

Abstract
Obesity and type II diabetes are leading causes of peripheral arterial disease (PAD), which is characterized by vascular insufficiency and ischemic damage in the limb skeletal muscle. Glycemic control is not sufficient to prevent progression of PAD, and molecular targets that can promote muscle neo‐angiogenesis in obesity and diabetes remain poorly defined. Here, we have investigated whether nuclear receptor estrogen‐related receptor alpha (ERRα) can promote ischemic revascularization in the skeletal muscles of diet‐induced obese (DIO) mice. Using muscle‐specific ERRα transgenic mice, we found that ERRα overexpression promotes revascularization, marked by increased capillary staining and muscle perfusion in DIO mice after hindlimb ischemic injury. Furthermore, ERRα facilitates repair and restoration of skeletal muscle myofiber size after limb ischemia in DIO mice. The ameliorative effects of ERRα overexpression did not involve the prevention of weight gain, hyperglycemia or glucose/insulin intolerance, suggesting a direct role for ERRα in promoting angiogenesis. Interestingly, levels of endogenous ERRα protein are suppressed in the skeletal muscles of DIO mice compared to lean controls, coinciding with the suppression of angiogenic gene expression, and reduced AMPK signaling in the DIO skeletal muscles. Upon further investigating the link between AMPK and ERRα, we found that AMPK activation increases the expression and recruitment of ERRα protein to specific angiogenic gene promoters in muscle cells. Further, the induction of angiogenic factors by AMPK activators in muscle cells is blocked by repressing ERRα. In summary, our results identify an AMPK/ERRα‐dependent angiogenic gene program in the skeletal muscle, which is repressed by DIO, and demonstrate that forced ERRα activation can promote ischemic revascularization and muscle recovery in obesity.
Keywords: AMPK, angiogenesis, ischemia, nuclear receptors, obesity, skeletal muscle
Abbreviations
- AMPK
5′
adenosine monophosphate‐activated protein kinase
- CD31
cluster of differentiation 31
- CycS
cytochrome c somatic
- DIO
diet induced obesity
- DLL4
delta‐like canonical notch ligand 4
- ERRα
estrogen‐related receptor alpha
- FGF1
fibroblast growth factor 1
- HIF
hypoxia inducible factor
- HLI
hindlimb ischemia
- PAD
peripheral arterial disease
- PPAR
peroxisome proliferator‐activated receptor
- TA
tibialis anterior
- Ucp3
uncoupling protein 3
- VEGF
vascular endothelial growth factor
1. INTRODUCTION
Obesity and type II diabetes are leading risk factors for cardiovascular disease, particularly known to increase the prevalence of peripheral arterial disease (PAD). 1 , 2 , 3 PAD is characterized by impaired blood supply to the limb musculature due to large artery occlusion or regression of local blood vessels in the limbs. The condition is exaggerated by obesity and diabetes‐mediated impairment in reparative angiogenesis, resulting in ischemic injury in limb musculature, leg pain, immobility and often limb amputation. 4 , 5 , 6 , 7 There is no effective treatment for PAD, and while individual angiogenic factors (e.g. Vegfa, Fgf1) have been explored for therapeutic purpose, they have not proved to be clinically effective in promoting limb reperfusion and repair. 8 , 9 Consequently, there is an unmet need to identify new molecular pathways for revascularization and recovery of the skeletal muscle after ischemia and for treatment of PAD. Particularly, studies in the context of diet‐induced obesity and type II diabetes to identify aberrant molecular pathways in the skeletal muscle associated with impaired angiogenesis are sparse and therefore warranted.
Estrogen‐related receptor alpha (ERRα) is an orphan nuclear receptor, belonging to the estrogen‐related receptor (ERR) sub‐family of nuclear receptors. ERRs are critical regulators of mitochondrial biogenesis, oxidative fatty acid metabolism and overall energy homeostasis through its transcriptional effects in metabolically active tissues such as adipose tissue, liver, and skeletal muscle. 10 , 11 , 12 ERRα and ERRγ are highly expressed sub‐types in the skeletal muscle, where ERRγ is preferentially expressed in oxidative slow‐twitch muscle, and ERRα is ubiquitously expressed in all muscle beds. 13 Both receptors are induced by exercise and are involved in the positive regulation of slow‐twitch myofibers, mitochondrial biogenesis, and oxidative metabolism, controlling muscle fitness and endurance. 13 , 14 , 15 , 16 , 17 ERRα and ERRγ have also been shown to play a role in muscle regeneration using murine cardiotoxin injury model or mdx dystrophic mice. 16 , 18 Studies using cultured myoblasts also indicate a direct role for ERRα and ERRγ in myogenesis. 19 , 20
ERRα and ERRγ have recently emerged as key regulators of angiogenesis. Both ERRα and ERRγ stimulate a paracrine angiogenic gene program in the skeletal muscle involving up‐regulation of secretory pro‐angiogenic factors including Vegfa and Fgf1. 15 , 21 Furthermore, both ERRα and ERRγ overexpression in the skeletal muscle stimulates basal vascularization, as well as ischemic revascularization in the skeletal muscles of lean non‐diabetic mice. 13 , 15 , 21 Other reports indicate that ERRα signaling in the endothelial cells may also contribute to regulation of angiogenesis. 22 , 23 In the skeletal muscle, ERRγ‐regulated angiogenic program seems to function independent of classical hypoxia/HIF1 pathway. 15 In contrast, ERRα is regulated by ischemia/hypoxia in the skeletal muscle in HIF1‐dependent fashion. 21 Based on these reports, ERRs are probable candidates for promoting ischemic neo‐angiogenesis and muscle recovery in diabetes, and consequently are of potential interest for treating diabetes‐associated PAD.
Diet‐induced obese (DIO) mice develop glucose and insulin intolerance, and exhibit impaired ischemic revascularization in the skeletal muscle closely mimicking the pathological impairment in hindlimb neo‐angiogenesis in diabetes‐associated PAD or critical limb ischemia (CLI). 24 , 25 , 26 In the present study, we have examined the effect of muscle ERRα overexpression on obesity and glucose/insulin tolerance, but more specifically on neo‐angiogenesis and skeletal muscle recovery after ischemic injury in DIO mice. We have also examined the impact of DIO on ERRα expression in the skeletal muscle, and simultaneously explored potential mechanism for the regulation of ERRα expression. Our results demonstrate that muscle‐specific overexpression of ERRα, while not preventing obesity and glucose/insulin intolerance, activates angiogenic genes in the skeletal muscle, promotes ischemic neo‐angiogenesis, and facilitates reperfusion and skeletal muscle recovery after hindlimb ischemia in DIO mice. ERRα and angiogenic factors along with AMPK signaling is suppressed in the DIO skeletal muscle. Furthermore, AMPK and ERRα are inter‐linked in a fashion that they transcriptionally regulate angiogenic genes in skeletal muscle.
2. MATERIALS AND METHODS
2.1. Mouse husbandry
The generation of muscle‐specific ERRα transgenic (TG) mice in our laboratory, on the C57Bl/6J background strain has been described previously. 21 In this transgenic line, the ERRα transgene is driven by a human alpha skeletal actin promotor to selectively overexpress ERRα in the mouse skeletal muscles. The TG mouse line has been maintained for several regenerations by crossing to C57Bl/6J mice from Jackson laboratory. Littermate wild type (WT) mice were used as controls. WT and TG mice were housed in a temperature‐controlled room (20–22°C) with ad libitum access to water and food (Pico Lab rodent diet 20; 13.2% fat) under a 12:12 h. light dark cycle. Experiments were performed in 4–5 months old male mice. Animals were maintained and treated in accordance with the U.S. National Institute of Health Guide for Care and Use of Laboratory Animals; and the procedures were approved by the Animal Welfare Committee at The University of Texas Health Science Center in Houston.
2.2. Diet‐induced obesity and glucose/insulin tolerance tests
WT (WT‐DIO) and TG (TG‐DIO) littermates were fed a high fat diet (Research diet Inc. D12492i—60% calories from fat) starting at 8 weeks of age. The mice were weighed every week until the experimental end‐point. Age and sex‐matched WT C57Bl6/J mice from Jackson laboratory obtained at 7 weeks of age and simultaneously included in the study cohort along with the aforementioned groups were used as non‐diabetic WT normal chow diet (WT‐NCD) (5% calories from fat) controls. After 14 weeks on high fat diet (HFD), the WT‐DIO and TG‐DIO mice, and the companion WT‐NCD control mice were subjected to glucose tolerance test (GTT) after 6 h of fasting, as we previously described. 27 , 28 For insulin tolerance test (ITT), mice were fasted for 4 h followed by the measurement of 0 min glucose measurement. Blood was also collected for measuring serum insulin levels, immediately following which IP injection of insulin (Sigma, I9278) was administered at 1 unit/kg body weight. Blood glucose was measured at 15, 30, 60, 90, and 120 min following the injection to measure insulin tolerance. The 0 min collected blood was allowed to clot at room temperature for 1 h and then centrifuged at 2000g for 20 min at 4°C to obtain serum. Serum was immediately used to measure fasting serum insulin levels using the ultra‐sensitive mouse Insulin ELISA Kit from Crystal Chem following the manufacturer's instructions. Mice were subjected to hindlimb ischemia 1 week after the confirmation of glucose and insulin intolerance. WT‐DIO and TG‐DIO mice were continued on HFD, whereas the WT‐NCD were continued on NCD during the course of the HLI experiment.
2.3. Tissue collection and preparation
Mice were euthanized by isoflurane inhalation followed by cervical dislocation and tissues were rapidly extracted. For RNA and protein analysis, muscles were freeze‐clamped in liquid nitrogen. For immunofluorescence, TA muscles were mounted in OCT and frozen on melting isopentane in liquid nitrogen.
