

Abstract
The lung epithelium forms the first barrier against respiratory pathogens and noxious chemicals; however, little is known about how more than 90% of this barrier, made of AT1 (alveolar type 1) cells, responds to injury. Using the Sendai virus to model natural infection in mice, we find evidence that AT1 cells have an intermediary role by persisting in areas depleted of AT2 cells, upregulating IFN responsive genes, and receding from invading airway cells. Sendai virus infection mobilizes airway cells to form alveolar SOX2+ (Sry-box 2+) clusters without differentiating into AT1 or AT2 cells. Large AT2 cell-depleted areas remain covered by AT1 cells, which we name “AT2-less regions”, and are replaced by SOX2+ clusters spreading both basally and luminally. AT2 cell proliferation and differentiation are largely confined to topologically distal regions and form de novo alveolar surface, with limited contribution to in situ repairs of AT2-less regions. Time-course single-cell RNA sequencing profiling and RNAscope validation suggest enhanced immune responses and altered growth signals in AT1 cells. Our comprehensive spatiotemporal and genomewide study highlights the hitherto unappreciated role of AT1 cells in lung injury–repair.
Keywords: lung viral injury, AT1 cells, single-cell RNA-seq
Individual cell types in a multicellular organ must act in concert to fulfill physiological functions and react likewise to injuries. This holistic reaction is expected to reach its full extent in response to naturally occurring pathogens, possibly because of evolutionary pressure, compared with engineered cell ablation using diphtheria toxin or adapted pathogens from noncognate species. Sendai virus (SeV), also known as murine parainfluenza virus type 1, is used for gene delivery and to model chronic lung diseases but also provides a natural injury–repair model (1–3). A comprehensive understanding of spatiotemporal and cell-type–specific responses to SeV, which is the focus of this study, will not only better establish a disease model for testing therapeutics but also shed light on natural infections in humans such as influenza and coronavirus disease (COVID-19).
As the first barrier against microbial and chemical agents, the otherwise quiescent lung epithelium has evolved robust repair mechanisms that, in the alveolar region, activate a facultative stem cell population, AT2 (alveolar type 2) cells, which have attracted the most attention in the field (4–6). In contrast, AT1 cells, constituting 95% of the alveolar surface, are often considered a passive structural component to be injured and then replaced by AT2 cells but are likely a key component of the lung’s holistic reaction to injury because of their sheer mass (7, 8). Our limited knowledge of AT1 cell biology in injury–repair stems partly from their expansive and ultrathin morphology that spatially uncouples the nucleus from cellular extensions such that cytokeratin, junctional, or nuclear staining alone is inadequate to study AT1 cells (9). Furthermore, the dense three-dimensional (3D) packing of alveolar tissues obscures the connection between topologically distal and proximal regions (important spatial attributes also missed in biochemical assays of whole organs). Conceptually, the alveolar epithelium can be unfurled into a two-dimensional surface (an imagery evoked when analogizing the human lung to a half tennis court) that is predominantly AT1 cell surface and sprinkled with AT2 cells (10). Accordingly, injury–repair could be 1) in situ healing similar to wound healing as a result of cell expansion, proliferation, and migration (a process also artificially invoked during reepithelialization of decellularized lungs (11); or 2) de novo growth around the border of the tennis court (a likely process during postpneumonectomy compensatory growth in which there is no direct injury to the existing surface) (7, 12). Regardless, it is integral to our understanding of lung injury–repair to delineate how the majority of the epithelial surface, or essentially the AT1 cell, responds and contributes.
In this study, we notice that upon SeV infection in mice, AT1 cells persist in large alveolar areas depleted of AT2 cells, providing an opportunity to ascertain the role of AT1 cells in injury–repair. Applying our knowledge of and tools for AT1 cells in the uninjured lung (9), we show that the surviving AT1 cells robustly upregulate IFN responsive genes, comparable to that of AT2 cells, and are replaced by dysplastic SOX2+ airway cells. In contrast, AT2 cells proliferate and differentiate into AT1 cells mostly in topologically distal regions and contribute minimally to in situ healing of damaged areas. Therefore AT1 cells have an intermediary role during SeV infection by temporarily covering the tissue surface and coordinating an immune response. The resulting transcriptomic profiling and cellular analysis of AT1 cells and their relationship with other epithelial and nonepithelial cells fill in a gap in our knowledge of lung injury–repair.
Some of the results of these studies have been previously reported in the form of a preprint (bioRxiv [04 August 2021] https://doi.org/10.1101/2021.08.04.455124).
Methods
Mouse Strains and SeV Infection
The following mouse strains were used: Sox2GFP (13), Sox2CreER (13), RosatdT (14), RosamTmG (15), RosaL10GFP (16), SftpcCreER (17), and Rtkn2CreER (18). Tamoxifen was administered intraperitoneally at least 3 weeks before infection at time points and doses specified in figure legends. Animal infections were performed as previously described (19, 20). Briefly, mice at least 8 weeks of age were anesthetized with isofluorane and suspended by the maxillary incisors on a board with a 60° incline. Mice were infected through oropharyngeal aspiration of a sublethal dose of 2.1 × 107 plaque-forming units of SeV (parainfluenza type 1) strain 52 (ATCC #VR-105, RRID:SCR_001672) in 40 μl PBS. Mice of both sexes and mixed genetic backgrounds were used. Investigators were not blind to the genotypes. All animal experiments were approved by the Institutional Animal Care and Use at Texas A&M Health Science Center Institute of Biosciences and Technology and MD Anderson Cancer Center.
Section and Wholemount Immunostaining
Lung harvest and immunostaining were performed as published (18, 21). Briefly, after anesthesia with Avertin (T48402, Sigma), lungs were perfused with PBS and gravity inflated with 0.5% paraformaldehyde (PFA; P6148, Sigma) in PBS at 25 cm H2O pressure, followed by submersion fixation in 0.5% PFA over 4 to 6 hours at room temperature. Lobes were cryoprotected in a mixture of 20% sucrose and 10% optimal cutting temperature compound (4583, Tissue-Tek) in PBS. The resulting frozen sections were blocked in PBS with 0.3% Triton X-100 and 5% normal donkey serum (017–000–121, Jackson ImmunoResearch) and incubated with primary antibodies (see Methods in the data supplement) overnight in a humidified chamber at 4°C, followed by donkey secondary antibodies (Jackson ImmunoResearch) diluted in PBS with 0.3% Triton X-100 plus DAPI (4’,6-diamidino-2-phenylindole) for 1 hour at room temperature. The slides were washed for 30 minutes in PBS after antibody incubation before being mounted in Aqua-Poly/Mount (18606, Polysciences) and imaged on a Nikon A1plus confocal microscope or an Olympus FV1000 confocal microscope. Cells were quantified manually with Image J.
For wholemount immunostaining, lobes were blocked and incubated with primary and secondary antibodies as above, except for being washed with PBS with 1% Triton X-100 and 1% Tween-20. Fresh or immunostained lobes were imaged with a Leica M205C fluorescence stereomicroscope or an optical projection tomography scanner (Bioptonics, 3001M) as published (21, 22).
RNAscope In Situ Hybridization
Lungs harvested and fixed as above were cryoprotected, without PBS wash, in a mixture of 20% sucrose, 10% optimal cutting temperature compound, and 0.5% PFA in PBS overnight. The resulting frozen sections were probed for Vegfa, Irf7, Ifit3, Icam1, and Oasl2 using the Multiplex Fluorescent Detection Kit v2 (Advanced Cell Diagnostics, 323,110) and OPAL 520/570/690 reagent packs (Akoya Biosciences, FP1487001KT/FP1488001KT/FP1497001KT).
Single-Cell RNA Sequencing (RNA-seq) and Data Analysis
See Methods and Files in the data supplement for experimental details and analysis code.
Results
Airway Cells Are Mobilized to Populate Damaged Alveolar Regions, with Minimal Alveolar Differentiation
Expecting SeV infection to induce an asynchronous, spatially heterogeneous response of injury–repair that needed an organ-level depiction, we used optical projection tomography to visualize whole lung lobes immunostained for the airway transcription factor SOX2 (Figure 1A). On Day 9 after infection (9 dpi [day postinfection]), SOX2 staining diminished in lobar and segmental airways but remained in terminal bronchioles, consistent with cell loss from a viral infection that spread distally. The most proximal airways near the lobe entrance had normal SOX2 staining and thus possibly had limited infection or had recovered (Figure 1A). By 28 dpi, all airways regained SOX2 staining and, strikingly, formed cauliflower-like SOX2+ clusters that extended beyond terminal bronchioles (Figure 1A). These outgrowing clusters were often well into the alveolar region and even reached the most distal mesothelium, although our 3D imaging showed that they were continuous with and likely originated from the airways (Figure 1A), as further tested by lineage tracing below. The SOX2+ clusters were visible in freshly dissected lungs using a Sox2GFP allele (13) and were mutually exclusive with AT2 cells that were genetically labeled before infection (an initial, to-be-explored clue for a limited contribution of AT2 cells to restoring these damaged regions) (Figure 1A).
Figure 1.
