

Abstract
Asthma is the most prevalent chronic respiratory disease worldwide. There is currently no cure, and it remains an important cause of morbidity and mortality. Here we report that lung-specific loss of function of the transcription factor Miz1 (c-Myc-interacting zinc finger protein-1) upregulates the pro–T-helper cell type 1 cytokine IL-12. Upregulation of IL-12 in turn stimulates a Th1 response, thereby counteracting T-helper cell type 2 response and preventing the allergic response in mouse models of house dust mite- and OVA (ovalbumin)-induced asthma. Using transgenic mice expressing Cre under a cell-specific promoter, we demonstrate that Miz1 acts in lung epithelial cells and dendritic cells in asthma. Chromatin immunoprecipitation coupled with high-throughput DNA sequencing or quantitative PCR reveals the binding of Miz1 on the Il12 promoter indicating direct repression of IL-12 by Miz1. In addition, HDAC1 (histone deacetylase 1) is recruited to the Il12 promoter in a Miz1-depdenent manner, suggesting epigenetic repression of Il12 by Miz1. Furthermore, Miz1 is upregulated in the lungs of asthmatic mice. Our data together suggest that Miz1 is upregulated during asthma, which in turn promotes asthma pathogenesis by preventing Th1 skewing through the transcriptional repression of IL-12.
Keywords: asthma, gene regulation, Th1/Th2 immunity, Miz1, IL-12
Asthma is one of the most prevalent chronic diseases worldwide and affects around 10% of adults and an even greater proportion of children (1, 2). Although recent advances in diagnosis and management have improved the quality of life for many patients with asthma, there is no cure, and asthma remains an important cause of morbidity and mortality.
Asthma is characterized by reversible airflow obstruction, bronchial hyperresponsiveness, and airway inflammation that is predominately characterized as type 2 immune response often accompanied by airway and peripheral eosinophilia. Both lung epithelial cells and dendritic cells (DCs) express pattern recognition receptors (PRRs) and can be activated directly by allergens. It is generally accepted that the allergic immune response is initiated by interactions of allergens, including house dust mites (HDMs), pollen, and fungal spores, with barrier epithelial cells (Ecs) through PRRs. This results in the release of the epithelial cytokines, including TSLP (thymic stromal lymphopoietin), IL-33, IL-25, and GM-CSF (granulocyte–macrophage colony–stimulating factor). TSLP mediates migration of DCs to local draining lymph nodes and their maturation via upregulation of OX40L. These mature DCs activate I CD4 T cells in lymph nodes to an IL-4–competent state. In turn, IL-4–competent T-helper (Th) cells in lymph nodes migrate to B-cell zones and differentiate into T-follicular-helper (TFH) cells or egress from the lymph nodes and enter the airway, where they mature into T-helper type 2 (Th2) cells. TFH cells in B-cell zones mediate IgE class switching in B cells, and IgE binds to mast cells and basophils, enhancing their survival, activation, and/or degranulation. Th2 cells produce large quantities of the type 2 cytokines, including IL-4, IL-5, and IL-13, which drive a cascade of downstream events, including IgE-triggered hypersensitivity to aeroallergens, activation of airway epithelial cells, alternative macrophage activation, chemo attraction, and/or survival of effector cells (mast cells, eosinophils, and basophils), and remodeling of the epithelium and subepithelial matrix (epithelial mucus metaplasia, smooth muscle hypertrophy, increased subepithelial fibrosis, enhanced deposition of subepithelial matrix glycoproteins, etc.). Type 2 cytokines are also produced by non-Th2 cells; for example, IL-33 stimulates IL-4 release from basophils, and IL-33 and IL-25 promote IL-13 and IL-5 production from IL25R+ group 2 innate lymphoid cell (ILC2) (3–7).
Human asthma is associated with airway infiltration of Th2 cells. It is generally recognized that a dominant Th2 cell activation and an inadequate Th1 cell response are responsible for the development of allergic asthma. Th1 cells are induced by IL-12 and express the transcription factor T-bet and the hallmark cytokine of Type 1 immunity, IFN-γ, which repress IL-4 and suppress eosinophil lung infiltration and airway hyperresponsiveness. Reduced expression of T-bet is observed in T cells from airways of patients with asthma compared with that from patients without asthma (8). T-bet deficiency spontaneously skews toward a Th2 phenotype and induces airway changes consistent with human asthma in mice (8, 9). Administration of IFN-γ inhibited allergic inflammation (10–12), and administration of IL-12 abolished the allergic response in an IFN-γ–dependent manner in mouse models (13), whereas blocking IL-12 during the sensitization process worsened allergic airway inflammation (14). Decreased IFN-γ+CD4+ cells and reduced IL-12 and IFN-γ concentrations have been detected in patients with allergic asthma (15, 16). All these together suggest that study of the pathway of Th1 skewing is important in understanding Th2 development in asthma and might therefore identify targets for therapy.
Miz1, also known as Zbtb17 (c-Myc-interacting zinc finger protein-1), is a transcription factor that mediates both transcriptional activation and repression. It has an amino-terminal POZ (poxvirus and zinc-finger) domain that is required for its transcriptional activity, and 13 zinc fingers at its carboxyl terminus (17, 18). Miz1 plays a critical role in regulating proliferation, differentiation, cell-cycle progression, and apoptosis through the transcriptional regulation of its target genes (17, 19–21). We previously reported that loss of function of Miz1 results in augmented and prolonged lung inflammation induced by LPS (major component of the outer membrane of the gram-negative bacteria) or enhanced innate immune response to the gram-negative bacterium Pseudomonas aeruginosa (P. aeruginosa) (22). Furthermore, we recently reported that lung epithelial cell–specific loss of function of Miz1 causes spontaneous, age-related progressive chronic obstructive pulmonary disease (COPD)-like phenotype in mice (23). Whether Miz1 plays a role in another inflammatory lung disease, asthma, is not known. Here we report that loss of function of Miz1 skews toward a Th1 response by augmenting IL-12 expression and thereby suppresses allergic asthma, in sharp contrast to its proinflammatory role in response to LPS and Pseudomonas pneumonia as well as in COPD (22, 23).
Methods
Mice
Miz1(POZ)fl/fl mice (C57BL/6 background) have been described as we previously reported (22). SPC-Cre mice were kindly provided by Brigid L. Hogan (24). CD11c-Cre and LysM-Cre mice were from Jackson Laboratories. The animal care and animal experiments were performed in compliance with the institutional and U.S. National Institutes of Health regulations and were approved by the Northwestern University Animal Care and Use Committee and the University of Illinois at Chicago Animal Care Committee.
Mouse Model of HDM-induced Allergic Airway Inflammation
HDM extract (Greer Laboratories) was dissolved in saline and delivered by intranasal instillation, with the mice anesthetized with 5% isoflurane. Mice were treated with saline or HDM (50 μg in 50 μl saline) every other weekday (Monday, Wednesday, and Friday) for three 3. Twenty-four hours after the last HDM or saline exposure, mice were harvested.
Mouse Model of OVA-induced Allergic Airway Inflammation
Mice were sensitized with intraperitoneal OVA (ovalbumin) (10 μg; Sigma-Aldrich) plus alum (2 mg/ml) as adjuvant in 0.1 ml saline on Days 0 and 14, followed by challenges by exposure for 30 minutes to nebulized 1% OVA or saline alone delivered via an ultrasonic nebulizer on Days 21, 22, and 23. Twenty-four hours after the last exposure, mice were harvested.
Intratracheal Adenovirus Instillation
Ad-null, Ad-Cre, Ad-Miz1(WT), and Ad-Miz1(V89M) were from ViraQuest. Mice were infected intratracheally with 1 × 109 plaque-forming units of adenovirus per mouse.
Multiplex Analysis of Cytokines and IgE ELISA
The concentration of cytokines, including IL-4, -5, and -13, TNF, and IFN-γ in the BAL fluid were quantified by multiplex (IL-4, EPX010–20613–901; IL-5, EPX010–20610–901; IL-13, EPX010–20615–901; TNF, EPX010–20607–901; IFNγ, EPX010–20606–901; eBioscience). Concentrations of IgE in sera were determined by ELISA (88–50460; eBioscience).
Antibodies for Western Blot
Miz-1 Antibody (H-190, 1:500): sc-22837 was used until it was discontinued by the vendor, and then Miz-1 Antibody (B-10, 1:250): sc-136985 was used in replacement of Miz-1 Antibody (H-190): sc-22837. β-actin antibody (A5441, 1:20,000; Sigma-Aldrich).
Histological Analysis, Trichrome and Periodic Acid–Schiff Staining
Tissue samples were fixed in 4% paraformaldehyde for 24 hours and embedded in paraffin. Tissue sections were prepared at 5 μm and mounted on slides for staining with hematoxylin and eosin, Masson’s trichrome stains, or periodic acid–Schiff (25). The staining was performed by the Northwestern University Mouse Histology and Phenotyping Laboratory. The images were captured with Nikon ECLIPSE E800, or with the TissueGnostics Tissue/Cell High Throughput Imaging Analysis System and TissueFAXS software (TissueGnostics) at the Northwestern University Cell Imaging Facility.
