

Abstract
Structural biology has the potential to illuminate the evolution of pathogen effectors and their commonalities that cannot be readily detected at the primary sequence level. Recent breakthroughs in protein structure modeling have demonstrated the feasibility to predict the protein folds without depending on homologous templates. These advances enabled a genome-wide computational structural biology approach to help understand proteins based on their predicted folds. In this study, we employed structure prediction methods on the secretome of the destructive fungal pathogen Magnaporthe oryzae. Out of 1,854 secreted proteins, we predicted the folds of 1,295 proteins (70%). We showed that template-free modeling by TrRosetta captured 514 folds missed by homology modeling, including many known effectors and virulence factors, and that TrRosetta generally produced higher quality models for secreted proteins. Along with sensitive homology search, we employed structure-based clustering, defining not only homologous groups with divergent members but also sequence-unrelated structurally analogous groups. We demonstrate that this approach can reveal new putative members of structurally similar MAX effectors and novel analogous effector families present in M. oryzae and possibly in other phytopathogens. We also investigated the evolution of expanded putative ADP-ribose transferases with predicted structures. We suggest that the loss of catalytic activities of the enzymes might have led them to new evolutionary trajectories to be specialized as protein binders. Collectively, we propose that computational structural genomics approaches can be an integral part of studying effector biology and provide valuable resources that were inaccessible before the advent of machine learning-based structure prediction.
Keywords: computational structural genomics, effectors, fungal effectors, fungus–plant interactions, genomics, Magnaporthe oryzae, phytopathogen, structure
Fungal phytopathogens encode a diverse set of effector proteins to infect and colonize plant hosts. Effectors include rapidly evolving or recently emerged proteins, the biological roles of which cannot be easily inferred with primary sequence information alone. On the other hand, structural biology has illuminated effector evolution through effectors’ structural commonality (Franceschetti et al. 2017). MAX effectors from Magnaporthe oryzae and Pyrenophora tritici-repentis are unrelated by their sequences but share a common β-sandwich fold (de Guillen et al. 2015). A few sequence-unrelated proteins form an analogous LARS effector family in Leptosphaeria maculans (Blondeau et al. 2015; Lazar et al. 2020). RALPH effectors commonly adopt the RNase fold despite highly divergent sequences in powdery mildews (Bauer et al. 2021; Pedersen et al. 2012; Pennington et al. 2019; Spanu 2017). Common structural folds without sequence similarity may be the outcome of convergent evolution or, alternatively, the loss of detectable homology (Andrie et al. 2008; de Guillen et al. 2015). Regardless of the true origin, their repeated appearance manifests their essential roles in virulence. However, because solved effector structures are limited, only a small number of common structural folds have been uncovered to date (Mukhi et al. 2020).
Structural genomics is an approach that aims to determine the structures for all proteins by experimental and prediction methods (Baker and Sali 2001). The application of structural genomics is valuable to elucidate effector biology; however, it has remained largely limited. Experimental determination of all effector structures would require immense time and community effort. Prediction is an attractive alternative. Nonetheless, predicting the folds of proteins has, until recently, been predominantly dependent on homology modeling that has obvious limitations: because of the lack of template structures and detectable homology between related effectors, most of the effector structures are not predictable.
The Critical Assessment of Structure Prediction (CASP) 13 in 2018 demonstrated the success of de novo folding algorithms. These algorithms, represented with AlphaFold, leverage machine learning to predict interresidue distances based on covariance inferred from a multiple sequence alignment (MSA) of the target and its homologous sequences (AlQuraishi 2019). Although structure prediction does not achieve atomic resolutions comparable with experimental structure determination techniques, the folds of unknown proteins were successfully predicted in the absence of homologous templates (Senior et al. 2020). Additionally, the expected quality score reported for predicted structures strongly correlates with the actual precision (Senior et al. 2020; Yang et al. 2020; Zhang 2008). Collectively, the ability to predict the structural folds and estimate the model quality enables us to apply structural prediction on phytopathogens’ secretomes.
In this study, we tested the computational structural genomics approach on 1,854 secreted proteins from destructive fungal phytopathogen M. oryzae (Dean et al. 2012; Wilson and Talbot 2009). We show that the template-free algorithm TrRosetta performs better than template-based modeler I-TASSER in predicting known and unknown secreted protein structures. Using structural information, we found analogous gene families and uncovered evidence of new common folds across phytopathogens. We propose that computational structural genomics can complement traditional genomic approaches in the analyses of effector evolution. The rapidly evolving protein structure field would help to guide the rational selection of conserved effector folds in phytopathogens for the development of new disease resistance strategies in near future.
RESULTS
Template-free modeling with TrRosetta outperforms homology modeling in predicting the folds of known effectors.
We assessed the performance of TrRosetta and I-TASSER on nonredundant 9 secreted and 15 nonsecreted Magnaporthe proteins available in the Protein Data Bank (PDB) (Supplementary Fig. S1; Supplementary Table S1) (Berman et al. 2000). We predicted their structures with each tool and superposed the computational models against the experimental ones with template modeling (TM)-align to measure actual precision as TM scores (Zhang and Skolnick 2005). A TM score >0.5 indicates that the two compared structures display approximately the same fold (Xu and Zhang 2010). Nonsecreted proteins with functional annotations were relatively easy targets for both methods, given the high precision of the predicted structures (Fig. 1A). TrRosetta produced structures with TM scores >0.5 for known effectors AvrPiz-t, Avr-Pia, Avr-Pib, and Avr-PikD, whereas I-TASSER only predicted the Avr-Pia structure (Supplementary Fig. S2) (De la Concepcion et al. 2018; Ose et al. 2015; Zhang et al. 2013; Zhang et al. 2018). This suggested that template-free modeling with TrRosetta outperforms homology-based modeling with I-TASSER in the selected effector structure modeling, successfully predicting the effector folds (TM score >0.5).
Fig. 1.
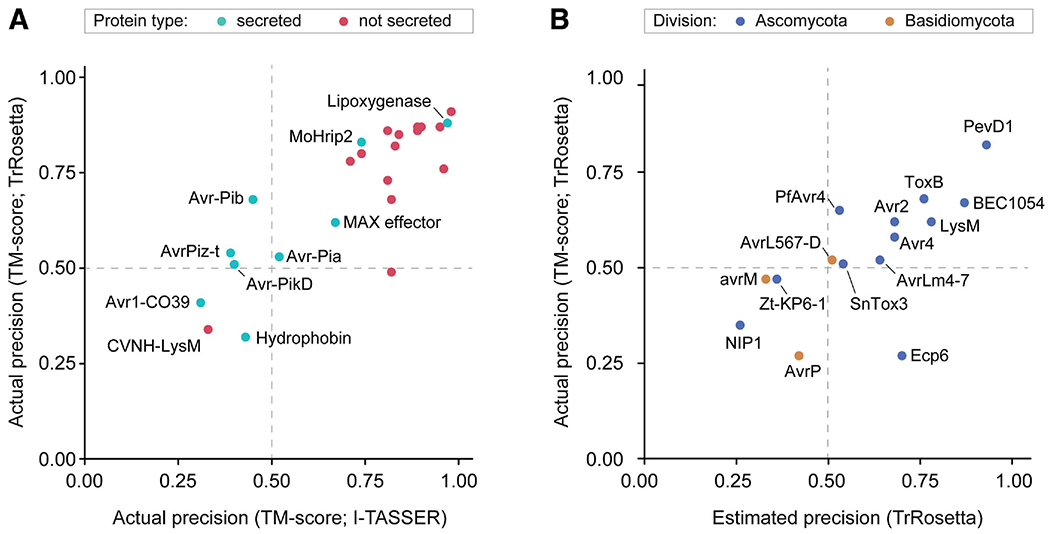
TrRosetta predicts the folds of many known effector structures. Statistics of protein structure prediction on A, 24 Magnaporthe proteins and B, 15 fungal effector proteins with solved structures available in the Protein Data Bank. The actual precision was obtained as template modeling (TM) scores by superposing the computational models from TrRosetta or I-TASSER against the experimental structures with TM-align. The estimated precision is a metric TrRosetta produces as a measure of the prediction quality and is reported to well correlate with actual precision. Actual precision (TM score) >0.5 indicates that the predicted and experimentally determined structures display approximately the same fold. Estimated precision >0.5 indicates that the fold of the predicted structures is likely correct.
We collected 15 available effector structures across other fungal phytopathogens and examined whether TrRosetta could predict their folds (Fig. 1B; Supplementary Table S2). The MSAs for carbohydrate-binding Avr4, LysM-containing Ecp6, and RNAse-like BEC1054 contained diverse homologs (>2,000) for coevolutionary inference and, accordingly, TrRosetta predicted their folds. In contrast, AvrL567-D, AvrLm4-7, Avr2, SnTox3, and ToxB had only a limited number of homologs collected (≤60). Nonetheless, their folds were still predicted, suggesting that TrRosetta can capture the overall folds of many effector structures when coevolutionary information is limited.
A combination of template-free and homology-based modeling resolves a large subset of secreted protein structures.
We next modeled 1,854 putative secreted proteins encoded in the M. oryzae genome. TrRosetta and I-TASSER use the average probability of the top L predicted long- plus medium-range contacts (|i – j| > 12) and the mean estimated TM score to estimate prediction qualities. These metrics, which we collectively termed as estimated precision, are reported to correlate well with actual precision, and the estimated precision >0.5 indicates that the fold of the predicted structures is likely correct (Yang et al. 2020; Zhang 2008). TrRosetta and I-TASSER predicted 627 structures in common with estimated precision >0.5 (Fig. 2A; Supplementary Table S3). Among them, 493 TrRosetta models displayed equal or higher expected precision. Each tool predicted additional 514 and 154 structures with estimated precision >0.5. Collectively, 70% of the secreted proteins were modeled by at least one of the methods with expected precision >0.5.
Fig. 2.