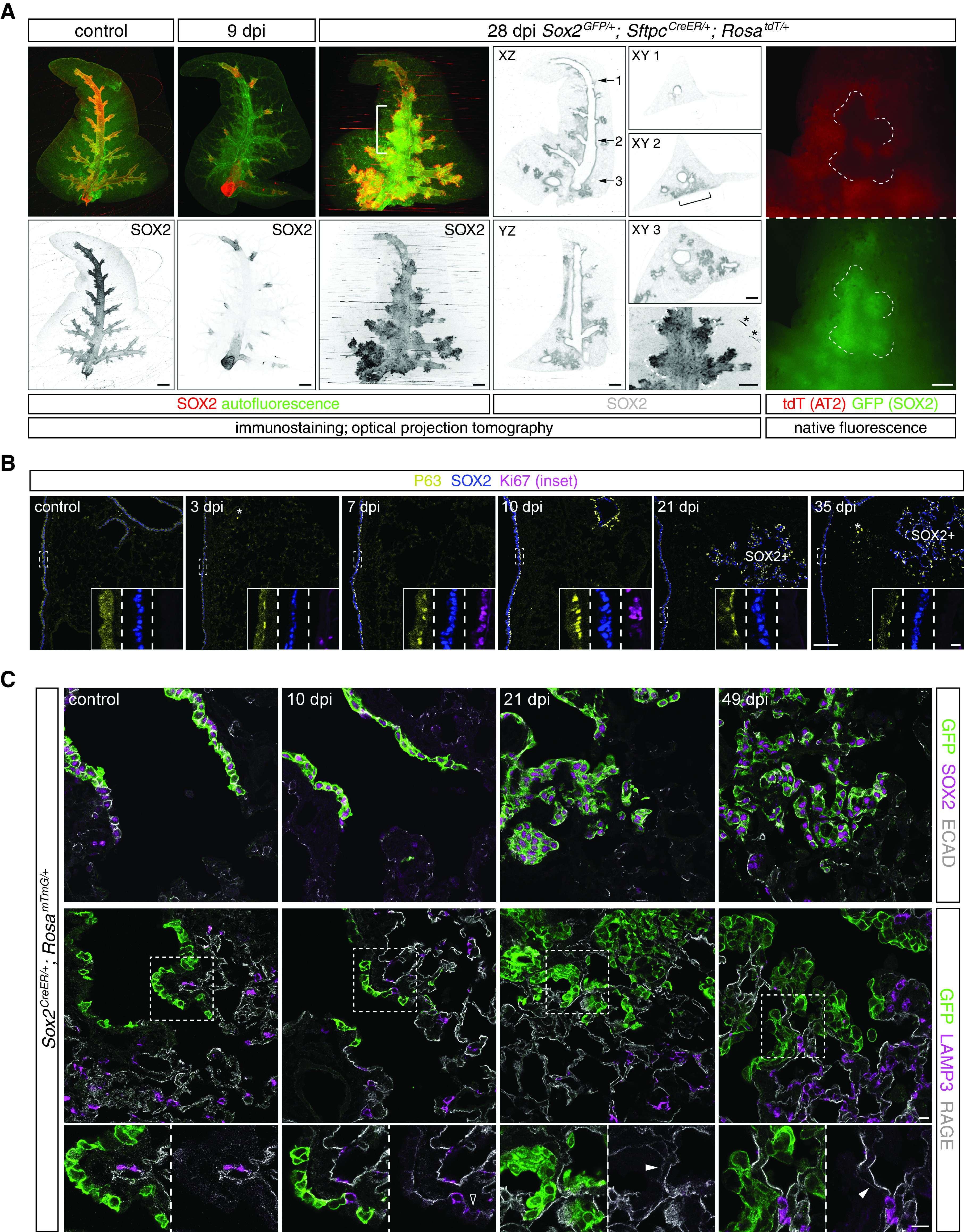
Airway cells are mobilized to populate damaged alveolar regions with minimal alveolar differentiation. (A) OPT images of wholemount immunostained accessory lobes from control and Sendai virus (SeV)-infected (dpi [day postinfection]) adult mouse lungs. Tissue autofluorescence highlights airways and macrovasculature and is more noticeable at 9 dpi when SOX2 (SRY-box 2) expression is lost from most airways. The recovery and cauliflower-like alveolar clusters of SOX2 expression at 28 dpi are apparent on XZ, YZ, and XY (at the numbered levels) optical planes. Alveolar SOX2 clusters can be found at lobe edges but are still continuous with the airways (bracket). The 28 dpi lung is from a Sox2GFP/+; SftpcCreER/+; RosatdT/+ mouse and received 2 mg tamoxifen 3 weeks before infection. Native fluorescence images before immunostaining in the bracketed region (rightmost column) show mutual exclusion of airway cells marked by Sox2GFP (the same contours outlined by dashes as those in the OPT image; asterisk, technical ring artifacts) and AT2 (alveolar type 2) cells marked by SftpcCreER and RosatdT. Scale bars, 500 um. Images are representative of at least three lungs (same in subsequent figures). (B) Section immunostaining images showing SeV-induced proliferation (Ki67 in insets for boxed regions), expansion, and persistence of P63+ basal-like cells as a subset of SOX2+ cells in airways and alveolar SOX2 clusters (SOX2+). Asterisk, nonspecific macrophage fluorescence. Scale bars, 100 um (10 um for insets). (C) Section immunostaining images of control and SeV-infected adult Sox2CreER/+; RosamTmG/+ lungs with 2 mg tamoxifen administered 3 weeks before infection. All lineage-labeled cells (GFP+) are positive for the airway marker SOX2 and cuboidal/columnar as outlined by E-cadherin (ECAD; top row) and negative for an AT2 cell marker (LAMP3 [lysosome-associated membrane glycoprotein 3]) except for rare cells transiently expressing LAMP3 (open arrowhead at 10 dpi; bottom row). Solid arrowhead: weak AT1 cell marker (RAGE [receptor for advanced glycation endproducts]) under GFP+ cells, which will be further examined later. Scale bars, 10 um. OPT = optical projection tomography.
The SOX2+ clusters were reminiscent of “pods” fueled by rare basal-like cells upon severe influenza infection (23–25). Indeed, basally located, proliferative P63+ cells first appeared at 3 dpi within airways and were readily found at 7 and 10 dpi (Figure 1B). Although no longer expressing Ki67, these airway P63+ cells persisted for at least 35 dpi (Figure 1B). Mirroring the observed pattern and kinetics of airway repair (Figure 1A), the wave of P63 expression extended to alveolar SOX2+ clusters and persisted (Figure 1B). Other basal cell markers KRT14 and KRT5 were expressed by most cells in SOX2+ clusters at 14 dpi and became more restricted at 42 dpi, especially for KRT5, possibly reflecting differentiation to lumenal cells (Figure E1A in the data supplement). The aberrant persistence of P63+ cells in bronchioles and alveoli reflected either a mimicry of larger airways that were normally populated by P63+ cells or a defect in terminating the injury–repair response, possibly contributing to the chronic abnormalities (26).
To ascertain the fate of alveolar SOX2+ cells, we used Sox2CreER (13) to label airway cells before infection and found that most, if not all, lineage-labeled cells, including those aberrantly present in the alveolar region, remained SOX2+ and cuboidal/columnar, as outlined by a cell junction protein E-cadherin, and did not express an AT1 marker RAGE (receptor for advanced glycation endproducts) nor an AT2 marker LAMP3 (lysosome-associated membrane glycoprotein 3) (Figures 1C and E1B). The dysplastic SOX2+ clusters often overlay a surface with weak RAGE staining (Figure 1C), suggesting the replacement of AT1 cells, as examined in detail later. We noted rare Sox2CreER-labeled LAMP3+ cells near terminal bronchioles in control lungs, possibly because of low or leaky expression of Sox2CreER, which could also exist or even expand in SeV lungs (Figure E1B). Our whole-lobe and cellular imaging, together with lineage tracing, captured mobilization of P63/SOX2 airway cells to repair damaged airway and alveolar regions without noticeable differentiation into alveolar cells.
AT2-less Regions Are Depleted of AT2 Cells, but Covered by AT1 Cells
The rapidly growing, dysplastic SOX2+ clusters prompted us to search for damaged alveolar regions that were presumably in need of repair. A timecourse survey of AT1 (marked by RAGE) and AT2 (marked by SFTPC [surfactant protein C]) cells revealed regions depleted of AT2 cells at 7 dpi and spanning hundreds of microns, comparable in size to future SOX2+ clusters (Figures 2A and E1C). Strikingly, these AT2-depleted regions were indistinguishable from the rest of the lung on the basis of RAGE staining, suggesting continued coverage with AT1 cells (Figure 2A). This uncoupling between AT1 and AT2 cells was confirmed on sequential sections with another set of AT1 and AT2 markers (PDPN [podoplanin] and LAMP3), although PDPN additionally marked P63+ cells at 14 dpi as expected for a basal cell marker (27) (Figure 2A). Besides their cell membranes, AT1 cell nuclei also persisted, as marked by the lung lineage transcription factor NKX2–1 [NK2 homeobox 1] without surrounding SFTPC (18, 28) (Figure 2B). To confirm that RAGE staining was from AT1 cells instead of aberrant gene expression upon infection, we used our recently characterized AT1 cell driver Rtkn2CreER (18) and labeled AT1 cells before infection. Indeed, regions depleted of AT2 cells were as fully covered with lineage-labeled AT1 cells as uninvolved regions (Figure 2C). Accordingly, we referred to these AT2 cell-depleted but AT1 cell-covered areas as “AT2-less regions”.
