

Abstract
Background
Brachydactyly type B is an autosomal dominant disorder that is characterized by hypoplasia of the distal phalanges and nails and can be divided into brachydactyly type B1 (BDB1) and brachydactyly type B2 (BDB2). BDB1 is the most severe form of brachydactyly and is caused by truncating variants in the receptor tyrosine kinase–like orphan receptor 2 (ROR2) gene.
Case presentation
Here, we report a five-generation Chinese family with brachydactyly with or without syndactyly. The proband and her mother underwent digital separation in syndactyly, and the genetic analyses of the proband and her parents were provided. The novel heterozygous frameshift variant c.1320dupG, p.(Arg441Alafs*18) in the ROR2 gene was identified in the affected individuals by whole-exome sequencing and Sanger sequencing. The c.1320dupG variant in ROR2 is predicted to produce a truncated protein that lacks tyrosine kinase and serine/threonine- and proline-rich structures and remarkably alters the tertiary structures of the mutant ROR2 protein.
Conclusion
The c.1320dupG, p.(Arg441Alafs*18) variant in the ROR2 gene has not been reported in any databases thus far and therefore is novel. Our study extends the gene variant spectrum of brachydactyly and may provide information for the genetic counselling of family members.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12887-022-03564-z.
Keywords: Brachydactyly type B1, ROR2, Whole-exome sequencing, Variant
Background
Brachydactyly (BD) is an autosomal dominant hand and foot malformation that is characterized by abnormally shortened or missing phalanges and/or metacarpals/metatarsals [1]. It may occur as an isolated trait or as part of a complex malformation syndrome [2]. According to the Bell classification, heritable isolated BDs have been classified into subtypes A to E according to their patterns of skeletal involvement [3]. Clinical features and pathogenic genes of isolated BDs are listed in Table 1 [4–20]. Brachydactyly type B1 (BDB1, OMIM 113,000) is the most severe form of brachydactyly and is characterized by shortening or hypoplasia of the distal and middle phalanges of digits 2 through 5 with or without nail dysplasia, fusion of the middle and distal phalanges, variable degrees of distal and proximal symphalangism, and a broad or bifid thumb. The feet are similarly but less severely affected [13–15].
Table 1.
Types of isolated brachydactyly
| Phenotype | Phenotype MIM number |
Clinical features | Pathogenic genes | Gene/Locus MIM number | Reference | |
|---|---|---|---|---|---|---|
| BDA1 | BDA1 | 112,500 | Short middle phalanges of all fingers and short proximal phalanx of the thumb. The middle phalanx may be absent in severe cases | IHH | 600,726 | [4] |
| BDA1B | 607,004 | 5p13.3-p13.2 | None | [5] | ||
| BDA1C | 615,072 | GDF5 | 601,146 | [6] | ||
| BDA1D | 616,849 | BMPR1B | 603,248 | [7] | ||
| BDA2 | 112,600 | Short middle phalanx with clinodactyly of the index fingers and second toes | BMPR1B | 603,248 | [8] | |
| GDF5 | 601,146 | [9] | ||||
| BMP2 | 112,261 | [10] | ||||
| BDA3 | 112,700 | Short middle phalanx of the little fingers with or without clinodactyly | HOXD13 | 142,989 | [11] | |
| BDA4 | 112,800 | Short middle phalanx of the index and little fingers. The middle phalanges of the lateral four toes are frequently involved | HOXD13 | 142,989 | [12] | |
| BDB1 | 113,000 | Hypoplastic or absent distal phalanges of digits 2–5 with or without nail dysplasia, the fusion of the middle and distal phalanges, abroad or bifid thumb, and sometimes distal and proximal symphalangism or syndactyly | ROR2 | 602,337 | [13–15] | |
| BDB2 | 611,377 | Hypoplasia of distal phalanges with distal and proximal symphalangism, fusion of carpal or tarsal bones, and partial cutaneous syndactyly | NOG | 602,991 | [16] | |
| BDC | 113,100 | Brachymesophalangy of the index, middle and little fingers with preservation of the ring finger. The index and middle fingers show hyperphalangism and their most proximal phalanges have abnormal configuration lending to ulnar deviation. The thumb metacarpals are slightly short | GDF5 | 601,146 | [17] | |
| BDD | 113,200 | Stub thumbs (short distal phalanges of the thumbs). The big toes may be similarly affected | HOXD13 | 142,989 | [18] | |
| BDE1 | 113,300 | Short metacarpal IV, with/without short metatarsal IV (possible involvement of an isolated metatarsal) | HOXD13 | 142,989 | [18] | |
| BDE2 | 613,382 | Short metacarpals IV and V (and metatarsals) with short distal phalanx of the thumb | PTHLH | 168,470 | [19] | |
| BDE3 | None | Short metacarpals without phalangeal involvement | None | None | [20] | |
BDB1 is usually caused by a heterozygous variant in the receptor tyrosine kinase (RTK)-like orphan receptor 2 (ROR2) gene (OMIM 602,337), which is located on chromosome 9q22.31 [14, 21]. The ROR2 gene contains nine exons and spans a genomic length of approximately 228 kb. The ROR2 protein contains 943 amino acids and belongs to the ROR family of RTKs. It consists of an extracellular region, a transmembrane section, and an intracellular region [22]. Heterozygous variants that truncate the intracellular portion of ROR2, either the N-terminal or C-terminal of the TK domain, are the leading causes of BDB1 [22, 23]. In addition, homozygous or compound heterozygous variants in the ROR2 gene are responsible for recessive Robinow syndrome [22].