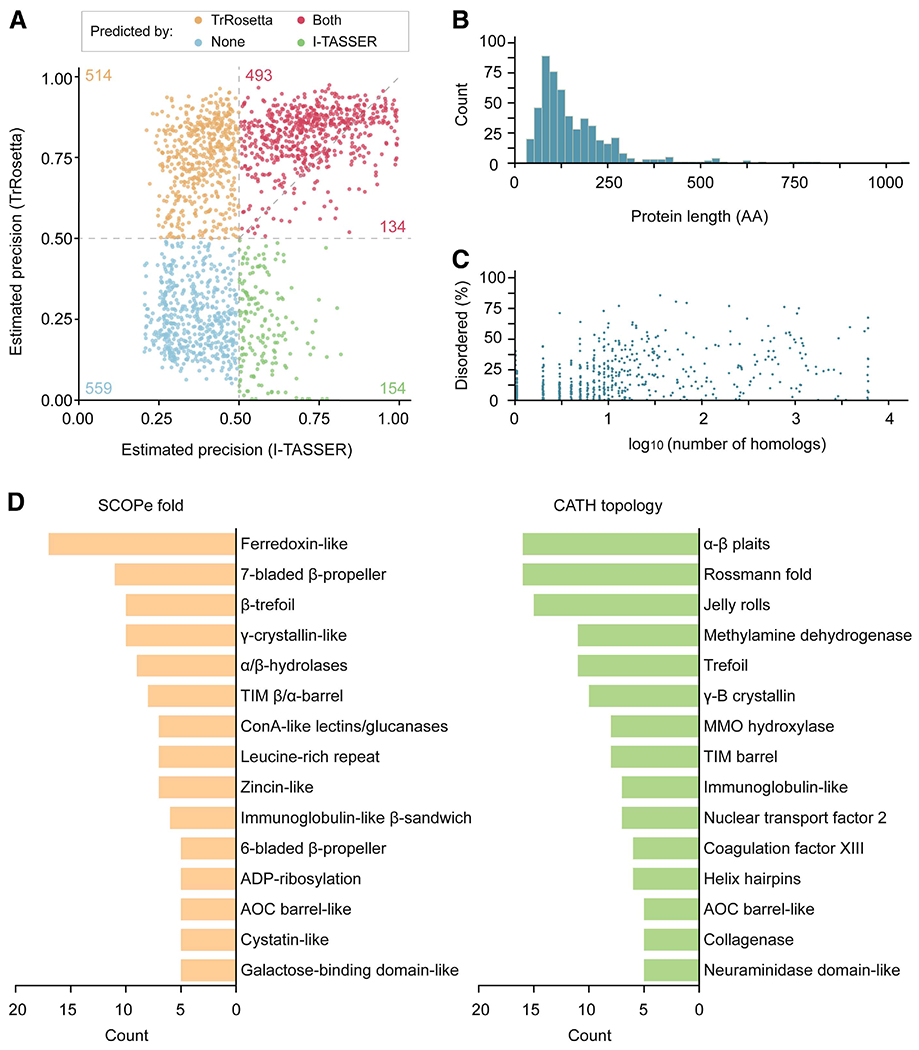
Structure prediction statistics on 1,854 Magnaporthe oryzae secreted proteins. A, Structure prediction statistics on 1,854 M. oryzae secreted proteins. The average probability of the top L predicted long- plus medium-range contacts (|i – j| >12) and the mean estimated template modeling (TM) score were used as the expected precision for TrRosetta and I-TASSER, respectively. These values are reported to well correlate with the actual precision (Yang et al. 2020; Zhang 2008). The number of proteins belonging to each section is indicated in the plot. B, Mature protein length distribution (AA = amino acids) and C, number of homologs collected for coevolutionary inference and the proportion of predicted disordered residues for 559 proteins that were not predicted by both TrRosetta and I-TASSER. D, Structure-based classification of 527 proteins without functional annotations. The 527 protein structures were assigned to SCOPe and CATH categories with RUPEE. The TM score cut-off >0.5 was required for the classification. The top 15 hits are reported.
The lack of homologous templates explains the relatively poor performance of homology modeling with I-TASSER. Many models exclusively modeled by I-TASSER included very short or highly disordered proteins that may not be trustworthy (Supplementary Fig. S3; Supplementary Table S3). The 559 proteins missed by both TrRosetta and I-TASSER were predominantly small (Fig. 2B) and did not have a sufficient number of homologs for coevolutionary inference, possibly attributed to the loss of detectable homology or recent origin (Fig. 2C). A subset of these proteins was likely intrinsically disordered, because the proportion of predicted disordered residues is high (Fig. 2C). These structures tend not to adopt a single, foldable confirmation and, therefore, their structures would not be able to be predicted.
We classified the 527 uncharacterized proteins without PFAM domains or with the domain of unknown functions by searching for similar structural folds and topologies in the SCOPe and CATH databases with RUPEE (Fig. 2D; Supplementary Table S4). The classification identified enzymes of diverse functions. These include hydrolytic enzymes adopting the α/β-hydrolase and Rossmann fold, glycosidases displaying the TIM (β/α) barrel structure, glucanases belonging to the concanavalin A-like lectins or glucanase and jelly roll structures, and metallopeptidase of the zincin-like and collagenase fold. Such results indicated that a subset of the unknown M. oryzae secretome contains divergently or rapidly evolving enzymes. Other structural folds and topologies were also present, which may supplement the infection mechanisms of M. oryzae (Supplementary Table S3).
Template-free modeling with TrRosetta captures the structures of previously identified effector proteins.
We predicted the folds of previously identified effectors from M. oryzae, the structures and functions of which are mostly unknown (Table 1). Among those with homologous structures, all were predicted, except for MoCDIP12, which has a homologous domain to Avr-Pik (Fig. 3A). In most cases, the TrRosetta models displayed higher expected precision, and structural comparisons supported the quality of the predicted models (Supplementary Fig. S4). Among the cloned effectors without homologous structures, TrRosetta could generate models with high estimated precision for MoNIS1, Avr-Pi54, EMP1, MoCDIP1, MoCDIP5, MoCDIP10, Avr-Pita1, and BAS4 (Fig. 3B). With lower estimated precision, the structures for PWL, BAS3, and MoCDIP3 were also predicted (Supplementary Fig. S5).
Table 1.
Collection of previously identified effectors in Magnaporthe oryzae and structure prediction statistics
| Protein ID | Protein name | Annotationa | Precb | Method | Matchc | Reference |
|---|---|---|---|---|---|---|
| MGG_08054T0 | MoChi1 | Chitinase 1 | 0.86 | TrRosetta | 5Y2A_A | Han et al. 2019 |
| MGG_08409T0 | MoCDIP4 | CGS protein | 0.85 | TrRosetta | 4B5Q_A | Chen et al. 2013 |
| MGG_02347T0 | MoNIS1 | PU protein | 0.97 | TrRosetta | – | Irieda et al. 2019 |
| MGG_03685T0 | Avr-Pi54 | PU protein | 0.94 | TrRosetta | – | Ray et al. 2016 |
| MGG_15022T0 | MoHrip1 | PU protein | 0.94 | TrRosetta | 5XMZ_A | Chen et al. 2012 |
| MGG_10914T0 | BAS4 | BAS protein 4 | 0.84 | TrRosetta | – | Mosquera et al. 2009 |
| MGG_02154T0 | SPD5 | PU protein | 0.87 | TrRosetta | – | Sharpee et al. 2017 |
| MGG_09378T0 | MoHEG13 | PU protein | 0.49 | I-TASSER | – | Mogga et al. 2016 |
| MGG_03356T0 | MoCDIP1 | Ricin B lectin | 0.91 | TrRosetta | – | Chen et al. 2013 |
| MGG_04546T0 | Nup3 | PU protein | 0.35 | I-TASSER | – | Dong et al. 2015 |
| MGG_10280T0 | MoHTR2 | PU protein | 0.43 | I-TASSER | – | Kim et al. 2020 |
| MGG_18041T0 | AvrPiz-t | PU protein | 0.8 | I-TASSER | 2LW6_A | Li et al. 2009 |
| MGG_12426T0 | Avr-Pib | PU protein | 0.59 | TrRosetta | – | Zhang et al. 2015 |
| MGG_05531T0 | MoCDIP2 | PU protein | 0.52 | I-TASSER | – | Chen et al. 2013 |
| MGG_12521T0 | MoCDIP11 | PU protein | 0.35 | I-TASSER | – | Guo et al. 2019 |
| MGG_07900T0 | Nup1 | PU protein | 0.43 | TrRosetta | – | Dong et al. 2015 |
| MGG_15370T0 | Avr-Pita1 | Metalloproteinase | 0.74 | TrRosetta | – | Dai et al. 2010 |
| MGG_10097T0 | SLP1 | IH protein 1 | 0.73 | TrRosetta | 4B8V_A | Mentlak et al. 2012 |
| MGG_16187T0 | MoHrip2 | PU protein | 0.96 | TrRosetta | 5FID_A | Chen et al. 2014 |
| MGG_05344T0 | MSP1; MgSM1 | SnodProt1 | 0.98 | I-TASSER | 3M3G_A | Jeong et al. 2007; Yang et al. 2009 |
| MGG_01173T0 | MHP1 | Hydrophobin | 0.89 | I-TASSER | 4AOG_A | Kim et al. 2005 |
| MGG_10532T0 | MoNLP | NEI peptide | 0.84 | TrRosetta | 3GNZ_P | Fang et al. 2017 |
| MGG_04301T0 | PWL1 | PU protein | 0.56 | TrRosetta | – | Kang et al. 1995 |
| MGG_13863T0 | PWL2 | PU protein | 0.56 | TrRosetta | – | Sweigard et al. 1995 |
| MGG_11610T0 | BAS3 | BAS protein 3 | 0.53 | TrRosetta | – | Mosquera et al. 2009 |
| MGG_09693T0 | BAS2 | BAS protein 2 | 0.51 | TrRosetta | – | Mosquera et al. 2009 |
| MGG_12655T0 | Avr-Pi9 | PU protein | 0.45 | I-TASSER | – | Wu et al. 2015 |
| MGG_00527T0 | EMP1 | PU protein | 0.8 | TrRosetta | – | Ahn et al. 2004 |
| MGG_08411T0 | MoCDIP9 | PU protein | 0.47 | I-TASSER | – | Guo et al. 2019 |
| MGG_12275T0 | MoCDIP10 | PU protein | 0.87 | TrRosetta | – | Guo et al. 2019 |
| MGG_10315T0 | MPG1 | MPG1 | 0.84 | I-TASSER | 2N4O_A | Talbot et al. 1993 |
| MGG_01532T0 | MoCDIP6 | PU protein | 0.3 | TrRosetta | – | Guo et al. 2019 |
| MGG_02848T0 | MC69 | PU protein | 0.54 | I-TASSER | – | Saitoh et al. 2012 |
| MGG_03354T0 | MoCDIP7 | PU protein | 0.38 | I-TASSER | – | Guo et al. 2019. |
| MGG_04795T0 | BAS1 | BAS protein 1 | 0.3 | I-TASSER | – | Mosquera et al. 2009 |
| MGG_07986T0 | MoCDIP3 | PU protein | 0.5 | TrRosetta | – | Chen et al. 2013 |
| MGG_08024T0 | Nup2 | PU protein | 0.45 | I-TASSER | – | Dong et al. 2015 |
| MGG_10234T0 | MoCDIP5 | Hypothetical protein | 0.84 | TrRosetta | – | Chen et al. 2013 |
| MGG_10276T0 | MoHTR1 | PU protein | 0.29 | I-TASSER | – | Kim et al. 2020 |
| MGG_13283T0 | MoCDIP12 | PU protein | 0.33 | TrRosetta | 6FUD_B | Guo et al. 2019 |
| MGG_14371T0 | MoCDIP13 | PU protein | 0.34 | I-TASSER | – | Guo et al. 2019 |
| MGG_15972T0 | Avr-PikD | PU protein | 0.77 | I-TASSER | 6R8M_C | Yoshida et al. 2009 |
Abbreviations: CGS = cellulose-growth-specific, PU = putative uncharacterized, BAS = biotrophy-associated secreted, IH = intracellular hyphae, and NEI = necrosis and ethylene inducing.
Estimated precision.
Homologous matches were identified by searching the M. oryzae effector sequences against the Protein Data Bank database with BLASTP.
Fig. 3.