Immunohistochemistry
Immunohistochemistry staining of eosinophils from mouse lungs was performed by the Northwestern University Mouse Histology and Phenotyping Laboratory. Briefly, mouse lungs were fixed in 4% paraformaldehyde for 24 hours and embedded in paraffin. Deparaffinized slides were incubated with eosinophil major basic protein antibody (Santa Cruz sc-33938; 1:1500) overnight at 4°, followed by detection with the biotin-streptavidin peroxidase system (Vector Lab). Nucleus was visualized concomitantly with DAPI using Aqua Mount.
Quantitative PCR, Chromatin Immunoprecipitation Assay and Sequencing
Quantitative PCR (qPCR) was performed using iQTM SYBR Green Supermix (BIO-RAD) on a CFX ConnectTM Real-Time PCR Dection System (BIO-RAD). mRNA expression of a specific gene was normalized to HPRT (hypoxanthine-guanine phosphoribosyltransferse). Primer sequences were listed in Table E1. For qRT-PCR, total RNA was extracted from lung tissues or isolated cells using NucleoSpin RNA kit (Macherey-Nagel). cDNA was synthesized using M-MuLV Reverse Transcriptase (NEB) according to the manufacturer’s instructions. Chromatin immunoprecipitation (ChIP) assays were performed as we previously reported (22). The following antibodies were used for immunoprecipitation: anti-Miz1 (H-190 X; Santa Cruz Biotechnology), anti-HDAC1 (05–100; Millipore), and RelA/p65 (sc-372X; Santa Cruz Biotechnology). Primer sequences for ChIP-qPCR were listed in Table E1. ChIP sequencing (ChIP-seq) was performed and analyzed as we previously reported (26). Briefly, DNA samples were quantitated with Qubit, and libraries were prepared using Illumina TruSEQ. Libraries were then sequenced on an Illumina NovaSEQ6000 (100 bp, paired-end) at University of Chicago Genomics Facility. Raw sequencing data were demultiplexed using Illumina bcl2fastq, and sequencing raw reads were aligned to the mouse genome mm10 using BWA MEM. PCR duplicates were removed using Picard MarkDuplicates so that downstream results were not influenced by PCR duplication artifacts. ChIP peaks were called relative to inputs using Macs2. Normalized bedgraph tracks were produced with the –SPMR flag and converted to bigWig tracks using UCSC tool bedGraphToBigWig.
Airway Hyperresponsiveness to Metacholine Administration
Mice were tracheotomized and mechanically ventilated on a computer-controlled piston ventilator (FlexiVent; SCIREQ) with a tidal volume of 10 ml/kg at 150 breaths per minute against 2–3 cm H2O positive end-expiratory pressure. A standard volume history was obtained with two total lung capacity operations. Two baseline assessments of respiratory system input impedance were then obtained. This was followed by an inhalation of either aerosolized methacholine or control saline for 40 seconds by directing the inspiratory flow from the ventilator through the nebulizer chamber. Each dose of methacholine aerosol was administrated in a dose–response manner (10, 25, 50, and 100 mg/ml). Respiratory system input impedance was then measured every 10 seconds for 3 minutes.
Fluorescence-activated Cell Sorting and Isolation of Bone Marrow–derived DCs
The lungs were digested with Dispase (Corning) and 0.1 mg/mL DNase I, followed by red blood cells removal with 1x BD Pharm Lyse solution (BD Biosciences). Cells were then washed in MACS buffer (Miltenyi Biotech) and the number of cells was counted with a Countess automated cell counter (Invitrogen). For isolation of AT2 cells and AMs, single cell suspensions were first incubated with 0.5 μg Fc Block (BD Biosciences) for 10 minutes at 4°C, then stained with FITC conjugated anti-mouse CD45 (eBioscience), APC conjugated anti-mouse EpCAM (eBioscience), PE conjugated anti-mouse CD31 (eBioscience), PE-CF594 conjugated anti-mouse SiglecF (BD Biosciences) and efluor450 conjugated anti-mouse CD11b (eBioscience). Cell sorting were performed with a BD FACSAria SORP 5-Laser instrument (BD Immunocytometry Systems) equipped with 355, 405, 488, 561 and 640 nm excitation lasers at the Northwestern University Flow Cytometry Core Facility. Data collection and sorting were implemented with BD FACS Diva software (BD Biosciences) and data analyses were performed with FlowJo software (Tree Star, Ashland, OR). Fluorescence minus one (FMO) controls were included for gating analyses to allocate positive staining cells from negative staining populations. Compensation was performed using single color controls prepared from BD Comp Beads (BD Biosciences). Compensation matrix was calculated by FlowJo software (Tree Star). Biexponential transformation was calibrated manually if needed. For isolation of AT2 together with other myeloid cells, after Fc blocking, cells were incubated with Biotin conjugated anti-mouse CD45 antibody for 10 minutes at room temperature. Cells were then separated using MagniSort Streptavidin Positive selection beads (eBioscience) according to the manufacturer’s instructions. From the CD45 negative fraction, AT2 cells were further purified by flow sorting using FITC conjugated anti-mouse CD45 (eBioscience), APC conjugated anti-mouse EpCAM (eBioscience) and PE conjugated anti-mouse CD31 (eBioscience). From the CD45 positive fraction, the myeloid cell populations were isolated by flow sorting using FITC conjugated anti-mouse CD45 (eBioscience), PerCPCy5.5 conjugated anti-mouse MHC II (BioLegend), eFluor450 conjugated anti-mouse Ly6C (eBioscience), APC conjugated anti-mouse CD24 (eBioscience), Alexa700 conjugated anti-mouse Ly6G (BD Biosciences), APCCy7 conjugated anti-mouse CD11b (BioLegend), PE conjugated anti-mouse CD64 (BioLegend), PE-CF594 conjugated anti-mouse SiglecF (BD Biosciences) and PECy7 conjugated anti-mouse CD11c (eBioscience)(27). For isolation of bone marrow–derived DCs (BMDCs), bone marrow (BM) cells were isolated and cultured with GM-CSF.
Statistical Analysis
Data were analyzed using an unpaired Student's t test, with the assumption of normal distribution with equal variance. Sample sizes were selected on the basis of preliminary results to ensure an adequate power.
Results
Lung-Specific Loss of Function of Miz1 Results in Reduced HDM-induced Airway Hyper Reactivity in Mice
To understand the role of Miz1 in a chronic inflammatory lung disease such as asthma, we used a mouse model of asthma induced by the clinically relevant allergen, HDM (28). The POZ domain of Miz1, which is essential for its transcriptional activity (17), was deleted in the lungs by intratracheal administration of an Ad/Cre (adenovirus encoding Cre recombinase) into Miz1(POZ)fl/fl mice, in which the coding exons of the POZ domain of Miz1 were flanked by loxP sites (22). An empty vector (Ad/null) was used as a control. We have reported lung-specific deletion of the Miz1 POZ domain with this approach (22). Thirty days after virus administration, mice were treated intranasally with saline or HDM every other weekday for three weeks (Figure E1 in the data supplement). Although loss of function of Miz1 augmented LPS- or P. aeruginosa–induced lung inflammation (22), it reduced the production of the Th2 cytokines, including IL-4, IL-5, and IL-13, in the BAL fluid in the HDM-induced asthma model (compare Ad/Cre-treated mice [Miz1ΔPOZ] to Ad/null-treated mice [control]) (Figure 1A). Consistently, the mRNA concentrations of these Th2 cytokines were decreased in whole-lung homogenates of Miz1ΔPOZ mice compared with Ad-Null control mice (Figure 1B). Accordingly, loss of the Miz1 POZ domain attenuated airway hyperresponsiveness in the HDM-challenged mice (Figure 1C). Loss of function of Miz1 also inhibited the systemic allergic response, as shown by reduced serum total IgE concentrations in HDM-treated mice (Figure 1D).
Figure 1.
