

Abstract
The broad clinical application of mRNA therapeutics has been hampered by a lack of delivery vehicles that induce protein expression in extrahepatic organs and tissues. Recently, it was shown that mRNA delivery to the spleen or lungs is possible upon the addition of a charged lipid to a standard four-component lipid nanoparticle formulation. This approach, while effective, further complicates an already complex drug formulation and has the potential to slow regulatory approval and adversely impact manufacturing processes. We were thus motivated to maintain a four-component nanoparticle system while achieving shifts in tropism. To that end, we replaced the standard helper lipid in lipidoid nanoparticles, DOPE, with one of eight alternatives. These lipids included the neutral lipids, DOPC, sphingomyelin, and ceramide; the anionic lipids, phosphatidylserine (PS), phosphatidylglycerol, and phosphatidic acid; and the cationic lipids, DOTAP and ethyl phosphatidylcholine. While neutral helper lipids maintained protein expression in the liver, anionic and cationic lipids shifted protein expression to the spleen and lungs, respectively. For example, replacing DOPE with DOTAP increased positive LNP surface charge at pH 7 by 5-fold and altered the ratio of liver to lung protein expression from 36:1 to 1:56. Similarly, replacing DOPE with PS reduced positive charge by half and altered the ratio of liver to spleen protein expression from 8:1 to 1:3. Effects were consistent across ionizable lipidoid chemistries. Regarding mechanism, nanoparticles formulated with neutral and anionic helper lipids best transfected epithelial and immune cells, respectively. Further, the lung-tropic effect of DOTAP was linked to reduced immune cell infiltration of the lungs compared to neutral or anionic lipids. Together, these data show that intravenous non-hepatocellular mRNA delivery is readily achievable while maintaining a four-component formulation with modified helper lipid chemistry.
Keywords: lipid nanoparticles, mRNA delivery, helper lipids, charge, extrahepatic, targeted delivery
Introduction
Lipid nanoparticles (LNPs) represent the only clinically approved form of non-viral RNA delivery[1], and their role in stemming the COVID-19 pandemic has underscored their relevance to modern medicine. These delivery vehicles are formulated with ionizable lipids that are typically uncharged at neutral pH and become cationic in the acidic endosome, enabling endosomal escape and successful mRNA translation [2, 3]. Although efficacious, LNPs have been limited in application because they primarily target cell populations in the liver [4]. While the chemical identity of the ionizable lipid influences which organs and cell types are transfected [5–7], general approaches to shift LNP targeting have been limited until recently.
Modulation of the LNP surface tends to alter nanoparticle biodistribution, given that the surface interacts directly with cellular receptors, serum proteins, and extracellular matrix components [8, 9]. The surface of nanoparticles can be decorated with antibodies, peptides, proteins, and other molecules to increase interactions with receptors on the surface of target cells [10–13]. While this “active” targeting approach is beneficial in some cases, it rarely results in substantial shifts in protein expression. Additionally, the complex reactions to conjugate some of these ligands, coupled with their high cost, render them less than ideal for widespread use [14].
An alternative strategy focuses on alteration of lipid nanoparticle chemistry and relative ratios of the ingredients [6, 15]. Such changes impact endogenous targeting, which is the LNP biodistribution that results from altered serum protein interactions in response to LNP surface chemistry11. Lipid nanoparticle chemistry is defined, in most cases, by four lipids: an ionizable lipid, a helper lipid, cholesterol, and lipid-anchored poly(ethylene glycol) (PEG) [16]. Of these components, cryo-transmission electron microscopy and molecular dynamic simulations have identified the helper lipid as the primary component of the LNP surface [17, 18]. Therefore, to alter the surface properties of LNPs, tuning the helper lipid is a rational choice.
For mRNA delivery, one commonly used helper lipid is 1,2-di-(9Z-octadecenoyl)-sn-glycero-3-phosphoethanolamine (DOPE) [19, 20]. Phosphatidylethanolamines tend to augment LNP-mediated mRNA transfection because they facilitate fusion of the nanoparticle membrane with the membrane of target cells [21–23]. LNPs generated with DOPE, which contains one anion and one cation, is net neutral and typically facilitates mRNA delivery to the liver [24]. Interestingly, several studies have shown that altering nanoparticle charge has the potential to shift RNA delivery from the liver to the spleen with negative charge or the lungs with positive charge [25–27]. More recently, Cheng et al. demonstrated that such a shift in LNP charge and resultant LNP tropism was achievable through the addition of charged helper lipid to the nanoparticle formulation [28]. This is an important finding that will facilitate the use of mRNA as a treatment for extrahepatocellular diseases.
We were motivated to expand upon this work for two reasons. First, the addition of a fifth lipid component to lipid nanoparticles further complicates what is already a complex formulation space. Such complexity may slow regulatory approval processes and/or cause manufacturing challenges due to GMP production of an additional lipid and a more complex final drug product. Second, we wondered if similar tropism-shifting effects could be achieved with a broader array of charged helper lipids. Accordingly, we examined eight alternative helper lipids for complete replacement of the standard DOPE lipid used in lipidoid nanoparticle formulations.
For the replacement lipids, we selected at least one member of all major naturally occurring classes of phospholipids as well as several non-natural cationic helper lipids. Apart from charge, helper lipids have a variety of biological signaling interactions, including detection of apoptotic cells and exosomes, immunostimulation, and membrane fusion [29–31]. Helper lipids such as phosphatidylserine, sphingomyelin, and ceramide have been shown to be enriched in exosomal membranes [32, 33]. Positively charged lipids, such as DOTAP, do not have native biological interactions, as they do not occur naturally in any known biological organism. Nonetheless, cationic lipids can aid in membrane fusion and endosomal escape, which can facilitate shifts in efficacy due to differences in organ susceptibility to fusion [34, 35]. The ability to control organ distribution by adjusting the helper lipid alone could be a robust and powerful method to expand the application of lipid nanoparticle technology.
Herein, we demonstrate that most neutral, anionic, and cationic helper lipids deliver mRNA to the liver, spleen, and lungs, respectively. This can be accomplished with a four-component lipid nanoparticle formulation. Further, we show that the magnitude of charge linearly increases this shift in specificity, suggesting that physical parameters of LNPs and resultant LNP-cell interactions play a key role in tissue specific mRNA delivery. These observations were not universal, however, indicating that the relationship between charge and specificity is more complex than cationic delivers to the lung and anionic delivers to the spleen.
Results
Helper lipid charge and concentration affected organ specificity
The goal of this study was to determine whether the tropism of mRNA-loaded lipid nanoparticles could be altered through helper lipid substitution in a four-component formulation. Although the effect of the chemistry and concentration of helper lipids has been described for some classes of ionizable materials, it has not been clear whether these trends extend to other materials. Accordingly, we first probed the impact of helper lipid chemistry on the in vivo performance of LNPs containing the class of ionizable lipidoids generated by our lab [36, 37]. As a benchmark, we used our standard formulation, which contains 35 mol% ionizable lipidoid, 46.5 mol% cholesterol, 16 mol% DOPE, and 2.5 mol% C14-PEG2000, with a lipidoid to mRNA mass ratio of 10:1. This formulation was identified in 2018 through a design of experiment optimization process [15] that we independently validated (data not shown). We focused on complete replacement of DOPE with an alternative helper lipid to maintain a four-component formulation.
For an initial experiment, we chose to work with the ionizable lipidoid 306O10 (Supplementary Figure 1A), which is potent both in vitro and in vivo [38]. We formulated it with one of three helper lipids (Figure 1A): DOPE (net neutral charge), PS (net negative charge), or DOTAP (positive charge). LNPs were loaded with mRNA encoding Firefly luciferase (mLuc) and injected intravenously into mice before resultant luciferase expression was measured three hours later.
Figure 1: Helper lipid charge influences the organ location of protein expression following mRNA delivery.
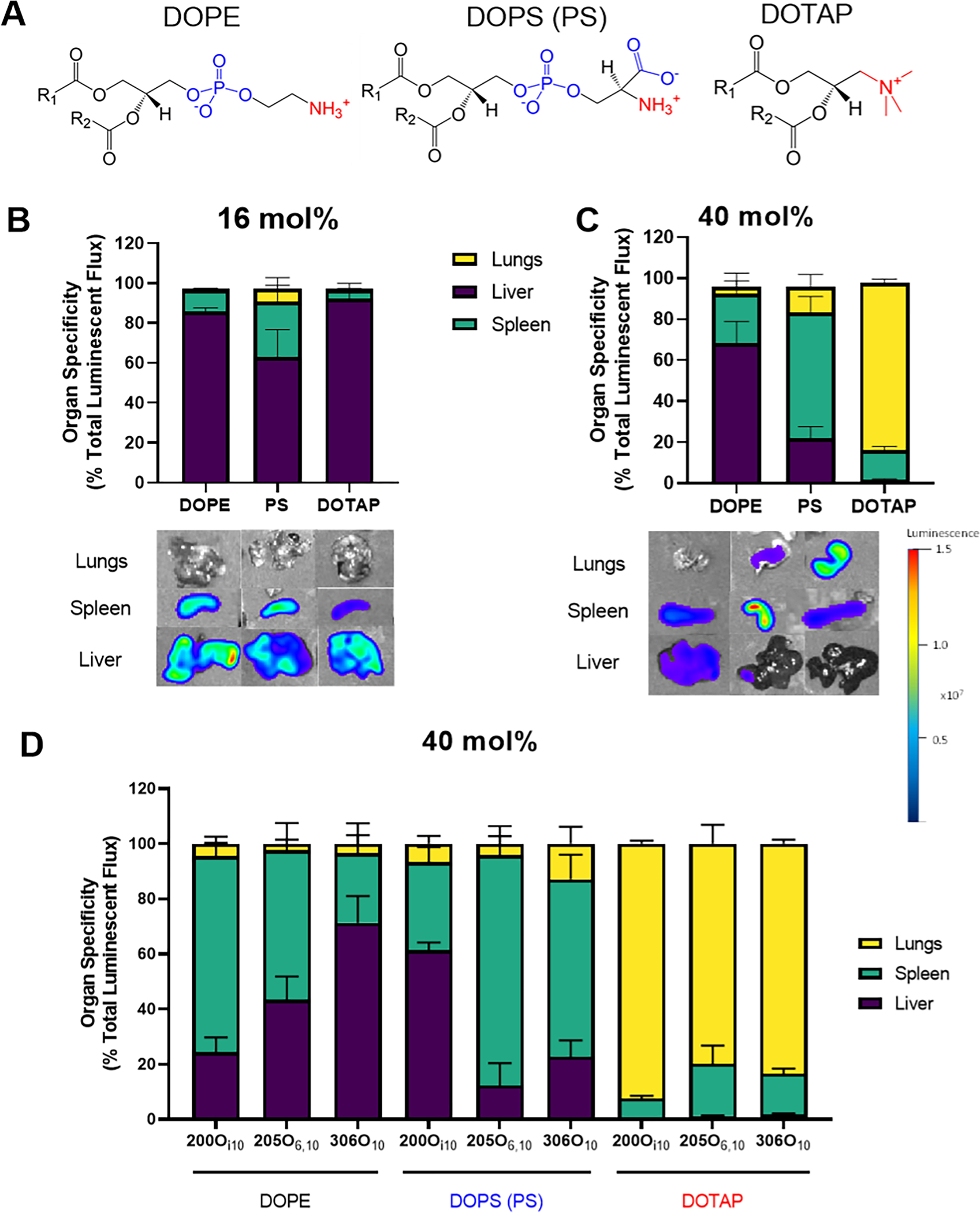
LNPs were formulated with one of three helper lipids: DOPE (net neutral charge), PS (net negative charge), or DOTAP (positive charge). Mice were injected intravenously with LNPs at a dose of 0.75 mg/kg of mRNA encoding Firefly luciferase. Luciferase signal was quantified three hours post-injection using an In Vivo Imaging System (IVIS). (A) Structures of the 3 helper lipids considered in this figure. (B) Luciferase expression occurred predominantly in the liver, regardless of helper lipid charge, for a standard LNP formulation (16 mol% helper lipid) incorporating the ionizable lipidoid 306O10. The 3 columns in the images represent the 3 helper lipids, and the scale bar is on the right. (C)Luciferase expression shifted from the liver to the spleen or lungs when LNPs were formulated with 40 mol% PS or DOTAP instead of DOPE. (D) Helper lipid charge altered the organ location of protein expression irrespective of ionizable lipidoid identity for LNPs. Data for three ionizable lipidoids (200Oi10, 205O6,10, and 306O10) are shown here. Data represent mean values. Error bars represent standard deviation (n = 3).