In recent years, whole-exome sequencing has become a routine strategy for discovering potential causal variants in inherited Mendelian disorders. It provides a cost-effective, fast-track approach to variant discovery and considerably increases the overall diagnostic rate. In this study, we identified the disease-causing variant in a five-generation Chinese family with BDB1 using whole-exome sequencing and Sanger sequencing. Pathogenicity was inferred by bioinformatic analysis.
Case presentation
A five-generation Chinese family with BDB1 was recruited at Central Hospital Affiliated to Shenyang Medical College. Clinical information of all family members and blood specimens from the proband and her parents were obtained. Among the 13 affected individuals, 10 had brachydactyly, and the remaining three had brachydactyly with syndactyly (Fig. 1). The 1-year-old female proband was physically examined and received a radiographic examination of the right hand before surgery. She exhibited bilateral shortening and hypoplasia of the distal and middle phalanges of digits 2 to 5 with cutaneous syndactyly of right fingers 3 to 4. The proband underwent digital separation on her right hand. Her mother underwent digital separation on both hands before enrolment in this study. Therefore, her phenotypic information was determined after reviewing her medical records and performing a physical examination. She had shortening/hypoplasia of the distal and middle phalanges of digits 2 to 5 with cutaneous syndactyly of fingers 3 to 4 on both hands, and she also had a broad hallux on both feet with nail dysplasia (Fig. 2). The phenotypic information of the other family members was provided by the proband’s mother.
Fig. 1.

Pedigree of the family with BDB1. Black shading on the left half represents brachydactyly, and grey shading on the right half represents syndactyly. The proband is marked by an arrow
Fig. 2.
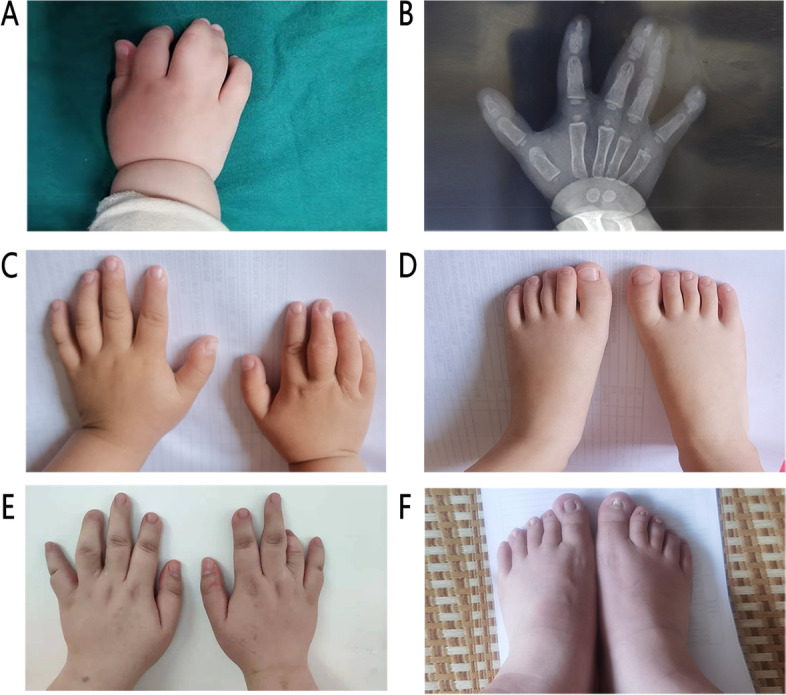
Phenotype of the proband and her mother. a A photograph of the proband's right hand before surgery. b An X-ray of the proband’s right hand before surgery. c A picture of the proband’s hands after the operation. d A photograph of the proband's feet. e A photograph of the mother’s hands after the operation. f A photograph of the mother’s feet
Genomic DNA was extracted from the peripheral venous blood of the proband using the QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s protocol and the AI Exome V1 Kit (iGeneTech, AIEV1). The extracted DNA was used for whole-exome capture and enrichment. Exome sequencing was performed on the NovaSeq 6000 sequencing platform (Illumina). Gene variants were initially annotated using the wANNOVAR tool (http://wannovar.wglab.org/). Those variants with minor allele frequencies greater than 0.01 according to public genomic databases, such as 1000Genomes, dbSNP, Exome Variant Server, gnomAD, and the Exome Aggregation Consortium were filtered out. The c.1320dupG, p.(Arg441Alafs*18) variant was identified in exon 8 of ROR2 by whole-exome sequencing and Sanger sequencing. This variant results in a frameshift at amino acid position 441. This frameshift creates a polypeptide chain of 17 new amino acids and a premature termination codon at amino acid position 458. To read the duplication sequencing chromatogram more clearly, the purified PCR fragment of the proband was inserted into the pMD-18 T vector. The selected clones were sequenced using the universal primers M13F and M13R (Fig. 3a). At the protein level, the c.1320dupG variant is predicted to produce a truncated protein that lacks tyrosine kinase and serine/threonine- and