2.4. Gene expression
Quantitative real‐time PCR (qPCR) analysis was performed using SYBR Green PCR Master Mix (Applied Biosystems) with a Bio‐Rad XF96 cycler (Bio‐Rad) using cDNA reverse transcribed from mRNA extracted from WT‐NCD, WT‐DIO, TG‐DIO gastrocnemius (N = 4–6), and C2C12 cells, as previously described. 21 Primer sequences are shown in Data Unit S1. All data were normalized to Tbp and Hsp90.
2.5. Western and Dot immunoblotting
Gastrocnemius from WT‐NCD, WT‐DIO, and TG‐DIO mice (N = 4–6) were homogenized in RIPA buffer (Thermo Scientific) using Tissue Lyser (Qiagen) at speed 50, for three cycles of 1 min followed by centrifugation at 12,000g for 15 min at 4°C. For C2C12 and murine embryonic fibroblast (MEF) cultures, cells were collected and lysed in RIPA buffer and centrifuged similarly. The Pierce BCA protein assay kit (Thermo Scientific) was used to quantify protein in supernatant, which was then stored at −80°C. Samples were separated by SDS‐PAGE, transferred onto nitrocellulose membrane, stained using Ponceau S, blocked with 5% milk in phosphate buffered saline with 0.1% Tween 20 (PBST), and incubated overnight at 4 °C with primary antibodies for t‐AMPK (#2532), p‐AMPK (#2535), ACC (#3676), p‐ACC (#11818), ERRα (# 13826), DLL4 (# 96406) at 1:1000 dilution (Cell signaling, Danvers, MA), and β actin at 1:1000 dilution (#A5441 from Sigma). Membranes were then washed with PBST and incubated with the appropriate secondary antibodies (Cell Signaling) for 1 h at room temperature, washed with PBST, and bands visualized using chemiluminescence western blotting detection reagents.
Proteome profiler mouse angiogenesis array kit (#ARY015 from Biotechne R&D Systems) was used to analyze protein levels of 53 different pro and anti‐angiogenic markers in 300 μg of WT‐DIO and TG‐DIO gastrocnemius muscle lysates using manufacturer's instructions.
2.6. Immunohistology
Cryosections (10 μM) were obtained from mid‐section of tibialis anterior (TA) muscle from WT‐NCD, WT‐DIO and TG‐DIO mice. Laminin (sarcolemmal marker) was stained using antibody L9393 from Sigma at 3.5 μg/ml dilution to visualize sarcolemma. Capillaries were stained using CD31 at 10 μg/ml (#MCA2388 Bio‐Rad). All primary antibodies were visualized using suitable Alexa Fluor® secondary antibodies from Molecular Probes, Eugene, OR. Nuclei were stained using DRAQ5 fluorescent probe 5 μM (Thermo Scientific). Immunostained sections were examined using confocal microscopy. Myofiber number was counted using Amira software version 6.3 from FEI Hillsboro, OR. CD31 and nuclei were counted using Image J.
2.7. Hindlimb ischemia, tissue collection, and laser Doppler blood flowmetry
Hindlimb ischemia (HLI) was induced in WT‐NCD, WT‐DIO and TG‐DIO mice (N = 8–10) by unilateral femoral vessel ligation and transection, as we previously described. 15 , 21 In this model, ischemia was induced in left hindlimb, and the contralateral right hindlimb served as the control. Mice were anesthetized, as described above, and blood flow was measured in both the contralateral and ischemic flexor digitorum brevis (FDB) muscle with a deep tissue laser Doppler flow probe (PeriFlux system 5000, Perimed). FDB was selected for the blood flow measurement as this is the distal most muscle to the site of ligation/transection, therefore, restoration of blood flow at this site is reflective of reperfusion at upstream sites. Furthermore, diabetic ischemia commonly causes severe injury and gangrene in the foot, leading to amputations in this region. The foot and the paw regions are also easily accessible for non‐invasive blood flow measurement. Blood perfusion in the ischemic hindlimb muscle is reported as the percent of perfusion in contralateral non‐ischemic hindlimb muscle (Ischemic limb perfusion × 100/contralateral limb perfusion). To measure vascular recovery in other muscles, TA muscles were harvested at indicated time‐points after the induction of ischemia and processed for immunohistology, as a representative muscle group.
2.8. Cell culture
Murine C2C12 muscle cells (CRL1772 from ATCC, Manassas) were cultured in DMEM with 10% FBS and normocin (Invitrogen). For the differentiation of C2C12 cells into myotubes, 10% FBS media was replaced with DMEM containing 2% horse serum when the cells were ~90% confluent. Four‐day differentiated C2C12 cells were treated with the following drugs and time durations: (i) 1 mM AICAR for 24 h, (ii) 200 μM compound C for 6 h, (iii) 1 μM XCT 790 for 24 h, (iv) 2.5 μM PF739 for 24 h, and (v) 25 μM PF991 for 24 h. The cells were subsequently collected and processed for RNA or protein expression analysis.
Wild‐type (Prkaa +/+ ) and Prkaa1 and Prkaa2 double knockout (Prkaa −/− ) murine embryonic fibroblasts (MEFs) were previously described (PMID: 16809770 PMCID: PMC1592699) and were originally generated and provided by L.C. Cantley (Weill Cornell Medical College). Primary Prkaa MEFs derived from 10.5‐day post‐coitum embryos were infected with retroviral particles prepared from 293 T cells expressing pBabe–H‐RasV12 (Addgene, #9051) and further grown, as previously described. 29 Cells were cultured up to 90% confluence then collected and processed for protein expression analysis.
2.9. Chromatin Immunoprecipitation
Four‐days differentiated C2C12 cells treated with or without 1 mM AICAR for 24 hr. were collected on day 5, as described above, and processed for chromatin immunoprecipitation (ChIP) using the Simple ChIP Enzymatic Chromatin IP Kit (# 9003S, Cell Signaling) following manufacturer's instructions. As described previously, 21 cells from two 15 cm plates were combined per sample and fixed, washed, and lysed using 40 strokes of Dounce homogenizer B. Chromatin was digested using 1 μl of enzyme per sample for 20 min at 37°C. It was further sonicated two times for 10 s each. Part of the digested chromatin was saved for 2% input and for confirming effective digestion. The remaining digested chromatin was split into two equal halves and incubated overnight at 4°C with 5 μg of either ERRα antibody (# 13826, Cell Signaling) or control rabbit IgG. This was followed by incubation with 45 μl of protein G magnetic beads. The beads were washed and further processing was carried out as per the kit protocol to obtain de‐crosslinked purified input, ChIP‐IgG, and ChIP‐ERRα chromatin. qPCR was performed for promoter region of several genes using primer pairs, as shown in Data Unit S1. Percent enrichment for ChIP‐IgG and ChIP‐ERRα was obtained relative to input. Data are represented as fold enrichment for ChIP‐ERRα relative to its ChIP‐IgG for comparison between AICAR treated and untreated groups.
2.10. Statistics
Unpaired Student's t‐test and one‐way ANOVA with appropriate post‐hoc tests were used, as indicated in the figure legends, to compare the different groups. Data in figures are presented as mean ± standard error of mean (SEM), and p < 0.05 is considered as statistically significant. Data were analyzed and plotted using GraphPad Prism 5.
3. RESULTS
3.1. High fat diet induces a comparable level of weight gain and glucose intolerance in muscle‐specific ERRα TG and WT mice
The design of the study is outlined in Figure 1A, where muscle‐specific ERRα TG and littermate WT mice were subjected to high fat diet (HFD) starting at 8 weeks of age. After 13 weeks of HFD, mice were administered GTT and ITT to ensure the onset of glucose and insulin intolerance. Subsequently, HLI was performed at 14 weeks (when the mice are at 22 weeks of age). Mice from each group were sacrificed at 18 and 30 days post‐HLI and tissues collected for analysis, to investigate the effect of muscle‐specific ERRα overexpression on ischemic recovery in DIO mice. WT and TG mice showed similar weight gain on HFD (Figure 1B ). GTT (Figure 1C), ITT (Figure 1D), fasting blood glucose (Figure 1E) and fasting serum insulin (Figure 1F) measurements revealed that compared to NCD‐fed WT mice, both HFD‐fed WT (WT‐DIO) and HFD‐fed TG (TG‐DIO) mice become comparably glucose and insulin intolerant. Therefore, ERRα overexpression does not seem to protect against diet‐induced obesity and glucose/insulin intolerance.
FIGURE 1.