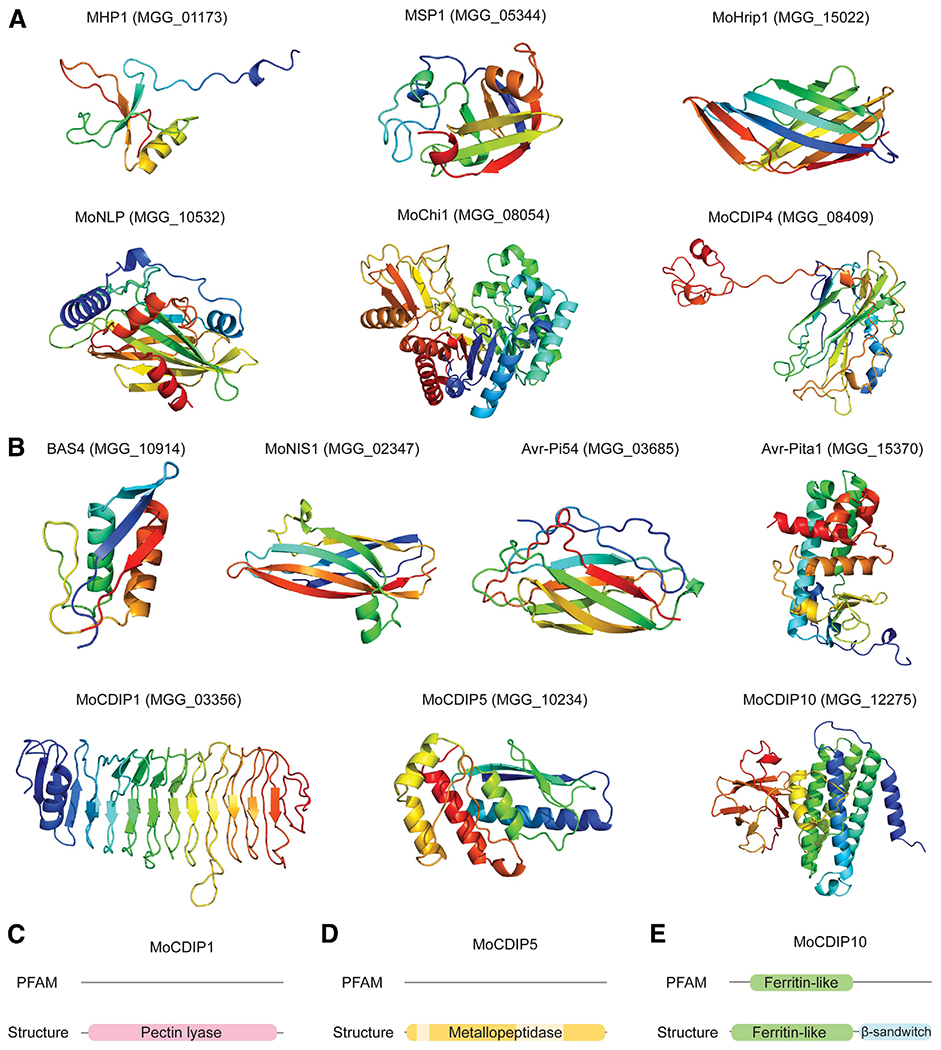
Predicted structures of known effectors in Magnaporthe oryzae. A, Predicted structures of known effectors that have easily identifiable template structures with BLASTP. B, Structures of known effectors predicted by TrRosetta that do not have easily identifiable templates. C to E, PFAM and structure-based annotations of MoCDIP1, MoCDIP5, and MoCDIP10. The PFAM domain search and structural similarity search with RUPEE against the SCOPe and CATH databases were used for the annotation. The structurally unaligned region of MoCDIP5 is indicated in lighter yellow (D).
We searched for structural matches of the effectors to reveal their putative biological roles. Although PFAM search did not uncover any conserved domains, structural comparability to pectin lyases and metallopeptidases existed for MoCDIP1 and MoCDIP5, suggesting that these cell-death-inducing proteins may be enzymes (Fig. 3C and D) (Chen et al. 2013). However, conserved catalytic residues found in structural matches were not present, indicating different modes of catalytic mechanisms (Supplementary Fig. S6) (Cho et al. 2001; Lenart et al. 2013). For MoCDIP10, ferritin-like domain and immunoglobulin-like β-sandwich fold were detected (Fig. 3E) (Guo et al. 2019). The newly detected β-sandwich fold resembled DNA-binding and other protein-binding domains, possibly suggesting that it may aid binding to host targets (Supplementary Fig. S6).
Sensitive sequence similarity search and structural comparisons define secretome classes in M. oryzae.
Structural comparisons together with sensitive sequence similarity searches can better unravel the interconnection between the secreted proteins in M. oryzae. Sequence-to-sequence similarity search with BLASTP revealed that 819 proteins had at least one other homolog present in the secretome (Fig. 4A; Supplementary Table S5). A more sensitive profile-to-sequence similarity search with HHblits linked an additional 186 sequences into homologous groups. Profile-to-profile similarity search with HHsearch and structural comparisons with TM-align added 49 and 69 proteins to the clusters, respectively. Eventually, 1,123 proteins (60%) had at least another sequence or structure-related protein in the secretome. In all, 731 proteins remained as singletons that lacked detectable paralogs and structural analogs in the secretome, and only 118 proteins had functional annotations (Fig. 4B). Of the 613 remaining proteins without PFAM annotations, 86 proteins had predicted structures with the estimated precision ≥ 0.6, which could be relatively reliably used for subsequent analyses.
Fig. 4.
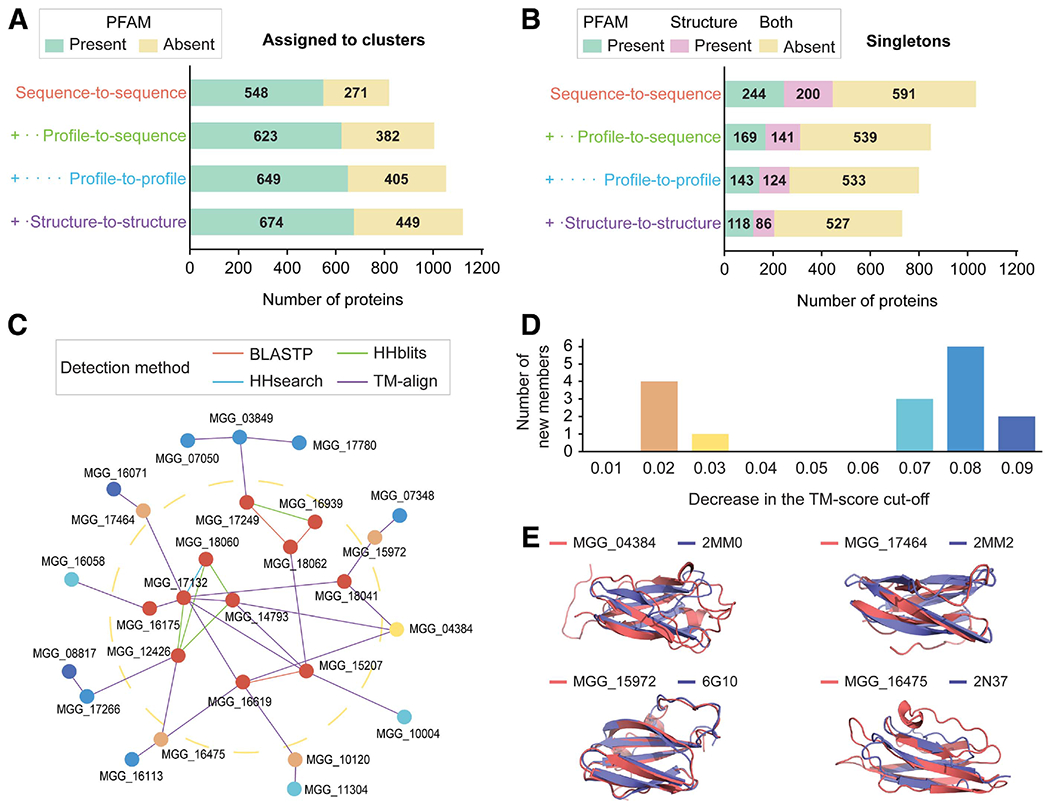
Statistics on secretome clustering and the MAX effector cluster. Four methods were sequentially used to reveal sequence-based and structure-based similarity. In all cases of the sequence-based methods, only the pairs with E-value < 10−4 and bidirectional coverage >50% were regarded as significant. A, The number of proteins found in clusters with at least another homolog or structural analog in the Magnaporthe oryzae secretome. B, The number of singletons without any homologs or analogs in the M. oryzae secretome. The number of proteins with meaningful PFAM domains, excluding domains of unknown functions, or structures predicted with estimated precision ≥ 0.6 are indicated. C and D, The network graph for MAX effectors and the number of newly retrieved singletons. C, Each node and edge represent a protein and similarity that can be detected by the method. The MAX effector cluster with 11 members (cluster 26) exists inside the yellow dotted ring. The newly retrieved singletons remain outside the ring. D, Criteria for the final model selection and the significant structural similarity were relaxed by the template modeling (TM) score of 0.01. The number of newly retrieved singleton members is indicated. Colors correspond in C and D. E, Structural superposition of the newly retrieved MAX effector members and their most similar structures available in the Protein Data Bank. The normalized TM scores, as a measure of similarity, were 0.47 and 0.63 for MGG_04384 and 2MM0 (ToxB), 0.47 and 0.46 for MGG_17464 and 2MM2 (ToxB), 0.81 and 0.89 for MGG_15972 and 6G10 (Avr-PikD), and 0.47 and 0.60 for MGG_16475 and 2N37 (Avr-Pia).
We specifically traced putative MAX effectors, because sensitive sequence similarity search and structural comparisons are required to reveal them (de Guillen et al. 2015). Among the final clusters was cluster 26, a group of 11 uncharacterized proteins, which includes homologs of Avr-Pib (MGG_12426) and AvrPiz-t (MGG_18041) (Fig. 4C; Supplementary Table S5). In sequence-to-sequence similarity search, these 11 proteins were separated into six singletons and two clusters. By the profile-to-sequence similarity search, two singletons and three individual clusters formed, which did not display sufficient sequence similarity to each other. However, the structure-to-structure comparison was able to link them all, eventually placing the Avr-Pib and AvrPiz-t homologs or analogs into a single cluster.
Many predicted MAX effector structures were barely modeled with a precision of 0.5 (Fig. 1A), and structural similarity of some solved MAX effector pairs displayed TM scores ≤ 0.5 (Supplementary Fig. S7). Therefore, the standard criterion of TM score >0.5 will likely miss putative MAX effector candidates in the final model selection or structural similarity comparison. We reduced the TM score cut-off by 0.01 in the two procedures and examined whether any singletons could be retrieved into this MAX effector cluster (Fig. 4D). By decreasing the cut-off by 0.03, we found five new members, four of which identified solved MAX effector structures as their most similar analogs (Fig. 4E). By 0.09, 11 new members were retrieved. However, only one of them (MGG_17266) displayed similarity to a MAX effector structure with an average TM score of 0.45, indicating that the new members are unlikely MAX effectors (Supplementary Table S5). Such outcomes suggested that relaxing the TM score cut-offs to some extent may be appropriate for putative candidate effector selection for a family of interest.
Structural clustering unravels novel families of sequence-unrelated structural analogs.
With the structure-based clustering, we could capture the presence of novel sequence-unrelated, structural analogs (Supplementary Table S5). An example is BAS4, which is highly expressed at the invasive hyphae and participates in the transition from biotrophy to necrotrophy (Mosquera et al. 2009; Wang et al. 2019). BAS4 exists in cluster 17, with 14 other members initially divided into five distinct groups based on homology (Fig. 5A). However, the predicted structures disclosed a common ferredoxin-like fold and α-β plait topology, linking the groups into a single cluster (Fig. 5B). Interestingly, the CATH classification assigns LARS effectors, including AvrLm4-7 (4FPR), to the α-β plait topology (Blondeau et al. 2015). In comparison with the members in cluster 17, AvrLm4-7 is larger and displays a difference in the secondary structure topology. Nevertheless, the structural superposition illustrated that MGG_01064 is roughly contained in AvrLm4-7, possibly suggesting unknown virulence functions associated with this topology that may be important for phytopathogens (Fig. 5C).