proline-rich structures, resulting in a loss of the whole intracellular region (Fig. 3b). Three-dimensional structures of wild-type and mutant ROR2 proteins were generated by the Robetta online server (http://robetta.org/) and validated using different parameters, such as the Ramachandran plot, ERRAT score, confidence and P value [24, 25]. The first two parameters were calculated using Structure Analysis and Verification Server (SAVES) version 6.0 (https://saves.mbi.ucla.edu/), and the last two parameters were determined using the ModFOLD Model Quality Assessment Server (https://www.reading.ac.uk/bioinf/ModFOLD/) [26–29]. Illustrations were prepared using VMD (www.ks.uiuc.edu/Research/vmd/). Molecular modelling shows that p.(Arg441Alafs*18) severely affects the tertiary structure of the remaining peptide chain of ROR2 (Fig. 4). The Ramachandran plot shows that the total number of residues in the most favoured regions for the wild-type and mutant ROR2 proteins was 688 (85.9%) and 330 (86.2%), respectively, and the corresponding number of residues in disallowed regions was 5 (0.6%) and 4 (1.0%), respectively. The ERRAT scores of wild-type and mutant ROR2 proteins were 89.732 and 86.9863, respectively. The confidence scores and P values of the wild-type (0.04122) and mutant proteins (0.05723) were less than 0.1 (Additional file 1). The above results indicate that the three-dimensional structures of the wild-type and mutant ROR2 proteins we predicted were reliable.
Fig. 3.

Gene variant analysis of a family with BDB1. a The novel heterozygous variant c.1320dupG, p.(Arg441Alafs*18) in ROR2 was verified by Sanger sequencing. The variant is marked by a red arrow. CS: Clone sequencing. b A schematic diagram showing the encoded domain structure of the ROR2 gene. A recurrent variant is marked in black, and the novel variant identified in this study is highlighted in red. This ROR2 variant results in the loss of the whole intracellular region
Fig. 4.
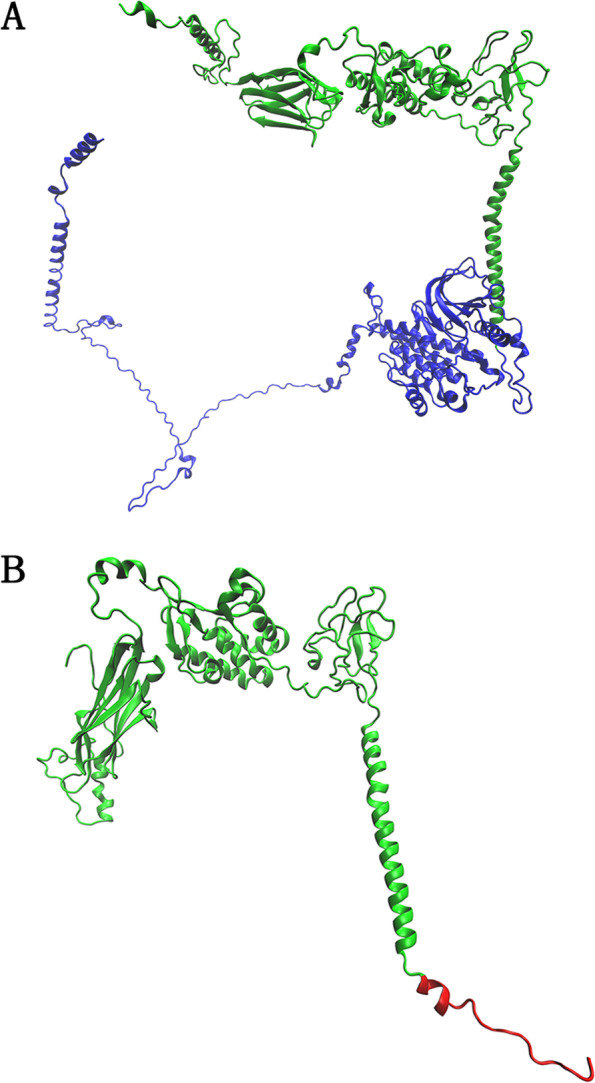
Three-dimensional structures of the wild-type and variant ROR2 proteins. Molecular modelling by Robetta showing that p.(Arg441Alafs*18) remarkably altered the tertiary structures of the remaining peptide chain of the ROR2 protein. a Wild-type; b Mutant type
Discussion and conclusion
ROR2 is a single-pass transmembrane protein. The domain structure of ROR2 is composed of an extracellular immunoglobulin-like domain (Ig, aa residues 55–145), frizzled-like cysteine-rich domain (FRZ or CRD, aa residues 169–303), Kringle domain (Kr, aa residues 316–394), transmembrane domain (TM, aa residues 404–424), and intracellular tyrosine kinase (TK, aa residues 473–746), and serine/threonine-rich (ST1, aa residues 753–782; ST2, aa residues 859–882) and proline-rich (PRD, aa residues 784–857) structures [22]. The extracellular region interacts with Wnt5a, which binds to the CRD region of ROR2 [30]. Both canonical and noncanonical Wnt5a/Ror2 signalling play an important role in limb skeletal development and morphogenesis [30–32]. The intracellular region of ROR2 is associated with many factors, such as 14–3-3 protein beta/alpha, nuclear receptor subfamily 2 group C member 2, proto-oncogene tyrosine-protein kinase Src, Wilms tumour protein 1-interacting protein, and SHC-transforming protein 1 [23, 30, 33–36]. Activation of ROR2 kinase requires the intracellular C-terminal region for recruitment of the nonreceptor kinase Src [34, 37].
