

Abstract
TLR7 (Toll-like receptor 7), the sensor for single-stranded RNA, contributes to systemic inflammation and mortality in murine polymicrobial sepsis. Recent studies show that extracellular miR-146a-5p serves as a TLR7 ligand and plays an important role in regulating host innate immunity. However, the role of miR-146a-5p and TLR7 signaling in pulmonary inflammation, endothelial activation, and sepsis-associated acute respiratory distress syndrome remains unclear. Here, we show that intratracheal administration of exogenous miR-146a-5p in mice evokes lung inflammation, activates endothelium, and increases endothelial permeability via TLR7-dependent mechanisms. TLR7 deficiency attenuates pulmonary barrier dysfunction and reduces lung inflammatory response in a murine sepsis model. Moreover, the impact of miR-146a-5p−TLR7 signaling on endothelial activation appears to be a secondary effect because TLR7 is undetectable in the human pulmonary artery and microvascular endothelial cells (ECs), which show no response to direct miR-146a-5p treatment in vitro. Both conditioned media of miR-146a-5p−treated macrophages (Mϕ) and septic sera of wild-type mice induce a marked EC barrier disruption in vitro, whereas Mϕ conditioned media or septic sera of TLR7−/− mice do not exhibit such effect. Cytokine array and pathway enrichment analysis of the Mϕ conditioned media and septic sera identify TNFα (tumor necrosis factor α) as the main downstream effector of miR-146a-5p−TLR7 signaling responsible for the EC barrier dysfunction, which is further supported by neutralizing anti-TNFα antibody intervention. Together, these data demonstrate that TLR7 activation elicits pulmonary inflammation and endothelial barrier disruption by sensing extracellular miR-146a-5p and contributes to sepsis-associated acute respiratory distress syndrome.
Keywords: sepsis, Toll-like receptors, acute respiratory distress syndrome, microRNAs, innate immunity
Acute respiratory distress syndrome (ARDS) is a life-threatening condition affecting 190,000 people annually in the United States, with 40–60% reported mortality (1). ARDS occurs in 25–40% of patients who are diagnosed with sepsis (2, 3). Patients with ARDS caused by sepsis are associated with higher mortality and worse clinical outcomes than ARDS caused by other diseases (4). Despite decades of effort in research, current proven and effective treatments of sepsis-associated ARDS are still limited to supportive therapy such as protective mechanical ventilation (5).
Excessive pulmonary inflammation and alveolar barrier hyperpermeability are the key pathophysiologic derangement of sepsis-associated ARDS (6, 7). In direct sepsis-associated ARDS, injury starts with pathogens entering the lung directly. The downstream pathways leading to epithelial injury are markedly activated. In contrast, indirect sepsis-associated ARDS begins with an infection outside the lung. Signals mediating endothelial cell (EC) activation and/or injury are disproportionally activated (8). A variety of effectors and mediators, such as inflammatory cytokines, serotonin, Ang2 (angiopoietin 2), and syndecan-1, have been implicated in endothelial activation and hyperpermeability during sepsis (7, 9, 10). They cause disruption of bonds between adjacent ECs and cytoskeleton contraction by activating intracellular signaling, including Ca2+, tyrosine kinase, protein kinase C, myosin light chain kinase, and small Rho GTPase. These kinases and GTPase phosphorylate and change the conformation of subcellular components such as VE-cadherin and cytoskeleton, leading to endothelial hyperpermeability (11).
Host responses initiated by pathogen-associated molecular patterns and aggravated by endogenous damage-associated molecular patterns are considered potent driving forces for sepsis-associated ARDS (12, 13). Extracellular RNA (ex-RNA), a recently discovered damage-associated molecular pattern, is released from injured or necrotic cells and reportedly mediates vascular dysfunction (14), coagulopathy (15), and inflammation (16–18). Fischer and coworkers report that ex-RNA plays a role in endothelial hyperpermeability by inducing adhesion and transmigration of leukocytes through activation of the vascular endothelial growth factor (VEGF) and VEGF–receptor-2 system (14). In addition, ex-RNA also functions as a procoagulant cofactor in the amplification of blood coagulation (15). We have shown that ex-RNA induces a robust cytokine response in macrophages, neutrophils, and cardiomyocytes and contributes to tissue inflammation and injury (16–18). Recent RNA sequencing reveals that microRNAs (miRNAs), small noncoding RNAs that function as intracellular gene regulators (19), are the most abundant plasma RNA species in both mice and humans and are differentially expressed during sepsis (20). Most importantly, certain uridine-rich extracellular miRNAs, such as miR-146a-5p, drive innate immune inflammatory response in vitro and in vivo by activating TLR7 (Toll-like receptor 7) signaling through its UU-containing motif (16, 17, 20). Finally, mice deficient in miR-146a or TLR7 show attenuated inflammation and improved survival compared with wild-type (WT) mice in sepsis (20–23).
In this study, we tested the hypothesis that TLR7 activation by miR-146a-5p in immune cells drives endothelial barrier disruption and lung inflammation and contributes to alveolar barrier hyperpermeability and lung inflammation in murine sepsis. Some of the results of these studies have been previously reported in the form of an abstract (24).
Methods
Animals and Model of Polymicrobial Sepsis
Male 8- to 16-week old WT (C57BL/6J), TLR7−/− (Tlr7tm1Flv/J), and miR-146−/− mice (Mir146tim.Bal) (Jackson Laboratories), both in C57BL/6J background, were used in this study approved by the Institutional Animal Care and Use Committee of the University of Maryland, Baltimore. A sepsis model was achieved by cecal ligation and puncture (CLP) as described previously (16).
Lung and Endothelial Permeability Assays
Lung permeability was assessed in vivo by intravenous injection of fluorescein isothiocyanate (FITC)-labeled dextran (40 kD, Sigma-Aldrich). The fluorescence ratio of BAL to plasma was used to evaluate alveolar permeability. EC permeability in vitro was measured by an electric cell-substrate impedance system (ECIS, Applied Biophysics) at a frequency of 4 kHz or visualized by XperT assay, in which permeability was quantified as a bulk amount of fluorescence-labeled ligand (FITC-avidin) bound to surface-immobilized biotin underneath the EC monolayer as described previously (25).
Intratracheal Delivery of miRNA Mimics
After anesthesia, mice were intratracheally administered either 20 μg single-stranded miR-146a-5p (Integrated DNA Technologies), its U→A mutant packed with lipofectamine 3000 (Thermo Fisher Scientific), or lipofectamine alone. At 18 hours, BAL fluid and lung tissue were collected for analysis.
Quantitative RT-PCR
Total RNA was reversely transcribed by M-MLV (Promega) or miRCURY LNA RT (Qiagen) kit, followed by detection using SYBR green PCR kits. The relative expression of miR-146a-5p or mRNAs was normalized by spike-in cel-miR-39 or GAPDH (glyceraldehyde 3-phosphate dehydrogenase), respectively, and calculated by the comparative Ct method (2-ΔΔCt).
Cell Cultures and Conditioned Media (CM)
Human pulmonary artery and microvascular ECs (HPAECs and HMVECs; Lonza) were cultured per the manufacturer’s instructions. Bone marrow-derived macrophages (Mϕs) were isolated and cultured as previously described (16). CM of Mϕs were collected 24 hours after treatments with miRNA mimics or mutants.
Western Blot
Cellular proteins were separated by SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) and immunoblotted with GAPDH and TLR7 antibodies from Cell Signaling Technology.
Cytokine Array and Pathway Analysis
Cytokines in Mϕs CM and pooled sera were profiled by Mouse XL Cytokine Array Kit (R&D Systems). KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis (http://www.genome.jp/kegg/) was applied to identify biological pathways through Metascape analysis (http://metascape.org) with the details in data supplements.
Flow Cytometry
BAL was collected, and cells were pelleted by centrifuge. The isolated cells were incubated with viability dye and Fc blocker followed by surface marker staining in PBS solution containing 5% FBS with the details in data supplements.
Enzyme-linked Immunosorbent Assay (ELISA)
IL-6, CXCL2, and intercellular adhesion molecule 1 (ICAM-1) in the BAL and media were analyzed using ELISA kits (R&D) per the manufacturer’s instructions. BAL protein was measured by the Pierce BCA protein assay kit (Thermo Fisher). Albumin ELISA kit was from Bethyl laboratories.
EC Immunofluorescent Staining
HPAECs in chamber slides were fixed and stained with VE-cadherin (Cayman), Phalloidin-Texas Red (Invitrogen), and DAPI (4′,6-diamidino-2-phenylindole; Sigma-Aldrich). Images were acquired by Nikon A1 or spin-disk confocal microscope.
Statistical Analysis
Statistical analysis was performed by GraphPad Prism 9 software (GraphPad). Data were analyzed by unpaired t test, one-way ANOVA, or two-way ANOVA. The null hypothesis was rejected for P < 0.05 with two tails.
Results
BAL miR-146a-5p Is Increased in Sepsis and Exogenous miR-146a-5p Triggers Lung Inflammation via TLR7 Signaling
To identify the role of miR-146a-5p in lung injury, we first tested the BAL miR-146a-5p expression at 24 hours after CLP. Consistent with the previous results from septic plasma (16), BAL miR-146a-5p was increased significantly in septic mice by approximately 14-fold compared with that of sham mice (Figure 1A). To determine if miR-146a-5p is sufficient to trigger lung inflammation, miR-146a-5p mimics or vehicle control group were intratracheally delivered into miR-146a−/− mice to avoid the potential confounder of endogenous miR-146a-5p. BAL albumin, a marker of alveolar–capillary permeability (26), was increased significantly at 18 hours after delivery of the miR-146a-5p mimics compared with the control group (Figure 1B). Intratracheal administration of miR-146a-5p also induced pulmonary inflammation, as evidenced by a marked increase in the proinflammatory cytokine IL-6 and TNFα (tumor necrosis factor α) production (Figure 1C) in the BAL and lung tissue and robust neutrophil infiltration to alveolar space (Figures 1D and 1E) on the basis of the flow cytometry gating strategy in Figure E1A in the data supplement. Of note, no significant difference was observed between WT and miR-146a−/− mice in the amount of albumin, cytokines, and leukocytes in the alveolar space (Figures E1B–E1D) after intratracheal delivery of lipofectamine control, suggesting that miR-146a deficiency does not impact lung baseline condition. Moreover, miR-146a-5p caused a similar increase in BAL albumin, IL-6, and TNFα in WT and miR-146a−/− mice (Figures E1B and E1C). However, the ability of miR-146a-5p to recruit leukocytes and neutrophils was partially dampened in the miR-146a−/− group compared with that of WT mice (Figure E1D). Together, these data suggest that exogenous miR-146a-5p is capable of inducing lung inflammation and increasing alveolar permeability.
