

Abstract
Clinical and molecular heterogeneity are common features of human disease. Understanding the basis for heterogeneity has led to major advances in therapy for many cancers and pulmonary diseases such as cystic fibrosis and asthma. Although heterogeneity of risk factors, disease severity, and outcomes in survivors are common features of the acute respiratory distress syndrome (ARDS), many challenges exist in understanding the clinical and molecular basis for disease heterogeneity and using heterogeneity to tailor therapy for individual patients. This report summarizes the proceedings of the 2021 Aspen Lung Conference, which was organized to review key issues related to understanding clinical and molecular heterogeneity in ARDS. The goals were to review new information about ARDS phenotypes, to explore multicellular and multisystem mechanisms responsible for heterogeneity, and to review how best to account for clinical and molecular heterogeneity in clinical trial design and assessment of outcomes. The report concludes with recommendations for future research to understand the clinical and basic mechanisms underlying heterogeneity in ARDS to advance the development of new treatments for this life-threatening critical illness.
Keywords: ARDS, biological mechanisms, clinical trials, critical care
Acute respiratory distress syndrome (ARDS) is characterized by severe abnormalities of pulmonary gas exchange not fully explained by cardiac failure or fluid overload (1). As a syndrome, ARDS is not a single disorder with a well-defined cause and a dominant pathophysiological pathway to guide the development of potential treatments, which creates major challenges for developing new clinical and pharmacological therapies. Although substantial progress has been made in understanding the clinical and molecular heterogeneity of ARDS, this progress has not yet been translated into new therapies, and many questions remain (2).
In 2021, the Aspen Lung Conference, entitled “ARDS in the 21st Century: New insights into clinical and mechanistic heterogeneity”, addressed emerging concepts of heterogeneity in ARDS and implications for new diagnostic and therapeutic approaches. The specific goals of the Conference were to address the importance of heterogeneity in ARDS with a focus on 1) understanding the presence and therapeutic significance of ARDS mechanistic and phenotypic subtypes; 2) exploring multicellular and multisystemic mechanisms responsible for this heterogeneity; and 3) determining how to best account for disease heterogeneity during clinical trial design and assessment of outcomes. In this summary, we highlight key points and new developments that were discussed and conclude with a discussion of areas for continuing research.
Historical Evolution of the Importance of Clinical and Molecular Heterogeneity in ARDS
The importance of clinical and pathophysiological heterogeneity has been recognized since the original description of ARDS by Ashbaugh and colleagues, and key milestones are shown in Table 1. The first report included 12 patients with acute lung injury from diverse etiologies, including trauma, pancreatitis, gastric aspiration, drug overdose, and suspected viral pneumonia (3). Mortality was 58% (7/12), and pathologic examination of the lungs suggested severe injury to the gas exchange parenchyma. Five patients were treated with positive end-expiratory pressure (PEEP) ranging from 5 to 10 cm H2O, with improvement in oxygenation, suggesting that PEEP might be an effective therapy, and lung samples from two patients who died had reduced surface tension lowering properties in vitro, suggesting that surfactant depletion might contribute to the pathophysiology, which was confirmed in subsequent studies (4, 5). The pathologic findings suggested that ARDS might be a common response to heterogeneous causes of lung injury.
Table 1.
Key Milestones in the History of Acute Respiratory Distress Syndrome
| Authors | Item | Sponsor | References |
|---|---|---|---|
| Pre–COVID-19 | |||
| Ashbaugh, et al. & Petty | Description of ARDS, with heterogeneous causes. | None | (3) |
| Staub, et al. | Mechanisms of edema formation and clearance; the concept of hydrostatic vs. increased permeability edema. | NHLBI | (6, 7) |
| Clements, et al. & Avery, et al. | Biology of lung surfactant; the importance of lung epithelium. | NHLBI | (13, 14) |
| Webb & Tierney | Demonstration that high VT injures rat lungs. | NHLBI | (26) |
| Bachofen & Weibel | Morphologic features of acute lung injury. | (17) | |
| Matthay, et al. | Physiology of alveolar fluid clearance. | NHLBI | (9, 10) |
| Wiener-Kronish, et al. | Differential responses of endothelial and epithelial barriers in the lungs. | NHLBI | (260) |
| Ware, et al. | Better alveolar fluid clearance in ARDS is associated with a better outcome, building on advances in the physiology of edema fluid clearance in the lungs. | NHLBI | (12) |
| ARDSnet ARMA | Improved outcome with low VT ventilation. Factorial trial design. |
NHLBI | (29) |
| Other trials of clinical care | Conservative fluid therapy, prone positioning, neuromuscular blockade, HFNO. | NHLBI and others | (53, 60, 62, 64) |
| Herridge, et al. | Long-term outcomes after ARDS. | (72, 73) | |
| NHLBI SCOR & SCCOR programs | Characterization of alveolar inflammation and repair before and after the onset of ARDS. | NHLBI | (36, 37, 40, 45) |
| Parsons, et al. | Use of plasma biomarkers to understand effects of interventions (low VT reduces biomarkers of inflammation and injury). | NHLBI | (30) |
| Calfee, et al. | Latent class analysis to define clinical subgroups. | NHLBI | (92) |
| NIH/NHLBI Petal Network | Treatment trials enrolling subjects before the ICU. | NHLBI | (261) |
| Frat, et al. (FLORALI group & REVA network) | HFNO Trial: changed practice before and after the onset of COVID-19. | REVA | (62) |
| COVID-19 era | |||
| RECOVERY investigators | Design of large open-label trials of potential therapies in the pandemic setting (e.g., dexamethasone). | UK | (65) |
| NIH, RECOVERY, REMAP-CAP | Large clinical trials with negative (hydroxychloroquine) and positive (dexamethasone, tocilizumab, and baricitinib) signals in moderate, severe, and critical COVID-19 illness. | NIAID, others | (65–67, 261) |
Definition of abbreviations: ARDSnet = Acute Respiratory Distress Syndrome Network; ARMA = Lower Tidal Volume Trial; COVID-19 = coronavirus disease; FLORALI = High-Flow Oxygen through Nasal Cannula in Acute Hypoxemic Respiratory Failure; HFNO = High-flow nasal oxygen; NHLBI = National Heart, Lung, and Blood Institute; NIAID = National Institute of Allergy and Infectious Diseases; NIH = National Institutes of Health; RECOVERY = Randomised evaluation of COVID-19 therapy; REMAP-CAP = Randomized Embedded Multifactorial Adaptive Platform for Community-acquired Pneumonia; REVA = Réseau Européen de Recherche en Ventilation Artificielle Network; SCOR = Specialized Centers of Research; SCORR = Specialized Centers of Clinically Oriented Research; VT = tidal volume.
Initial progress in understanding acute lung injury was on the basis of identifying mechanisms of edema formation and clearance in the lungs, the importance of the lung surfactant system, inflammatory mechanisms in injured lungs, and the consequences of ventilator-induced mechanical forces across alveolar walls (Table 1). Norman Staub used Starling’s equation to define mechanisms responsible for edema formation in the lungs, leading to the critical distinction between edema caused by increased hydrostatic pressure versus increased vascular permeability (6). A sampling of undiluted pulmonary edema fluid by direct endobronchial aspiration in newly intubated patients showed that most patients with clinically defined acute lung injury had high permeability protein edema, defined by a high edema fluid to plasma protein ratio (7).
The critical role of the lung epithelium in reabsorbing edema fluid was shown in a series of studies by Matthay and colleagues showing active fluid and protein clearance from the airspaces of intact lungs. Alveolar fluid reabsorption is driven by active sodium and chloride transfer through specific epithelial sodium and chloride channels (8–10), whereas alveolar proteins are absorbed by separate transporters (11). Alveolar fluid is reabsorbed more rapidly across the epithelium than protein, causing the edema fluid protein concentration to rise with time in normal lungs. The clinical relevance of this experimental finding was shown in an important human study by Ware and colleagues (12). When lung edema fluid from patients with acute lung injury was sampled two times soon after intubation, the patients in whom the edema fluid protein concentration rose had a better prognosis, suggesting that relatively preserved alveolar epithelial function is an important determinant of the outcome of acute lung injury in humans.
The important role of lung surfactant was established in studies by Clements and colleagues, who showed that alveolar type II cells produce surfactant that stabilizes alveolar units at low lung volumes, and by Avery and colleagues, who showed that surfactant replacement could save newborns with infant respiratory distress syndrome (13, 14). Petty’s original observation about abnormal surfactant function in injured lungs led to trials of various surfactant replacement strategies in adults with ARDS, but these were unsuccessful (15, 16).
The classic ultrastructural studies of Bachofen and Weibel showed that ARDS is characterized by extensive injury to lung microvascular endothelial cells and alveolar type I cells, microvascular thrombosis, prominent neutrophilic infiltrates in the alveolar spaces, denuded alveolar basement membranes and areas of reepithelialization by type II cells, suggesting areas of active epithelial repair (17). The prominence of neutrophils in the inflammatory alveolar infiltrates and the known capacity of neutrophils to produce oxidants and release proteolytic enzymes led to the hypothesis that neutrophils were the primary drivers of the acute lung injury process; however, additional studies showed that neutrophil migration does not necessarily injure tissue and that acute lung injury also occurs in neutropenic adults and children, so neutrophils are not absolutely required (18–22). These observations supported the concept of heterogeneous mechanisms causing acute lung injury. Microvascular thrombosis was identified in early autopsy studies and also is a prominent feature of lung injury because of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (23–25). Thus, severe hypoxemia in patients with ARDS is explained largely by mismatching and shunting because of alveolar exudates, the collapse of alveoli because of surfactant depletion, and microvascular abnormalities.
The recognition that some patients with ARDS required high inspiratory pressures to support ventilation led to the hypothesis that high distending pressures might injure relatively normal areas of injured lungs. In a classic study, Webb and Tierney found that ventilation of rats with high inspiratory pressures caused hemorrhagic lung injury and that low concentrations of PEEP were protective (26). These findings were confirmed and extended by Parker and colleagues and Dreyfuss and colleagues and formed the rationale for the NIH (National Institutes of Health)-sponsored ARMA (Lower Tidal Volume Trial) that showed that ventilating patients with relatively low tidal volumes (6 ml/kg vs. 12 ml/kg predicted body weight) and a plateau pressure limit of 30 cm H2O was associated with a significant reduction in mortality in ARDS (27–29). Importantly, the lung-protective strategy was associated with a significant reduction in plasma biomarkers of inflammation as well as endothelial and alveolar epithelial injury, suggesting that mechanical forces across the injured alveolar epithelium were driving some of the alveolar inflammation (30, 31). This landmark study changed the standard of practice for how to ventilate patients with ARDS.
Although the mortality from acute lung injury was high after the initial description of ARDS, many patients survived, raising important questions about how injured lungs are repaired. Henson and colleagues were instrumental in identifying the cellular processes involved in the resolution of inflammation, including the important role of macrophages in the phagocytosis of neutrophils and clearance of cellular debris from the airspaces (32–34). Studies by Snyder and colleagues and Clark and colleagues showed that markers of fibroblast proliferation and collagen production are detectable in lung fluids soon after the onset of ARDS, which made the important point that repair processes begin very soon after the onset of injury (35–37). A persistent fibroproliferative response in the lungs occurs in some survivors and has an adverse effect on long-term outcomes (38).
The Lung Division of the National Heart, Lung, and Blood Institute of the NIH has had an important role in advancing basic and clinical studies of the mechanisms and treatment of ARDS through the funding of Specialized Centers of Research (SCORs) and Specialized Centers of Clinically Oriented Research (SCCORs) and Clinical Trial Networks (ARDSnet, PETAL [Prevention and Early Treatment of Acute Lung Injury]). The SCOR and SCCOR programs contributed a great deal to understanding inflammation and repair in the lungs of patients with ARDS and also supported small pilot studies of potential new therapies. BAL was shown to be a safe procedure in critically ill patients (39), and sequential BAL sampling during the course of ARDS showed important differences in the evolution of lung inflammatory responses in patients with sepsis versus trauma, reflecting heterogeneity by etiology (40). Subsequent studies described components of the lung inflammatory response, including cytokines and cytokine balance, early markers of repair, the importance of cell death pathways, and other pathophysiological events (41–46). These studies, along with studies of pulmonary edema fluid obtained by direct endotracheal aspiration, showed the value of studying samples obtained directly from the site of injury in the lungs (12, 47). The SCCOR programs also conducted proof-of-concept studies of potential new therapies, including granulocyte-macrophage colony stimulating factor (GM-CSF) (i.v.), activated protein C (i.v.), and enteral ω3 fatty acids, but none of these pilot studies suggested a benefit (48–50).
The Clinical Trial Networks established by the NHLBI (National Heart, Lung, and Blood Institute) Lung Division (ARDSnet, PETAL) have had key roles in sponsoring clinical studies in ARDS (51, 52). Importantly, the investigators in these networks had the foresight to collect plasma samples from enrolled subjects for the study of biological mechanisms that might be reflected in plasma, adding a great deal of value to these clinical trials. The second of the ARDSnet trials (ARMA) used an innovative 2 × 2 factorial design to study the antiinflammatory effects of ketoconazole together with the effect of lowering tidal volume during mechanical ventilation. Although the drug treatment arm with ketoconazole showed no benefit, the mechanical ventilation arm showed a significant and clinically important reduction in mortality, as noted above (29). A subsequent trial used a similar factorial design to test the effects of liberal versus conservative fluid therapy in one arm and two different clinical monitoring strategies (pulmonary artery catheters vs. central venous catheters) in the other (53). This second landmark trial showed that the fluid conservative strategy led to a shorter time on mechanical ventilation, whereas using pulmonary artery catheters for patient monitoring did not provide a measurable clinical benefit. Each of these results regarding clinical management changed clinical practice, although lack of uniform application in “real world” use could contribute to heterogeneity in clinical outcomes. Additional trials of pharmacological treatments for ARDS have not shown consistent benefits, including statins, enteral omega-3 fatty acid and antioxidant supplementation, inhaled albuterol, corticosteroids, and others (54–59).
Other international networks, including the French Réseau Européen de Recherche en Ventilation Artificielle Network (REVA), also have performed important clinical trials of therapies for all-cause ARDS. The PROSEVA (Proning Severe ARDS Patients) trial was based on earlier studies showing that prone positioning can redistribute lung fluid density on computed tomography scanning and showed that a protocol of intermittent prone positioning improved short- and long-term outcomes in ventilated patients with moderate or severe ARDS (P/F < 150) (60, 61). The High-Flow Oxygen through Nasal Cannula in Acute Hypoxemic Respiratory Failure (FLORALI) trial of different methods of oxygenation support in patients with nonhypercapnic respiratory failure showed a beneficial effect of high-flow nasal oxygen on 90-day mortality and reduced need for mechanical ventilation in the subgroup with moderate to severe ARDS (P/F ⩽ 200 mm Hg) (62). Neuromuscular blockade in patients with severe ARDS was shown to have a beneficial effect in a French trial, but this was not reproduced in the NIH Network ROSE (Reevaluation of Systemic Early Neuromuscular Blockade) trial that enrolled similar patients (63, 64). International networks organized to study coronavirus disease (COVID-19)-associated ARDS have identified new therapies with positive signals, including dexamethasone, tocilizumab, and baricitinib, but whether these will translate into successful treatments for all-cause ARDS remains to be seen (65–67).
A great deal of progress also has been made in understanding the epidemiology and outcomes of ARDS, and the concept of heterogeneity has been important. A comprehensive survey of mechanically ventilated hospitalized patients in King County, Washington, showed that the incidence of acute lung injury was approximately 86/100,000 per year, amounting to 190,000 cases annually in the United States, with a mortality of 24% in patients 15–19 years old rising to over 40% in patients 65 years of age or older (68). A subsequent study using ICD9 (International Classification of Diseases, Ninth Revision) codes for hospitalized patients produced higher annual estimates (69). The LUNGSafe Study (Large observational study to UNderstand the Global impact of Severe Acute respiratory FailurE) was an international cross-sectional study of all patients in 459 ICUs in 50 countries over a 1-month period (70). This study showed that ARDS is still underrecognized, even in high-quality ICUs, and provided mortality data for categories of ARDS severity defined by the Berlin Consensus Conference criteria. Hospital mortality was 40% overall and ranged from 34.9% to 40.3% to 46.1% for mild, moderate, and severe ARDS, respectively. The COVID-19 pandemic has greatly increased the number of cases of ARDS worldwide (Figure 1) (71).
Figure 1.

Effect of the coronavirus disease (COVID-19) pandemic on numbers of ARDS cases. COVID-19 acute respiratory distress syndrome (ARDS) cases (940,000) are estimated from COVID-19 deaths in the United States from March 2020 through February 2022, assuming 80% of deaths had severe pneumonia/ARDS and mortality of 20% from COVID-19 ARDS. ARDS cases before COVID-19 (380,000) are estimated for a similar period from the annual incidence data of Rubenfeld and colleagues. (68). Modified with permission from reference (71).
The work of Herridge and colleagues provided new insights into the long-term outcomes of patients who survived ARDS before the COVID-19 pandemic at 1 and 5 years after hospital discharge (72, 73). Although lung function improved by 1 year in most survivors, persistent neuromuscular and psychological dysfunction were identified as major determinants of long-term disability. Prehospital health status and comorbidities also have emerged as important risk factors for events after hospitalization (74–76).
As noted, ARDS is a clinical syndrome associated with heterogeneous clinical events, and underlying host factors are likely to be important contributors to heterogeneity in clinical outcomes. For the most part, the clinical features of COVID-19 ARDS are very similar to those of all ARDS (77). In most cases, the initial pathophysiological event appears to be the activation of innate immune mechanisms in the lungs and systemic organs, triggered by infectious agents (eg, microbes and viruses) and/or primary tissue damage from noninfectious causes (eg, acid aspiration or physical trauma). In COVID-19 ARDS, both the innate and the adaptive immune systems have important roles, and both are likely to be involved in non–COVID-19 ARDS as well (78, 79). Differences in the severity of the initial event, as well as individual differences in host inflammatory responses, are likely to underlie heterogeneity in the clinical severity of the disease and response to therapy.
Understanding molecular heterogeneity has been fundamental in developing new treatments for cancer as well as pulmonary diseases like cystic fibrosis and asthma, but less progress has been made in understanding molecular heterogeneity in ARDS. The discovery of the specific molecular abnormalities in cystic fibrosis led to the development of effective medications for patients with molecular variants in the cystic fibrosis conductance regulator protein (80). Identification of different clinical phenotypes of asthma and the importance of the IL-5 and IL-4/IL-13 pathways (Th2 phenotype) has led to effective new molecular therapies for the subgroup of patients with severe eosinophilic asthma (81–84). Similar advances are needed for ARDS to improve patient-centered treatment and move toward precision medicine (2, 71).
Clinical and Molecular Dimensions of Heterogeneity in ARDS
Carolyn Calfee reviewed major advances in understanding the clinical heterogeneity of ARDS. The importance of clinical and pathophysiological heterogeneity in ARDS has been recognized since Steinberg and colleagues showed that hospital mortality was higher and lung inflammatory responses were more persistent in patients with sepsis-associated vs those with trauma-associated ARDS (40). The NIH ARDSnet low tidal volume study collected plasma samples to use circulating biomarkers to investigate the biological basis for the beneficial effect of low tidal volume ventilation. Improved survival in the low tidal volume (VT) group was associated with lower plasma concentrations of IL-8, IL-6, and sTNFR1 (soluble tumor necrosis factor receptor-1), suggesting reduced systemic inflammation, whereas lower plasma concentrations of SP-D (surfactant protein D) and RAGE (receptor for advanced glycation end-products) suggested less alveolar injury (30, 85–87). These and other plasma biomarkers of endothelial (e.g., von Willebrand factor and ANG2 [angiopoietin 2]) and epithelial (SP-D and RAGE) injury have been evaluated as tools for predicting outcome (42, 88–91).
A major conceptual advance occurred when Calfee and colleagues used latent class analysis (LCA) to identify subgroups of patients with ARDS with different outcomes (92). LCA is a validated statistical method that uses baseline parameters in an unbiased computational approach to determine whether there are distinct subgroups at baseline within a potentially heterogenous population. With this approach, two subclasses (latent phenotypes) of patients were identified using clinical, laboratory, and biomarker data from two large NHLBI-sponsored clinical trials, ARMA (lower versus higher VT) and ALVEOLI (Assessment of Low Tidal Volume and Elevated End-Expiratory Lung Volume to Obviate Lung Injury) (lower versus higher PEEP). Patients in Class 2 (Phenotype 2, 31% of patients) included patients with more severe clinical characteristics, higher plasma concentrations of inflammatory biomarkers, and worse outcomes, whereas patients in Class 1 (Phenotype 1, 71% of patients) had less severe illness, lower concentrations of inflammatory biomarkers, and better outcome. For example, patients with Phenotype 2 (“hyperinflammatory”) had significantly more vasopressor use, lower plasma bicarbonate and protein C concentrations, and higher plasma concentrations of IL-8, IL-6, and TNFR1. In the ARMA cohort, mortality was 44% for Phenotype 2 versus 23% for Phenotype 1 (P = 0.006), and there were significantly fewer ventilator-free and organ failure-free days. Importantly, these findings were present in both the ARMA and ALVEOLI cohorts and were not explained by the overall severity of illness scores. In analyses of data from eight separate randomized controlled trials, mortality in patients with the hyperinflammatory phenotype (Phenotype 2) was consistently higher, ranging from 38–60% for Phenotype 2 versus 19–33% for Phenotype 1 (Figure 2). These two ARDS phenotypes appeared to be stable over the first 3 days of mechanical ventilation and were associated with different responses to PEEP and fluid administration (92–94). By contrast, different treatment responses were not observed in subgroups defined by overall severity of illness scores.