Up to the time of this article’s publication, a total of 15 different variants in the ROR2 gene were found to be associated with BDB1 (Table 2). All the documented BDB1-causing ROR2 variants were nonsense or frameshift variants and were clustered in the last two exons (exons 8 and 9) or in the last intron (intron 8). Thus, the mutant mRNAs were able to escape degradation by nonsense-mediated decay, resulting in a partial or complete truncation of the intracellular portion of the ROR2 protein [38]. These mutations are believed to have a specific gain-of-function effect, not a simple haploinsufficiency [39]. The BDB1-related variants can be divided into distal and proximal variants. The distal variant that is located after TK produces a truncated protein that lacks the ST1, ST2, and PRD domains, leading to a more severe phenotype [14, 40]. In contrast, the proximal variant results in a loss of the whole intracellular region, causing a less severe but more variable phenotype [37].
Table 2.
Clinical manifestation of ROR2 causing BDB1
| Number | Nucleotide change | Predicted amino acid change | Clinical features | Reference |
|---|---|---|---|---|
| 1 | c.1324C > T | p.(Arg442*) | Homozygous individuals exhibited features of recessive Robinow syndrome in conjunction with severe recessive brachydactyly | [39] |
| 2 | c.2246G > A | p.(Trp749*) | The patients have classical BDB1 with bilateral symmetry of the deformities | [41] |
| 3 | c.2247G > A | p.(Trp749*) | An English family with typical BDB1. In addition, they have a short philtrum and a prominent nose with a high bridge and bulbous tip; A Chinese family had classical BDB1 | [42, 43] |
| 4 | c.2265C > A | p.(Tyr755*) | These families displayed classical BDB1, with (or without) a prominent nose with a bulbous tip, wide-spaced eyes, and a short philtrum | [13, 40, 41, 44] |
| 5 | c.2273C > A | p.(Ser758*) | Classical BDB1 | [37] |
| 6 | c.2278C > T | p.(Gln760*) | The proband lacked distal phalanges and nails and had hypoplastic middle phalanges of digits 2–5 | [14] |
| 7 | c.1321_1325delCGGCG | p.(Arg441Thrfs*16) | Heterozygous individuals exhibited classical BDB1, whereas homozygous individuals showed severe skeletal defects, primarily affecting the distal limbs and the spine | [14] |
| 8 | c.1394_1395delTC | p.(Leu465Glnfs*59) | No detailed clinical description | [45] |
| 9 | c.1397_1398delAA | p.(Lys466Argfs*58) | Variable degrees of hypoplastic or shortened distal phalanges on digits 2–5 of the hands. Very few of the toes were affected | [46] |
| 10 | c.1937_1943delACAAGCT | p.(Tyr646Cysfs*57) | Homozygous individuals exhibited features of recessive Robinow syndrome in conjunction with severe brachydactyly | [47] |
| 11 | c.2244delC | p.(Trp749Glyfs*25) | The patients exhibited atypical BDB1 and cutaneous syndactyly of varying degrees | [38] |
| 12 | c.2249delG | p.(Gly750Alafs*24) | The morphologic abnormalities were more severe, such as a bifid thumb and syndactyly of the central digits (digits 2 and 4) | [41] |
| 13 | c.1366dupC | p.(Leu456Profs*3) | The patients exhibited bilateral or unilateral fourth finger distal symphalangism with (or without) absence of the distal phalanx or fusion of the phalangeal bones on the hand radiograph | [48] |
| 14 | c.1398dupA | p.(Glu467Argfs*58) | They exhibited the absence of distal phalanges of the 4th finger and hypoplasia of distal phalanges of fingers 2, 3, and 5, with or without nail dysplasia | [14] |
| 15 | c.1386 + 3_1386 + 5delCTCins19 | p.(Ala463fs*64) | No detailed clinical description | [14] |
