

Keywords: gluconeogenesis, glycolysis, oxidative phosphorylation, proximal tubule, sodium-glucose cotransporters
Abstract
Renal blood flow represents >20% of total cardiac output and with this comes the great responsibility of maintaining homeostasis through the intricate regulation of solute handling. Through the processes of filtration, reabsorption, and secretion, the kidneys ensure that solutes and other small molecules are either returned to circulation, catabolized within renal epithelial cells, or excreted through the process of urination. Although this occurs throughout the renal nephron, one segment is tasked with the bulk of solute reabsorption—the proximal tubule. Among others, the renal proximal tubule is entirely responsible for the reabsorption of glucose, a critical source of energy that fuels the body. In addition, it is the only other site of gluconeogenesis outside of the liver. When these processes go awry, pathophysiological conditions such as diabetes and acidosis result. In this review, we highlight the recent advances made in understanding these processes that occur within the renal proximal tubule. We focus on the physiological mechanisms at play regarding glucose reabsorption and glucose metabolism, emphasize the conditions that occur under diseased states, and explore the emerging class of therapeutics that are responsible for restoring homeostasis.
INTRODUCTION
Renal tissue is among some of the most intricate and essential in the human body as its functions of filtration, glucose handling, and water balance serve to maintain homeostasis on several fronts. The kidneys receive blood flow that is ∼20% of total cardiac output (1). The journey of fluid through the nephron units begins with the glomerulus, followed by the proximal (convoluted) tubule (PT) where reabsorption of solutes occurs early on to prevent buildup and nephrotoxicity (2, 3). The fluid continues through the loop of Henle, the distal convoluted tubule, and finally the collecting duct, where, through the coordinated processes of reabsorption and secretion, urine is concentrated, acid-base balance is maintained, and sodium and phosphate levels are fine-tuned. Disruptions to any of these processes can be detected in the final urine. The functions of the kidney, and in particular the PT, are diverse and play important roles in the maintenance of homeostasis. In this review, we will home in on just one of these processes—renal glucose handling—and will highlight recent advances in our understanding of glucose reabsorption and metabolism in both health and disease.
THE RENAL PROXIMAL TUBULE
The PT is made up of three types of epithelial cells with subtle yet important distinctions (4). The S1 and S2 PT is found within the outer cortex that is highly convoluted. The S3 cells make up the straight PT and can be found deeper in the cortex heading toward the medulla. Proteomic analysis of the PT revealed that all three segments share the majority of their proteins (5). However, the S1 and S2 segments have a significantly larger abundance of mitochondrial proteins and are said to be more metabolically active. Conversely, the S3 segment is more highly enriched in peptidases and metalloproteases (5). Interestingly, albeit not entirely well characterized, some transcriptome analyses have revealed that there are upward of 10 different clusters of PT cells based on slight differences in protein expression profiles (6).
The renal PT is designed for transport. At the cellular level, the PT is composed of polarized epithelial cells, with a luminal side known as the brush border, and a basolateral membrane facing the interstitial fluid that is adjacent to peritubular capillaries (7). Transcriptome analyses of such cells reveal the brush border, packed with microvilli, as a location of secretion for protective factors and proinflammatory cytokines (8, 9). The brush border greatly enhances total membrane surface area that facilitates transporter abundance on the apical membrane that are responsible for, among others, the reabsorption of glucose, sodium, phosphate, iron, chloride, and bicarbonate (10–12). These cells also contain the highest concentration of mitochondria within the kidney that allows for ATP generation to promote active transport (13). As a result, the PT is designed to reabsorb most of what is filtered and maintains a delicate solute balance both within the urine and the bloodstream. When this balance is impacted by disorders that increase a solute beyond handling capacity, such as the hyperglycemia associated with diabetes mellitus, inflammatory responses can occur.