Figure 2.
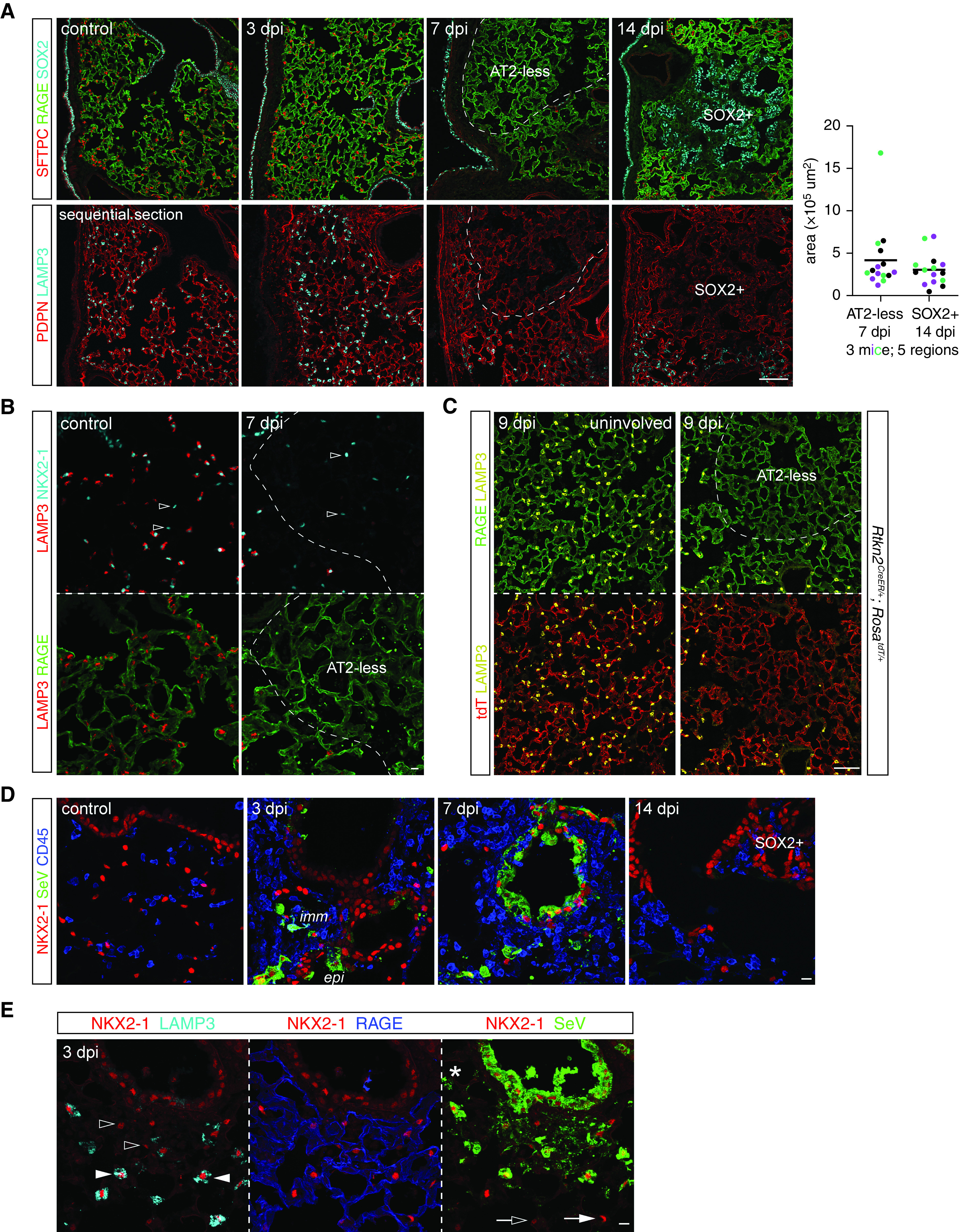
AT2-less regions are depleted of AT2 cells but covered by AT1 cells. (A) Sequential section immunostaining images showing SeV infection depletes dashed areas of AT2 cell markers (SFTPC [surfactant protein C] and LAMP3) without affecting AT1 cell markers (RAGE and PDPN [podoplanin]) at 7 dpi. These AT2 cell-depleted (AT2-less) regions are similar in size and location to SOX2+ clusters at 14 dpi. Area quantification from three mice (color-coded) and five regions; each mouse shows no significant difference between 7 dpi AT2-less regions and 14 dpi SOX2+ clusters. See Figure E1C for quantification methods. PDPN, a basal cell marker, also marks P63+ basal-like cells. Scale bar, 100 um. (B) Section immunostaining images showing AT1 cell nuclei (NKX2–1+ [NK2 homeobox 1+] LAMP3−; open arrowheads) are intermingled with AT2 cell nuclei (NKX2–1+ LAMP3+) in the control lung but are the only nuclei present in AT2-less regions (dashes). Scale bar, 10 um. (C) Section immunostaining images of Rtkn2CreER/+; RosatdT/+ lungs with recombination induced at P5 with 500 ug tamoxifen to efficiently label AT1 cells. At 9 dpi, both uninvolved (left) and AT2-less (right; depleted of LAMP3) regions are covered by tdT+ RAGE+ AT1 cells. Scale bar, 100 um. (D) Section immunostaining images showing SeV viral proteins (SeV) in immune cells (CD45+) and epithelial cells (NKX2–1+) at 3 and 7 dpi and cleared by 14 dpi. Scale bar, 10 um. (E) Section immunostaining images showing both thin AT1 cells (open arrowheads; RAGE+ NKX2–1+) and cuboidal AT2 cells (solid arrowheads; LAMP3+ NKX2–1+) are filled with SeV viral proteins and thus infected, in comparison with their uninfected neighbors (open and solid arrows). Asterisk, an infected macrophage. Scale bar, 10 um. epi = epithelial cells; imm = immune cells.
A possible explanation for the preferential depletion of AT2 cells was differential viral infection or clearance of infected cells, although the receptor for SeV is sialic acid and is widely present (29). Examining cell-type infection in vivo was challenging, and an underestimate because of the transient nature of infected/dying cells compounded by the likely concomitant decrease of cellular markers. Nevertheless, abundant SeV proteins were readily detected in NKX2–1+ epithelial cells and CD45+ immune cells at 3 dpi and spread to adjacent cells at 7 dpi before largely disappearing at 14 dpi (Figure 2D). Infected airway and AT2 cells were identifiable by their anatomic location, cell shape as filled by SeV proteins, and the AT2 marker LAMP3 (Figure 2E). Although infrequent, infected AT1 cells were identifiable when the SeV protein staining outlined both the NKX2–1+ nucleus and RAGE+ cellular extensions (Figure 2E). Therefore, although AT1 cells had a larger surface area accessible to SeV and could be infected, AT2 cells either were more easily infected or survived less, leading to AT2-less regions; the possibility of AT2 cell depletion via their differentiation into AT1 cells was tested and excluded below. As a result, the exposed tissue surface from dying AT1 and AT2 cells was expected to be resealed by the remaining AT1 cells, as supported by the continuous RAGE staining (Figure 2).
AT2 Cells Differentiate into AT1 Cells in Topologically Distal Regions but Contribute Minimally to In Situ Repair of Damaged Alveolar Surface
As stem cells, AT2 cells were known to proliferate and differentiate into AT1 cells and were often assumed to do so to repair damaged regions (6). However, as introduced earlier, AT2 cells could also be activated upon pneumonectomy without direct injury, and such activation was more apparent near the tissue edge, which was topologically most distal and had more space to expand (30, 31). The AT2-less regions in the SeV model provided a landmark to locate activated AT2 cells. At 14 dpi, expanding SOX2+ clusters were still bordered by regions without AT2 cells, let alone activated ones (Figure 3A). Instead, clusters of AT2 cells formed further away (near the organ edge or major blood vessels and airways, both of which were the most topologically distal part of the respiratory tree even when embedded in the center of the lung) (Figure 3A). These aberrant AT2 cell clusters expressed Ki67 (Figure E2) and were intermingled with small, LAMP3-negative AT1 cells (Figure 3A), an arrangement reminiscent of that in nascent alveolar sacs during development (9), and were still present at 42 dpi when the AT2-less regions had disappeared (Figure 3A). This AT2 cell depletion followed by hyperplasia was evident when tracking the ratio of AT2 to AT1 cells over time and was specific to damaged regions (Figure 3B). Therefore, AT2 cells clustered de novo near vessels, airways, and the edges of the tissue but at a distance from AT2-less regions (Figure 3C).
Figure 3.