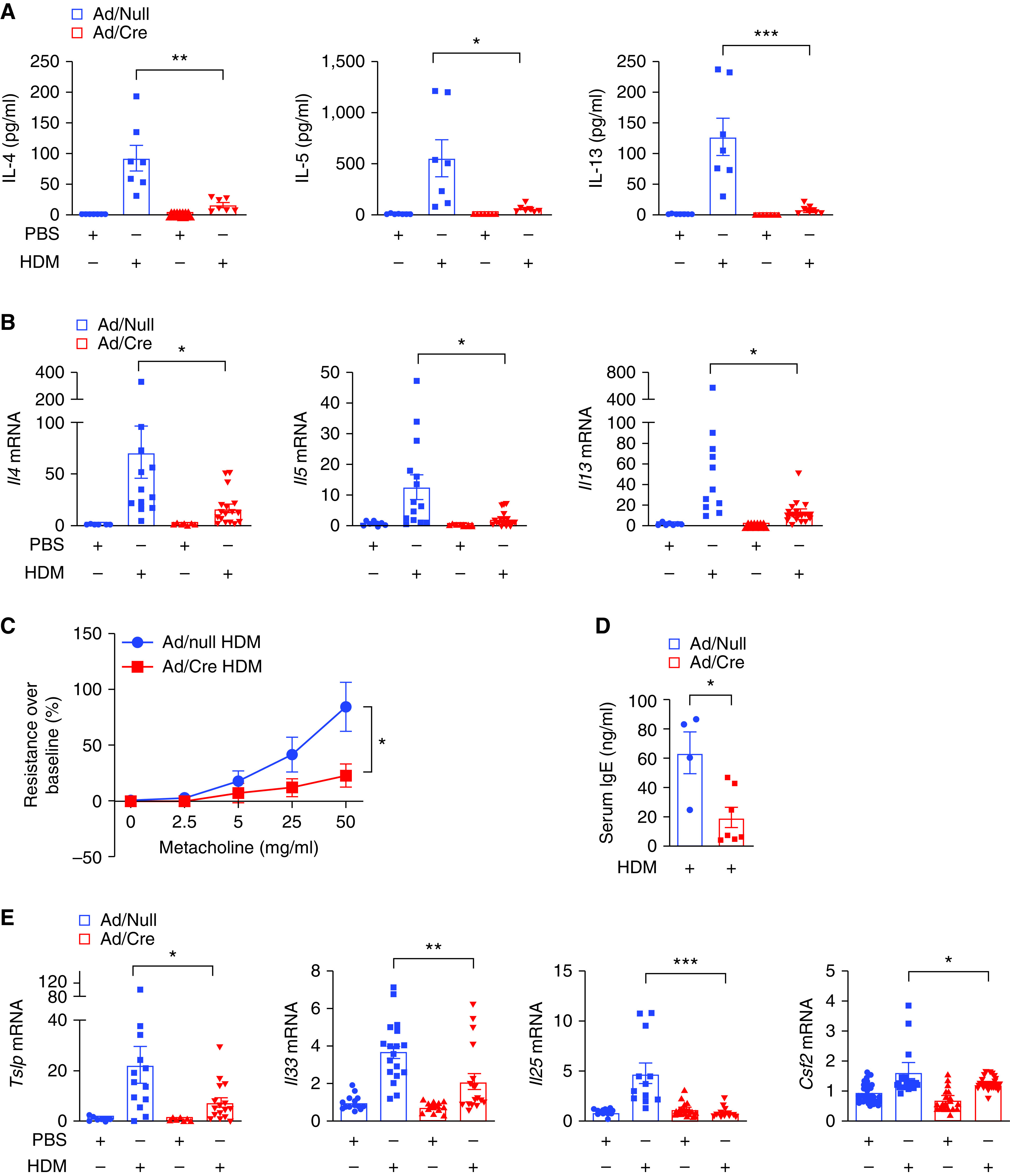
Lung-specific loss of function of Miz1 (c-Myc-interacting zinc finger protein-1) inhibits house dust mite (HDM)-induced allergic asthma in mice. Miz1(POZ)fl/fl mice were intratracheally instilled with Ad/Null or Ad/Cre. Thirty days later, allergic asthma was induced in mice with HDM. (A–E) Production of Th2 cytokines in the BAL fluid (A), or mRNA concentrations of cytokines in whole-lung homogenates as indicated (B and E), or airway hyperresponsiveness to aerosolized metacholine in increasing concentrations (C), or serum total IgE concentrations (D) from Ad/Null- or Ad/Cre-pretreated, HDM-challenged Miz1(POZ)fl/fl mice. n = 5–7 for A, C, and D. n = 5–7 with technical replicates for B and E. Values represent the mean ± SEM. Unpaired Student’s t test was used. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Airway epithelial–derived cytokines, including IL-25, IL-33, TSLP, and GM-CSF, which are rapidly released in exposure to allergens, activate and potentiate innate and adaptive immunity in allergic responses (29). On the other hand, Th2 cytokines can act on epithelial cells as a feedback mechanism to induce the production of TSLP, GM-CSF, IL-25, and IL-33 to perpetuate dendritic cell–driven Th2 cell development (30–33). Consistently, the mRNA concentrations of epithelial-derived cytokines, including Il33, Csf2 (encoding GM-CSF), Il25, and Tslp, were also decreased in HDM-treated Miz1ΔPOZ mice compared with HDM-treated control mice (Figure 1E).
Lung-Specific Loss of Function of Miz1 Also Results in Suppression of OVA-induced Allergic Asthma in Mice
To further validate our observations in HDM-induced asthma model, we used another mouse model of asthma induced by OVA (34, 35). Mice were sensitized with OVA plus aluminum hydroxide (36) and challenged as indicated (Figure E2). Similar to the HDM model, loss of the Miz1 POZ domain also reduced the Th2 response in the OVA model. The observed effects included decreased production and mRNA expression of the Th2 cytokines, including IL-4, IL-5, and IL-13 (Figures 2A and 2B), attenuated airway hyperresponsiveness (Figure 2C), and decreased expression of epithelial-derived cytokines, including Tslp, Il33, Il25, and Csf2 (Figure 2D). Taken together, these data suggest that lung-specific loss of function of Miz1 results in reduced Th2 response in HDM- or OVA-induced mouse model of asthma.
Figure 2.
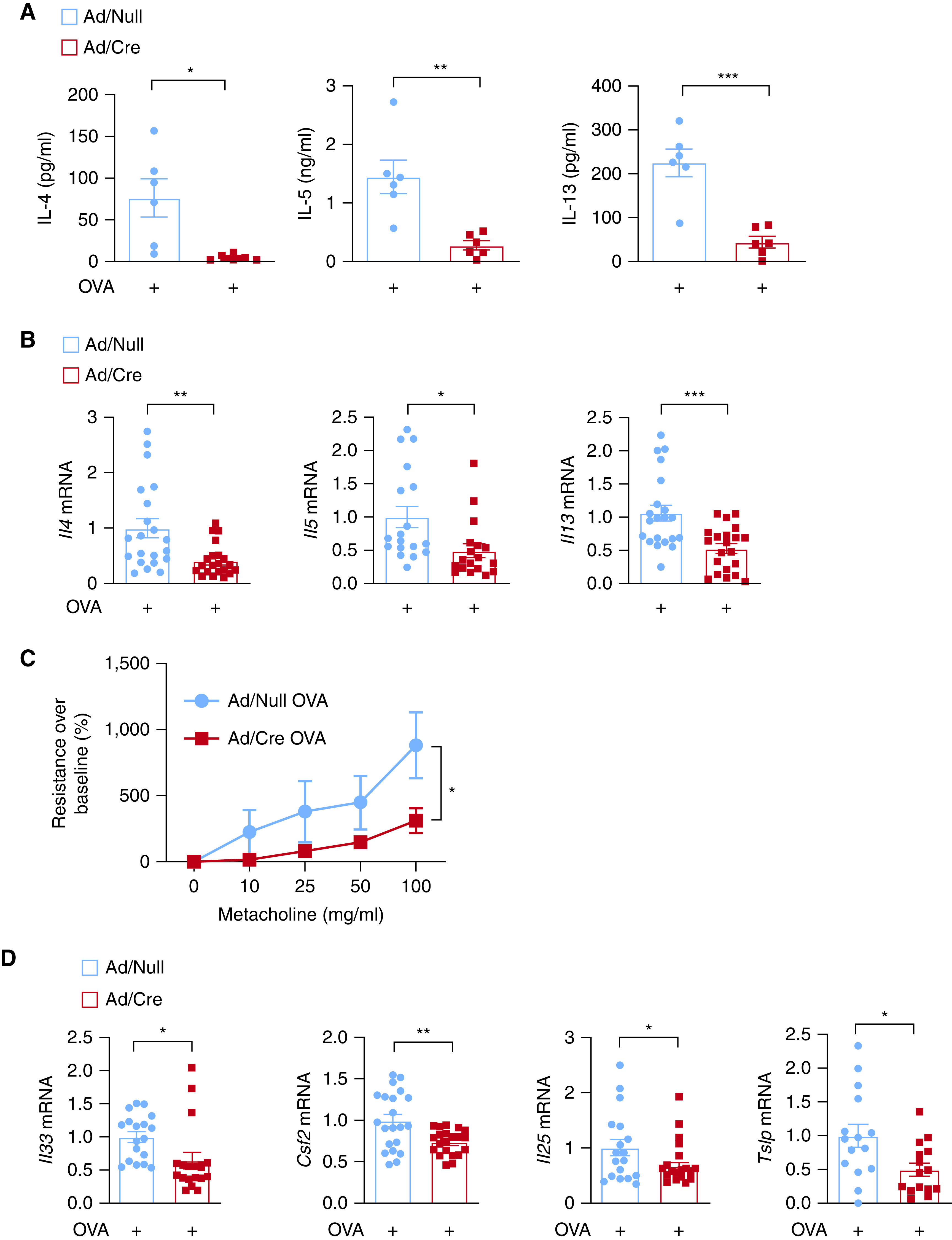
Lung-specific loss of function of Miz1 suppresses OVA (ovalbumin)-induced allergic asthma in mice. Miz1(POZ)fl/fl mice were intratracheally instilled with Ad/Null or Ad/Cre. Thirty days later, allergic asthma was induced in mice with OVA/alum (40). (A–D) Production of T-helper cell type 2 (Th2) cytokines in the BAL fluid (A), or mRNA concentrations of cytokines in whole-lung homogenates as indicated (B and D), or airway hyperresponsiveness to aerosolized metacholine in increasing concentrations (C) from Ad/Null- or Ad/Cre-pretreated, OVA-challenged Miz1(POZ)fl/fl mice. n = 5–7 for (A and C). n = 5–7 with technical replicates for (B and D). Values represent the mean ± SEM. Unpaired Student's t test was used. *P < 0.05, **P < 0.01, and ***P < 0.001.