In this study, the novel proximal frameshift variant c.1320dupG, p.(Arg441Alafs*18) in the ROR2 gene was identified in a Chinese family with BDB1. This variant is predicted to produce a premature termination codon at amino acid position 458 and a new polypeptide consisting of 17 amino acids at the variant position. The location of this variant was very similar to the locations of c.1321_1325delCGGCG, p.(Arg441Thrfs*16), and c.1324C > T, p.(Arg442*), but their phenotypes were different. Individuals heterozygous for c.1321_1325delCGGCG, p.(Arg441Thrfs*16) exhibited bilateral hypoplasia of the distal and middle phalanges of the fingers and toes 2 to 5 to varying degrees, with or without hypoplasia of the nails. In contrast, individuals homozygous for c.1321_1325delCGGCG, p.(Arg441Thrfs*16), showed severe skeletal defects that primarily affected the distal limbs and the spine [14]. Interestingly, a heterozygous c.1324C > T, p.(R442*) carrier had a normal limb phenotype, and a homozygous c.1324C > T, p.(R442*) individual exhibited features of recessive Robinow syndrome in conjunction with severe recessive brachydactyly [39]. The c.1320dupG, p.(Arg441Alafs*18) variant in the ROR2 gene reported by our study caused bilateral or unilateral shortening/hypoplasia of the distal and middle phalanges of digits 2 to 5 with or without cutaneous syndactyly of fingers 3 to 4. The relationship between the ROR2 gene variants and phenotypes is not completely clear. Previous studies suggest that BDB1 variants result in a gain in function, whereas Robinow syndrome variants result in a loss of function [14, 49]. Further research should investigate the reasons for the different phenotypes resulting from these similar gene variants.
In conclusion, we report the novel variant c.1320dupG, p.(Arg441Alafs*18) in the ROR2 gene in a Chinese family with BDB1. Our study extends the gene variant spectrum of BDB1 and provides information for the genetic counselling of family members.
Supplementary Information
Additional file 1. The results of three-dimensional structures of wild-type and mutant ROR2 proteins were validated using different parameters.
Acknowledgements
We thank all of the family members for their contributions to this work.
Abbreviations
- BD
Brachydactyly
- BDB1
Brachydactyly type B1
- RTK
Receptor tyrosine kinase
- Ig
Immunoglobulin-like domain
- FRZ or CRD
Frizzled-like cysteine-rich domain
- Kr
Kringle domain
- TM
Transmembrane domain
- TK
Intracellular tyrosine kinase
- ST1 ST2
Serine/threonine-rich
- PRD
Proline-rich
Authors’ contributions
LHC designed the study and prepared the article. YL and JZ obtained the clinical information. JQS performed the experiments. SYZ and WSS analyzed experimental data. All authors read and approved the final manuscript.
Funding
This work was supported by the Natural Science Foundation of China (81670896). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
The datasets generated and/or analysed during the current study are available in the NCBI Sequence Read Archive (SRA) repository (accession number: SRP390837).
Declarations
Ethics approval and consent to participate
This study adhered to the tenets of the Declaration of Helsinki and was approved by the Central Hospital Affiliated to Shenyang Medical College Institutional Review Board. Written informed consent was obtained from all participants.
Consent for publication
Written informed consent for publication was obtained from patient’s parents, also the individual information of the family members of this case report.
Competing interests
All authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jiaqi Shao, Email: sjq15542472657@163.com.
Yue Liu, Email: hahaliuyue@126.com.
Shuyang Zhao, Email: overmind_del@163.com.
Weisheng Sun, Email: 13654916921@163.com.
Jie Zhan, Email: adam971111@sina.com.
Lihua Cao, Email: lhcao@cmu.edu.cn.