When working with our standard formulation of 16 mol% helper lipid, substitution of DOPE with PS or DOTAP did not cause a statistically significant shift in luciferase expression away from the liver (Figure 1B). Since helper lipids effects here were minimal, we performed concentration gradient experiments in vitro (Supplementary Figure 2). These experiments identified 40 mol% helper lipid as the concentration at which helper lipid variation produced the greatest effects, in agreement with previous work [28]. To accomplish this, we reduced the mol% of cholesterol since its concentration has less of an effect on LNP efficacy and circulation time compared to the ionizable lipidoid or PEG-lipid [39, 40].
In contrast to the 16 mol% data, intravenous administration of LNPs at 40 mol% helper lipid significantly altered the organ specificity of mRNA expression (Figure 1B,C). Specifically, the negatively charged PS and positively charged DOTAP particles shifted luciferase expression from the liver to the spleen and lungs, respectively. Altered tropism did come at some cost, as the total luminescent signal produced by the LNPs formulated at 40 mol% helper lipid was roughly half of those formulated at 16 mol% (Supplementary Figure 3A,B). Moving forward, we chose to examine only formulations containing 40% helper lipid.
We next asked whether these results extended to LNPs formulated with ionizable lipidoids that are not exclusively liver-tropic. For these experiments, LNPs were formulated with DOPE, PS, or DOTAP and one of three efficacious lipidoids: 200Oi10, 205O6,10, and 306O10 (Supplementary Figure 1). When formulated with 40 mol% DOPE, these formulations had varied tropism for the liver and spleen, with spleen: liver ratios of 3:1, ~1:1, and 0.3:1, respectively (Figure 1D). PS formulations shifted specificity to the spleen, except for 200Oi10. Because all LNPs formulated with 200Oi10 had reduced efficacy, it is possible that they were poorly compatible with the PS helper lipid (Supplementary Figure 3C). LNPs formulated with any of the three ionizable lipidoids and DOTAP shifted specificity to the lungs, with varying degrees of efficacy. These data support the concept of anionic helper lipids shifting protein expression to the spleen and cationic helper lipids transfecting primarily the lung. However, the 200Oi10 data suggest that both helper lipid and the specific ionizable lipidoid contribute to alterations in tropism.
The effect of helper lipid charge was conserved for different phospholipid chemistries
We next sought to understand whether these trends extend to alternative classes of helper lipids. These included the neutrally charged ethanolamine (PE), phosphatidylcholine (DOPC), sphingomyelin (SM), and ceramide; the negatively charged phosphatidylserine (PS), phosphatidic acid (PA), and phosphatidylglycerol (PG); and the positively charged DOTAP and ethyl phosphatidylcholine (EPC). The ionizable lipidoid 306O10 was used in all formulations. As with DOPE, the net neutral helper lipids SM and ceramide produced luminescence primarily in the liver (Figure 2). In comparison, all anionic and cationic helper lipids shifted LNP specificity to the spleen and lungs, respectively. Unfortunately, the helper lipids PC, SM, and PA extinguished LNP efficacy in vivo (Supplementary Figure 3D), so they are unlikely to be good choices when formulating LNPs. Overall, DOTAP and PS best maintained LNP efficacy (total signal in the organ) while shifting specificity (percentage of total signal in the organ) to the lungs and spleen, respectively.
Figure 2: The effect of helper lipid charge on the location of protein expression holds is conserved across helper lipid chemistries.
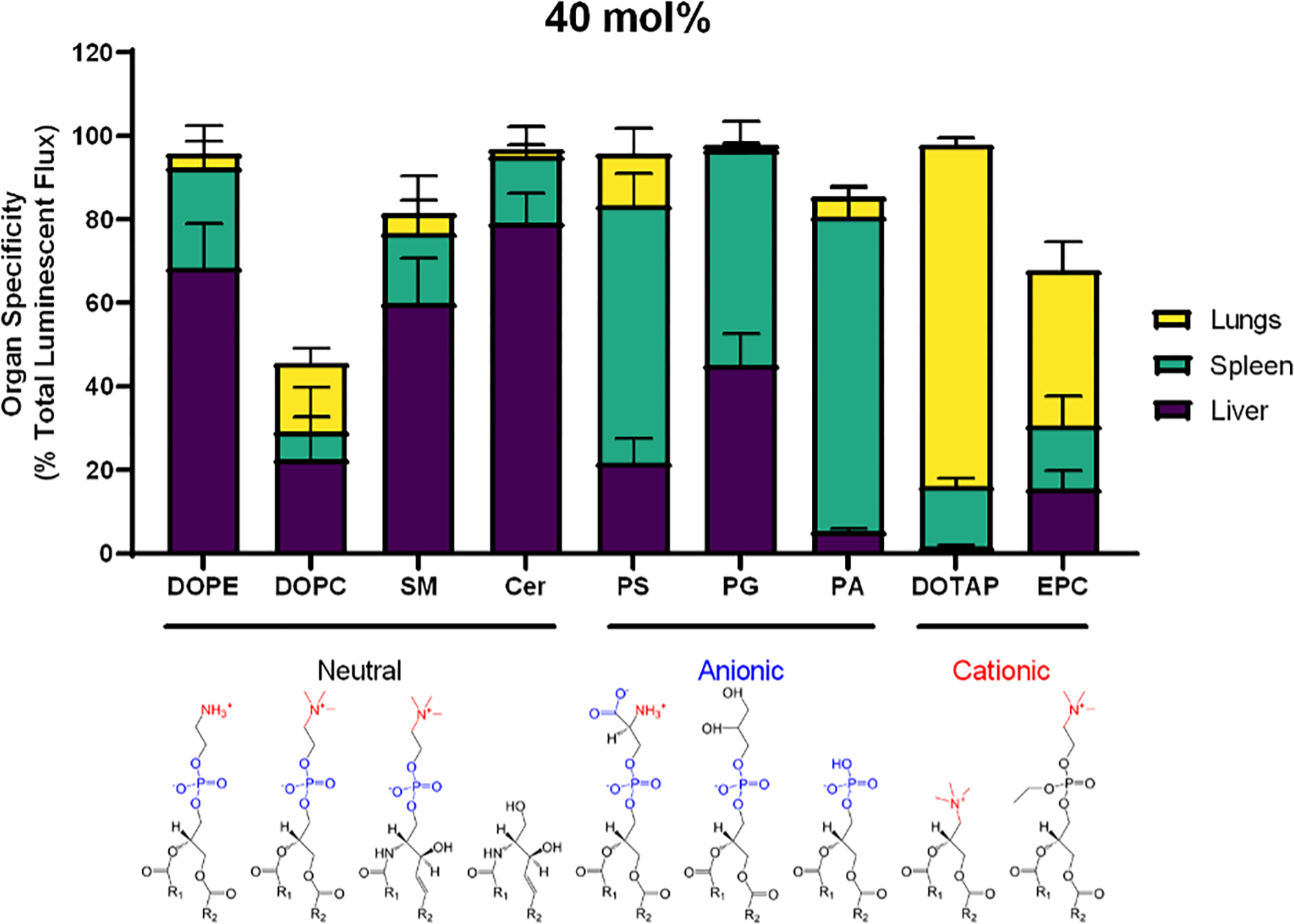
LNPs were formulated with the ionizable lipidoid 306O10 and 40 mol% helper lipid and IV injected into mice (0.75 mg/kg mRNA). Luciferase signal was measured three hours later. In addition to DOPE, neutral lipids 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), sphingomyelin (SM), and ceramide (Cer) induced protein expression primarily in the liver. Anionic lipids, including PS, phosphatidylglycerol (PG) and phosphatidic acid (PA) shifted expression to the spleen, and the cationic lipids DOTAP and ethyl phosphatidylcholine (EPC) shifted expression to the lungs. Luminescence values as a percentage of total luminescence of all organs (liver, lungs, spleen, kidneys, intestines, pancreas, and heart) are plotted above. Mean values shown. Error bars represent standard deviation (n = 3).
Changes in LNP charge and ionization correlated with in vivo specificity results
Based on these results, we investigated how formulation of LNPs with different helper lipids affected physical properties. We wanted to understand if these properties were responsible for the altered in vivo specificity and efficacy. LNP size was determined using dynamic light scattering (DLS). All LNPs remained a similar size except DOTAP, which increased in diameter by 50% compared to DOPE. In vivo lung specificity correlated with LNP size (R2 = 0.83, p = 0.0005) and ionization at pH 7 (R2 = 0.89, p<0.0001) and pH 5 (R2 = 0.85, p = 0.0003) (Figure 3A–C). The increased correlation from efficacy to specificity also suggests that increased ionization (positive charge) causes more effects on specificity than on efficacy. These trends transcended lipidoid chemistry and were similar for LNPs generated with the ionizable lipids shown in Supplementary Figure 1B and C (data not shown).
Figure 3: In vivo lung and spleen specificity correlated with LNP physical properties.
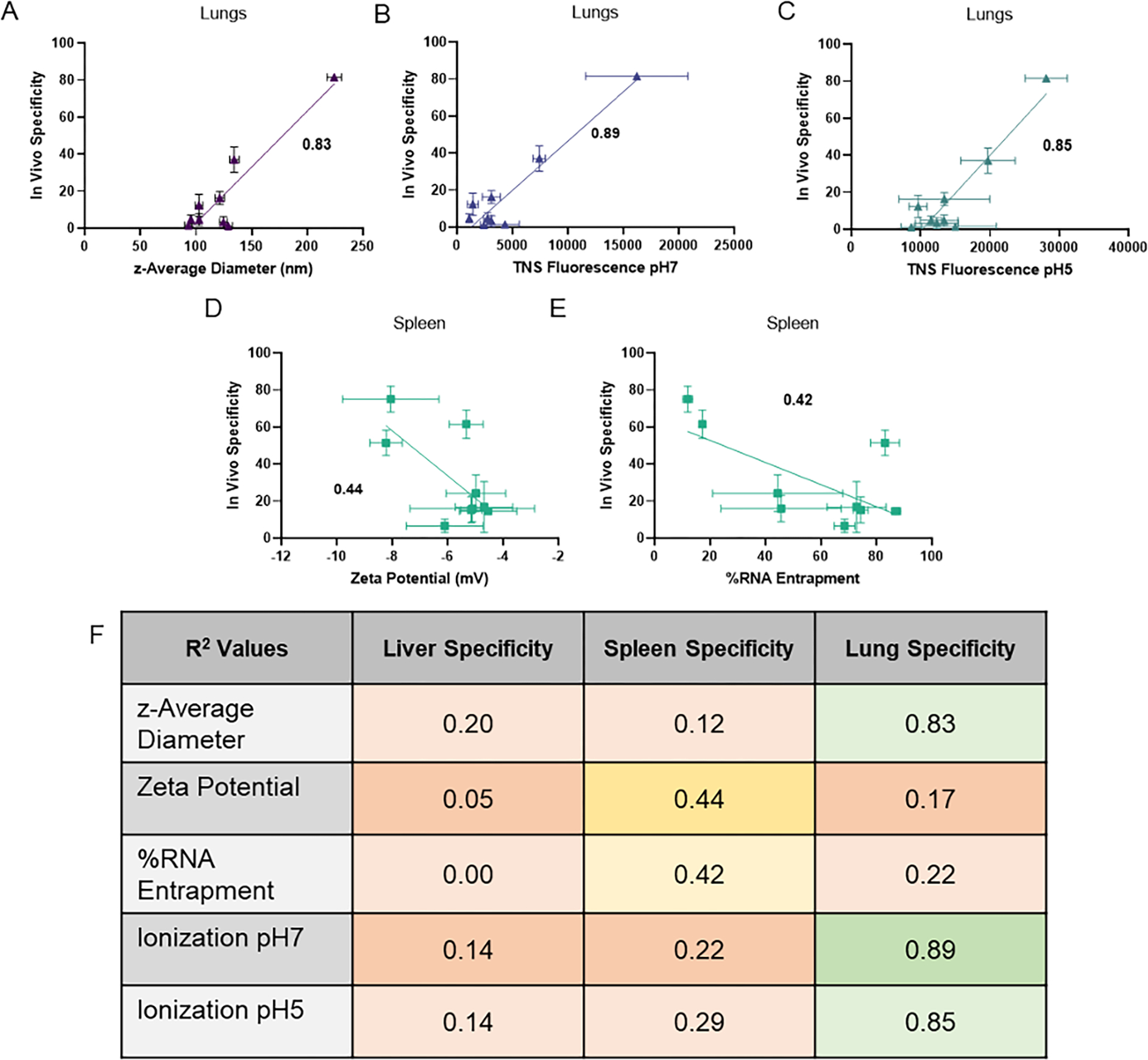
The physical properties that correlated with lung specificity were (A) z-average size, (B) TNS fluorescence (a surrogate for LNP surface ionization) at pH 7 and (C) pH 5. The physical properties (D) zeta potential and (E) RNA entrapment correlated negatively with in vivo spleen specificity. (F) The correlation values of physical properties (z-average diameter, zeta potential, RNA entrapment and TNS ionization) were calculated for liver, spleen, and lung efficacy in vivo. Data shown represents mean. R-squared values are Pearson correlation coefficients. Boxes were shaded with colors indicating degree of correlation with <0.3 (low correlation) shaded red, 0.3–0.6 shaded yellow, and >0.6 (high correlation) shaded green. Error bars indicate s.d. (n = 6). R-squared values are shown on each plot.