Lung Epithelial Cell-Specific Loss of Function of Miz1 Results in Reduced HDM-induced Airway Hyperreactivity in Mice
To determine the cell types in the mouse lung in which Miz1 promotes the allergic response, we crossed transgenic mice expressing Cre driven by the human SPC promoter (SPC-Cre mice) (37) with Miz1(POZ)fl/fl mice as we recently reported (23). As SPC is expressed early in lung epithelial progenitors, developmental expression of SPC-Cre results in Cre-mediated recombination in the airway and alveolar epithelium that persists over the lifespan of nonconditional SPC-Cre mice (37). Note, although SPC is an AT2 (alveolar type 2) cell marker, it is expressed in endodermal lung progenitors, which generate all distal conducting airway and alveolar cell types. In addition, SPC-driven recombination is also observed within epithelial cells of more proximal conducting airway epithelium because progeny of early lung progenitor cells contribute to these structures (38). We recently reported that the Miz1(POZ) was partially deleted (∼50%) in primary AT2 cells flow-sorted from heterozygous SPC-Cre+/Miz1(POZ)+/f littermates, and there was no deletion of Miz1(POZ) in alveolar macrophages (AMs) from heterozygous mutant mice (23), indicating the specificity of the Miz1(POZ) deletion in lung epithelial cells. We also reported that although homozygous SPC-Cre+/Miz1(POZ)f/f mice exhibited age-related spontaneous COPD-like phenotype starting from a young age (<4 mo), heterozygous SPC-Cre+/Miz1(POZ)+/f mice appeared histologically normal at a relatively young age (<6 mo) compared with control Miz1(POZ)fl/fl mice (also see Figure E3A). Therefore, for all the experiments here, we used heterozygous SPC-Cre+/Miz1(POZ)+/f mice at a much younger age (8–12 wk). When challenged with HDM, SPC-Cre+/Miz1(POZ)+/f mice exhibited attenuated airway inflammation (Figure 3A), decreased eosinophil infiltration and mucus secretion in the airways (Figures 3B and 3C), reduced production of IL-5 in the BAL fluid (Figure 3D), and reduced Tslp mRNA concentrations from whole-lung homogenates (Figure 3E). Together, these data suggest that Miz1 in lung epithelial cells contributes to HDM-induced allergic response.
Figure 3.
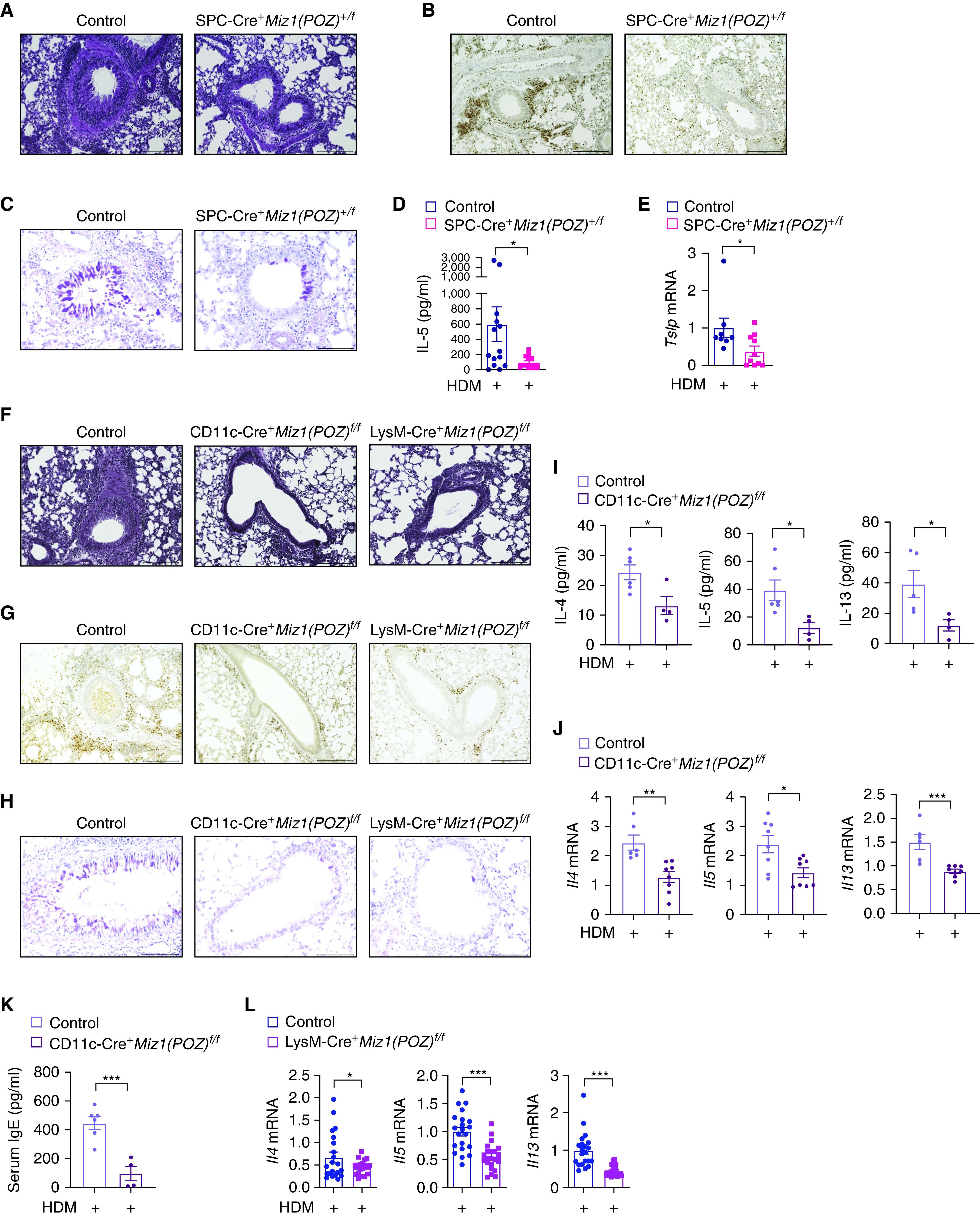
Miz1 in lung epithelial cells or dendritic/myeloid cells contributes to HDM-induced allergic asthma. (A–C) Lung histology by hematoxylin and eosin staining (A), eosinophil major basic protein (EMBP) staining of eosinophils (B), or Periodic Acid–Schiff (PAS) staining (C) in the airways of HDM-treated control Miz1(POZ)fl/fl or heterozygous SPC-Cre+/Miz1(POZ)+/f mice. Eosinophils are stained brown/dark brown by EMBP staining. The mucin produced by goblet cells is stained a purple-magenta color by PAS staining. The nuclei are stained blue. (D and E) Production of IL-5 in the BAL fluid (D) and mRNA concentrations of Tslp in whole-lung homogenates (E) as indicated in HDM-treated control Miz1(POZ)fl/fl or heterozygous SPC-Cre+/Miz1(POZ)+/f mice. n = 5 with technical replicates. (F–H) Lung histology of the airways by hematoxylin and eosin staining (F), EMBP staining of eosinophils (G), or PAS staining (H) in the airways of HDM-treated control Miz1(POZ)fl/fl or CD11c-Cre/Miz1(POZ)fl/fl or LysM-Cre/Miz1(POZ)fl/fl mice. Eosinophils are stained brown/dark brown by EMBP staining. The mucin produced by goblet cells is stained a purple-magenta color by PAS staining. The nuclei are stained blue. (I–K) Production of the Th2 cytokines in the BAL fluid (I), mRNA concentrations of the Th2 cytokines in whole-lung homogenates (J), or serum total IgE concentration (K) from HDM-treated control Miz1(POZ)fl/fl (n = 3) or CD11c-Cre/Miz1(POZ)fl/fl mice (n = 4). (L) mRNA concentrations of the Th2 cytokines from whole-lung homogenates of HDM-treated control Miz1(POZ)fl/fl or LysM-Cre/Miz1(POZ)fl/fl mice (n = 7 with technical replicates). Values represent the mean ± SEM. Unpaired Student's t test was used. *P < 0.05 and ***P < 0.001.