Historically, study of the PT fell to isolated tubule studies and in vivo and ex vivo experimentation (14–30). More recently, mechanistic studies have been expanded upon with the use of different in vitro cell lines (Table 1). PT cell lines are varied in origin, benefit, and longevity but all are potential research tools depending on the type of project being conducted. It should be noted, however, that none of the available cell lines are known to completely recapitulate the native environment of the PT (32). Researchers should thoroughly characterize their chosen cell line before initiating studies as recent RNA sequencing has revealed a lack of expression of key enzymes/proteins in several of these lines (32). Although the in vitro cell lines do not fully recapitulate all of PT function, studies have also shown that some of the cells can be “PT enhanced” through the addition of flow or shear stress that better mimics the native environment of the epithelium (33–35). In addition to standard in vitro culturing conditions, efforts have been made to develop more PT-like environments. This includes generating “organs on a chip” where cells are grown in a microfluidic chamber and exposed to constant flow to recapitulate the in vivo environment of the kidney (3, 36). This technology has allowed for cells to develop a polarized phenotype complete with a primary cilia and enhanced transport function. Similarly, researchers are also starting to use three-dimensional models that mimic tubule and vascular interactions (37). This model involves co-printing PT epithelial cells alongside vascular endothelial cells within a modified extracellular matrix and has been successfully used to mimic glucose and protein reabsorption as well as cell repair (38). Finally, with the understanding that PT function is intrinsically linked to the rest of the nephron, technology has advanced to generate organoids using nephron progenitor cells (31, 39–41). Human organoids have the power to redevelop all segments of the renal nephron ranging from the glomeruli to the distal convoluted tubule and/or the collecting duct. Depending on precursor cells used, the technology can also incorporate the interstitium and endothelial cells that have the power to influence the physiological functions of the kidney epithelium including the renal PT. All of these model systems have shed light on a variety of pathophysiological conditions including diabetes, inflammation, oxidative stress, and ferroptosis (42–44), and when thoroughly characterized, have the ability to move the renal field forward to promote more mechanistic studies to assist in the analysis of the phenomena discussed later.
Table 1.
Commonly used renal proximal tubule cell lines
| Glucose Transport |
Gluconeogenesis |
Glycolysis |
TCA Cycle |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell Line | Species | Slc5a2 | Slc5a1 | Slc2a1 | Slc2a2 | Pck1 | Fbp1 | G6p | HK | Pfk1 | Pfkp | Pkm | Ldh3a | Ldh3b |
| HK2 | Human | − | − | UNK | UNK | − | − | − | + | + | + | + | + | + |
| OK | Opossum | + | − | UNK | UNK | + | + | + | + | + | + | + | + | + |
| LLCPK1 | Porcine | + | + | UNK | UNK | − | − | − | + | + | + | + | + | + |
| MDCK | Canine | − | − | UNK | UNK | − | + | − | + | + | + | − | + | − |
| NRK | Rat | − | − | UNK | UNK | − | − | − | + | + | − | − | + | + |
Expression data obtained from Takasato et al. (31). TCA, tricarboxylic acid; +, expressed; −, not expressed; UNK, not examined.
PROXIMAL TUBULE GLUCOSE TRANSPORT
Glucose Filtration
Taking a simplistic view, the main function of the kidney is to excrete toxins through the production of urine. This begins at the level of the glomerulus where the interdigitated and selective filtration barrier exists forming a giant sieve surrounding the capillary bed (45, 46). As a result, water and small solutes leave the blood and enter the lumen of the nephron. Included in this ultrafiltrate is glucose, the uncharged small molecule that serves as the major energy source for the human body. If left to flow through the nephron, this molecule would end up in the final urine leading to wasting, anxiety, fatigue, seizures, visual problems, and more. For all of these reasons, glucose must find its way back to the blood and this challenge falls to the first tubular section of the nephron, the renal PT.