Figure 1.
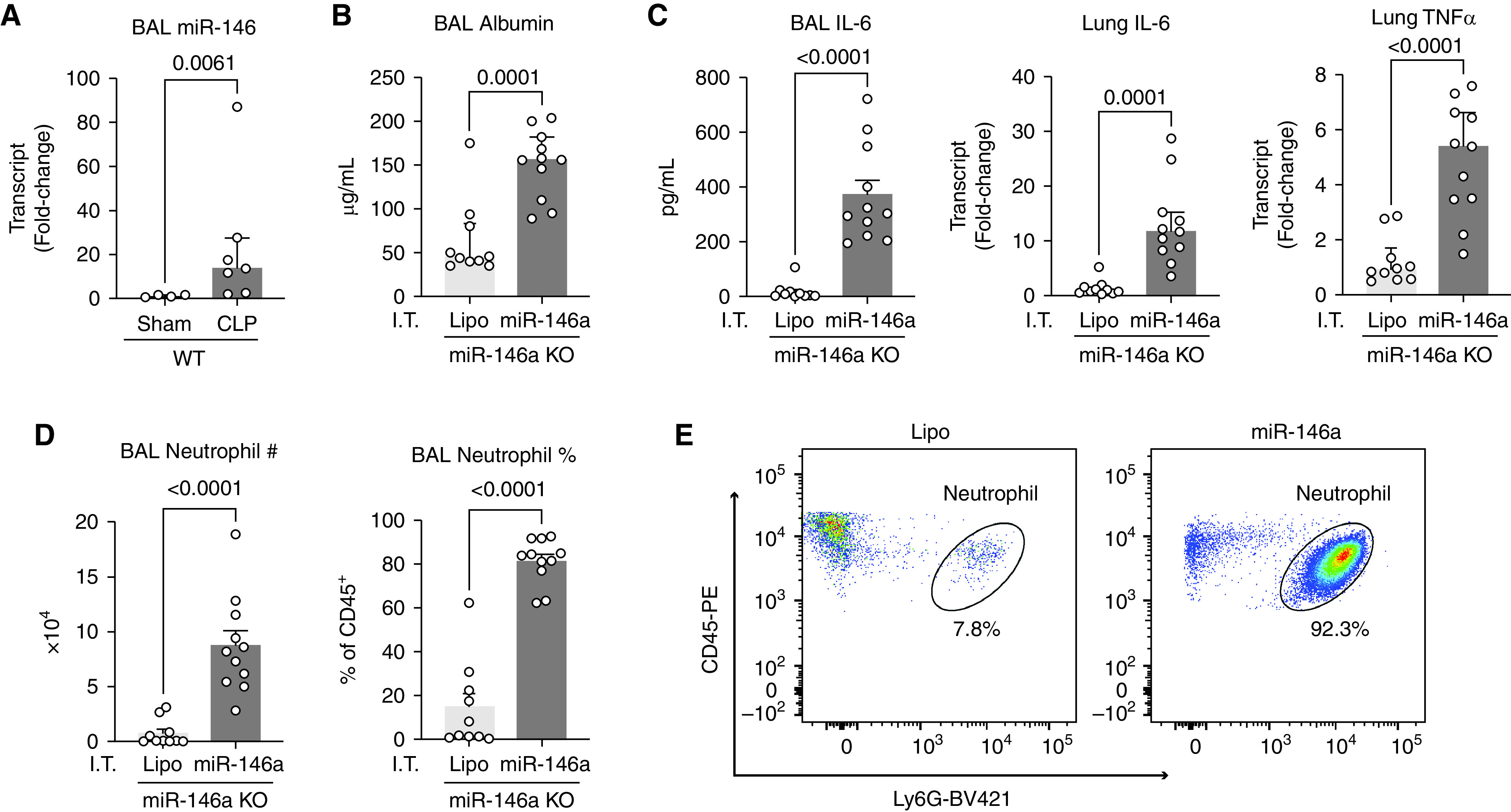
Exogenous miR-146a-5p disrupts barrier integrity and evokes lung inflammation. (A) miR-146a-5p expression in the BAL was measured at 24 hours after sham (n = 4) or CLP (n = 6) surgeries. (B–E) BAL and lung tissue from miR-146a knockout mice were collected and analyzed 18 hours after intratracheal injection of lipofectamine (Lipo) or miR-146a-5p (miR-146a). n = 10–11 per group. (B) BAL albumin concentration. (C) Enzyme-linked immunosorbent assay (ELISA) and quantitative RT-PCR analysis of cytokine in the BAL and lung tissue. (D) Neutrophils (Ly6G+CD45+) infiltration was assessed by flow cytometry. (E) Representative flow cytometry plots gated for Ly6G+ and CD45+. Data were presented as mean ± SEM and analyzed with an unpaired t test. CLP = cecal ligation and puncture; TNF = tumor necrosis factor.
To dissect the downstream signaling pathways of exogenous miR-146a-5p in the lung, we employed mice deficient of TLR7, a sensor for single-stranded RNA (27). miR-146a-5p mimics or its inactive U→A mutant were administered intratracheally in both WT and TLR7−/− mice. Lung permeability and inflammation were assessed at 18 hours. Consistent with what we observed in miR-146a−/− mice, intratracheal delivery of miR-146a-5p, but not its inactive mutant, caused lung injury in WT mice as evidenced by an increase in BAL albumin, IL-6, and TNFα (Figures 2A and 2B), an increase in lung tissue TNFα, IL-1β, and CXCL1 gene expression (Figure 2C), as well as an increase in total BAL cell counts, leukocytes, and neutrophils to alveolar space (Figures 2D, 2E, and E1E). The absence of TLR7 significantly dampened the injurious effect of exogenous miR-146a-5p in the lung with diminished BAL albumin leaking, lung cytokine production, and leukocyte recruitment. These data indicate that exogenous miR-146a-5p elicits lung inflammation by activating TLR7 signaling.
Figure 2.
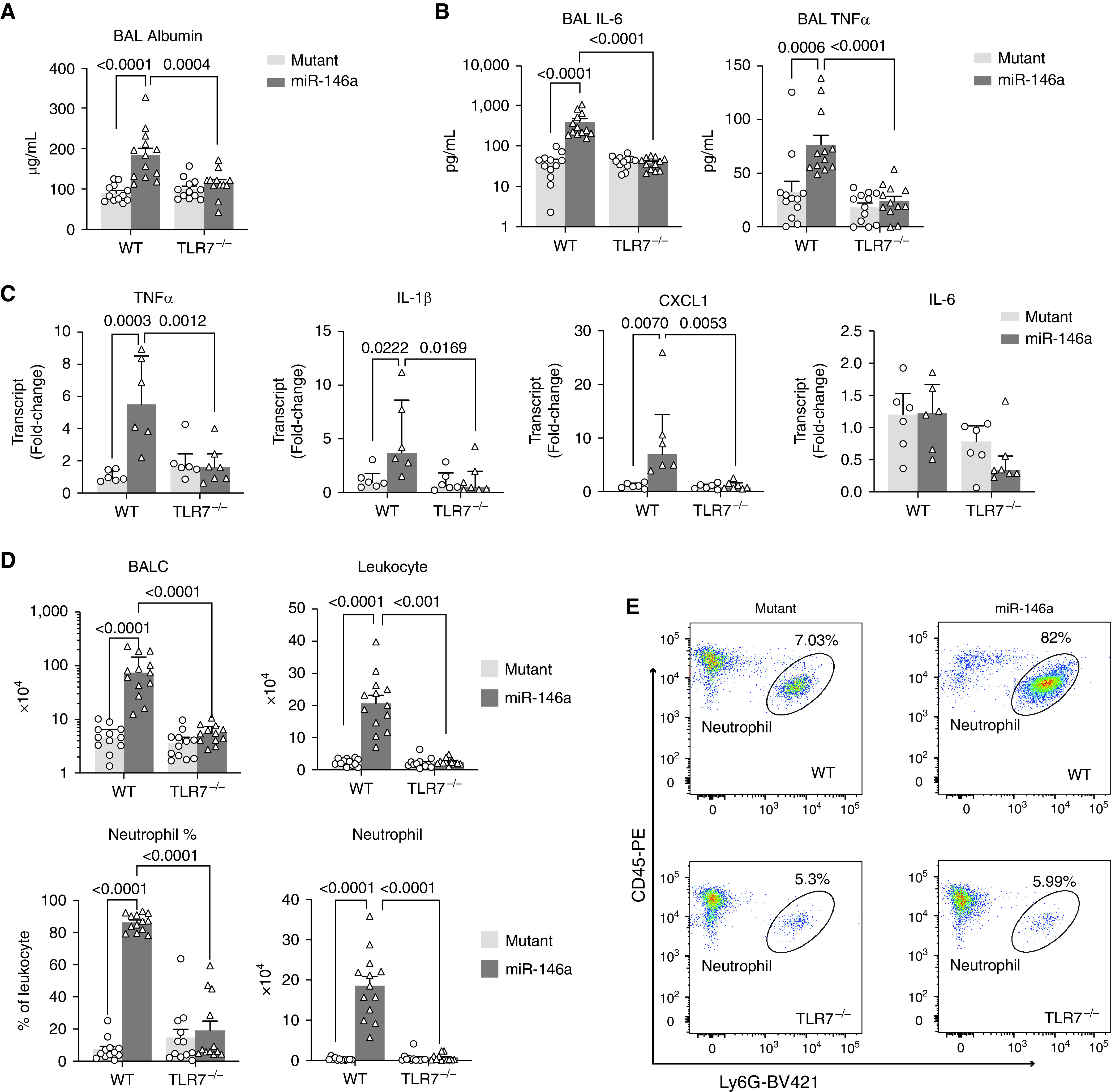
TLR7 (Toll-like receptor 7) deficiency protects against miR-146a-5p–induced lung inflammation. BAL and lung tissue were collected and analyzed in both wild-type (WT) and TLR7−/− mice at 18 hours after intratracheal delivery of miR-146a-5p or U→A mutant at a dose of 20 μg. (A) Albumin concentration in the BAL. (B) IL-6 and TNFα (tumor necrosis factor α) in the BAL were measured by ELISA. (C) TNFα, IL-1β, CXCL1, and IL-6 transcripts in the lung were tested by qRT-PCR. (D) The number of total BALC, leukocytes (CD45+), and neutrophils (Ly6G+CD45+) in the BAL were analyzed by flow cytometry. (E) Representative flow cytometry plot gated for Ly6G+ and CD45+. Data were presented as mean ± SEM or median with interquartile range. n = 6–13 mice per group. Data were analyzed by two-way ANOVA with Bonferroni’s post hoc test. BALC = BAL cells.