Figure 2.

Mortality in subclasses is defined by latent class analysis in eight different clinical trials of ARDS. The numbers at the bottom of each column show the percentage of patients in each subclass in the clinical trial population. The proportion in subclass 2 (hyperinflammatory) ranged from 26–40% across trials. Data are from references (92, 94–96, 100, 104).
When these phenotypes were used to evaluate response to therapy in a trial of simvastatin in ARDS, that showed no overall effect, the patients with the hyperinflammatory phenotype appeared to have benefitted from simvastatin therapy (95), raising the possibility that this approach to phenotyping patients at baseline might be a useful way to manage heterogeneity in clinical trials of new therapies. However, a similar retrospective analysis of these phenotypes in the NHLBI trial of rosuvastatin in ARDS did not show a differential response to treatment (96). When long-term outcomes were evaluated by phenotype, delirium was more common during the ICU stay in Phenotype 2, but there were no differences in patient-reported outcomes or functional status in survivors after hospital discharge (97). More information is needed about factors during critical illness that contribute to heterogeneity in long-term outcomes.
These phenotypes also have been identified in patients with COVID-19– associated ARDS, suggesting that they are not unique to specific etiologies of lung injury (98). In one study, patients with COVID-19 with the more hyperinflammatory Phenotype 2 had a better treatment response to corticosteroids, although the steroid treatment was not controlled or randomized (99).
Importantly, the LCA-defined phenotypes also have been identified in several different large observational cohorts of ARDS, including the Validating Acute Lung Injury markers for Diagnosis cohort at Vanderbilt and the Early Assessment of Renal and Lung Injury cohort at the University of California, San Francisco (100). Similar phenotypes (termed “reactive” and “uninflamed”) have been identified in the Molecular Diagnosis and Risk Stratification for Sepsis cohort in Europe (101). Taken together, the hyper- and hypoinflammatory phenotypes identified by LCA analysis have been identified in over 4,000 patients in randomized and observational cohorts. Recent evidence also suggests that the hyper- and hypoinflammatory phenotypes identified in patients with ARDS are relevant in patients who are mechanically ventilated but do not meet the criteria for ARDS, as well as the broader population of patients with hypoxemic respiratory failure (102, 103).
An important question is whether these clinical phenotypes can be identified rapidly and prospectively so that patients might be stratified by phenotype when they enter clinical trials. Sinha and colleagues used a machine learning approach to identify important variables in three NIH cohorts and then validated the models in three independent ARDS cohorts, using LCA as the gold standard (104). Six classifier variables were identified, including five serum or plasma markers (IL-6, IL-8, TNFR1, HCO3−, Protein C) and one clinical variable (vasopressor use). Models combining 3 or 4 of these accurately classified the LCA phenotypes, suggesting that as technology for rapid measurements improves, prospective classification of patients by phenotype at baseline will become feasible.
The biological basis for the differences in the two clinical phenotypes identified by LCA is not yet clear. The variables that are most different between Phenotypes 1 and 2 suggest that Phenotype 2 has more intense systemic inflammation at baseline. Whether this difference is because of differences in the intensity of the initial stimulus (e.g., an inoculum of microbial agent), differences in individual host responses to the stimulus, or both, is not clear. Transcriptomic analysis of blood samples from patients with ARDS has identified differences in upregulated and downregulated genes in patients classified as reactive versus uninflamed, an important first step in understanding pathways that are differentially regulated (105). As an alternative approach to blood sampling, Sarma and colleagues performed a transcriptomic analysis using tracheal aspirate samples derived from the lung compartment to identify biological differences between COVID-19 and non–COVID-19 ARDS (106). This more lung-specific approach could lead to a better understanding of the key events and molecular pathways in the lungs that characterize the hyper- and hypoinflammatory phenotypes.
Overall, the use of LCA has been a major advance in understanding heterogeneity in ARDS, but several key issues need to be considered in using this approach (Table 2).
Table 2.
Key Questions About Latent Phenotypes
| Generalizability | Are ARDS phenotypes also present in nonrandomized trials or in “real-world” patients? |
| Feasibility | Can ARDS phenotypes be identified in real-time to allow use in the stratification of subjects? |
| Specificity | Are the identified phenotypes specific for ARDS, or are they applicable more broadly to critical illness? |
| Mechanism | What are the biological determinants of differences and outcomes in each phenotype? |
| Approach | How should clinical trials incorporate clinical phenotypes? |
Definition of abbreviation: ARDS = acute respiratory distress syndrome.
Cellular and Molecular Heterogeneity in ARDS
Nuala Meyer reviewed new approaches to understanding molecular heterogeneity in critical illness, focusing on baseline genetic determinants, gene expression in peripheral blood mononuclear cells, and plasma proteomics. SNPs in genes involved in inflammatory responses and tissue injury have been used to identify heterogeneity in risk and treatment effects in critical illness and identify possible causal contributors (107). For example, SNPs in the IL-1RA gene are associated with higher plasma IL-1RA, lower risk of ARDS, and lower mortality in sepsis (108, 109). A retrospective subgroup analysis of a trial of IL-1RA in sepsis showed that the risk of death was related to baseline plasma concentrations of IL-1RA and that treatment with IL-1RA improved survival in patients with high baseline IL-1RA concentrations (110). A recent trial of anakinra (ILβ-1 receptor antagonist) in patients with moderate to severe COVID-19 illness selected for higher risk on the basis of high baseline concentrations of plasma soluble urokinase/plasminogen activator receptor, a biomarker of inflammation in COVID-19, showed significant benefit in reducing mortality and hospital stay (111). On the other hand, genetic variants associated with higher plasma concentrations of an endothelial marker, ANG2, and an epithelial marker, sRAGE (soluble RAGE), are strongly associated with the onset of ARDS, implicating ANG2 and sRAGE as contributors to ARDS risk (89, 112).
SNPs in a number of other genes have been associated with outcomes in ARDS, but these have not been tested prospectively because of the difficulty in performing sequencing fast enough to classify patients at baseline. In general, single SNPs explain only a small part of the variability in critically ill patient populations, but gene- or protein-controlling SNPs could be more informative, particularly when combined with clinical variables, such as those identified using LCA.
Peripheral blood leukocytes provide an opportunity to study the heterogeneity of “immunophenotypes” of patients with a critical illness. In patients with COVID-19 illness, an analysis using single-cell mRNA profiling identified three dominant patterns of lymphocyte responses (113). Patients with immunotype 1 had highly activated CD4 and CD8 cells; patients with immunotype 2 had highly active T-bet+ CD4 and CD8 cells plus activation of memory B cells; whereas patients with immunotype 3 had little activation of CD4 or CD8 cells. More information is needed about whether this approach could be useful in the broader population of patients with ARDS, in whom bacterial infections predominate. As with genetic phenotyping, immunophenotyping is not routinely available but is helpful in studying the underlying pathophysiology of illness, including responses to therapy and potential new biomarkers for rapid classification of patients at baseline.
Proteomics approaches have been explored in ARDS and applied to understanding the pathogenesis of COVID-19 illness (114–117). A small study comparing BAL fluid from normal volunteers and patients with ARDS identified protein networks associated with inflammation, infection, and tissue injury in the lungs (115). In COVID-19 illness, plasma proteomics analysis identified protein patterns associated with the initial severity of illness, including 27 new candidates for biomarkers of disease severity (116). In a separate study of over 700 different plasma proteins in 161 patients with moderate versus severe COVID-19 illness, Cosgriff and colleagues identified a combination of 9 proteins representing different dysregulated pathways that separated patients with moderate versus severe disease (118).
Although technological advances have greatly reduced the time needed for proteomics analyses, the methodology remains too complex to be used in a prospective manner to classify patients at baseline. However, the proteomics approach is useful in understanding pathways involved in the pathogenesis of disease and identifying new biomarkers.
Computational Approaches to Phenotyping Patients with Disease
Lisa Bastarache reviewed new computational approaches to extracting information from electronic health records (EHRs) that are not well-organized for research (119). These new approaches can facilitate research about a range of primary diseases that may or may not have been identified as known ARDS risk factors and the modifying effects of comorbid conditions. Similarly, the risks of different outcomes can be evaluated for an index disease, such as ARDS or critical illness. Combining data from EHRs in different medical centers that use identical or similar EHRs greatly expands the database of patients at risk or with established ARDS.
There are numerous ways to extract patient-level phenotype data from the EHR. Phenotype algorithms can be used to define cases and controls for a single disease outcome by combining various data elements (e.g., laboratory results, medication orders, ICD codes, etc.) using a series of predefined rules (120). These algorithms can achieve high accuracy across institutions but may take considerable time to develop and implement (121).
An alternative approach to EHR phenotyping is to use “phecodes” that combine related ICD-9 and ICD-10 billing codes into higher-level disease-related codes. Phecodes were originally developed to conduct phenomewide association studies to scan for phenotypic associations with common genetic variants, but they also have been used to study genetic and nongenetic risk factors for numerous chronic and acute conditions (122). ICD-based phecodes are powerful because they allow researchers to rapidly ascertain case or control status for diseases across the medical phenome. Moreover, they can aggregate data across institutions because of the relative standardization of ICD codes. Although there is heterogeneity in the way ICD codes are assigned, phecodes reduce heterogeneity by combining related ICD codes into clinically relevant groups to facilitate clinical research. Importantly, in many EHRs, ICD diagnosis codes capture outpatient as well as inpatient data, so phecodes have the potential to provide an innovative way to study clinical events before, during, and after critical illness.
Heterogeneity of Outcomes in ARDS and Critical Illness
Catherine Hough reviewed advances and challenges in understanding the heterogeneity of outcomes in survivors of ARDS and critical illness. Initial studies showed that survivors left the hospital with restrictive pulmonary function abnormalities that tended to resolve over 6 months, but they often had important physical and neuropsychiatric disabilities and impaired overall health status (123–126). In a comparison of outcomes in matched groups of critical patients with or without ARDS, long-term survival was similar whether or not patients had a diagnosis of ARDS (127). Long-term outcome was determined in large part by age, an underlying risk factor for ARDS and baseline comorbidities, and older patients with sepsis had the worst long-term survival. This study provided an important clue that outcomes after ARDS are heterogeneous and are associated with many factors other than ARDS, including baseline age, health status and comorbid disease, ARDS risk factors, and more.
The landmark studies of Herridge and colleagues of survivors 1 and 5 years after recovery from ARDS showed that the most common disability at 1 year was related to physical functioning (72). At 5 years, many survivors had persistent physical and psychological sequelae and higher overall healthcare costs (73). A high proportion of caregivers for survivors of critical illness also report depressive symptoms (128).
The recognition that critical illness by itself is an important determinant of outcome led to a series of studies showing a strong relationship between critical illness, cognitive function, and physical function after hospital discharge (75, 129–131). The term “Post-Intensive Care Syndrome” was coined to describe this collection of abnormalities in survivors of critical illness, and the COVID-19 pandemic has amplified its importance (132–134). In observational studies, risk factors for cognitive and physical impairment have included ICU delirium, older age, longer ICU stay, and immobility, among others (135–137). Importantly, in two different studies, functional independence before the ICU stay predicted better long-term functioning (135, 136). Other parameters related to the critical illness, including the underlying cause, the “hyperinflammatory” phenotype, plasma markers of inflammation and coagulation, and medications such as corticosteroids and neuromuscular blockers have not had consistent relationships to long-term outcomes across studies (64, 97, 138, 139). More information is needed about risk factors, including treatments that can be modified during the hospital stay and the pathophysiological mechanisms that affect long-term outcomes.
One important possibility is that heterogeneity in patient outcomes might obscure subgroups that could benefit from specific approaches to modify or treat risk factors. Gandotra and colleagues applied LCA to data from a large trial of rehabilitation of survivors of acute respiratory failure to identify patient subgroups with different outcomes and the characteristics of patients in each group (140). Four subgroups were identified that differed in the degree and rate of recovery of physical functioning. The group with the best recovery included younger females with a shorter length of stay and fewer days of sedation, whereas the group with the slowest recovery included older patients with a longer length of ICU stay and more sedation days.
Although knowledge has improved about the domains of disability and the effects of patient heterogeneity on overall outcomes after hospitalization, specific approaches have not had major effects on long-term physical or cognitive function (141–144), mental health (145–147), or overall health-related quality of life in survivors (148–150).
Major challenges remain in understanding the underlying pathophysiology of long-term outcomes of critical illness. Table 3 lists some of the elements in a roadmap for improving long-term outcomes. In particular, better methods are needed to assess health status before hospitalization and health trajectory before critical illness. Larger prospective observational cohorts are needed in which subgroups with different health outcomes can be identified and evaluated. Improved specific and measurable intermediate outcomes are needed for mechanistic and interventional studies, along with better ways to minimize bias and identify competing risks. Increasing collaboration across the spectrum of healthcare providers, caregivers, and survivors of critical illness will be important for identifying ways to improve long-term outcomes.
Table 3.
Strategies to Improve Outcomes after Critical Illness
|
Clinical Trials in the Age of Heterogeneity
Taylor Thompson reviewed clinical trial designs in the age of heterogeneity (Table 4), recognizing that achieving the goal of “precision medicine” means matching new therapies to individuals or subgroups of patients who are most likely to benefit (2, 71). Precision medicine is most easy to achieve when the molecular cause of a disease is known and a specific targeted therapy exists, as in cystic fibrosis (80). “Relative” precision medicine can be achieved when clinical subgroups of a disease have been defined, the pathways responsible for those subgroups have been defined, and a therapeutic agent has been developed that targets key nodes in those pathways, as in asthma (82–84). However, ARDS as a disease entity is not yet in either of these categories.
Table 4.
Selected Enrichment Strategies Used or Proposed for Acute Respiratory Distress Syndrome Clinical Trials
| Trial/Author | Enrichment Strategy | Intervention | Findings/Rationale |
|---|---|---|---|
| ACURASYS, Papazian (63) ROSE, PETAL Network (64) |
ARDS Severity P/F < 120–150 P/F < 120 |
Early neuromuscular blockade | ACURASYS demonstrated higher placebo mortality in and benefits limited to the P/F < 120 subsets (prognostic and predictive enrichment, respectively). Did not replicate in ROSE. |
| PROSEVA, Guerin (60) | ARDS Severity P/F < 150 |
Prone positioning | Large treatment effect in moderate to severe ARDS concordant with prior metanalyses suggesting predictive enrichment. |
| LASRS, Steinberg (58) | ARDS for 7–28 d | Methylprednisolone | Attempted to enrich for a steroid-responsive phase of ARDS (fibro-proliferation). Late steroids (>14 d) may be harmful. |
| Wilson (262) Spragg (16) |
Direct vs. indirect injury | Surfactant replacement | Benefit with pediatric direct lung injury. Did not replicate in adults. |
| Constatin (263) | Focal vs. diffuse ARDS | Personalized ventilator strategy: higher Vt and lower PEEP for focal versus lower Vt and higher PEEP for diffuse ARDS | No difference in mortality; high rates of misclassification and higher mortality if a strategy is applied to the incorrect subgroup. |
| Calfee (264) | Trauma vs. nontrauma | Reduce heterogeneity by studying traumatic ARDS separately | Lower mortality is not explained by baseline clinical factors; biomarker profiles suggest the differing extent of epithelial and endothelial injury. |
| Villar (265) Goligher (266) |
Evaluate stability on standardized ventilator settings Assess physiologic responsiveness during a run-in period |
Enroll only persistent ARDS Randomize to higher vs. lower PEEP in PEEP responders only |
Reevaluation after 24 h enriches for higher mortality. Analysis of PEEP responsiveness in RCTs suggests a potential for predictive and prognostic enrichment. |
| Gattinoni (267) Goligher (268, 269) |
Match lung-protective intervention to physiology to optimize benefit/risk | Assess for recruitability or lung weight (CT) ECCO2R for subset likely to have a ⩾ 5 cm H2O drop in driving pressure Titration of tidal volume to elastance |
Modeling and observational data suggest potential for both prognostic and predictive enrichment. |
| Calfee (95) | ARDS subclass | Simvastatin for Class 2 (“Hyperinflammatory”) ARDS (see text) | Post hoc analysis of RCT demonstrates mortality benefit limited to Class 2 ARDS. |
| Lai (270) Sinha (271) |
Markers of dysregulated coagulation, high dead space fraction or ventilatory ratio, and RV function by cardiac ultrasound | Anticoagulants or pulmonary vascular targeted therapies | Identify subsets with or at risk for microvascular thrombi, vascular remodeling, pulmonary hypertension, or adverse outcomes. |
Definition of abbreviations: ACURASYS = ARDS et Curarisation Systematique; ARDS = acute respiratory distress syndrome; ECCO2R = extracorporeal CO2 removal; LASRS = Late Steroid Rescue Study; PEEP = positive end-expiratory pressure; PETAL = Prevention and Early Treatment of Acute Lung Injury; PROSEVA = Proning Severs ARDS Patients; RCT = Randomized Clinical Trial; ROSE = Reevaluation of Systemic Early Neuromuscular Blockade; Vt = tidal volume.
Therapeutic progress in ARDS has been on the basis of large controlled randomized clinical trials and low tidal volume ventilation, and conservative fluid management and prone positioning are now part of the standard of care (29, 53, 60). These trials compared interventional groups with comparison groups in a randomized manner, although they were not blinded because the treatments were obvious. The large sizes suggested the results would be generalizable to similar patient populations and the statistically significant differences between groups suggested that the results were not because of chance. The ARMA Trial of low tidal volume ventilation and FACTT (Fluids and Catheters Treatment Trial) of fluid and catheter management used factorial designs to test more than one therapy, thereby increasing efficiency and enrolled unselected participants with ARDS, unlike the PROSEVA trial of prone ventilation that focused on the subset of patients with ARDS with moderate to severe ARDS, as defined by baseline PaO2/FiO2 (P/F) < 150. This decision was on the basis of prior trials suggesting benefit in this subset, illustrating predictive enrichment by selecting a subset most likely to benefit, as well as a subset anticipated to have higher mortality, illustrating prognostic enrichment, which increases power, reduces the sample size, and improves risk-to-benefit ratio.
Despite these positive trials of clinical management, virtually all clinical trials of new pharmacological therapies for ARDS have failed to show significant effects, and attempts at predictive or prognostic enrichment have been limited to clinical features and not predictive biomarkers tightly linked to the drug target. In addition to severity by initial P/F ratio, these include the cause or timing of ARDS (e.g., trauma versus nontrauma, direct vs. indirect, early vs. late). Prospective enrichment by ARDS subclass assignment as defined by LCA shows great promise (92).
Several new approaches to improving clinical trial efficiency include the use of platform designs incorporating master protocols, the use of plausible biomarkers to prospectively enrich patient populations at baseline for treatment-responsive subsets, and/or to allow biomarker-adaptive designs and the use of adaptive-intervention trials (151–153). The COVID-19 pandemic has shown the value of large platform trials that use master protocols to test multiple agents simultaneously or in parallel. Platform trials may use placebo-controlled “efficacy” designs suitable for testing new agents to provide rigorous data for registration of new drugs or biologics or pragmatic unblinded randomized trials to examine repurposing existing drugs for new indications. The randomised evaluation of COVID-19 therapy (RECOVERY) platform trials have shown that it is possible to use a platform design to enroll large numbers of patients with a range of illness severity and test multiple proposed therapies in a very short period of time (154). Success in RECOVERY with dexamethasone in critically ill patients with COVID-19 illness led to a rapid change in clinical care because of the low cost and widespread availability of the drug (65). Platforms testing similar interventions have joined forces to hasten discovery, such as the ACTIV-4a (Accelerating COVID-19 Therapeutic Interventions and Vaccines), REMAP-CAP (Randomized Embedded Multifactorial Adaptive Platform for Community-acquired Pneumonia), and ATTACC (Antithrombotic Therapy to Ameliorate Complications of COVID-19) studies of anticoagulation for COVID-19 (155, 156). The I-SPY-COVID platform trial is modeled after the successful I-SPY 2 (Investigation of Serial Studies to Predict Your Therapeutic Response With Imaging And moLecular Analysis 2) trial in breast cancer and is designed to rapidly screen potential new treatments for large effects using small clinical trial populations (157, 158). I-SPY 2 identified two pathway-specific treatments for breast cancers with distinct molecular signatures. Whether or not these promising approaches will succeed in COVID-19 illness or in all-cause ARDS, in which key molecular pathways have not been identified, remains to be seen. However, the overall COVID-19 experience clearly demonstrates the feasibility, flexibility, and efficiency of platform designs, and such designs could be the key to advancing precision medicine objectives for the treatment of ARDS (2).
Although a number of different plausible biomarkers have been identified and tested retrospectively in clinical trials of ARDS, none have been used prospectively to stratify patient populations, in part because of the difficulty in measuring baseline blood biomarkers quickly enough to enable them to be used in real-time to stratify patients. Candidate biomarkers include markers of epithelial injury in the lungs (e.g., RAGE and SP-D) and microvascular integrity or activation (e.g., von Willebrand Factor or ANG2) (86, 87, 89, 159). An alternative to single blood biomarkers is to identify phenotypes of illness using clinical and laboratory parameters and use the phenotypes as biomarkers, as in the work of Calfee and colleagues (92).