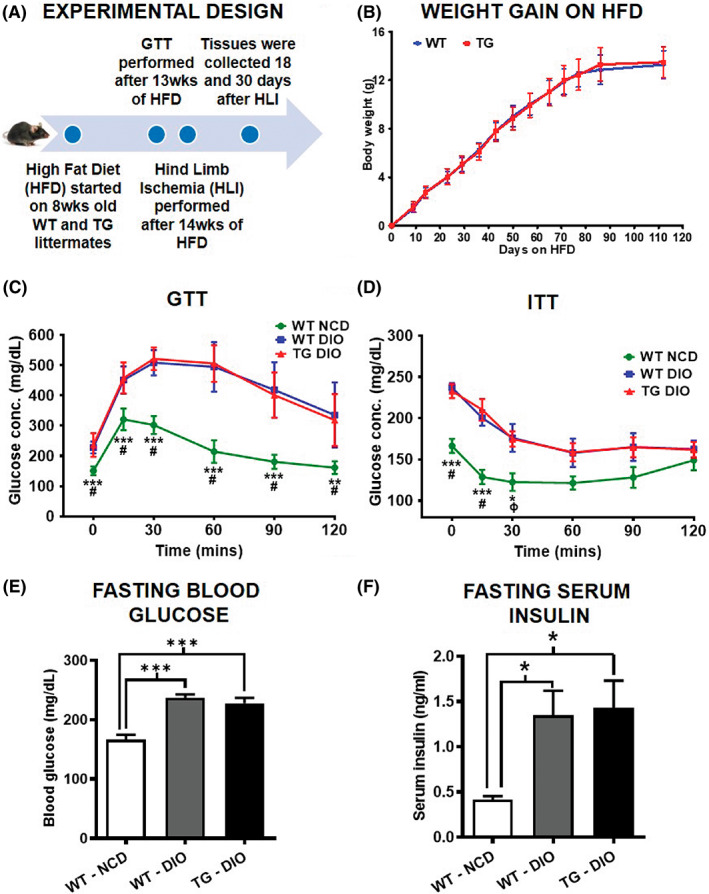
Muscle‐specific ERR⍺ overexpression does not affect weight gain and glucose tolerance in diet‐induced obese (DIO) mice. (A) Schematic diagram showing the experimental design for investigating the effect of muscle‐specific ERRα overexpression on recovery after hindlimb ischemia (HLI) in high fat diet (HFD)‐fed transgenic (TG) and wild type (WT) mice. (B) The body weight gained on HFD over the course of the study in WT and TG mice (N = 8 mice). (C) Glucose tolerance test (GTT) in WT‐DIO and TG‐DIO mice compared to normal chow diet (NCD)‐fed WT mice (N = 8 mice). (D) Insulin tolerance test (ITT) in WT‐DIO and TG‐DIO mice compared to NCD‐fed WT mice (N = 8 mice). (E) The fasting blood glucose after 6 h of fasting in WT‐DIO and TG‐DIO mice compared to NCD‐fed WT mice (N = 8 mice). (F) The fasting serum insulin in WT‐DIO and TG‐DIO mice compared to NCD fed WT mice (N = 8 mice). One‐way ANOVA followed by Tukey's post‐hoc test. (C) p < 0.001 = # for WT‐NCD versus WT‐DIO comparison; and p < 0.01 = ** and p < 0.001 = *** for WT‐NCD versus TG‐DIO comparison. (D) p < 0.001 = # and p < 0.05 = φ for WT‐NCD versus WT‐DIO comparison; and p < 0.01 = * and p < 0.001 = *** for WT‐NCD versus TG‐DIO comparison.
3.2. Targeted overexpression of ERR⍺ induces angiogenic factors and promotes neo‐angiogenesis in skeletal muscles of DIO mice
Before conducting the HLI experiments, we measured the expression of angiogenesis growth factor genes in the skeletal muscle of WT‐DIO and TG‐DIO mice. Note that the fold increase in muscle ERRα gene expression in TG‐DIO vs. WT‐DIO mice was 15.63 ± 1.42 (Figure 2A) even after HFD, which is comparable to fold increase (17.48 ± 2.48) in muscle ERRα gene expression in TG‐NCD versus WT‐NCD mice on normal chow diet. We found that compared to WT‐DIO mice, gastrocnemius muscles of TG‐DIO mice have significantly higher expression of pro‐angiogenic genes (Angpt1, Egf, Fgf1, Epas1, Vegfa, Vegf121, Vegf165, Vegf189, Vegfb), and a significantly lower expression of anti‐angiogenic genes [Col18a1 (endostatin) and Cxcl4] (Figure 2A). Dot blot protein analysis confirmed this effect at the protein level, where we see a statistically higher expression of pro‐angiogenic proteins VEGFA and FGF1 in TG‐DIO compared to WT‐DIO tibialis anterior (TA) muscle (Figure 2B —red box). Conversely, TG‐DIO TA has significantly lower expression of Cxcl4 and serpin1 anti‐angiogenic proteins compared to WT‐DIO TA (Figure 2B—blue box). These data demonstrate that despite the induction of obesity, the TG‐DIO muscles express a gene signature representative of a higher angiogenic potential compared to WT‐DIO muscle. Therefore, to test the neo‐angiogenic potential of muscle ERRα overexpression in DIO mice, we subjected the TG‐DIO and WT‐DIO mice to HLI. After 18 days of HLI in WT‐DIO and TG‐DIO mice, we analyzed TA cryosections for capillary immunostaining (Figure 2C), and found that the ratio of capillary to fiber number was significantly higher in both the contralateral and ischemic TA of TG‐DIO compared to WT‐DIO mice (Figure 2D). Notably, the ischemic TG‐DIO TA showed the maximum increase in capillaries to fiber ratio after 18 days of HLI; whereas, in comparison the WT‐DIO ischemic muscle did not exhibit significant increase in the ratio (Figure 2D). These data show at the histological level that muscle ERRα overexpression facilitates ischemic muscle neo‐angiogenesis in the DIO mice, which agrees with the higher angiogenic gene and protein expression in TG‐DIO vs. WT‐DIO muscle (Figure 2A,B).
FIGURE 2.
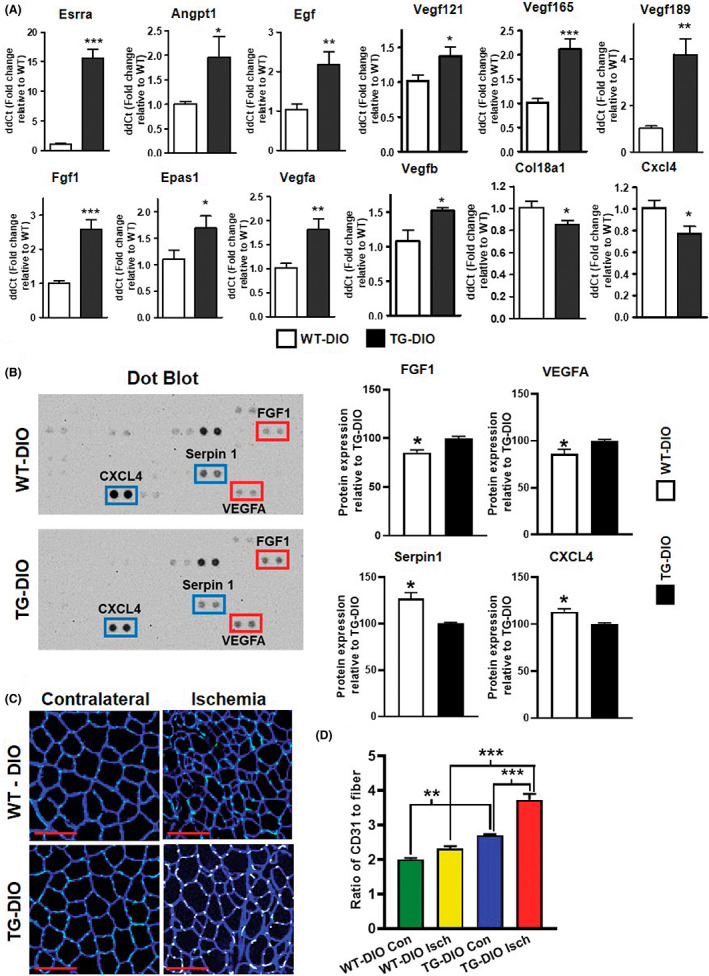
Muscle‐specific ERR⍺ overexpression induces angiogenic factors and promotes neo‐angiogenesis in skeletal muscles of DIO mice. (A) qPCR analysis measuring expression of pro‐angiogenic and anti‐angiogenic gene expression in gastrocnemius muscle of diet‐induced obese (DIO) TG compared to WT mice (N = 5–6 mice). (B) Representative dot blot of DIO TG and WT tibialis anterior (TA) muscles showing expression of pro‐angiogenic (red boxes) and anti‐angiogenic (blue boxes) proteins, as well as densitometric quantification showing significantly lower expression of pro‐angiogenic proteins (FGF1 and VEGFA) and higher expression of anti‐angiogenic proteins (CXCL4 and serpin1) in WT‐DIO TA relative to TG‐DIO TA (N = 3 mice). (C) Laminin (myofiber sarcolemma) and CD31 (capillary) immunostaining of TA cross‐sections to measure neo‐angiogenesis in TG‐DIO vs WT‐DIO muscles in response to 18 days of HLI in contralateral and ischemic limbs. (D) Quantification of (C) as capillary (CD31 staining) to fiber ratio. Red scale bar = 100 μm (N = 4 mice). Unpaired Student's test and one‐way ANOVA followed by Tukey's post‐hoc test. p < 0.05 = *, p < 0.01 = **, and p < 0.001 = ***.
3.3. Muscle ERR⍺ overexpression promotes muscle ischemic revascularization in DIO mice
For further in‐depth analysis of the effect of muscle ERR⍺ overexpression on revascularization and recovery after HLI in DIO mice, we performed analysis over a 30‐day period after HLI in contralateral and ischemic hindlimbs of TG‐DIO and WT‐DIO mice. Here, we included age and sex‐matched WT‐NCD mice as lean controls. Analysis of capillary immunostaining (CD31) in TA was performed in WT‐NCD, WT‐DIO, and TG‐DIO mice (Figure 3A,B) at the end of 30 days. Compared to contralateral TA, there is a significant increase in capillary to fiber ratio in ischemic TA of WT‐NCD. Notably, WT‐DIO ischemic TA did not show a similar increase in capillary to fiber ratio compared to WT‐DIO contralateral muscle, exhibiting impaired ischemic neo‐angiogenesis in DIO muscle. The ischemia‐induced neo‐angiogenesis was restored in the TG‐DIO muscles, as demonstrated by an increase in capillary to fiber ratio in ischemic compared to the contralateral non‐ischemic limb. Notably, the capillary to fiber ratio in TG‐DIO ischemic TA was the highest and significantly greater than the ratio in WT‐DIO ischemic TA.
FIGURE 3.