Exogenous miR-146a-5p Indirectly Induces Endothelial Cell Dysfunction Through Macrophages
Because endothelial activation and injury are pivotal components contributing to pulmonary barrier dysfunction in sepsis, we next tested the effect of miR-146a-5p on endothelial barrier function. Although we initially postulated that miR-146a-5p would induce barrier disruption through directly activating ECs, we surprisingly found that neither miR-146a-5p nor R837 (TLR7 ligand) had any impact on the permeability of HPAECs as assessed by an ECIS system (Figure 3A). In contrast, VEGF, a positive control, caused a time-dependent barrier dysfunction in the same HPAEC cultures. We further confirmed the result using an XperT assay, an in vitro permeability imaging assay using FITC-avidin as a tracer that can be quantified by the percentage of the leaky area (green fluorescence) within the total area. As shown in Figure E2A, the area of green fluorescence was comparable between lipofectamine- and miR-146a-5p–treated HPAECs, whereas TNFα treatment caused dramatically increased leakage. Similarly, direct treatment of miR-146a-5p did not change barrier function in HMVECs (Figure E2B). This lack of direct response to miR-146a-5p was most likely because of the lack or low expression of TLR7 in HPAECs, as demonstrated in Figures 3B and 3C. Neither TLR7 mRNA nor protein expression was detected in the ECs, whereas TLR7 was clearly present in HEK293 cells expressing human TLR7. Inflammatory stimulation with LPS or septic sera did not induce any TLR7 mRNA expression in both HPAECs and HMVECs (Figure E2C). Given these findings and the expected role of innate immune cells in sepsis-induced lung injury, we hypothesized that miR-146a-5p indirectly induces endothelial barrier dysfunction through a mechanism involving innate immune cells. To test this hypothesis, we treated WT or TLR7−/− Mϕ with or without lipofectamine, miR-146a-5p (5, 10, 20, and 50 nM), or miR-210-3p (5 and 50 nM), and collected culture media at 24 hours. The CM were then applied to HPAEC at 20% of the final concentrations (vol/vol) and tested for their effect on EC barrier function and soluble ICAM-1 production (Figure 3D). As shown in Figure 3E, the HPAEC resistance in the ECIS assay was decreased in a time- and dose-dependent manner in response to the CM collected from miR-146a-5p–treated Mϕs, but not from lipofectamine or no treatment group. Interestingly, CM of miR-210–treated Mϕs, a miRNA possessing no inflammatory effect (23), failed to disrupt the EC barrier function (Figure 3F). In comparison to CM from miR-146a-5p–treated Mϕ, CM collected from TLR7−/− Mϕ cultures treated with miR-146a-5p failed to reduce HPAEC resistance (Figure 3G), suggesting that miR-146a-5p−TLR7 signaling in Mϕ plays an essential role in the CM-induced EC barrier disruption. Consistent with the permeability change, CM from miR-146a-5p–, but not miR-210-3p–treated cells, also led to a marked increase in the release of soluble ICAM-1, an EC activation marker, in the EC cultures (Figure 3H). Similarly, the property of CM on ICAM-1 release was lost in TLR7−/− Mϕ subjected to the same miR-146a-5p treatment (Figure 3I). Furthermore, we also tested the effect of CM from miR-146a-5p–treated Mϕ in HMVECs and observed a similar response (Figures E2D and E2E) as HPAECs. All together, this evidence implicates that miRNA-TLR7 of innate immune cells plays a potential role in EC activation and barrier disruption.
Figure 3.
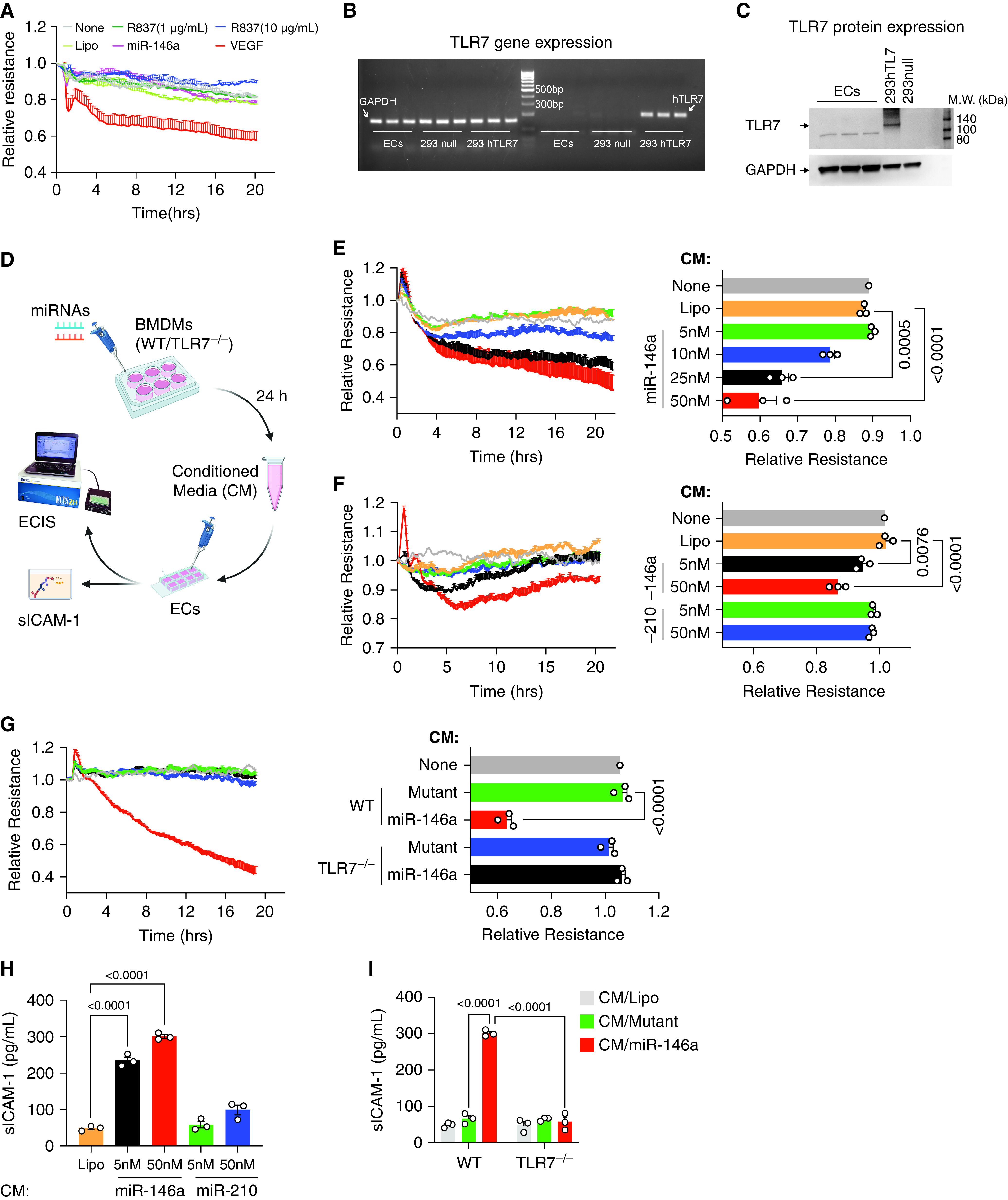
miR-146a-5p increases endothelial permeability via macrophage TLR7. (A) Neither miR-146a-5p (50 nM) nor TLR7 agonist R837 increased permeability in endothelial cells (ECs) tested by electric cell-substrate impedance system (ECIS). Vascular endothelial growth factor (VEGF, 200 ng/ml) was used as a positive control. The ECIS curves are from duplicate samples. The experiment was repeated once in triplicates with similar results. (B–C) No TLR7 gene and protein expression was detected in endothelial cells. HEK293 null cell (293 null) served as a negative control, and HKE293 overexpressing human TLR7 (293 hTLR7) served as a positive control. (D) Schematic experiment flowchart. ECs were treated with 20% conditioned media (CM) collected from macrophages (BMDMs) (WT and TLR7−/−) incubated with either Lipofectamine (Lipo), miR-146a-5p, or miR-210-3p at various dosages for 24 hours. Resistance was measured by ECIS, and sICAM-1 (secreted intercellular adhesion molecule 1) was tested using ELISA. (E–G) Conditioned media from miR-146a-5p (proinflammatory miRNA) but not that of miR-210 (nonproinflammatory miRNA) increased EC permeability via macrophage TLR7. Resistance at 10 hours was quantified for statistical analysis and presented as a bar graph. (H) sICAM-1 concentrations were measured by ELISA after designated CM treatment in (F) and (G). Data were presented as mean ± SEM. Data of (E) were analyzed by one-way ANOVA with Tukey post hoc test. Data of (F), (G), and (H) were analyzed using two-way ANOVA with Bonferroni’s post hoc test. BMDM = bone marrow-derived macrophages; miRNA = microRNA.
TLR7 Deficiency Attenuates Acute Lung Injury in Murine Sepsis
To further determine whether TLR7 sensing is involved in acute lung injury in sepsis, WT and TLR7−/− mice were subjected to sham or CLP procedure, a clinically relevant model of polymicrobial sepsis. Twenty-four hours after CLP, there was a significant increase in cytokines (IL-6), chemokine (CXCL2), and adhesion molecule (ICAM-1) in the BAL and/or lung tissue from septic WT mice (Figures 4A and 4B). In comparison, septic TLR7−/− mice exhibited significantly reduced amounts of these proinflammatory molecules. Moreover, we measured vascular barrier function in the lung by measuring leakage of FITC-labeled dextran from circulation to bronchoalveolar space. Dextran leakage was found significantly higher in the WT septic lung compared with that of sham mice (BAL/plasma ratio: 0.098 ± 0.004 vs. 0.20 ± 0.04 sham vs. CLP; P = 0.0371) (Figure 4C), implicating alveolar capillary barrier disruption. TLR7−/− septic mice had preserved endothelial barrier function as evidenced by less dextran leakage into the lung alveolar space (BAL/plasma ratio: 0.109 ± 0.023 vs. 0.20 ± 0.04, TLR7−/− vs. WT; P = 0.037). Collectively, these loss-of-function studies demonstrate the importance of TLR7 signaling in mediating lung barrier dysfunction and inflammation during extrapulmonary sepsis.
