Grant/Award Numbers: PI19/01310, PI16/01048.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its Additional files.
Declarations
Ethics approval and consent to participate
Written informed consent was obtained from the patient’s parents for participation in the study. All procedures were performed under the ethical standards of good clinical practice and were in accordance with the Declaration of Helsinki. This study was approved by the Research Ethics Committee of Córdoba.
Consent for publication
Written informed consent was obtained from the parents of our patient for the publication of this study and the use of the video.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014;13:397–406. doi: 10.1074/mcp.M113.035600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al Mutairi F, Shamseldin HE, Alfadhel M, Rodenburg RJ, Alkuraya FS. A lethal neonatal phenotype of mitochondrial short-chain enoyl-CoA hydratase-1 deficiency: a lethal phenotype of SCEH deficiency. Clin Genet. 2017;91:629–633. doi: 10.1111/cge.12891. [DOI] [PubMed] [Google Scholar]
- 3.Sharpe A, McKenzie M. Mitochondrial fatty acid oxidation disorders associated with short-chain enoyl-CoA hydratase (ECHS1) deficiency. Cells. 2018;7:46. doi: 10.3390/cells7060046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haack TB, Jackson CB, Murayama K, Kremer LS, Schaller A, Kotzaeridou U, et al. Deficiency of ECHS1 causes mitochondrial encephalopathy with cardiac involvement. Ann Clin Transl Neurol. 2015;2:492–509. doi: 10.1002/acn3.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wanders RJA, Duran M, Loupatty FJ. Enzymology of the branched-chain amino acid oxidation disorders: the valine pathway. J Inherit Metab Dis. 2012;35:5–12. doi: 10.1007/s10545-010-9236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peters H, Ferdinandusse S, Ruiter JP, Wanders RJA, Boneh A, Pitt J. Metabolite studies in HIBCH and ECHS1 defects: Implications for screening. Mol Genet Metab. 2015;115:168–173. doi: 10.1016/j.ymgme.2015.06.008. [DOI] [PubMed] [Google Scholar]
- 7.Peters H, Buck N, Wanders R, Ruiter J, Waterham H, Koster J, et al. ECHS1 mutations in Leigh disease: a new inborn error of metabolism affecting valine metabolism. Brain. 2014;137:2903–2908. doi: 10.1093/brain/awu216. [DOI] [PubMed] [Google Scholar]
- 8.Masnada S, Parazzini C, Bini P, Barbarini M, Alberti L, Valente M, et al. Phenotypic spectrum of short-chain enoyl-Coa hydratase-1 (ECHS1) deficiency. Eur J Paediatr Neurol. 2020;28:151–158. doi: 10.1016/j.ejpn.2020.07.007. [DOI] [PubMed] [Google Scholar]
- 9.Marti-Sanchez L, Baide-Mairena H, Marcé-Grau A, Pons R, Skouma A, López-Laso E, et al. Delineating the neurological phenotype in children with defects in the ECHS1 or HIBCH gene. J Inherit Metab Dis. 2021;44:401–414. doi: 10.1002/jimd.12288. [DOI] [PubMed] [Google Scholar]
- 10.Balasubramaniam S, Riley LG, Bratkovic D, Ketteridge D, Manton N, Cowley MJ, et al. Unique presentation of cutis laxa with Leigh-like syndrome due to ECHS1 deficiency. J Inherit Metab Dis. 2017;40:745–747. doi: 10.1007/s10545-017-0036-4. [DOI] [PubMed] [Google Scholar]
- 11.Ferdinandusse S, Waterham HR, Heales SJR, Brown GK, Hargreaves IP, Taanman J-W, et al. HIBCH mutations can cause Leigh-like disease with combined deficiency of multiple mitochondrial respiratory chain enzymes and pyruvate dehydrogenase. Orphanet J Rare Dis. 2013;8:188. doi: 10.1186/1750-1172-8-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olgiati S, Skorvanek M, Quadri M, Minneboo M, Graafland J, Breedveld GJ, et al. Paroxysmal exercise-induced dystonia within the phenotypic spectrum of ECHS1 deficiency. Mov Disord. 2016;31:1041–1048. doi: 10.1002/mds.26610. [DOI] [PubMed] [Google Scholar]