The biomarker strategy must be matched to the strength of evidence for the biomarker to define the disease and/or predict the outcome (Figure 3) (160). If the biomarker evidence is strong, patients might be entered into the trial on the basis of the positive biomarker so that a drug is tested only in patients who are biomarker-positive. If the biomarker evidence is less strong, then the drug effects must be tested prospectively in biomarker-positive as well as biomarker-negative patients to define predictive validity. A biomarker-adaptive threshold design allows the evaluation of an overall treatment effect in a study population in combination with a predefined cut-point of the biomarker to identify subsets with a greater treatment effect (161). This approach addresses the concept of heterogeneity of treatment effect, in which a positive result is obscured by baseline heterogeneity of risk in the population (107, 162, 163). In this way, subgroup(s) of patients can be identified who might have a positive benefit/risk ratio in a trial in which they otherwise might be unrecognized because of the overall heterogeneity of baseline risk for the outcome of interest.
Figure 3.
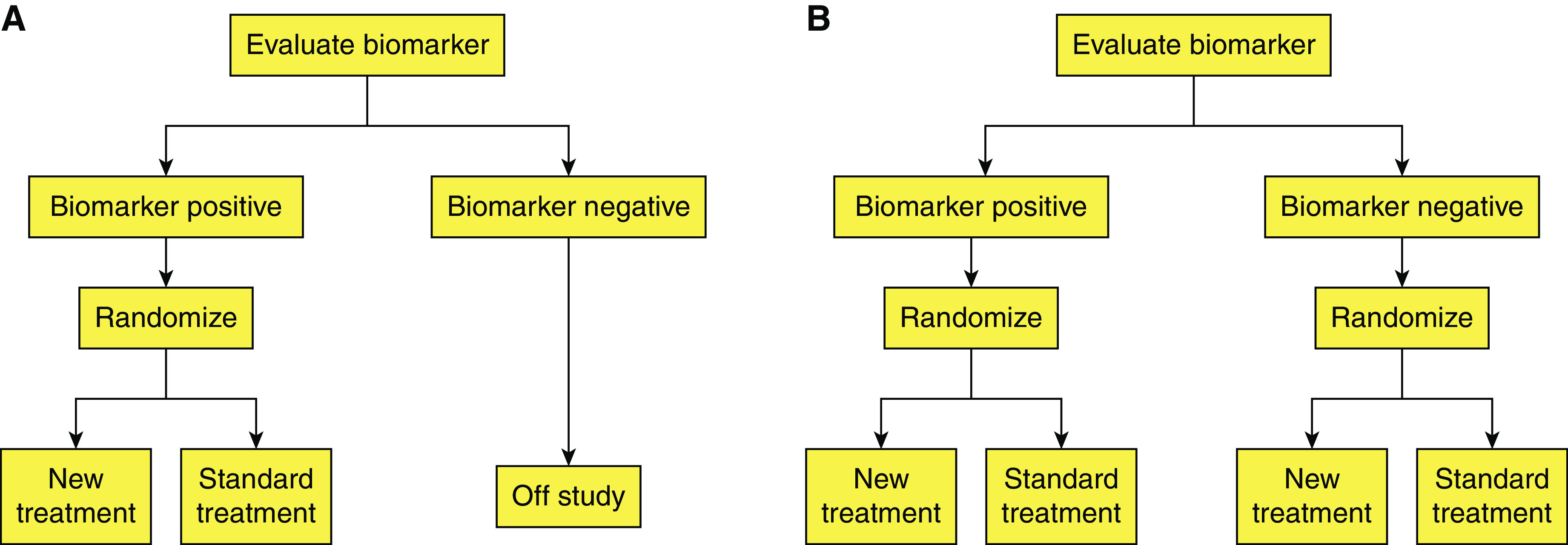
Biomarker strategies in clinical trial designs. (A) Biomarker enrichment designs use a biomarker to select a subpopulation for inclusion. (B) Biomarker stratified designs evaluate a treatment in biomarker-positive and biomarker-negative populations so that a treatment effect occurs in biomarker-positive but not in biomarker-negative participants. Reprinted by permission from reference 160.
Lastly, adaptive intervention trials using sequential randomization could be used for testing several new therapies for ARDS in the same trial. Adaptive intervention trials first randomize participants to one of two potential treatments, then look for prespecified treatment responses (164). Responders continue on the initial treatment to the prespecified end of the trial. Nonresponders in each treatment group are rerandomized to a second treatment that can be given either alone or in combination with the original treatment, depending on the trial design rules. This is an improved use of clinical trial participants, provided that the initial treatment does not modify responsiveness to the second treatment and the characteristics of the underlying illness are reasonably stable over time. This latter factor could be very challenging for critical care trials. An overall approach to new clinical trials in ARDS has been summarized in a recent NIH/NHLBI Workshop Report on precision medicine in ARDS (2).
Pathogenesis
Monocyte/Macrophage Dynamics
Susanne Herold reviewed new information about macrophage dynamics and functional heterogeneity in lung injury and repair. Macrophage heterogeneity is determined in part by their origin (tissue-resident vs. recruited) and by programming (polarization phenotype) imprinted by signals received during recruitment into the lungs, as well as the inflamed or resolving microenvironment (summarized in Figure 4). Macrophage polarization phenotypes can be proinflammatory and damaging or injury-resolving and epithelial-protective (165–168).
Figure 4.
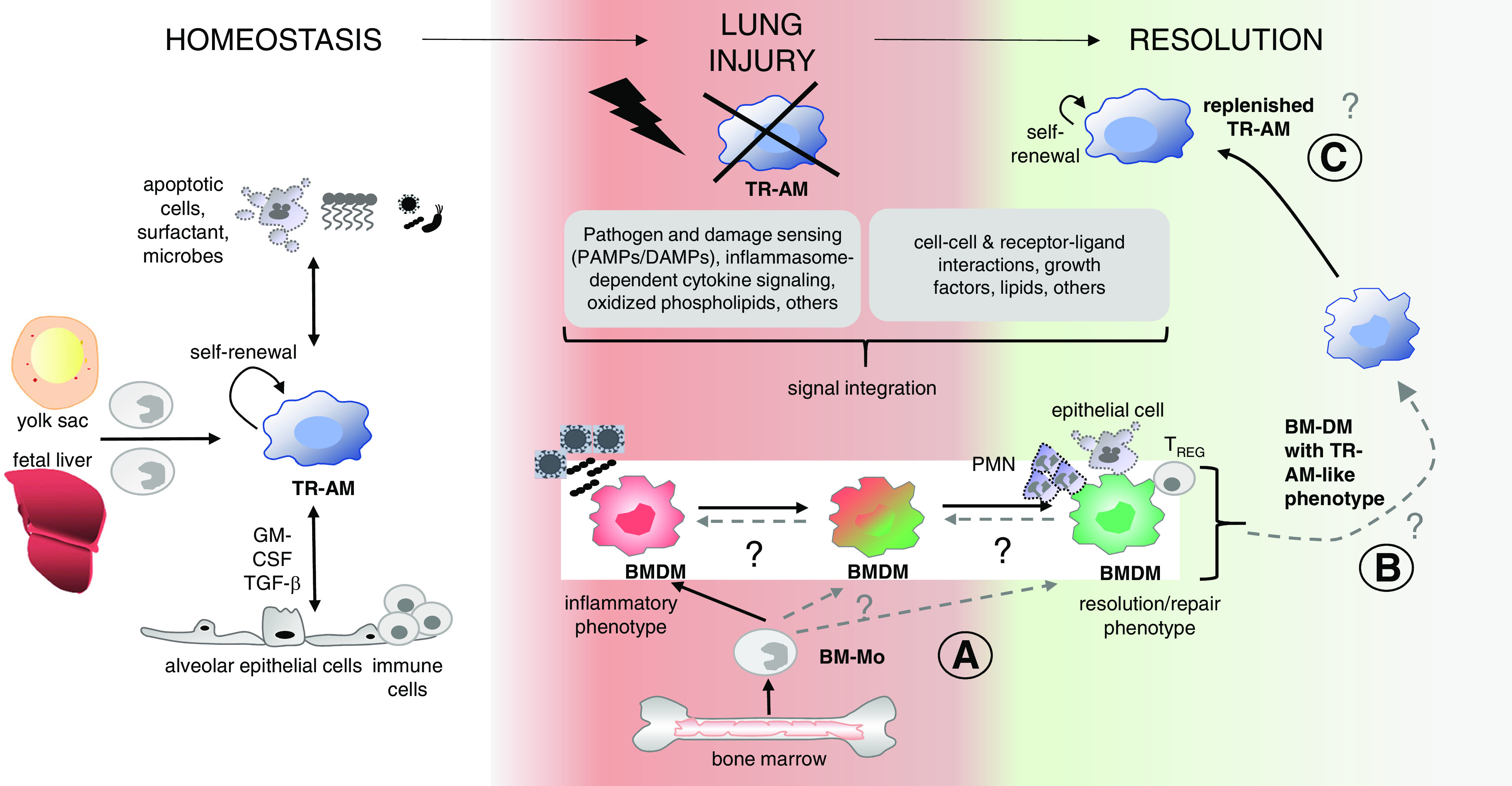
Lung macrophages: ontogeny and mechanisms of functional programming in injury and repair. Tissue-resident alveolar macrophages (TR-AMs) originate from the yolk sac and fetal liver monocytes during lung development. In homeostasis, TR-AMs are replenished by self-renewal, and the phenotype is determined by interaction with niche cells and niche-specific factors (e.g., surfactant, phagocytosed apoptotic cells, local microbes, and regulatory cytokines [e.g., epithelial GM-CSF and TGF-β]). In lung injury, TR-AMs are depleted, and BM-Mos enter the lung and differentiate into bone marrow-derived macrophages (BMDMs). BMDMs integrate diverse signals that create BMDM phenotypes in a spatially resolved and time-dependent manner. These phenotypes comprise a spectrum from the inflammatory BMDM that contributes to epithelial cell injury yet also has host defense functions and the resolution/repair BMDM that resolves alveolar inflammation and drives tissue repair. During resolution, BMDMs replenish depleted TR-AMs. These newly appearing TR-AMs often retain transcriptomic and epigenetic signatures different from the initial “homeostatic” TR-AMs, creating “innate immune memory”. Areas of uncertainty (A, B, and C) are shown with dashed lines and (?) and relate to questions about whether defined polarization phenotypes of BMDMs give rise to TR-AMs in a disease-specific context and whether and how innate memory functions of replenished TR-AMs relate to precursor BMDM polarization phenotypes. Modified with permission from reference 168; permission conveyed through Copyright Clearance Center, Inc. BM-Mo = bone marrow-derived monocytes.
Recent studies using cell surface markers and single-cell RNA sequencing have identified subtypes of airspace leukocytes in normal volunteers and critically ill patients (169–172). The lungs of healthy volunteers contain two major macrophage subsets defined by gene expression patterns and a minor population of mononuclear cells that resemble blood mononuclear cells and dendritic cells, implying low steady-state trafficking of blood monocytes into the airspaces during homeostasis (172). In the setting of acute lung inflammation, blood monocytes are recruited into the lungs, creating a mixed population of resident and recruited mononuclear cells with gene expression profiles that reflect their origins (171, 173–175).
Single-cell RNA sequencing analysis of alveolar myeloid cells from patients with ARDS has revealed considerable diversity compared with normal volunteers or mechanically ventilated patients without ARDS, including enrichment of subtypes that may regulate inflammation and repair (170). Notably, the expression of PD-L1 that inhibits the proinflammatory activity of CD4+ and CD8+ T cells was significantly lower on alveolar macrophages from patients with ARDS who experienced prolonged mechanical ventilation compared with normal subjects or patients on mechanical ventilation without ARDS. In patients with ARDS who survived and were extubated by Day 28, the temporal gene expression pattern in macrophages showed enrichment of innate immune inflammatory profiles on Day 1, followed by downregulation on Days 4 and 8 after onset, whereas in patients who had died or were still intubated on Day 28 there was progressive upregulation of proinflammatory programs (176). These data suggest that reprogramming of airspace macrophages into less inflammatory phenotypes is a crucial step in resolving lung injury in patients with ARDS and highlights lung macrophages as major orchestrators of inflammation and resolution processes.
Lung macrophages also have important interactions with lymphocyte subpopulations and can form feedback loops that sustain alveolar inflammation. Studies of lung cells from patients with SARS-CoV-2 infection show that lung macrophages infected with the virus produce lymphocyte chemoattractants that recruit T cells into the lungs, where they produce IFN-γ and drive macrophage-dependent cellular inflammation (177, 178). Recent single-cell/single-nuclei RNA sequencing and proteomics data from two cohorts of severely ill patients with COVID-19–ARDS revealed a recruited macrophage subset with a distinct proinflammatory and profibrotic profile that could contribute to fibrotic lung remodeling (179).
Taken together, these and other studies are consistent with the paradigm shown in Figure 4. In normal homeostasis, lung macrophages constitute a slowly self-renewing population whose primary function is noninflammatory clearance of inhaled particulates, apoptotic cells, proteins, and other airspace constituents. When airspace macrophages recognize pathogen- or damage-associated molecular patterns, they trigger innate immune–inflammatory mechanisms with the recruitment of bone marrow-derived monocytes into the airspaces that can have either proinflammatory or regenerative phenotypes, depending on the local context (165, 166, 168).
Recent groundbreaking studies have used three-dimensional lung organoids seeded with alveolar macrophages of different phenotypes to study their role in development, antiviral immune response, and regeneration. Coculture of murine bronchioalveolar stem cells with lung mesenchymal cells results in highly branched structures composed of bronchi and alveoli containing the respective epithelial cell types (basal, ciliated, club, and alveolar type I/II cells), as well as differentiated mesenchyme (180–182). When the lung organoids were infected with influenza virus (H7N7 or H1N1) via the bronchial tree, the virus was detectable initially in airway epithelial cells, then later in alveolar epithelial cells. The airway epithelial cells produced IFN-β, confirming a relevant host response to the viral infection. Microinjection of tissue macrophages into the organoids resulted in the local release of proinflammatory cytokines such as TNF-α, IL-1β, and IL-6 in infected but not uninfected organoids, showing that this complex experimental system recapitulates key immunological events in normal lungs, including macrophage–epithelial crosstalk. This model makes it possible to study the specific effects of distinct macrophage subtypes in the alveolar microenvironment and the biological importance of lung macrophage heterogeneity in homeostasis and disease.
Neutrophils, Platelets, and Neutrophil Extracellular Traps (NETs) in ARDS
Mark Looney reviewed new information about neutrophils, platelets, and NETs in inflammation and ARDS. Neutrophil accumulation in the airspaces is a prominent feature of ARDS, but there is little information about the biological or temporal heterogeneity of neutrophils in the blood or airspaces. Evidence from a small study of patients with COVID-19 illness suggests that in patients with mild-to-moderate disease, blood neutrophils have upregulated IFN-stimulated gene signatures, whereas these are not present in blood neutrophils from patients with more severe diseases, including COVID-19–ARDS (183). Whether this reflects individual patient responses or the intensity of COVID infection is unclear. More information is needed about the biology of neutrophils and other leukocytes in the blood and lungs of patients with critical illness and ARDS.
One aspect of neutrophil function that has attracted considerable attention is the formation of NETs (184). NETs are web-like structures of uncoiled nuclear and mitochondrial DNA that also contain cytosolic and granular proteins. They can trap and neutralize bacteria, fungi, viruses, and parasites and prevent bacterial dissemination, all of which benefit host antimicrobial defenses (185). NET release occurs by NETosis, which can occur through two processes (Figure 5). Lytic NETosis leads to slow cell death, beginning with the disassembly of the nuclear envelope and ending with plasma membrane rupture and release of uncoiled nuclear chromatin. Nonlytic NETosis is rapid and is associated with abrupt degranulation and expulsion of nuclear chromatin and associated proteins into the surrounding environment, resulting in anucleate cytoplasts that remain capable of phagocytosis (186). A variety of microbial and host stimuli, including microbes, immune complexes, crystals, and others, cause NETosis by triggering cell surface receptors and activating intracellular signaling pathways. NET formation is regulated in part by host deoxyribonucleases (DNases) that degrade extracellular DNA (187).
Figure 5.
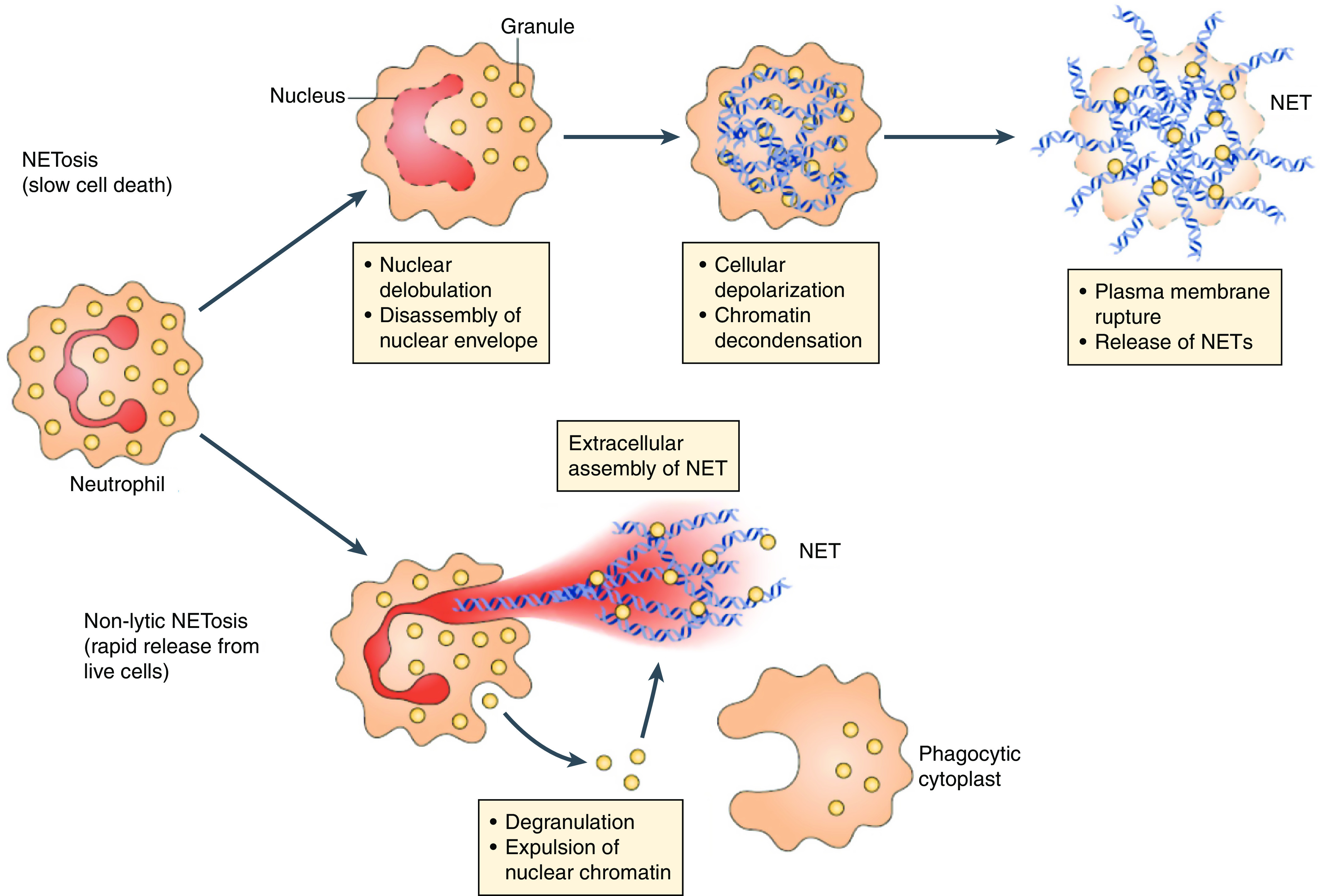
Neutrophil extracellular trap (NET) formation pathways. NETosis is slow and begins with nuclear delobulation and disassembly of the nuclear envelope, followed by loss of cellular polarization, chromatin decondensation, plasma membrane rupture, and cell death. Nonlytic NETosis can occur independently of cell death and involves the secreted expulsion of nuclear chromatin and granule proteins, leaving anucleated cytoplasts that retain phagocytic capacity. Reprinted with permission from reference 184.
Beyond their role in antimicrobial defenses, NETs also have roles in acute and chronic inflammation and may cause tissue damage (188). For example, NETs can damage the pulmonary epithelium in bacterial infections and the lung endothelium in transfusion-related lung injury and may be involved in bacterial sepsis (189–192). In addition, NET formation is associated with vascular thrombosis in mice (193). NETs were shown to be pathogenic in two different models of lung transplantation and were more abundant in BAL fluid of lung transplant recipients with severe primary graft dysfunction (194). Furthermore, treatment of mice with primary graft dysfunction with endobronchial DNase I to lyse NETs improved oxygenation and reduced parameters of lung injury. NET formation can be driven by platelet-neutrophil interactions and contributes to the pathogenesis of transfusion-related lung injury in animal models and humans (190). NET formation also has a role in asthma, as patients with asthma with high concentrations of extracellular DNA and NETs in sputum comprise a subset of patients with lower asthma control scores and more frequent corticosteroid use, and neutrophil-derived NETs are directly toxic to airway epithelial cells in vitro (195). NETs have been proposed as drivers of lung and systemic inflammation in COVID-19 illness (196–198).