Figure 4.
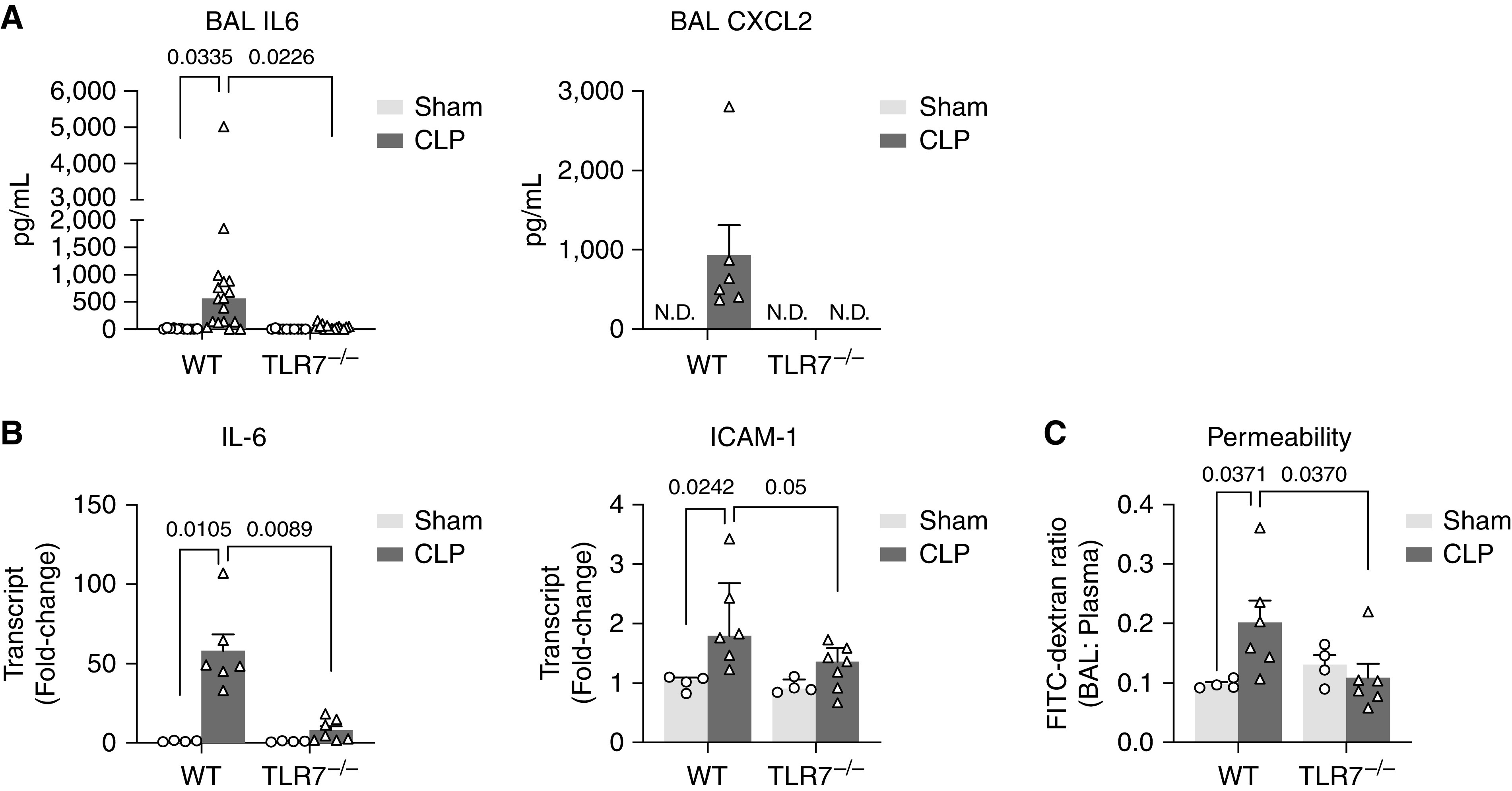
TLR7 deficiency attenuates lung injury in murine sepsis. Twenty-four hours after sham or CLP procedure, BAL, plasma, and lung were collected from both WT and TLR7−/− mice. (A) IL-6 and CXCL2 in the BAL. (B) IL-6 and ICAM-1 gene expression in the lung tissue. (C) Alveolar–capillary permeability was measured by leakage of fluorescein isothiocyanate (FITC)–dextran (40 kD) from blood circulation to alveolar space presented as the ratio of FITC fluorescence in BAL to plasma. n = 4–6/group. Data were presented as mean ± SEM and analyzed by two-way ANOVA with Bonferroni’s post hoc test.
TLR7-dependent Serum Factor Causes EC Barrier Disruption
To explore the mechanism by which TLR7 signaling mediates endothelial barrier dysfunction in CLP, we next tested the ability of septic serum to interrupt EC barrier function. To do that, we used an ex vivo model by treating HPAECs with 1% serum collected from WT or TLR7−/− mice at 24 hours after sham or CLP to simulate the in vivo scenario in which lung endothelium is exposed to mediators in blood circulation. As shown in Figure 5A, after being incubated with serum for 24 hours, EC permeability was measured by XperT assay together with VE-cadherin and F-actin imaging and soluble ICAM-1 ELISA. Comparable green fluorescence of approximately 5.2% was observed in sham sera-treated and no-treatment cells (Figure E3A). ECs treated with sera from WT-CLP mice showed a dramatical increase in green fluorescence along the cell border compared with those treated with WT-sham sera (percentage, 5.2 ± 0.9 vs. 22.4 ± 3.7, sham vs.CLP; P = 0.0139) (Figure E3A), indicating that circulating factors from septic sera disrupt the EC function. In comparison to WT CLP sera-treated ECs, permeability was preserved when ECs were exposed to CLP sera from TLR7−/− mice (percentage, 31.7 ± 2.8 vs. 5.1 ± 1.1, WT vs. TLR7−/−; P < 0.0001) (Figures 5B and 5C), strongly suggesting that TLR7-dependent circulating factors mediate septic serum-induced EC disruption.
Figure 5.
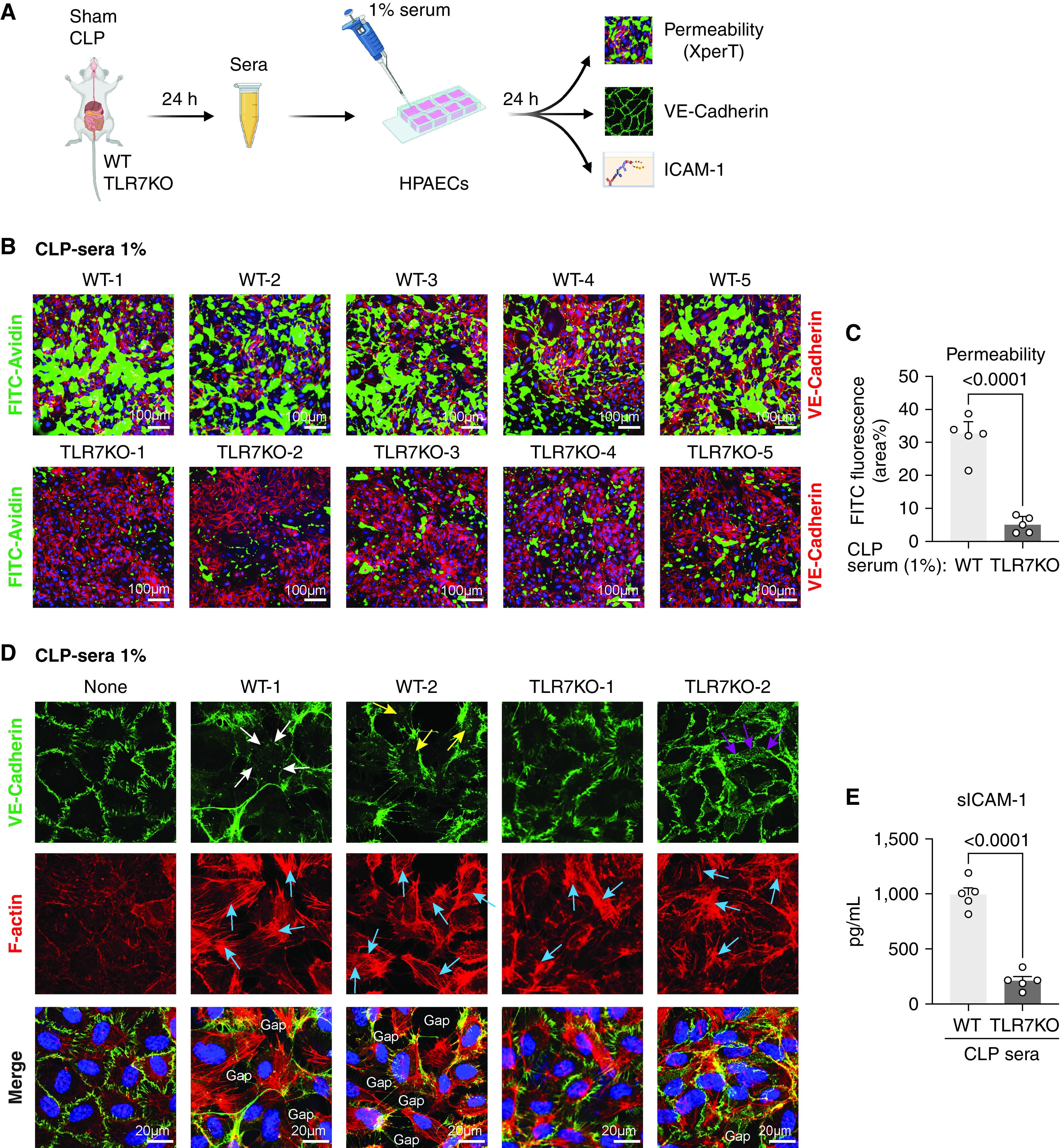
The absence of TLR7 prevents EC barrier disruption by septic sera. (A) Schematic experimental flowchart. ECs were treated with 1% sera collected 24 hours after sham or CLP surgery in WT or TLR7−/− mice. After 24 hours of incubation, permeability was measured by XperT assay, VE-cadherin and F-actin were stained by immunofluorescence, and media ICAM-1 was tested by ELISA. (B–C) Endothelial permeability visualized by XperT and quantified by the area percentage of FITC. n = 5 per group. (D) VE-cadherin and stress fiber formation in ECs treated with 1% sera. White arrowheads: disrupted VE-cadherin; yellow arrowheads: internalized VE-cadherin; purple arrowheads: VE-cadherin network; blue arrowheads: stress fiber. (E) sICAM-1 in the media from treated ECs. n = 5 per group. Data were presented by mean ± SEM and analyzed by unpaired t test.