Mice with Dendritic Cell- or Myeloid Cell-Specific Loss of Function of Miz1 Have Reduced HDM-induced Airway Hyperreactivity
DCs play an important role in determining Th1 and Th2 differentiation and also in maintaining effector Th2 response in asthma. We sought to determine whether Miz1 in DCs is important for asthma promotion. We crossed Miz1(POZ)fl/fl mice with transgenic CD11c-Cre mice as we recently reported (23). CD11c-Cre induces efficient Cre-mediated recombination in DCs and AMs (39). We have reported a significant deletion of the Miz1 POZ domain in CD11C+ DCs and AMs from the lungs of CD11c-Cre/Miz1(POZ)fl/fl mice, and that at a younger age (<6 mo), CD11c-Cre/Miz1(POZ)fl/fl mice were indistinguishable from the control Miz1(POZ)fl/fl mice (also see Figure E3B) (23). Reduced allergic response was observed in HDM-treated CD11c-Cre/Miz1(POZ)fl/fl mice, as shown by attenuated airway inflammation and reduced eosinophil infiltration and mucus production in the airways (Figures 3F–3H), decreased production and mRNA expression of the Th2 cytokines (Figures 3I and 3J), and decreased serum total IgE concentrations (Figure 3K). These data suggest that Miz1 in DCs and/or AMs contributes to allergic asthma. We also generated mice with myeloid cell–specific deletion of the Miz1 POZ domain [LysM-Cre/Miz1(POZ)fl/fl]. There was a significant deletion of the Miz1 POZ domain in DCs, AMs, neutrophils, monocytes, and AT2 cells from the lungs of LysM-Cre/Miz1(POZ)fl/fl mice (Figure E3C). At a younger age (<6 mo), LysM-Cre/Miz1(POZ)fl/fl mice were indistinguishable from the control Miz1(POZ)fl/fl mice, as analyzed by lung histology (Figure E3B). Similar to CD11c-Cre/Miz1(POZ)fl/fl mice, LysM-Cre/Miz1(POZ)fl/fl mice also showed a reduced allergic response to HDM, including attenuated airway inflammation, and reduced eosinophil infiltration and mucus production in the airways (Figures 3F–3H), as well as decreased mRNA expression of the Th2 cytokines (Figure 3L).
Miz1 in Dendritic or Myeloid Cells Also Contributes to OVA-induced Asthma
Similar to the HDM-induced asthma model, CD11c-Cre/Miz1(POZ)fl/fl or LysM-Cre/Miz1(POZ)fl/fl mice also had reduced OVA-induced allergic asthma, as shown by decreased Th2 cytokine production or mRNA expression (Figures 4A and 4B), and attenuated airway inflammation (Figure 4C). These data suggest that Miz1 in dendritic or myeloid cells contributes to the pathogenesis of asthma.
Figure 4.

Miz1 in dendritic or myeloid cells contributes to OVA-induced asthma. Control Miz1(POZ)fl/fl, CD11c-Cre/Miz1(POZ)fl/fl, or LysM-Cre/Miz1(POZ)fl/fl mice were subjected to OVA/alum-induced allergic asthma. (A) Production of the Th2 cytokines in the BAL fluid of OVA-treated mice as indicated. n = 5 with technical replicates. (B) mRNA concentrations of the Th2 cytokines in whole-lung homogenates of OVA-treated mice as indicated. n = 5 with technical replicates. (C) Lung histology by hematoxylin and eosin staining in the airways of OVA-treated mice as indicated. Values represent the mean ± SEM. Unpaired Student's t test was used. *P < 0.05, **P < 0.01, and ***P < 0.001.
Loss of Function of Miz1 Results in Th1 Skewing
Because Th1 cells antagonize Th2 cell functions (41, 42), the decreased Th2 response by loss of the Miz1 POZ domain we observed in the mouse asthma models may be owing to an increase in the Th1 response. Indeed, mRNA concentrations (Figure 5A) and secretion (Figure 5B) of the Th1 cytokines, including IFN-γ and TNF, were increased from the lungs of HDM-treated Miz1ΔPOZ mice compared with HDM-treated control mice. Consistently, the expression of Rantes (regulated upon activation, normal T cell expressed and presumably secreted), which is induced by the Th1 cytokines, was increased by loss of the Miz1 POZ domain in the HDM model (Figure 5C). IL-12, which is predominantly produced by activated antigen-presenting cells such as DCs and also by lung epithelial cells, is pivotal for Th1 differentiation and is among the most critical factors skewing the immune response toward a Th1 cytokine profile (high IFN-γ and low IL-4) (26, 43). We found that the mRNA concentrations of Il12 were augmented in the lung by loss of the Miz1 POZ domain in response to HDM (Figure 5D). Similar results were obtained in the OVA model—increased Ifng, Tnf, and Il12 expression by loss of function of Miz1 (Figure 5E). Similar to Miz1ΔPOZ mice, heterozygous SPC-Cre+/Miz1(POZ)+/f, CD11c-Cre/Miz1(POZ)fl/fl, or LysM-Cre/Miz1(POZ)fl/fl mice also showed increased expression of IL-12 or IFN-γ or TNF in the lung in exposure to HDM or OVA (Figures 5F–5J). These data collectively suggest that Miz1 suppresses the Th1 response, thereby promoting the Th2 response in the asthma models.
Figure 5.
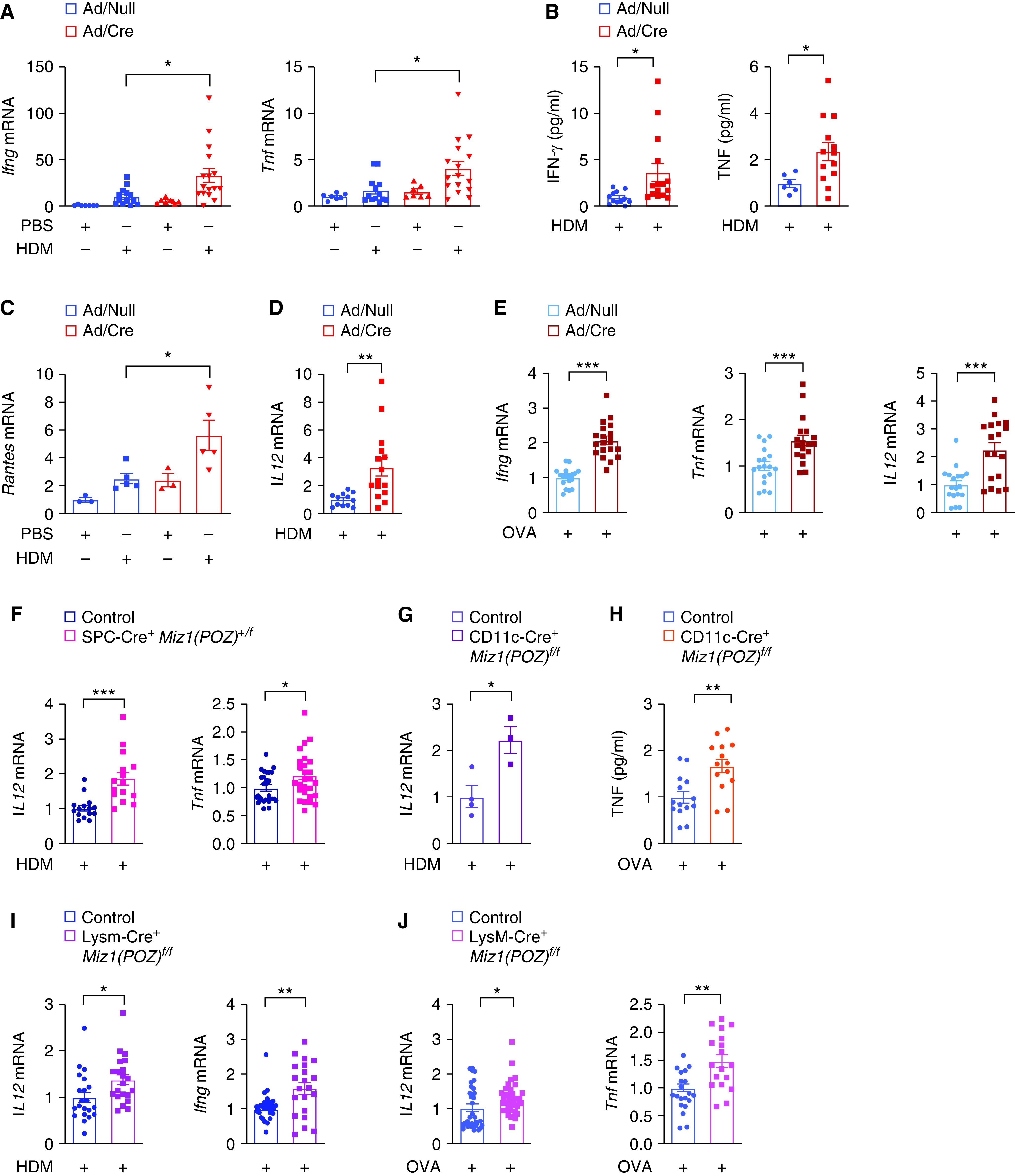
Loss of function of Miz1 results in Th1 skewing. Ad/null or Ad/Cre-preinfected Miz1(POZ)fl/fl mice, or control Miz1(POZ)fl/fl, heterozygous SPC-Cre+/Miz1(POZ)+/f, CD11c-Cre/Miz1(POZ)fl/fl, or LysM-Cre/Miz1(POZ)fl/fl mice were subjected to HDM- or OVA-induced allergic asthma as indicated. (A, C–G, I, and J) mRNA concentrations of Ifng, Tnf, Rantes, or Il12 in whole-lung homogenates of HDM- or OVA-treated mice as indicated. (B and H) Production of IFNγ and TNF in the BAL fluid from HDM- or OVA-treated mouse as indicated. n = 5–7 except in PBS groups in C (n = 3) and in G (3–4). Values represent the mean ± SEM. Unpaired Student's t test was used. *P < 0.05, **P < 0.01, and ***P < 0.001.