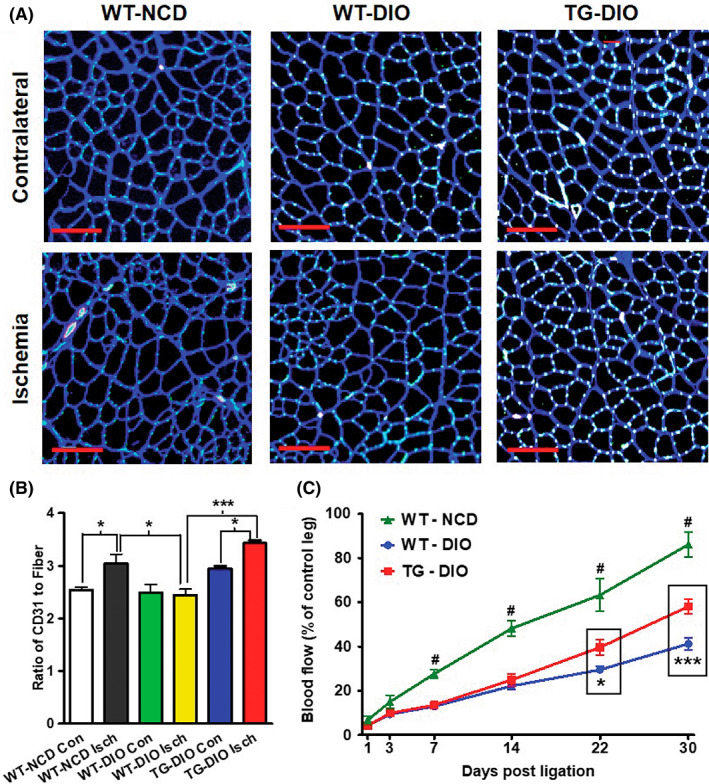
ERR⍺ overexpression promotes ischemic revascularization in the skeletal muscle of DIO mice. (A) Laminin (myofiber sarcolemma) and CD31 (capillary) immunostaining of contralateral and ischemic TA cross‐sections 30 days after HLI in WT‐NCD, WT‐DIO, and TG‐DIO mice. Red scale bar = 100 μm. (B) Quantification of (A) represented as capillary to myofiber ratio (index of neo‐angiogenesis) in ischemic versus contralateral WT‐NCD, WT‐DIO, and TG‐DIO TA (N = 4–6 mice). (C) Laser Doppler flowmetry measuring reperfusion in WT‐NCD, WT‐DIO and TG‐DIO mice after HLI (N = 8 mice). One‐way ANOVA followed by Tukey's post‐hoc test. p < 0.05 = *, p < 0.001 = ***, and p < 0.001 = #. The black rectangle highlights difference between WT‐DIO and TG‐DIO groups, statistical significance between these groups is represented by ‘*’. ‘#’ represents the difference between WT‐NCD and both the DIO groups.
Next, we monitored blood flow recovery over 30 days in FDB muscle (the distal most muscle from ligation site) in contralateral and ischemic hindlimbs using laser Doppler flowmetry in the three groups of mice. Compared to the WT‐NCD muscle, which had the highest rate of blood flow recovery (Figure 3C), the muscle blood flow recovery was significantly impaired in WT‐DIO mice. However, compared to WT‐DIO mice muscle‐specific ERRα overexpression in the TG‐DIO mice, significantly improved blood reperfusion to the muscle (statistically significant differences highlighted by black rectangles in Figure 3C), albeit not to the same level as in the lean mice.
3.4. Muscle ERR⍺ overexpression promotes post‐ischemic muscle recovery in DIO mice
To measure the effect of ERRα overexpression on myofiber recovery after HLI, we performed immunostaining of TA cryosections from contralateral and ischemic limbs for muscle sarcolemma (laminin) and nuclei, 30 days after ischemia (Figure 4A). Qualitative inspection of the immunostained sections showed that the contralateral TAs across the three groups were undamaged and similar in architecture (Figure 4A, Upper panel). As expected, the ischemic TA of the WT‐DIO mice qualitatively exhibit more small and non‐uniformly shaped myofibers compared to WT‐NCD indicating muscle damage, and this pathological effect was reduced in TG‐DIO ischemic TA overexpressing ERRα (Figure 4A, Lower panel). Fiber area distribution analysis (Figure 4B) revealed that there is a higher percentage of smaller sized fibers in ischemic (red bars) compared to the contralateral hindlimb muscle (blue bars) in all three groups. The leftward shift towards smaller fiber size (below 750 μm2) is significantly higher in WT‐DIO ischemic TA (34.57 ± 3.01% of fibers below 250 μm2) compared to WT‐NCD ischemic TA (13.95 ± 3.680% of fibers below 250 μm2) indicating greater muscle damage in WT‐DIO ischemic muscle (Figure 4B and Data Unit S2A). However, the leftward shift was less in ischemic TG‐DIO TA (22.85 ± 6.129% of fibers below 250 μm2) and compared to WT‐NCD ischemic TA (13.95 ± 3.680% of fibers below 250 μm2), and the difference between the two groups was statistically insignificant (Figure 4B and Data Unit S2B). Furthermore, quantification of total fiber number revealed that WT‐DIO ischemic TA had higher myofiber number compared to WT‐NCD ischemic TA, an effect which was mitigated in TG‐DIO ischemic TA (Figure 4C). These data suggest that WT‐DIO ischemic TA have many more regenerating fibers majority of which are below 500 μm2, a sign of incomplete repair at the 30‐day post‐ischemia time point. Percentage of central nuclei is also an indicator of the regenerative state of the muscle. Here too, we see a trend that WT‐DIO ischemic TA has the higher percentage of central nuclei (Figure 4D) compared to WT‐NCD, which is reduced in TG‐DIO TA. However, the data did not reach statistical significance. Collectively, these data indicate that the post‐ischemic muscle recovery is significantly higher in TG‐DIO mice compared to WT‐DIO mouse muscle, which agrees with the faster blood flow recovery in the TG‐DIO vs. the WT‐DIO (Figure 3).
FIGURE 4.
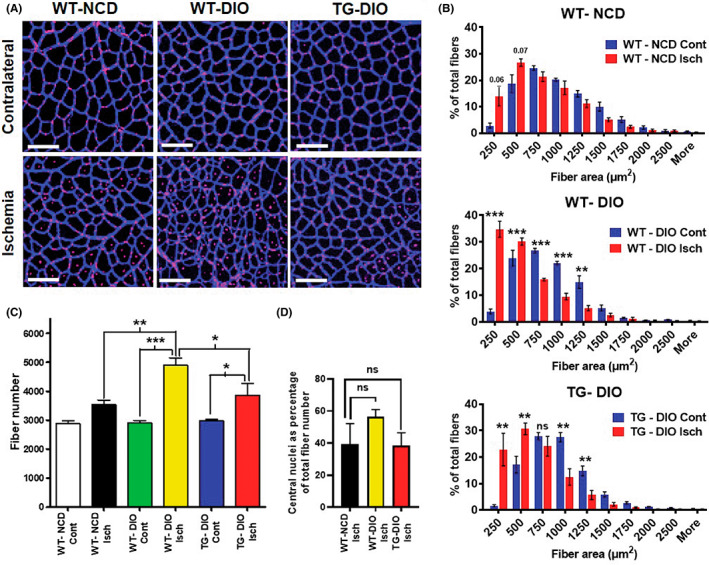
Muscle ERR⍺ overexpression stimulates post‐ischemic muscle recovery in DIO mice. (A) Laminin (myofiber sarcolemma) and nuclei staining of contralateral and ischemic TA cross‐sections 30 days after HLI in WT‐NCD, WT‐DIO, and TG‐DIO mice. White scale bar = 100 μm. (B) Myofiber size distribution in TA cross‐sections of WT‐NCD (top panel), WT‐DIO (middle panel) and TG‐DIO (bottom panel) mice, with blue bars representing contralateral and red bars representing ischemic hindlimb TA. N = 4–6 mice. (C) Total myofiber number in the WT‐NCD, WT‐DIO and TG‐DIO contralateral and ischemic TA (N = 4–6 mice). (D) Percentage of central nuclei in ischemic limb TAs of WT‐NCD, WT‐DIO, and TG‐DIO TA (N = 4–6 mice). One‐way ANOVA followed by Tukey's post‐hoc test p < 0.05 = *, p < 0.01 = **, and p < 0.001 = ***. NS=Not significant.
Overall, ERRα overexpression in the muscle facilitates vascular as well as myocellular recovery in response to ischemic hindlimb injury in the DIO mice. As expected vascular as well as myocellular regeneration are impaired in obesity.
3.5. Muscle ERR⍺ is repressed in the skeletal muscles of DIO mice
The observation that DIO impairs ischemic revascularization, and that muscle‐specific ERRα overexpression stimulates ischemic muscle recovery in DIO mice, compelled us to examine the effect of DIO on ERRα expression in the skeletal muscles. In addition, we also measured the expression of phospho and total‐AMPK (alpha catalytic subunit), known to be involved in muscle angiogenesis and repair. 30 , 31 , 32 Strikingly, we found that the baseline ERRα protein expression is repressed in WT‐DIO compared to WT‐NCD TA (Figure 5A). The phospho‐and total‐AMPK levels were not different between WT‐NCD and WT‐DIO TA muscle. However, AMPK target phospho‐ACC was repressed in the DIO skeletal muscle (Figure 5A), suggesting that AMPK signaling may be decreased. More importantly, the gene expression of pro‐angiogenic regulators VEGFA and FGF1 was decreased in the skeletal muscle of DIO vs. NCD WT mice (Figure 5B ).
FIGURE 5.