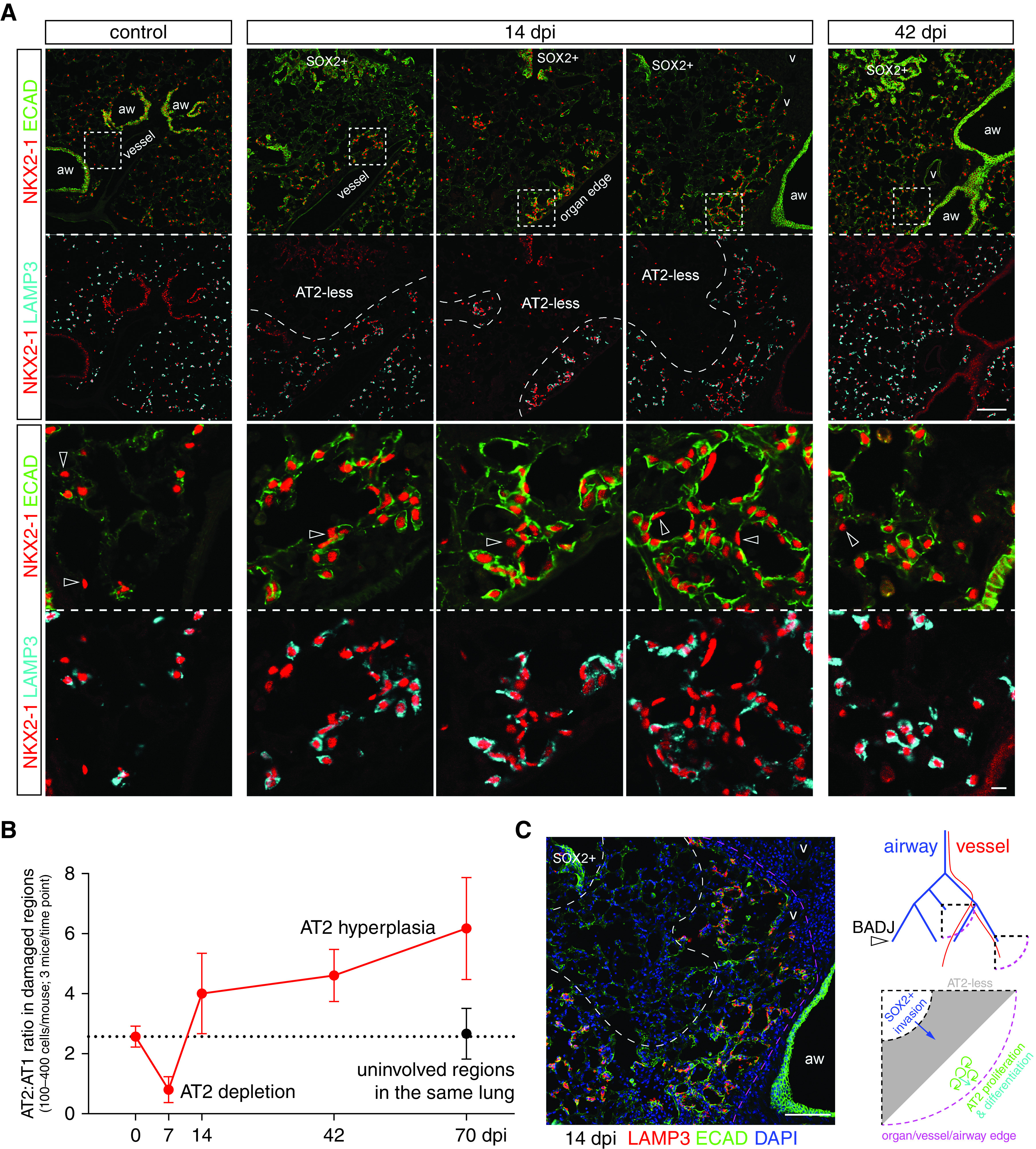
AT2 cells form clusters in topologically distal regions near the organ edge or major blood vessels and airways. (A) Section immunostaining images with boxed topologically distal regions enlarged. AT2 cells are LAMP3+ and cuboidal, as outlined by ECAD, as opposed to AT1 cells (open arrowheads). Three examples of 14 dpi lung sections show that AT2 cells cluster around a nascent airspace lumen away from AT2-less regions and SOX2 clusters, but near the organ edge or major blood vessels and airways, a pattern only partially resolved at 42 dpi. Scale bars, 10 um. (B) Quantification of the ratio of AT2 to AT1 cells supports AT2 cell depletion at 7 dpi and subsequent hyperplasia and clustering. Uninvolved regions in infected lungs have a normal cell ratio. Error bars correspond to standard deviations. (C) An image from a 14 dpi lung in (A) overlaid with DAPI (4’,6-diamidino-2-phenylindole) to demonstrate topologically distal regions beyond a SOX2 cluster and an AT2-less region corresponding to the center quarter circle in the diagram that is topologically equivalent to the quarter circle near the organ edge. Only arterial vessels are drawn for clarity. It is predicted and subsequently tested that AT2-less regions are invaded by SOX2 cells, whereas AT2 cell proliferation and differentiation occur further distally. aw = airways; BADJ = bronchoalveolar duct junction; v = vessels.
This conclusion was substantiated by lineage-tracing experiments showing that AT2 cell clusters, including adjacent AT1 cells, arose from preexisting AT2 cells that were labeled before infection, whereas no expansion of labeled AT2 cells nor their differentiation into AT1 cells was observed in AT2-less regions (Figure 4). Taken together, at least in the SeV model, AT2 cell-mediated tissue growth occurred in topologically distal regions and away from obviously damaged regions, possibly to avoid generating excessive AT1 cells, given the persistence of AT1 cells (Figure 2). Our analysis also highlighted the importance of spatial information in distinguishing de novo growth versus in situ repair.
Figure 4.
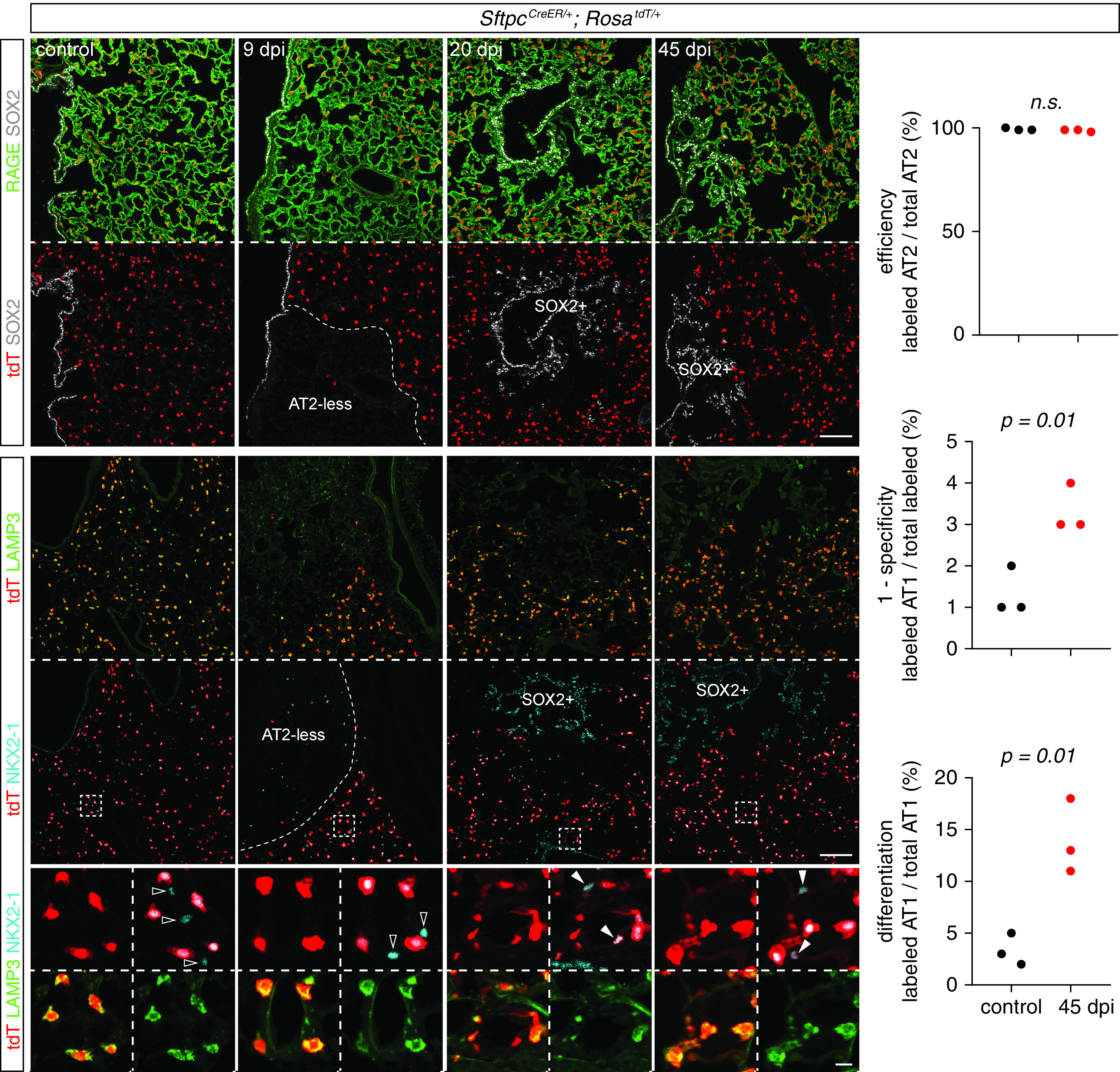
AT2 cells differentiate into AT1 cells in topologically distal regions but not AT2-less regions. Section immunostaining images from control and SeV-infected lungs of SftpcCreER/+; RosatdT/+ mice that received 2 mg tamoxifen 3 weeks before infection. Lineage-labeled AT2 cells are excluded from AT2-less regions (RAGE+ LAMP3−; 9 dpi) and later SOX2 clusters (20 and 45 dpi) but are present and differentiate into AT1 cells (open arrowheads; NKX2–1+ LAMP3−) in topologically distal regions (boxed) as explained in Figure 3. Scale bars, 100 um (10 um for enlarged boxed regions). Quantification shows near-complete labeling of AT2 cells (top graph) and their increased differentiation into AT1 cells on infection (middle graph; “specificity” refers to the SftpcCreER driver, and “1-specificity” was used to show the low percentages without truncating the y-axis), constituting 10–20% AT1 cells in topologically distal regions (bottom graph). Labeling of AT1 cells in the control subjects is because of cumulative baseline cell turnover that is additionally amplified by tamoxifen-independent, leaky recombination of SftpcCreER.