Fig. 5.
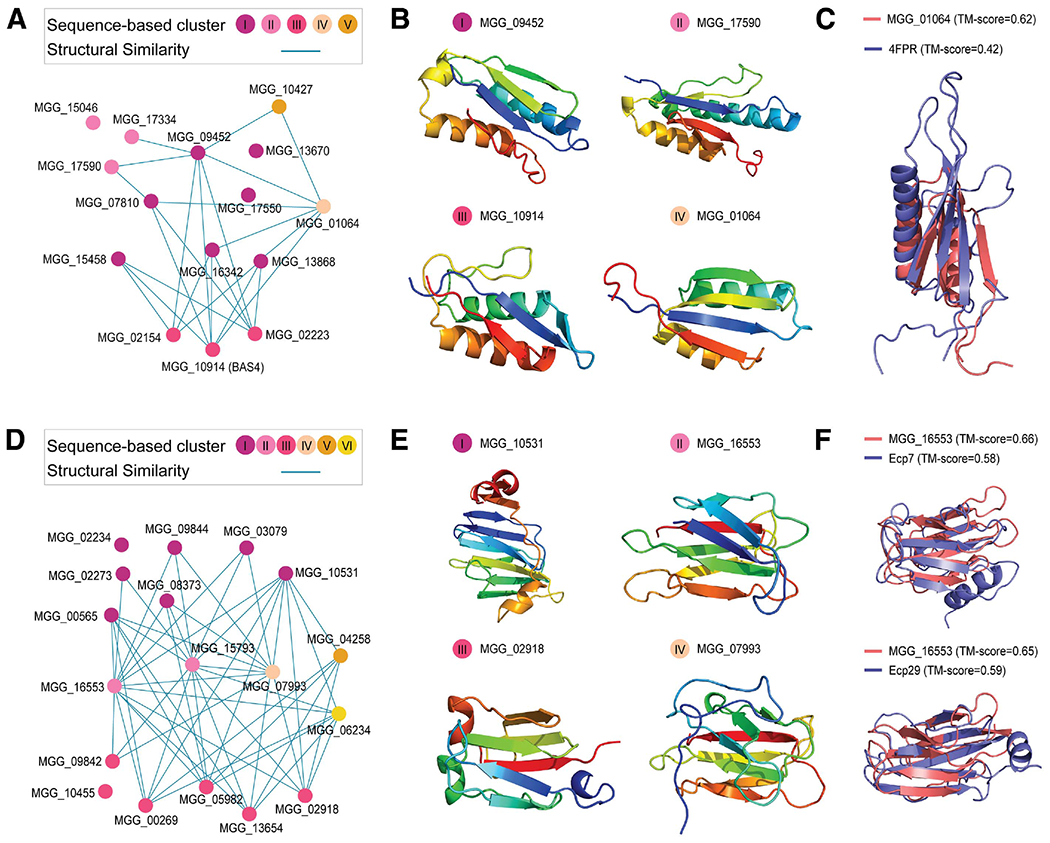
Large clusters of sequence-unrelated structurally similar effectors and the appearance of structural analogs in other phytopathogens. A and D, Network graphs for clusters 17 and 14, respectively. The node represents a member in the cluster and is colored according to its membership based on profile-to-profile and profile-to-sequence similarity search for clusters 17 and 14, respectively. The edge indicates detectable structural similarity. If such similarity is present between two proteins with different membership, their nodes are connected. B and E, Representative structures for clusters 17 and 14, respectively. Structures predicted with the highest expected precision were selected from each sequence-based cluster. C, Structural superposition between the predicted MGG_01064 and LARS effector protein 4FPR. The template modeling (TM) score, as a measure of similarity, normalized for each structure is indicated in the parentheses. F, Structural superposition between the MGG_16533 model and Ecp7 as well as the C-terminal region of Ecp29 (158-266) predicted by TrRosetta.
Structural comparisons revealed another fold comprising the effector family in M. oryzae and its analogs in a phytopathogen. The 18 members in cluster 14 are divergent, and the profile-to-sequence similarity searches only revealed three homologous groups and three singletons (Fig. 5D; Supplementary Fig. S8). Yet all of them share the γ-crystallin-like fold and, thus, are structurally related (Fig. 5E). Lineage-specific presence and absence variations, the exclusive appearance of some homolog in cereal-infecting fungi, and a high expression during the transition to biotrophy all indicate that this group of secreted proteins likely represents true effectors (Gardiner et al. 2012; Torres et al. 2016; Zhong et al. 2016). It was also suggested that four extracellular effectors (Ecp4, Ecp7, Ecp29, and Ecp30) from Cladosporium fulvum would adopt a β/γ-crystallin fold (Mesarich et al. 2018). Consistently, TrRosetta predicted the expected fold for the Ecps with the estimated precision >0.5 (Supplementary Fig. S9). Indeed, the Ecp proteins displayed noticeable similarity to the M. oryzae effectors, collectively indicating that structural analogs may play a significant role across phytopathogens (Fig. 5F).
Secretome clustering and structure-based functional inference lead to new hypotheses for the infection mechanism of M. oryzae.
Secretome clustering and structure-based annotation could help formulate new hypotheses. For instance, the structure prediction and comparisons identified a small RNase cluster in the M. oryzae secretome (cluster 74). The predicted structures belong to the T1 family with similarity to the Blumeria graminis RNase-like effector (6FMB), pointing to the possible existence of a common mechanism in the two distant phytopathogens (Supplementary Fig. S10). Another example is proteins containing the necrosis-inducing factor domain in cluster 33 (Hec2 domain; PF14856), the structures of which were predicted with high estimated precision (Stergiopoulos et al. 2010). The predicted model identified glycan-binding protein Y3 isolated from fungus Coprinus comatus (5V6J) as the closest analog and was linked to seven other sequence-unrelated members by structural analogy (Supplementary Table S5) (Zhang et al. 2017). The structural similarity to Y3 is limited to the fold level. However, because the previous study identified cooccurrence of the Hce2 domain with other glycan-binding LysM domains, chitinbinding modules, and chitinase, the Hec2 domain may be possibly involved in their common biological goal of glycan binding and processing (Stergiopoulos et al. 2012).
A group of secreted pseudoADP-ribose transferases may have evolved from canonical ADP-ribose transferases to serve as specialized binders.
One of the largest clusters, cluster 8, includes 27 members, 10 of which possessed predicted structures with estimated precision > 0.75 and the ADP-ribosylation fold (Fig. 1C; Supplementary Table S5). ADP-ribose transferases (ARTs) in plant pathogenesis are well represented with type III effectors and Scabin toxins (Feng et al. 2016; Fu et al. 2007; Lyons et al. 2016; Singer et al. 2004); however, their role is largely unknown in fungal pathogens. To elucidate how this clade evolved, we employed Shannon’s entropy on the 10 core ART members of this cluster (Prigozhin and Krasileva 2021; Seong et al. 2020). Well-conserved residues measured with entropy < 1 contained some known catalytic residues and residues around them (Fig. 6A) (Aravind et al. 2014; Katoh and Standley 2013). These residues primarily appeared in proximity in the three-dimensional structure (Fig. 6B), composing a highly similar structural core among paralogs (Fig. 6C) to which the NAD+ molecule was predicted to dock (Supplementary Fig. S11) (W. Zhang et al. 2020). Conversely, sequence variations correlated with structural deviations for other residues, suggesting that the core ARTs have evolved divergently while maintaining their core structures and functions.
Fig. 6.
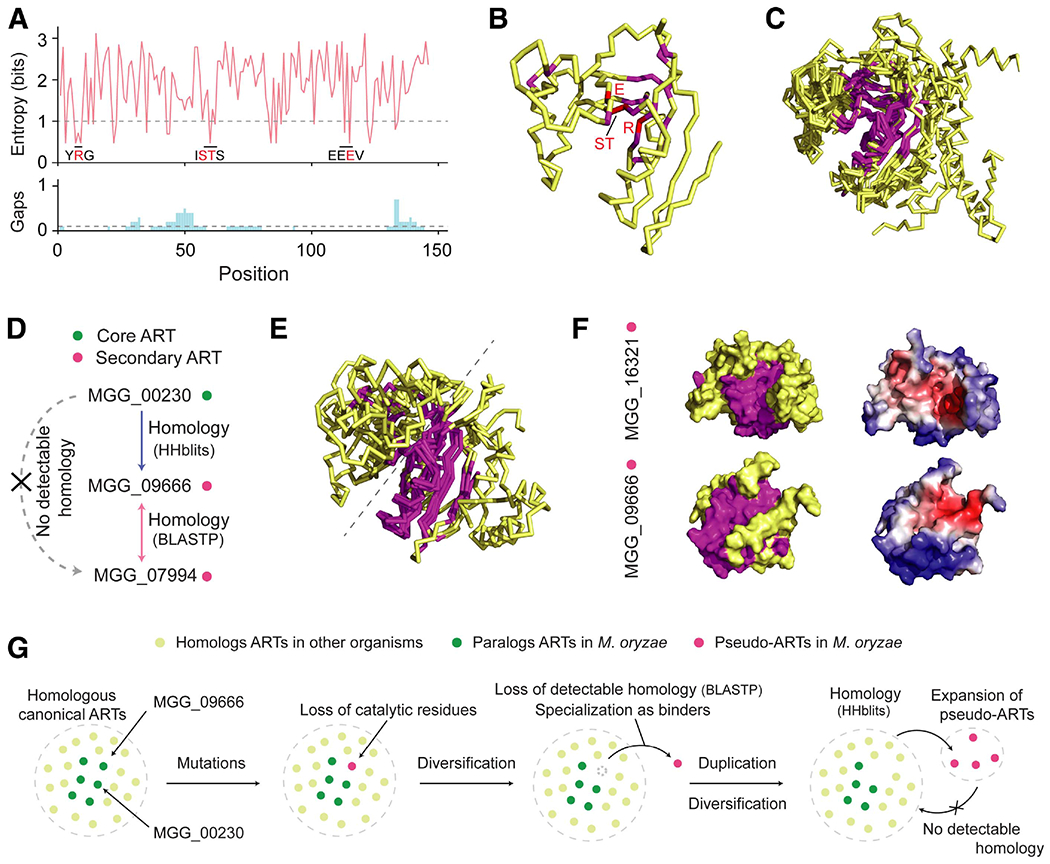
Evolution of putative ADP-ribose transferases (ARTs) informed through structural comparison. A, Entropy plot for the core putative ARTs with structures predicted with estimated precision > 0.75 and the ADP-ribosylation fold. The 10 sequences were aligned with MAFFT, and Shannon’s entropy was calculated for the columns that contain MGG_00230 sequences. The gap was ignored in the entropy calculation but the proportion of gap characters is indicated below the entropy plot. The cut-off for conserved residues was entropy < 1 and the proportion of gap < 0.1. Known catalytic residues (red) and residues around them (black) are indicated in the entropy plot. B, Ribbon structure of MGG_00230 with annotated conserved and catalytic residues in magnate and red, respectively. C, Structural superposition of the 10 core ARTs and Scabin toxin generated with multiple template modeling (mTM)-align (Dong et al. 2015). The structural core measured with the maximum pairwise residue distance < 4Å is indicated in magnate. D, Homology relationship between the core and secondary ARTs in cluster 8. The arrow indicates the direction of homology detection. For instance, MGG_00230 → MGG_09666 denotes that homology to MGG_09666 is detectable when MGG_00230 is used as a query. HHblits was used as a more sensitive sequence similarity method. E, Structural superposition of the four secondary ARTs and a core ART, MGG_00230, generated with mTM-align. The structural core is in magnate. F, Surface structures of secondary ARTs MGG_16321 and MGG_09666. The left models indicate the conserved structural core in magnate. The right models display interaction interfaces in red predicted by MaSIF. G, A proposed mechanism of the ART evolution in Magnaporthe oryzae.