References
- 1.Temtamy SA, Aglan MS. Brachydactyly. Orphanet J Rare Dis. 2008;3:15. doi: 10.1186/1750-1172-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.David A, Vincent M, Quéré MP, Lefrançois T, Frampas E, David A. Isolated and syndromic brachydactylies: diagnostic value of hand X-rays. Diagn Interv Imaging. 2015;96(5):443–448. doi: 10.1016/j.diii.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 3.Bell J. On brachydactyly and symphalangism. In: Penrose LS, editor. Treasury of Human Inheritance. London: Cambridge University Press; 1951. pp. 191–201. [Google Scholar]
- 4.Gao B, Guo J, She C, Shu A, Yang M, Tan Z, et al. Mutations in IHH, encoding Indian hedgehog, cause brachydactyly type A-1. Nat Genet. 2001;28(4):386–388. doi: 10.1038/ng577. [DOI] [PubMed] [Google Scholar]
- 5.Armour CM, McCready ME, Baig A, Hunter AG, Bulman DE. A novel locus for brachydactyly type A1 on chromosome 5p13.3-p13.3.2. J Med Genet. 2002;39(3):186–8. doi: 10.1136/jmg.39.3.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byrnes AM, Racacho L, Nikkel SM, Xiao F, MacDonald H, Underhill TM, et al. Mutations in GDF5 presenting as semidominant brachydactyly A1. Hum Mutat. 2010;31(10):1155–1162. doi: 10.1002/humu.21338. [DOI] [PubMed] [Google Scholar]
- 7.Racacho L, Byrnes AM, MacDonald H, Dranse HJ, Nikkel SM, Allanson J, et al. Two novel disease-causing variants in BMPR1B are associated with brachydactyly type A1. Eur J Hum Genet. 2015;23(12):1640–1645. doi: 10.1038/ejhg.2015.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lehmann K, Seemann P, Boergermann J, Morin G, Reif S, Knaus P, et al. A novel R486Q mutation in BMPR1B resulting in either a brachydactyly type C/symphalangism-like phenotype or brachydactyly type A2. Eur J Hum Genet. 2006;14(12):1248–1254. doi: 10.1038/sj.ejhg.5201708. [DOI] [PubMed] [Google Scholar]
- 9.Seemann P, Schwappacher R, Kjaer KW, Krakow D, Lehmann K, Dawson K, et al. Activating and deactivating mutations in the receptor interaction site of GDF5 cause symphalangism or brachydactyly type A2. J Clin Invest. 2005;115(9):2373–2381. doi: 10.1172/JCI25118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dathe K, Kjaer KW, Brehm A, Meinecke P, Nürnberg P, Neto JC, et al. Duplications involving a conserved regulatory element downstream of BMP2 are associated with brachydactyly type A2. Am J Hum Genet. 2009;84(4):483–492. doi: 10.1016/j.ajhg.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang M, Lu L, Wei B, Zhang Y, Li X, Shi Y, et al. Brachydactyly type A3 is caused by a novel 13 bp HOXD13 frameshift deletion in a Chinese family. Am J Med Genet A. 2020;182(10):2432–2436. doi: 10.1002/ajmg.a.61788. [DOI] [PubMed] [Google Scholar]
- 12.Zhao X, Sun M, Zhao J, Leyva JA, Zhu H, Yang W, et al. Mutations in HOXD13 underlie syndactyly type V and a novel brachydactyly-syndactyly syndrome. Am J Hum Genet. 2007;80(2):361–371. doi: 10.1086/511387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oldridge M, Temple IK, Santos HG, Gibbons RJ, Mustafa Z, Chapman KE, et al. Brachydactyly type B: linkage to chromosome 9q22 and evidence for genetic heterogeneity. Am J Hum Genet. 1999;64(2):578–585. doi: 10.1086/302255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwabe GC, Tinschert S, Buschow C, Meinecke P, Wolff G, Gillessen-Kaesbach G, et al. Distinct mutations in the receptor tyrosine kinase gene ROR2 cause brachydactyly type B. Am J Hum Genet. 2000;67(4):822–831. doi: 10.1086/303084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mundlos S. The brachydactylies: a molecular disease family. Clin Genet. 2009;76(2):123–136. doi: 10.1111/j.1399-0004.2009.01238.x. [DOI] [PubMed] [Google Scholar]
- 16.Lehmann K, Seemann P, Silan F, Goecke TO, Irgang S, Kjaer KW, et al. A new subtype of brachydactyly type B caused by point mutations in the bone morphogenetic protein antagonist NOGGIN. Am J Hum Genet. 2007;81(2):388–396. doi: 10.1086/519697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faryal S, Farooq M, Abdullah U, Ali Z, Saadi SM, Ullah F, et al. A GDF5 frameshift mutation segregating with Grebe type chondrodysplasia and brachydactyly type C+ in a 6 generations family: Clinical report and mini review. Eur J Med Genet. 2021;64(7):104226. doi: 10.1016/j.ejmg.2021.104226. [DOI] [PubMed] [Google Scholar]