Invading Airway Cells Displace AT1 Cells Via Basal and Luminal Spreading
As AT2-less regions, without being repaired by AT2 cells, disappeared with the same kinetics as the expansion of SOX2+ clusters (Figure 3A), we posited that the persistent and then disappearing AT1 cells were displaced by the invading SOX2+ cells. Indeed, at 12 dpi, when SOX2+ clusters were first detected consistently in the alveolar region, SOX2+ cells were in three conformations relative to RAGE+ AT1 cell extensions: 1) basal, in which SOX2+ cells were underneath an intact RAGE membrane and embedded in a collagen matrix; 2) luminal I, in which 2–3 SOX2+ cells gained access to the airspace, accompanied by weakened or gapped RAGE staining; and 3) luminal II, in which tens of SOX2+ cells accumulated in the airspace and spilled over AT1 cell surfaces with normal RAGE staining, possibly causing further AT1 cell displacement (Figure 5A). Although live imaging of this displacement was not available, SOX2+ cells of the two luminal conformations were connected by adherens junctions and continuous with those embedded in the tissue, suggesting that the three conformations represented snapshots of a continuous, if not sequential, process (Figure 5B).
Figure 5.
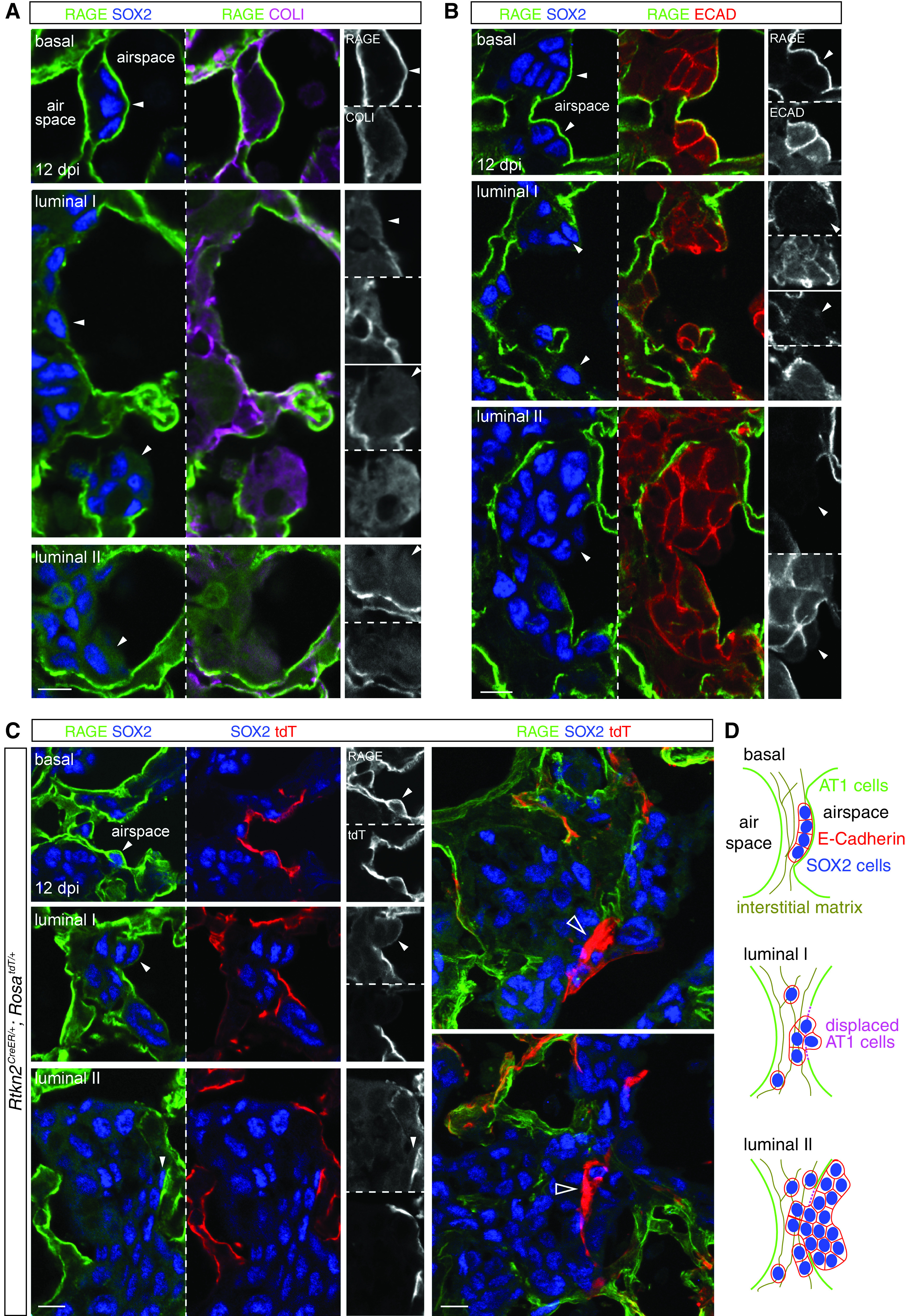
Invading airway cells displace AT1 cells via basal and luminal spreading. Section immunostaining images of lungs at 12 dpi when SOX2 clusters are reliably detected. (A) Invading SOX2+ cells (arrowheads) are found embedded in the interstitial matrix (COLI [collagen I]) and basal to the AT1 cell membrane (RAGE), gaining access to the airspace and covered by reduced RAGE (luminal I), or accumulated in the airspace and spreading over neighboring RAGE membrane (luminal II). (B) Invading SOX2+ cells of the three conformations form continuous cell junctions (ECAD). (C) Left, RAGE staining is from AT1 cells lineage-labeled at P5 with 500 ug tamoxifen. Right, AT1 cells surrounded by SOX2+ cells accumulate brighter tdT (tandem dimer Tomato; open arrowheads), possibly amid cell displacement. (D) Schematics showing the relationship of SOX2+ cells and AT1 cells. Scale bars, 10 um.
Lineage labeling using the AT1 cell driver Rtkn2CreER (18) confirmed that the displaced RAGE staining was from AT1 cells and that SOX2+ cells existed basal or luminal to AT1 cells (Figure 5C). Strikingly, at their interface, in which AT1 cell displacement was hypothesized to occur, shrunken fragments of AT1 cells accumulated an excessive amount of the lineage reporter, perhaps because of cell condensation before its shedding and clearance (Figures 5C and E3). Our observations were consistent with a model in which AT1 cells in AT2-less regions temporarily sealed the alveolar surface before being displaced by airway-originated SOX2+ clusters spreading both basally and luminally (Figure 5D).
AT1 Cells, as well as AT2 Cells, Acutely Upregulate IFN Responsive Genes, as Revealed by Timecourse Single-Cell RNA-seq
To examine cellular and molecular dynamics upon SeV infection in an unbiased manner, we profiled all major lung cell types using single-cell RNA-seq at 7, 14, and 49 dpi, representing the injury, repair, and chronic phases, respectively (Figures 6A and E4). Several infection-induced cell clusters were readily identified (Figures 6A and 6B) and included 1) Trp63+ basal-like cells at 14 and 49 dpi (cluster 4), corresponding to the invading SOX2+ clusters (Figure 1); 2) Ifi27l2a+ cells mainly at 7 dpi for both AT1 (clusters 14 and 15) and AT2 (clusters 6 and 11) cells, corresponding to an acute IFN response to SeV; and 3) Mki67+ proliferative cells that were present as a result of homeostatic cell turnover but increased upon infection at 14 dpi, consistent with de novo clustering and hyperplasia of AT2 cells (Figures 3 and 4). Intriguingly, at 14 dpi, AT1 cells included a cell cluster 12 that shared genes with normal AT2 cells, such as Napsa and H2-Aa; such residual expression of AT2 genes raised the possibility that they arose from AT2 cells, as observed in regions of de novo growth (Figure 4). Direct comparison of AT1 cells at each time point revealed induction of additional IFN pathway genes predominantly at 7 dpi, including those for antigen processing and presentation (Psmb8/9/10, B2 m, Cd74, H2-K1, H2-D1, and H2-Q6), antiviral effectors (Ifit1, Oasl2, Rsad2, and Rtp4), and IFN pathway transcription factors (Irf1 and Irf7) (Figure 6C and Tables E1–E3). Upregulation of Irf7, Ifit3, Icam1, and Oasl2 in AT1 cells, marked by Vegfa (9, 32), was confirmed using RNAscope (Figure 6D). In contrast, AT1 cell-enriched genes, including Vegfa and Abca1, were reduced, suggesting the reduction of angiogenic and cholesterol transport functions upon infection (32, 33), although Vegfa reduction was not obvious by RNAscope, possibly because of assay sensitivity or AT1 cell heterogeneity (Figure 6D).