The effect of helper lipid charge on LNP surface charge was measured via zeta potential and cationic ionization of the LNPs through the TNS fluorescence assay. The zeta potential of LNPs at 16 mol% of helper lipids did not show any obvious trends (Supplementary Figure 5A). Ionization was measured using the TNS fluorescence assay at pH 7 and pH 5 to mimic physiological and endosomal conditions, respectively. The TNS molecule is known to integrate into lipid membranes and become more fluorescent when exposed to more positive charge [41]. At 16 mol% helper lipid, replacement of DOPE with anionic helper lipids caused no significant changes in ionization, but there was an increase in ionization with cationic helper lipids (Supplementary Figure 5C, E). At 40 mol% helper lipid, the cationic helper lipids DOTAP and EPC both exhibited higher ionization via TNS assay at both pH 7 and pH 5 (Supplementary Figure 5D, F). This suggests that higher concentrations of helper lipid have more pronounced changes in overall nanoparticle charge. Specificity in the spleen in vivo correlated moderately and negatively with zeta potential (R2 = 0.47, p =0.04) and RNA entrapment (R2 = 0.45, p = 0.048), suggesting that negative charge leads to preferential effects in the spleen (Figure 5D, E). We confirmed that these correlation values were similar for LNPs formulated with the ionizable lipidoids in Supplementary Figure 1B and C (data not shown).
Figure 5: Organ specificity was linked to immune cell infiltration patterns.
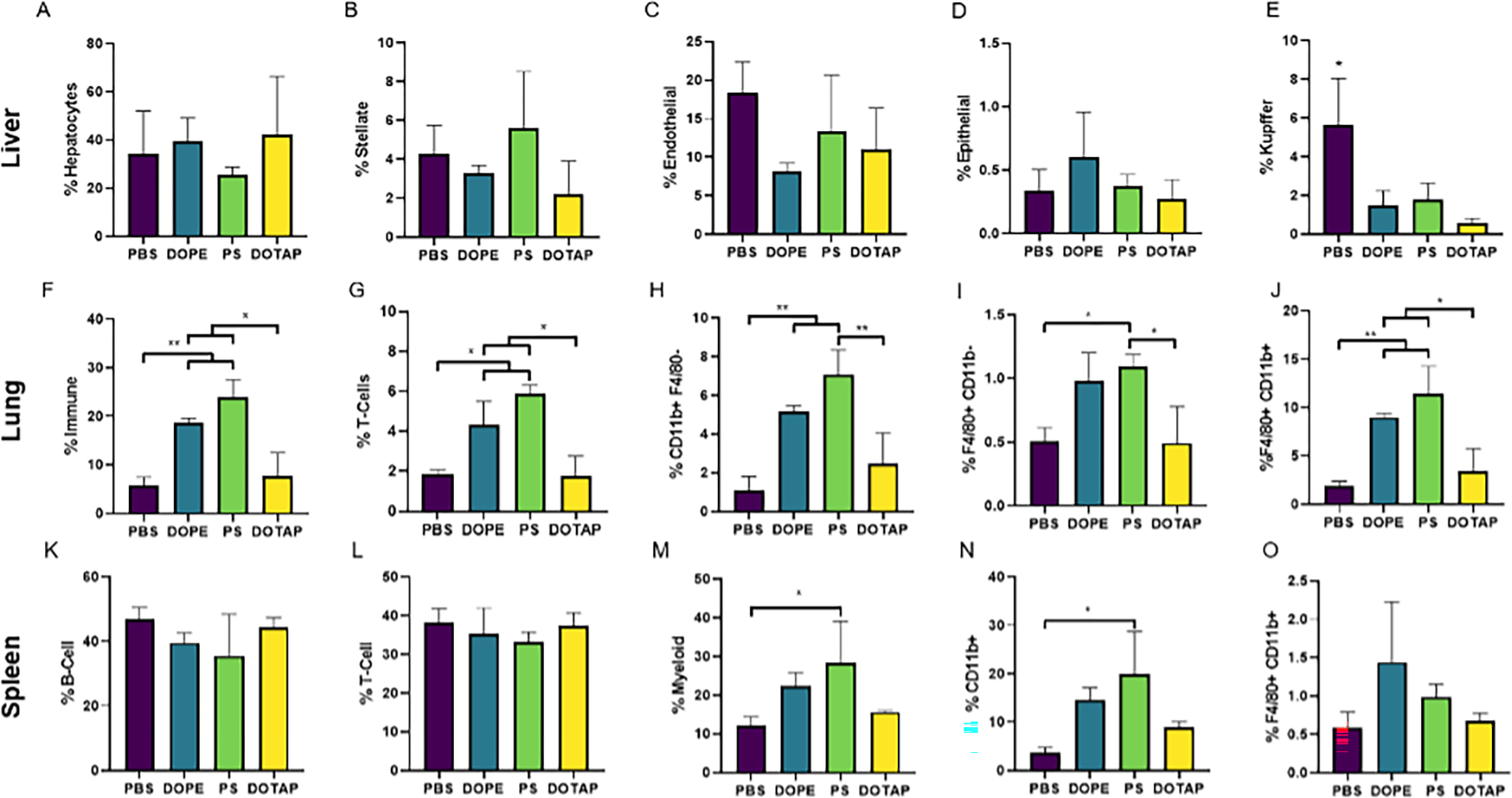
LNPs were formulated with the lipidoid 306O10 and 40 mol% DOPE, PS, or DOTAP. Three hours after LNP injection via tail vein at an mLuc dose of 0.75 mg/kg, cell populations in the liver, lungs, and spleen were analyzed via flow cytometry. Cell types are identified on the y-axes. Total immune cells were identified as CD45+, T cells as CD3+, B-cells as CD19+ with F4/80+ and CD11b+ labeled as such as further markers are needed for distinct cell definition. (A-E) LNP formulations did not alter the proportion of most cell types in the liver. (F-J) LNPs formulated with DOPE or PS increased the proportion of immune cells, and, specifically, myeloid cells in the lungs. DOTAP LNPs did not have an effect. (K-O) LNPs formulated with PS increased the proportion of myeloid cells in the spleen. Mean values are shown with error bars indicating s.d. (n = 3), 2-way ANOVA Tukey Test with * and ** representing p ≤ 0.05 and p ≤ 0.01, respectively.
We also noted that negatively charged PS and PA reduced RNA entrapment, which can be visualized in Figure 3E by noting the formulations with highest spleen efficacy (PS and PA) had the lowest entrapment. This could be due to poor interactions between the negatively charged RNA and the negatively charged helper lipids. The negative correlation with RNA entrapment could also suggest that anionic helper lipids lead to externalization of mRNA onto the surface of LNPs, causing a negative surface charge. All in vivo specificity correlations with LNP physical properties are summarized in Figure 3F.
Helper lipid choice caused differential efficacy in adherent and immune cell lines
Next, we asked if helper lipids shifted in vivo organ efficacy because they have inherent cellular specificity. For these experiments, LNPs were formulated with DOPE or one of the eight alternative helper lipids and delivered to cells in culture. We used cell lines representative of the major populations of the liver, lung, and spleen. Liver was represented by hepatocyte (HepG2), macrophage (RAW 264.7), fibroblast (NIH/3T3), and endothelial (HULEC-5a) cell lines. Major lung cell types were represented by lung epithelial cells (A549), endothelial cells (HULEC-5a), fibroblasts (NIH/3T3), and macrophages (RAW 264.7). Finally, the spleen was represented by B cells (RajiB), T cells (Jurkat), and macrophages (RAW 264.7). HeLa cells were included as they are commonly used for in vitro investigations of nanoparticle efficacy [36].
Results revealed that the use of different helper lipids’ effect on transfection efficacy was due to both cellular specificity and helper lipid concentration. At 40 mol% helper lipid, DOPE produced the highest efficacy in HeLa, HepG2 hepatocytes, NIH3T3 fibroblasts, A549 lung epithelial cells and HULEC-5a endothelial cells (Figure 4A–E). These cells are most abundant in the liver and lungs. Immune cells had significantly lower transfection levels compared to other cell types investigated (Figure 4F–H). In RAW macrophages, ceramide performed similarly to DOPE, while PG was the most potent helper lipid (Figure 4F). In Jurkat T cells, DOPE, SM, ceramide, and PS all performed similarly (Figure 4G). In Raji B cells, PG then PS were the most effective helper lipids while DOPE was only mildly effective, representing the only cell type in which DOPE was not a top performer (Figure 4H). This illustrates that the optimal helper lipid in an LNP formulation can vary with cell type. Of note, similar trends of helper lipid effect on cell line transfection efficacy were not observed for LNPs formulated with only 16 mol% helper lipid (Supplementary Figure 6). This further illustrates that higher concentrations of helper lipid are required to affect nanoparticle tropism.
Figure 4: Adherent and immune cell lines were best transfected by neutral and anionic helper lipids, respectively.