To decipher the molecular mechanism responsible for TLR7-mediated endothelial barrier disruption and activation, we stained VE-cadherin and F-actin. VE-cadherin is an endothelial-specific adherent junction protein that connects the adjacent ECs and helps maintain their integrity (28). Endocytosis and degradation of VE-cadherin in response to inflammatory stimuli lead to increased endothelial permeability through impaired endothelial junctions (29, 30). In ECs treated with or without sham sera, VE-cadherin appeared as an intercellular continuous lining along cell–cell borders (Figure E3B). However, when ECs were exposed to 1% sera from CLP mice, the continuity of VE-cadherin was disrupted and internalized to cytoplasm, as highlighted in white or yellow arrowheads in Figure E3B and Figure 5D. In comparison, there was a continuous expression of VE-cadherin in the ECs treated with 1% TLR7−/− CLP sera (Figure 5D). Of note, a reticular network of VE-cadherin, referred to as junction-associated intermittent lamellipodia (JAIL), a subcellular structure indicating a process that restores weak or lost VE-cadherin adhesion (31), was observed in TLR7−/− CLP serum-treated EC (Figure 5D, purple arrowheads).
Endothelial actin cytoskeleton is linked to adherent junctions to provide mechanical stability. The formation of actin stress fiber generates a tensile force that pulls ECs apart to form paracellular gaps (32). Under normal conditions, F-actin staining displays thin filaments along the cell borders and inside of the cells (Figure E3B, none; Figure 5D, none). WT CLP sera led to the development of thick actin filament with strong red fluorescence inside cells, indicating the formation of stress fiber (Figure E3B; CLP-1, CLP-2). However, the difference in the stress fiber formation between WT in TLR7−/− CLP serum-treated ECs was not dramatic and, at this time, difficult to quantify (Figure 5D). As a result of changes of VE-cadherin in WT CLP serum-treated ECs, multiple gaps formed between adjacent cells, which did not show in ECs treated with TLR7−/− CLP sera. Consistent with these observations, soluble ICAM-1 (Figure E3C) dropped significantly in the EC media treated with TLR7−/− CLP sera compared with that of WT CLP (994 ± 61 vs. 211 ± 37 pg/ml, WT vs. TLR7−/−; P < 0.0001) (Figure 5E), indicating an attenuation of proinflammatory EC phenotype.
TNFα Is the Downstream Mediator of miR-146a-5p–TLR7 Signaling Responsible for Endothelial Barrier Dysfunction in Sepsis
To identify the downstream mediator of TLR7 signaling responsible for the observed EC barrier disruption induced by either Mϕ CM or septic sera, we performed cytokine arrays. Among 111 cytokines tested, 30 cytokines were found to be markedly increased in miR-146a-5p–treated CM compared with its inactive mutant (Figures 6A and 6B). Among the upregulated cytokines, 15 cytokines appeared to be TLR7-dependent (Figure 6B), including TNFα, IL-6, CXCL2, IL-12, MIP-1α, IL-27, CXCL16, and IL1Rα. Similarly, cytokine arrays revealed that over 70 cytokines were differentially expressed in sera between WT CLP and TLR7−/− CLP mice, including the proinflammatory cytokines IL-6, TNFα, ICAM-1, and E-selectin (Figures 6C and 6D). For this set of data, statistical analysis was not applied as the cytokine array was performed in pooled sera. To identify the candidates for TLR7-mediated endothelial hyperpermeability in murine sepsis, the differently expressed serum cytokines were subjected to bioinformatics analysis using the KEGG pathway enrichment program. Two criteria were applied for the enrichment search: 1) cytokines related to the TLR7 pathway and 2) cytokines related to endothelial adhesion. Six cytokines met both criteria and thus were selected for their potential role in TLR7-mediated endothelial hyperpermeability, which were CCL2, TNFα, CCL5, CXCL2, E-selectin, and ICAM-1 (Figure 6E). Among them, TNFα signaling was among the top-10 ranked pathways (Figure 6F) and thus suggested as the most promising mediator responsible for increased EC permeability and regulated by miR-146a-5p−TLR7 signaling.
Figure 6.
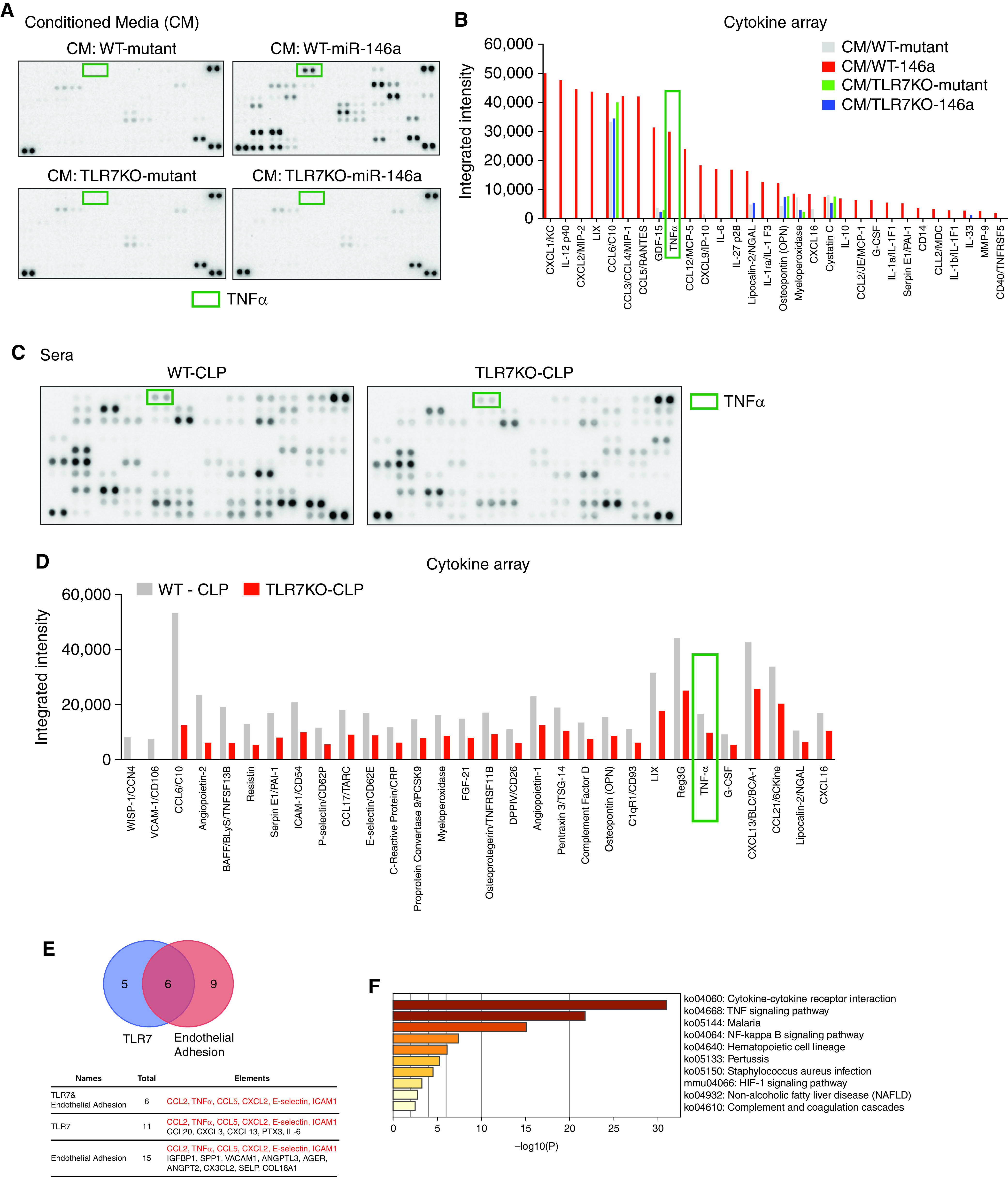
Cytokine array and pathway enrichment analysis identify TNFα as a possible effector responsible for TLR7-dependent endothelial injury. (A–B) Immune blot (A) and quantification of integrated intensity (B) of cytokine array in CM collected 24 hours from mutant- (50 nM) or miR-146a-5p– (50 nM) treated macrophages (WT and TLR7−/−). (C–D) Cytokine array analysis from pooled sera (five mice in each strain) collected 24 hours after CLP procedure from WT and TLR7−/− mice. (C) Immune blot and (D) quantification of the integrated intensity of top 30 differential cytokines. (E) TNFα is related to both TLR7 and endothelial injury. (F) Top-10 ranked pathways analyzed by KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis.
Next, we tested the possible role of TNFα in mediating the CM– or CLP–serum-induced EC barrier disruption. Exogenously added TNFα increased HPAEC permeability in a dose-dependent manner as measured by ECIS (Figure E4A). We also tested the other cytokines (IL-6, CXCL2, IL-12, MIP-1α, IL-27, CXCL16, and IL-1Rα) upregulated in the miR-146a-5p–treated CM as noted above. None of them had any effect on EC permeability (Figures E4B, E4C, and E5). Pretreatment with anti-TNFα (1 μg/ml) of the CM collected from miR-146a-5p–treated Mϕ cultures, but not control IgG, in part attenuated the CM-induced EC dysfunction (Figure 7A) and alleviated release of ICAM-1 (Figure 7B). Barrier function was also preserved when ECs were exposed to WT-CLP sera preincubated with anti-TNFα (10 ng/ml), as evidenced by an approximately 87.5% drop in FITC fluorescence shown in Figures 7C and 7D, a similar amount of that seen in TLR7−/− CLP serum (Figures 7C and 7D). The benefit of blocking TNFα against WT CLP serum-induced permeability changes seemed to be attributed to the structure change of VE-cadherin and actin cytoskeleton. VE-cadherin of EC showed a discontinued membrane structure and internalization, as well as accumulated stress fibers in the WT CLP serum group treated with IgG. Blocking TNFα in WT CLP sera restored the majority of VE-cadherin organization located on the cell surface with less stress fibers, as shown in Figure 7E. After preincubation with anti-TNFα of WT serum or incubation with TLR7−/− serum, VE-cadherin appeared well-organized with thin actin filament in ECs, which was similar to those images of cells without treatment (Figure 5D). These data demonstrate that plasma TNFα is the downstream effector of miR-146a-5p−TLR7 signaling responsible for CM- or sepsis serum-induced EC barrier dysfunction.