The other members in this cluster are highly divergent from the core ARTs in primary sequences and, therefore, we designate them as secondary ARTs. The homology from core ARTs to the secondary ARTs is not always obviously detectable (Supplementary Fig. S12). Homology detection is often unidirectional, and sequence similarity to an intermediate sequence (e.g., MGG_09666) is required to infer homology between two members (Fig. 6D) (Park et al. 1997). To understand how the secondary ARTs may evolve, we focused on the four members with predicted structures (Supplementary Table S5). Upon superpositioning their structures against a core ART, noticeable differences in evolutionary patterns appeared in sequences and structures. First, the catalytic residues were absent, implying that these proteins may be pseudoenzymes incapable of mediating NAD+-dependent ADP-ribosylation (Supplementary Fig. S13) (Waterhouse et al. 2009). Second, structural conservation is skewed toward one side of the core ART (Fig. 6E). Because the conserved region may play a functional role, we employed MaSIF to predict the protein interaction interface (Gainza et al. 2020). The comparison of the surface structures revealed that the predicted interaction interface overlaps with the structurally conserved regions, while the overall fingerprints of the paralogs are distinct (Fig. 6F). This possibly indicates that the structural core may constitute the backbone of the interface, whereas the other sequence around the core may determine the shape complementarity for substrates or protein targets.
The data suggest a hypothesis for the ART evolution in M. oryzae. Canonical, bifunctional ARTs, which bind to possibly essential host targets and transfer an ADP moiety to alter their cellular activities, first emerged and formed the core ART cluster by frequent duplication and diversification (Fig. 6G). One of the paralogs lost catalytic residues necessary to metabolize NAD+ and began to deviate from the canonical ARTs in evolution. Instead of being selected against, this paralog evolved the remaining protein–protein interaction interface and became specialized as host protein binders. Eventually, additional pseudoARTs arose by duplication events, and they subsequently diversified for different host proteins.
DISCUSSION
Although sequencing the genomes of new pathogen strains became common practice (Islam et al. 2016; Tembo et al. 2021; Win et al. 2021), elucidating the functions and structures of secreted proteins remains challenging. Additionally, primary sequences alone are typically insufficient to infer the roles of effectors, necessitating a novel approach to tackle the problem. Computational structural genomics has been a nearly infeasible concept with homology-based structural modeling. However, we show that template-free modeling enables us to apply this methodology to study pathogen proteins. The accuracy of predicted structures is not yet comparable with experimentally determined structures. Nonetheless, analyses such as structural classification, comparisons, and clustering can be adequately conducted with the predicted structures. It is also advantageous that such analyses are performed at the secretome-wide level, which would not be possible with experimentally determined structures because of their limited availability. With the new layer of information, computational structural genomics opens new possibilities in data analyses of pathogen secretomes.
Comparative genomics with predicted structures may enhance our understanding of effector evolution.
Computational structural genomics allows us to pinpoint unknown proteins that would adopt similar folds of known effector structures such as MAX effectors. However, whether the evolution of the new candidates would resemble that of the known effectors is unclear. Comparative genomics has provided insights about effector evolution, revealing lineage-specific presence or absence variations and sequence diversification (Kim et al. 2019). Comparative genomics should include predicted structures to examine evolutionary mechanisms that may govern effector evolution in analogous families.
Comparative computational structural genomics may reveal commonly used effector folds for immune receptor engineering.
Our computational structural genomics approach revealed novel effector families that display the α-β plait topology or the γ-crystallin-like folds. More importantly, structural analogy to other sequence-unrelated effectors in different phytopathogens was present, suggesting that phytopathogens may commonly employ effectors with similar folds. Therefore, we propose that effectorome-wide structure prediction for diverse phytopathogens and comparative computational structural genomic analyses should be followed. Such studies may also provide a new path for nucleotide binding leucine-rich repeat immune receptor engineering to improve plant immunity. In a recent study, the authors demonstrated that allelic intracellular MLA receptors recognize structurally similar RNase-like effectors through their polymorphic C-terminal leucine-rich repeats (Bauer et al. 2021). If phytopathogens share effectors with a common structural fold, with immune receptors that can recognize such an effector, we may be able to rationally design or directly evolve variants that can target other structurally similar effectors.
Secretome-wide structural clustering helps to prioritize effectors for experimental structural determination and functional elucidation.
The structure-based functional inference is analogous to sequence-based functional annotation transfer. Both require not only predicted structures or sequences of good quality but also the existence of references with known roles and functions. The scope of solved effector structures and our understanding of their functions are currently limited. Therefore, molecular and structural biology work remains critical, regardless of the improvement in structure prediction algorithms. Our work provides a unified structural genomics resource that can be used to group and prioritize candidate effectors for further analyses.
Rapidly improving protein structure prediction algorithms are offering solutions to the current challenges.
Approximately 30% of the secreted proteins could not be predicted by either TrRosetta or I-TASSER. Some of these proteins are predicted to be largely disordered and would likely be unfoldable and, therefore, protein structure prediction may not provide any useful information about their folds and functions. Others typically failed to retrieve a sufficient number of diverse homologs necessary for coevolutionary inference, likely attributed to homology detection failure or recent origins of effectors. There are a few available options to predict the folds of these proteins. First, other state-of-the-art prediction tools such as RaptorX and RoseTTAFold could be additionally utilized (Baek et al. 2021; Xu et al. 2021). Second, prediction performance with small MSAs could be improved through, for instance, MSA dropout and consistency learning (Liu et al. 2021). Third, more intensive structural refinement with DeepAccNet may be able to correct the inaccurate original structures (Hiranuma et al. 2021). Finally, AlphaFold 2 demonstrated exceptional success in the CASP 14 (Callaway 2020). Algorithms with a similar level of performance could become available in the near future, and we expect that they will be able to predict the folds of many proteins missed in this study.
The emergence of pseudoenzymes—Can this be a common theme in effector evolution?
We proposed the emergence of pseudoenzymes and their subsequent diversification in the ART evolution (Fig. 6G). Although the discussion about this notion is scarce as yet in effector evolution, it is not entirely new. B. graminis secretes a significant number of effectors that adopt the RNase fold; however, many of these effectors lack essential residues for catalytic activities, suggesting that they may be pseudoenzymes (Pedersen et al. 2012). Recent studies demonstrated that RNase-like effectors without a canonical enzymatic activity have a functional role in pathogenesis and are targeted by immune receptors (Bauer et al. 2021; Pennington et al. 2019).
The expansion of pseudoenzymes and the validation work of their function raise an intriguing perspective in effector evolution. For both M. oryzae pseudoARTs and B. graminis RNase-like effectors, the ancestral, canonical protein would likely have properties to bind to important host targets. Loss of catalytic activities would not be evolutionarily deleterious to the effector, if its binding to the host targets was sufficient for virulence. Instead, this event would relax evolutionary constraints to maintain the enzymatic functions in both sequence and structure, eventually opening new paths in evolutionary trajectories that canonical ARTs or RNAse effectors could not access. Therefore, the ancestor could become specialized as a binder, and frequent duplication and diversification could subsequently allow the paralogous proteins to follow other accessible evolutionary paths, eventually leading to an expanded protein family that may target other host molecules.
Future perspective.
Genome-scale protein structure prediction is still time consuming and computationally intensive. However, with the advances in machine learning and parallel computing, the field of protein structure prediction is rapidly evolving to challenge this problem. The growing structural data will shift our perspectives on evolution toward three-dimensional space, unrestricted to primary sequences. We also foresee that computational structural genomics will be applied to much larger proteomes of the plant hosts. Such datasets will facilitate our understanding of the interplay between effectors and host proteins and coevolution stemming from this interaction.
MATERIALS AND METHODS
Secretome prediction.
The 12,755 protein sequences of M. oryzae strain 70-15 were downloaded from Ensembl (Dean et al. 2005). We utilized the neural network of SignalP v3.0, one of the most sensitive to defect fungal effectors, to identify putative secreted proteins (Bendtsen et al. 2004; Sperschneider et al. 2015). SignalP initially predicted 2,348 secreted proteins with a D-score ≥ 0.43. 119 possible false positives were removed because their predicted signal peptides overlapped with PFAM domains annotated with InterProScan v5.30-69.0 over ≥10 amino acids (Quevillon et al. 2005). Following the predicted cleavage site based on the Y-score from SignalP, mature protein sequences were generated. We used TMHMM v2.0 to eliminate 344 putative integral membrane proteins with one or more transmembrane helices in the mature proteins (Krogh et al. 2001). Finally, 25 mature proteins with ≥1,000 amino acids were removed, and only the longest isoform was selected. In total, 1,854 proteins remained for the analysis.
Gene prediction for the Magnaporthales.
We obtained 248 genome assemblies for the organisms in the order Magnaporthales from the NCBI. Because the primary purpose of the genome annotation was to collect homologs of the predicted secreted proteins of M. oryzae, we mainly relied on Liftoff v1.3.0 to transfer existing annotations of M. oryzae 70-15 to a target genome (Shumate and Salzberg 2021). We then utilized BRAKER v2.1.5 to predict additional gene models uncaptured by Liftoff (Brůna et al. 2020, 2021; Buchfink et al. 2015; Gotoh et al. 2014; Iwata and Gotoh 2012; Lomsadze et al. 2005; Stanke et al. 2006). For 222 M. oryzae genomes, for instance, the reference annotation of M. oryzae 70-15 was initially mapped into each target genome with Minimap v2.17-r974-dirty (Li 2016). The gene models with ≥98% coverage and ≥40% sequence identity were annotated by Liftoff and collected if no premature stop codon existed (−a 0.98 −s 0.4 −sc 0.4 −copies −exclude_partial). Prior to running BRAKER, the repeat library for the reference genome was generated by RepeatModeler v2.0.1 (Flynn et al. 2020). RepeatMasker v4.1.0 was used to soft-mask each target genome (Smit et al. 2013). The fungal proteomes collected from OrthoDB v10 and the available genome annotations for the order Magnaporthales were used as protein evidence for BRAKER (Kim et al. 2019; Kriventseva et al. 2019). Among the final annotations, we selected all the gene models not overlapping with those previously predicted by Liftoff. For other species in Magnaporthales, we used the annotation sets of their closely related species as the reference and followed the annotation pipeline.
Generation of multiple sequence alignments.