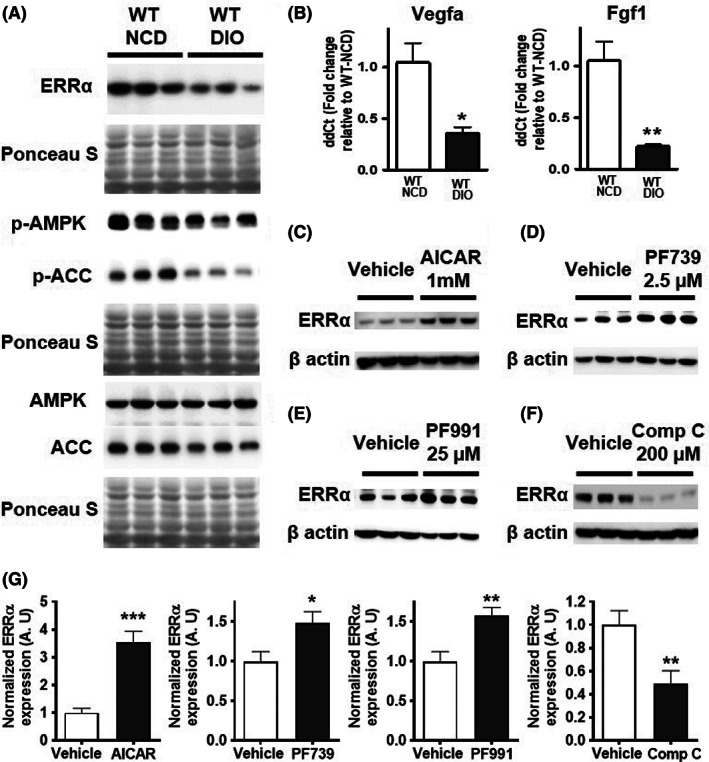
Regulation of ERR⍺ by AMPK in muscle. (A) Protein expression of ERRα, phospho‐AMPK, total‐AMPK, phospho‐ACC, and ACC in WT‐NCD vs. WT‐DIO mouse TA (N = 6 mice). (B) Gene expression of angiogenic genes Vegfa and Fgf1 (N = 4). (C‐F) Representative images of Western blots showing ERRα expression in (C) vehicle versus AICAR (1 mM) AMPK‐treated, (D) vehicle versus PF739 (2.5 μM)‐treated, (E) vehicle versus PF991 (25 μM)‐treated, and (F) vehicle versus compound C (200 μM)‐treated C2C12 myotubes (N = 6). Ponceau S staining was used as loading control. (G) Densitometric quantification for 5C‐F. Unpaired Student's t test with p < 0.05 = *, p < 0.01 = **, and p < 0.001 = ***.
3.6. AMPK activators and inhibitors regulate ERRα expression
We investigated whether different AMPK activators can induce ERRα expression in the skeletal muscle cells. To do so, we conducted cell culture experiments in murine muscle cell line (C2C12 cells). We found that treatment of 4 day differentiated myotubes with AMPK activators, 1 mM AICAR (Figure 5C), 2.5 μM PF739 (Figure 5D), and 25 μM PF991 (Figure 5E) for 24 h resulted in a robust induction of ERRα protein expression in C2C12 myotubes. Effect of these compounds on AMPK and ACC was confirmed in C2C12 myotubes, by showing the induction of phospho‐AMPK and phospho‐ACC levels (Data unit S3A–C). Conversely, when 4‐day differentiated myotubes are treated with 200 μM compound C (AMPK inhibitor) for 6 h, it resulted in a robust downregulation of ERRα protein expression (Figure 5F). The downregulation of phospho‐AMPK and phospho‐ACC in response to compound C treatment in these cells is also shown (Data Unit S3D). In addition, using MEF isolated from AMPK α1/α2 double knockout and wild type murine embryos, we found that ERRα expression is downregulated in cells lacking AMPK catalytic subunits, further confirming that ERRα expression is regulated by AMPK activity Data Unit S3E). Therefore, AMPK is a potent regulator of ERRα expression in cultured cells.
3.7. ERR⍺ is involved in AMPK mediated expression of angiogenic genes in muscle cells
We next asked whether AMPK and ERRα are linked in the regulation of angiogenic gene expression in muscle cells. To answer this question, we used C2C12 cells and combined treatments with AICAR, PF739, PF991 and XCT 790 (reverse agonist/inhibitor of ERR⍺), as described in the figure legend. We discovered that XCT 790 not only downregulates ERR⍺ protein expression at baseline, but it also blocks the AICAR‐mediated induction of ERRα in C2C12 cells (Figure 6A). We confirmed the activation of AMPK pathway in these cells by measuring protein expression of phospho‐AMPK and phospho‐ACC, and found that AICAR increased the expression of p‐AMPK and p‐ACC even in the presence of XCT 790 (Data Unit S4A). AICAR induces the expression of several angiogenic genes (Vegfa, Vegfb, and Egf) in muscle cells, and this induction by AICAR is significantly inhibited in the presence of XCT 790 (Figure 6B). Note that qPCR analysis showed that mRNA expression of ERR⍺ is induced by AICAR, however XCT 790 does not affect ERR⍺ at the gene expression level (Figure 6B). These data suggest that ERR⍺ is involved in the AMPK‐mediated induction of key angiogenic genes. We further confirmed this by looking for potential ERR⍺ response elements (EREs) in a 2000 bp region upstream of the transcription start site for these genes. Two of the genes, Egf and Vegfa, have EREs as shown in the schematic (Figure 6C). We conducted ERR⍺ ChIP‐qPCR in the presence and absence of AICAR in C2C12 cells and found that while ERR⍺ is enriched at these sites, this enrichment is enhanced upon the stimulation of muscle cells with AICAR (Figure 6D). Interestingly, a similar analysis for oxidative metabolic genes, which are classical targets of ERR⍺, show that ERRα is also involved in AMPK‐mediated induction of Cycs, Pdk4, and myoglobin (Data Unit S5A), and its enrichment at the EREs of these genes (Data Unit S5B) is also regulated by AICAR (Data Unit S5C). We further confirmed the regulation of ERRα protein expression by AMPK activator PF739 and PF991 in the presence of XCT 790 (Figure 7A and Data Unit S4B‐C). XCT 790 not only downregulates ERR⍺ protein expression at baseline, but it also blocks the PF739 or PF991‐mediated induction of ERRα in C2C12cells, while not affecting the phosphorylation status of AMPK and ACC (Data Unit S4B,C). Additionally, we measured the effect of PF739 on angiogenic gene expression in presence and absence of XCT 790, and found that PF739‐mediated induction of angiogenic genes (Egf and Vegfb) is significantly inhibited by XCT 790 (Figure 7B). These data show that AMPK stimulation promotes angiogenic gene expression in muscle cells via up‐regulation of ERR⍺ expression and enrichment of the receptor at angiogenic gene promoters.
FIGURE 6.
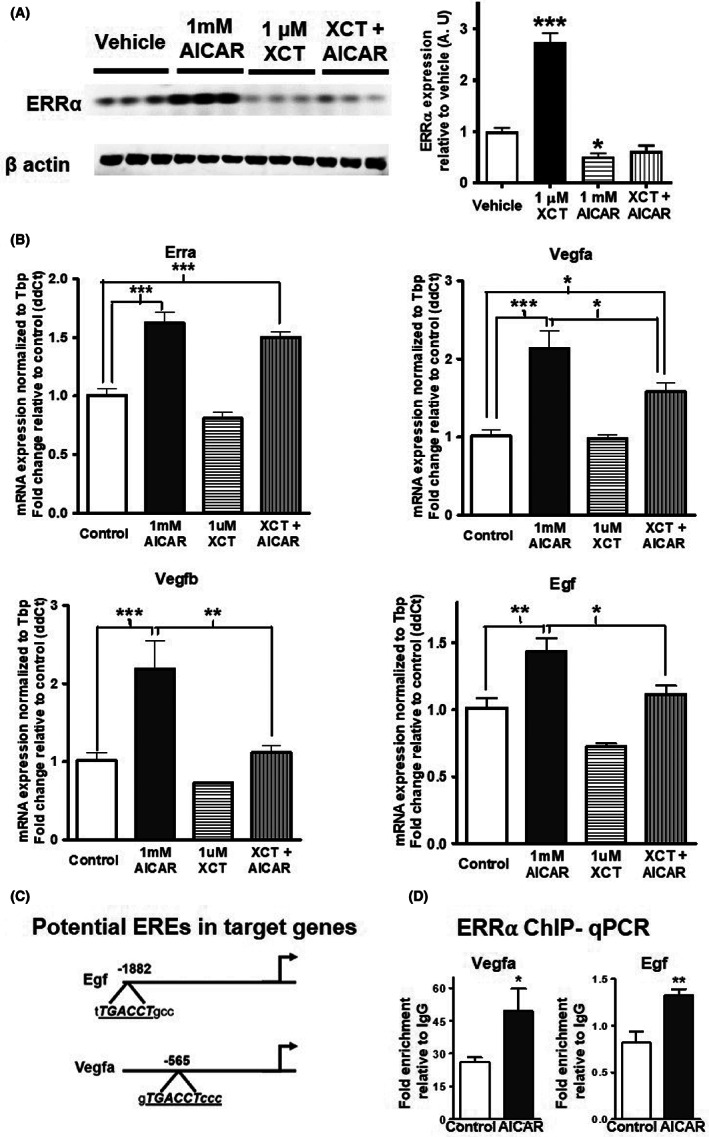
ERR⍺ is involved in AICAR‐mediated induction of angiogenic genes. (A) Representative Western blots showing the effect of XCT 790 (XCT) (1 μM) on baseline and AICAR (1 mM) induced ERRα expression in C2C12 myotubes (N = 6). (B) mRNA expression of Esrra (ERRα) and angiogenic genes (Vegfa, Vegfb, and Egf) in AICAR (1 mM) and XCT 790 (1 μM) treated C2C12 myotubes. (C) Schematic diagram showing potential ERRα response elements (EREs) in the angiogenic target genes of AMPK (Egf and Vegfa). (D) ERRα ChIP‐qPCR showing enrichment of ERRα at angiogenic genes (Vegfa and Egf) in vehicle and AICAR (1 mM) treated C2C12 myotubes (N = 3). Unpaired Student's t‐test and one‐way ANOVA followed by Tukey's post hoc test p < 0.05 = *, p < 0.01 = **, and p < 0.001 = ***.
FIGURE 7.