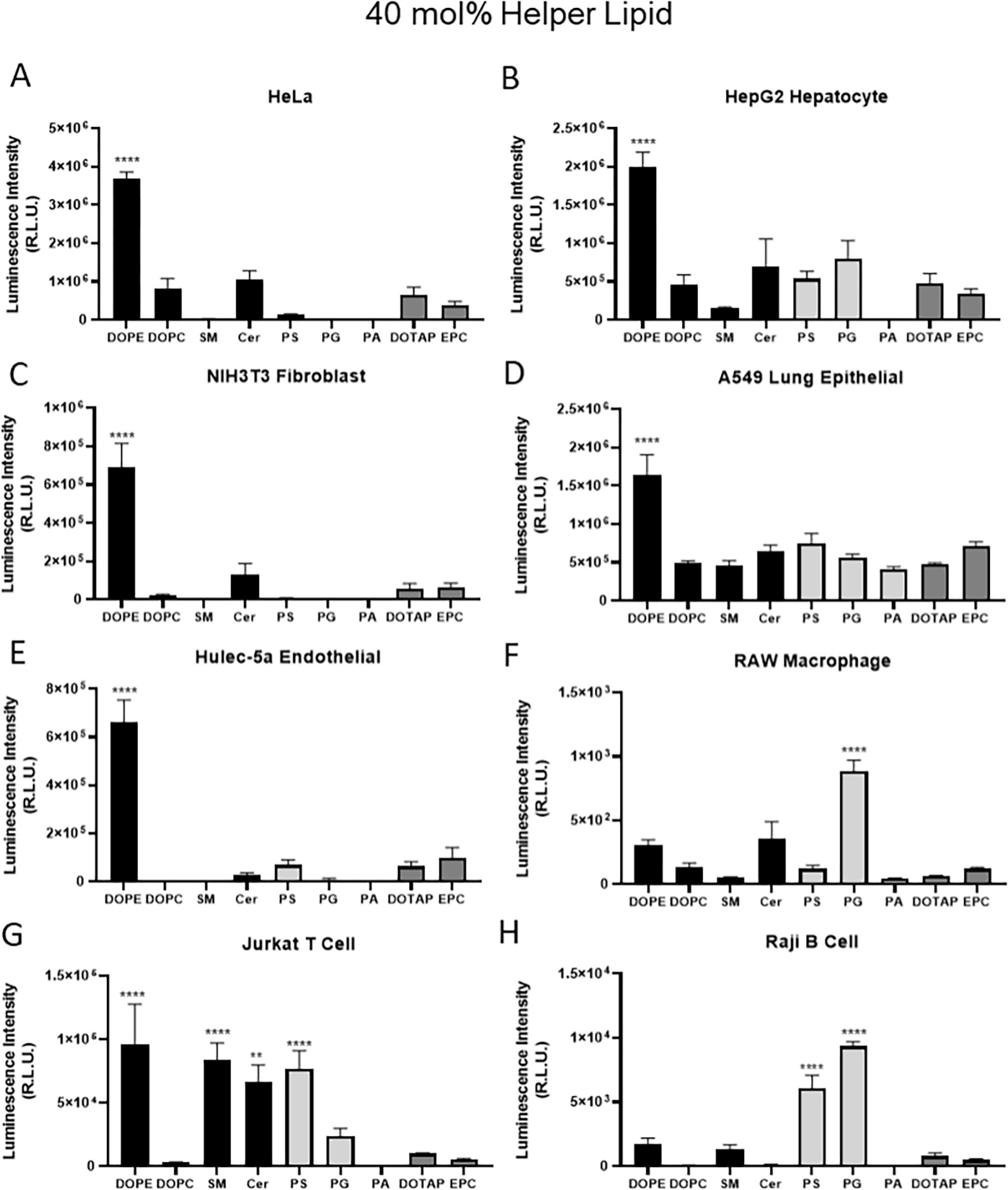
Lipid nanoparticles formulated with 40 mol% helper lipid and mLuc were incubated with eight cell lines for 24 hours at 100 ng/well prior to measurement of luciferase signal. Cell lines included HeLa (human cervical), HepG2 (human hepatocytes), NIH 3T3 (mouse fibroblasts), A549 (human lung epithelial), Hulec-5a (human lung endothelial), RAW 264.7 (mouse macrophage), Jurkat (human T) and Raji B (human B). Data shown represent mean values. Error bars indicate standard deviation, 2-way ANOVA Tukey Test where *, **, *** and **** represent p ≤ 0.05, p ≤ 0.01, p ≤ 0.001 and p ≤ 0.001 respectively.
Helper lipids altered acute inflammatory responses in vivo
Given that we identified the organs to which alternative helper lipids shifted expression, we next asked how helper lipids affected cells within these targeted organs. The helper lipids PS and DOTAP both are known to elicit cellular responses, with PS being an apoptotic signal to immune cells and DOTAP causing toxicity [4, 42]. To characterize the cellular response to intravenous administration of these different LNP formulations, we performed flow cytometric analysis on the liver, lungs, and spleen following LNP administration. LNPs formulated with the lipidoid 306O10 and either DOPE, PS, or DOTAP were delivered to C57BL/6 mice at an mFLuc dose of 0.75 mg/kg.
Choice of helper lipid did not affect the relative ratios of resident liver cell types (Figure 5A–D). LNPs also did not change these ratios compared to control, except for Kupffer cells (F4/80+). LNP treatment resulted in 3–10x fewer Kupffer cells in the liver compared to control animals (Figure 5E), indicating an acute response from these liver-resident immune cells.
In the lung, the presence of various myeloid and immune cell populations changed in response to lipid nanoparticles and the choice of helper lipid (Figure 5F – J). Specifically, treatment with LNPs containing DOPE and PS caused an infiltration of several types of immune cells, including T-cells (CD3+) and F4/80+CD11b+ macrophages. There were no changes in epithelial or endothelial cell populations in the lung, with each representing ~30% of the total population (data not shown).
In general, splenic immune cell (CD45+) population were consistent regardless of LNP formulation (Figure 5K–O). For example, B-cell and T-cell populations remained at 35–40% across treatment groups. Only PS altered relative myeloid cell populations, likely driven by a corresponding increase in CD11b+ cells. When considering all the organs, LNP administration caused a host response that resulted in changes in CD11b+ and F4/80+ myeloid cell populations.
LNP formulations did not cause acute organ damage
Finally, we investigated the effects of LNP administration upon the pathology of the organs they target. Following LNP administration, the liver, lungs, and spleen of mice were fixed in formalin and embedded for frozen sectioning. Sections of mouse organs were stained with hematoxylin and eosin for histologic analysis (Figure 6). Upon evaluation under brightfield microscopy, there were no obvious signs of gross pathological changes suggestive of tissue damage in the liver, lungs, or spleen. Investigation of all the other helper lipids tested via histology similarly did not reveal any gross changes in tissue morphology (Supplementary Figure 7). There was higher cellularity in the lungs in mice administered with PS LNPs and slightly with DOPE LNPs, suggesting an acute inflammatory response similar to our results from flow cytometry. However, further investigation of tissue damage at acute as well as several chronic timepoints would be necessary to determine the safety of these new formulations.
Figure 6: Helper lipid identity affected inflammatory state and cell-specific location of protein expression within organs.
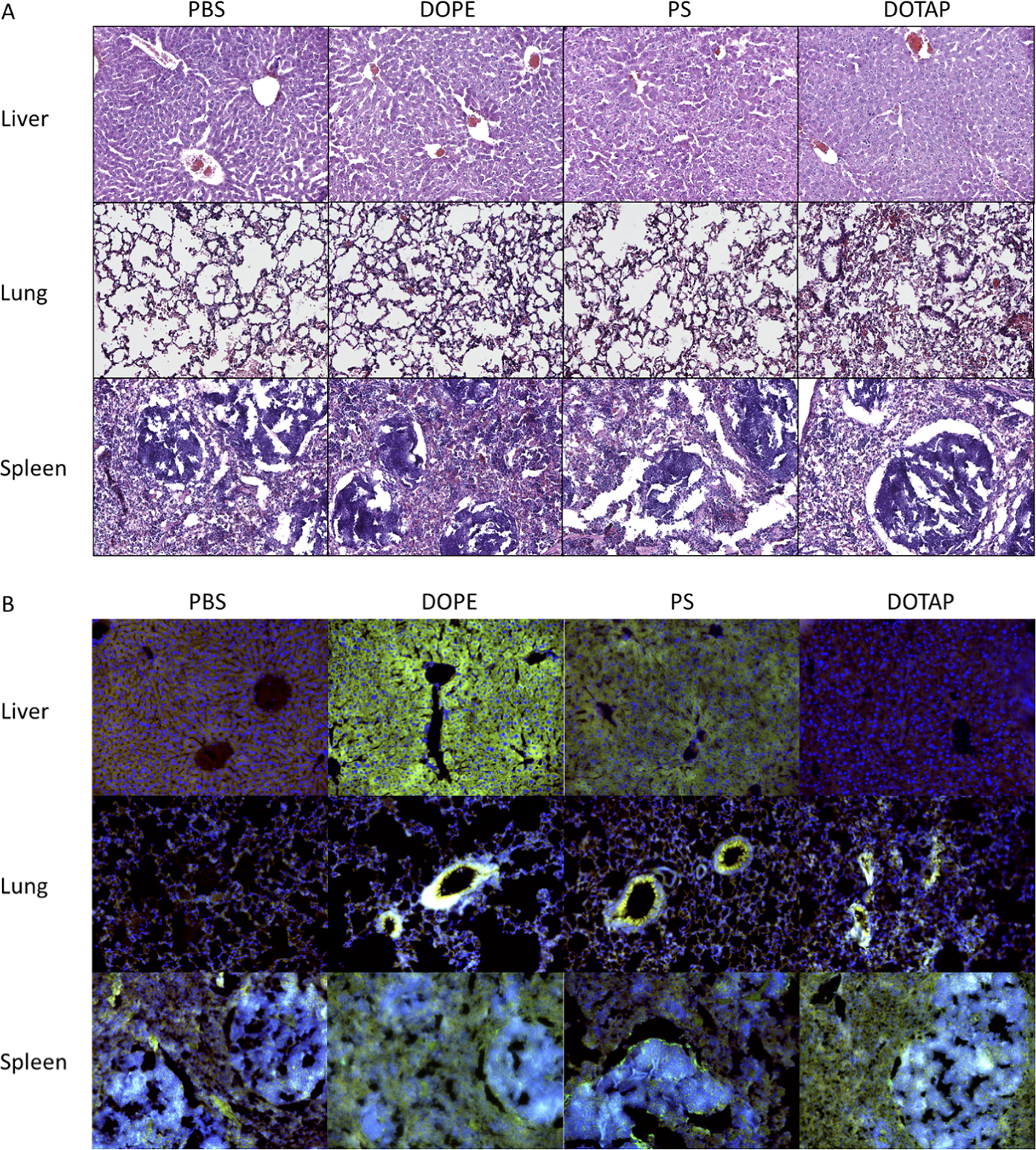
LNPs were formulated with the lipidoid 306O10 and at a helper lipid concentration of 40 mol%. (A) Three hours following intravenous LNP administration in mice, organs were harvested and prepared for hematoxylin & eosin staining. Hypercellularity was observed in the lungs of mice treated with DOTAP LNPs, suggesting the host response to DOTAP LNPs may contribute to the shift in efficacy to the lungs. (B) Tissues sections were also stained with antibodies against Firefly luciferase (green), phalloidin (red) and Hoescht (blue). Staining suggested that DOPE LNPs transfected the entirety of the liver emanating from liver sinusoidal endothelial cells. Transfection in the lungs appeared localized to the blood vessels. Splenic tissue sections suggested that transfection was throughout the spleen in DOPE and DOTAP LNP-treated mice, while PS LNP treated mice had more transfection in the white pulp marginal zones which contain adaptive immune cells such as B cells.
Comparison with SORT method
To address the anticipated comparison with the SORT method described by Cheng and colleagues for shifting organ specificity, we performed a head-to-head study of our four-component method versus SORT method [28]. Here, we used the lipidoid, helper lipids and helper lipid concentration optimized for our four-component method for consistency. Characterization data between the two methods were similar. Regarding LNP size, SORT LNPs were somewhat larger than four-component LNPs (Supplementary Figure 9A). Both four-component and SORT LNPs identically altered surface charge as assessed by zeta potential (Supplementary Figure 9B). Incorporation of DOTAP into four-component and SORT methods resulted in nearly identical shifts in ionization as assessed by TNS at pH7 and pH5 (Supplementary Figure 9C,D). DOPS resulted in reduced ionization at pH5 in both four-component and SORT methods, while four-component resulted in higher ionization within each helper lipid group at pH5 (Supplementary Figure 9D).