NETosis is likely to have a very important role in pneumonia and acute lung injury. In two different mouse models of severe bacterial pneumonia and lung injury because of intratracheal instillation of either methicillin-resistant Staphylococcus aureus or Pseudomonas aeruginosa, NETs were detectable in the airspaces, and pulmonary microvasculature beginning approximately 3 hours after infection and became prominent by 24 hours after infection (188). Bacteria colocalized with NETs in the airspaces. Lung injury was reduced in mice with deficient NET formation because of knockout of the PAD4−/− (protein arginine deiminase 4 gene), but bacterial recovery increased, showing the dual roles of NET formation in injury and host defense. Treatment of mice with intratracheal DNase I reduced lung NET formation without impairing bacterial clearance, and improved physiology and survival, suggesting a new treatment approach. In a cohort of patients with pneumonia and sepsis who were monitored for development of ARDS, NETs were identified in the plasma of patients with ARDS, increased with severity, and were highest in those who died. Plasma DNase I is the major endonuclease found in blood, and a low concentration at presentation was associated with the development of ARDS in patients who were septic (188). Although plasma DNase I concentrations did not differentiate patients with or without ARDS, the plasma NET/DNase I ratio was higher in patients with pneumonia and ARDS (because of low DNase I concentrations) and was associated with ARDS severity and mortality.
Although existing preclinical and clinical data suggest that NETs may have an important role in acute lung injury, particularly in patients with sepsis or primary lung infections, relatively little is known about individual heterogeneity in NET formation in health and disease or heterogeneity that might occur during the course of lung injury. Increased susceptibility to NET formation might be an additional biological factor that contributes to patient subgroups identified by LCA. NET formation in the lungs and microvasculature offers a new target for therapy (e.g., with intravenous or inhaled recombinant DNase I), but the appropriate criteria for patient selection and the optimal timing of therapy remain important questions that need to be answered.
Angiopoietins and Tie-2 in Vascular Injury
Samir Parikh reviewed the Tie-2/angiopoietin axis in normal vascular homeostasis and injury (Figure 6) (199, 200). The angiopoietins comprise a group of secreted growth factors that are important in vascular development, angiogenesis, and homeostasis of mature blood vessels. Angiopoietins (Angpt) engage the Tie-2 receptor on endothelial cells to regulate vascular development and integrity. Tie-2 is a transmembrane tyrosine kinase that functions as a signaling receptor, whereas Tie-1 does not bind angiopoietins directly but still modulates Tie-2 signaling (201). Angpt-1 is a direct agonist of the Tie-2 receptor, which signals through PI3K/AKT1 to promote endothelial cell spreading and enhance barrier function. Tie-2 activation also blocks NFκB activation, thereby downregulating endothelial proinflammatory signals. Angpt-2 is a context-dependent antagonist of Tie-2 and promotes vascular leakage during inflammation. Angpt-2 is stored in the endothelial cell Weibel-Palade bodies and is released with endothelial activation by thrombin, TNFα, turbulent flow, and other proinflammatory factors (202–204). Reduction in Tie-2 signaling shifts the balance toward endothelial activation, with an increase in endothelial permeability, expression of leukocyte adhesion molecules (e.g., ICAM1 and VCAM1), and enhancement of procoagulant proteins at the endothelial cell surface. Other important pathways also contribute to vascular integrity, including vascular endothelial growth factor (VEGF) and its receptors, VEGFR-1 and VEGFR-2, sphingosine-1 kinase and its receptors, and mechanical forces such as shear stress generated by blood flow (200).
Figure 6.
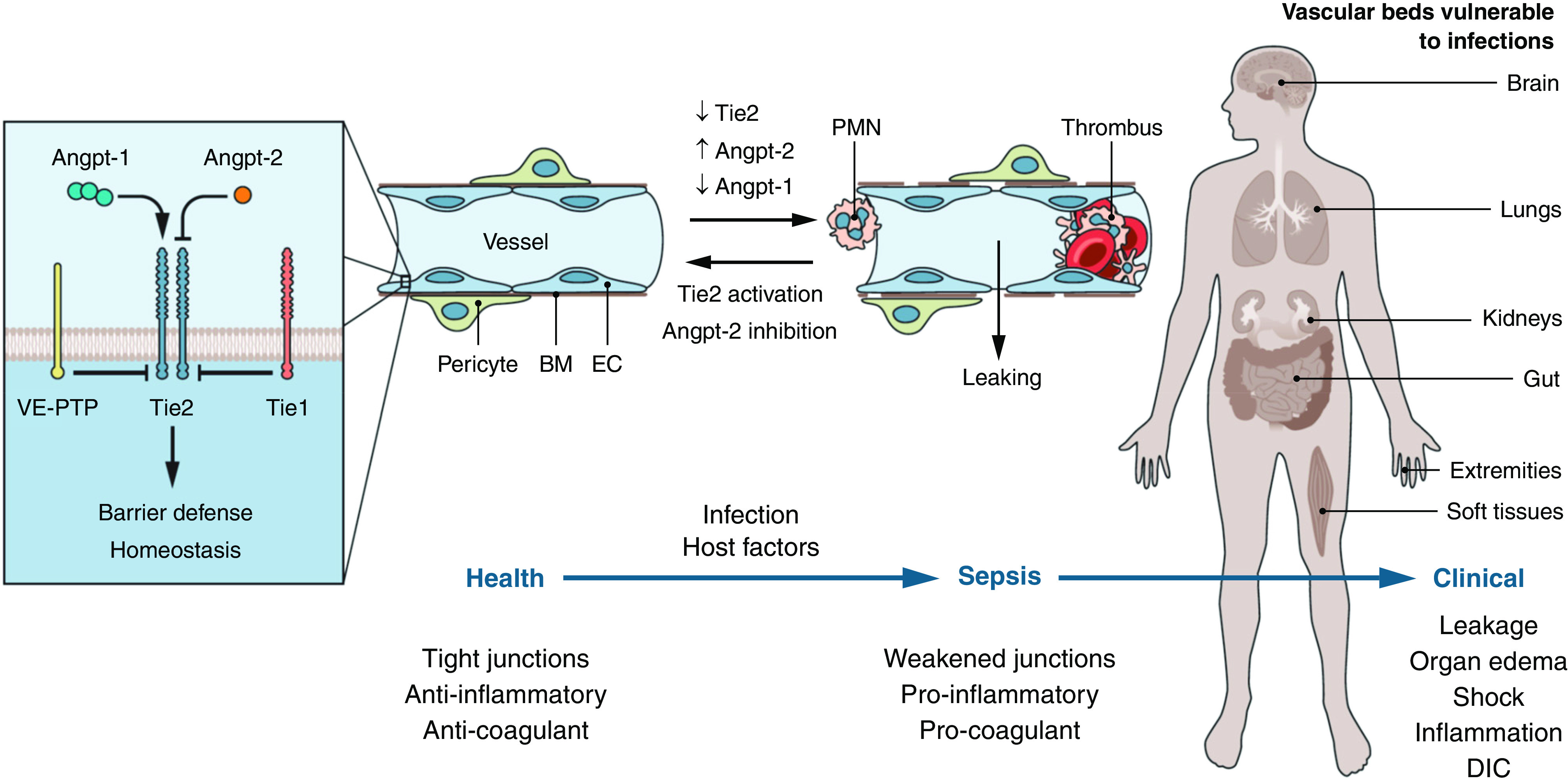
TIE2 and vascular responses. (Left) Tie2 is activated by Angpt-1 (angiopoietin-1) and antagonized by Angpt-2. The orphan receptor Tie1 antagonizes Tie2 signaling, as does vascular endothelial protein tyrosine phosphatase VE-PTP. Active Tie2 maintains vascular barrier function. (Middle) In sepsis, coordinated changes in Tie proteins lead to signaling inhibition, promoting vascular leakage, inflammation, and thrombosis. (Right) Organs affected by vascular leakage during sepsis. The host vascular response contributes to systemic inflammation, disseminated intravascular coagulation, and multiorgan dysfunction. Modified with permission from reference 199. BM = basement membrane; EC = endothelial cell; PMN = polymorphonuclear leukocyte, VE-PTP = vascular endothelial protein tyrosine phosphatase.
Preclinical models and observational studies in humans suggest that the angiopoietin/Tie2 axis has an important role in endothelial responses in sepsis, which is characterized by a marked increase in vascular permeability throughout the body. In a variety of animal models, Tie-2 activation by Angpt-1 prevents vascular leakage, reduces thrombus formation and cellular inflammation, and improves survival, whereas Tie-2 antagonism by Angpt-2 is deleterious (Table 5). Beneficial effects of Tie-2 activation or Angpt-2 antagonism have been shown with endotracheal and intravenous endotoxin, cecal ligation and puncture (CLP), hyperoxia, phosgene, monocrotaline, and others (205–209). Activation of Tie-2 by overexpression of Angpt-1 improved organ function and survival in endotoxic shock (210), and treatment of mice with recombinant human Angpt-1 improved survival in sepsis induced by CLP (206). Inactivation of one of the two Tie-2 alleles in transgenic mice worsened mortality from endotoxin as well as CLP (205). In addition, treatment with Angpt-1 improves long-term outcomes in models of acute kidney injury (211, 212).
Table 5.
Animal Models that Improve with Tie-2 Activation
| Animal Species | Model | Stimulus | (References) |
|---|---|---|---|
| Infection/Inflammation | |||
| Mouse | Lung inflammation | LPS, intratracheal | (272) |
| Mouse | Systemic inflammation | LPS, parenteral | (210, 273, 274) |
| Mouse | Lung injury | LPS or CLP (polymicrobial sepsis) | (205–209) |
| Biological/chemical | |||
| Mouse | Lung injury | Phosgene, inhalation | (275, 276) |
| Mouse | Lung injury | Anthrax toxin i.v. | (205, 277) |
| Other | |||
| Mouse | Lung injury | Hyperoxia, 100% O2 up to 72 h | (278) |
| Mouse | Pulmonary hypertension | Serotonin, s.c. daily for 7 d or IL-6, s.c. daily for 14 d | (279) |
| Mouse | Pulmonary hypertension | Monocrotaline, i.p. | (279, 280) |
Definition of abbreviations: CLP = cecal ligation and puncture; i.p. = intraperitoneal; LPS = lipopolysaccharide; s.c. = subcutaneous.
In humans, high circulating concentrations of Angpt-2 are detectable in septic patients and decline as the severity of illness improves (213–215). A number of studies have identified Angpt-2 as a risk factor for the onset and outcome of ARDS (89, 216–219). The ratio between circulating Angpt-2 and Angpt-1 increases in sepsis, suggesting that the ratio may be a better marker than either ligand alone (215, 220). Genetic polymorphisms associated with Angpt-2 overproduction confer increased risk for ARDS (89, 221). Angpt-2 is also strongly associated with markers of disseminated intravascular coagulation and accurately predicted mortality in two large independent cohorts with disseminated intravascular coagulation (222).
Because of the prominent role of the Angpt/Tie-2 axis in animal models and human disease, several different approaches to therapy for sepsis and critical illness are in development, including administration of recombinant human Angpt-1 derivatives, inhibitory antibodies against Angpt-2 and other activators of Tie-2, including agonistic antibodies or inhibitors of the Tie2-inhibiting phosphatase VE-PTP (vascular endothelial protein tyrosine phosphatase). Enhanced signaling through the Tie-2 receptor may improve vascular barrier function, reduce thrombosis and inflammation, and improve essential organ function.
Lung Endothelial–Brain Interactions
Troy Stevens presented new information about lung endothelial tau and amyloid proteins and interactions between the lungs, brain, and other organs during lung infection (Figure 7). Survivors of ARDS and critical illness are at high risk for significant cognitive dysfunction, but the biological mechanisms responsible are unclear (74, 131, 223, 224). A series of experimental studies have linked cytotoxic variants of amyloid and tau proteins with cognitive dysfunction in animals with bacterial pneumonia (225, 226). Amyloid proteins are diverse and include polypeptides that polymerize to form cross-β sheet structures inside and outside of cells (227). Tau proteins comprise a group of six protein isoforms encoded by the MAPT (microtubule-associated protein tau) gene that was originally identified in neurons as proteins that stabilized microtubules (228). Although normally very soluble, hyperphosphorylated forms of tau are insoluble and form intracellular aggregates that can impair cellular function. Neurofibrillary tangles containing tau have been identified in the brains of patients with neurodegenerative diseases and are thought to contribute to pathology (229). Tau proteins also are found in endothelial cells in the lungs and elsewhere, where soluble tau monomers stabilize microtubules (226, 230).
Figure 7.
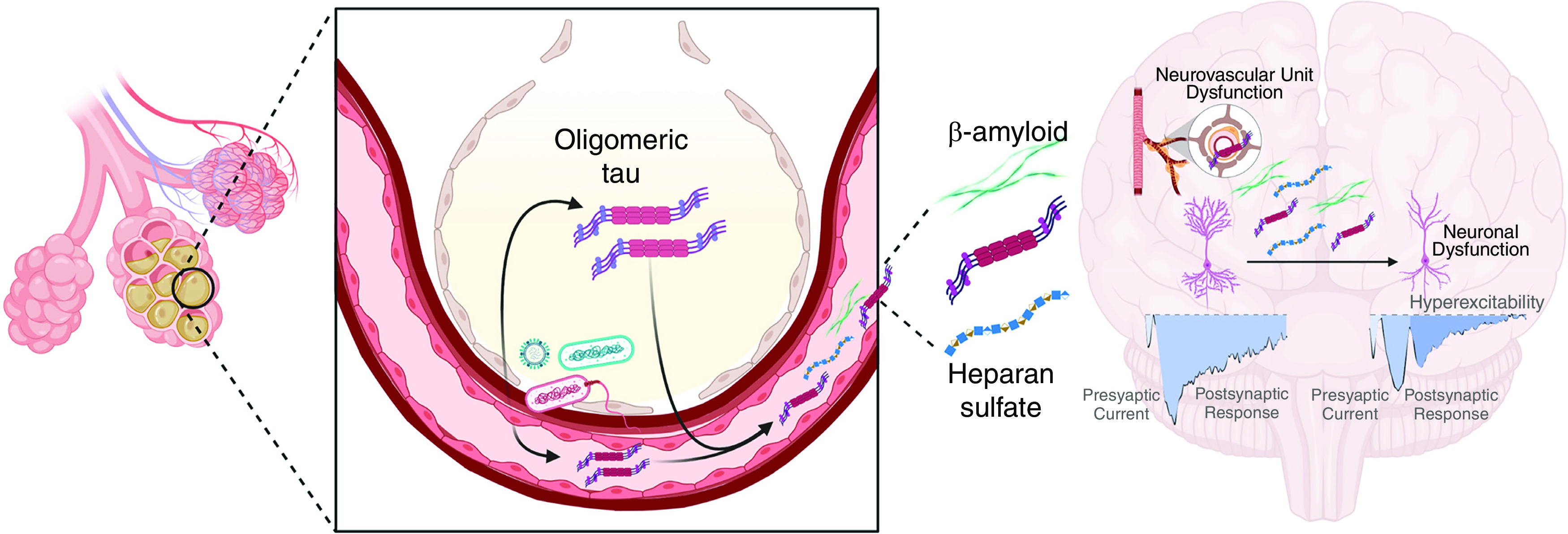
Proposed relationship between lung infection and neurovascular dysfunction. Infection causes the production of cytotoxic tau variants in lung capillary endothelium that disseminate through the circulation and access the brain, where they disrupt hippocampal neuronal information processing. Heparan sulfate from lung matrix degradation and β amyloid also increase in the circulation and interfere with neural information processing. It was modified with permission from reference 225.
Bacterial infections in the lungs of critically ill patients and animals cause the formation of high–molecular-weight aggregates of tau that are cytotoxic (226, 231). P. aeruginosa type III secretion system effectors, including exoenzymes U and Y, caused time-dependent intracellular gap formation in pulmonary microvascular endothelial cells and the extracellular release of tau aggregates (232). Transfer of the high–molecular-weight tau aggregates to normal endothelial cells caused the appearance of intracellular tau aggregates and endothelial cell injury, suggesting a transmissible proteinopathy. Mice treated with intratracheal instillations of P. aeruginosa and then studied 1 week later had accumulations of tau aggregates in the brain. Mice treated with intraventricular injections of tau protein aggregates derived from supernatants of cultured microvascular endothelial cells showed learning and memory deficits (226). Recordings from hippocampal slices treated with endothelial supernatants containing tau aggregates showed disruption of postsynaptic synaptic responses, suggesting that the endothelium-derived tau aggregates were neurotoxic.
A subsequent study of mice with P. aeruginosa pneumonia showed that the infection elicited the release of cytotoxic aggregates of tau and amyloid proteins into the lungs and the circulation. In addition, they were detectable in the hippocampus, where they caused tau-dependent inhibition of neural long-term potentiation (225). Injection of cytotoxic tau variants purified from the blood of infected animals into the blood of naive animals acutely and chronically impaired hippocampal information processing, an effect that was not seen in tau knockout mice.
Cytotoxic tau and amyloid species also have been recovered from the blood of critically ill patients with bacterial pneumonia, and the instillation of these cytotoxic variants into the lungs of mice caused lung injury and impaired neural long-term potentiation (225, 231). Taken together, these data suggest that bacterial pneumonia can induce what the authors called a protein “tauopathy” that connects the lung infection with significant neurocognitive impairment (225).
In related studies, Hippensteel and colleagues found that sulfated variants of heparan sulfate are additional candidates that might link sepsis with neurocognitive dysfunction (233). Highly sulfated heparan sulfate fragments accumulate in the plasma of humans and animals with sepsis because of degradation of the endothelial glycocalyx (234, 235). Mice surviving endotoxemia had increased plasma concentrations of heparan sulfate, impaired memory function, and loss of long-term potentiation (LTP) in hippocampal slices (233). In a sepsis model, LTP also was impaired in mice 7 days after recovery from CLP. Labeled heparan sulfate fragments crossed the blood–brain barrier, were detectable in the hippocampus, and were associated with impaired LTP. High concentrations of heparan sulfate fragments were identified in the plasma of patients with sepsis, 50% of whom also had ARDS, and patients with the highest concentrations of heparan sulfate fragments at baseline had moderate or severe cognitive impairment 2 weeks after ICU discharge (233).
Further prospective studies are needed that assess plasma tau, amyloid, and glycocalyx fragments at the onset and during the evolution of critical illness and relate the findings to long-term cognitive and functional outcomes. In addition, more information is needed about whether interactions between these mediators and circulating proinflammatory cytokines also might affect long-term neurocognitive outcomes.
Alveolar Repair
Alveolar Epithelial Progenitor Cells in Lung Homeostasis and Injury
Tushar Desai summarized new information about progenitor cells in lung homeostasis and repair, focusing on AEC2 (alveolar epithelial Type 2) progenitor cells and the role of Wnt signaling in AEC2 biology. The classical studies of Adamson and Bowden showed that AEC2 cells repopulate alveolar walls after alveolar epithelial injury (236). More recent studies show that alveoli are maintained by AEC2 cells during homeostasis by intermittent activation of a minor subset of AEC2 cells that are long-lived, self-renewing, and display active canonical Wnt signaling (237–240). These Axin2+ stem cells can be isolated by unique surface markers and have distinct transcriptomic, epigenomic, and functional phenotypes (239). They tend to be perivascular or subpleural, and each AEC2 stem cell abuts a platelet-derived growth factor receptor-α+ fibroblast that expresses Wnt genes, thereby creating a supportive niche (240). Wnt signaling not only endows AEC2 stem cells with proliferative potential but also actively maintains their AEC2 phenotype, as acute loss of signaling induces AEC1 differentiation (Figure 8) (240). The addition of purified EGF ligands stimulates AEC2 replication in culture, and conditional activation of the downstream Kras pathway (KrasLSL-G12D) in AEC2 cells drives adenoma formation in vivo, supporting EGF as an important proliferative cue (238).
Figure 8.

Paradigm for epithelial regeneration by AEC2 cells (alveolar epithelial type 2) showing crucial roles of Wnt signaling and TGFβ (transforming growth factor β) activity. (A) Lineage tracing showing alveolar stem cells (AEC2, red) and their progeny (AEC2 lineage, green). When Wnt/βcatenin activity is lost (top, middle panel), AEC2 cells differentiate into flattened AEC1 cells. When Wnt/βcatenin is constitutively active, AEC2 cells maintain the AEC2 phenotype, and AEC1 differentiation is inhibited (top, third panel). (B) After lung injury, AEC2 cells proliferate, then assume a transitional cell state characterized by partially flattened morphology and a unique gene expression signature regulated by TGFβ. With downregulation of TGFβ signaling, transitional cells mature into AEC1 cells to repopulate the alveolar walls, whereas, with persistent TGFβ signaling, transitional cells fail to mature into AEC1, and fibrosis may follow. The first panel is modified with permission from reference 240.