Figure 7.
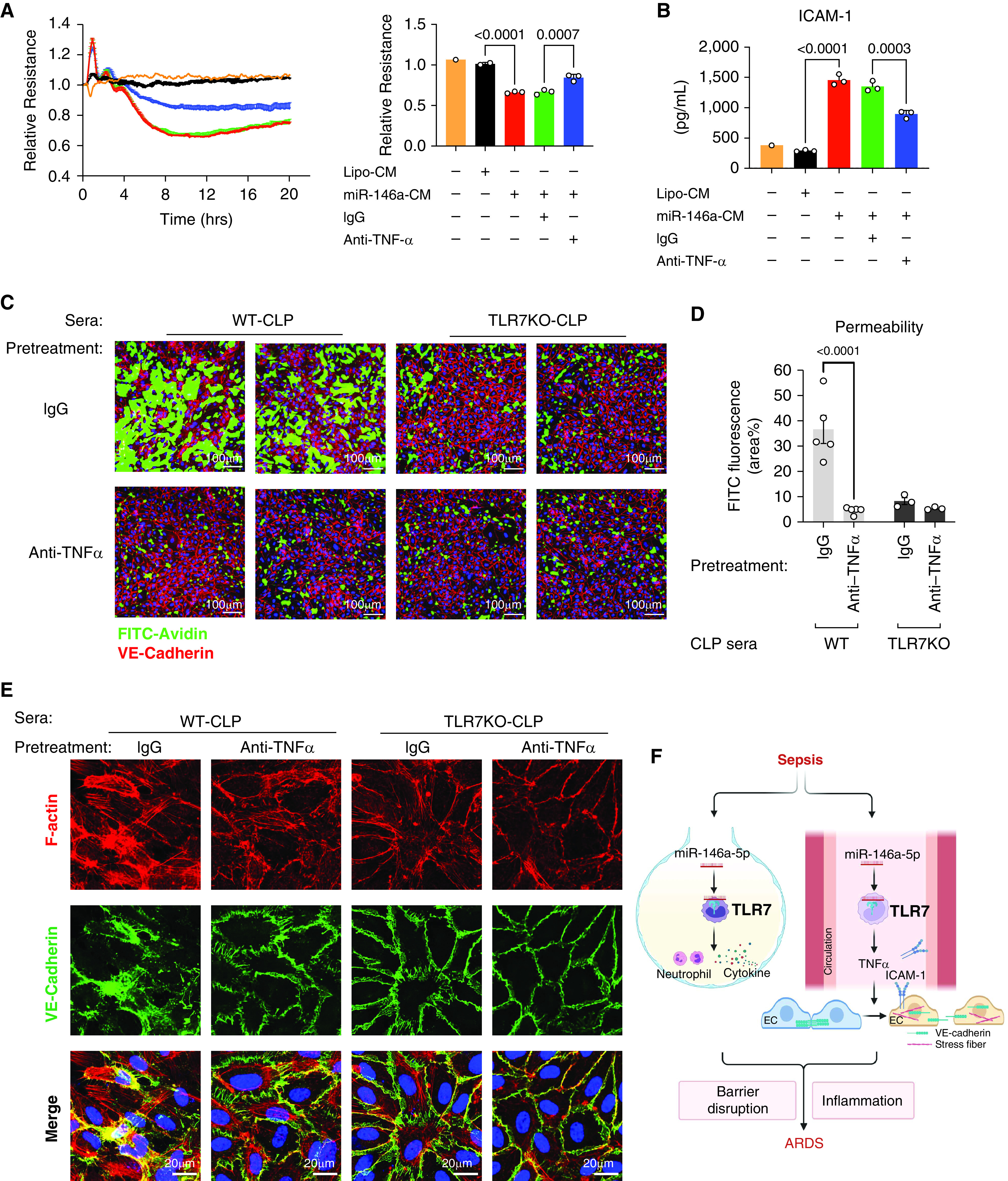
Block TNFα reduces permeability induced by conditioned media and septic sera in ECs. CM or septic sera were incubated with anti-TNFα antibody or control IgG for 45 minutes at room temperature before the treatment. (A) Resistance of ECs was measured and quantified at 10 hours as a bar graph after treatment of CM pretreated with anti-TNFα (1 μg/ml) or IgG (1 μg/ml). Experiments were performed in duplicates or triplicates and repeated at least twice. (B) Media ICAM-1. (C) Representative XperT images of ECs after treatment of sera pretreated with anti-TNFα (10 ng/ml) or IgG (10 ng/ml). (D) Endothelial permeability was quantified by the area percentage of FITC in XperT images. n = 5 per group. (E) VE-cadherin disruption and stress fiber formation in ECs. (F) Schematic illustration of how TLR7 mediates sepsis-induced acute respiratory distress syndrome (ARDS). Cellular miR-146a-5p is released into the alveolar space and circulation after CLP-induced sepsis, which then binds to and activates TLR7 signaling. TLR7 stimulation in the alveolar space induces neutrophil accumulation and inflammatory cytokine production. On the other hand, TLR7 activation in the blood causes endothelial activation and hyperpermeability via its downstream effector TNFα by modulating the structure of VE-cadherin and the formation of stress fiber. Barrier disruption and inflammation then lead to ARDS. Graph was created with BioRender.com. Data were presented as mean ± SEM and analyzed by one-way ANOVA with Tukey’s post hoc test (A–B) or two-way ANOVA (D) with Bonferroni’s post hoc test.
Discussion
In the current study, we identified that one of the G (Guanosine) U (Uridine)-rich miRNAs, miR-146a-5p, was released into the lung alveolar space in a murine sepsis model. Intratracheal administration of exogenous miR-146a-5p mimic evoked an acute innate immune response in the lung and pulmonary hyperpermeability through TLR7-dependent signaling. Furthermore, in a mouse model of sepsis-associated indirect lung injury, TLR7 deficiency led to reduced lung inflammation and preserved barrier function. Barrier disruption in sepsis-associated indirect lung injury, however, was probably not a direct consequence of miR-146a-5p−TLR7 activation in ECs because TLR7 was not detected in human pulmonary artery and microvascular ECs. Cytokine array and bioinformatic analysis from both in vitro cell systems and in vivo murine sepsis models identified TNFα as a critical downstream effector of TLR7 activation leading to EC barrier interruption likely by modulating EC VE-cadherin and cytoskeleton.
miR-146a is one of the widely studied miRNAs likely involved in the pathogenesis of sepsis and ARDS (33). It is expressed in various cell types in the lung, including endothelium (34), epithelial cells (35), alveolar macrophage (36), and monocytes (37). Previous research has been primarily focused on its role as a gene regulator that negatively modulates the inflammatory response in the lung (33). Cellular miR-146a is reportedly upregulated in response to bacterial components such as LPS or proinflammatory cytokines such as IL-1β (37). These intracellularly elevated miR-146a function to limit the exacerbation of inflammation initiated by injurious stimuli as a compensatory mechanism (37). Overexpression of intracellular miR-146a represses inflammatory response in the lung by downregulating its target genes and pathways, such as RNA-binding protein human antigen R (34, 38), IRAK-1 (interleukin-1 receptor-associated kinase 1), and TRAF6 (TNF receptor-associated factor 6) (36), endothelial nitric oxide synthase (34), and NF-κB pathway (39). In addition, we find that cellular miR-146a-5p is released to extracellular space during sepsis in mice and humans (20) and elicits strong proinflammatory responses in immune and nonimmune cells (16, 20, 23). Extracellular vesicles released during sepsis mediate inflammatory responses in the immune cells via carried miRNA cargo and TLR7 signaling (40). In the current study, we discovered that intratracheal administration of exogenous miR-146a-5p inflamed the lung by eliciting lung cytokines, increasing alveolar–capillary permeability, and recruiting neutrophils. These data suggest that extracellular miR-146a-5p in the lung is sufficient to induce pulmonary inflammation and injury. Furthermore, blocking TLR7 activation by replacing all uridine with adenosine in miR-146a-5p (U→A mutant) or using TLR7 knockout mice attenuated miR-146a-5p–induced lung inflammation, suggesting that extracellular miR-146a-5p functions as a TLR7 activator in triggering lung inflammation. Because both alveolar macrophages and bronchial/alveolar epithelial cells express TLR7 (41, 42), the inflammatory response to miR-146a-5p may be a synergistic result of the activation of both cell types. Furthermore, systemic delivery of miR-146a-5p induces pulmonary inflammation, and deficiency of miR-146a reduces the inflammatory response in the alveolar space in polymicrobial sepsis (20). Taken together, these data suggest that miR-146a-5p is a possible host-derived danger molecule capable of inducing lung inflammation via TLR7 signaling.
Taking a loss-of-function approach, our in vivo data suggest that TLR7 signaling contributes to lung inflammation, endothelial activation, and alveolar–capillary barrier disruption in a mouse model of polymicrobial sepsis. We initially hypothesized that proinflammatory miRNAs, such as miR-146a-5p, would cause EC activation and barrier disruption by driving TLR7 signaling in ECs. Surprisingly, we found that treatment with miR-146a-5p did not cause any changes in endothelial permeability in HPAECs or HMVECs, in which TLR7 is undetectable by PCR or Western blot both at the basal and under inflammatory conditions. We also found that CM from miR-146a-5p–treated TLR7 knockout Mϕ or TLR7 knockout septic sera preserved EC permeability by maintaining VE-cadherin lining. Together, these facts support the notion that the contribution of TLR7 to the EC injury we observed in murine sepsis is a secondary effect via downstream mediators of TLR7 activation in cells other than ECs. Cytokine arrays and bioinformatic analysis from CM and sera implicate TNFα as a potential downstream effector of TLR7 signaling in activating ECs. The findings that the TNFα-neutralizing antibody partially blocked CM-induced EC barrier dysfunction in the ECIS assay and reversed septic serum-induced EC hyperpermeability strongly suggest that TNFα is indeed the downstream effector of miR-146a-5p–TLR7 signaling leading to EC activation and barrier disruption. In line with our findings, Evankovich and colleagues (43) observed that extracted RNA fractions, most likely miRNAs, from the plasma of patients with ARDS, can activate human TLR8, a functional homolog of murine TLR7. Together, these studies demonstrate an important role of the nucleic acid sensor by circulating RNA ligands such as miRNAs in innate immune response and endotheliopathy, the hallmark of sepsis-induced indirect lung injury.