Miz1 Binds to the Promoter of Il12 and Represses Its Transcription
Our data showed that loss of function of Miz1 resulted in a decreased Th2 response and increased expression of the Th1-skewing cytokine IL-12 in HDM- and OVA-induced asthma models. We sought to determine whether Miz1 directly represses IL-12 expression. We generated BMDCs by isolating BM cells from CD11c-Cre/Miz1(POZ)fl/fl or control Miz1(POZ)fl/fl mice and treated them with GM-CSF. Expression of Il12 was augmented by loss of the Miz1 POZ domain in INF-γ– or OVA-treated, LPS-primed BMDCs (Figures 6A and 6B). Note, OVA- or HDM-treated, LPS-primed BMDCs are commonly used for in vitro experimental model to study asthma in DCs, in which LPS is commonly used to induce DC “maturation” (44). These data suggest that Miz1 inhibits Il12 expression in DCs.
Figure 6.

Miz1 binds to the promoter of il12 and represses its transcription. (A and B) mRNA concentrations of Il12 in IFNγ- (A) or OVA-treated (B) LPS-primed bone marrow–derived dendritic cells (BMDCs) isolated from control Miz1(POZ)fl/fl or CD11c-Cre/Miz1(POZ)fl/fl mice. LPS, 0.1ng/ml; IFNγ, 10ng/ml; OVA, 10 μg/ml. (C) mRNA concentrations of Il12 in whole-lung homogenates from control Miz1(POZ)fl/fl or heterozygous SPC-Cre+/Miz1(POZ)+/f mice. n = 4–6. (D) Chromatin immunoprecipitation (ChIP) analyses of Miz1 binding on the Il12 promoter in HDM-treated MLE-12/Miz1(WT) or MLE-12/Miz1(ΔPOZ) cells. (E–G) ChIP-seq traces of Miz1, HDAC1, or RelA proteins on the promoter of Il12a in MLE-12/Miz1(WT) or MLE-12/Miz1(ΔPOZ) cells. Input is shown as control. Top line indicates the Il12a gene (exons were shown with blue boxes and introns were shown with line). Black arrow indicates transcription start site (TSS). Red columns indicate peaks callings, which are enlarged for viewing in E. Values represent the mean ± SEM. Unpaired Student's t test was used. *P < 0.05, **P < 0.01, and ***P < 0.001.
IL-12 is also produced by lung epithelial cells (45). The lungs from SPC-Cre+/Miz1(POZ)f/f mice showed increased Il12 expression compared with the control Miz1(POZ)f/f mice (Figure 6C). To determine whether Miz1 directly represses Il12 transcription by binding to its promoter, we used stable murine type II–like lung epithelial cells (MLE-12–Miz1[ΔPOZ] or MLE12–Miz1[WT]), in which endogenous Miz1 was stably knocked down by specific short hairpin RNA (shRNA) and simultaneously the exogenous Miz1(ΔPOZ) mutant or wild-type Miz1, which contains silent mutations making it resistant to shRNA against endogenous Miz1, was stably expressed in amounts similar to the endogenous protein, as we previously reported (22). Chromatin immunoprecipitation assays showed that Miz1 was recruited to the Il12 promoter in HDM-treated MLE12–Miz1(WT) cells, while the recruitment of the Miz1(ΔPOZ) mutant was reduced (Figure 6D). ChIP sequencing (ChIP-seq) also revealed Miz1 binding on the Il12 promoter, which was almost diminished by the Miz1(ΔPOZ) mutation (Figure 6E). These data suggest that Miz1 binds to the Il12 promoter and represses its transcription. We previously reported that Miz1 epigenetically represses gene transcription through histone deacetylation via recruiting HDAC1 (histone deacetylase 1) (22). ChIP-seq showed recruitment of HDAC1 to the Il12 promoter in Miz1 POZ domain–dependent manner (Figure 6F), suggesting that Miz1 epigenetically represses Il12 through histone deacetylation. We previously also reported that Miz1 represses NF-κB–dependent gene through histone deacetylation-mediated exclusion of NF-κB binding (22). IL-12 is an NF-κB target gene (46, 47). ChIP-seq showed enhanced recruitment of RelA (also called p65), the predominant transactivating submit of the NF-κB family, on the Il12 promoter by loss of function of Miz1 (Figure 6G). These data suggest that Miz1 recruits HDAC1 to the Il12 promoter, resulting in histone deacetylation of the promoter and thereby excluding NF-κB binding. Altogether, our data suggest that Miz1 represses Il12 transcription in monocyte derived DCs as well as in lung epithelial cells, thereby promoting the Th2 response in asthma.
Miz1 Is Upregulated in Asthmatic Mouse Lungs
We sought to determine whether Miz1 expression is subjected to regulation during asthma. Miz1 protein concentrations were increased in whole-lung homogenates from HDM- (Figure 7A) or OVA-treated mice (Figure 7B). Miz1 mRNA expression was also upregulated in lung epithelial cells flow-sorted from HDM-treated mice (Figure 7C). These data suggest that Miz1 is upregulated during asthma, which in turn promotes asthma pathogenesis.
Figure 7.

Miz1 is upregulated in asthmatic mice. (A and B) Miz1 protein expression in whole-lung homogenates from PBS- or HDM- (A) or OVA-treated mice (B). (C) Miz1 mRNA expression in AT2 cells flow sorted from PBS- or HDM-treated mice. n = 3. Unpaired Student's t test was used. *P < 0.05.
Discussion
It is generally considered that type 1 immunity contributes to the eradication of intracellular pathogens, whereas type 2 immunity is predominantly responsible for the pathogenesis of allergic diseases such as asthma and is also important for protection against extracellular organisms such as parasites. Overactivation of either pathway may lead to detrimental inflammation, and therefore, the type 1/type 2 balance is important to maintain immune homeostasis. We previously reported that Miz1 suppresses lung inflammation induced by LPS (a major component of the outer membrane of the gram-negative bacteria) or the immune response to gram-negative bacterium P. aeruginosa. Moreover, we recently reported that lung epithelial cell–specific loss of function of Miz1 causes spontaneous, age-related progressive COPD-like phenotype in mice (23). Here we report that Miz1 inhibits type 1 immunity, resulting in promotion of type 2 immune response in allergic asthma. Our observations are in line with the prevailing paradigm regarding the Th1/Th2 counterregulation.
In this report, we provide evidence that loss of function of Miz1 skews toward a Th1 response by upregulating IL-12, thereby counteracting Th2 response and protecting against asthma. Our findings are consistent with a growing body of evidence from patients with asthma, which supports the inverse relationship between the concentrations of IL-12 and the development of asthma (48–52). Causal studies in animal models of asthma show that the administration of IL-12 or IL-12–producing agents prevent allergen-induced Th2 response and airway eosinophilia (53–60), whereas blocking IL-12 during the sensitization process worsened allergic airway inflammation (14). Intriguingly, a recent genome-wide association study identified a SNP (rs4410871) in the Miz1 interacting protein Myc, whose transcriptional repression is mediated by Miz1 (61), as being associated with asthma (62). Most importantly, the same SNP was reported to be associated with protection against multiple sclerosis, with the risk allele (T) in asthma being the protective allele for multiple sclerosis, a Th1 disease (63), implying a crucial role for this SNP in the Th1/Th2 balance.
Our data show that Miz1 acts to shift the balance of cytokines toward a Th2 phenotype in multiple cell populations in the lung including epithelial, dendritic, and myeloid cells, indicating a general mechanism of Miz1 action in different cell types. This is consistent with our previous report that Miz1 in both epithelial cells and hematopoietic-derived cells is important to suppress LPS-induced lung inflammation. The lung epithelium was initially viewed to function only as a physical barrier in asthma; however, it is now considered to play a central role in Th2 sensitization by inducing the activation and Th2 cell–polarizing capacity of DCs through the production of cytokines, including TSLP, GM-CSF, IL-33, and IL-25 (30). Our data that Miz1 in lung epithelial cells is as important as in DCs in asthma are in line with this notion. The Th1/Th2 paradigm has been extended to several of other T-cell subsets, including Th17 cells, Th9 cells, and Treg cells, as well as ILC2 cells, all of which may contribute to the development of asthma in subsets of patients. Future studies will investigate whether Miz1 is engaged in regulation of these subsets of cells.