- 18.Johnson D, Kan SH, Oldridge M, Trembath RC, Roche P, Esnouf RM, et al. Missense mutations in the homeodomain of HOXD13 are associated with brachydactyly types D and E. Am J Hum Genet. 2003;72(4):984–997. doi: 10.1086/374721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas-Teinturier C, Pereda A, Garin I, Diez-Lopez I, Linglart A, Silve C, et al. Report of two novel mutations in PTHLH associated with brachydactyly type E and literature review. Am J Med Genet A. 2016;170(3):734–742. doi: 10.1002/ajmg.a.37490. [DOI] [PubMed] [Google Scholar]
- 20.Pereda A, Garin I, Garcia-Barcina M, Gener B, Beristain E, Ibañez AM, et al. Brachydactyly E: isolated or as a feature of a syndrome. Orphanet J Rare Dis. 2013;8:141. doi: 10.1186/1750-1172-8-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gong Y, Chitayat D, Kerr B, Chen T, Babul-Hirji R, Pal A, et al. Brachydactyly type B: clinical description, genetic mapping to chromosome 9q, and evidence for a shared ancestral mutation. Am J Hum Genet. 1999;64(2):570–577. doi: 10.1086/302249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Afzal AR, Jeffery S. One gene, two phenotypes: ROR2 mutations in autosomal recessive robinow syndrome and autosomal dominant brachydactyly type B. Hum Mutat. 2003;22(1):1–11. doi: 10.1002/humu.10233. [DOI] [PubMed] [Google Scholar]
- 23.Stricker S, Rauschenberger V, Schambony A. ROR-family receptor tyrosine kinases. Curr Top Dev Biol. 2017;123:105–142. doi: 10.1016/bs.ctdb.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004;32:W526–W531. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021;373(6557):871–876. doi: 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gurung AB. In silico structure modelling of SARS-CoV-2 Nsp13 helicase and Nsp14 and repurposing of FDA approved antiviral drugs as dual inhibitors. Gene Rep. 2020;21:100860. doi: 10.1016/j.genrep.2020.100860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maghrabi AHA, McGuffin LJ. ModFOLD6: an accurate web server for the global and local quality estimation of 3D protein models. Nucleic Acids Res. 2017;45(W1):W416–W421. doi: 10.1093/nar/gkx332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGuffin LJ, Shuid AN, Kempster R, Maghrabi AHA, Nealon JO, Salehe BR, et al. Accurate template-based modeling in CASP12 using the IntFOLD4-TS, ModFOLD6, and ReFOLD methods. Proteins. 2018;86(Suppl 1):335–344. doi: 10.1002/prot.25360. [DOI] [PubMed] [Google Scholar]
- 29.McGuffin LJ, Aldowsari FMF, Alharbi SMA, Adiyaman R. ModFOLD8: accurate global and local quality estimates for 3D protein models. Nucleic Acids Res. 2021;49(W1):W425–W430. doi: 10.1093/nar/gkab321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minami Y, Oishi I, Endo M, Nishita M. Ror-family receptor tyrosine kinases in noncanonical Wnt signaling: their implications in developmental morphogenesis and human diseases. Dev Dyn. 2010;239(1):1–15. doi: 10.1002/dvdy.21991. [DOI] [PubMed] [Google Scholar]
- 31.Bernatik O, Radaszkiewicz T, Behal M, Dave Z, Witte F, Mahl A, et al. A novel role for the BMP antagonist noggin in sensitizing cells to non-canonical Wnt-5a/Ror2/disheveled pathway activation. Front Cell Dev Biol. 2017;5:47. doi: 10.3389/fcell.2017.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kamizaki K, Endo M, Minami Y, Kobayashi Y. Role of noncanonical Wnt ligands and Ror-family receptor tyrosine kinases in the development, regeneration, and diseases of the musculoskeletal system. Dev Dyn. 2021;250(1):27–38. doi: 10.1002/dvdy.151. [DOI] [PubMed] [Google Scholar]
- 33.Liu Y, Ross JF, Bodine PV, Billiard J. Homodimerization of Ror2 tyrosine kinase receptor induces 14–3-3(beta) phosphorylation and promotes osteoblast differentiation and bone formation. Mol Endocrinol. 2007;21(12):3050–3061. doi: 10.1210/me.2007-0323. [DOI] [PubMed] [Google Scholar]
- 34.Akbarzadeh S, Wheldon LM, Sweet SM, Talma S, Mardakheh FK, Heath JK. The deleted in brachydactyly B domain of ROR2 is required for receptor activation by recruitment of Src. PLoS ONE. 2008;3(3):e1873. doi: 10.1371/journal.pone.0001873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feike AC, Rachor K, Gentzel M, Schambony A. Wnt5a/Ror2-induced upregulation of xPAPC requires xShcA. Biochem Biophys Res Commun. 2010;400(4):500–506. doi: 10.1016/j.bbrc.2010.08.074. [DOI] [PubMed] [Google Scholar]