Figure 6.
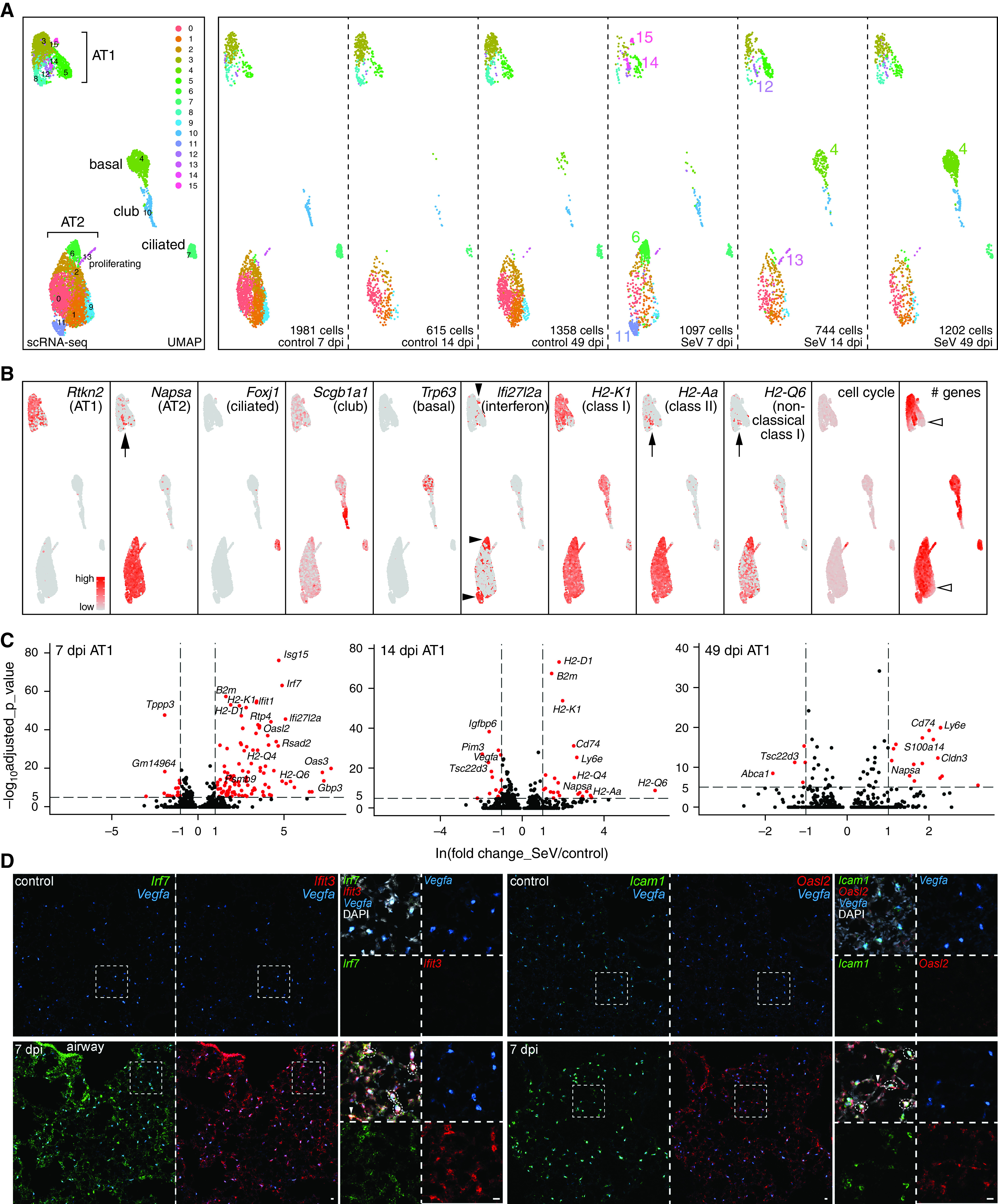
Time-course single-cell RNA sequencing reveals IFN and signaling responses in AT1 cells. (A) Merged and split UMAP (uniform manifold approximation and projection) graphs of epithelial cells from paired control and SeV-infected lungs at 7, 14, and 49 dpi, color-coded for computer-identified clusters with differential clusters numbered in the split UMAP graphs. Cluster 4 corresponds to persistent basal-like cells. Clusters 6, 11, 14, and 15 are prominent for 7 dpi SeV lungs; clusters 13 and 12 are predicted as proliferating and differentiating AT2 cells, respectively. (B) Feature plots of cell-type–specific markers, supporting cell type names in (A). Cluster 12 (arrows) expresses markers of AT2 cells, including Napsa, an MHC class II gene H2-Aa, and an MHC nonclassical I gene H2-Q6, the last of which has the highest expression in clusters 6 and 11. An IFN signature gene Ifi27I2a is highest (solid arrowheads) in clusters 6, 11, and 15 (lower in 14) at 7 dpi. The cell-cycle gene score marks the proliferative cluster 13. Additional heterogeneity arises from the total number of genes detected (open arrowheads). (C) Volcano plots comparing AT1 cells from control and infected lungs at 7, 14, and 49 dpi. See Tables E1–E3 for complete data. (D) Section RNAscope in situ hybridization showing upregulation of Irf7, Ifit3, Icam1, and Oasl2 in AT1 cells (dash oval; marked by Vegfa) in SeV-infected lungs at 7 dpi. Non-AT1 cells (arrowhead) also upregulate Irf7, Ifit3, and Oasl2. Scale bars, 10 um. MHC = major histocompatibility complex.
To place these differentially expressed genes in the context of all lung cell types and focus on intercellular signaling, we applied our interactome algorithm (19) to identify outlying changes in ligand–receptor interactions with AT1 cells receiving or sending signals. Pairwise comparison of data from 7, 14, and 49 dpi highlighted altered interactions unique and common to two or all time points (Figure E5 and Tables E4 and E5), supporting an increased recognition of AT1 cells by NK and T cells and a decreased AT1 cell-derived angiogenic signal toward the endothelium. Intriguingly, AT1 cells at 14 dpi upregulated Hbegf, a possible mitotic signal for AT2 cells or invading airway cells (34). Increased Icam1 expression in AT1 cells might facilitate immune cell extravasation (35). With respect to AT1 cells receiving signals, a number of matrix genes, including Tnc, Fn1, Fbn1, Col1a1, and Col5a2, were upregulated in various mesenchymal cells, potentially affecting tissue stiffness and integrin-mediated adhesion of epithelial cells; whereas Il18 from immune cells could stimulate inflammation via Il18r1 in epithelial cells (36) (Figure E5 and Tables E5 and E6). Taken together, our timecourse single-cell RNA-seq profiling confirmed our imaging-based molecular and cellular characterization and implicated active signaling roles of AT1 cells in countering SeV infection.
Discussion
In this study, we use 3D imaging, lineage tracing, and single-cell transcriptomics to delineate the spatiotemporal and cellular heterogeneity of lung response to natural viral infection. As diagramed in Figure 7, the lung epithelium, when viewed as a two-dimensional surface, is predominantly made of AT1 cells with minor contributions from SOX2+ airway cells and AT2 cells. SeV infection causes widespread loss of airway and AT2 cells, but most AT1 cells persist, forming AT2-less regions. SOX2+/P63+ basal-like cells expand to heal the airways and aberrantly extend to the AT2-less regions, displacing AT1 cells without differentiating into alveolar cells. AT2 cells form hyperplastic clusters and undergo AT1 cell differentiation in topologically distal regions, generating a de novo alveolar surface with limited contribution to healing the AT2-less regions. AT1 cells have intermediary structural and signaling roles by sealing the alveolar surface temporarily and mounting an antiviral interferon response, and possibly modulating growth signals such as Vegfa and Hbegf. Such molecular and cellular understanding is necessary to dissect associated mechanisms and test therapies using the SeV or other models of lung injury–repair.
Figure 7.