In summary, our studies demonstrate that Miz1 promotes Th2-mediated allergic asthma by counteracting Th1 response. Additionally, Miz1 is upregulated in asthmatic mice. These studies directly implicate MIZ1 in asthma pathogenesis.
Acknowledgments
Acknowledgment
The authors thank Zhengdeng Lei, Pinal Kanabar, and Mark Maienschein-Cline (Research Informatics Core, Research Resources Center, University of Illinois at Chicago) for the ChIP-seq analyses. The authors thank Lin Li and the Northwestern University Mouse Histology and Phenotyping Laboratory for the assistance with immunohistochemistry and histopathology. Imaging was performed at the Northwestern University Cell Imaging Facility, supported by NCI CCSG P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center. All data needed to evaluate the conclusions in the paper are present in the paper and/or the data supplement.
Footnotes
Supported by the National Heart, Lung, and Blood Institute grants HL114763 and HL141459 (to J.L.) and American Asthma Foundation Scholar Award (13-0114 to J.L.). G.R.S.B. is supported by U.S. National Institutes of Health grants ES013995, HL071643, AG049665, and AI135964; Veterans Administration grant BX000201; and U.S. Department of Defense grant PR141319.
Author Contributions: H.C.D.-U., C.C., and Q.Z. performed experiments. R.P.S. and G.R.S.B. provided reagents and suggestions. J.L. contributed to hypothesis generation, experimental design, study supervision, and manuscript preparation.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2022-0135OC on July 14, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Beasley R, Semprini A, Mitchell EA. Risk factors for asthma: is prevention possible? Lancet . 2015;386:1075–1085. doi: 10.1016/S0140-6736(15)00156-7. [DOI] [PubMed] [Google Scholar]
- 2. GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet . 2017;390:1211–1259. doi: 10.1016/S0140-6736(17)32154-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol . 2015;16:45–56. doi: 10.1038/ni.3049. [DOI] [PubMed] [Google Scholar]
- 4. Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med . 2009;15:410–416. doi: 10.1038/nm.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Locksley RM. Asthma and allergic inflammation. Cell . 2010;140:777–783. doi: 10.1016/j.cell.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fahy JV. Type 2 inflammation in asthma--present in most, absent in many. Nat Rev Immunol . 2015;15:57–65. doi: 10.1038/nri3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gandhi NA, Bennett BL, Graham NM, Pirozzi G, Stahl N, Yancopoulos GD. Targeting key proximal drivers of type 2 inflammation in disease. Nat Rev Drug Discov . 2016;15:35–50. doi: 10.1038/nrd4624. [DOI] [PubMed] [Google Scholar]
- 8. Finotto S, Neurath MF, Glickman JN, Qin S, Lehr HA, Green FH, et al. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science . 2002;295:336–338. doi: 10.1126/science.1065544. [DOI] [PubMed] [Google Scholar]
- 9. Hwang ES, Szabo SJ, Schwartzberg PL, Glimcher LH. T helper cell fate specified by kinase-mediated interaction of T-bet with GATA-3. Science . 2005;307:430–433. doi: 10.1126/science.1103336. [DOI] [PubMed] [Google Scholar]
- 10. Mitchell C, Provost K, Niu N, Homer R, Cohn L. IFN-γ acts on the airway epithelium to inhibit local and systemic pathology in allergic airway disease. J Immunol . 2011;187:3815–3820. doi: 10.4049/jimmunol.1100436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Iwamoto I, Nakajima H, Endo H, Yoshida S. Interferon gamma regulates antigen-induced eosinophil recruitment into the mouse airways by inhibiting the infiltration of CD4+ T cells. J Exp Med . 1993;177:573–576. doi: 10.1084/jem.177.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol . 2003;21:713–758. doi: 10.1146/annurev.immunol.21.120601.140942. [DOI] [PubMed] [Google Scholar]
- 13. Gavett SH, O’Hearn DJ, Li X, Huang SK, Finkelman FD, Wills-Karp M. Interleukin 12 inhibits antigen-induced airway hyperresponsiveness, inflammation, and Th2 cytokine expression in mice. J Exp Med . 1995;182:1527–1536. doi: 10.1084/jem.182.5.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meyts I, Hellings PW, Hens G, Vanaudenaerde BM, Verbinnen B, Heremans H, et al. IL-12 contributes to allergen-induced airway inflammation in experimental asthma. J Immunol . 2006;177:6460–6470. doi: 10.4049/jimmunol.177.9.6460. [DOI] [PubMed] [Google Scholar]
- 15. Berker M, Frank LJ, Geßner AL, Grassl N, Holtermann AV, Höppner S, et al. Allergies - a T cells perspective in the era beyond the TH1/TH2 paradigm. Clin Immunol . 2017;174:73–83. doi: 10.1016/j.clim.2016.11.001. [DOI] [PubMed] [Google Scholar]
- 16. van der Pouw Kraan TC, Boeije LC, de Groot ER, Stapel SO, Snijders A, Kapsenberg ML, et al. Reduced production of IL-12 and IL-12-dependent IFN-gamma release in patients with allergic asthma. J Immunol . 1997;158:5560–5565. [PubMed] [Google Scholar]
- 17. Peukert K, Staller P, Schneider A, Carmichael G, Hänel F, Eilers M. An alternative pathway for gene regulation by Myc. EMBO J . 1997;16:5672–5686. doi: 10.1093/emboj/16.18.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bardwell VJ, Treisman R. The POZ domain: a conserved protein-protein interaction motif. Genes Dev . 1994;8:1664–1677. doi: 10.1101/gad.8.14.1664. [DOI] [PubMed] [Google Scholar]
- 19. Wanzel M, Kleine-Kohlbrecher D, Herold S, Hock A, Berns K, Park J, et al. Akt and 14-3-3eta regulate Miz1 to control cell-cycle arrest after DNA damage. Nat Cell Biol . 2005;7:30–41. doi: 10.1038/ncb1202. [DOI] [PubMed] [Google Scholar]
- 20. Herold S, Wanzel M, Beuger V, Frohme C, Beul D, Hillukkala T, et al. Negative regulation of the mammalian UV response by Myc through association with Miz-1. Mol Cell . 2002;10:509–521. doi: 10.1016/s1097-2765(02)00633-0. [DOI] [PubMed] [Google Scholar]
- 21. Kosan C, Saba I, Godmann M, Herold S, Herkert B, Eilers M, et al. Transcription factor miz-1 is required to regulate interleukin-7 receptor signaling at early commitment stages of B cell differentiation. Immunity . 2010;33:917–928. doi: 10.1016/j.immuni.2010.11.028. [DOI] [PubMed] [Google Scholar]
- 22. Do-Umehara HC, Chen C, Urich D, Zhou L, Qiu J, Jang S, et al. Suppression of inflammation and acute lung injury by Miz1 via repression of C/EBP-δ. Nat Immunol . 2013;14:461–469. doi: 10.1038/ni.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Do-Umehara HC, Chen C, Zhang Q, Misharin AV, Abdala-Valencia H, Casalino-Matsuda SM, et al. Epithelial cell-specific loss of function of Miz1 causes a spontaneous COPD-like phenotype and up-regulates Ace2 expression in mice. Sci Adv . 2020;6:eabb7238. doi: 10.1126/sciadv.abb7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA . 2011;108:E1475–E1483. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Correia CN, Nalpas NC, McLoughlin KE, Browne JA, Gordon SV, MacHugh DE, et al. Circulating microRNAs as potential biomarkers of infectious disease. Front Immunol . 2017;8:118. doi: 10.3389/fimmu.2017.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang J, Perez EA, Hou C, Zhang P, Van Scoyk M, Winn RA, et al. Identification of the SARS-CoV-2 entry receptor ACE2 as a direct target for transcriptional repression by Miz1. Front Immunol . 2021;12:648815. doi: 10.3389/fimmu.2021.648815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol . 2013;49:503–510. doi: 10.1165/rcmb.2013-0086MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Collison A, Hatchwell L, Verrills N, Wark PA, de Siqueira AP, Tooze M, et al. The E3 ubiquitin ligase midline 1 promotes allergen and rhinovirus-induced asthma by inhibiting protein phosphatase 2A activity. Nat Med . 2013;19:232–237. doi: 10.1038/nm.3049. [DOI] [PubMed] [Google Scholar]
- 29. Lambrecht BN, Hammad H, Fahy JV. The cytokines of asthma. Immunity . 2019;50:975–991. doi: 10.1016/j.immuni.2019.03.018. [DOI] [PubMed] [Google Scholar]
- 30. Hammad H, Lambrecht BN. Dendritic cells and epithelial cells: linking innate and adaptive immunity in asthma. Nat Rev Immunol . 2008;8:193–204. doi: 10.1038/nri2275. [DOI] [PubMed] [Google Scholar]