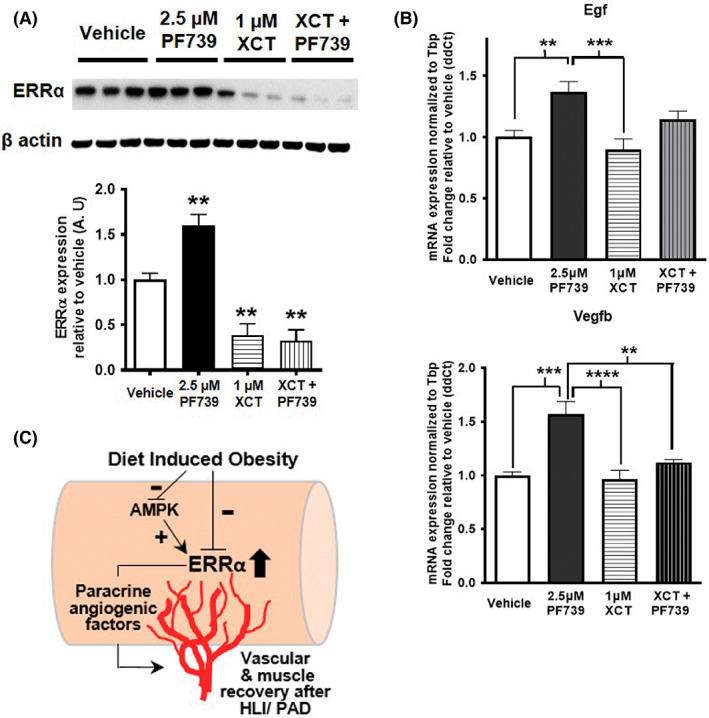
ERR⍺ is involved in PF739‐mediated induction of angiogenic genes. (A) Representative Western blots showing the effect of XCT 790 (XCT) (1 μM) on baseline and PF739 (2.5 μM) induced ERRα expression in C2C12 myotubes (N = 6). Beta‐actin is used as the loading control (N = 6). Densitometric quantification is shown in the lower panel. (B) mRNA expression of angiogenic genes (Egf and Vegfb) in PF739 (2.5 μM) and XCT 790 (1 μM) treated C2C12 myotubes. (C) Scheme summarizing that AMPK/ERRα angiogenic signaling pathway is suppressed in the skeletal muscle in DIO, and forced ERRα activation in the skeletal muscle promotes recovery after HLI in DIO mice. One‐way ANOVA followed by Tukey's post hoc test p < 0.01 = ** and p < 0.001 = ***.
4. DISCUSSION
PAD and CLI are a major complication of obesity and type II diabetes, associated with impaired angiogenesis and limb ischemia. 4 , 5 , 6 , 7 Molecular pathways for enhancing angiogenesis and revascularization in diabetic limb ischemia are poorly defined. Here, we show that ERRα overexpression in the skeletal muscle activates a paracrine angiogenic gene program that promotes ischemic revascularization and muscle repair in DIO mice. These effects of ERRα overexpression are independent of any effect on weight control or glucose/insulin homeostasis in DIO mice. We further identify AMPK to ERRα signaling in the skeletal muscle that regulates the expression of angiogenesis growth factor genes in muscle cells. This pathway is repressed in skeletal muscle of DIO mice and could therefore be a contributing factor to impaired muscle angiogenesis and PAD in obesity and type II diabetes. A scheme summarizing our findings in provided in Figure 7C.
Recently, we showed that ERRα overexpression in the skeletal muscle drives muscle remodeling to increase aerobic slow‐twitch myofibers and oxidative capacity. 21 While such aerobic remodeling raised the possibility that muscle ERRα overexpression might prevent weight gain and promote glucose/insulin tolerance in DIO diabetic mice, we did not observe any protection against obesity or glucose/insulin intolerance in muscle‐specific ERRα transgenic mice. In similar studies with muscle‐specific overexpression of ERRγ or ERR coactivator PGC1α, 28 , 33 , 34 , 35 neither of these factors protected against weight gain and glucose intolerance in diet‐induced obesity, despite fiber‐type and aerobic metabolic remodeling similar to ERRα. Our previous study of ERRγ transgenic mice suggests that while these transcriptional factors may increase the sheer number of mitochondria in the skeletal muscle, they remain a relatively ‘inactive reserve’, without further boosting myocellular metabolism or preventing the accumulation of triglycerides, toxic ceramides, and diacylglycerols that cause skeletal muscle insulin resistance in DIO. 28 Nevertheless, with regards to the primary focus of this investigation on ischemic complications, our data suggest that the ameliorative effects of ERRα on ischemic recovery is independent of glycemic control in DIO mice. This is noteworthy because clinical control of glycemia is not sufficient to prevent cardiovascular complications such as PAD in chronic diabetes, requiring a direct intervention at the level of the ischemic tissue. 3 , 4
In our recent work, RNA‐sequencing analysis of ERRα transcriptome revealed that angiogenic gene program is qualitatively and quantitatively the top‐most pathway activated by ERRα overexpression in the skeletal muscle. The ‘ERRα angiogenic transcriptome’ included a cohort of paracrine/secretory angiogenic growth factors that are induced by the receptor in the skeletal muscle. We found that the stimulatory effect of ERRα on paracrine angiogenic growth factor gene expression in the skeletal muscle is retained in mice subjected to DIO and glucose intolerance. Some of the pro‐angiogenic genes elevated by ERRα (e.g., Angpt1, Vegfa isoforms, Vegfb, Egf) in the diabetic skeletal muscle are known for their paracrine activation of endothelial cells and stimulation of angiogenesis. 9 , 36 We found that ERRα also repressed anti‐angiogenic factor expression in the muscle. Endostatin is one of the most potent inhibitors of angiogenesis through binding to VEGFR1 and 2 receptors for VEGF. 37 Similarly, PEDF decreases VEGF signaling by binding to PEDF‐R and initiating a series of events that lead to reduced expression of VEGFR1. 38 In addition, CXCL4 also inhibits neo‐angiogenesis by inhibiting endothelial cell proliferation and migration by interfering with VEGF165 and 121. 39 These powerful anti‐angiogenic factors that are suppressed by ERRα are known to directly interfere with VEGF signaling. This effect coupled with the induction of VEGF expression by ERRα likely primes the muscles of the TG mice for mounting a robust response to HLI in DIO. Indeed, the application of HLI resulted in a greater activation of muscle recovery in DIO mice overexpressing ERRα in the skeletal muscle. First, compared to the non‐obese mice, both induction of neo‐angiogenesis and ischemic reperfusion was dramatically impaired in the skeletal muscle of DIO mice, as has been previously reported. 24 , 25 , 26 Overexpression of ERRα significantly stimulated ischemic neo‐angiogenesis in the skeletal muscles of DIO mice. However, the post‐ischemic reperfusion was not completely restored to the same level as non‐obese mice in the time course of the study. There could be several reasons for this. Although the capillary density is restored in the TG‐DIO TA, return to normal hemodynamics may not be complete due to other effects of diet‐induced obesity (e.g., changes in vascular tone). Another reason might be the selective activation of ERRα in the skeletal muscle, and not in other cell types such as endothelial cells, pericytes, and immune cells, which are also involved in regulating angiogenesis in limb ischemia, 40 and where ERRα could orchestrate a more global angiogenic control. Another reason might be that the overexpressed ERRα is an apo receptor, which as much as is constitutively active, can be further stimulated with co‐factors or potential ligands to elicit full transcriptional activation. 12 This has been demonstrated at least in culture, where co‐activators such as PGC1α enhance the ability of ERRα to transcriptionally induce angiogenic factor Vegfa. 41 Finally, it is possible that constitutive overexpression of ERRα and its angiogenic targets prior to HLI may somewhat blunt the ischemic neo‐angiogenic response that could otherwise be achieved by post‐ischemic inducible activation of ERRα.
The sub‐maximal ischemic revascularization was sufficient to facilitate muscle regeneration and restoration, as indicated by our observation that TG‐DIO ischemic muscle has fiber number and size distribution that is closer to that of WT‐NCD ischemic muscle compared to WT‐DIO ischemic muscle. The accelerated restoration of muscle size could be secondary to enhancement of vascular reperfusion after ischemic injury in the obese mice by ERRα. ERRα could also directly promote muscle regeneration after ischemic injury. Previous study using murine cardiotoxin injury model has shown that ERRα can promote skeletal muscle regeneration. 18 Angiogenic factors themselves can also stimulate muscle regeneration. 42 , 43 Therefore, ERRα may promote muscle recovery in hindlimb ischemia indirectly through myocellular paracrine signaling and/or potentially directly through muscle stem cell regulation. The relative contribution of ERRα to different aspects of ischemic recovery in hindlimbs (e.g. angiogenesis, muscle stem cell activation, regeneration) will be an interesting area of future study particularly in the context of diabetic PAD, where mechanisms of regeneration after ischemic injury are poorly defined.