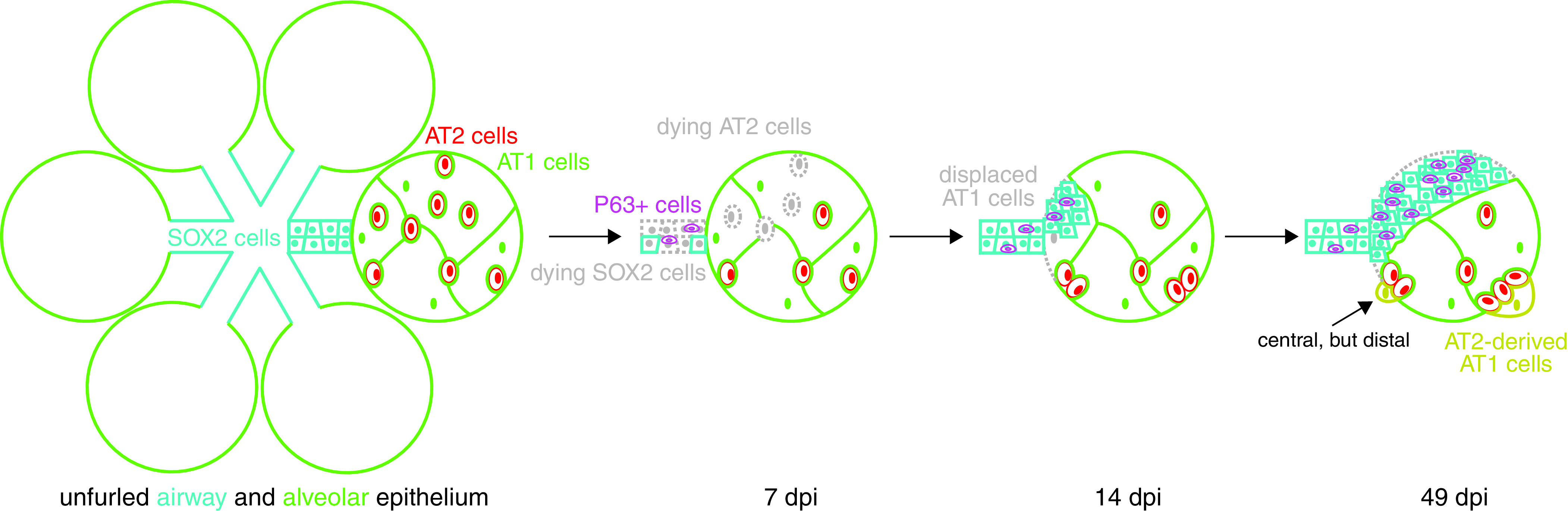
Diagram illustrating cellular changes in the lung epithelium upon SeV infection. Unfurled lung epithelium consists of proximal SOX2+ columnar airway cells and distal intermingled flat AT1 and cuboidal AT2 cells in which a central region can be topologically distal. Upon SeV infection, loss of airway cells amplifies P63+ basal-like cells, whereas loss of AT2 cells, but not AT1 cells, forms AT2-less regions. Subsequently, SOX2+ airway cells, including P63+ basal-like cells, invade alveoli and displace AT1 cells; AT2 cells proliferate in topologically distal regions and differentiate into AT1 cells to form de novo alveolar surface.
A notable feature in the SeV model is the formation of AT2-less regions in which AT2 cells are lost, whereas AT1 cells are spared. The initial gaps left by disappearing AT2 cells can be readily sealed by AT1 cells, given their ability to expand their surface by over 10-fold during development (9). The resulting AT2-less surface must be sensed and then induce the invasion of SOX2+/P63+ cells. Loss of AT2 cells is unlikely the only trigger as genetic ablation of AT2 cells stimulates clonal expansion of remaining AT2 cells but not airway cell invasion (5). Hypoxia and fibroblast growth factor signaling pathways have been implicated in influenza and bleomycin models (25, 37), although it will be necessary to map their activation spatiotemporally and determine their sufficiency in providing a directional cue. The vascular and mesenchymal cells underneath the AT2-less surface are likely also altered, possibly compromising gas exchange efficiency or even recruiting SOX2+ cells.
The correlation between AT2-less regions and SOX2+ clusters in their size, location, and kinetics, as well as their physical interactions (Figures 1, 2, and 5), supports the replacement of AT1 cells by SOX2+ cells, possibly via integrin-mediated cell adhesion (Table E6). However, we cannot rule out that areas bordering the AT2-less regions could be populated by spared AT2 cells nearby, although this scenario predicts that AT2 cells migrate across AT1 cells for dispersion as they do not differentiate into AT1 cells in large numbers (Figure 4). Future live imaging experiments will provide definitive evidence for the cellular sources and interactions during the repair of AT2-less regions. It also remains to be determined if similar AT2-less regions form in other microbial and chemical injury models.
The existence of AT2-less regions also highlights the uncoupling of AT2 cell activation and in situ healing of preexisting-and-then-damaged alveolar surfaces. We notice AT2 cell proliferation and differentiation in topologically distal regions close to tissue edges, airways, or vessels (Figures 3 and 4), the last of which has been suggested as a niche for AT2 cells partly because of their spatial proximity (38, 39). This de novo formation of the alveolar surface is intuitive during compensatory growth induced by pneumonectomy but is less appreciated in models of chemical and microbial injury. Nevertheless, to counter life-threatening damage, the lung is as likely to mobilize multiple stem and progenitor cell populations as to evoke both in situ healing and de novo growth. These two repair processes may involve distinct mechanisms in which in situ healing is similar to wound healing, whereas de novo growth forms nascent alveolar lumen, reminiscent of developmental sacculation. Distinguishing these two processes will allow further understanding of recently identified KRT8+ intermediate AT2 cells (40–42).
The persistence of SOX2+ clusters with minimal differentiation into alveolar cells (Figure 1C) supports the idea that P63+ basal-like cells repair the airways properly but are resorted to for dysplastic alveolar repair only during severe injury, as seen in the influenza model as well as patients with COVID-19 (23–25, 43). The resulting ectopic airway differentiation in the alveolar region could form excessive mucus-producing cells or tuft cells (20, 44), contributing to the asthma phenotype independent of a deviated immune environment (26). The SOX2+ clusters could also associate with mesenchymal cells of the airways, such as those in the bronchovascular bundles, which might produce more matrix proteins than those of the alveoli, offering a potential link between pulmonary fibrosis and alveolar bronchiolization in fibrotic patient lungs (45–48). The seemingly irreversible nature of the SOX2+ clusters and associated P63+ cells (Figures 1 and 6) prompts future studies of their responses to repeated injuries and the cumulative consequences of seasonal flu in humans. It is worth noting that other injury models, including influenza and bleomycin, additionally mobilize another pool of Sox2CreER- or Scgb1a1CreER-traced cells, possibly corresponding to distal club cells or bronchioalveolar stem cells, which differentiate into AT1 and AT2 cells (25, 37, 49–51). The observed inability of Sox2CreER-traced cells to contribute to alveolar cells in the Sendai model might be because of selective suppression or removal of non-P63 progenitors, especially those in AT2-less regions that are near the bronchioalveolar duct junction. Given the functional difference between the resulting alveolar and basal-like cells, future studies are needed to identify the molecular nature and activation signals of diverse pools of lung progenitor and stem cells.
Footnotes
Supported by the National Heart, Lung, and Blood Institute (R01HL130129 and R01HL153511 [J.C.]) and the University of Texas MD Anderson Cancer Center Start-up and Retention Fund. The University of Texas MD Anderson Cancer Center DNA Analysis Facility and Flow Cytometry and Cellular Imaging Core Facility are supported by the Cancer Center Support Grant (CA #16672).
Author Contributions: B.J.H., M.P.C., and J.C. designed and performed research and wrote the paper. A.M.L., J.R.F., M.J.T., and B.F.D. performed research. All authors read and approved the paper.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2021-0421OC on June 9, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Faísca P, Desmecht D. Sendai virus, the mouse parainfluenza type 1: a longstanding pathogen that remains up-to-date. Res Vet Sci . 2007;82:115–125. doi: 10.1016/j.rvsc.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 2. Holtzman MJ, Tyner JW, Kim EY, Lo MS, Patel AC, Shornick LP, et al. Acute and chronic airway responses to viral infection: implications for asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc . 2005;2:132–140. doi: 10.1513/pats.200502-015AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wu K, Kamimoto K, Zhang Y, Yang K, Keeler SP, Gerovac BJ, et al. Basal epithelial stem cells cross an alarmin checkpoint for postviral lung disease. J Clin Invest . 2021;131:e149336. doi: 10.1172/JCI149336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal, and cancer. Nature . 2014;507:190–194. doi: 10.1038/nature12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest . 2013;123:3025–3036. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hogan BL, Barkauskas CE, Chapman HA, Epstein JA, Jain R, Hsia CC, et al. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell . 2014;15:123–138. doi: 10.1016/j.stem.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vila Ellis L, Chen J. A cell-centric view of lung alveologenesis. Dev Dyn . 2021;250:482–496. doi: 10.1002/dvdy.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen J. Origin and regulation of a lung repair kit. Nat Cell Biol . 2017;19:885–886. doi: 10.1038/ncb3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang J, Hernandez BJ, Martinez Alanis D, Narvaez del Pilar O, Vila-Ellis L, Akiyama H, et al. The development and plasticity of alveolar type 1 cells. Development . 2016;143:54–65. doi: 10.1242/dev.130005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weibel ER. What makes a good lung? Swiss Med Wkly . 2009;139:375–386. doi: 10.4414/smw.2009.12270. [DOI] [PubMed] [Google Scholar]
- 11. Uriarte JJ, Uhl FE, Rolandsson Enes SE, Pouliot RA, Weiss DJ. Lung bioengineering: advances and challenges in lung decellularization and recellularization. Curr Opin Organ Transplant . 2018;23:673–678. doi: 10.1097/MOT.0000000000000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee JH, Rawlins EL. Developmental mechanisms and adult stem cells for therapeutic lung regeneration. Dev Biol . 2018;433:166–176. doi: 10.1016/j.ydbio.2017.09.016. [DOI] [PubMed] [Google Scholar]