Glucose Reabsorption
Virtually all (>99%) of all filtered glucose is recovered by the PT. Fitting with the active transport that occurs within the S1 and S2 segments, most of the glucose is reabsorbed here. There are two main transporters that are responsible for glucose uptake: the Slc2 family that are ion-independent sugar carriers and the Slc5 family that mediates sodium-dependent sugar transport (47–52) (Fig. 1). The Slc5 transporters move glucose into the cell by taking advantage of the sodium gradient that is built because of the basolateral Na+/K+ ATPase. Within the early PT, the 14-transmembrane domain-containing sodium-glucose cotransporter 2 (SGLT2; gene—Slc5a2) sits on the apical membrane. This transporter mediates the reabsorption of >90% of all glucose in a 1:1 stoichiometry with sodium. PT cells do not have the ability to metabolize glucose via glycolysis as they lack the rate-limiting enzyme hexokinase (see glucose metabolism in the kidney more details) (53). Thus, the glucose that enters the cells via SGLT2 exits the cells in a facilitative manner to return to the peritubular capillaries. Within the S1 segment, the facilitative transport of glucose falls to GLUT2 (gene: Slc2a2), a 12-membrane spanning transporter. In the S2 segment, transcript and protein levels in both mouse and human kidneys point toward GLUT1 (gene: Slc2a1) being the major transporter that completes the glucose reabsorption. Although ∼90% of all glucose is recovered in the convoluted tubule segments, SGLT1 (gene: Slc5a1) is found primarily within the later, straight PT where it participates in the remaining glucose recovery in a 2:1 sodium:glucose stoichiometry. From there, the glucose will traverse the basolateral membrane to return to the peritubular capillaries via GLUT1. Upon return to the peritubular capillaries, the newly received glucose can either re-enter circulation or be delivered to the distal tubule where it can serve as a substrate for energy production via glycolysis (54). Under conditions of SGLT2 inhibition, SGLT1 has been shown to partially compensate by reabsorbing close to 20% of luminal glucose (50, 54).
Figure 1.
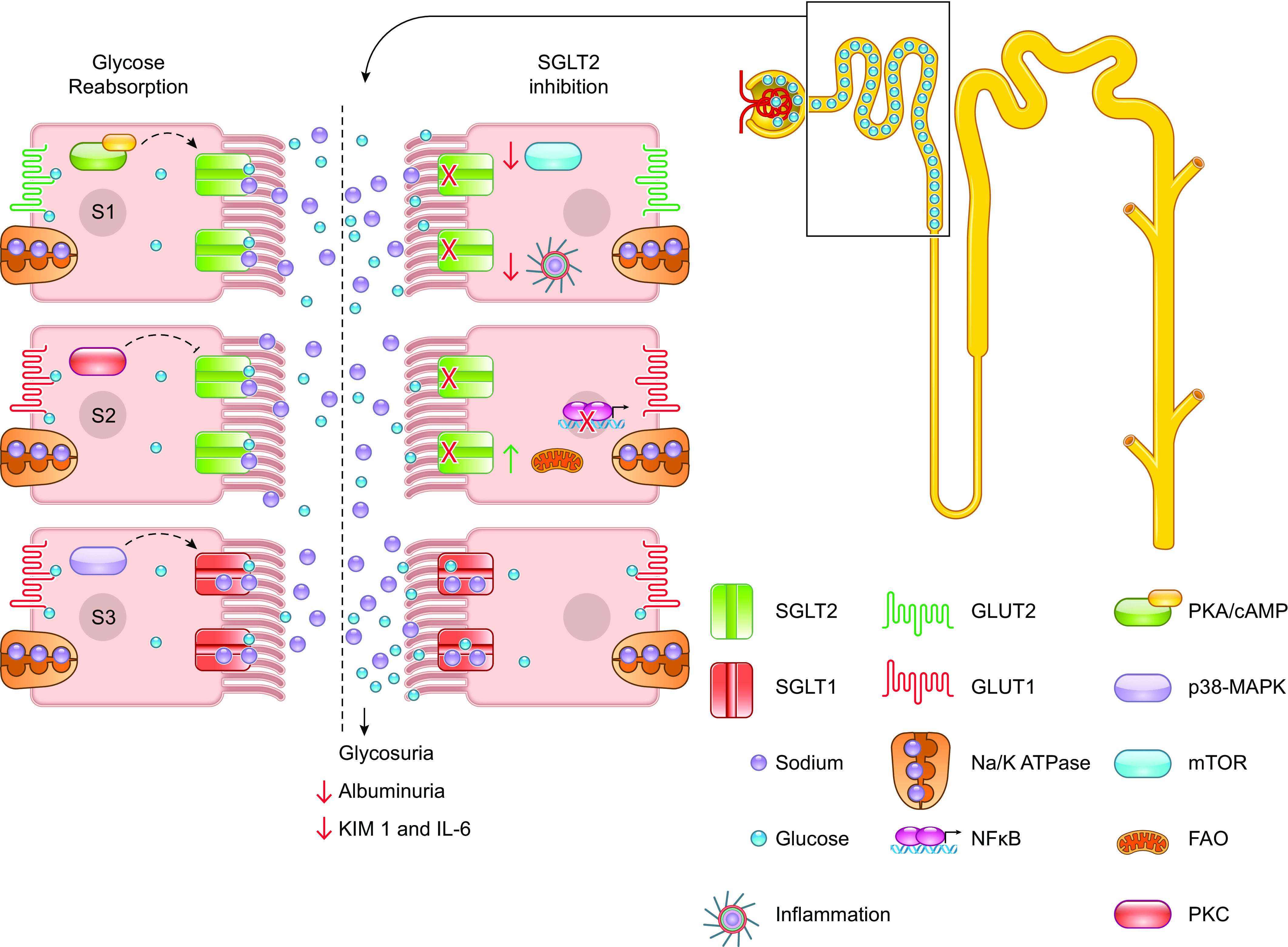