Differences in efficacy between our four-component system and SORT were determined in vivo by injecting LNPs intravenously at 0.25 mg/kg mLuciferase for 3 hours, followed by IVIS imaging. Four-component and SORT LNPs behaved comparable with DOPE and DOTAP formulations (Supplementary Figure 9E–G). The effect of adding DOPS, however, differed between methods. Here, DOPS four-component and SORT formulations provided a ~2.5x increase and ~4x decrease in efficacy, respectively, compared to DOPE formulations. These overall efficacy differences were driven by efficacy increases in the spleen for four-component DOPS LNPs and decreases in liver expression for SORT LNPs. Despite differences in efficacy, the four-component system and SORT resulted in similar shifts in organ specificity (Supplementary Figure 9I–L). The only difference was a reduced shift to the spleen for DOPS for SORT compared to four-component systems. We discuss the limitations of these comparisons below.
Discussion
Here, we present alternative four-component mRNA lipid nanoparticle formulations that alter protein expression patterns in mice following IV administration. Although LNPs are highly efficacious delivery vehicles, their delivery capabilities are typically localized to the liver with minimal spleen or lung efficacy [4]. This is due to the control of the mononuclear phagocytic system (MPS), which involves phagocytes such as macrophages in the liver and spleen rapidly taking up nanoparticles to clear them from the body [43]. Some lipid nanoparticles can also preferentially enter liver hepatocytes and macrophages due to LNP binding to the protein ApoE in the blood [11].
The targeting of specific organs is one of the biggest hurdles facing delivery of mRNA therapeutics [36]. Although there has been some success targeting extrahepatic organs using alternative administration routes (e.g., intraperitoneal, oral, intramyocardial), these are not ideal for clinical translation [44–46]. In this study, we show that the organ specificity of LNP formulations can be altered by tuning the chemistry and concentration of the helper lipid. This effect, which shifted delivery from the liver to the spleen or lungs, was achieved via complete replacement of the helper lipid with anionic or cationic lipids to induce negative or positive charge in the lipid nanoparticle.
Several previous reports have shown that imbuing a positive or negative charge shifts lipid nanoparticle tropism to the lungs or spleens, respectively. For example, Kranz et al. showed LNP surface charge can be altered by varying the ratio of cationic lipid to the anionic mRNA cargo [26]. However, this approach is problematic with lipidoid nanoparticles, as low mRNA or low lipidoid concentrations significantly reduces the efficacy of these particles. Another paper by Chen et al. demonstrated that alteration of lipid nanoparticle charge by swapping DC-cholesterol (cationic) for cholesterol or using phosphatidylglycerol (PG) as the helper lipid altered the protein corona that coated the LNPs [27]. Other reports have shown that cationic liposomes cause efficacy in the lung and can even shift the efficacy of adenoviral vectors to the lungs [47, 48]. Our approach differs from a recently reported method that incorporates an additional lipid, termed a SORT lipid, to achieve similar effects [28]. Although SORT LNPs are quite effective, the additional lipid in the formulation has the potential to complicate regulatory and manufacturing processes. We were thus motivated to extend those results to “simpler”, four component nanoparticles. Together, four- and five-component extrahepatic lipid nanoparticle formulations offer a breadth of formulation space to meet the needs of specific products in terms of specificity, potency, immunogenicity, stability, and more.
The ability of anionic and cationic lipids to shift LNP specificity while maintaining efficacy appears linked to the chemistry of helper lipid functional groups. Phosphatidylserine’s higher efficacy than the other anionic lipids may be related to pKa properties, with PS’s carboxylic acid functional group yielding a pKa value of roughly 5.5, whereas the phosphate groups in PG and PA acid have pKa values around 3 [49]. Therefore, PS is more likely to have a protonated carboxylic acid residue and a net neutral charge in the acidic environments of the endosome, which may aid endosomal escape. Similarly, the cationic lipids DOTAP and EPC differentially impacted efficacy despite their common net positive charge. The head group of both DOTAP and EPC contain a quaternary nitrogen, so their pKa values should be similar. However, the ethyl modification in EPC prevents this lipid from making hydrogen bonds, which could affect its membrane properties [50].
The reduction of cholesterol concentration from 46.5 mol% to 22.5 mol% when increasing helper lipid concentrations from 16 to 40 mol% also contributes to shifted organ specificity away from the liver. Cholesterol is a critical LNP component because it aids nanoparticle stability, RNA encapsulation, and cell uptake via interaction with lipid rafts in cellular membranes [39, 40, 51, 52]. Further, native cholesterol lipoparticles undergo receptor-mediated uptake by hepatocytes in the liver through LDL receptor and scavenger receptors [53, 54]. All 40 mol% helper lipid formulations reduced liver specificity, with their only commonality being a roughly 2-fold reduction in cholesterol concentration compared to the standard lipidoid formulation (16 mol% helper). This suggests that cholesterol within an LNP formulation augments liver specificity. This concept is supported by studies that examined the inclusion of cholesterol analogues in LNP formulations [55, 56], one of which reported efficacy shifts from hepatocytes to other liver cells. Substitution of cholesterol for analogues could reduce LNP interactions with LDL, after which scavenger receptors on immune cells could aid in shifting LNP specificity away from the liver.
Apart from imbuing surface charge to lipid nanoparticles, helper lipids have physiological roles that may determine their fate upon intravenous administration. The ethanolamine, DOPE, is the most common choice for a helper lipid in lipidoid nanoparticle formulations [57]. The success of this helper lipid is attributed to its support of a non-bilayer hexagonal II (HII) phase, which forms during membrane fusion and disruption [58]. This has led to DOPE being described as a fusogenic lipid as it helps nanoparticles fuse with the endosomal membrane, leading to more efficient endosomal escape [59]. Phosphatidylcholines such as DOPC are the most common lipids in mammalian lipid membranes [60]. This lipid class provides membrane stability by favoring the lipid bilayer formation [61]. Unfortunately, the stability that DOPC lipids provide to lipid nanoparticles may also cause reduced endosomal escape based upon on the observed reduction in efficacy of these LNPs.
Several lipid classes investigated in this study are enriched in exosomal membranes. PS, SM, ceramide, and PA, in particular, are found in elevated levels in exosomes while PE, PC, and PG are reduced in level [62]. Phosphatidylserine is involved in recognition of apoptotic cells - normally maintained on the inner leaflet of cells through transmembrane lipid transporter proteins called flippases, they present externally only upon cell death [63]. Upon intravenous administration, the marginal zone macrophages of the spleen could facilitate uptake of PS LNPs via the PS-specific receptor Tim4, leading to shifts in efficacy [64]. Our flow cytometry results showed increased levels of CD11b+ myeloid cells in the lungs and spleen and CD3+ T cells in the lungs, suggesting PS LNPs are immunostimulatory in this setting.
Sphingomyelin and ceramide have a special relationship in their role during exosome budding. Sphingomyelin, like phosphatidylcholine, is maintained on the outer membrane of cells and acts as a stabilizing lipid [65]. Sphingomyelin also plays a role in the stabilization of cholesterol in lipid rafts due to strong interactions between the two which could lead to reduced transfection efficacy. The cleavage of sphingomyelin into ceramide is involved in the budding off of exosomes, as inhibition of sphingomyelinases prevented exosome formation [32]. Therefore, ceramide is naturally enriched in exosomes and could be used as a signaling molecule for cell-to-cell communication.
Phosphatidylglycerol has unique signaling properties of its own which may contribute to its distinct efficacy profile. Interestingly, PG has been shown to activate RNA synthesis, which could be beneficial for applications of mRNA delivery [66]. Phosphatidylglycerol was also found to inhibit TLR2 and TLR4 activation in the context of damage-associated molecular pattern signaling [67]. This could also reduce immune cell uptake of PG LNPs, which may be responsible for the reduced shift to the spleen observed in this study.
The cationic helper lipids, DOTAP and EPC, showed specificity shifts to the lungs. Apart from the role of protein corona, which was discussed above, there are several other potential mechanisms that could be responsible for lung specificity. It has been previously shown that cationic DOTAP liposomes interact with heparan sulfate proteoglycans in the glycocalyx around the alveolar endothelial cells due to electrostatic interactions enabling lung specific delivery [47, 48, 68, 69]. It is possible that the cationic helper lipids cause similar association with the negatively charged proteoglycans [70]. The increased lung efficacy could also be due to lung cells being more susceptible to the membrane destabilization caused by cationic lipids. If the lungs are more susceptible to these cationic lipids, there may also be more damage being caused by these formulations [71]. Further studies investigating the acute and chronic inflammatory responses to both these cationic and other LNP formulations to understand the immune response which could affect the viability of their clinical translation.
The unique specificity shifts of LNPs formulated with different lipidoids suggests that lipidoids have differentially interact with charged helper lipids. This could be due to the natural charge of each lipidoid and/or their unique chemistries causing different ionic interactions with helper lipid functional groups. The removal of cholesterol could also have caused specificity shifts as cholesterol facilitates liver uptake through cholesterol receptors. A larger screen of lipidoids is still necessary to observe the strength of this effect.
Charge seemed to be the most predictive indicator of fate in vivo for the LNP formulations investigated in this paper. The charge of LNPs was investigated using both zeta potential measurements and the TNS fluorescence assay for ionization. Zeta potential surface charge relies on applying an electric field to nanoparticle suspensions and measuring the trafficking of particles to one electrode or the other. This electric field to lipid nanoparticles causes other complications as there are several ionizable groups including the lipidoid and sometimes the helper lipid, which may cause drifts in charge or rearrangement of the LNP structure. This unreliability may be responsible for the lack of substantial shifts in zeta potential when formulating LNPs with cationic or anionic helper lipids.
The fluorescent molecule 6-(p-Toluidino)-2-naphthalenesulfonic acid (TNS) is a negatively charged molecule that increases in fluorescence when it interacts with positively charged lipids [72]. As this technique does not apply the external force of an electric field like zeta potential, it may alter LNPs less and give a better representation of their natural state. Further investigation with a larger sample of LNP formulations is necessary to understand if this correlation holds and would be useful for screening LNPs.
Flow cytometric analysis in both lungs and spleen indicated there was inflammatory activation of monocytes and macrophages [73]. It is unclear if this phenomenon, observed most in PS formulations, is due to recruitment of CD11b+ monocytes, neutrophils, or eosinophils into the lungs and spleen, or if it is due to activation of tissue-resident macrophage populations [74]. As discussed previously, phosphatidylserine is recognized by and activates immune cell populations so these formulations could be leading to recruitment via activation of blood- and tissue-derived immune populations. As there was a similar increase in lung and spleen immune cell populations with PS LNP administration, it is unclear why there was not a similar functional signal in the lungs as assessed by IVIS imaging.
Flow cytometry on liver samples revealed that LNP treatment led to a significant reduction in F4/80+ Kupffer/macrophages. Lipidoid nanoparticles are chiefly made of lipids and cholesterol, both of which are taken up and metabolized by the liver [75]. Analysis of high fat- or cholesterol diets induce an anti-inflammatory response in liver macrophages, as opposed to in bone-marrow derived macrophages [34]. This anti-inflammatory activation appears to be mediated by liver X receptors (LXRs) which regulate lipid homeostasis in the liver [76]. Further investigation is necessary to confirm this relationship; however, it makes sense that this pathway is involved in the liver response to nanoparticles chiefly composed of lipids.
Interestingly, DOTAP did not elicit many of the inflammatory changes that DOPE and PS LNPs did. One potential reason is that DOTAP, being an artificial lipid, does not have any natural receptor recognition [77]. Liposomes containing DOTAP and other cationic lipids have been shown to elicit pro-inflammatory responses, but these liposomes are much larger than lipid nanoparticles (1 μm liposomes vs 100 nm lipid nanoparticles) [78]. The lack of detection of a cellular response to DOTAP LNP formulations suggest that the shifts in organ functional efficacy may be due only to physical interactions such as alterations in protein corona or interaction with the negatively charged lung glycocalyx. Further analysis of the immune response is necessary as the cellular analysis was limited and cytokine expression was not measured.