To generate an MSA of a target protein and its homologs, DeepMSA was used (C. Zhang et al. 2020). DeepMSA iteratively searches the Uniclust30, Uniref90, and Metaclust databases to construct an MSA (Mirdita et al. 2017; Steinegger and Söding 2018; Suzek et al. 2015). We supplemented the Metaclust database with additional fungal protein sequences to facilitate collecting diverse fungal homologs. The fungal datasets consisted of 1,689 annotated proteomes from the Joint Genome Institute and the 248 Magnaporthales annotations we produced (Grigoriev et al. 2014). The two datasets were concatenated, and gene models with premature stop codons were removed. The 25,077,589 gene models were clustered with Linclust from MMseqs2 to reduce redundancy (−min-seq-id 0.95 −cov-mode 1 −c 0.99) (Hauser et al. 2016; Mirdita et al. 2017). The resulting 17,679,966 representative gene models were appended to the Metaclust database. DeepMSA was run on these databases to generate an MSA for each secreted protein without the final filtering step.
Protein structure modeling and final structure selection.
The protein structures of all the candidate secreted proteins were predicted with homology modeling by I-TASSER v5.1 and template-free modeling by TrRosetta (Yang et al. 2015, 2020). For I-TASSER, the template library was downloaded from the I-TASSER server (24 August 2020). I-TASSER was run in the light mode with the MSA from the previous step converted into the PSI-BLAST-readable format using the a3m2mtx script in the DeepMSA package (−a3m −neff 7). For the benchmarking set, any homologous templates with ≥70% sequence identity were excluded (−homoflag benchmark −idcut 0.7). We used the estimated TM score of the first model as a measure of precision, and the predicted structures with the mean TM score >0.5 were considered reliable.
For TrRosetta, we selectively filtered the MSAs, limiting their sizes to 6,000 sequences to prevent unnecessarily deep, large MSAs. If the MSA from DeepMSA was larger than the limit, sequences with ≥75% of the query coverage were first sampled from high to low sequence identity over the aligned regions. If necessary, sequences with ≥50% of the query coverage were additionally sampled. TrRosetta was run with the filtered MSAs to predict interresidue orientations and distances, and we generated five full-atom models with PyRosetta (Chaudhury et al. 2010). The model with the lowest energy score was chosen as a final model. We used the average probability of the top L predicted long- plus medium-range contacts (|i – j| > 12) estimated by the top_prob.py script in the TrRosetta suite as precision measurement. A structure with the probability >0.5 was considered reliable. Among the outputs from I-TASSER and TrRosetta, the structure with higher estimated precision was selected as a final model. We used PyMOL v2.4.0 (Schrödinger, LLC) to visualize the structures.
Functional and structural annotations of the secretome.
We employed InterProScan v5.30-69.0 to identify homologous PFAM v31.0 entries (El-Gebali et al. 2019). PaperBLAST and PHI-base were used to search for M. oryzae effectors and their homologs in other pathogens (Price and Arkin 2017; Urban et al. 2020). The secondary structures and disordered residues were predicted with PSIPRED v4.0 and DISOPRED v3.16 (McGuffin et al. 2000; Ward et al. 2004). The predicted structures were compared with RUPEE against the SCOPe v2.07 and CATH v4.2.0 databases as well as the PDB chains (TOP_A-LIGNED, FULL_LENGTH) (Ayoub and Lee 2019; Berman et al. 2000; Fox et al. 2014; Sillitoe et al. 2019).
Network analysis using homology and structures.
We identified homology using sequence-to-sequence search with BLASTP v2.7.1, profile-to-sequence search with HHblits v3.1.0, and profile-to-profile search with HHsearch v3.1.0 (Camacho et al. 2009; Remmert et al. 2012). The MSAs generated in the previous step represented the profiles. In all cases, only the pairs with E-value < 10−4 and bidirectional coverage >50% were regarded as significant. The structural similarities were compared with TM-align by superposing each pair of structures predicted with the estimated precision >0.5 by TrRosetta or >0.55 by I-TASSER. We increased the cut-off for I-TASSER because the structures with the estimated precision of approximately 0.5 introduced spurious clustering, and the sources of their selected templates were often distantly related organisms like humans. We also did not use any proteins with ≥35% disordered residues for structural comparisons, because these proteins tended to be modeled by I-TASSER with similar homologous templates and, thus, introduced biases in the similarity search. The two compared structures were considered similar if the TM score was >0.5 for both structures or >0.6 and >0.4 for each. We used the igraph package v1.2.4.1 in R v3.6.1 for the network analyses and to reveal the cluster membership of secreted proteins (Csardi and Nepusz 2006; Ihaka and Gentleman 1996).
Supplementary Material
ACKNOWLEDGMENTS
We thank B. Staskawicz for access to the computational resources; J. Yang for his advice on structure prediction; C. L. Shaw, D. M Prigozhin, E. L Baggs, and P. Joubert for the critical review of the manuscript; S. Kamoun, M. Banfield, and P. Gladieux for their thoughtful comments on the manuscript; and anonymous reviewers for their constructive comments.
Funding:
This research relied on the Savio computational cluster resource provided by the Berkeley Research Computing program at the University of California, Berkeley. K. Seong is supported by the Berkeley Fellowship. K. V. Krasileva is supported by the Gordon and Betty Moore Foundation (grant number 8802) as well as by the joint funding from the Foundation for Food and Agriculture and 2Blades (CA19-SS-0000000046) and the Innovative Genomics Institute.
Footnotes
AUTHOR-RECOMMENDED INTERNET RESOURCES
Schrödinger, LLC: https://pymol.org/2
The author(s) declare no conflict of interest.
Data availability.
The genome annotations, MSAs. and structures generated for the secreted proteins, and the data used for network analyses are available to download in Zenodo.
LITERATURE CITED
- Ahn N, Kim S, Choi W, Im K-H, and Lee Y-H 2004. Extracellular matrix protein gene, EMP1, is required for appressorium formation and pathogenicity of the rice blast fungus, Magnaporthe grisea. Mol. Cells 17:166–173. [PubMed] [Google Scholar]
- AlQuraishi M 2019. AlphaFold at CASP13 A. Bioinformatics 35:4862–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrie RM, Schoch CL, Hedges R, Spatafora JW, and Ciuffetti LM 2008. Homologs of ToxB, a host-selective toxin gene from Pyrenophora tritici-repentis, are present in the genome of sister-species Pyrenophora bromi and other members of the Ascomycota. Fungal Genet. Biol 45:363–377. [DOI] [PubMed] [Google Scholar]
- Aravind L, Zhang D, de Souza RF, Anand S, and Iyer LM 2014. The natural history of ADP-ribosyltransferases and the ADP-ribosylation system. Pages 3–32 in: Endogenous ADP-Ribosylation. Current Topics in Microbiology and Immunology, 384. Koch-Nolte F, ed. Springer International Publishing, Cham, Switzerland. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoub R, and Lee Y 2019. RUPEE: A fast and accurate purely geometric protein structure search. PLoS One 14:e0213712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD, Millán C, Park H, Adams C, Glassman CR, DeGiovanni A, Pereira JH, Rodrigues AV, van Dijk AA, Ebrecht AC, Opperman DJ, Sagmeister T, Buhlheller C, Pavkov-Keller T, Rathinaswamy MK, Dalwadi U, Yip CK, Burke JE, Garcia KC, Grishin NV, Adams PD, Read RJ, and Baker D 2021. Accurate prediction of protein structures and interactions using a 3-track network. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D, and Sali A 2001. Protein structure prediction and structural genomics. Science 294:93–96. [DOI] [PubMed] [Google Scholar]
- Bauer S, Yu D, Lawson AW, Saur IML, Frantzeskakis L, Kracher B, Logemann E, Chai J, Maekawa T, and Schulze-Lefert P 2021. The leucine-rich repeats in allelic barley MLA immune receptors define specificity towards sequence-unrelated powdery mildew avirulence effectors with a predicted common RNase-like fold B. PLoS Pathog. 17:e1009223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, von Heijne G, and Brunak S 2004. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol 340: 783–795. [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, and Bourne PE 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondeau K, Blaise F, Graille M, Kale SD, Linglin J, Ollivier B, Labarde A, Lazar N, Daverdin G, Balesdent M-H, Choi DHY, Tyler BM, Rouxel T, van Tilbeurgh H, and Fudal I 2015. Crystal structure of the effector AvrLm4-7 of Leptosphaeria maculans reveals insights into its translocation into plant cells and recognition by resistance proteins. Plant J. 83:610–624. [DOI] [PubMed] [Google Scholar]
- Brůna T, Hoff KJ, Lomsadze A, Stanke M, and Borodovsky M 2021. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genomics Bioinf. 3:lqaa108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brůna T, Lomsadze A, and Borodovsky M 2020. GeneMark-EP+: Eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics Bioinf. 2:lqaa026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B, Xie C, and Huson DH 2015. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12:59–60. [DOI] [PubMed] [Google Scholar]
- Callaway E 2020. ‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures. Nature 588:203–204. [DOI] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, and Madden TL 2009. BLAST+: Architecture and applications. BMC Bioinf. 10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury S, Lyskov S, and Gray JJ 2010. PyRosetta: A script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics 26:689–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Zeng H, Qiu D, Guo L, Yang X, Shi H, Zhou T, and Zhao J 2012. Purification and characterization of a novel hypersensitive response-inducing elicitor from Magnaporthe oryzae that triggers defense response in rice. PLoS One 7:e37654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Zhang C, Zi Q, Qiu D, Liu W, and Zeng H 2014. A novel elicitor identified from Magnaporthe oryzae triggers defense responses in tobacco and rice. Plant Cell Rep. 33:1865–1879. [DOI] [PubMed] [Google Scholar]
- Chen S, Songkumarn P, Venu RC, Gowda M, Bellizzi M, Hu J, Liu W, Ebbole D, Meyers B, Mitchell T, and Wang G-L 2013. Identification and characterization of in planta-expressed secreted effector proteins from Magnaporthe oryzae that induce cell death in rice. Mol. Plant-Microbe Interact 26:191–202. [DOI] [PubMed] [Google Scholar]
- Cho SW, Lee S, and Shin W 2001. The X-ray structure of Aspergillus aculeatus polygalacturonase and a modeled structure of the polygalacturonase-octagalacturonate complex. J. Mol. Biol 311:863–878. [DOI] [PubMed] [Google Scholar]
- Csardi G, and Nepusz T 2006. The igraph software package for complex network research. InterJournal Complex Syst. 1695. [Google Scholar]
- Dai Y, Jia Y, Correll J, Wang X, and Wang Y 2010. Diversification and evolution of the avirulence gene AVR-Pita1 in field isolates of Magnaporthe oryzae. Fungal Genet. Biol 47:973–980. [DOI] [PubMed] [Google Scholar]