- 31. Kato A, Favoreto S, Jr, Avila PC, Schleimer RP. TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J Immunol . 2007;179:1080–1087. doi: 10.4049/jimmunol.179.2.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kuperman DA, Huang X, Koth LL, Chang GH, Dolganov GM, Zhu Z, et al. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med . 2002;8:885–889. doi: 10.1038/nm734. [DOI] [PubMed] [Google Scholar]
- 33. Kuperman D, Schofield B, Wills-Karp M, Grusby MJ. Signal transducer and activator of transcription factor 6 (Stat6)-deficient mice are protected from antigen-induced airway hyperresponsiveness and mucus production. J Exp Med . 1998;187:939–948. doi: 10.1084/jem.187.6.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wilson RH, Maruoka S, Whitehead GS, Foley JF, Flake GP, Sever ML, et al. The Toll-like receptor 5 ligand flagellin promotes asthma by priming allergic responses to indoor allergens. Nat Med . 2012;18:1705–1710. doi: 10.1038/nm.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ge MQ, Ho AW, Tang Y, Wong KH, Chua BY, Gasser S, et al. NK cells regulate CD8+ T cell priming and dendritic cell migration during influenza A infection by IFN-γ and perforin-dependent mechanisms. J Immunol . 2012;189:2099–2109. doi: 10.4049/jimmunol.1103474. [DOI] [PubMed] [Google Scholar]
- 36. Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer . 2003;3:459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 37. Okubo T, Knoepfler PS, Eisenman RN, Hogan BL. Nmyc plays an essential role during lung development as a dosage-sensitive regulator of progenitor cell proliferation and differentiation. Development . 2005;132:1363–1374. doi: 10.1242/dev.01678. [DOI] [PubMed] [Google Scholar]
- 38. Hashimoto S, Chen H, Que J, Brockway BL, Drake JA, Snyder JC, et al. β-Catenin-SOX2 signaling regulates the fate of developing airway epithelium. J Cell Sci . 2012;125:932–942. doi: 10.1242/jcs.092734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med . 2017;214:2387–2404. doi: 10.1084/jem.20162152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu D, Mamorska-Dyga A. Syk inhibitors in clinical development for hematological malignancies. J Hematol Oncol . 2017;10:145. doi: 10.1186/s13045-017-0512-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Neurath MF, Finotto S, Glimcher LH. The role of Th1/Th2 polarization in mucosal immunity. Nat Med . 2002;8:567–573. doi: 10.1038/nm0602-567. [DOI] [PubMed] [Google Scholar]
- 42. Romagnani S. The Th1/Th2 paradigm. Immunol Today . 1997;18:263–266. doi: 10.1016/s0167-5699(97)80019-9. [DOI] [PubMed] [Google Scholar]
- 43. Magram J, Connaughton SE, Warrier RR, Carvajal DM, Wu CY, Ferrante J, et al. IL-12-deficient mice are defective in IFN gamma production and type 1 cytokine responses. Immunity . 1996;4:471–481. doi: 10.1016/s1074-7613(00)80413-6. [DOI] [PubMed] [Google Scholar]
- 44. Helft J, Böttcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, et al. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c(+)MHCII(+) macrophages and dendritic cells. Immunity . 2015;42:1197–1211. doi: 10.1016/j.immuni.2015.05.018. [DOI] [PubMed] [Google Scholar]
- 45. Walter MJ, Kajiwara N, Karanja P, Castro M, Holtzman MJ. Interleukin 12 p40 production by barrier epithelial cells during airway inflammation. J Exp Med . 2001;193:339–351. doi: 10.1084/jem.193.3.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Homma Y, Cao S, Shi X, Ma X. The Th2 transcription factor c-Maf inhibits IL-12p35 gene expression in activated macrophages by targeting NF-kappaB nuclear translocation. J Interferon Cytokine Res . 2007;27:799–808. doi: 10.1089/jir.2007.0006. [DOI] [PubMed] [Google Scholar]
- 47. Murphy TL, Cleveland MG, Kulesza P, Magram J, Murphy KM. Regulation of interleukin 12 p40 expression through an NF-kappa B half-site. Mol Cell Biol . 1995;15:5258–5267. doi: 10.1128/mcb.15.10.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Naseer T, Minshall EM, Leung DY, Laberge S, Ernst P, Martin RJ, et al. Expression of IL-12 and IL-13 mRNA in asthma and their modulation in response to steroid therapy. Am J Respir Crit Care Med . 1997;155:845–851. doi: 10.1164/ajrccm.155.3.9117015. [DOI] [PubMed] [Google Scholar]
- 49. Tomita K, Lim S, Hanazawa T, Usmani O, Stirling R, Chung KF, et al. Attenuated production of intracellular IL-10 and IL-12 in monocytes from patients with severe asthma. Clin Immunol . 2002;102:258–266. doi: 10.1006/clim.2001.5176. [DOI] [PubMed] [Google Scholar]
- 50. Prescott SL, Taylor A, King B, Dunstan J, Upham JW, Thornton CA, et al. Neonatal interleukin-12 capacity is associated with variations in allergen-specific immune responses in the neonatal and postnatal periods. Clin Exp Allergy . 2003;33:566–572. doi: 10.1046/j.1365-2222.2003.01659.x. [DOI] [PubMed] [Google Scholar]
- 51. Reider N, Reider D, Ebner S, Holzmann S, Herold M, Fritsch P, et al. Dendritic cells contribute to the development of atopy by an insufficiency in IL-12 production. J Allergy Clin Immunol . 2002;109:89–95. doi: 10.1067/mai.2002.120556. [DOI] [PubMed] [Google Scholar]
- 52. Hamid QA, Schotman E, Jacobson MR, Walker SM, Durham SR. Increases in IL-12 messenger RNA+ cells accompany inhibition of allergen-induced late skin responses after successful grass pollen immunotherapy. J Allergy Clin Immunol . 1997;99:254–260. doi: 10.1016/s0091-6749(97)70106-4. [DOI] [PubMed] [Google Scholar]
- 53. Kips JC, Brusselle GJ, Joos GF, Peleman RA, Tavernier JH, Devos RR, et al. Interleukin-12 inhibits antigen-induced airway hyperresponsiveness in mice. Am J Respir Crit Care Med . 1996;153:535–539. doi: 10.1164/ajrccm.153.2.8564093. [DOI] [PubMed] [Google Scholar]
- 54. Sur S, Lam J, Bouchard P, Sigounas A, Holbert D, Metzger WJ. Immunomodulatory effects of IL-12 on allergic lung inflammation depend on timing of doses. J Immunol . 1996;157:4173–4180. [PubMed] [Google Scholar]
- 55. Kuribayashi K, Kodama T, Okamura H, Sugita M, Matsuyama T. Effects of post-inhalation treatment with interleukin-12 on airway hyper-reactivity, eosinophilia and interleukin-18 receptor expression in a mouse model of asthma. Clin Exp Allergy . 2002;32:641–649. doi: 10.1046/j.0954-7894.2002.01346.x. [DOI] [PubMed] [Google Scholar]
- 56. Gerhold K, Blümchen K, Bock A, Seib C, Stock P, Kallinich T, et al. Endotoxins prevent murine IgE production, T(H)2 immune responses, and development of airway eosinophilia but not airway hyperreactivity. J Allergy Clin Immunol . 2002;110:110–116. doi: 10.1067/mai.2002.125831. [DOI] [PubMed] [Google Scholar]
- 57. Erb KJ, Holloway JW, Sobeck A, Moll H, Le Gros G. Infection of mice with Mycobacterium bovis-Bacillus Calmette-Guérin (BCG) suppresses allergen-induced airway eosinophilia. J Exp Med . 1998;187:561–569. doi: 10.1084/jem.187.4.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Major T, Wohlleben G, Reibetanz B, Erb KJ. Application of heat killed Mycobacterium bovis-BCG into the lung inhibits the development of allergen-induced Th2 responses. Vaccine . 2002;20:1532–1540. doi: 10.1016/s0264-410x(01)00496-0. [DOI] [PubMed] [Google Scholar]
- 59. Pochard P, Gosset P, Grangette C, Andre C, Tonnel AB, Pestel J, et al. Lactic acid bacteria inhibit TH2 cytokine production by mononuclear cells from allergic patients. J Allergy Clin Immunol . 2002;110:617–623. doi: 10.1067/mai.2002.128528. [DOI] [PubMed] [Google Scholar]
- 60. Wohlleben G, Müller J, Tatsch U, Hambrecht C, Herz U, Renz H, et al. Influenza A virus infection inhibits the efficient recruitment of Th2 cells into the airways and the development of airway eosinophilia. J Immunol . 2003;170:4601–4611. doi: 10.4049/jimmunol.170.9.4601. [DOI] [PubMed] [Google Scholar]
- 61. Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol . 2005;6:635–645. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- 62. Bønnelykke K, Matheson MC, Pers TH, Granell R, Strachan DP, Alves AC, et al. AAGC Meta-analysis of genome-wide association studies identifies ten loci influencing allergic sensitization. Nat Genet . 2013;45:902–906. doi: 10.1038/ng.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, et al. International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium 2 Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature . 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]