Glucose reabsorption and SGLT2 inhibition. Under healthy conditions (left side of inset), glucose is completely reabsorbed by sodium-glucose cotransporter 2 (SGLT2) and sodium-glucose cotransporter 1 (SGLT1) along the length of the proximal tubule. These transporters are known to be regulated by different signaling pathways and protein kinases depicted in each of the epithelial cells. On the right side of the inset, this process is being blocked using SGLT2 inhibitors. Consequently, this promotes glycosuria and has some beneficial consequences on inflammation and renal injury. FAO, fatty acid oxidation; mTOR, mammalian target of rapamycin.
Although it is generally agreed that the S1 and S2 mediate most of the glucose reabsorption via SGLT2, there is some discrepancy in transporter localization based on methodology used, model system examined, and the sex of the organism. The “gold standard” for localization is the use of validated protein-specific antibodies. Using these tools, SGLT2 was found in the convoluted PT within mouse and specifically within the S1 and S2 segments in human kidney (50, 52). For SGLT1, in the human it appears to be confined to the S3 segment with no overlapping stain noted with SGLT2 (52). In the rat, there is a gradient of detection that increases in intensity from S1 → S2 → S3, and in the mouse, it is primarily found in the straight PT (50, 51). One caveat to antibody staining is the loss of detection in low-expressing nephron segments. With the advent of single-segment and single-cell transcriptomics, more refined localization patterns are beginning to emerge. Using these methodologies, Slc5a2 gene expression (SGLT2) was overwhelmingly expressed in the S1 segment of mouse and within the S1 and S2 segments of rat (55, 56). There was also some detection in the thick ascending limb and the distal convoluted tubule. For Slc5a1, there was highest expression within the murine S3 PT but also in the thick ascending limb (55). In the rat, gene expression was noted in both the S2 and S3 segments as well as the thick ascending limb (56). Although the nuances of transporter expression vary based on sample, method, and sex, what is clear is that collectively, SGLT1 and SGLT2 are said to account for all glucose reabsorption in the PT (49).