Disruption of the alveolar epithelium with diphtheria toxin in mice expressing the diphtheria toxin receptor throughout the lung epithelium caused nearly all surviving AEC2 cells to proliferate, suggesting that AEC2 cells that are not stem cells also can activate Wnt signaling, proliferate, and contribute to alveolar repair after significant alveolar epithelial injury (240). Interestingly, acute injury induced a transient Wnt niche “switch” in which, rather than depending on exogenous Wnt from adjacent fibroblasts, AEC2 cells themselves began producing and receiving Wnt in an autocrine manner until the repair was complete. In this model, administration of diphtheria toxin in the setting of pan inhibition of Wnt signaling significantly reduced proliferation and repair, supporting a role for Wnt as a proliferative “gatekeeper” for AEC2 cells.
Thus, the alveolar epithelium is maintained by a molecularly distinct subset of Wnt-dependent AEC2 stem cells that can differentiate into “bulk” AEC2 cells during normal homeostasis or into AEC1 cells as needed to repopulate alveolar walls after injury.
Heterogeneity in Alveolar Epithelial Repair after Injury
Rachel Zemans reviewed information about alveolar epithelial repair in normal homeostasis in the setting of acute or chronic lung injury (34, 241, 242). Consistent with the work of Desai and others, Zemans and colleagues used an inhibitor of Wnt/β-catenin signaling to demonstrate the essential role of this pathway in AEC2 cell proliferation during repair after lung injury (239, 240, 243–245). AEC2 cell proliferation is further supported by KGF (keratinocyte growth factor), HGF (hepatocyte growth factor), GM-CSF, FOXM1 (forkhead box protein M1), and STAT3 (signal transducer and activator of transcription 3) signaling (246–249). Moreover, in normal regeneration after lung injury, AEC2 cells proliferate, then exit the cell cycle and enter a transitional state characterized by markers of cell cycle arrest, downregulation of AEC2 cell markers, modest upregulation of AEC1 cell markers, TGFβ (transforming growth factor β) activation, and expression of a cluster of signature genes that are not highly expressed by either AEC2 or AEC1 cells (250). These transitional cells ultimately differentiate into AEC1 cells and restore normal alveolar architecture and barrier and gas exchange function (251, 252).
In chronic fibrotic lung diseases and fibroproliferative ARDS, epithelial cells persist in this transitional state with impaired AEC1 cell differentiation (251–256). Although causality has not been established, this suggests that persistence of the transitional state with impaired AEC1 cell differentiation is the critical regenerative defect underlying the pathogenesis of pulmonary fibrosis. These cells are likely responsible for driving fibrogenesis through TGFβ activation (250, 257). Thus, transitional cells differentiate into AEC1 cells in mouse models of alveolar regeneration but appear to persist and promote fibrogenesis in humans with pulmonary fibrosis.
Recent data suggest, however, that transitional cells are not pathognomonic of fibrosis in humans. In lung specimens from patients with early ARDS without fibrosis, AEC2 cells appeared to proliferate and assume the transitional state (258). In these cases, transitional cells appeared in a monolayer with increasingly spread morphologies, filling gaps on structurally normal alveolar septa denuded of AEC1 cells, suggesting that they were in the process of AEC1 cell differentiation and restoration of normal alveolar structure and function. However, AEC1 cell differentiation from the transitional state was incomplete, and this may contribute to ongoing vascular leak with persistent pulmonary edema and in some cases, death from acute hypoxemic respiratory failure (258).
A longstanding important and clinically relevant question is why some patients with ARDS repair with normal lung structure and function, whereas others develop fibrosis. On a cellular degree, it appears that this may be because of divergent fates of alveolar epithelial transitional cells that in some cases differentiate into AEC1 cells and restore normal lung epithelial structure and function but in other cases persist and possibly drive fibrosis. However, it remains unclear why transitional cells retain the capacity for AEC1 cell differentiation and restoration of normal alveolar structure and function in mouse models of physiologic regeneration and early human ARDS but seem to lose the capacity for AEC1 cell differentiation with ensuing fibrosis in fibroproliferative ARDS and chronic fibrotic lung diseases. Recent data from mouse models of physiologic regeneration and humans with early ARDS suggest that AEC2 cells exit the cell cycle and assume a transient state of cell cycle arrest in anticipation of AEC1 cell differentiation, whereas in fibroproliferative ARDS and chronic fibrotic lung disease, these transitional cells evolve into a permanent state of cell cycle arrest or senescence, losing the capacity for AEC1 cell differentiation, with ensuing fibrosis (258). Conversion of transitional cells from a transient state of cell cycle arrest to permanent senescence may be the key “switch” or “point of no return” past which transitional cells lose the capacity for AEC1 cell differentiation and perhaps cause fibrosis (258, 259) (Figure 8).
Conclusions and Future Directions
Understanding the clinical and molecular mechanisms of heterogeneity in critical illness and ARDS is an important priority in ARDS research. A significant amount of progress has been made in understanding heterogeneity in patient populations at the onset of ARDS using clinical classifications on the basis of LCA, but major challenges remain. The subgroups identified using LCA have consistently different outcomes across studies, but the hyperinflammatory subgroup with the highest mortality only includes approximately 30% of the patient population, and the larger hypoinflammatory subgroup has a mortality of approximately 20%, so targeting the hyperinflammatory subgroup with a new therapy could exclude many patients with ARDS who need better treatments. More progress is needed to develop rapid methods using the electronic medical record and baseline biomarker analyses to categorize patients immediately before or very early after the onset of ARDS so that patients can be selected appropriately for new treatments or entered into stratified clinical trials.
Progress has been made in understanding the cellular and molecular heterogeneity underlying patient responses at the onset and during the course of ARDS using powerful new technologies for single-cell analysis, genomics, proteomics, and metabolomics. These approaches are helping to understand pathophysiology, but they are not yet ready for the bedside. Importantly, the key interacting “nodes” in the pathways regulating acute lung injury and repair have not been identified, and the relevance of basic pathophysiological advances for long-term outcome research needs to be established.
New approaches to clinical trial design that recognize patient heterogeneity are now in use. Although the placebo-controlled blinded randomized clinical trial will remain the gold standard for therapeutic evidence, the COVID-19 pandemic has shown the value of large international platform trials that use simplified data collection instruments and outcome measurements. Innovative trial designs are available that could facilitate testing of multiple agents and appropriate biomarkers in single large trials. The COVID-19 pandemic has shown how rapidly large multicenter/multinational clinical trials can be organized and how well academia, government sponsors, the pharmaceutical industry, and regulatory agencies can work together to accelerate clinical research in times of great need.
Understanding clinical and basic heterogeneity in other lung diseases, such as cancer, asthma, and cystic fibrosis has led to important new treatments and significant improvements in clinically important patient outcomes. ARDS is a complex critical illness with many contributing factors, and critical care research is challenging because of the compressed time scale for identifying patients and beginning experimental treatments. Nevertheless, mortality remains very high, and the need for new approaches remains acute. Improving understanding of clinical and molecular heterogeneity in ARDS should lead to new ways to improve immediate and long-term outcomes and advance precision medicine for patients with this important critical illness.
Footnotes
Supported by the U.S. National Library of Medicine (R01-LM010685 [L.B.]); the National Institute on Aging (RO1AG049493 [S.M.P.]); U.S. Department of Veterans Affairs (VA 1 I01 BX003471 [D.W.H.R.]); Stanford Medical Scientist Training Program (T.J.D.); National Heart, Lung, and Blood Institute (1UO1HL123009 [B.T.T.]); German Ministry of Education and Research (BMBF and IPSELON [S.H.]); National Institute of Allergy and Infectious Diseases (AI160167 [M.R.L.]); Deutsche Forschungsgemeinschaft (Germany) (KFO309/P2,8, SFB1021/C5, and SFB-TRR84/B9 [S.H.]); National Institute of General Medical Sciences (GM125095 [E.P.S.]); Pandemeinetzwerk Hessen (Germany) (LOEWE Research Clusters Diffusible Signals and iCanX [S.H.]); U.S. Department of Defense (A130219 [L.B.W.]); and grants HL147920 and HL131608 (R.L.Z.); HL103836, HL126176, and HL135849 (L.B.W.); NHLBI 2020 R13 HL152479 and NHLBI 1R01 HL140595 (D.W.H.R.); R35 HL140026 (C.S.C.); NHLBI 5R01HL14254902 (T.J.D.); German Research Foundation (EXC2026 [S.H.]); K24HL141526 and RO1HL132232 (C.L.H.); HL134828, HL126456, and HL140026, HL143896 (M.A.M.); HL137006, HL137915, and HL155804 (N.L.M.); R35 HL139424 (S.M.P.); R35 HL161241 (M.R.L.); and HL66299, HL148069, and HL140182 (T.S.).
Originally Published in Press as DOI: 10.1165/rcmb.2022-0089WS on June 9, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. ARDS Definition Task Force Acute respiratory distress syndrome: the Berlin definition. JAMA . 2012;307:2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 2. Beitler JR, Thompson BT, Baron RM, Bastarache JA, Denlinger LC, Esserman L, et al. Advancing precision medicine for acute respiratory distress syndrome. Lancet Respir Med . 2022;10:107–120. doi: 10.1016/S2213-2600(21)00157-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults Lancet 1967. 2 319 323 4143721 [Google Scholar]
- 4. Petty TL, Reiss OK, Paul GW, Silvers GW, Elkins ND. Characteristics of pulmonary surfactant in adult respiratory distress syndrome associated with trauma and shock. Am Rev Respir Dis . 1977;115:531–536. doi: 10.1164/arrd.1977.115.3.531. [DOI] [PubMed] [Google Scholar]
- 5. Petty TL, Silvers GW, Paul GW, Stanford RE. Abnormalities in lung elastic properties and surfactant function in adult respiratory distress syndrome. Chest . 1979;75:571–574. doi: 10.1378/chest.75.5.571. [DOI] [PubMed] [Google Scholar]
- 6. Staub NC. Pulmonary edema. Physiol Rev . 1974;54:678–811. doi: 10.1152/physrev.1974.54.3.678. [DOI] [PubMed] [Google Scholar]
- 7. Fein A, Grossman RF, Jones JG, Overland E, Pitts L, Murray JF, et al. The value of edema fluid protein measurement in patients with pulmonary edema. Am J Med . 1979;67:32–38. doi: 10.1016/0002-9343(79)90066-4. [DOI] [PubMed] [Google Scholar]
- 8. Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, et al. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature . 1994;367:463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 9. Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev . 2002;82:569–600. doi: 10.1152/physrev.00003.2002. [DOI] [PubMed] [Google Scholar]
- 10. Matthay MA. Resolution of pulmonary edema. Thirty years of progress. Am J Respir Crit Care Med . 2014;189:1301–1308. doi: 10.1164/rccm.201403-0535OE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Berthiaume Y, Albertine KH, Grady M, Fick G, Matthay MA. Protein clearance from the air spaces and lungs of unanesthetized sheep over 144 h. J Appl Physiol (1985) . 1989;67:1887–1897. doi: 10.1152/jappl.1989.67.5.1887. [DOI] [PubMed] [Google Scholar]
- 12. Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med . 2001;163:1376–1383. doi: 10.1164/ajrccm.163.6.2004035. [DOI] [PubMed] [Google Scholar]
- 13. Clements JA. Lung surfactant: a personal perspective. Annu Rev Physiol . 1997;59:1–21. doi: 10.1146/annurev.physiol.59.1.1. [DOI] [PubMed] [Google Scholar]
- 14. Avery ME, Merritt TA. Surfactant-replacement therapy. N Engl J Med . 1991;324:910–912. doi: 10.1056/NEJM199103283241308. [DOI] [PubMed] [Google Scholar]
- 15. Anzueto A, Baughman RP, Guntupalli KK, Weg JG, Wiedemann HP, Raventós AA, et al. Exosurf Acute Respiratory Distress Syndrome Sepsis Study Group Aerosolized surfactant in adults with sepsis-induced acute respiratory distress syndrome. N Engl J Med . 1996;334:1417–1421. doi: 10.1056/NEJM199605303342201. [DOI] [PubMed] [Google Scholar]
- 16. Spragg RG, Taut FJ, Lewis JF, Schenk P, Ruppert C, Dean N, et al. Recombinant surfactant protein C-based surfactant for patients with severe direct lung injury. Am J Respir Crit Care Med . 2011;183:1055–1061. doi: 10.1164/rccm.201009-1424OC. [DOI] [PubMed] [Google Scholar]
- 17. Bachofen M, Weibel ER. Alterations of the gas exchange apparatus in adult respiratory insufficiency associated with septicemia. Am Rev Respir Dis . 1977;116:589–615. doi: 10.1164/arrd.1977.116.4.589. [DOI] [PubMed] [Google Scholar]
- 18. Martin TR, Pistorese BP, Chi EY, Goodman RB, Matthay MA. Effects of leukotriene B4 in the human lung. Recruitment of neutrophils into the alveolar spaces without a change in protein permeability. J Clin Invest . 1989;84:1609–1619. doi: 10.1172/JCI114338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burns AR, Smith CW, Walker DC. Unique structural features that influence neutrophil emigration into the lung. Physiol Rev . 2003;83:309–336. doi: 10.1152/physrev.00023.2002. [DOI] [PubMed] [Google Scholar]
- 20. Ognibene FP, Martin SE, Parker MM, Schlesinger T, Roach P, Burch C, et al. Adult respiratory distress syndrome in patients with severe neutropenia. N Engl J Med . 1986;315:547–551. doi: 10.1056/NEJM198608283150904. [DOI] [PubMed] [Google Scholar]
- 21. Sivan Y, Mor C, al-Jundi S, Newth CJ. Adult respiratory distress syndrome in severely neutropenic children. Pediatr Pulmonol . 1990;8:104–108. doi: 10.1002/ppul.1950080208. [DOI] [PubMed] [Google Scholar]
- 22. Maunder RJ, Hackman RC, Riff E, Albert RK, Springmeyer SC. Occurrence of the adult respiratory distress syndrome in neutropenic patients. Am Rev Respir Dis . 1986;133:313–316. doi: 10.1164/arrd.1986.133.2.313. [DOI] [PubMed] [Google Scholar]
- 23. Pannone G, Caponio VCA, De Stefano IS, Ramunno MA, Meccariello M, Agostinone A, et al. Lung histopathological findings in COVID-19 disease - a systematic review. Infect Agent Cancer . 2021;16:34. doi: 10.1186/s13027-021-00369-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Parra-Medina R, Herrera S, Mejia J. Systematic review of microthrombi in COVID-19 autopsies. Acta Haematol . 2021;144:476–483. doi: 10.1159/000515104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peiris S, Mesa H, Aysola A, Manivel J, Toledo J, Borges-Sa M, et al. Pathological findings in organs and tissues of patients with COVID-19: a systematic review. PLoS One . 2021;16:e0250708. doi: 10.1371/journal.pone.0250708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Webb HH, Tierney DF. Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures. Protection by positive end-expiratory pressure. Am Rev Respir Dis . 1974;110:556–565. doi: 10.1164/arrd.1974.110.5.556. [DOI] [PubMed] [Google Scholar]
- 27. Parker JC, Townsley MI, Rippe B, Taylor AE, Thigpen J. Increased microvascular permeability in dog lungs due to high peak airway pressures. J Appl Physiol . 1984;57:1809–1816. doi: 10.1152/jappl.1984.57.6.1809. [DOI] [PubMed] [Google Scholar]
- 28. Dreyfuss D, Saumon G. What is the mechanism of pulmonary edema during high volume ventilation? Am Rev Respir Dis . 1991;143:1198–1200. doi: 10.1164/ajrccm/143.5_Pt_1.1198a. [DOI] [PubMed] [Google Scholar]
- 29. Brower RG, Matthay MA, Morris A, Schoenfeld D, Thompson BT, Wheeler A, Acute Respiratory Distress Syndrome Network Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med . 2000;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 30. Parsons PE, Eisner MD, Thompson BT, Matthay MA, Ancukiewicz M, Bernard GR, et al. NHLBI Acute Respiratory Distress Syndrome Clinical Trials Network Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Crit Care Med . 2005;33:1–6, discussion 230–232. doi: 10.1097/01.ccm.0000149854.61192.dc. [DOI] [PubMed] [Google Scholar]
- 31. Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest . 2012;122:2731–2740. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest . 1989;83:865–875. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Henson PM, Tuder RM. Apoptosis in the lung: induction, clearance, and detection. Am J Physiol Lung Cell Mol Physiol . 2008;294:L601–L611. doi: 10.1152/ajplung.00320.2007. [DOI] [PubMed] [Google Scholar]
- 34. Zemans RL, Henson PM, Henson JE, Janssen WJ. Conceptual approaches to lung injury and repair. Ann Am Thorac Soc . 2015;12:S9–S15. doi: 10.1513/AnnalsATS.201408-402MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Snyder LS, Hertz MI, Harmon KR, Bitterman PB. Failure of lung repair following acute lung injury. Regulation of the fibroproliferative response (Part 1) Chest . 1990;98:733–738. doi: 10.1378/chest.98.3.733. [DOI] [PubMed] [Google Scholar]
- 36. Clark JG, Milberg JA, Steinberg KP, Hudson LD. Type III procollagen peptide in the adult respiratory distress syndrome. Association of increased peptide levels in bronchoalveolar lavage fluid with increased risk for death. Ann Intern Med . 1995;122:17–23. doi: 10.7326/0003-4819-122-1-199501010-00003. [DOI] [PubMed] [Google Scholar]
- 37. Chesnutt AN, Matthay MA, Tibayan FA, Clark JG. Early detection of type III procollagen peptide in acute lung injury. Pathogenetic and prognostic significance. Am J Respir Crit Care Med . 1997;156:840–845. doi: 10.1164/ajrccm.156.3.9701124. [DOI] [PubMed] [Google Scholar]
- 38. Burnham EL, Janssen WJ, Riches DW, Moss M, Downey GP. The fibroproliferative response in acute respiratory distress syndrome: mechanisms and clinical significance. Eur Respir J . 2014;43:276–285. doi: 10.1183/09031936.00196412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Steinberg KP, Mitchell DR, Maunder RJ, Milberg JA, Whitcomb ME, Hudson LD. Safety of bronchoalveolar lavage in patients with adult respiratory distress syndrome. Am Rev Respir Dis . 1993;148:556–561. doi: 10.1164/ajrccm/148.3.556. [DOI] [PubMed] [Google Scholar]
- 40. Steinberg KP, Milberg JA, Martin TR, Maunder RJ, Cockrill BA, Hudson LD. Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am J Respir Crit Care Med . 1994;150:113–122. doi: 10.1164/ajrccm.150.1.8025736. [DOI] [PubMed] [Google Scholar]
- 41. Goodman RB, Strieter RM, Martin DP, Steinberg KP, Milberg JA, Maunder RJ, et al. Inflammatory cytokines in patients with persistence of the acute respiratory distress syndrome. Am J Respir Crit Care Med . 1996;154:602–611. doi: 10.1164/ajrccm.154.3.8810593. [DOI] [PubMed] [Google Scholar]
- 42. Greene KE, Wright JR, Steinberg KP, Ruzinski JT, Caldwell E, Wong WB, et al. Serial changes in surfactant-associated proteins in lung and serum before and after onset of ARDS. Am J Respir Crit Care Med . 1999;160:1843–1850. doi: 10.1164/ajrccm.160.6.9901117. [DOI] [PubMed] [Google Scholar]
- 43. Madtes DK, Rubenfeld G, Klima LD, Milberg JA, Steinberg KP, Martin TR, et al. Elevated transforming growth factor-alpha levels in bronchoalveolar lavage fluid of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med . 1998;158:424–430. doi: 10.1164/ajrccm.158.2.9711112. [DOI] [PubMed] [Google Scholar]
- 44. Martin TR, Rubenfeld G, Steinberg KP, Hudson LD, Raghu G, Moriarty AM, et al. Endotoxin, endotoxin-binding protein, and soluble CD14 are present in bronchoalveolar lavage fluid of patients with adult respiratory distress syndrome. Chest . 1994;105(3)(Suppl):55S–56S. doi: 10.1378/chest.105.3.55s. [DOI] [PubMed] [Google Scholar]
- 45. Park WY, Goodman RB, Steinberg KP, Ruzinski JT, Radella F, II, Park DR, et al. Cytokine balance in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med . 2001;164:1896–1903. doi: 10.1164/ajrccm.164.10.2104013. [DOI] [PubMed] [Google Scholar]
- 46. Sittipunt C, Steinberg KP, Ruzinski JT, Myles C, Zhu S, Goodman RB, et al. Nitric oxide and nitrotyrosine in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med . 2001;163:503–510. doi: 10.1164/ajrccm.163.2.2004187. [DOI] [PubMed] [Google Scholar]