- Dean R, Van Kan JAL, Pretorius ZA, Hammond-Kosack KE, Di Pietro A, Spanu PD, Rudd JJ, Dickman M, Kahmann R, Ellis J, and Foster GD 2012. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol 13:414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean RA, Talbot NJ, Ebbole DJ, Farman ML, Mitchell TK, Orbach MJ, Thon M, Kulkarni R, Xu J-R, Pan H, Read ND, Lee Y-H, Carbone I, Brown D, Oh YY, Donofrio N, Jeong JS, Soanes DM, Djonovic S, Kolomiets E, Rehmeyer C, Li W, Harding M, Kim S, Lebrun M-H, Bohnert H, Coughlan S, Butler J, Calvo S, Ma L-J, Nicol R, Purcell S, Nusbaum C, Galagan JE, and Birren BW 2005. The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 434:980–986. [DOI] [PubMed] [Google Scholar]
- de Guillen K, Ortiz-Vallejo D, Gracy J, Fournier E, Kroj T, and Padilla A 2015. Structure analysis uncovers a highly diverse but structurally conserved effector family in phytopathogenic fungi. PLoS Pathog. 11:e1005228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Concepcion JC, Franceschetti M, Maqbool A, Saitoh H, Terauchi R, Kamoun S, and Banfield MJ 2018. Polymorphic residues in rice NLRs expand binding and response to effectors of the blast pathogen. Nat. Plants 4:576–585. [DOI] [PubMed] [Google Scholar]
- Dong Y, Li Y, Zhao M, Jing M, Liu X, Liu M, Guo X, Zhang X, Chen Y, Liu Y, Liu Y, Ye W, Zhang H, Wang Y, Zheng X, Wang P, and Zhang Z 2015. Global genome and transcriptome analyses of Magnaporthe oryzae epidemic isolate 98-06 uncover novel effectors and pathogenicity-related genes, revealing gene gain and lose dynamics in genome evolution. PLoS Pathog. 11:e1004801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, Sonnhammer ELL, Hirsh L, Paladin L, Piovesan D, Tosatto SCE, and Finn RD 2019. The Pfam protein families database in 2019. Nucleic Acids Res. 47:D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y-L, Peng Y-L, and Fan J 2017. The Nep1-like protein family of Magnaporthe oryzae is dispensable for the infection of rice plants. Sci. Rep 7:4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B, Liu C, Shan L, and He P 2016. Protein ADP-ribosylation takes control in plant–bacterium interactions. PLoS Pathog. 12:e1005941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, and Smit AF 2020. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. U.S.A 117:9451–9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox NK, Brenner SE, and Chandonia J-M 2014. SCOPe: Structural classification of proteins–extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res. 42:D304–D309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschetti M, Maqbool A, Jiménez-Dalmaroni MJ, Pennington HG, Kamoun S, and Banfield MJ 2017. Effectors of filamentous plant pathogens: Commonalities amid diversity. Microbiol. Mol. Biol. Rev 81:e00066–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu ZQ, Guo M, Jeong BR, Tian F, Elthon TE, Cerny RL, Staiger D, and Alfano JR 2007. A type III effector ADP-ribosylates RNA-binding proteins and quells plant immunity. Nature 447:284–288. [DOI] [PubMed] [Google Scholar]
- Gainza P, Sverrisson F, Monti F, Rodolè E, Boscaini D, Bronstein MM, and Correia BE 2020. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat. Methods 17:184–192. [DOI] [PubMed] [Google Scholar]
- Gardiner DM, McDonald MC, Covarelli L, Solomon PS, Rusu AG, Marshall M, Kazan K, Chakraborty S, McDonald BA, and Manners JM 2012. Comparative pathogenomics reveals horizontally acquired novel virulence genes in fungi infecting cereal hosts. PLoS Pathog. 8:e1002952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh O, Morita M, and Nelson DR 2014. Assessment and refinement of eukaryotic gene structure prediction with gene-structure-aware multiple protein sequence alignment. BMC Bioinf. 15:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, Riley R, Salamov A, Zhao X, Korzeniewski F, Smirnova T, Nordberg H, Dubchak I, and Shabalov I 2014. MycoCosm portal: Gearing up for 1000 fungal genomes. Nucleic Acids Res. 42:D699–D704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Zhong D, Xie W, He Y, Zheng Y, Lin Y, Chen Z, Han Y, Tian D, Liu W, Wang F, Wang Z, and Chen S 2019. Functional identification of novel cell death-inducing effector proteins from Magnaporthe oryzae. Rice (N. Y.) 12:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Song L, Peng C, Liu X, Liu L, Zhang Y, Wang W, Zhou J, Wang S, Ebbole D, Wang Z, and Lu GD 2019. A Magnaporthe chitinase interacts with a rice jacalin-related lectin to promote host colonization. Plant Physiol. 179:1416–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser M, Steinegger M, and Söding J 2016. MMseqs software suite for fast and deep clustering and searching of large protein sequence sets. Bioinformatics 32:1323–1330. [DOI] [PubMed] [Google Scholar]
- Hiranuma N, Park H, Baek M, Anishchenko I, Dauparas J, and Baker D 2021. Improved protein structure refinement guided by deep learning based accuracy estimation. Nat. Commun 12:1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihaka R, and Gentleman R 1996. R: A language for data analysis and graphics. J. Comput. Graph. Stat 5:299–314. [Google Scholar]
- Irieda H, Inoue Y, Mori M, Yamada K, Oshikawa Y, Saitoh H, Uemura A, Terauchi R, Kitakura S, Kosaka A, Singkaravanit-Ogawa S, and Takano Y 2019. Conserved fungal effector suppresses PAMP-triggered immunity by targeting plant immune kinases. Proc. Natl. Acad. Sci. U.S.A 116:496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MT, Croll D, Gladieux P, Soanes DM, Persoons A, Bhattacharjee P, Hossain MS, Gupta DR, Rahman MM, Mahboob MG, Cook N, Salam MU, Surovy MZ, Sancho VB, Maciel JL, NhaniJúnior A, Castroagudín VL, de Assis Reges JT, Ceresini PC, Ravel S, Kellner R, Fournier E, Tharreau D, Lebrun M-H, McDonald BA, Stitt T, Swan D, Talbot NJ, Saunders DGO, Win J, and Kamoun S 2016. Emergence of wheat blast in Bangladesh was caused by a South American lineage of Magnaporthe oryzae. BMC Biol. 14:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata H, and Gotoh O 2012. Benchmarking spliced alignment ?show $132#?>programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic Acids Res. 40:e161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong JS, Mitchell TK, and Dean RA 2007. The Magnaporthe grisea snodprot1 homolog, MSP1, is required for virulence. FEMS Microbiol. Lett 273:157–165. [DOI] [PubMed] [Google Scholar]
- Kang S, Sweigard JA, and Valent B 1995. The PWL host specificity gene family in the blast fungus Magnaporthe grisea. Mol. Plant-Microbe Interact 8:939–948. [DOI] [PubMed] [Google Scholar]
- Katoh K, and Standley DM 2013. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K-T, Ko J, Song H, Choi G, Kim H, Jeon J, Cheong K, Kang S, and Lee Y-H 2019. Evolution of the genes encoding effector candidates within multiple pathotypes of Magnaporthe oryzae. Front. Microbiol 10:2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Ahn I-P, Rho H-S, and Lee Y-H 2005. MHP1, a Magnaporthe grisea hydrophobin gene, is required for fungal development and plant colonization. Mol. Microbiol 57:1224–1237. [DOI] [PubMed] [Google Scholar]
- Kim S, Kim C-Y, Park S-Y, Kim K-T, Jeon J, Chung H, Choi G, Kwon S, Choi J, Jeon J, Jeon J-S, Khang CH, Kang S, and Lee Y-H 2020. Two nuclear effectors of the rice blast fungus modulate host immunity via transcriptional reprogramming. Nat. Commun 11:5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriventseva EV, Kuznetsov D, Tegenfeldt F, Manni M, Dias R, Simão FA, and Zdobnov EM 2019. OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 47:D807–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, and Sonnhammer ELL 2001. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol 305:567–580. [DOI] [PubMed] [Google Scholar]
- Lazar N, Mesarich CH, Petit-Houdenot Y, Talbi N, Li de la Sierra-Gallay I, Zélie E, Blondeau K, Gracy J, Ollivier B, Blaise F, Rouxel T, Balesdent MH, Idnurm A, van Tilbeurgh H, and Fudal I 2020. A new family of structurally conserved fungal effectors displays epistatic interactions with plant resistance proteins. bioRxiv. 10.1101/2020.12.17.423041v1.full [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenart A, Dudkiewicz M, Grynberg M, and Pawłowski K 2013. CLCAs—A family of metalloproteases of intriguing phylogenetic distribution and with cases of substituted catalytic Sites. PLoS One 8:e62272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H 2016. Minimap and miniasm: Fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32:2103–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Wang B, Wu J, Lu G, Hu Y, Zhang X, Zhang Z, Zhao Q, Feng Q, Zhang H, Wang Z, Wang G, Han B, Wang Z, and Zhou B 2009. The Magnaporthe oryzae avirulence gene AvrPiz-t encodes a predicted secreted protein that triggers the immunity in rice mediated by the blast resistance gene Piz-t. Mol. Plant-Microbe Interact 22:411–420. [DOI] [PubMed] [Google Scholar]
- Liu X, Jin L, Gao S, and Zhao S 2021. Protein contact map prediction using multiple sequence alignment dropout and consistency learning for sequences with fewer homologs. bioRxiv. 10.1101/2021.05.12.443740v3.full [DOI] [Google Scholar]
- Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, and Borodovsky M 2005. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33:6494–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons B, Ravulapalli R, Lanoue J, Lugo MR, Dutta D, Carlin S, and Merrill AR 2016. Scabin, a Novel DNA-acting ADP-ribosyltransferase from Streptomyces scabies. J. Biol. Chem 291:11198–11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin LJ, Bryson K, and Jones DT 2000. The PSIPRED protein structure prediction server. Bioinformatics 16:404–405. [DOI] [PubMed] [Google Scholar]
- Mentlak TA, Kombrink A, Shinya T, Ryder LS, Otomo I, Saitoh H, Terauchi R, Nishizawa Y, Shibuya N, Thomma BPHJ, and Talbot NJ 2012. Effector-mediated suppression of chitin-triggered immunity by Magnaporthe oryzae is necessary for rice blast disease. Plant Cell 24:322–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesarich CH, Ökmen B, Rovenich H, Griffiths SA, Wang C, Karimi Jashni M, Mihajlovski A, Collemare J, Hunziker L, Deng CH, van der Burgt A, Beenen HG, Templeton MD, Bradshaw RE, and de Wit PJGM 2018. Specific hypersensitive response-associated recognition of new apoplastic effectors from Cladosporium fulvum in wild tomato. Mol. Plant-Microbe Interact 31:145–162. [DOI] [PubMed] [Google Scholar]
- Mirdita M, von den Driesch L, Galiez C, Martin MJ, Söding J, and Steinegger M 2017. Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 45:D170–D176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogga V, Delventhal R, Weidenbach D, Langer S, Bertram PM, Andresen K, Thines E, Kroj T, and Schaffrath U 2016. Magnaporthe oryzae effectors MoHEG13 and MoHEG16 interfere with host infection and MoHEG13 counteracts cell death caused by Magnaporthe-NLPs in tobacco. Plant Cell Rep. 35:1169–1185. [DOI] [PubMed] [Google Scholar]
- Mosquera G, Giraldo MC, Khang CH, Coughlan S, and Valent B 2009. Interaction transcriptome analysis identifies Magnaporthe oryzae BAS1-4 as Biotrophy-associated secreted proteins in rice blast disease. Plant Cell 21:1273–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhi N, Gorenkin D, and Banfield MJ 2020. Exploring folds, evolution and host interactions: Understanding effector structure/function in disease and immunity. New Phytol. 227:326–333. [DOI] [PubMed] [Google Scholar]
- Ose T, Oikawa A, Nakamura Y, Maenaka K, Higuchi Y, Satoh Y, Fujiwara S, Demura M, Sone T, and Kamiya M 2015. Solution structure of an avirulence protein, AVR-Pia, from Magnaporthe oryzae. J. Biomol. NMR 63:229–235. [DOI] [PubMed] [Google Scholar]
- Park J, Teichmann SA, Hubbard T, and Chothia C 1997. Intermediate sequences increase the detection of homology between sequences. J. Mol. Biol 273:349–354. [DOI] [PubMed] [Google Scholar]