- 13.Pajares S, López RM, Gort L, Argudo-Ramírez A, Marín JL, González de Aledo-Castillo JM, et al. An incidental finding in newborn screening leading to the diagnosis of a patient with ECHS1 mutations. Mol Genet Metab Rep. 2020;22:100553. doi: 10.1016/j.ymgmr.2019.100553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huffnagel IC, Redeker EJW, Reneman L, Vaz FM, Ferdinandusse S, Poll-The BT. Mitochondrial encephalopathy and transient 3-methylglutaconic aciduria in ECHS1 deficiency: long-term follow-up. JIMD Rep. 2018;39:83–87. doi: 10.1007/8904_2017_48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uesugi M, Mori J, Fukuhara S, Fujii N, Omae T, Sasai H, et al. Short-chain enoyl-CoA hydratase deficiency causes prominent ketoacidosis with normal plasma lactate levels: a case report. Mol Genet Metab Rep. 2020;25:100672. doi: 10.1016/j.ymgmr.2020.100672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simon MT, Eftekharian SS, Ferdinandusse S, Tang S, Naseri T, Reupena MS, et al. ECHS1 disease in two unrelated families of Samoan descent: common variant-rare disorder. Am J Med Genet A. 2021;185:157–167. doi: 10.1002/ajmg.a.61936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nair P, Hamzeh AR, Mohamed M, Malik EM, Al-Ali MT, Bastaki F. Novel ECHS1 mutation in an Emirati neonate with severe metabolic acidosis. Metab Brain Dis. 2016;31:1189–1192. doi: 10.1007/s11011-016-9842-x. [DOI] [PubMed] [Google Scholar]
- 18.Kerr DS. Treatment of mitochondrial electron transport chain disorders: a review of clinical trials over the past decade. Mol Genet Metab. 2010;99:246–255. doi: 10.1016/j.ymgme.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 19.Abdenur JE, Sowa M, Simon M, Steenari M, Skaar J, Eftekharian S, et al. Medical nutrition therapy in patients with HIBCH and ECHS1 defects: clinical and biochemical response to low valine diet. Mol Genet Metab Rep. 2020;24:100617. doi: 10.1016/j.ymgmr.2020.100617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parikh S, Saneto R, Falk MJ, Anselm I, Cohen BH, Haas R, et al. A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol. 2009;11:414–430. doi: 10.1007/s11940-009-0046-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF. Treatment for mitochondrial disorders. Cochrane Database Syst Rev. 2012;4:CD004426. doi: 10.1002/14651858.CD004426.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gillingham MB, Scott B, Elliott D, Harding CO. Metabolic control during exercise with and without medium-chain triglycerides (MCT) in children with long-chain 3-hydroxy acyl-CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiency. Mol Genet Metab. 2006;89:58–63. doi: 10.1016/j.ymgme.2006.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spiekerkoetter U, Bastin J, Gillingham M, Morris A, Wijburg F, Wilcken B. Current issues regarding treatment of mitochondrial fatty acid oxidation disorders. J Inherit Metab Dis. 2010;33:555–561. doi: 10.1007/s10545-010-9188-1. [DOI] [PubMed] [Google Scholar]
- 24.Bedoyan JK, Yang SP, Ferdinandusse S, Jack RM, Miron A, Grahame G, DeBrosse SD, Hoppel CL, Kerr DS, Wanders RJA. Lethal neonatal case and review of primary short-chain enoyl-CoA hydratase (SCEH) deficiency associated with secondary lymphocyte pyruvate dehydrogenase complex (PDC) deficiency. Mol Genet Metab. 2017;120:342–349. doi: 10.1016/j.ymgme.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferdinandusse S, Friederich MW, Burlina A, Ruiter JPN, Coughlin CR, 2nd, Dishop MK, et al. Clinical and biochemical characterization of four patients with mutations in ECHS1. Orphanet J Rare Dis. 2015;10:79. doi: 10.1186/s13023-015-0290-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ronchi D, Monfrini E, Bonato S, Mancinelli V, Cinnante C, Salani S, et al. Dystonia-ataxia syndrome with permanent torsional nystagmus caused by ECHS1 deficiency. Ann Clin Transl Neurol. 