- 36.Dickinson SC, Sutton CA, Brady K, Salerno A, Katopodi T, Williams RL, et al. The Wnt5a receptor, receptor tyrosine kinase-like orphan receptor 2, is a predictive cell surface marker of human mesenchymal stem cells with an enhanced capacity for Chondrogenic differentiation. Stem Cells. 2017;35(11):2280–91. doi: 10.1002/stem.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong S, Wang Y, Tao S, Zheng F. Mutation screening in candidate genes in four Chinese brachydactyly families. Ann Clin Lab Sci. 2015;45(1):94–99. [PubMed] [Google Scholar]
- 38.Lv D, Luo Y, Yang W, Cao L, Wen Y, Zhao X, et al. A novel single-base deletion in ROR2 causes atypical brachydactyly type B1 with cutaneous syndactyly in a large Chinese family. J Hum Genet. 2009;54(7):422–425. doi: 10.1038/jhg.2009.48. [DOI] [PubMed] [Google Scholar]
- 39.Schwarzer W, Witte F, Rajab A, Mundlos S, Stricker S. A gradient of ROR2 protein stability and membrane localization confers brachydactyly type B or Robinow syndrome phenotypes. Hum Mol Genet. 2009;18(21):4013–4021. doi: 10.1093/hmg/ddp345. [DOI] [PubMed] [Google Scholar]
- 40.Hamamy H, Saleh N, Oldridge M, Al-Hadidy A, Ajlouni K. Brachydactyly type B1: report of a family with de novo ROR2 mutation. Clin Genet. 2006;70(6):538–540. doi: 10.1111/j.1399-0004.2006.00719.x. [DOI] [PubMed] [Google Scholar]
- 41.Oldridge M, Fortuna AM, Maringa M, Propping P, Mansour S, Pollitt C, et al. Dominant mutations in ROR2, encoding an orphan receptor tyrosine kinase, cause brachydactyly type B. Nat Genet. 2000;24(3):275–278. doi: 10.1038/73495. [DOI] [PubMed] [Google Scholar]
- 42.Bacchelli C, Wilson LC, Cook JA, Winter RM, Goodman FR. ROR2 is mutated in hereditary brachydactyly with nail dysplasia, but not in Sorsby syndrome. Clin Genet. 2003;64(3):263–265. doi: 10.1034/j.1399-0004.2003.00139.x. [DOI] [PubMed] [Google Scholar]
- 43.Qin L, Lou G, Guo L, Zhang Y, Wang H, Wang L, et al. Targeted next-generation sequencing-based molecular diagnosis of congenital hand malformations in Chinese population. Sci Rep. 2018;8(1):12721. doi: 10.1038/s41598-018-30940-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li CM, Wang FY, Sun WW, Han SL, Chang MX, Feng HG. Mutation analysis of the pathogenic gene in a Chinese family with Brachydactyly type B1. Yi Chuan. 2011;33(2):147–152. doi: 10.3724/sp.j.1005.2011.00147. [DOI] [PubMed] [Google Scholar]
- 45.Jourdain AS, Petit F, Odou MF, Balduyck M, Brunelle P, Dufour W, et al. Multiplex targeted high-throughput sequencing in a series of 352 patients with congenital limb malformations. Hum Mutat. 2020;41(1):222–239. doi: 10.1002/humu.23912. [DOI] [PubMed] [Google Scholar]
- 46.Huang D, Jiang S, Zhang Y, Liu X, Zhang J, He R. A new mutation in the gene ROR2 causes brachydactyly type B1. Gene. 2014;547(1):106–110. doi: 10.1016/j.gene.2014.06.035. [DOI] [PubMed] [Google Scholar]
- 47.Tufan F, Cefle K, Türkmen S, Türkmen A, Zorba U, Dursun M, et al. Clinical and molecular characterization of two adults with autosomal recessive Robinow syndrome. Am J Med Genet A. 2005;136(2):185–189. doi: 10.1002/ajmg.a.30785. [DOI] [PubMed] [Google Scholar]
- 48.Kjaer KW, Tiner M, Cingoz S, Karatosun V, Tommerup N, Mundlos S, et al. A novel subtype of distal symphalangism affecting only the 4th finger. Am J Med Genet A. 2009;149A(7):1571–1573. doi: 10.1002/ajmg.a.32905. [DOI] [PubMed] [Google Scholar]
- 49.Kirat E, Mutlu Albayrak H, Sahinoglu B, Gurler AI, Karaer K. Autosomal recessive Robinow syndrome with novel ROR2 variants: distinct cases exhibiting the clinical variability. Clin Dysmorphol. 2020;29(3):137–140. doi: 10.1097/mcd.0000000000000319. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. The results of three-dimensional structures of wild-type and mutant ROR2 proteins were validated using different parameters.
Data Availability Statement
The datasets generated and/or analysed during the current study are available in the NCBI Sequence Read Archive (SRA) repository (accession number: SRP390837).