Model for Glucose Transport
SGLT1 was first cloned from the intestine in 1987 by Ernest Wright and his team (57). Over the next 30 years, they and others would go to identify and characterize the class of SGLTs in multiple species and begin to elucidate the transport pathway as it exists on the molecular level. Using Xenopus oocytes, electrophysiology was used to first elucidate the kinetics of sodium and glucose transport (58, 59). In the absence of substrate, the SGLTs exhibit a sodium leak; in the presence of glucose, sodium was thought to bind first when the transporter exists in its outward facing conformation. This subsequently allows for sugar binding that traps the glucose molecule within the protein. A conformational change occurs to switch the orientation of the transporter to expose the inward facing gate. From there, the glucose molecule is thought to be released first into the cytoplasm followed by the sodium ion. Using enhanced molecular modeling and crystallization, more recent studies have suggested that the sodium ion can disengage before glucose (60). In the most recent model analysis, the events of the molecule exit appear to be unordered (60, 61) that may have therapeutic implications (refer to glucose handling in disease and therapeutics). In either case, the binding and transport of sodium and glucose via SGLT1 and SGLT2 allow for the reabsorption of these key solutes and greatly impact homeostasis. This process is regulated by the transport maximum of these proteins. On average, the maximal transport capacity for glucose is 300–350 g/min for healthy females and 430–500 g/day for males (54). If blood glucose levels exceed this maximal transport capacity, glucose will escape the PT and be excreted in the final urine. This condition, known as glycosuria, is a hallmark characteristic of uncontrolled diabetes (62). It is also observed upon inhibition of SGLT2, either through genetic mutations as observed in renal familial glycosuria or through intentional inhibition (63), the latter of which will be described in more detail later.
Regulation of Glucose Transporters
With the emerging appreciation for the role that the renal glucose transporters play in both health and disease, it is no wonder that recent literature has turned its attention to determining whether transporter expression and/or regulation varies under different pathological conditions. Thus far, no definitive answer exists: several studies report that under conditions of hyperglycemia, SGLT1 and/or SGLT2 expression is decreased. In contrasting studies, others find that mRNA or protein expression is increased (54, 64–67). These conflicting reports are confounded by using different expression analysis, model systems, and modes of diabetes induction. Recently, in the largest clinical analysis involving human kidney samples from healthy controls and patients with diabetic kidney disease and glomerulonephritis (NEPTUNE study), the authors found that mRNA expression of SGLT2 is decreased under states of diabetic kidney disease whereas there were no changes in SGLT1 expression relative to the healthy control kidneys (66). This decreased expression may be a mechanism by which the renal PT protects itself against glucotoxicity (68). Although there is no clear answer as to how blood glucose levels affect SGLT expression, it can be appreciated that these renal transporters are not static. Indeed, their expression profiles are altered based on changing intracellular and extracellular conditions.
SGLT1 and SGLT2 are expressed on the apical plasma membrane, and like all newly synthesized membrane transporters, these proteins must traverse the biosynthetic pathway to find its home on the membrane. What governs its trafficking, and whether or not their expression is regulated by posttranslational modifications and cellular signaling, remains to be determined. Recent reports suggest that membrane expression of both SGLT1 and SGLT2 are modulated by the second messenger cAMP (Fig. 1). Under conditions of increased cAMP, SGLT2 membrane expression appears to be enhanced in cell culture models. This aligns with previous reports that cAMP enhances sodium-glucose transport (69). cAMP is derived from ATP by adenylyl cyclase, a membrane-bound enzyme that is commonly activated by members of the G protein-coupled receptor (GPCR) family. GPCRs are the largest gene family in the human body and are also the most therapeutically targeted class of receptors for drug discovery (70, 71). Indeed, we recently identified an olfactory receptor, a member of the GPCR family, that appears to modulate expression of murine SGLT1 and SGLT2 within the kidney (72–74). The “drugability” of this family suggests that indirect modulation of these glucose transporters may be possible using GPCR agonists and/or antagonists.