Given the similarity of our approach to SORT, we performed a direct comparison between the two methods. Our approach replaces a standard helper lipid such as DOPE completely and reduces the amount of cholesterol to increase the molarity of the helper lipid. The SORT method, in contrast, adds a charged helper lipid in addition to DOPE. Both methods produced nanoparticles that similarly shifted organ specificity to the spleen or lungs. While our method produced higher efficacy compared to SORT in this comparison, direct comparisons of the two methods are difficult to perform. Each method will have unique optimizations based on the helper lipid(s) and lipidoids used. Furthermore, the inclusion of a standard helper lipid in the SORT method will affect the overall charge of the lipid nanoparticle. For instance, inclusion of phosphatidic acid (PA) in our method produced poor results but was one of the more successful anionic lipids tested in the SORT method. Regardless of the method used to achieve organ specificity shifts, formulation optimization will be necessary to achieve application-specific tropism and efficacy.
Conclusion
This paper demonstrates that the complete replacement of a conventional helper lipid with an alternative anionic or cationic lipid causes a pronounced and consistent shift of lipid nanoparticle specificity in vivo to the spleen or lungs, respectively. These formulations contained only four lipid components, reducing their complexity compared to alternative, five component formulations with similar organ-shifting properties. The specificity shift effect was consistent across phospholipid classes and lipidoids, the mechanism of which may be tied to acute immune cell trafficking patterns that occur without overt signs of tissue damage. In summary, the four-component lipid nanoparticle formulations reported here for extrahepatocellular mRNA delivery enhance the formulation toolbox for distinct, organ- and disease-specific applications with specific product requirements.
Materials & Methods
Materials
CleanCap® Firefly Luciferase mRNA (L-7602) was purchased from TriLink Biotechnologies. 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) (850725P), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), L-α-phosphatidylserine (Brain, Porcine) (sodium salt) (840032P), 1,2-dioleoyl-sn-glycero-3-phosphate (sodium salt) (PA) (840875P), 1,2-dioleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (sodium salt) (PG) (840475P), Sphingomyelin (Brain, Porcine) (860062P), N-oleoyl-D-erythro-sphingosine (Ceramide) (860519P), 1,2-dioleoyl-3-trimethylammonium-propane (chloride salt) (DOTAP) (890890P) and 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt) (C14-PEG2000) (880150P) were obtained from Avanti Polar Lipids. Cholesterol (C8667), sodium citrate monobasic (71497) and Sodium 2-(p-toluidino)-6-naphthalenesulfonic acid (T9792) were purchased from Sigma Aldrich. Quant-iT™ RiboGreen™ RNA Assay Kit (R11490) was sourced from ThermoFisher Scientific.
Lipid Nanoparticle Formulation
Lipidoids, helper lipids, cholesterol, and C14-PEG2000 were dissolved in reagent grade ethanol at 1–10 mg/mL. Firefly luciferase mRNA was dissolved in 10 mM sodium citrate monobasic. Lipid solutions were mixed at a 35:16:46.5:2.5 or 35:40:22.5:2.5 lipidoid to helper lipid to cholesterol to PEG molar ratio. Citrate buffer was added to the lipid solutions at 1: 10 volume ratio. The resultant lipid solution was then added to an equal volume of RNA solution at a 10:1 lipidoid:mRNA mass ratio, followed by thorough mixing by vortex. Finally, an equal volume of phosphate-buffered saline (PBS) was added to the ethanol-citrate mixture, and this was mixed thoroughly with vortexing. Lipid nanoparticles for in vitro and in vivo studies were formulated at final mRNA concentrations of 5 μg/mL and 90 μg/mL, respectively. Lipid nanoparticles for in vivo studies were dialyzed against 2 L of PBS in 3 kDa molecular weight cut off dialysis cassettes for 1 hour prior to use.
Lipid Nanoparticle Characterization
Lipid nanoparticles were characterized for size and surface zeta potential using a Malvern ZetaSizer Nano (Malvern Instruments). Prior to analysis by ZetaSizer, the LNPs were diluted ten-fold in PBS to a concentration of 0.5 μg/mL mRNA. Three technical replicates were conducted on each sample for both size and surface zeta potential. To measure RNA entrapment, intact and lysed nanoparticles were measured for RNA content using a Quant-iT™ RiboGreen™ RNA Assay Kit according to manufacturer instructions. Briefly, LNPs were diluted in equal volumes of Tris-EDTA buffer or 2% Triton X-100 in Tris-EDTA buffer. Then, an equal volume of RiboGreen™ reagent was added to each sample and incubated at 37 °C for 15 minutes. The fluorescence (ex/em 480/520 nm) was read on a Tecan Spark® Multimode Microplate Reader. To measure Sodium 2-(p-toluidino)-6-naphthalenesulfonic acid (TNS) ionization a surrogate measure of LNP surface charge, 5 μL nanoparticles were diluted in 250 μL TNS assay buffer (20 mM sodium phosphate tribasic, 25 mM ammonium citrate dibasic, 20 mM ammonium acetate, 150 mM sodium chloride) at pH 7.4 or pH 5103. Then, 5 μL of a 0.16 mM stock solution of 2-(p-toluidinyl)naphthalene-6-sulphonic acid (TNS, Sigma Aldrich) in DI water was added to each well. The TNS fluorescence was read on a Tecan Spark® (ex/em 320/430 nm). Metrics of LNP physical properties were plotted against the specificity (percentage of total in vivo efficacy) in liver, lung or spleen (Figure 3). Pearson correlation constants were calculated for correlation between physical LNP properties and LNP efficacy and specificity in the liver, spleen and lungs in vivo
Cell Culture
HeLa, HepG2, NIH3T3, RAW264.7, A549, Jurkat, Raji B, and HULEC-5a cells were obtained from ATCC. HeLa, HepG2, NIH3T3, and RAW264.7 cells were cultured in DMEM, A549 were cultured in F12 and, Jurkat and Raji B cells were cultured in RPMI as the basal media. All media were supplemented with FBS (10% v/v) and Penicillin/Streptomycin (1% v/v). HULEC-5a cells were cultured in MCDB131 supplemented with FBS (10% v/v), Penicillin/Streptomycin (1% v/v), EGF (10 ng/mL), hydrocortisone (1 μg/mL) and glutamine (10 mM). All cells were maintained at 37 °C and 5% CO2 in a humidified incubator. For in vitro transfection assays, RAW, HepG2, A549, Jurkat and Raji B were seeded at 100,000 cells per well, NIH-3T3 at 50,000 cells/well, HULEC-5a at 25,000 cells per well, and HeLa cells at 15,000 cells per well. Cells were allowed to adhere or settle for 24 hours prior to LNP administration. Twenty microliters of LNPs per well at an original concentration of 5 μg/mL Firefly luciferase mRNA (as described above) were added into cell culture media then allowed to incubate for 24 hours. Transfection efficiency was measured using Bright-Glo™ Luciferase Assay System (Promega). Briefly, BrightGlo reagent was diluted 1:4 in PBS then 50 μL was added to each well. After a 7 minute incubation in the dark, luminescence intensity was read on a Tecan Spark plate reader.
In Vivo Intravenous LNP Administration
Female C57BL/6NCrl (Charles River) mice of at least 6 weeks of age were used for all in vivo experiments. Animal experiments were approved by the Carnegie Mellon University IACUC prior to work and conformed to all federal, state, and local regulations. Mice received tail vein injections of Firefly luciferase mRNA-containing LNPs at an mRNA dose of 0.75 mg/kg. Three hours later, mice were injected intraperitoneally with 130 μL of 30 mg/mL D-luciferin. Fifteen minutes following luciferin administration, mice were euthanized via CO2 asphyxiation and secondary cervical dislocation. Organs were removed, excess blood was blotted off, and organs were placed on black construction paper. Luminescent signal was measured using an In Vivo Imaging System (Perkin Elmer), and luminescent images were juxtaposed with brightfield images. Total luminescent flux (p/s) was calculated for each organ using Living Image® software. Organs used for histology were immediately placed in 10% neutral buffered formalin and stored at 4 °C for 4 days before washing in PBS and storing in 70% ethanol at 4 °C. Organs used for flow cytometric analysis were placed into cold DMEM during transport to the cell culture room for processing.
Histologic Analysis
Following fixation, organs were dissected and placed in optimal cutting temperature (OCT) in histology molds and frozen at −80 °C. Frozen tissue sections were sliced at 10 μm and mounted on glass slides and stored at −80 °C until used for staining. Slides were stained for hematoxylin & eosin to evaluate gross morphological changes. Slides were thawed and then rehydrated in PBS. Sections were then stained in Harris hematoxylin for 8 minutes, washed twice in deionized water, differentiated in 5% glacial acetic acid for 1 minute, then washed again twice in deionized water. Hematoxylin was blued using Scott’s tap water for 15 seconds then washed again twice in deionized water. Sections were stained with Alcoholic Eosin Y for 2 minutes. Following eosin staining, slides were rapidly dehydrated in 95% and 100% ethanol then cleared with xylenes. Slides were mounted using a toluene-based mounting media (Shandon ClearVue) and allowed to dry overnight. Sections were imaged on an Invitrogen EVOS™ FL Auto 2 Imaging System using a 10X objective.
Flow Cytometric Analysis
Livers, lungs, and spleens from three mice injected with 40 mol% DOPE, PS or DOTAP LNPs were analyzed for cell populations and transfection using flow cytometry. Organs were digested using Miltenyi Tissue Dissociation Kits for liver (130–105-807), lung (130–095-927), and spleen (130–095-926) according to manufacturer instructions.
For liver dissociation, 200 μL Enzyme D, 100 μL Enzyme R, and 20 μL Enzyme A were added to 4.7 mL DMEM and warmed to 37 °C for 30 minutes. Livers were rinsed with DMEM and transferred to a Miltenyi C Tube containing the liver enzyme mix. Livers were dissociated using the gentleMACS dissociator (Miltenyi) using a custom liver program set to 20 RPM for 30 min at 37 °C. Liver samples were resuspended with 5 mL flow buffer (PBS/10% FBS) and then centrifuged at 300 RCF for 5 minutes. Pellets were resuspended in 10 mL flow buffer and debris was removed using a 70 μm cell strainer. Following centrifugation, red blood cells were lysed by resuspending pellets in 3 mL RBC lysis buffer (Biolegend) for 3 min. Lysis was stopped by adding 7 mL flow buffer. Cells were then centrifuged again at 300 RCF for 5 minutes. The cell pellet was resuspended in flow buffer and then stained and analyzed as described below.
For lung dissociation, 100 μL Enzyme D and 15 μL Enzyme A were added to 2.4 mL Buffer S (Miltenyi). After the lungs were washed in DMEM, individual lobes were dissected and added to a gentleMACS™ C tube containing the enzyme mix. Tubes were placed on the gentleMACS™ Dissociator, and the program 37C_m_LDK_1 was run. Following dissociation, lung samples were resuspended with 7.5 mL flow buffer and centrifuged at 300 RCF for 10 minutes. Pellets were resuspended in 10 mL flow buffer, and debris was removed using a 70 μm cell strainer. Following centrifugation, red blood cells were lysed by resuspending pellets in 3 mL RBC lysis buffer for 3 min. Lysis was stopped by adding 7 mL flow buffer, then cells were centrifuged again. Cells were resuspended in flow buffer, and then staining and analysis was performed as described below.
For spleen dissociation, spleens were washed in DMEM and manually dissociated by forcing spleens through a 70 μm cell strainer. The strainer was then washed with DMEM. Cells were centrifuged at 300 RCF for 5 minutes then red blood cells were lysed by adding 3 mL RBC lysis buffer to pellets for 3 minutes. Lysis was stopped by adding 7 mL flow buffer, and then cells were centrifuged again. Cells were resuspended in flow buffer, and then staining and analysis was performed as described below.