DIO represses ERRα protein expression in the skeletal muscle. This coincides not only with decreased expression of angiogenic factors in the skeletal muscle but also impaired ischemic revascularization. Interestingly, skeletal muscle master‐regulator AMPK signaling is decreased in DIO mice. Pharmacological activation of AMPK has been shown to promote ischemic revascularization in hindlimb ischemia. 44 , 45 The pro‐angiogenic effects of AMPK are postulated to be via activation of the kinase signaling in endothelial cells, particularly in response to stimuli such as hypoxia, Fgf2, Vegfa and apalin‐2. 30 , 46 , 47 Studies using LKB1, AMPKα1 and AMPKα2 knockout mice also suggests a critical need for endothelial AMPK in regulating angiogenesis, as angiogenesis and ischemic revascularization are impaired in these mutant mice. 31 , 48 Here, the downregulation of AMPK signaling in the DIO skeletal muscles suggests that myocyte AMPK may contribute to paracrine angiogenic regulation. Indeed, hindlimb ischemic revascularization in response to AICAR and adiponectin seems to be in part mediated by muscle AMPK signaling. 32 , 49 Here, we found that AMPK activator AICAR and PF739 induces pro‐angiogenic genes in cultured muscle cells. Notably, ERRα is also induced by different AMPK activators such as AICAR, PF739 and PF991 in muscle cells. Furthermore, the AICAR and PF739‐mediated induction of pro‐angiogenic genes is blocked by ERRα inverse agonist/inhibitor XCT 790. Therefore, ERRα could be the transcriptional mediator of the effects of AMPK on angiogenic genes. As mentioned above, transcriptional co‐activator PGC1α is an allosteric activator of ERRα transcriptional activity in muscle cells. 41 AMPK can activate PGC1α directly via phosphorylation or indirectly via activation of histone deacetylase and PGC1α activator SIRT1, and in turn control the expression of metabolic genes in the skeletal muscle. 50 , 51 Whether AMPK/PGC1α/SIRT1 complex also regulates angiogenic genes potentially via ERRα remains to be seen. Nevertheless, our experiments reveal a direct link between AMPK and angiogenic genes, through transcriptional induction of ERRα. Notably, induction of ERRα by AMPK has been previously described including in different cell types. 18 , 52 , 53 In addition to gene/protein induction, ERRα regulation by AMPK via post‐translational modifications and protein stabilization may also be relevant, as has been reported for other regulatory factors such as Nrf‐1, Insig‐1 and PPARδ. 54 , 55 , 56 Overall, our combined in vivo and in vitro findings highlight an AMPK‐ERRα paracrine angiogenic pathway in muscle cells which is repressed by DIO in mice, and which is a likely contributing factor to impaired ischemic revascularization. The expression of AMPK subunits or baseline activity is not downregulated in the skeletal muscles of human type 2 diabetic patients; nevertheless, its activation can lead to beneficial effects in the skeletal muscle. 57 , 58 , 59 Currently, there is no clear evidence whether ERRα expression or its transcriptional activity is affected in the skeletal muscles of type 2 diabetes patients. Furthermore, the expression of both AMPK and ERRα in muscles from type 2 diabetic patients with PAD complication has not been measured. However, a clinically approved AMPK activator metformin has been shown to promote ischemic revascularization in models of ischemia. 45 , 60 , 61 Further, metformin treatment is associated with a lower risk of cardiovascular events in diabetic patients with PAD, 62 and a clinical trial (PERMET, #NCT03054519) investigating the effects of metformin in PAD patients is currently ongoing. Therefore, pathways described in this study are of potential clinical relevance.
Our work opens new avenues of query in the area of diabetic PAD complication. While constitutive expression of ERRα promotes ischemic muscle revascularization in DIO mice, whether this can be translated into therapeutics will require additional studies. Several new tools such as inducible transgenic mice, AAV9‐ERRα delivery vectors or synthetic ligands for ERRα would need to be developed and investigated for post‐ischemic delivery/activation and repair. Muscle‐specific ERRα and ERRγ single/compound knockout mice studies could be instrumental in defining the role of endogenous ERRs in ischemic muscle recovery. Fundamentally, the link between AMPK and ERRα will need to be further explored. How does AMPK induce ERRα expression in the skeletal muscle? What is the comprehensive paracrine angiogenic ‘transcriptome’ activated by AMPK in the skeletal muscle cells, and to what extent is this dependent on ERRα? In summary, our findings identify the AMPK‐ERRα paracrine angiogenic pathway in the skeletal muscle as a potential target for neo‐angiogenesis and ischemic recovery in diabetic PAD, and likely also in other diabetic cardiovascular complications.
AUTHOR CONTRIBUTIONS
The project was conceptualized and experiments designed by D.H.S, A.K., and V.A.N. All the experiments were performed by D.H.S with help from A.R. Wild type and AMPK α1/α2 KO mouse embryonic fibroblast cells were provided by M.S. and were grown, harvested and processed for protein expression analysis by M.P. Overall, data analysis was performed by D.H.S., A.R., and V.A.N and data interpretation was performed by D.H.S, A.K., and V.A.N. Manuscript was written by D.H.S, A.K., and V.A.N.
FUNDING INFORMATION
This research was supported in parts by NIH/NHLBI grants (R01HL152108, R01HL129191), American Heart Association Transformational Research Award (20TPA35410038), and Hamman Foundation Endowment in Cardiovascular Research to V.A.N. D.H.S. was supported by a post‐doctoral fellowship from American Heart Association (19POST34380771). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Supporting information
Appendix S1
Appendix S2
Appendix S3
Appendix S4
Appendix S5
ACKNOWLEDGMENTS
We thank Dr. Zhengmei Mao in the UTHealth Institute of molecular medicine microscopy core facility for assistance with microscopy, imaging and analysis.
Sopariwala DH, Rios AS, Park MK, Song MS, Kumar A, Narkar VA. Estrogen‐related receptor alpha is an AMPK‐regulated factor that promotes ischemic muscle revascularization and recovery in diet‐induced obese mice. FASEB BioAdvances. 2022;4(9):602‐618. doi: 10.1096/fba.2022-00015
Andrea S. Rios is formerly Andrea S. Guzman.
DATA AVAILABILITY STATEMENT
Data supporting the conclusions of this manuscript are presented in the form of the main or supplemental figures and tables. Raw data supporting the findings of the study and used in the generation of the figures are available on request from the corresponding author.
REFERENCES
- 1. Belch JJ, Topol EJ, Agnelli G, et al. Critical issues in peripheral arterial disease detection and management: a call to action. Arch Intern Med. 2003;163:884‐892. [DOI] [PubMed] [Google Scholar]
- 2. Korthuis, R. J. (2011) Skeletal Muscle Circulation. Morgan and Claypool Publishers. [PubMed] [Google Scholar]
- 3. Wong KL, Nather A, Liang S, Chang Z, Wong TT, Lim CT. Clinical outcomes of below knee amputations in diabetic foot patients. Ann Acad Med Singapore. 2013;42:388‐394. [PubMed] [Google Scholar]
- 4. Jude EB, Eleftheriadou I, Tentolouris N. Peripheral arterial disease in diabetes—a review. Diabet Med. 2010;27:4‐14. [DOI] [PubMed] [Google Scholar]
- 5. Owings, M. F. , and Kozak, L. J. (1998) Ambulatory and inpatient procedures in the United States, 1996. Vital and health statistics. Series 13, Data from the National Health Survey, 1‐119. [PubMed]
- 6. Roger, V. L. , Go, A. S. , Lloyd‐Jones, D. M. , et al., American Heart Association Statistics, C , and Stroke Statistics, S (2012) Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation 125, e2‐e220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ziegler‐Graham K, MacKenzie EJ, Ephraim PL, Travison TG, Brookmeyer R. Estimating the prevalence of limb loss in the United States: 2005 to 2050. Arch Phys Med Rehabil. 2008;89:422‐429. [DOI] [PubMed] [Google Scholar]
- 8. Annex BH. Therapeutic angiogenesis for critical limb ischaemia. Nat Rev Cardiol. 2013;10:387‐396. [DOI] [PubMed] [Google Scholar]
- 9. Cooke JP, Losordo DW. Modulating the vascular response to limb ischemia: angiogenic and cell therapies. Circ Res. 2015;116:1561‐1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Audet‐Walsh E, Giguere V. The multiple universes of estrogen‐related receptor alpha and gamma in metabolic control and related diseases. Acta Pharmacol Sin. 2015;36:51‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dufour CR, Wilson BJ, Huss JM, et al. Genome‐wide orchestration of cardiac functions by the orphan nuclear receptors ERRalpha and gamma. Cell Metab. 2007;5:345‐356. [DOI] [PubMed] [Google Scholar]
- 12. Giguere V. Transcriptional control of energy homeostasis by the estrogen‐related receptors. Endocr Rev. 2008;29:677‐696. [DOI] [PubMed] [Google Scholar]
- 13. Narkar VA, Fan W, Downes M, et al. Exercise and PGC‐1alpha‐independent synchronization of type I muscle metabolism and vasculature by ERRgamma. Cell Metab. 2011;13:283‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gan Z, Rumsey J, Hazen BC, et al. Nuclear receptor/microRNA circuitry links muscle fiber type to energy metabolism. J Clin Invest. 2013;123:2564‐2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matsakas A, Yadav V, Lorca S, Evans RM, Narkar VA. Revascularization of ischemic skeletal muscle by estrogen‐related receptor‐gamma. Circ Res. 2012;110:1087‐1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Matsakas A, Yadav V, Lorca S, Narkar V. Muscle ERRgamma mitigates Duchenne muscular dystrophy via metabolic and angiogenic reprogramming. FASEB J. 2013;27:4004‐4016. [DOI] [PubMed] [Google Scholar]