- 47. Hendrickson CM, Abbott J, Zhuo H, Liu KD, Calfee CS, Matthay MA, NHLBI ARDS Network Higher mini-BAL total protein concentration in early ARDS predicts faster resolution of lung injury measured by more ventilator-free days. Am J Physiol Lung Cell Mol Physiol . 2017;312:L579–L585. doi: 10.1152/ajplung.00381.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu KD, Levitt J, Zhuo H, Kallet RH, Brady S, Steingrub J, et al. Randomized clinical trial of activated protein C for the treatment of acute lung injury. Am J Respir Crit Care Med . 2008;178:618–623. doi: 10.1164/rccm.200803-419OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stapleton RD, Martin TR, Weiss NS, Crowley JJ, Gundel SJ, Nathens AB, et al. A phase II randomized placebo-controlled trial of omega-3 fatty acids for the treatment of acute lung injury. Crit Care Med . 2011;39:1655–1662. doi: 10.1097/CCM.0b013e318218669d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Paine R, III, Standiford TJ, Dechert RE, Moss M, Martin GS, Rosenberg AL, et al. A randomized trial of recombinant human granulocyte-macrophage colony-stimulating factor for patients with acute lung injury. Crit Care Med . 2012;40:90–97. doi: 10.1097/CCM.0b013e31822d7bf0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.NHLBI ARDS Network. 2022. http://www.ardsnet.org/studies.shtml
- 52.NHLBI PETAL Network; National Institutes of Health. 2022. https://petalnet.org/
- 53. Wiedemann HP, Wheeler AP, Bernard GR, Thompson BT, Hayden D, deBoisblanc B, et al. National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network Comparison of two fluid-management strategies in acute lung injury. N Engl J Med . 2006;354:2564–2575. doi: 10.1056/NEJMoa062200. [DOI] [PubMed] [Google Scholar]
- 54. McAuley DF, Laffey JG, O’Kane CM, Perkins GD, Mullan B, Trinder TJ, et al. HARP-2 Investigators; Irish Critical Care Trials Group Simvastatin in the acute respiratory distress syndrome. N Engl J Med . 2014;371:1695–1703. doi: 10.1056/NEJMoa1403285. [DOI] [PubMed] [Google Scholar]
- 55. Truwit JD, Bernard GR, Steingrub J, Matthay MA, Liu KD, Albertson TE, et al. National Heart, Lung, and Blood Institute ARDS Clinical Trials Network Rosuvastatin for sepsis-associated acute respiratory distress syndrome. N Engl J Med . 2014;370:2191–2200. doi: 10.1056/NEJMoa1401520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rice TW, Wheeler AP, Thompson BT, deBoisblanc BP, Steingrub J, Rock P, NIH NHLBI Acute Respiratory Distress Syndrome Network of Investigators Enteral omega-3 fatty acid, gamma-linolenic acid, and antioxidant supplementation in acute lung injury. JAMA . 2011;306:1574–1581. doi: 10.1001/jama.2011.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Matthay MA, Brower RG, Carson S, Douglas IS, Eisner M, Hite D, et al. National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network Randomized, placebo-controlled clinical trial of an aerosolized β2-agonist for treatment of acute lung injury. Am J Respir Crit Care Med . 2011;184:561–568. doi: 10.1164/rccm.201012-2090OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Steinberg KP, Hudson LD, Goodman RB, Hough CL, Lanken PN, Hyzy R, et al. National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med . 2006;354:1671–1684. doi: 10.1056/NEJMoa051693. [DOI] [PubMed] [Google Scholar]
- 59. Villar J, Ferrando C, Martínez D, Ambrós A, Muñoz T, Soler JA, et al. dexamethasone in ARDS network Dexamethasone treatment for the acute respiratory distress syndrome: a multicentre, randomised controlled trial. Lancet Respir Med . 2020;8:267–276. doi: 10.1016/S2213-2600(19)30417-5. [DOI] [PubMed] [Google Scholar]
- 60. Guérin C, Reignier J, Richard JC, Beuret P, Gacouin A, Boulain T, et al. PROSEVA Study Group Prone positioning in severe acute respiratory distress syndrome. N Engl J Med . 2013;368:2159–2168. doi: 10.1056/NEJMoa1214103. [DOI] [PubMed] [Google Scholar]
- 61. Gattinoni L, Tognoni G, Pesenti A, Taccone P, Mascheroni D, Labarta V, et al. Prone-Supine Study Group Effect of prone positioning on the survival of patients with acute respiratory failure. N Engl J Med . 2001;345:568–573. doi: 10.1056/NEJMoa010043. [DOI] [PubMed] [Google Scholar]
- 62. Frat JP, Thille AW, Mercat A, Girault C, Ragot S, Perbet S, et al. FLORALI Study Group; REVA Network High-flow oxygen through nasal cannula in acute hypoxemic respiratory failure. N Engl J Med . 2015;372:2185–2196. doi: 10.1056/NEJMoa1503326. [DOI] [PubMed] [Google Scholar]
- 63. Papazian L, Forel JM, Gacouin A, Penot-Ragon C, Perrin G, Loundou A, et al. ACURASYS Study Investigators Neuromuscular blockers in early acute respiratory distress syndrome. N Engl J Med . 2010;363:1107–1116. doi: 10.1056/NEJMoa1005372. [DOI] [PubMed] [Google Scholar]
- 64. Moss M, Huang DT, Brower RG, Ferguson ND, Ginde AA, Gong MN, et al. National Heart, Lung, and Blood Institute PETAL Clinical Trials Network Early neuromuscular blockade in the acute respiratory distress syndrome. N Engl J Med . 2019;380:1997–2008. doi: 10.1056/NEJMoa1901686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L, et al. RECOVERY Collaborative Group Dexamethasone in hospitalized patients with COVID-19. N Engl J Med . 2021;384:693–704. doi: 10.1056/NEJMoa2021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gordon AC, Mouncey PR, Al-Beidh F, Rowan KM, Nichol AD, Arabi YM, et al. REMAP-CAP Investigators Interleukin-6 receptor antagonists in critically ill patients with COVID-19. N Engl J Med . 2021;384:1491–1502. doi: 10.1056/NEJMoa2100433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kalil AC, Patterson TF, Mehta AK, Tomashek KM, Wolfe CR, Ghazaryan V, et al. ACTT-2 Study Group Members Baricitinib plus remdesivir for hospitalized adults with COVID-19. N Engl J Med . 2021;384:795–807. doi: 10.1056/NEJMoa2031994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, et al. Incidence and outcomes of acute lung injury. N Engl J Med . 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 69. Eworuke E, Major JM, Gilbert McClain LI. National incidence rates for acute respiratory distress syndrome (ARDS) and ARDS cause-specific factors in the United States (2006–2014) J Crit Care . 2018;47:192–197. doi: 10.1016/j.jcrc.2018.07.002. [DOI] [PubMed] [Google Scholar]
- 70. Bellani G, Laffey JG, Pham T, Fan E, LUNG SAFE Investigators and the ESICM Trials Group The LUNG SAFE study: a presentation of the prevalence of ARDS according to the Berlin definition! Crit Care . 2016;20:268. doi: 10.1186/s13054-016-1443-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wick KD, McAuley DF, Levitt JE, Beitler JR, Annane D, Riviello ED, et al. Promises and challenges of personalized medicine to guide ARDS therapy. Crit Care . 2021;25:404. doi: 10.1186/s13054-021-03822-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Herridge MS, Cheung AM, Tansey CM, Matte-Martyn A, Diaz-Granados N, Al-Saidi F, et al. Canadian Critical Care Trials Group One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med . 2003;348:683–693. doi: 10.1056/NEJMoa022450. [DOI] [PubMed] [Google Scholar]
- 73. Herridge MS, Tansey CM, Matté A, Tomlinson G, Diaz-Granados N, Cooper A, et al. Canadian Critical Care Trials Group Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med . 2011;364:1293–1304. doi: 10.1056/NEJMoa1011802. [DOI] [PubMed] [Google Scholar]
- 74. Ferrante LE, Murphy TE, Leo-Summers LS, Gahbauer EA, Pisani MA, Gill TM. The combined effects of frailty and cognitive impairment on post-ICU disability among older ICU survivors. Am J Respir Crit Care Med . 2019;200:107–110. doi: 10.1164/rccm.201806-1144LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ferrante LE, Pisani MA, Murphy TE, Gahbauer EA, Leo-Summers LS, Gill TM. Functional trajectories among older persons before and after critical illness. JAMA Intern Med . 2015;175:523–529. doi: 10.1001/jamainternmed.2014.7889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ferrante LE, Murphy TE, Gahbauer EA, Leo-Summers LS, Pisani MA, Gill TM. Pre-intensive care unit cognitive status, subsequent disability, and new nursing home admission among critically ill older adults. Ann Am Thorac Soc . 2018;15:622–629. doi: 10.1513/AnnalsATS.201709-702OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fan E, Beitler JR, Brochard L, Calfee CS, Ferguson ND, Slutsky AS, et al. COVID-19-associated acute respiratory distress syndrome: is a different approach to management warranted? Lancet Respir Med . 2020;8:816–821. doi: 10.1016/S2213-2600(20)30304-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Diamond MS, Kanneganti TD. Innate immunity: the first line of defense against SARS-CoV-2. Nat Immunol . 2022;23:165–176. doi: 10.1038/s41590-021-01091-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Breton G, Mendoza P, Hägglöf T, Oliveira TY, Schaefer-Babajew D, Gaebler C, et al. Persistent cellular immunity to SARS-CoV-2 infection. J Exp Med . 2021;218:e20202515. doi: 10.1084/jem.20202515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Elborn JS. Cystic fibrosis. Lancet . 2016;388:2519–2531. doi: 10.1016/S0140-6736(16)00576-6. [DOI] [PubMed] [Google Scholar]
- 81. Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med . 2009;180:388–395. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pavord ID, Korn S, Howarth P, Bleecker ER, Buhl R, Keene ON, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet . 2012;380:651–659. doi: 10.1016/S0140-6736(12)60988-X. [DOI] [PubMed] [Google Scholar]
- 83. Castro M, Corren J, Pavord ID, Maspero J, Wenzel S, Rabe KF, et al. Dupilumab efficacy and safety in moderate-to-severe uncontrolled asthma. N Engl J Med . 2018;378:2486–2496. doi: 10.1056/NEJMoa1804092. [DOI] [PubMed] [Google Scholar]
- 84. Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med . 2012;18:716–725. doi: 10.1038/nm.2678. [DOI] [PubMed] [Google Scholar]
- 85. Parsons PE, Matthay MA, Ware LB, Eisner MD, National Heart, Lung, Blood Institute Acute Respiratory Distress Syndrome Clinical Trials Network Elevated plasma levels of soluble TNF receptors are associated with morbidity and mortality in patients with acute lung injury. Am J Physiol Lung Cell Mol Physiol . 2005;288:L426–L431. doi: 10.1152/ajplung.00302.2004. [DOI] [PubMed] [Google Scholar]
- 86. Calfee CS, Ware LB, Eisner MD, Parsons PE, Thompson BT, Wickersham N, et al. NHLBI ARDS Network Plasma receptor for advanced glycation end products and clinical outcomes in acute lung injury. Thorax . 2008;63:1083–1089. doi: 10.1136/thx.2008.095588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Eisner MD, Parsons P, Matthay MA, Ware L, Greene K, Acute Respiratory Distress Syndrome Network Plasma surfactant protein levels and clinical outcomes in patients with acute lung injury. Thorax . 2003;58:983–988. doi: 10.1136/thorax.58.11.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Briot R, Frank JA, Uchida T, Lee JW, Calfee CS, Matthay MA. Elevated levels of the receptor for advanced glycation end products, a marker of alveolar epithelial type I cell injury, predict impaired alveolar fluid clearance in isolated perfused human lungs. Chest . 2009;135:269–275. doi: 10.1378/chest.08-0919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Reilly JP, Wang F, Jones TK, Palakshappa JA, Anderson BJ, Shashaty MGS, et al. Plasma angiopoietin-2 as a potential causal marker in sepsis-associated ARDS development: evidence from Mendelian randomization and mediation analysis. Intensive Care Med . 2018;44:1849–1858. doi: 10.1007/s00134-018-5328-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ware LB, Koyama T, Billheimer DD, Wu W, Bernard GR, Thompson BT, et al. NHLBI ARDS Clinical Trials Network Prognostic and pathogenetic value of combining clinical and biochemical indices in patients with acute lung injury. Chest . 2010;137:288–296. doi: 10.1378/chest.09-1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ware LB, Conner ER, Matthay MA. von Willebrand factor antigen is an independent marker of poor outcome in patients with early acute lung injury. Crit Care Med . 2001;29:2325–2331. doi: 10.1097/00003246-200112000-00016. [DOI] [PubMed] [Google Scholar]
- 92. Calfee CS, Delucchi K, Parsons PE, Thompson BT, Ware LB, Matthay MA, NHLBI ARDS Network Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med . 2014;2:611–620. doi: 10.1016/S2213-2600(14)70097-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Delucchi K, Famous KR, Ware LB, Parsons PE, Thompson BT, Calfee CS, ARDS Network Stability of ARDS subphenotypes over time in two randomised controlled trials. Thorax . 2018;73:439–445. doi: 10.1136/thoraxjnl-2017-211090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Famous KR, Delucchi K, Ware LB, Kangelaris KN, Liu KD, Thompson BT, et al. ARDS Network Acute respiratory distress syndrome subphenotypes respond differently to randomized fluid management strategy. Am J Respir Crit Care Med . 2017;195:331–338. doi: 10.1164/rccm.201603-0645OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Calfee CS, Delucchi KL, Sinha P, Matthay MA, Hackett J, Shankar-Hari M, et al. Irish Critical Care Trials Group Acute respiratory distress syndrome subphenotypes and differential response to simvastatin: secondary analysis of a randomised controlled trial. Lancet Respir Med . 2018;6:691–698. doi: 10.1016/S2213-2600(18)30177-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sinha P, Delucchi KL, Thompson BT, McAuley DF, Matthay MA, Calfee CS, NHLBI ARDS Network Latent class analysis of ARDS subphenotypes: a secondary analysis of the statins for acutely injured lungs from sepsis (SAILS) study. Intensive Care Med . 2018;44:1859–1869. doi: 10.1007/s00134-018-5378-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hashem MD, Hopkins RO, Colantuoni E, Dinglas VD, Sinha P, Aronson Friedman L, et al. Six-month and 12-month patient outcomes based on inflammatory subphenotypes in sepsis-associated ARDS: secondary analysis of SAILS-ALTOS trial. Thorax . 2022;77:22–30. doi: 10.1136/thoraxjnl-2020-216613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sinha P, Calfee CS, Cherian S, Brealey D, Cutler S, King C, et al. Prevalence of phenotypes of acute respiratory distress syndrome in critically ill patients with COVID-19: a prospective observational study. Lancet Respir Med . 2020;8:1209–1218. doi: 10.1016/S2213-2600(20)30366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Sinha P, Furfaro D, Cummings MJ, Abrams D, Delucchi K, Maddali MV, et al. Latent class analysis reveals COVID-19-related ARDS subgroups with differential responses to corticosteroids. Am J Respir Crit Care Med . 2021;204:1274–1285. doi: 10.1164/rccm.202105-1302OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sinha P, Delucchi KL, Chen Y, Zhuo H, Abbott J, Wang C, et al. Latent class analysis-derived subphenotypes are generalisable to observational cohorts of acute respiratory distress syndrome: a prospective study. Thorax . 2022;77:13–21. doi: 10.1136/thoraxjnl-2021-217158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Bos LD, Schouten LR, van Vught LA, Wiewel MA, Ong DSY, Cremer O, et al. MARS consortium Identification and validation of distinct biological phenotypes in patients with acute respiratory distress syndrome by cluster analysis. Thorax . 2017;72:876–883. doi: 10.1136/thoraxjnl-2016-209719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Heijnen NFL, Hagens LA, Smit MR, Cremer OL, Ong DSY, van der Poll T, et al. Biological subphenotypes of acute respiratory distress syndrome show prognostic enrichment in mechanically ventilated patients without acute respiratory distress syndrome. Am J Respir Crit Care Med . 2021;203:1503–1511. doi: 10.1164/rccm.202006-2522OC. [DOI] [PubMed] [Google Scholar]
- 103. Sathe NA, Zelnick LR, Mikacenic C, Morrell ED, Bhatraju PK, McNeil JB, et al. Identification of persistent and resolving subphenotypes of acute hypoxemic respiratory failure in two independent cohorts. Crit Care . 2021;25:336. doi: 10.1186/s13054-021-03755-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sinha P, Delucchi KL, McAuley DF, O’Kane CM, Matthay MA, Calfee CS. Development and validation of parsimonious algorithms to classify acute respiratory distress syndrome phenotypes: a secondary analysis of randomised controlled trials. Lancet Respir Med . 2020;8:247–257. doi: 10.1016/S2213-2600(19)30369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Bos LDJ, Scicluna BP, Ong DSY, Cremer O, van der Poll T, Schultz MJ. Understanding heterogeneity in biologic phenotypes of acute respiratory distress syndrome by leukocyte expression profiles. Am J Respir Crit Care Med . 2019;200:42–50. doi: 10.1164/rccm.201809-1808OC. [DOI] [PubMed] [Google Scholar]
- 106. Sarma A, Christenson SA, Byrne A, Mick E, Pisco AO, DeVoe C, et al. COMET Consortium Tracheal aspirate RNA sequencing identifies distinct immunological features of COVID-19 ARDS. Nat Commun . 2021;12:5152. doi: 10.1038/s41467-021-25040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kent DM, Paulus JK, van Klaveren D, D’Agostino R, Goodman S, Hayward R, et al. The predictive approaches to treatment effect heterogeneity (PATH) statement. Ann Intern Med . 2020;172:35–45. doi: 10.7326/M18-3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Meyer NJ, Feng R, Li M, Zhao Y, Sheu CC, Tejera P, et al. IL1RN coding variant is associated with lower risk of acute respiratory distress syndrome and increased plasma IL-1 receptor antagonist. Am J Respir Crit Care Med . 2013;187:950–959. doi: 10.1164/rccm.201208-1501OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Meyer NJ, Ferguson JF, Feng R, Wang F, Patel PN, Li M, et al. A functional synonymous coding variant in the IL1RN gene is associated with survival in septic shock. Am J Respir Crit Care Med . 2014;190:656–664. doi: 10.1164/rccm.201403-0586OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Meyer NJ, Reilly JP, Anderson BJ, Palakshappa JA, Jones TK, Dunn TG, et al. Mortality benefit of recombinant human interleukin-1 receptor antagonist for sepsis varies by initial interleukin-1 receptor antagonist plasma concentration. Crit Care Med . 2018;46:21–28. doi: 10.1097/CCM.0000000000002749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kyriazopoulou E, Poulakou G, Milionis H, Metallidis S, Adamis G, Tsiakos K, et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: a double-blind, randomized controlled phase 3 trial. Nat Med . 2021;27:1752–1760. doi: 10.1038/s41591-021-01499-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Jones TK, Feng R, Kerchberger VE, Reilly JP, Anderson BJ, Shashaty MGS, et al. Plasma sRAGE acts as a genetically regulated causal intermediate in sepsis-associated acute respiratory distress syndrome. Am J Respir Crit Care Med . 2020;201:47–56. doi: 10.1164/rccm.201810-2033OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Mathew D, Giles JR, Baxter AE, Oldridge DA, Greenplate AR, Wu JE, et al. UPenn COVID Processing Unit Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science . 2020;369:eabc8511. doi: 10.1126/science.abc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Bowler RP, Duda B, Chan ED, Enghild JJ, Ware LB, Matthay MA, et al. Proteomic analysis of pulmonary edema fluid and plasma in patients with acute lung injury. Am J Physiol Lung Cell Mol Physiol . 2004;286:L1095–L1104. doi: 10.1152/ajplung.00304.2003. [DOI] [PubMed] [Google Scholar]
- 115. Chang DW, Hayashi S, Gharib SA, Vaisar T, King ST, Tsuchiya M, et al. Proteomic and computational analysis of bronchoalveolar proteins during the course of the acute respiratory distress syndrome. Am J Respir Crit Care Med . 2008;178:701–709. doi: 10.1164/rccm.200712-1895OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Messner CB, Demichev V, Wendisch D, Michalick L, White M, Freiwald A, et al. Ultra-high-throughput clinical proteomics reveals classifiers of COVID-19 infection. Cell Syst . 2020;11:11–24.e4. doi: 10.1016/j.cels.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Demichev V, Tober-Lau P, Lemke O, Nazarenko T, Thibeault C, Whitwell H, et al. PA-COVID-19 Study group A time-resolved proteomic and prognostic map of COVID-19. Cell Syst . 2021;12:780–794.e7. doi: 10.1016/j.cels.2021.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Cosgriff CV, Giannini HM, Mathew D, Anderson BJ, Jones TK, Ittner C, et al. Proteomic analysis of COVD-19 plasma reveals dysregulated TREM-1, IL-17, and tumor microenvironment pathways associated with disease severity. Am J Respir Crit Care Med . 2021;203:A2532. [Google Scholar]