Cells used for antibody staining were fixed with Flow Cytometry Fixation Buffer (R&D Systems) for 10 minutes at 4°C. Fixation was quenched by adding flow buffer at a volumetric ratio of 9:1 in relation to fixation buffer. Cells were then centrifuged and resuspended in flow buffer to wash. Following centrifugation, cells were permeabilized, and Fc receptors were blocked by resuspending cells in Flow Cytometry Permeabilization/Wash Buffer I (R&D Systems) containing 1:100 anti-CD16/32 antibodies for 10 minutes at 4 °C. Following permeabilization and Fc blocking, cells were stained with antibodies diluted 1:100 in Permabilization/Wash Buffer for 45 minutes at 4 °C. Liver cells were stained with BV421 anti-ASGR1, BV605 anti-EpCAM, BV785 anti-F4/80, PE anti-Firefly Luciferase, AF647 anti-GFAP, and PerCP/Cyanine5.5 anti-CD31. Lung cells were stained with BV421 anti-EpCAM, BV570 anti-CD11b, BV650 anti-F4/80, BV785 anti-CD31, PE anti-Firefly Luciferase, PerCP anti-CD45, and APC anti-CD3. Spleen cells were stained with BV421 anti-CD19, BV570 anti-CD11b, BV650 anti-F4/80, PE anti-Firefly Luciferase, PerCP anti-CD45, and APC anti-CD3 (all purchased from Biolegend). Following antibody staining, cells were centrifuged and washed with permeabilization/wash buffer once, centrifuged again, resuspended in flow buffer, and run on the flow cytometer. Unstained and single stain controls were performed for all antibodies and a fluorescence minus one (FMO) control was run for Firefly Luciferase to accurately quantify transfection. Flow cytometry data was compensated and analyzed using NovoExpress® software.
Our liver gating analysis identified Kupffer cells (liver macrophages) as F4/80+/GFAP− and stellate cells (liver fibroblasts) as GFAP+/F4/80− (Supplementary Figure 8A). From the F4/80−/GFAP− population, hepatocytes were identified as ASGR1+/CD31− and endothelial cells were identified as CD31+/ASGR1− (Supplementary Figure 8A). Our spleen gating analysis identified T cells as CD3+/CD19− and B cells as CD19+CD3− (Supplementary Figure 8B). From the CD3−/CD19− population, populations were analyzed for being F4/80+ and/or CD11b+ with F4/80+/CD11b+ identified as macrophages (Supplementary Figure B). For our lung gating analysis, we identified T cells as CD3+/CD45+ (Supplementary Figure C). From CD45+/CD3− population, immune cells were further characterized as F4/80+ and/or CD11b+ with F4/80+/CD11b+ cells characterized as macrophages. Endothelial cells were identified as CD31+/CD45− and epithelial cells were identified as EpCAM+/CD45− (Supplementary Figure 8C).
Statistical Analysis
Data were analyzed for statistical significance using one-way ANOVA (comparison between helper lipids) or two-way ANOVA (comparison between lipidoids and helper lipids) with Tukey post-hoc analysis using GraphPad Prism software. Correlation coefficients were calculated as Pearson R2 values assuming a linear regression with an estimated intercept term.
Supplementary Material
Acknowledgements
We thank our funding sources that made this work possible, including the Defense Advanced Research Projects Agency (DARPA) grant number HR0011–19-2–0007, National Institutes of Health grant number DP2-HD098860, and the Wadhwani Foundation. M.L.A. was supported by an NSF Graduate Research Fellowship Program award (number DGE1745016).
References
- [1].Kowalski PS, Rudra A, Miao L, Anderson DG, Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery, Mol Ther, 27 (2019) 710–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Basha G, Novobrantseva T, Rosin N, Tam Y, Hafez I, Wong M, Sugo T, Ruda V, Qin J, Klebanov B, Ciufolini M, Akinc A, Tam Y, Hope M, Cullis P, Influence of Cationic Lipid Composition on Gene Silencing Properties of Lipid Nanoparticle Formulations of siRNA in Antigen-Presenting Cells, Molecular therapy : the journal of the American Society of Gene Therapy, 19 (2011) 2186–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Patel S, Ashwanikumar N, Robinson E, DuRoss A, Sun C, Murphy-Benenato KE, Mihai C, Almarsson Ö, Sahay G, Boosting Intracellular Delivery of Lipid Nanoparticle-Encapsulated mRNA, Nano letters, 17 (2017) 5711–5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Blanco E, Shen H, Ferrari M, Principles of nanoparticle design for overcoming biological barriers to drug delivery, Nat Biotechnol, 33 (2015) 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Miller JB, Kos P, Tieu V, Zhou K, Siegwart DJ, Development of Cationic Quaternary Ammonium Sulfonamide Amino Lipids for Nucleic Acid Delivery, ACS Appl Mater Interfaces, 10 (2018) 2302–2311. [DOI] [PubMed] [Google Scholar]
- [6].Kauffman KJ, Dorkin JR, Yang JH, Heartlein MW, DeRosa F, Mir FF, Fenton OS, Anderson DG, Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs, Nano Lett, 15 (2015) 7300–7306. [DOI] [PubMed] [Google Scholar]
- [7].Sago CD, Lokugamage MP, Islam FZ, Krupczak BR, Sato M, Dahlman JE, Nanoparticles That Deliver RNA to Bone Marrow Identified by in Vivo Directed Evolution, J Am Chem Soc, 140 (2018) 17095–17105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Li M, Du C, Guo N, Teng Y, Meng X, Sun H, Li S, Yu P, Galons H, Composition design and medical application of liposomes, Eur J Med Chem, 164 (2019) 640–653. [DOI] [PubMed] [Google Scholar]
- [9].Kobayashi K, Wei J, Iida R, Ijiro K, Niikura K, Surface engineering of nanoparticles for therapeutic applications, Polymer Journal, 46 (2014) 460–468. [Google Scholar]
- [10].Yang X, Koh CG, Liu S, Pan X, Santhanam R, Yu B, Peng Y, Pang J, Golan S, Talmon Y, Jin Y, Muthusamy N, Byrd JC, Chan KK, Lee LJ, Marcucci G, Lee RJ, Transferrin receptor-targeted lipid nanoparticles for delivery of an antisense oligodeoxyribonucleotide against Bcl-2, Mol Pharm, 6 (2009) 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Akinc A, Querbes W, De S, Qin J, Frank-Kamenetsky M, Jayaprakash KN, Jayaraman M, Rajeev KG, Cantley WL, Dorkin JR, Butler JS, Qin L, Racie T, Sprague A, Fava E, Zeigerer A, Hope MJ, Zerial M, Sah DW, Fitzgerald K, Tracy MA, Manoharan M, Koteliansky V, Fougerolles A, Maier MA, Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms, Mol Ther, 18 (2010) 1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ritz S, Schöttler S, Kotman N, Baier G, Musyanovych A, Kuharev J, Landfester K, Schild H, Jahn O, Tenzer S, Mailänder V, Protein Corona of Nanoparticles: Distinct Proteins Regulate the Cellular Uptake, Biomacromolecules, 16 (2015) 1311–1321. [DOI] [PubMed] [Google Scholar]
- [13].Chai Z, Ran D, Lu L, Zhan C, Ruan H, Hu X, Xie C, Jiang K, Li J, Zhou J, Wang J, Zhang Y, Fang RH, Zhang L, Lu W, Ligand-Modified Cell Membrane Enables the Targeted Delivery of Drug Nanocrystals to Glioma, ACS Nano, 13 (2019) 5591–5601. [DOI] [PubMed] [Google Scholar]
- [14].Yoo J, Park C, Yi G, Lee D, Koo H, Active Targeting Strategies Using Biological Ligands for Nanoparticle Drug Delivery Systems, Cancers (Basel), 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fenton OS, Kauffman KJ, McClellan RL, Kaczmarek JC, Zeng MD, Andresen JL, Rhym LH, Heartlein MW, DeRosa F, Anderson DG, Customizable Lipid Nanoparticle Materials for the Delivery of siRNAs and mRNAs, Angew Chem Int Ed Engl, 57 (2018) 13582–13586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hajj KA, Whitehead KA, Tools for translation: non-viral materials for therapeutic mRNA delivery, Nature Reviews Materials, 2 (2017) 17056. [Google Scholar]
- [17].Kulkarni JA, Darjuan MM, Mercer JE, Chen S, van der Meel R, Thewalt JL, Tam YYC, Cullis PR, On the Formation and Morphology of Lipid Nanoparticles Containing Ionizable Cationic Lipids and siRNA, ACS Nano, 12 (2018) 4787–4795. [DOI] [PubMed] [Google Scholar]
- [18].Leung AK, Hafez IM, Baoukina S, Belliveau NM, Zhigaltsev IV, Afshinmanesh E, Tieleman DP, Hansen CL, Hope MJ, Cullis PR, Lipid Nanoparticles Containing siRNA Synthesized by Microfluidic Mixing Exhibit an Electron-Dense Nanostructured Core, J Phys Chem C Nanomater Interfaces, 116 (2012) 18440–18450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lokugamage MP, Gan Z, Zurla C, Levin J, Islam FZ, Kalathoor S, Sato M, Sago CD, Santangelo PJ, Dahlman JE, Mild Innate Immune Activation Overrides Efficient Nanoparticle-Mediated RNA Delivery, Adv Mater, 32 (2020) e1904905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Miller JB, Zhang S, Kos P, Xiong H, Zhou K, Perelman SS, Zhu H, Siegwart DJ, Non-Viral CRISPR/Cas Gene Editing In Vitro and In Vivo Enabled by Synthetic Nanoparticle Co-Delivery of Cas9 mRNA and sgRNA, Angew Chem Int Ed Engl, 56 (2017) 1059–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Du Z, Munye MM, Tagalakis AD, Manunta MD, Hart SL, The role of the helper lipid on the DNA transfection efficiency of lipopolyplex formulations, Sci Rep, 4 (2014) 7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cavalcanti RRM, Lira RB, Riske KA, Study of the Fusion Mechanism of Fusogenic Cationic Liposomes with Anionic Model Membranes, Biophysical Journal, 114 (2018) 606a. [Google Scholar]
- [23].Kolašinac R, Kleusch C, Braun T, Merkel R, Csiszár A, Deciphering the Functional Composition of Fusogenic Liposomes, Int J Mol Sci, 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ball RL, Hajj KA, Vizelman J, Bajaj P, Whitehead KA, Lipid Nanoparticle Formulations for Enhanced Co-delivery of siRNA and mRNA, Nano Lett, 18 (2018) 3814–3822. [DOI] [PubMed] [Google Scholar]
- [25].Ke X, Howard GP, Tang H, Cheng B, Saung MT, Santos JL, Mao HQ, Physical and chemical profiles of nanoparticles for lymphatic targeting, Adv Drug Deliv Rev, 151-152 (2019) 72–93. [DOI] [PubMed] [Google Scholar]
- [26].Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, Grunwitz C, Vormehr M, Hüsemann Y, Selmi A, Kuhn AN, Buck J, Derhovanessian E, Rae R, Attig S, Diekmann J, Jabulowsky RA, Heesch S, Hassel J, Langguth P, Grabbe S, Huber C, Türeci Ö, Sahin U, Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy, Nature, 534 (2016) 396–401. [DOI] [PubMed] [Google Scholar]
- [27].Chen D, Parayath N, Ganesh S, Wang W, Amiji M, The role of apolipoprotein- and vitronectin-enriched protein corona on lipid nanoparticles for in vivo targeted delivery and transfection of oligonucleotides in murine tumor models, Nanoscale, 11 (2019) 18806–18824. [DOI] [PubMed] [Google Scholar]
- [28].Cheng Q, Wei T, Farbiak L, Johnson LT, Dilliard SA, Siegwart DJ, Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing, Nat Nanotechnol, 15 (2020) 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lee SH, Meng XW, Flatten KS, Loegering DA, Kaufmann SH, Phosphatidylserine exposure during apoptosis reflects bidirectional trafficking between plasma membrane and cytoplasm, Cell Death Differ, 20 (2013) 64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Teissier E, Pécheur EI, Lipids as modulators of membrane fusion mediated by viral fusion proteins, Eur Biophys J, 36 (2007) 887–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bernardi S, Marcuzzi A, Piscianz E, Tommasini A, Fabris B, The Complex Interplay between Lipids, Immune System and Interleukins in Cardio-Metabolic Diseases, Int J Mol Sci, 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brügger B, Simons M, Ceramide triggers budding of exosome vesicles into multivesicular endosomes, Science, 319 (2008) 1244–1247. [DOI] [PubMed] [Google Scholar]