Although our work reveals the role of TLR7 in sepsis-induced indirect lung injury, the underlying molecular mechanism remains unclear. In a mouse model, Wang and colleagues (44) report that circulating miR-122 released from the injured liver is transported to the lung, where it induces lung inflammation by activating TLR7 signaling in alveolar macrophages. Moreover, we report that extracellular RNAs, such as miRNAs, induce cytokines and complement production in cardiomyocytes, neutrophils, and macrophages via TLR7 (16, 18, 23, 45). Given the various cell types involved in sepsis pathogenesis, we speculate that extracellular miR-146a-5p in the plasma or alveolar space is released from multiple cell types and interacts with immune and nonimmune cells via TLR7 and unleashes the downstream cascade such as TNFα expression, which can impair EC integrity and contribute to ARDS.
VE-cadherin, a major gap junction component specifically expressed in vascular ECs, plays a critical role in maintaining vascular barrier function in response to a variety of environmental stimuli (28). The cleavage/shedding, internalization, and degradation of VE-cadherin are crucial mechanisms in forming paracellular junctions that account for increased permeability. In an ex-vivo assay, we show that the VE-cadherin lining is disrupted and internalized, and stress fiber is formed in ECs after treatment with WT septic sera. TNFα appears to be responsible for this effect as neutralizing TNFα restores the continuity of VE-cadherin and attenuates the formation of stress fiber. Interestingly, we observe a special structure of VE-cadherin in ECs treated with TLR7−/− septic sera, named JAIL (31). The formation and termination of JAIL are thought to initiate new VE-cadherin clusters that regulate endothelial polarization and angiogenesis, contributing to junction remodeling (31). The presence of a reticular network of VE-cadherin in TLR7−/− sepsis serum-treated ECs may indicate a new molecular mechanism in regulating junction remodeling and barrier function, which warrants future study.
It is known that TNF blockade has failed in several clinical sepsis efficacy trials, but the cause might be multifactorial. The time of intervention and patient heterogeneity were considered to play critical roles (46). Early death from sepsis is caused by a pronounced hyperinflammatory response to the pathogen or danger signals (12). When applying interventions aiming at blocking profound inflammatory responses such as anti-TNF, the timing is critical (46, 47). Study shows that early intervention of anti-TNF therapy prevents patients with severe sepsis from progressing to septic shock (48). Even though TNF blockade failed to demonstrate significant improvement in survival rate in multiple trials, the anti-TNF treatment showed a beneficial trend in those patients with shock by meta-analysis (49). This evidence suggests that anti-TNF therapy might provide maximum benefit if enrolled patients could be carefully stratified and interventions start earlier. We recently demonstrated that plasma miR-146a-5p was positively correlated with several sepsis severity parameters, including complement activation in mice (16), blood lactate concentrations, and severity of coagulopathy in humans (20). Furthermore, TNFα is demonstrated as an effector of miR-146a-5p−TLR7 signaling, contributing to endothelial injury, a hallmark of sepsis pathogenesis. Hence, we speculate that plasma miR-146a-5p may serve as a biomarker that could stratify patients for anti-TNF therapy in the future. Given the fact that blocking TNFα in miR-146a-5p−treated CM only provides partial protection (Figure 7A), we speculate the possibility that different cytokines other than TNFα, such as IL-6, are also involved. Both our data (Figure E4B) and published work (50) suggested that IL-6 alone has little effect. Instead, IL-6 may require the addition of its soluble receptor (IL-6R) to cause permeability changes in vitro (50). IL-6/IL-6R complexes exhibit a higher inflammatory potential than their single form (51). Taken together, the mediators responsible for miR-146a-5p−TLR7 signaling on EC injury may be multiple, but TNFα proves to be a major player.
The dose of miR-146a-5p used in the current study is higher than what we detected in the plasma at an average of 33 fmol/dl in mice at CLP 24 hours and 2.69 fmol/dl in humans with abdominal infection and SOFA (Sequential Organ Failure Assessment) scores between 7 and 12 (data not shown). Of note, it is technically challenging to measure the exact concentration of miR-146a in the extracellular space because of the loss of RNA during extraction. In addition, the release of miR-146a is likely a continuous process as we found that miR-146a expression was sustained in the plasma of CLP mice for up to 7 days (17) and continued delivery of miR-146a subcutaneously for 3 days with plasma concentration around 400 fmol/dl significantly increased lung IL-6 and IL-1β expression (20). In the current study, we took a proof-of-concept approach to establish the contribution of miR-146a-5p−TLR7 signaling to lung inflammation and EC injury. We propose that the continued activation of miR-146a-5p−TLR7 signaling during sepsis serves as a constant proinflammatory stimulator that contributes to lung inflammation and injury.
There are limitations in the current study. We performed in vitro testing by incubating murine macrophage CM and sera with ECs of human lung arterial and microvascular origins. Therefore, we cannot rule out a potential species-specific difference. Furthermore, whereas our data clearly demonstrate the absence of TLR7 expression and lack of direct effect of miR-146a-5p mimic on human ECs in vitro, we could not completely rule out the possibility of direct effects via miR-146a-5p−TLR7 signaling on lung endothelium in vivo, which may be studied in the future by employing EC-specific TLR7 knockout mice via Cre-LoxP approach.
Conclusions
As summarized in Figure 7F, we demonstrate that miR-146a-5p is increased in the lung alveolar space in murine sepsis and that direct intratracheal administration of exogenous miR-146a-5p induces robust lung inflammation and barrier interruption via TLR7-dependent mechanisms. Compared with WT mice, TLR7−/− mice exhibit significant attenuation of acute lung injury as evidenced by reduced pulmonary cytokine production and alleviated barrier loss in sepsis. Both miR-146a-5p–treated macrophage CM and septic sera induce a marked EC barrier disruption in vitro. Cytokine array and bioinformatic analysis suggest TNFα as the key downstream effector of TLR7 signaling in the circulation responsible for the EC barrier dysfunction. Neutralizing the anti-TNFα antibody eliminates the detrimental effect of both miR-146a-5p–treated CM and septic sera on endothelial permeability. Our data provide new insight into the role and molecular mechanism of TLR7 sensing in acute lung injury during sepsis.
Acknowledgments
Acknowledgment
The authors thank Andrew P. Ziman, Advanced Imaging Specialist from Nikon Instruments, for the help in fluorescence imaging. The authors thank the University of Maryland School of Medicine Center for Innovative Biomedical Resources Core Confocal Facility—Baltimore, Maryland, for the assistance in fluorescence imaging. The authors thank Jessica Neder for the help in cell culture and Xiaoxuan Fan from the University of Maryland School of Medicine Center for Innovative Biomedical Resources, Flow Cytometry Core (Baltimore, Maryland) for the assistance in flow cytometry. Additional support was provided by the University of Maryland School of Pharmacy Mass Spectrometry Center (SOP1841-IQB2014). The authors also thank Laurence Wechsler and Gillian Choquette for their diligent proofreading of the manuscript.
Footnotes
Supported by Frontiers in Anesthesia Research Award from International Anesthesia Research Society (W.C.), Faculty Research Award from Shock Society (L.Z.), the Foundation for the National Institutes of Health (R35GM124775 [L.Z.]; R01GM117233, R01GM122908, and R35GM140822 [W.C.]; R01NS110567 [W.C. and L.Z.]; R01HL155051 [A.A.B.]; and R01HL076259 [K.G.B.]), and an S10 grant (S10 OD026698).