- 119. Bastarache L. Using phecodes for research with the electronic health record: from PheWAS to PheRS. Annu Rev Biomed Data Sci . 2021;4:1–19. doi: 10.1146/annurev-biodatasci-122320-112352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Kirby JC, Speltz P, Rasmussen LV, Basford M, Gottesman O, Peissig PL, et al. PheKB: a catalog and workflow for creating electronic phenotype algorithms for transportability. J Am Med Inform Assoc . 2016;23:1046–1052. doi: 10.1093/jamia/ocv202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Newton KM, Peissig PL, Kho AN, Bielinski SJ, Berg RL, Choudhary V, et al. Validation of electronic medical record-based phenotyping algorithms: results and lessons learned from the eMERGE network. J Am Med Inform Assoc . 2013;20:e147–e154. doi: 10.1136/amiajnl-2012-000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Denny JC, Bastarache L, Ritchie MD, Carroll RJ, Zink R, Mosley JD, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol . 2013;31:1102–1110. doi: 10.1038/nbt.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. McHugh LG, Milberg JA, Whitcomb ME, Schoene RB, Maunder RJ, Hudson LD. Recovery of function in survivors of the acute respiratory distress syndrome. Am J Respir Crit Care Med . 1994;150:90–94. doi: 10.1164/ajrccm.150.1.8025779. [DOI] [PubMed] [Google Scholar]
- 124. Hopkins RO, Weaver LK, Pope D, Orme JF, Bigler ED, Larson-LOHR V. Neuropsychological sequelae and impaired health status in survivors of severe acute respiratory distress syndrome. Am J Respir Crit Care Med . 1999;160:50–56. doi: 10.1164/ajrccm.160.1.9708059. [DOI] [PubMed] [Google Scholar]
- 125. Davidson TA, Caldwell ES, Curtis JR, Hudson LD, Steinberg KP. Reduced quality of life in survivors of acute respiratory distress syndrome compared with critically ill control patients. JAMA . 1999;281:354–360. doi: 10.1001/jama.281.4.354. [DOI] [PubMed] [Google Scholar]
- 126. Angus DC, Musthafa AA, Clermont G, Griffin MF, Linde-Zwirble WT, Dremsizov TT, et al. Quality-adjusted survival in the first year after the acute respiratory distress syndrome. Am J Respir Crit Care Med . 2001;163:1389–1394. doi: 10.1164/ajrccm.163.6.2005123. [DOI] [PubMed] [Google Scholar]
- 127. Davidson TA, Rubenfeld GD, Caldwell ES, Hudson LD, Steinberg KP. The effect of acute respiratory distress syndrome on long-term survival. Am J Respir Crit Care Med . 1999;160:1838–1842. doi: 10.1164/ajrccm.160.6.9903058. [DOI] [PubMed] [Google Scholar]
- 128. Cameron JI, Chu LM, Matte A, Tomlinson G, Chan L, Thomas C, et al. RECOVER Program Investigators (Phase 1: towards RECOVER); Canadian Critical Care Trials Group One-year outcomes in caregivers of critically ill patients. N Engl J Med . 2016;374:1831–1841. doi: 10.1056/NEJMoa1511160. [DOI] [PubMed] [Google Scholar]
- 129. Feemster LC, Cooke CR, Rubenfeld GD, Hough CL, Ehlenbach WJ, Au DH, et al. The influence of hospitalization or intensive care unit admission on declines in health-related quality of life. Ann Am Thorac Soc . 2015;12:35–45. doi: 10.1513/AnnalsATS.201404-172OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ehlenbach WJ, Hough CL, Crane PK, Haneuse SJ, Carson SS, Curtis JR, et al. Association between acute care and critical illness hospitalization and cognitive function in older adults. JAMA . 2010;303:763–770. doi: 10.1001/jama.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA . 2010;304:1787–1794. doi: 10.1001/jama.2010.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Needham DM, Davidson J, Cohen H, Hopkins RO, Weinert C, Wunsch H, et al. Improving long-term outcomes after discharge from intensive care unit: report from a stakeholders’ conference. Crit Care Med . 2012;40:502–509. doi: 10.1097/CCM.0b013e318232da75. [DOI] [PubMed] [Google Scholar]
- 133. Azoulay E, Vincent JL, Angus DC, Arabi YM, Brochard L, Brett SJ, et al. Recovery after critical illness: putting the puzzle together-a consensus of 29. Crit Care . 2017;21:296. doi: 10.1186/s13054-017-1887-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Arbov E, Tayara A, Wu S, Rich TC, Wagener BM. COVID-19 and long-term outcomes: lessons from other critical care illnesses and potential mechanisms. Am J Respir Cell Mol Biol . 2022 doi: 10.1165/rcmb.2021-0374PS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Needham DM, Wozniak AW, Hough CL, Morris PE, Dinglas VD, Jackson JC, et al. National Institutes of Health NHLBI ARDS Network Risk factors for physical impairment after acute lung injury in a national, multicenter study. Am J Respir Crit Care Med . 2014;189:1214–1224. doi: 10.1164/rccm.201401-0158OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Brown SM, Wilson E, Presson AP, Zhang C, Dinglas VD, Greene T, et al. with the National Institutes of Health NHLBI ARDS Network Predictors of 6-month health utility outcomes in survivors of acute respiratory distress syndrome. Thorax . 2017;72:311–317. doi: 10.1136/thoraxjnl-2016-208560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Kress JP, Hall JB. ICU-acquired weakness and recovery from critical illness. N Engl J Med . 2014;370:1626–1635. doi: 10.1056/NEJMra1209390. [DOI] [PubMed] [Google Scholar]
- 138. Hough CL, Steinberg KP, Taylor Thompson B, Rubenfeld GD, Hudson LD. Intensive care unit-acquired neuromyopathy and corticosteroids in survivors of persistent ARDS. Intensive Care Med . 2009;35:63–68. doi: 10.1007/s00134-008-1304-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Brummel NE, Hughes CG, Thompson JL, Jackson JC, Pandharipande P, McNeil JB, et al. Inflammation and coagulation during critical illness and long-term cognitive impairment and disability. Am J Respir Crit Care Med . 2021;203:699–706. doi: 10.1164/rccm.201912-2449OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Gandotra S, Lovato J, Case D, Bakhru RN, Gibbs K, Berry M, et al. Physical function trajectories in survivors of acute respiratory failure. Ann Am Thorac Soc . 2019;16:471–477. doi: 10.1513/AnnalsATS.201806-375OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Denehy L, Skinner EH, Edbrooke L, Haines K, Warrillow S, Hawthorne G, et al. Exercise rehabilitation for patients with critical illness: a randomized controlled trial with 12 months of follow-up. Crit Care . 2013;17:R156. doi: 10.1186/cc12835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Moss M, Nordon-Craft A, Malone D, Van Pelt D, Frankel SK, Warner ML, et al. A randomized trial of an intensive physical therapy program for patients with acute respiratory failure. Am J Respir Crit Care Med . 2016;193:1101–1110. doi: 10.1164/rccm.201505-1039OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Walsh TS, Salisbury LG, Merriweather JL, Boyd JA, Griffith DM, Huby G, et al. RECOVER Investigators Increased hospital-based physical rehabilitation and information provision after intensive care unit discharge: the RECOVER randomized clinical trial. JAMA Intern Med . 2015;175:901–910. doi: 10.1001/jamainternmed.2015.0822. [DOI] [PubMed] [Google Scholar]
- 144. Brummel NE, Girard TD, Ely EW, Pandharipande PP, Morandi A, Hughes CG, et al. Feasibility and safety of early combined cognitive and physical therapy for critically ill medical and surgical patients: the activity and cognitive therapy in ICU (ACT-ICU) trial. Intensive Care Med . 2014;40:370–379. doi: 10.1007/s00134-013-3136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Cox CE, Hough CL, Carson SS, White DB, Kahn JM, Olsen MK, et al. Effects of a telephone- and web-based coping skills training program compared with an education program for survivors of critical illness and their family members. A randomized clinical trial. Am J Respir Crit Care Med . 2018;197:66–78. doi: 10.1164/rccm.201704-0720OC. [DOI] [PubMed] [Google Scholar]
- 146. Wade DM, Mouncey PR, Richards-Belle A, Wulff J, Harrison DA, Sadique MZ, et al. POPPI Trial Investigators Effect of a nurse-led preventive psychological intervention on symptoms of posttraumatic stress disorder among critically ill patients: a randomized clinical trial. JAMA . 2019;321:665–675. doi: 10.1001/jama.2019.0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Garrouste-Orgeas M, Flahault C, Vinatier I, Rigaud JP, Thieulot-Rolin N, Mercier E, et al. Effect of an ICU diary on posttraumatic stress disorder symptoms among patients receiving mechanical ventilation: a randomized clinical trial. JAMA . 2019;322:229–239. doi: 10.1001/jama.2019.9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Cuthbertson BH, Rattray J, Campbell MK, Gager M, Roughton S, Smith A, et al. PRaCTICaL study group The PRaCTICaL study of nurse-led, intensive care follow-up programmes for improving long-term outcomes from critical illness: a pragmatic randomised controlled trial. BMJ . 2009;339:b3723. doi: 10.1136/bmj.b3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Elliott D, McKinley S, Alison J, Aitken LM, King M, Leslie GD, et al. Health-related quality of life and physical recovery after a critical illness: a multi-centre randomised controlled trial of a home-based physical rehabilitation program. Crit Care . 2011;15:R142. doi: 10.1186/cc10265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Schmidt K, Worrack S, Von Korff M, Davydow D, Brunkhorst F, Ehlert U, et al. SMOOTH Study Group Effect of a primary care management intervention on mental health-related quality of life among survivors of sepsis: a randomized clinical trial. JAMA . 2016;315:2703–2711. doi: 10.1001/jama.2016.7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Woodcock J, LaVange LM. Master protocols to study multiple therapies, multiple diseases, or both. N Engl J Med . 2017;377:62–70. doi: 10.1056/NEJMra1510062. [DOI] [PubMed] [Google Scholar]
- 152. Saville BR, Berry SM. Efficiencies of platform clinical trials: a vision of the future. Clin Trials . 2016;13:358–366. doi: 10.1177/1740774515626362. [DOI] [PubMed] [Google Scholar]
- 153. Berry SM, Connor JT, Lewis RJ. The platform trial: an efficient strategy for evaluating multiple treatments. JAMA . 2015;313:1619–1620. doi: 10.1001/jama.2015.2316. [DOI] [PubMed] [Google Scholar]
- 154.Randomised evaluation of COVID-19 therapy. 2022. https://www.recoverytrial.net
- 155. Goligher EC, Bradbury CA, McVerry BJ, Lawler PR, Berger JS, Gong MN, et al. REMAP-CAP Investigators; ACTIV-4a Investigators; ATTACC Investigators Therapeutic anticoagulation with heparin in critically ill patients with COVID-19. N Engl J Med . 2021;385:777–789. doi: 10.1056/NEJMoa2103417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Lawler PR, Goligher EC, Berger JS, Neal MD, McVerry BJ, Nicolau JC, et al. ATTACC Investigators; ACTIV-4a Investigators; REMAP-CAP Investigators Therapeutic anticoagulation with heparin in noncritically ill patients with COVID-19. N Engl J Med . 2021;385:790–802. doi: 10.1056/NEJMoa2105911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Harrington D, Parmigiani G. I-SPY 2--a glimpse of the future of phase 2 drug development? N Engl J Med . 2016;375:7–9. doi: 10.1056/NEJMp1602256. [DOI] [PubMed] [Google Scholar]
- 158. I-SPY COVID Consortium. Clinical trial design during and beyond the pandemic: the I-SPY COVID trial. Nat Med . 2022;28:9–11. doi: 10.1038/s41591-021-01617-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Jabaudon M, Blondonnet R, Pereira B, Cartin-Ceba R, Lichtenstern C, Mauri T, et al. Plasma sRAGE is independently associated with increased mortality in ARDS: a meta-analysis of individual patient data. Intensive Care Med . 2018;44:1388–1399. doi: 10.1007/s00134-018-5327-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Freidlin B, Korn EL. Biomarker enrichment strategies: matching trial design to biomarker credentials. Nat Rev Clin Oncol . 2014;11:81–90. doi: 10.1038/nrclinonc.2013.218. [DOI] [PubMed] [Google Scholar]
- 161. Jiang W, Freidlin B, Simon R. Biomarker-adaptive threshold design: a procedure for evaluating treatment with possible biomarker-defined subset effect. J Natl Cancer Inst . 2007;99:1036–1043. doi: 10.1093/jnci/djm022. [DOI] [PubMed] [Google Scholar]
- 162. Kent DM, Nelson J, Dahabreh IJ, Rothwell PM, Altman DG, Hayward RA. Risk and treatment effect heterogeneity: re-analysis of individual participant data from 32 large clinical trials. Int J Epidemiol . 2016;45:2075–2088. doi: 10.1093/ije/dyw118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Iwashyna TJ, Burke JF, Sussman JB, Prescott HC, Hayward RA, Angus DC. Implications of heterogeneity of treatment effect for reporting and analysis of randomized trials in critical care. Am J Respir Crit Care Med . 2015;192:1045–1051. doi: 10.1164/rccm.201411-2125CP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Almirall D, Nahum-Shani I, Sherwood NE, Murphy SA. Introduction to SMART designs for the development of adaptive interventions: with application to weight loss research. Transl Behav Med . 2014;4:260–274. doi: 10.1007/s13142-014-0265-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Herold S, Mayer K, Lohmeyer J. Acute lung injury: how macrophages orchestrate resolution of inflammation and tissue repair. Front Immunol . 2011;2:65. doi: 10.3389/fimmu.2011.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Herold S, Becker C, Ridge KM, Budinger GR. Influenza virus-induced lung injury: pathogenesis and implications for treatment. Eur Respir J . 2015;45:1463–1478. doi: 10.1183/09031936.00186214. [DOI] [PubMed] [Google Scholar]
- 167. Peteranderl C, Morales-Nebreda L, Selvakumar B, Lecuona E, Vadász I, Morty RE, et al. Macrophage-epithelial paracrine crosstalk inhibits lung edema clearance during influenza infection. J Clin Invest . 2016;126:1566–1580. doi: 10.1172/JCI83931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Watanabe S, Alexander M, Misharin AV, Budinger GRS. The role of macrophages in the resolution of inflammation. J Clin Invest . 2019;129:2619–2628. doi: 10.1172/JCI124615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Morrell ED, Radella F, II, Manicone AM, Mikacenic C, Stapleton RD, Gharib SA, et al. Peripheral and distress alveolar cell transcriptional programs are distinct in acute respiratory syndrome. Am J Respir Crit Care Med . 2018;197:528–532. doi: 10.1164/rccm.201703-0614LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Morrell ED, Wiedeman A, Long SA, Gharib SA, West TE, Skerrett SJ, et al. Cytometry TOF identifies alveolar macrophage subtypes in acute respiratory distress syndrome. JCI Insight . 2018;3:e99281. doi: 10.1172/jci.insight.99281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Mould KJ, Jackson ND, Henson PM, Seibold M, Janssen WJ. Single-cell RNA sequencing identifies unique inflammatory airspace macrophage subsets. JCI Insight . 2019;4:e126556. doi: 10.1172/jci.insight.126556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Mould KJ, Moore CM, McManus SA, McCubbrey AL, McClendon JD, Griesmer CL, et al. Airspace macrophages and monocytes exist in transcriptionally distinct subsets in healthy adults. Am J Respir Crit Care Med . 2021;203:946–956. doi: 10.1164/rccm.202005-1989OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Rosseau S, Hammerl P, Maus U, Walmrath HD, Schütte H, Grimminger F, et al. Phenotypic characterization of alveolar monocyte recruitment in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol . 2000;279:L25–L35. doi: 10.1152/ajplung.2000.279.1.L25. [DOI] [PubMed] [Google Scholar]
- 174. Brittan M, Barr L, Conway Morris A, Duffin R, Rossi F, Johnston S, et al. A novel subpopulation of monocyte-like cells in the human lung after lipopolysaccharide inhalation. Eur Respir J . 2012;40:206–214. doi: 10.1183/09031936.00113811. [DOI] [PubMed] [Google Scholar]
- 175. Mould KJ, Barthel L, Mohning MP, Thomas SM, McCubbrey AL, Danhorn T, et al. Cell origin dictates programming of resident versus recruited macrophages during acute lung injury. Am J Respir Cell Mol Biol . 2017;57:294–306. doi: 10.1165/rcmb.2017-0061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Morrell ED, Bhatraju PK, Mikacenic CR, Radella F, II, Manicone AM, Stapleton RD, et al. Alveolar macrophage transcriptional programs are associated with outcomes in acute respiratory distress syndrome. Am J Respir Crit Care Med . 2019;200:732–741. doi: 10.1164/rccm.201807-1381OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Grant RA, Morales-Nebreda L, Markov NS, Swaminathan S, Querrey M, Guzman ER, et al. NU SCRIPT Study Investigators Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature . 2021;590:635–641. doi: 10.1038/s41586-020-03148-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Mould KJ, Janssen WJ. Signs of self-sustained inflammatory circuits in severe COVID pneumonia. Nature . 2021;590:553–554. doi: 10.1038/d41586-021-00296-5. [DOI] [PubMed] [Google Scholar]
- 179. Wendisch D, Dietrich O, Mari T, von Stillfried S, Ibarra IL, Mittermaier M, et al. Deutsche COVID-19 OMICS Initiative (DeCOI) SARS-CoV-2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell . 2021;184:6243–6261.e27. doi: 10.1016/j.cell.2021.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Vazquez-Armendariz AI, Heiner M, El Agha E, Salwig I, Hoek A, Hessler MC, et al. Multilineage murine stem cells generate complex organoids to model distal lung development and disease. EMBO J . 2020;39:e103476. doi: 10.15252/embj.2019103476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Salwig I, Spitznagel B, Vazquez-Armendariz AI, Khalooghi K, Guenther S, Herold S, et al. Bronchioalveolar stem cells are a main source for regeneration of distal lung epithelia in vivo. EMBO J . 2019;38:e102099. doi: 10.15252/embj.2019102099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Vazquez-Armendariz AI, Herold S. From clones to buds and branches: the use of lung organoids to model branching morphogenesis ex vivo. Front Cell Dev Biol . 2021;9:631579. doi: 10.3389/fcell.2021.631579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Combes AJ, Courau T, Kuhn NF, Hu KH, Ray A, Chen WS, et al. UCSF COMET Consortium Global absence and targeting of protective immune states in severe COVID-19. Nature . 2021;591:124–130. doi: 10.1038/s41586-021-03234-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol . 2018;18:134–147. doi: 10.1038/nri.2017.105. [DOI] [PubMed] [Google Scholar]
- 185. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science . 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 186. Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med . 2012;18:1386–1393. doi: 10.1038/nm.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Jiménez-Alcázar M, Rangaswamy C, Panda R, Bitterling J, Simsek YJ, Long AT, et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science . 2017;358:1202–1206. doi: 10.1126/science.aam8897. [DOI] [PubMed] [Google Scholar]
- 188. Lefrançais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight . 2018;3:1–15. doi: 10.1172/jci.insight.98178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Thomas GM, Carbo C, Curtis BR, Martinod K, Mazo IB, Schatzberg D, et al. Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood . 2012;119:6335–6343. doi: 10.1182/blood-2012-01-405183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest . 2012;122:2661–2671. doi: 10.1172/JCI61303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe . 2012;12:324–333. doi: 10.1016/j.chom.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 192. Pulavendran S, Prasanthi M, Ramachandran A, Grant R, Snider TA, Chow VTK, et al. Production of neutrophil extracellular traps contributes to the pathogenesis of Francisella tularemia. Front Immunol . 2020;11:679. doi: 10.3389/fimmu.2020.00679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K, De Meyer SF, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost . 2012;10:136–144. doi: 10.1111/j.1538-7836.2011.04544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Sayah DM, Mallavia B, Liu F, Ortiz-Muñoz G, Caudrillier A, DerHovanessian A, et al. Lung Transplant Outcomes Group Investigators Neutrophil extracellular traps are pathogenic in primary graft dysfunction after lung transplantation. Am J Respir Crit Care Med . 2015;191:455–463. doi: 10.1164/rccm.201406-1086OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Lachowicz-Scroggins ME, Dunican EM, Charbit AR, Raymond W, Looney MR, Peters MC, et al. Extracellular DNA, neutrophil extracellular traps, and inflammasome activation in severe asthma. Am J Respir Crit Care Med . 2019;199:1076–1085. doi: 10.1164/rccm.201810-1869OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J Exp Med . 2020;217:e20200652. doi: 10.1084/jem.20200652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil extracellular traps in COVID-19. JCI Insight . 2020;5:e138999. doi: 10.1172/jci.insight.138999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood . 2020;136:1169–1179. doi: 10.1182/blood.2020007008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Parikh SM. Targeting Tie2 and the host vascular response in sepsis. Sci Transl Med . 2016;8:335fs9. doi: 10.1126/scitranslmed.aaf5537. [DOI] [PubMed] [Google Scholar]
- 200. Sack KD, Kellum JA, Parikh SM. The angiopoietin-tie2 pathway in critical illness. Crit Care Clin . 2020;36:201–216. doi: 10.1016/j.ccc.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Korhonen EA, Lampinen A, Giri H, Anisimov A, Kim M, Allen B, et al. Tie1 controls angiopoietin function in vascular remodeling and inflammation. J Clin Invest . 2016;126:3495–3510. doi: 10.1172/JCI84923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Fiedler U, Scharpfenecker M, Koidl S, Hegen A, Grunow V, Schmidt JM, et al. The Tie-2 ligand angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel-Palade bodies. Blood . 2004;103:4150–4156. doi: 10.1182/blood-2003-10-3685. [DOI] [PubMed] [Google Scholar]