- 13. Arnold K, Sarkar A, Yram MA, Polo JM, Bronson R, Sengupta S, et al. Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell . 2011;9:317–329. doi: 10.1016/j.stem.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci . 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis . 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 16. Liu J, Krautzberger AM, Sui SH, Hofmann OM, Chen Y, Baetscher M, et al. Cell-specific translational profiling in acute kidney injury. J Clin Invest . 2014;124:1242–1254. doi: 10.1172/JCI72126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA . 2011;108:E1475–E1483. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Little DR, Lynch AM, Yan Y, Akiyama H, Kimura S, Chen J. Differential chromatin binding of the lung lineage transcription factor NKX2-1 resolves opposing murine alveolar cell fates in vivo. Nat Commun . 2021;12:2509. doi: 10.1038/s41467-021-22817-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cain MP, Hernandez BJ, Chen J. Quantitative single-cell interactomes in normal and virus-infected mouse lungs. Dis Model Mech . 2020;13:dmm044404. doi: 10.1242/dmm.044404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goldblatt DL, Flores JR, Valverde Ha G, Jaramillo AM, Tkachman S, Kirkpatrick CT, et al. Inducible epithelial resistance against acute Sendai virus infection prevents chronic asthma-like lung disease in mice. Br J Pharmacol . 2020;177:2256–2273. doi: 10.1111/bph.14977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chang DR, Martinez Alanis D, Miller RK, Ji H, Akiyama H, McCrea PD, et al. Lung epithelial branching program antagonizes alveolar differentiation. Proc Natl Acad Sci USA . 2013;110:18042–18051. doi: 10.1073/pnas.1311760110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alanis DM, Chang DR, Akiyama H, Krasnow MA, Chen J. Two nested developmental waves demarcate a compartment boundary in the mouse lung. Nat Commun . 2014;5:3923. doi: 10.1038/ncomms4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kumar PA, Hu Y, Yamamoto Y, Hoe NB, Wei TS, Mu D, et al. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell . 2011;147:525–538. doi: 10.1016/j.cell.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kanegai CM, Xi Y, Donne ML, Gotts JE, Driver IH, Amidzic G, et al. Persistent pathology in influenza-infected mouse lungs. Am J Respir Cell Mol Biol . 2016;55:613–615. doi: 10.1165/rcmb.2015-0387LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xi Y, Kim T, Brumwell AN, Driver IH, Wei Y, Tan V, et al. Local lung hypoxia determines epithelial fate decisions during alveolar regeneration. Nat Cell Biol . 2017;19:904–914. doi: 10.1038/ncb3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Walter MJ, Morton JD, Kajiwara N, Agapov E, Holtzman MJ. Viral induction of a chronic asthma phenotype and genetic segregation from the acute response. J Clin Invest . 2002;110:165–175. doi: 10.1172/JCI14345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kanner WA, Galgano MT, Atkins KA. Podoplanin expression in basal and myoepithelial cells: utility and potential pitfalls. Appl Immunohistochem Mol Morphol . 2010;18:226–230. doi: 10.1097/PAI.0b013e3181c65141. [DOI] [PubMed] [Google Scholar]
- 28. Little DR, Gerner-Mauro KN, Flodby P, Crandall ED, Borok Z, Akiyama H, et al. Transcriptional control of lung alveolar type 1 cell development and maintenance by NK homeobox 2-1. Proc Natl Acad Sci USA . 2019;116:20545–20555. doi: 10.1073/pnas.1906663116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Markwell MA, Paulson JC. Sendai virus utilizes specific sialyloligosaccharides as host cell receptor determinants. Proc Natl Acad Sci USA . 1980;77:5693–5697. doi: 10.1073/pnas.77.10.5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Filipovic N, Gibney BC, Kojic M, Nikolic D, Isailovic V, Ysasi A, et al. Mapping cyclic stretch in the postpneumonectomy murine lung. J Appl Physiol (1985) . 2013;115:1370–1378. doi: 10.1152/japplphysiol.00635.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu Z, Wu H, Jiang K, Wang Y, Zhang W, Chu Q, et al. MAPK-mediated YAP activation controls mechanical-tension-induced pulmonary alveolar regeneration. Cell Rep . 2016;16:1810–1819. doi: 10.1016/j.celrep.2016.07.020. [DOI] [PubMed] [Google Scholar]
- 32. Vila Ellis L, Cain MP, Hutchison V, Flodby P, Crandall ED, Borok Z, et al. Epithelial VEGFA specifies a distinct endothelial population in the mouse lung. Dev Cell . 2020;52:617–630.e6. doi: 10.1016/j.devcel.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bates SR, Tao JQ, Collins HL, Francone OL, Rothblat GH. Pulmonary abnormalities due to ABCA1 deficiency in mice. Am J Physiol Lung Cell Mol Physiol . 2005;289:L980–L989. doi: 10.1152/ajplung.00234.2005. [DOI] [PubMed] [Google Scholar]
- 34. Leslie CC, McCormick-Shannon K, Shannon JM, Garrick B, Damm D, Abraham JA, et al. Heparin-binding EGF-like growth factor is a mitogen for rat alveolar type II cells. Am J Respir Cell Mol Biol . 1997;16:379–387. doi: 10.1165/ajrcmb.16.4.9115748. [DOI] [PubMed] [Google Scholar]
- 35. Gahmberg CG. Leukocyte adhesion: CD11/CD18 integrins and intercellular adhesion molecules. Curr Opin Cell Biol . 1997;9:643–650. doi: 10.1016/s0955-0674(97)80117-2. [DOI] [PubMed] [Google Scholar]
- 36. Krásná E, Kolesár L, Slavcev A, Valhová S, Kronosová B, Jaresová M, et al. IL-18 receptor expression on epithelial cells is upregulated by TNF alpha. Inflammation . 2005;29:33–37. doi: 10.1007/s10753-006-8967-1. [DOI] [PubMed] [Google Scholar]
- 37. Yuan T, Volckaert T, Redente EF, Hopkins S, Klinkhammer K, Wasnick R, et al. FGF10-FGFR2B signaling generates basal cells and drives alveolar epithelial regeneration by bronchial epithelial stem cells after lung injury. Stem Cell Reports . 2019;12:1041–1055. doi: 10.1016/j.stemcr.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee JH, Bhang DH, Beede A, Huang TL, Stripp BR, Bloch KD, et al. Lung stem cell differentiation in mice directed by endothelial cells via a BMP4-NFATc1-thrombospondin-1 axis. Cell . 2014;156:440–455. doi: 10.1016/j.cell.2013.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science . 2018;359:1118–1123. doi: 10.1126/science.aam6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Strunz M, Simon LM, Ansari M, Kathiriya JJ, Angelidis I, Mayr CH, et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat Commun . 2020;11:3559. doi: 10.1038/s41467-020-17358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jiang P, Gil de Rubio R, Hrycaj SM, Gurczynski SJ, Riemondy KA, Moore BB, et al. Ineffectual type 2-to-type 1 alveolar epithelial cell differentiation in idiopathic pulmonary fibrosis: persistence of the KRT8hi transitional state. Am J Respir Crit Care Med . 2020;201:1443–1447. doi: 10.1164/rccm.201909-1726LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kobayashi Y, Tata A, Konkimalla A, Katsura H, Lee RF, Ou J, et al. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat Cell Biol . 2020;22:934–946. doi: 10.1038/s41556-020-0542-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Delorey TM, Ziegler CGK, Heimberg G, Normand R, Yang Y, Segerstolpe Å, et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature . 2021;595:107–113. doi: 10.1038/s41586-021-03570-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rane CK, Jackson SR, Pastore CF, Zhao G, Weiner AI, Patel NN, et al. Development of solitary chemosensory cells in the distal lung after severe influenza injury. Am J Physiol Lung Cell Mol Physiol . 2019;316:L1141–L1149. doi: 10.1152/ajplung.00032.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cassandras M, Wang C, Kathiriya J, Tsukui T, Matatia P, Matthay M, et al. Gli1+ mesenchymal stromal cells form a pathological niche to promote airway progenitor metaplasia in the fibrotic lung. Nat Cell Biol . 2020;22:1295–1306. doi: 10.1038/s41556-020-00591-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tsukui T, Sun KH, Wetter JB, Wilson-Kanamori JR, Hazelwood LA, Henderson NC, et al. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat Commun . 2020;11:1920. doi: 10.1038/s41467-020-15647-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moiseenko A, Vazquez-Armendariz AI, Kheirollahi V, Chu X, Tata A, Rivetti S, et al. Identification of a repair-supportive mesenchymal cell population during airway epithelial regeneration. Cell Rep . 2020;33:108549. doi: 10.1016/j.celrep.2020.108549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dahlgren MW, Jones SW, Cautivo KM, Dubinin A, Ortiz-Carpena JF, Farhat S, et al. Adventitial stromal cells define group 2 innate lymphoid cell tissue niches. Immunity . 2019;50:707–722.e6. doi: 10.1016/j.immuni.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell . 2005;121:823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 50. Kathiriya JJ, Brumwell AN, Jackson JR, Tang X, Chapman HA. Distinct airway epithelial stem cells hide among club cells but mobilize to promote alveolar regeneration. Cell Stem Cell . 2020;26:346–358.e4. doi: 10.1016/j.stem.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu K, Tang M, Liu Q, Han X, Jin H, Zhu H, et al. Bi-directional differentiation of single bronchioalveolar stem cells during lung repair. Cell Discov . 2020;6:1. doi: 10.1038/s41421-019-0132-8. [DOI] [PMC free article] [PubMed] [Google Scholar]