Author Contributions: H.H. designed and performed most experiments, analyzed the data, prepared figures, and drafted the manuscript. L.G. performed in vivo experiments, qRT-PCR, ELISA, and data analysis. X.F., S.-H.L., and Y.Y. performed intratracheal injection, flow cytometry, and data analysis. S.W. performed cytokine array, contributed to in vitro ECIS experiments, and analyzed data. J.Z. performed mouse surgeries, cell cultures, qRT-PCR, ELISA, and ECIS experiments. P.C. performed mouse surgeries, assisted with flow cytometry, and contributed to data analysis. J.H. performed mouse surgeries and detected miRNA in BAL. A.S., B.K.S., and B.W. contributed to the experimental design. W.H., M.A.K., and H.H. contributed to bioinformatics analysis. A.A.B., K.G.B., Y.K., and C.-o.Z. contributed to ECIS and XperT assays. K.G.B. contributed to the experimental design and carefully reviewed the manuscript. W.C. supervised the research project and edited the manuscript. L.Z. designed the project, analyzed the data, prepared figures, and finalized the manuscript.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2021-0551OC on June 9, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Tsushima K, King LS, Aggarwal NR, De Gorordo A, D’Alessio FR, Kubo K. Acute lung injury review. Intern Med . 2009;48:621–630. doi: 10.2169/internalmedicine.48.1741. [DOI] [PubMed] [Google Scholar]
- 2. Fein AM, Calalang-Colucci MG. Acute lung injury and acute respiratory distress syndrome in sepsis and septic shock. Crit Care Clin . 2000;16:289–317. doi: 10.1016/s0749-0704(05)70111-1. [DOI] [PubMed] [Google Scholar]
- 3. Iscimen R, Cartin-Ceba R, Yilmaz M, Khan H, Hubmayr RD, Afessa B, et al. Risk factors for the development of acute lung injury in patients with septic shock: an observational cohort study. Crit Care Med . 2008;36:1518–1522. doi: 10.1097/CCM.0b013e31816fc2c0. [DOI] [PubMed] [Google Scholar]
- 4. Sheu CC, Gong MN, Zhai R, Chen F, Bajwa EK, Clardy PF, et al. Clinical characteristics and outcomes of sepsis-related vs non-sepsis-related ARDS. Chest . 2010;138:559–567. doi: 10.1378/chest.09-2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fan E, Brodie D, Slutsky AS. Acute respiratory distress syndrome: advances in diagnosis and treatment. JAMA . 2018;319:698–710. doi: 10.1001/jama.2017.21907. [DOI] [PubMed] [Google Scholar]
- 6. Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest . 2012;122:2731–2740. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Joffre J, Hellman J, Ince C, Ait-Oufella H. Endothelial responses in sepsis. Am J Respir Crit Care Med . 2020;202:361–370. doi: 10.1164/rccm.201910-1911TR. [DOI] [PubMed] [Google Scholar]
- 8. Englert JA, Bobba C, Baron RM. Integrating molecular pathogenesis and clinical translation in sepsis-induced acute respiratory distress syndrome. JCI Insight . 2019;4:124061. doi: 10.1172/jci.insight.124061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Opal SM, van der Poll T. Endothelial barrier dysfunction in septic shock. J Intern Med . 2015;277:277–293. doi: 10.1111/joim.12331. [DOI] [PubMed] [Google Scholar]
- 10. Dolmatova EV, Wang K, Mandavilli R, Griendling KK. The effects of sepsis on endothelium and clinical implications. Cardiovasc Res . 2021;117:60–73. doi: 10.1093/cvr/cvaa070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kumar P, Shen Q, Pivetti CD, Lee ES, Wu MH, Yuan SY. Molecular mechanisms of endothelial hyperpermeability: implications in inflammation. Expert Rev Mol Med . 2009;11:e19. doi: 10.1017/S1462399409001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent JL. Sepsis and septic shock. Nat Rev Dis Primers . 2016;2:16045. doi: 10.1038/nrdp.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xiang M, Fan J. Pattern recognition receptor-dependent mechanisms of acute lung injury. Mol Med . 2010;16:69–82. doi: 10.2119/molmed.2009.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fischer S, Gerriets T, Wessels C, Walberer M, Kostin S, Stolz E, et al. Extracellular RNA mediates endothelial-cell permeability via vascular endothelial growth factor. Blood . 2007;110:2457–2465. doi: 10.1182/blood-2006-08-040691. [DOI] [PubMed] [Google Scholar]
- 15. Kannemeier C, Shibamiya A, Nakazawa F, Trusheim H, Ruppert C, Markart P, et al. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci USA . 2007;104:6388–6393. doi: 10.1073/pnas.0608647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zou L, Feng Y, Xu G, Jian W, Chao W. Splenic RNA and microRNA mimics promote complement factor b production and alternative pathway activation via innate immune signaling. J Immunol . 2016;196:2788–2798. doi: 10.4049/jimmunol.1502106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zou L, He J, Gu L, Shahror RA, Li Y, Cao T, et al. Brain innate immune response via miRNA-TLR7 sensing in polymicrobial sepsis. Brain Behav Immun . 2022;100:10–24. doi: 10.1016/j.bbi.2021.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Feng Y, Chen H, Cai J, Zou L, Yan D, Xu G, et al. Cardiac RNA induces inflammatory responses in cardiomyocytes and immune cells via Toll-like receptor 7 signaling. J Biol Chem . 2015;290:26688–26698. doi: 10.1074/jbc.M115.661835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol . 2014;15:509–524. doi: 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- 20. Wang S, Yang Y, Suen A, Zhu J, Williams B, Hu J, et al. Role of extracellular microRNA-146a-5p in host innate immunity and bacterial sepsis. iScience . 2021;24:103441. doi: 10.1016/j.isci.2021.103441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Williams B, Neder J, Cui P, Suen A, Tanaka K, Zou L, et al. Toll-like receptors 2 and 7 mediate coagulation activation and coagulopathy in murine sepsis. J Thromb Haemost . 2019;17:1683–1693. doi: 10.1111/jth.14543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jian W, Gu L, Williams B, Feng Y, Chao W, Zou L. Toll-like receptor 7 contributes to inflammation, organ injury, and mortality in murine sepsis. Anesthesiology . 2019;131:105–118. doi: 10.1097/ALN.0000000000002706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Feng Y, Zou L, Yan D, Chen H, Xu G, Jian W, et al. Extracellular microRNAs induce potent innate immune responses via tlr7/myd88-dependent mechanisms. J Immunol . 2017;199:2106–2117. doi: 10.4049/jimmunol.1700730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Feng X, Gu L, Zhu J, Wang S, Yang Y, Ke Y, et al. Mir-146a induced acute lung injury via Toll-like receptor 7. Shock . 2020;53:139–145. [Google Scholar]
- 25. Dubrovskyi O, Birukova AA, Birukov KG. Measurement of local permeability at subcellular level in cell models of agonist- and ventilator-induced lung injury. Lab Invest . 2013;93:254–263. doi: 10.1038/labinvest.2012.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, et al. Acute Lung Injury in Animals Study Group An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol . 2011;44:725–738. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci USA . 2004;101:5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vestweber D. VE-cadherin: the major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler Thromb Vasc Biol . 2008;28:223–232. doi: 10.1161/ATVBAHA.107.158014. [DOI] [PubMed] [Google Scholar]
- 29. Orsenigo F, Giampietro C, Ferrari A, Corada M, Galaup A, Sigismund S, et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun . 2012;3:1208. doi: 10.1038/ncomms2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiong S, Hong Z, Huang LS, Tsukasaki Y, Nepal S, Di A, et al. IL-1β suppression of VE-cadherin transcription underlies sepsis-induced inflammatory lung injury. J Clin Invest . 2020;130:3684–3698. doi: 10.1172/JCI136908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cao J, Schnittler H. Putting VE-cadherin into JAIL for junction remodeling. J Cell Sci . 2019;132:jcs222893. doi: 10.1242/jcs.222893. [DOI] [PubMed] [Google Scholar]
- 32. Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol (1985) . 2001;91:1487–1500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 33. Lee LK, Medzikovic L, Eghbali M, Eltzschig HK, Yuan X. The role of microRNAs in acute respiratory distress syndrome and sepsis, from targets to therapies: a narrative review. Anesth Analg . 2020;131:1471–1484. doi: 10.1213/ANE.0000000000005146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cheng HS, Sivachandran N, Lau A, Boudreau E, Zhao JL, Baltimore D, et al. MicroRNA-146 represses endothelial activation by inhibiting proinflammatory pathways. EMBO Mol Med . 2013;5:1017–1034. doi: 10.1002/emmm.201202318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perry MM, Moschos SA, Williams AE, Shepherd NJ, Larner-Svensson HM, Lindsay MA. Rapid changes in microRNA-146a expression negatively regulate the IL-1beta-induced inflammatory response in human lung alveolar epithelial cells. J Immunol . 2008;180:5689–5698. doi: 10.4049/jimmunol.180.8.5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bobba CM, Fei Q, Shukla V, Lee H, Patel P, Putman RK, et al. Nanoparticle delivery of microRNA-146a regulates mechanotransduction in lung macrophages and mitigates injury during mechanical ventilation. Nat Commun . 2021;12:289. doi: 10.1038/s41467-020-20449-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA . 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Comer BS, Camoretti-Mercado B, Kogut PC, Halayko AJ, Solway J, Gerthoffer WT. MicroRNA-146a and microRNA-146b expression and antiinflammatory function in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol . 2014;307:L727–L734. doi: 10.1152/ajplung.00174.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cowan C, Muraleedharan CK, O’Donnell JJ, III, Singh PK, Lum H, Kumar A, et al. MicroRNA-146 inhibits thrombin-induced NF-κB activation and subsequent inflammatory responses in human retinal endothelial cells. Invest Ophthalmol Vis Sci . 2014;55:4944–4951. doi: 10.1167/iovs.13-13631. [DOI] [PubMed] [Google Scholar]
- 40. Xu J, Feng Y, Jeyaram A, Jay SM, Zou L, Chao W. Circulating plasma extracellular vesicles from septic mice induce inflammation via microRNA- and tlr7-dependent mechanisms. J Immunol . 2018;201:3392–3400. doi: 10.4049/jimmunol.1801008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roberts AW, Lee BL, Deguine J, John S, Shlomchik MJ, Barton GM. Tissue-resident macrophages are locally programmed for silent clearance of apoptotic cells. Immunity . 2017;47:913–927.e6. doi: 10.1016/j.immuni.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shikhagaie MM, Andersson CK, Mori M, Kortekaas Krohn I, Bergqvist A, Dahl R, et al. Mapping of TLR5 and TLR7 in central and distal human airways and identification of reduced TLR expression in severe asthma. Clin Exp Allergy . 2014;44:184–196. doi: 10.1111/cea.12176. [DOI] [PubMed] [Google Scholar]
- 43. Evankovich J, Lear T, Baldwin C, Chen Y, White V, Villandre J, et al. Toll-like receptor 8 stability is regulated by ring finger 216 in response to circulating microRNAs. Am J Respir Cell Mol Biol . 2020;62:157–167. doi: 10.1165/rcmb.2018-0373OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang Y, Liang H, Jin F, Yan X, Xu G, Hu H, et al. Injured liver-released miRNA-122 elicits acute pulmonary inflammation via activating alveolar macrophage TLR7 signaling pathway. Proc Natl Acad Sci USA . 2019;116:6162–6171. doi: 10.1073/pnas.1814139116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shimada BK, Yang Y, Zhu J, Wang S, Suen A, Kronstadt SM, et al. Extracellular mir-146a-5p induces cardiac innate immune response and cardiomyocyte dysfunction. Immunohorizons . 2020;4:561–572. doi: 10.4049/immunohorizons.2000075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Grau GE, Maennel DN. TNF inhibition and sepsis -- sounding a cautionary note. Nat Med . 1997;3:1193–1195. doi: 10.1038/nm1197-1193. [DOI] [PubMed] [Google Scholar]
- 47. Cohen J, Heumann D, Glauser MP. Do monoclonal antibodies and anticytokines still have a future in infectious diseases? Am J Med . 1995;99:45S–52S, discussion 52S–53S. doi: 10.1016/s0002-9343(99)80286-1. [DOI] [PubMed] [Google Scholar]
- 48. Abraham E, Glauser MP, Butler T, Garbino J, Gelmont D, Laterre PF, et al. p55 Tumor necrosis factor receptor fusion protein in the treatment of patients with severe sepsis and septic shock. A randomized controlled multicenter trial. Ro 45-2081 study group. JAMA . 1997;277:1531–1538. [PubMed] [Google Scholar]
- 49. Qiu P, Cui X, Sun J, Welsh J, Natanson C, Eichacker PQ. Antitumor necrosis factor therapy is associated with improved survival in clinical sepsis trials: a meta-analysis. Crit Care Med . 2013;41:2419–2429. doi: 10.1097/CCM.0b013e3182982add. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Goldman JL, Sammani S, Kempf C, Saadat L, Letsiou E, Wang T, et al. Pleiotropic effects of interleukin-6 in a “two-hit” murine model of acute respiratory distress syndrome. Pulm Circ . 2014;4:280–288. doi: 10.1086/675991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pallua N, Low JF, von Heimburg D. Pathogenic role of interleukin-6 in the development of sepsis. Part II: significance of anti–interleukin-6 and antisoluble interleukin-6 receptor–alpha antibodies in a standardized murine contact burn model. Crit Care Med . 2003;31:1495–1501. doi: 10.1097/01.CCM.0000065725.80882.BD. [DOI] [PubMed] [Google Scholar]