- 203. Oh H, Takagi H, Suzuma K, Otani A, Matsumura M, Honda Y. Hypoxia and vascular endothelial growth factor selectively up-regulate angiopoietin-2 in bovine microvascular endothelial cells. J Biol Chem . 1999;274:15732–15739. doi: 10.1074/jbc.274.22.15732. [DOI] [PubMed] [Google Scholar]
- 204. Goettsch W, Gryczka C, Korff T, Ernst E, Goettsch C, Seebach J, et al. Flow-dependent regulation of angiopoietin-2. J Cell Physiol . 2008;214:491–503. doi: 10.1002/jcp.21229. [DOI] [PubMed] [Google Scholar]
- 205. Ghosh CC, David S, Zhang R, Berghelli A, Milam K, Higgins SJ, et al. Gene control of tyrosine kinase TIE2 and vascular manifestations of infections. Proc Natl Acad Sci USA . 2016;113:2472–2477. doi: 10.1073/pnas.1519467113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. David S, Park JK, Meurs Mv, Zijlstra JG, Koenecke C, Schrimpf C, et al. Acute administration of recombinant Angiopoietin-1 ameliorates multiple-organ dysfunction syndrome and improves survival in murine sepsis. Cytokine . 2011;55:251–259. doi: 10.1016/j.cyto.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 207. Stiehl T, Thamm K, Kaufmann J, Schaeper U, Kirsch T, Haller H, et al. Lung-targeted RNA interference against angiopoietin-2 ameliorates multiple organ dysfunction and death in sepsis. Crit Care Med . 2014;42:e654–e662. doi: 10.1097/CCM.0000000000000524. [DOI] [PubMed] [Google Scholar]
- 208. Han S, Lee SJ, Kim KE, Lee HS, Oh N, Park I, et al. Amelioration of sepsis by TIE2 activation-induced vascular protection. Sci Transl Med . 2016;8:335ra55. doi: 10.1126/scitranslmed.aad9260. [DOI] [PubMed] [Google Scholar]
- 209. Kumpers P, Gueler F, David S, Slyke PV, Dumont DJ, Park JK, et al. The synthetic tie2 agonist peptide vasculotide protects against vascular leakage and reduces mortality in murine abdominal sepsis. Crit Care . 2011;15:R261. doi: 10.1186/cc10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Witzenbichler B, Westermann D, Knueppel S, Schultheiss HP, Tschope C. Protective role of angiopoietin-1 in endotoxic shock. Circulation . 2005;111:97–105. doi: 10.1161/01.CIR.0000151287.08202.8E. [DOI] [PubMed] [Google Scholar]
- 211. Kim W, Moon SO, Lee SY, Jang KY, Cho CH, Koh GY, et al. COMP-angiopoietin-1 ameliorates renal fibrosis in a unilateral ureteral obstruction model. J Am Soc Nephrol . 2006;17:2474–2483. doi: 10.1681/ASN.2006020109. [DOI] [PubMed] [Google Scholar]
- 212. Lee S, Kim W, Kim DH, Moon SO, Jung YJ, Lee AS, et al. Protective effect of COMP-angiopoietin-1 on cyclosporine-induced renal injury in mice. Nephrol Dial Transplant . 2008;23:2784–2794. doi: 10.1093/ndt/gfn168. [DOI] [PubMed] [Google Scholar]
- 213. Parikh SM, Mammoto T, Schultz A, Yuan HT, Christiani D, Karumanchi SA, et al. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med . 2006;3:e46. doi: 10.1371/journal.pmed.0030046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Orfanos SE, Kotanidou A, Glynos C, Athanasiou C, Tsigkos S, Dimopoulou I, et al. Angiopoietin-2 is increased in severe sepsis: correlation with inflammatory mediators. Crit Care Med . 2007;35:199–206. doi: 10.1097/01.CCM.0000251640.77679.D7. [DOI] [PubMed] [Google Scholar]
- 215. Ong T, McClintock DE, Kallet RH, Ware LB, Matthay MA, Liu KD. Ratio of angiopoietin-2 to angiopoietin-1 as a predictor of mortality in acute lung injury patients. Crit Care Med . 2010;38:1845–1851. doi: 10.1097/CCM.0b013e3181eaa5bf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Li F, Yin R, Guo Q. Circulating angiopoietin-2 and the risk of mortality in patients with acute respiratory distress syndrome: a systematic review and meta-analysis of 10 prospective cohort studies. Ther Adv Respir Dis . 2020;14:1753466620905274. doi: 10.1177/1753466620905274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Calfee CS, Gallagher D, Abbott J, Thompson BT, Matthay MA, NHLBI ARDS Network Plasma angiopoietin-2 in clinical acute lung injury: prognostic and pathogenetic significance. Crit Care Med . 2012;40:1731–1737. doi: 10.1097/CCM.0b013e3182451c87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Zinter MS, Spicer A, Orwoll BO, Alkhouli M, Dvorak CC, Calfee CS, et al. Plasma angiopoietin-2 outperforms other markers of endothelial injury in prognosticating pediatric ARDS mortality. Am J Physiol Lung Cell Mol Physiol . 2016;310:L224–L231. doi: 10.1152/ajplung.00336.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Agrawal A, Matthay MA, Kangelaris KN, Stein J, Chu JC, Imp BM, et al. Plasma angiopoietin-2 predicts the onset of acute lung injury in critically ill patients. Am J Respir Crit Care Med . 2013;187:736–742. doi: 10.1164/rccm.201208-1460OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Giuliano JS, Jr, Lahni PM, Harmon K, Wong HR, Doughty LA, Carcillo JA, et al. Admission angiopoietin levels in children with septic shock. Shock . 2007;28:650–654. [PMC free article] [PubMed] [Google Scholar]
- 221. Su L, Zhai R, Sheu CC, Gallagher DC, Gong MN, Tejera P, et al. Genetic variants in the angiopoietin-2 gene are associated with increased risk of ARDS. Intensive Care Med . 2009;35:1024–1030. doi: 10.1007/s00134-009-1413-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Higgins SJ, De Ceunynck K, Kellum JA, Chen X, Gu X, Chaudhry SA, et al. Tie2 protects the vasculature against thrombus formation in systemic inflammation. J Clin Invest . 2018;128:1471–1484. doi: 10.1172/JCI97488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Pandharipande PP, Girard TD, Jackson JC, Morandi A, Thompson JL, Pun BT, et al. BRAIN-ICU Study Investigators Long-term cognitive impairment after critical illness. N Engl J Med . 2013;369:1306–1316. doi: 10.1056/NEJMoa1301372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Hopkins RO, Weaver LK, Collingridge D, Parkinson RB, Chan KJ, Orme JF., Jr Two-year cognitive, emotional, and quality-of-life outcomes in acute respiratory distress syndrome. Am J Respir Crit Care Med . 2005;171:340–347. doi: 10.1164/rccm.200406-763OC. [DOI] [PubMed] [Google Scholar]
- 225. Balczon R, Lin MT, Lee JY, Abbasi A, Renema P, Voth SB, et al. Pneumonia initiates a tauopathy. FASEB J . 2021;35:e21807. doi: 10.1096/fj.202100718R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Balczon R, Pittet JF, Wagener BM, Moser SA, Voth S, Vorhees CV, et al. Infection-induced endothelial amyloids impair memory. FASEB J . 2019;33:10300–10314. doi: 10.1096/fj.201900322R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Fändrich M. On the structural definition of amyloid fibrils and other polypeptide aggregates. Cell Mol Life Sci . 2007;64:2066–2078. doi: 10.1007/s00018-007-7110-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron . 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 229. Lei P, Ayton S, Finkelstein DI, Adlard PA, Masters CL, Bush AI. Tau protein: relevance to Parkinson’s disease. Int J Biochem Cell Biol . 2010;42:1775–1778. doi: 10.1016/j.biocel.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 230. Balczon R, Morrow KA, Zhou C, Edmonds B, Alexeyev M, Pittet JF, et al. Pseudomonas aeruginosa infection liberates transmissible, cytotoxic prion amyloids. FASEB J . 2017;31:2785–2796. doi: 10.1096/fj.201601042RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Lin MT, Balczon R, Pittet JF, Wagener BM, Moser SA, Morrow KA, et al. Nosocomial pneumonia elicits an endothelial proteinopathy: evidence for a source of neurotoxic amyloids in critically ill patients. Am J Respir Crit Care Med . 2018;198:1575–1578. doi: 10.1164/rccm.201801-0060LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Morrow KA, Ochoa CD, Balczon R, Zhou C, Cauthen L, Alexeyev M, et al. Pseudomonas aeruginosa exoenzymes U and Y induce a transmissible endothelial proteinopathy. Am J Physiol Lung Cell Mol Physiol . 2016;310:L337–L353. doi: 10.1152/ajplung.00103.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Hippensteel JA, Anderson BJ, Orfila JE, McMurtry SA, Dietz RM, Su G, et al. Circulating heparan sulfate fragments mediate septic cognitive dysfunction. J Clin Invest . 2019;129:1779–1784. doi: 10.1172/JCI124485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Schmidt EP, Li G, Li L, Fu L, Yang Y, Overdier KH, et al. The circulating glycosaminoglycan signature of respiratory failure in critically ill adults. J Biol Chem . 2014;289:8194–8202. doi: 10.1074/jbc.M113.539452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Yang Y, Haeger SM, Suflita MA, Zhang F, Dailey KL, Colbert JF, et al. Fibroblast growth factor signaling mediates pulmonary endothelial glycocalyx reconstitution. Am J Respir Cell Mol Biol . 2017;56:727–737. doi: 10.1165/rcmb.2016-0338OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Adamson IY, Bowden DH, Cote MG, Witschi H. Lung injury induced by butylated hydroxytoluene: cytodynamic and biochemical studies in mice. Lab Invest . 1977;36:26–32. [PubMed] [Google Scholar]
- 237. Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest . 2013;123:3025–3036. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal, and cancer. Nature . 2014;507:190–194. doi: 10.1038/nature12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Zacharias WJ, Frank DB, Zepp JA, Morley MP, Alkhaleel FA, Kong J, et al. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature . 2018;555:251–255. doi: 10.1038/nature25786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science . 2018;359:1118–1123. doi: 10.1126/science.aam6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Aspal M, Zemans RL. Mechanisms of ATII-to-ATI cell differentiation during lung regeneration. Int J Mol Sci . 2020;21:21. doi: 10.3390/ijms21093188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Wu H, Tang N. Stem cells in pulmonary alveolar regeneration. Development . 2021;148:3188. doi: 10.1242/dev.193458. [DOI] [PubMed] [Google Scholar]
- 243. Flozak AS, Lam AP, Russell S, Jain M, Peled ON, Sheppard KA, et al. Beta-catenin/T-cell factor signaling is activated during lung injury and promotes the survival and migration of alveolar epithelial cells. J Biol Chem . 2010;285:3157–3167. doi: 10.1074/jbc.M109.070326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Zemans RL, Briones N, Campbell M, McClendon J, Young SK, Suzuki T, et al. Neutrophil transmigration triggers repair of the lung epithelium via beta-catenin signaling. Proc Natl Acad Sci USA . 2011;108:15990–15995. doi: 10.1073/pnas.1110144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Tanjore H, Degryse AL, Crossno PF, Xu XC, McConaha ME, Jones BR, et al. β-catenin in the alveolar epithelium protects from lung fibrosis after intratracheal bleomycin. Am J Respir Crit Care Med . 2013;187:630–639. doi: 10.1164/rccm.201205-0972OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Panos RJ, Rubin JS, Csaky KG, Aaronson SA, Mason RJ. Keratinocyte growth factor and hepatocyte growth factor/scatter factor are heparin-binding growth factors for alveolar type II cells in fibroblast-conditioned medium. J Clin Invest . 1993;92:969–977. doi: 10.1172/JCI116673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Cakarova L, Marsh LM, Wilhelm J, Mayer K, Grimminger F, Seeger W, et al. Macrophage tumor necrosis factor-alpha induces epithelial expression of granulocyte-macrophage colony-stimulating factor: impact on alveolar epithelial repair. Am J Respir Crit Care Med . 2009;180:521–532. doi: 10.1164/rccm.200812-1837OC. [DOI] [PubMed] [Google Scholar]
- 248. Liu Y, Sadikot RT, Adami GR, Kalinichenko VV, Pendyala S, Natarajan V, et al. FoxM1 mediates the progenitor function of type II epithelial cells in repairing alveolar injury induced by Pseudomonas aeruginosa. J Exp Med . 2011;208:1473–1484. doi: 10.1084/jem.20102041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Zepp JA, Zacharias WJ, Frank DB, Cavanaugh CA, Zhou S, Morley MP, et al. Distinct mesenchymal lineages and niches promote epithelial self-renewal and myofibrogenesis in the lung. Cell . 2017;170:1134–1148.e10. doi: 10.1016/j.cell.2017.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Riemondy KA, Jansing NL, Jiang P, Redente EF, Gillen AE, Fu R, et al. Single-cell RNA sequencing identifies TGFβ as a key regenerative cue following LPS-induced lung injury. JCI Insight . 2019;5:1–18. doi: 10.1172/jci.insight.123637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Kobayashi Y, Tata A, Konkimalla A, Katsura H, Lee RF, Ou J, et al. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat Cell Biol . 2020;22:934–946. doi: 10.1038/s41556-020-0542-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Choi J, Park JE, Tsagkogeorga G, Yanagita M, Koo BK, Han N, et al. Inflammatory signals induce AT2 cell-derived damage-associated transient progenitors that mediate alveolar regeneration. Cell Stem Cell . 2020;27:366–382.e7. doi: 10.1016/j.stem.2020.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Jiang P, Gil de Rubio R, Hrycaj SM, Gurczynski SJ, Riemondy KA, Moore BB, et al. Ineffectual type 2-to-type 1 alveolar epithelial cell differentiation in idiopathic pulmonary fibrosis: persistence of the KRT8hi transitional state. Am J Respir Crit Care Med . 2020;201:1443–1447. doi: 10.1164/rccm.201909-1726LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Strunz M, Simon LM, Ansari M, Kathiriya JJ, Angelidis I, Mayr CH, et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat Commun . 2020;11:3559. doi: 10.1038/s41467-020-17358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med . 2019;199:1517–1536. doi: 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Wu H, Yu Y, Huang H, Hu Y, Fu S, Wang Z, et al. Progressive pulmonary fibrosis is caused by elevated mechanical tension on alveolar stem cells. Cell . 2021;184:845–846. doi: 10.1016/j.cell.2021.01.020. [DOI] [PubMed] [Google Scholar]
- 257. Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell . 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 258. Ting C, Aspal M, Vaishampayan N, Huang SK, Riemondy KA, Wang F, et al. Fatal COVID-19 and non-COVID-19 acute respiratory distress syndrome is associated with incomplete alveolar type 1 epithelial cell differentiation from the transitional state without fibrosis. Am J Pathol . 2022;192:454–467. doi: 10.1016/j.ajpath.2021.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Auyeung VC, Sheppard D. Stuck in a moment: does abnormal persistence of epithelial progenitors drive pulmonary fibrosis? Am J Respir Crit Care Med . 2021;203:667–669. doi: 10.1164/rccm.202010-3898ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Wiener-Kronish JP, Albertine KH, Matthay MA. Differential responses of the endothelial and epithelial barriers of the lung in sheep to Escherichia coli endotoxin. J Clin Invest . 1991;88:864–875. doi: 10.1172/JCI115388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Self WH, Semler MW, Leither LM, Casey JD, Angus DC, Brower RG, et al. National Heart, Lung, and Blood Institute PETAL Clinical Trials Network Effect of hydroxychloroquine on clinical status at 14 days in hospitalized patients with COVID-19: a randomized clinical trial. JAMA . 2020;324:2165–2176. doi: 10.1001/jama.2020.22240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Willson DF, Thomas NJ, Markovitz BP, Bauman LA, DiCarlo JV, Pon S, et al. Pediatric Acute Lung Injury and Sepsis Investigators Effect of exogenous surfactant (calfactant) in pediatric acute lung injury: a randomized controlled trial. JAMA . 2005;293:470–476. doi: 10.1001/jama.293.4.470. [DOI] [PubMed] [Google Scholar]
- 263. Constantin JM, Jabaudon M, Lefrant JY, Jaber S, Quenot JP, Langeron O, et al. AZUREA Network Personalised mechanical ventilation tailored to lung morphology versus low positive end-expiratory pressure for patients with acute respiratory distress syndrome in France (the LIVE study): a multicentre, single-blind, randomised controlled trial. Lancet Respir Med . 2019;7:870–880. doi: 10.1016/S2213-2600(19)30138-9. [DOI] [PubMed] [Google Scholar]
- 264. Calfee CS, Eisner MD, Ware LB, Thompson BT, Parsons PE, Wheeler AP, et al. Acute Respiratory Distress Syndrome Network, National Heart, Lung, and Blood Institute Trauma-associated lung injury differs clinically and biologically from acute lung injury due to other clinical disorders. Crit Care Med . 2007;35:2243–2250. doi: 10.1097/01.ccm.0000280434.33451.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Villar J, Pérez-Méndez L, López J, Belda J, Blanco J, Saralegui I, et al. HELP Network An early PEEP/FIO2 trial identifies different degrees of lung injury in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med . 2007;176:795–804. doi: 10.1164/rccm.200610-1534OC. [DOI] [PubMed] [Google Scholar]
- 266. Goligher EC, Kavanagh BP, Rubenfeld GD, Ferguson ND. Physiologic responsiveness should guide entry into randomized controlled trials. Am J Respir Crit Care Med . 2015;192:1416–1419. doi: 10.1164/rccm.201410-1832CP. [DOI] [PubMed] [Google Scholar]
- 267. Gattinoni L, Marini JJ, Quintel M. Recruiting the acutely injured lung: how and why? Am J Respir Crit Care Med . 2020;201:130–132. doi: 10.1164/rccm.201910-2005ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Goligher EC, Amato MBP, Slutsky AS. Applying precision medicine to trial design using physiology. Extracorporeal CO2 removal for acute respiratory distress syndrome. Am J Respir Crit Care Med . 2017;196:558–568. doi: 10.1164/rccm.201701-0248CP. [DOI] [PubMed] [Google Scholar]
- 269. Goligher EC, Costa ELV, Yarnell CJ, Brochard LJ, Stewart TE, Tomlinson G, et al. Effect of lowering Vt on mortality in acute respiratory distress syndrome varies with respiratory system elastance. Am J Respir Crit Care Med . 2021;203:1378–1385. doi: 10.1164/rccm.202009-3536OC. [DOI] [PubMed] [Google Scholar]
- 270. Lai PS, Mita C, Thompson BT. What is the clinical significance of pulmonary hypertension in acute respiratory distress syndrome? A review. Minerva Anestesiol . 2014;80:574–585. [PMC free article] [PubMed] [Google Scholar]
- 271. Sinha P, Calfee CS, Beitler JR, Soni N, Ho K, Matthay MA, et al. Physiologic analysis and clinical performance of the ventilatory ratio in acute respiratory distress syndrome. Am J Respir Crit Care Med . 2019;199:333–341. doi: 10.1164/rccm.201804-0692OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. McCarter SD, Mei SH, Lai PF, Zhang QW, Parker CH, Suen RS, et al. Cell-based angiopoietin-1 gene therapy for acute lung injury. Am J Respir Crit Care Med . 2007;175:1014–1026. doi: 10.1164/rccm.200609-1370OC. [DOI] [PubMed] [Google Scholar]
- 273. Mammoto T, Parikh SM, Mammoto A, Gallagher D, Chan B, Mostoslavsky G, et al. Angiopoietin-1 requires p190 RhoGAP to protect against vascular leakage in vivo. J Biol Chem . 2007;282:23910–23918. doi: 10.1074/jbc.M702169200. [DOI] [PubMed] [Google Scholar]
- 274. Frye M, Dierkes M, Küppers V, Vockel M, Tomm J, Zeuschner D, et al. Interfering with VE-PTP stabilizes endothelial junctions in vivo via Tie-2 in the absence of VE-cadherin. J Exp Med . 2015;212:2267–2287. doi: 10.1084/jem.20150718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Shen J, Wang J, Shao YR, He DK, Zhang L, Nadeem L, et al. Adenovirus-delivered angiopoietin-1 treatment for phosgene-induced acute lung injury. Inhal Toxicol . 2013;25:272–279. doi: 10.3109/08958378.2013.777820. [DOI] [PubMed] [Google Scholar]
- 276. Shao Y, Shen J, Zhou F, He D. Mesenchymal stem cells overexpressing Ang1 attenuates phosgene-induced acute lung injury in rats. Inhal Toxicol . 2018;30:313–320. doi: 10.1080/08958378.2018.1521483. [DOI] [PubMed] [Google Scholar]
- 277. Ghosh CC, Mukherjee A, David S, Knaus UG, Stearns-Kurosawa DJ, Kurosawa S, et al. Impaired function of the Tie-2 receptor contributes to vascular leakage and lethality in anthrax. Proc Natl Acad Sci USA . 2012;109:10024–10029. doi: 10.1073/pnas.1120755109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Bhandari V, Choo-Wing R, Lee CG, Zhu Z, Nedrelow JH, Chupp GL, et al. Hyperoxia causes angiopoietin 2-mediated acute lung injury and necrotic cell death. Nat Med . 2006;12:1286–1293. doi: 10.1038/nm1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Kugathasan L, Ray JB, Deng Y, Rezaei E, Dumont DJ, Stewart DJ. The angiopoietin-1-Tie2 pathway prevents rather than promotes pulmonary arterial hypertension in transgenic mice. J Exp Med . 2009;206:2221–2234. doi: 10.1084/jem.20090389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Zhao YD, Campbell AI, Robb M, Ng D, Stewart DJ. Protective role of angiopoietin-1 in experimental pulmonary hypertension. Circ Res . 2003;92:984–991. doi: 10.1161/01.RES.0000070587.79937.F0. [DOI] [PubMed] [Google Scholar]