- Pedersen C, Ver Loren van Themaat E, McGuffin LJ, Abbott JC, Burgis TA, Barton G, Bindschedler LV, Lu X, Maekawa T, Wessling R, Cramer R, Thordal-Christensen H, Panstruga R, and Spanu PD 2012. Structure and evolution of barley powdery mildew effector candidates. BMC Genomics 13:694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington HG, Jones R, Kwon S, Bonciani G, Thieron H, Chandler T, Luong P, Morgan SN, Przydacz M, Bozkurt T, Bowden S, Craze M, Wallington EJ, Garnett J, Kwaaitaal M, Panstruga R, Cota E, and Spanu PD 2019. The fungal ribonuclease-like effector protein CSEP0064/BEC1054 represses plant immunity and interferes with degradation of host ribosomal RNA. PLoS Pathog. 15:e1007620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, and Arkin AP 2017. PaperBLAST: Text mining papers for information about homologs. mSystems 2:e00039–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigozhin DM, and Krasileva KV 2021. Analysis of intraspecies diversity reveals a subset of highly variable plant immune receptors and predicts their binding sites. Plant Cell 33:998–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, and Lopez R 2005. InterProScan: Protein domains identifier. Nucleic Acids Res. 33:W116–W120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray S, Singh PK, Gupta DK, Mahato AK, Sarkar C, Rathour R, Singh NK, and Sharma TR 2016. Analysis of Magnaporthe oryzae genome reveals a fungal effector, which is able to induce resistance response in transgenic rice line containing resistance gene, Pi54. Front. Plant Sci 7:1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmert M, Biegert A, Hauser A, and Söding J 2012. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 9:173–175. [DOI] [PubMed] [Google Scholar]
- Saitoh H, Fujisawa S, Mitsuoka C, Ito A, Hirabuchi A, Ikeda K, Irieda H, Yoshino K, Yoshida K, Matsumura H, Tosa Y, Win J, Kamoun S, Takano Y, and Terauchi R 2012. Large-scale gene disruption in Magnaporthe oryzae identifies MC69, a secreted protein required for infection by monocot and dicot fungal pathogens. PLoS Pathog. 8:e1002711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, Qin C, Žídek A, Nelson AWR, Bridgland A, Penedones H, Petersen S, Simonyan K, Crossan S, Kohli P, Jones DT, Silver D, Kavukcuoglu K, and Hassabis D 2020. Improved protein structure prediction using potentials from deep learning. Nature 577:706–710. [DOI] [PubMed] [Google Scholar]
- Seong K, Seo E, Witek K, Li M, and Staskawicz B 2020. Evolution of NLR resistance genes with noncanonical N-terminal domains in wild tomato species. New Phytol. 227:1530–1543. [DOI] [PubMed] [Google Scholar]
- Sharpee W, Oh Y, Yi M, Franck W, Eyre A, Okagaki LH, Valent B, and Dean RA 2017. Identification and characterization of suppressors of plant cell death (SPD) effectors from Magnaporthe oryzae. Mol. Plant Pathol 18:850–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumate A, and Salzberg SL 2021. Liftoff: Accurate mapping of gene annotations. Bioinformatics 8:1639–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillitoe I, Dawson N, Lewis TE, Das S, Lees JG, Ashford P, Tolulope A, Scholes HM, Senatorov I, Bujan A, Ceballos Rodriguez-Conde F, Dowling B, Thornton J, and Orengo CA 2019. CATH: Expanding the horizons of structure-based functional annotations for genome sequences. Nucleic Acids Res. 47:D280–D284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer AU, Desveaux D, Betts L, Chang JH, Nimchuk Z, Grant SR, Dangl JL, and Sondek J 2004. Crystal structures of the type III effector protein AvrPphF and its chaperone reveal residues required for plant pathogenesis. Structure 12:1669–1681. [DOI] [PubMed] [Google Scholar]
- Smit AF, Hubley R, and Green P 2013. RepeatMasker Open-4.0. https://www.repeatmasker.org/
- Spanu PD 2017. Cereal immunity against powdery mildews targets RNase-like proteins associated with haustoria (RALPH) effectors evolved from a common ancestral gene. New Phytol. 213:969–971. [DOI] [PubMed] [Google Scholar]
- Sperschneider J, Williams AH, Hane JK, Singh KB, and Taylor JM 2015. Evaluation of secretion prediction highlights differing approaches needed for oomycete and fungal effectors. Front. Plant Sci 6:1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Keller O, Gunduz I, Hayes A, Waack S, and Morgenstern B 2006. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 34:W435–W439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinegger M, and Söding J 2018. Clustering huge protein sequence sets in linear time. Nat. Commun 9:2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stergiopoulos I, Kourmpetis YAI, Slot JC, Bakker FT, De Wit PJGM, and Rokas A 2012. In silico characterization and molecular evolutionary analysis of a novel superfamily of fungal effector proteins. Mol. Biol. Evol 29:3371–3384. [DOI] [PubMed] [Google Scholar]
- Stergiopoulos I, van den Burg HA, Okmen B, Beenen HG, van Liere S, Kema GHJ, and de Wit PJGM 2010. Tomato Cf resistance proteins mediate recognition of cognate homologous effectors from fungi pathogenic on dicots and monocots. Proc. Natl. Acad. Sci. U.S.A 107:7610–7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH, and the UniProt Consortium. 2015. UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31:926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweigard JA, Carroll AM, Kang S, Farrall L, Chumley FG, and Valent B 1995. Identification, cloning, and characterization of PWL2, a gene for host species specificity in the rice blast fungus. Plant Cell 7:1221–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot NJ, Ebbole DJ, and Hamer JE 1993. Identification and characterization of MPG1, a gene involved in pathogenicity from the rice blast fungus Magnaporthe grisea. Plant Cell 5:1575–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tembo B, Mahmud NU, Paul SK, Asuke S, Harant A, Langner T, Reyes-Avila CS, Chanclud E, Were V, Sichilima S, Mulenga RM. Gupta DR, Mehebub MS. Muzahid ANM, Rabby MF, Singh PK, Bentley A, Tosa Y, Croll D, Lamour K, Islam T, Talbot NJ, Kamoun S, and Win J 2021. Multiplex amplicon sequencing dataset for genotyping pandemic populations of the wheat blast fungus. https://zenodo.org/record/4605959 [Google Scholar]
- Torres MF, Ghaffari N, Buiate EAS, Moore N, Schwartz S, Johnson CD, and Vaillancourt LJ 2016. A Colletotrichum graminicola mutant deficient in the establishment of biotrophy reveals early transcriptional events in the maize anthracnose disease interaction. BMC Genomics 17:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban M, Cuzick A, Seager J, Wood V, Rutherford K, Venkatesh SY, De Silva N, Martinez MC, Pedro H, Yates AD, Hassani-Pak K, and Hammond-Kosack KE 2020. PHI-base: The pathogen–host interactions database. Nucleic Acids Res. 48:D613–D620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Liu Y, Liu L, Wang Y, Yan J, Wang C, Li C, and Yang J 2019. The biotrophy-associated secreted protein 4 (BAS4) participates in the transition of Magnaporthe oryzae from the biotrophic to the necrotrophic phase. Saudi J. Biol. Sci 26:795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JJ, McGuffin LJ, Bryson K, Buxton BF, and Jones DT 2004. The DISOPRED server for the prediction of protein disorder. Bioinformatics 20:2138–2139. [DOI] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, and Barton GJ 2009. Jalview version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RA, and Talbot NJ 2009. Under pressure: Investigating the biology of plant infection by Magnaporthe oryzae. Nat. Rev. Microbiol 7:185–195. [DOI] [PubMed] [Google Scholar]
- Win J, Malmgren A, Langner T, and Kamoun S 2021. A pandemic clonal lineage of the wheat blast fungus. Zenodo. https://zenodo.org/record/4618522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Kou Y, Bao J, Li Y, Tang M, Zhu X, Ponaya A, Xiao G, Li J, Li C, Song MY, Cumagun CJR, Deng Q, Lu G, Jeon JS, Naqvi NI, and Zhou B 2015. Comparative genomics identifies the Magnaporthe oryzae avirulence effector AvrPi9 that triggers Pi9-mediated blast resistance in rice. New Phytol. 206:1463–1475. [DOI] [PubMed] [Google Scholar]
- Xu J, McPartlon M, and Li J 2021. Improved protein structure prediction by deep learning irrespective of co-evolution information. Nat. Mach. Intell 3:601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, and Zhang Y 2010. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics 26:889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Anishchenko I, Park H, Peng Z, Ovchinnikov S, and Baker D 2020. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. U.S.A 117:1496–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Yan R, Roy A, Xu D, Poisson J, and Zhang Y 2015. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 12:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Zhang H, Li G, Li W, Wang X, and Song F 2009. Ectopic expression of MgSM1, a Cerato-platanin family protein from Magnaporthe grisea, confers broad-spectrum disease resistance in Arabidopsis. Plant Biotechnol. J 7:763–777. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Saitoh H, Fujisawa S, Kanzaki H, Matsumura H, Yoshida K, Tosa Y, Chuma I, Takano Y, Win J, Kamoun S, and Terauchi R 2009. Association genetics reveals three novel avirulence genes from the rice blast fungal pathogen Magnaporthe oryzae. Plant Cell 21:1573–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Zheng W, Mortuza SM, Li Y, and Zhang Y 2020. DeepMSA: Constructing deep multiple sequence alignment to improve contact prediction and fold-recognition for distant-homology proteins. Bioinformatics 36:2105–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Li K, Yang G, Xia C, Polston JE, Li G, Li S, Lin Z, Yang LJ, Bruner SD, and Ding Y 2017. Cytotoxic protein from the mushroom Coprinus comatus possesses a unique mode for glycan binding and specificity. Proc. Natl. Acad. Sci. U.S.A 114:8980–8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Wang L, Wu W, He L, Yang X, and Pan Q 2015. Function and evolution of Magnaporthe oryzae avirulence gene AvrPib responding to the rice blast resistance gene Pib. Sci. Rep 5:11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Bell EW, Yin M, and Zhang Y 2020. EDock: Blind protein-ligand docking by replica-exchange Monte Carlo simulation. J. Cheminf 12:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, He D, Zhao Y, Cheng X, Zhao W, Taylor IA, Yang J, Liu J, and Peng Y-L 2018. A positive-charged patch and stabilized hydrophobic core are essential for avirulence function of AvrPib in the rice blast fungus. Plant J. 96:133–146. [DOI] [PubMed] [Google Scholar]
- Zhang Y 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinf. 9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, and Skolnick J 2005. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33:2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z-M, Zhang X, Zhou Z-R, Hu H-Y, Liu M, Zhou B, and Zhou J 2013. Solution structure of the Magnaporthe oryzae avirulence protein AvrPiz-t. J. Biomol. NMR 55:219–223. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Norvienyeku J, Chen M, Bao J, Lin L, Chen L, Lin Y, Wu X, Cai Z, Zhang Q, Lin X, Hong Y, Huang J, Xu L, Zhang H, Chen L, Tang W, Zheng H, Chen X, Wang Y, Lian B, Zhang L, Tang H, Lu G, Ebbole DJ, Wang B, and Wang Z 2016. Directional Selection from Host Plants Is a Major Force Driving Host Specificity in Magnaporthe Species. Sci. Rep 6:25591. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genome annotations, MSAs. and structures generated for the secreted proteins, and the data used for network analyses are available to download in Zenodo.