- [33].Elsherbini A, Bieberich E, Ceramide and Exosomes: A Novel Target in Cancer Biology and Therapy, Adv Cancer Res, 140 (2018) 121–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Fröhlich E, The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles, Int J Nanomedicine, 7 (2012) 5577–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lv H, Zhang S, Wang B, Cui S, Yan J, Toxicity of cationic lipids and cationic polymers in gene delivery, J Control Release, 114 (2006) 100–109. [DOI] [PubMed] [Google Scholar]
- [36].Whitehead KA, Dorkin JR, Vegas AJ, Chang PH, Veiseh O, Matthews J, Fenton OS, Zhang Y, Olejnik KT, Yesilyurt V, Chen D, Barros S, Klebanov B, Novobrantseva T, Langer R, Anderson DG, Degradable lipid nanoparticles with predictable in vivo siRNA delivery activity, Nat Commun, 5 (2014) 4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Knapp CM, Guo P, Whitehead KA, Lipidoid Tail Structure Strongly Influences siRNA Delivery Activity, Cellular and Molecular Bioengineering, 9 (2016) 305–314. [Google Scholar]
- [38].Hajj KA, Ball RL, Deluty SB, Singh SR, Strelkova D, Knapp CM, Whitehead KA, Branched-Tail Lipid Nanoparticles Potently Deliver mRNA In Vivo due to Enhanced Ionization at Endosomal pH, Small, 15 (2019) 1805097. [DOI] [PubMed] [Google Scholar]
- [39].Kulkarni JA, Witzigmann D, Leung J, Tam YYC, Cullis PR, On the role of helper lipids in lipid nanoparticle formulations of siRNA, Nanoscale, 11 (2019) 21733–21739. [DOI] [PubMed] [Google Scholar]
- [40].Cheng X, Lee RJ, The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery, Adv Drug Deliv Rev, 99 (2016) 129–137. [DOI] [PubMed] [Google Scholar]
- [41].Shobaki N, Sato Y, Harashima H, Mixing lipids to manipulate the ionization status of lipid nanoparticles for specific tissue targeting, Int J Nanomedicine, 13 (2018) 8395–8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Baratta JL, Ngo A, Lopez B, Kasabwalla N, Longmuir KJ, Robertson RT, Cellular organization of normal mouse liver: a histological, quantitative immunocytochemical, and fine structural analysis, Histochem Cell Biol, 131 (2009) 713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Song G, Petschauer JS, Madden AJ, Zamboni WC, Nanoparticles and the mononuclear phagocyte system: pharmacokinetics and applications for inflammatory diseases, Curr Rheumatol Rev, 10 (2014) 22–34. [DOI] [PubMed] [Google Scholar]
- [44].Turnbull IC, Eltoukhy AA, Fish KM, Nonnenmacher M, Ishikawa K, Chen J, Hajjar RJ, Anderson DG, Costa KD, Myocardial Delivery of Lipidoid Nanoparticle Carrying modRNA Induces Rapid and Transient Expression, Molecular Therapy, 24 (2016) 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ball RL, Bajaj P, Whitehead KA, Oral delivery of siRNA lipid nanoparticles: Fate in the GI tract, Sci Rep, 8 (2018) 2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hajj KA, Melamed JR, Chaudhary N, Lamson NG, Ball RL, Yerneni SS, Whitehead KA, A Potent Branched-Tail Lipid Nanoparticle Enables Multiplexed mRNA Delivery and Gene Editing In Vivo, Nano Letters, 20 (2020) 5167–5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ma Z, Mi Z, Wilson A, Alber S, Robbins PD, Watkins S, Pitt B, Li S, Redirecting adenovirus to pulmonary endothelium by cationic liposomes, Gene Ther, 9 (2002) 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bragonzi A, Boletta A, Biffi A, Muggia A, Sersale G, Cheng SH, Bordignon C, Assael BM, Conese M, Comparison between cationic polymers and lipids in mediating systemic gene delivery to the lungs, Gene Ther, 6 (1999) 1995–2004. [DOI] [PubMed] [Google Scholar]
- [49].Tsui FC, Ojcius DM, Hubbell WL, The intrinsic pKa values for phosphatidylserine and phosphatidylethanolamine in phosphatidylcholine host bilayers, Biophys J, 49 (1986) 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Koynova R, MacDonald RC, Cationic O-ethylphosphatidylcholines and their lipoplexes: phase behavior aspects, structural organization and morphology, Biochimica et Biophysica Acta (BBA) - Biomembranes, 1613 (2003) 39–48. [DOI] [PubMed] [Google Scholar]
- [51].Simons K, Ehehalt R, Cholesterol, lipid rafts, and disease, The Journal of clinical investigation, 110 (2002) 597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yang S-T, Kreutzberger AJB, Lee J, Kiessling V, Tamm LK, The role of cholesterol in membrane fusion, Chemistry and physics of lipids, 199 (2016) 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Goldstein JL, Brown MS, The LDL Receptor, Arteriosclerosis, Thrombosis, and Vascular Biology, 29 (2009) 431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Silver DL, Wang N, Xiao X, Tall AR, High density lipoprotein (HDL) particle uptake mediated by scavenger receptor class B type 1 results in selective sorting of HDL cholesterol from protein and polarized cholesterol secretion, J Biol Chem, 276 (2001) 25287–25293. [DOI] [PubMed] [Google Scholar]
- [55].Paunovska K, Da Silva Sanchez AJ, Sago CD, Gan Z, Lokugamage MP, Islam FZ, Kalathoor S, Krupczak BR, Dahlman JE, Nanoparticles Containing Oxidized Cholesterol Deliver mRNA to the Liver Microenvironment at Clinically Relevant Doses, Adv Mater, 31 (2019) e1807748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Patel S, Ashwanikumar N, Robinson E, Xia Y, Mihai C, Griffith JP 3rd, Hou S, Esposito AA, Ketova T, Welsher K, Joyal JL, Almarsson Ö, Sahay G, Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA, Nat Commun, 11 (2020) 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ciani L, Ristori S, Salvati A, Calamai L, Martini G, DOTAP/DOPE and DC-Chol/DOPE lipoplexes for gene delivery: zeta potential measurements and electron spin resonance spectra, Biochimica et Biophysica Acta (BBA) - Biomembranes, 1664 (2004) 70–79. [DOI] [PubMed] [Google Scholar]
- [58].Hattori Y, Suzuki S, Kawakami S, Yamashita F, Hashida M, The role of dioleoylphosphatidylethanolamine (DOPE) in targeted gene delivery with mannosylated cationic liposomes via intravenous route, J Control Release, 108 (2005) 484–495. [DOI] [PubMed] [Google Scholar]
- [59].Cervia LD, Chang CC, Wang L, Yuan F, Distinct effects of endosomal escape and inhibition of endosomal trafficking on gene delivery via electrotransfection, PLoS One, 12 (2017) e0171699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lagace TA, Phosphatidylcholine: Greasing the Cholesterol Transport Machinery, Lipid Insights, 8 (2015) 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Thewalt JL, Bloom M, Phosphatidylcholine: cholesterol phase diagrams, Biophys J, 63 (1992) 1176–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Skotland T, Sandvig K, Llorente A, Lipids in exosomes: Current knowledge and the way forward, Prog Lipid Res, 66 (2017) 30–41. [DOI] [PubMed] [Google Scholar]
- [63].Segawa K, Nagata S, An Apoptotic ‘Eat Me’ Signal: Phosphatidylserine Exposure, Trends Cell Biol, 25 (2015) 639–650. [DOI] [PubMed] [Google Scholar]
- [64].Rodriguez-Manzanet R, DeKruyff R, Kuchroo VK, Umetsu DT, The costimulatory role of TIM molecules, Immunological reviews, 229 (2009) 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Gulshan K, Brubaker G, Wang S, Hazen SL, Smith JD, Sphingomyelin depletion impairs anionic phospholipid inward translocation and induces cholesterol efflux, The Journal of biological chemistry, 288 (2013) 37166–37179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hirai H, Natori S, Sekimizu K, Reversal by phosphatidylglycerol and cardiolipin of inhibition of transcription and replication by histones in vitro, Arch Biochem Biophys, 298 (1992) 458–463. [DOI] [PubMed] [Google Scholar]
- [67].Choudhary V, Uaratanawong R, Patel RR, Patel H, Bao W, Hartney B, Cohen E, Chen X, Zhong Q, Isales CM, Bollag WB, Phosphatidylglycerol Inhibits Toll-Like Receptor-Mediated Inflammation by Danger-Associated Molecular Patterns, J Invest Dermatol, 139 (2019) 868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Uyechi LS, Gagné L, Thurston G, Szoka FC, Mechanism of lipoplex gene delivery in mouse lung: binding and internalization of fluorescent lipid and DNA components, Gene Therapy, 8 (2001) 828–836. [DOI] [PubMed] [Google Scholar]
- [69].Rolland A, Pharmaceutical Gene Delivery Systems, CRC Press, 2003. [Google Scholar]
- [70].Atukorale PU, Yang YS, Bekdemir A, Carney RP, Silva PJ, Watson N, Stellacci F, Irvine DJ, Influence of the glycocalyx and plasma membrane composition on amphiphilic gold nanoparticle association with erythrocytes, Nanoscale, 7 (2015) 11420–11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Scheule RK, George JAS, Bagley RG, Marshall J, Kaplan JM, Akita GY, Wang KX, Lee ER, Harris DJ, Jiang C, Yew NS, Smith AE, Cheng SH, Basis of Pulmonary Toxicity Associated with Cationic Lipid-Mediated Gene Transfer to the Mammalian Lung, Human Gene Therapy, 8 (1997) 689–707. [DOI] [PubMed] [Google Scholar]
- [72].Zhang J, Fan H, Levorse DA, Crocker LS, Ionization behavior of amino lipids for siRNA delivery: determination of ionization constants, SAR, and the impact of lipid pKa on cationic lipid-biomembrane interactions, Langmuir, 27 (2011) 1907–1914. [DOI] [PubMed] [Google Scholar]
- [73].Duan M, Steinfort DP, Smallwood D, Hew M, Chen W, Ernst M, Irving LB, Anderson GP, Hibbs ML, CD11b immunophenotyping identifies inflammatory profiles in the mouse and human lungs, Mucosal Immunol, 9 (2016) 550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GRS, Perlman H, Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung, American journal of respiratory cell and molecular biology, 49 (2013) 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Nguyen P, Leray V, Diez M, Serisier S, Le Bloc’h J, Siliart B, Dumon H, Liver lipid metabolism, J Anim Physiol Anim Nutr (Berl), 92 (2008) 272–283. [DOI] [PubMed] [Google Scholar]
- [76].Endo-Umeda K, Nakashima H, Komine-Aizawa S, Umeda N, Seki S, Makishima M, Liver X receptors regulate hepatic F4/80+CD11b+ Kupffer cells/macrophages and innate immune responses in mice, Scientific Reports, 8 (2018) 9281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].de Groot AM, Thanki K, Gangloff M, Falkenberg E, Zeng X, van Bijnen DCJ, van Eden W, Franzyk H, Nielsen HM, Broere F, Gay NJ, Foged C, Sijts A, Immunogenicity Testing of Lipidoids In Vitro and In Silico: Modulating Lipidoid-Mediated TLR4 Activation by Nanoparticle Design, Mol Ther Nucleic Acids, 11 (2018) 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Filion MC, Phillips NC, Toxicity and immunomodulatory activity of liposomal vectors formulated with cationic lipids toward immune effector cells, Biochim Biophys Acta, 1329 (1997) 345–356. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.