2020;7:839–845. doi: 10.1002/acn3.51025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loupatty FJ, Clayton PT, Ruiter JPN, Ofman R, Ijlst L, Brown GK, et al. Mutations in the gene encoding 3-hydroxyisobutyryl-CoA hydrolase results in progressive infantile neurodegeneration. Am J Hum Genet. 2007;80:195–199. doi: 10.1086/510725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soler-Alfonso C, Enns GM, Koenig MK, Saavedra H, Bonfante-Mejia E, Northrup H. Identification of HIBCH gene mutations causing autosomal recessive leigh syndrome: a gene involved in valine metabolism. Pediatr Neurol. 2015;52:361–365. doi: 10.1016/j.pediatrneurol.2014.10.023. [DOI] [PubMed] [Google Scholar]
- 29.Shayota BJ, Soler-Alfonso C, Bekheirnia MR, Mizerik E, Boyer SW, Xiao R, et al. Case report and novel treatment of an autosomal recessive Leigh syndrome caused by short-chain enoyl-CoA hydratase deficiency. Am J Med Genet A. 2019;179:803–807. doi: 10.1002/ajmg.a.61074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato-Shirai I, Ogawa E, Arisaka A, Osaka H, Murayama K, Kuwajima M, et al. Valine-restricted diet for patients with ECHS1 deficiency: divergent clinical outcomes in two Japanese siblings. Brain Dev. 2021;43:308–313. doi: 10.1016/j.braindev.2020.10.003. [DOI] [PubMed] [Google Scholar]
- 31.Burgin HJ, McKenzie M. Understanding the role of OXPHOS dysfunction in the pathogenesis of ECHS1 deficiency. FEBS Lett. 2020;594(4):590–610. doi: 10.1002/1873-3468.13735. [DOI] [PubMed] [Google Scholar]
- 32.Fitzsimons PE, Alston CL, Bonnen PE, Hughes J, Crushell E, Geraghty MT, Tetreault M, O’Reilly P, Twomey E, Sheikh Y, et al. (2018) Clinical, biochemical, and genetic features of four patients with short-chain enoyl-CoA hydratase (ECHS1) deficiency. Am J Med Genet A. 2018;176:1115–1127. doi: 10.1002/ajmg.a.38658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferdinandusse S, Friederich MW, Burlina A, Ruiter JP, Coughlin CR, II, Dishop MK, Gallagher RC, Bedoyan JK, Vaz FM, Waterham HR, et al. Clinical and biochemical characterization of four patients with mutations in ECHS1. Orphanet J Rare Dis. 2015;10:79. doi: 10.1186/s13023-015-0290-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mahajan A, Constantinou J, Sidiropoulos C. ECHS1 deficiency-associated paroxysmal exercise-induced dyskinesias: case presentation and initial benefit of intervention. J Neurol. 2017;264:185–187. doi: 10.1007/s00415-016-8381-z. [DOI] [PubMed] [Google Scholar]
- 35.Yamada K, Aiba K, Kitaura Y, Kondo Y, Nomura N, Nakamura Y, et al. Clinical, biochemical and metabolic characterisation of a mild form of human short-chain enoyl-CoA hydratase deficiency: significance of increased N-acetyl-S-(2-carboxypropyl)cysteine excretion. J Med Genet. 2015;52:691–698. doi: 10.1136/jmedgenet-2015-103231. [DOI] [PubMed] [Google Scholar]
- 36.Jensen RG, Ferris AM, Lammi-Keefe CJ. The composition of milk fat. J Daiy Sci. 1991;74:3228–3243. doi: 10.3168/jds.S0022-0302(91)78509-3. [DOI] [PubMed] [Google Scholar]
- 37.Fontecha J, Calvo MV, Juarez M, Gil A, Martínez-Vizcaino V. Milk and dairy product consumption and cardiovascular diseases: An overview of systematic reviews and meta-analyses. Adv Nutr. 2019;10(Suppl 2):164–189. doi: 10.1093/advances/nmy099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schönfeld P, Wojtczak L. Short- and medium-chain fatty acids in energy metabolism: the cellular perspective. J Lipid Res. 2016;57:943–954. doi: 10.1194/jlr.R067629. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. This video shows the clinical evolution of the patient with ECHS1 deficiency before and 3 years after treatment with a low-valine and low-fat diet. One can observe how after treatment he shows better stability, less walking and turning difficulties, an improvement in fine motor skills and tremors.
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its Additional files.