- 17. Rangwala SM, Wang X, Calvo JA, et al. Estrogen‐related receptor gamma is a key regulator of muscle mitochondrial activity and oxidative capacity. J Biol Chem. 2010;285:22619‐22629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. LaBarge S, McDonald M, Smith‐Powell L, Auwerx J, Huss JM. Estrogen‐related receptor‐alpha (ERRalpha) deficiency in skeletal muscle impairs regeneration in response to injury. FASEB J. 2014;28:1082‐1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Murray J, Auwerx J, Huss JM. Impaired myogenesis in estrogen‐related receptor gamma (ERRgamma)‐deficient skeletal myocytes due to oxidative stress. FASEB J. 2013;27:135‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Murray J, Huss JM. Estrogen‐related receptor alpha regulates skeletal myocyte differentiation via modulation of the ERK MAP kinase pathway. Am J Physiol Cell Physiol. 2011;301:C630‐C645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sopariwala DH, Likhite N, Pei G, et al. Estrogen‐related receptor alpha is involved in angiogenesis and skeletal muscle revascularization in hindlimb ischemia. FASEB J. 2021;35:e21480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Likhite N, Yadav V, Milliman EJ, et al. Loss of estrogen‐related receptor alpha facilitates angiogenesis in endothelial cells. Mol Cell Biol. 2019;39:e00411‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang LD, Chen L, Zhang M, et al. Downregulation of ERRalpha inhibits angiogenesis in human umbilical vein endothelial cells through regulating VEGF production and PI3K/Akt/STAT3 signaling pathway. Eur J Pharmacol. 2015;769:167‐176. [DOI] [PubMed] [Google Scholar]
- 24. Chen YL, Chang CL, Sun CK, et al. Impact of obesity control on circulating level of endothelial progenitor cells and angiogenesis in response to ischemic stimulation. J Transl Med. 2012;10:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li YJ, Guan H, Hazarika S, Liu CW, Annex BH. Impaired angiogenesis following hind‐limb ischemia in diabetes mellitus mice. Chin Med Sci J. 2007;22:232‐237. [PubMed] [Google Scholar]
- 26. Kito T, Shibata R, Kondo M, et al. Nifedipine ameliorates ischemia‐induced revascularization in diet‐induced obese mice. Am J Hypertens. 2012;25:401‐406. [DOI] [PubMed] [Google Scholar]
- 27. Badin PM, Sopariwala DH, Lorca S, Narkar VA. Muscle Arnt/Hif1beta is dispensable in myofiber type determination, vascularization and insulin sensitivity. PLoS One. 2016;11:e0168457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Badin PM, Vila IK, Sopariwala DH, et al. Exercise‐like effects by estrogen‐related receptor‐gamma in muscle do not prevent insulin resistance in db/db mice. Sci Rep. 2016;6:26442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vila IK, Yao Y, Kim G, et al. A UBE2O‐AMPKalpha2 Axis that promotes tumor initiation and progression offers opportunities for therapy. Cancer Cell. 2017;31:208‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nagata D, Mogi M, Walsh K. AMP‐activated protein kinase (AMPK) signaling in endothelial cells is essential for angiogenesis in response to hypoxic stress. J Biol Chem. 2003;278:31000‐31006. [DOI] [PubMed] [Google Scholar]
- 31. Ohashi K, Ouchi N, Higuchi A, Shaw RJ, Walsh K. LKB1 deficiency in Tie2‐Cre‐expressing cells impairs ischemia‐induced angiogenesis. J Biol Chem. 2010;285:22291‐22298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ouchi N, Shibata R, Walsh K. AMP‐activated protein kinase signaling stimulates VEGF expression and angiogenesis in skeletal muscle. Circ Res. 2005;96:838‐846. [DOI] [PubMed] [Google Scholar]
- 33. Wong KE, Mikus CR, Slentz DH, et al. Muscle‐specific overexpression of PGC‐1alpha does not augment metabolic improvements in response to exercise and caloric restriction. Diabetes. 2015;64:1532‐1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Summermatter S, Shui G, Maag D, Santos G, Wenk MR, Handschin C. PGC‐1alpha improves glucose homeostasis in skeletal muscle in an activity‐dependent manner. Diabetes. 2013;62:85‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Choi CS, Befroy DE, Codella R, et al. Paradoxical effects of increased expression of PGC‐1alpha on muscle mitochondrial function and insulin‐stimulated muscle glucose metabolism. Proc Natl Acad Sci USA. 2008;105:19926‐19931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Saharinen P, Eklund L, Alitalo K. Therapeutic targeting of the angiopoietin‐TIE pathway. Nat Rev Drug Discov. 2017;16:635‐661. [DOI] [PubMed] [Google Scholar]
- 37. Walia A, Yang JF, Huang YH, Rosenblatt MI, Chang JH, Azar DT. Endostatin's emerging roles in angiogenesis, lymphangiogenesis, disease, and clinical applications. Biochim Biophys Acta. 2015;1850:2422‐2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cai J, Jiang WG, Grant MB, Boulton M. Pigment epithelium‐derived factor inhibits angiogenesis via regulated intracellular proteolysis of vascular endothelial growth factor receptor 1. J Biol Chem. 2006;281:3604‐3613. [DOI] [PubMed] [Google Scholar]
- 39. Wang Z, Huang H. Platelet factor‐4 (CXCL4/PF‐4): an angiostatic chemokine for cancer therapy. Cancer Lett. 2013;331:147‐153. [DOI] [PubMed] [Google Scholar]
- 40. Magenta A, Florio MC, Ruggeri M, Furgiuele S. Autologous cell therapy in diabetesassociated critical limb ischemia: from basic studies to clinical outcomes (review). Int J Mol Med. 2021;48:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Arany Z, Foo SY, Ma Y, et al. HIF‐independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC‐1alpha. Nature. 2008;451:1008‐1012. [DOI] [PubMed] [Google Scholar]
- 42. Karkkainen AM, Kotimaa A, Huusko J, et al. Vascular endothelial growth factor‐D transgenic mice show enhanced blood capillary density, improved postischemic muscle regeneration, and increased susceptibility to tumor formation. Blood. 2009;113:4468‐4475. [DOI] [PubMed] [Google Scholar]
- 43. Arsic N, Zacchigna S, Zentilin L, et al. Vascular endothelial growth factor stimulates skeletal muscle regeneration in vivo. Mol Ther. 2004;10:844‐854. [DOI] [PubMed] [Google Scholar]
- 44. Yu JW, Deng YP, Han X, Ren GF, Cai J, Jiang GJ. Metformin improves the angiogenic functions of endothelial progenitor cells via activating AMPK/eNOS pathway in diabetic mice. Cardiovasc Diabetol. 2016;15:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Takahashi N, Shibata R, Ouchi N, Sugimoto M, Murohara T, Komori K. Metformin stimulates ischemia‐induced revascularization through an eNOS dependent pathway in the ischemic hindlimb mice model. J Vasc Surg. 2015;61:489‐496. [DOI] [PubMed] [Google Scholar]
- 46. Dai Q, Fan X, Meng X, et al. FGF21 promotes ischaemic angiogenesis and endothelial progenitor cells function under diabetic conditions in an AMPK/NAD+‐dependent manner. J Cell Mol Med. 2021;25:3091‐3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang X, Zhu W, Zhang P, et al. Apelin‐13 stimulates angiogenesis by promoting crosstalk between AMP‐activated protein kinase and Akt signaling in myocardial microvascular endothelial cells. Mol Med Rep. 2014;9:1590‐1596. [DOI] [PubMed] [Google Scholar]
- 48. Xu MJ, Song P, Shirwany N, et al. Impaired expression of uncoupling protein 2 causes defective postischemic angiogenesis in mice deficient in AMP‐activated protein kinase alpha subunits. Arterioscler Thromb Vasc Biol. 2011;31:1757‐1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ouchi N, Kobayashi H, Kihara S, et al. Adiponectin stimulates angiogenesis by promoting cross‐talk between AMP‐activated protein kinase and Akt signaling in endothelial cells. J Biol Chem. 2004;279:1304‐1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Canto C, Gerhart‐Hines Z, Feige JN, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056‐1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jager S, Handschin C, St‐Pierre J, Spiegelman BM. AMP‐activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC‐1alpha. Proc Natl Acad Sci USA. 2007;104:12017‐12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim SY, Yang CS, Lee HM, et al. ESRRA (estrogen‐related receptor alpha) is a key coordinator of transcriptional and post‐translational activation of autophagy to promote innate host defense. Autophagy. 2018;14:152‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hu X, Xu X, Lu Z, et al. AMP activated protein kinase‐alpha2 regulates expression of estrogen‐related receptor‐alpha, a metabolic transcription factor related to heart failure development. Hypertension. 2011;58:696‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ding J, Gou Q, Jia X, et al. AMPK phosphorylates PPARdelta to mediate its stabilization, inhibit glucose and glutamine uptake and colon tumor growth. J Biol Chem. 2021;297:100954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Han Y, Hu Z, Cui A, et al. Post‐translational regulation of lipogenesis via AMPK‐dependent phosphorylation of insulin‐induced gene. Nat Commun. 2019;10:623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Joo MS, Kim WD, Lee KY, Kim JH, Koo JH, Kim SG. AMPK facilitates nuclear accumulation of Nrf2 by phosphorylating at serine 550. Mol Cell Biol. 2016;36:1931‐1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen, M. B. , McAinch, A. J. , Macaulay, S. L. , et al. (2005) Impaired activation of AMP‐kinase and fatty acid oxidation by globular adiponectin in cultured human skeletal muscle of obese type 2 diabetics. J Clin Endocrinol Metab 90, 3665–3672 [DOI] [PubMed] [Google Scholar]
- 58. Steinberg GR, Smith AC, Van Denderen BJ, et al. AMP‐activated protein kinase is not down‐regulated in human skeletal muscle of obese females. J Clin Endocrinol Metab. 2004;89:4575‐4580. [DOI] [PubMed] [Google Scholar]
- 59. Hojlund K, Mustard KJ, Staehr P, et al. AMPK activity and isoform protein expression are similar in muscle of obese subjects with and without type 2 diabetes. Am J Physiol Endocrinol Metab. 2004;286:E239‐E244. [DOI] [PubMed] [Google Scholar]
- 60. Venna VR, Li J, Hammond MD, Mancini NS, McCullough LD. Chronic metformin treatment improves post‐stroke angiogenesis and recovery after experimental stroke. Eur J Neurosci. 2014;39:2129‐2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jin Q, Cheng J, Liu Y, et al. Improvement of functional recovery by chronic metformin treatment is associated with enhanced alternative activation of microglia/macrophages and increased angiogenesis and neurogenesis following experimental stroke. Brain Behav Immun. 2014;40:131‐142. [DOI] [PubMed] [Google Scholar]
- 62. Khan SZ, Rivero M, Nader ND, et al. Metformin is associated with improved survival and decreased cardiac events with no impact on patency and limb salvage after revascularization for peripheral arterial disease. Ann Vasc Surg. 2019;55:63‐77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Appendix S2
Appendix S3
Appendix S4
Appendix S5
Data Availability Statement
Data supporting the conclusions of this manuscript are presented in the form of the main or supplemental figures and tables. Raw data supporting the findings of the study and used in the generation of the figures are available on request from the corresponding author.