Downstream of cAMP are protein kinases; most prominently, cAMP activates protein kinase A (PKA) that in turn phosphorylates a host of other cellular factors to regulate gene expression and protein trafficking. PKA has also been shown to increase cellular glucose uptake and both SGLT1 and SGLT2 expression (66, 75–77). In Chinese hamster ovary cells, PKA activation has also been shown to promote phosphorylation of SGLT1 that promotes greater transport affinity for glucose when it is localized to the apical membrane (75). Apart from PKA, other downstream signaling pathways have been implicated in SGLT1 and SGLT2 expression (Fig. 1). Older studies have highlighted protein kinase C (PKC) in their regulation with studies conducted in Xenopus oocytes indicating a reciprocal regulation between PKA and PKC (78, 79). cAMP is also known to activate components of the mitogen-activated protein kinase (MAPK) pathway, and this too has been linked to SGLT expression and localization changes. Recently, inhibition of p38 MAPK has been shown to decrease membrane expression of SGLT1 and SGLT2 (77). Moreover, a study by Lee et al. (80) showed that SGLT1 and SGLT2 are expressed in caveolin-1-expressing lipid rafts. Their localization appears to be driven by cAMP-induced activation of PKA and p38 MAPK, and disruption of lipid rafts decreases membrane expression (80). Intriguingly, lipid rafts are cholesterol-rich membrane compartments. Given the link between cholesterol and glucose, it is tempting to investigate whether or not the lipid raft containing SGLTs are altered under conditions of impaired glucose and cholesterol homeostasis. However, the physiological relevance of this study remains in question. Healthy PT epithelial cells do not contain caveolae, indicating that the localization to caveolin-expressing lipid rafts may be an artifact of cell culture (81, 82). Further investigation is required to determine if the SGLTs are found in non-caveolin detergent resistance membranes that have been noted on the brush border of epithelium (83).
Other Glucose Transporters in the Kidney
Although SGLT1, SGLT2, GLUT1, and GLUT2 function as the primary glucose transporters involved in PT reabsorption, several other Slc2 and Slc5 transporters exist within the kidney including in the PT itself. There are a total of 13 GLUT transporters in mice and 10 in humans. Although GLUT1 is found in the PT (S2, S3), its highest levels of expression are found along the distal portion of the nephron. Therefore, it is believed that GLUT1-mediate glucose uptake facilitates delivery to epithelial cells that can metabolize glucose via glycolysis (84). Except for the PT, GLUT4 mRNA has been found throughout the nephron including within the glomerulus, ascending limb of the loop of Henle, the distal tubule, and collecting duct. However, gene expression does not always translate to protein, and this appears to be the case for GLUT4, at least in the mouse where proteomic analysis revealed little to no protein product (85). Whether it is expressed in the kidney or not, it is appreciated that GLUT4 is an insulin-regulated glucose transporter. Finally, GLUT5 has been observed on the apical membrane of the S3 segment of the PT where it serves as a fructose (not glucose) transporter. Once inside the cell, fructose can be converted to glucose and then effluxed via GLUT1.
There are a total of 12 Slc5 transporters found within the human transcriptome. Although SGLT2 is confined to the renal PT, SGLT1 has been found along the thick ascending limb and the macula densa (85). SGLT8, -9, -10, and -11 have all been found within the S2 and S3 segment of the PT although functional analysis is lacking (85). In addition, renal expression has been described for SGLT3, -4, and -5. SGLT3 does not appear to transport glucose itself but does increase intracellular sodium in the presence of the sugar (86, 87). Thus, it may serve as a glucose sensor (as opposed to a transporter). SGLT4 has a higher affinity for mannose and SGLT5 serves as a fructose transporter (85).
GLUCOSE METABOLISM IN THE KIDNEY
Gluconeogenesis in the Proximal Tubule
The healthy PT has limited ability to perform glycolysis (53). Thus, beyond its role in glucose reabsorption, the PT also contributes to glucose homeostasis through the production of glucose via gluconeogenesis. Although the kidney only contributes around 10% of the body’s total gluconeogenesis under postprandial conditions, this proportion reaches around 40%–50% in fasting conditions. In fact, the kidney’s capacity for gluconeogenesis exceeds that of the liver when normalized to tissue weight (88). In short, gluconeogenesis is a metabolic pathway that builds glucose from noncarbohydrate precursors such as lactate or glycerol (88) (Fig. 2). In the PT, where gluconeogenesis is highly active, the cells largely rely on the amino acid glutamine as its precursor (89). These precursors are used to create pyruvate that in turn is converted into glucose through a series of additional reactions that ultimately concludes with the dephosphorylation of glucose 6-phosphate by glucose 6-phosphatase (G6P). Pyruvate and other precursors can also be converted to oxaloacetate; however, oxaloacetate is also useful for several other cellular metabolic pathways, so its subsequent conversion to phosphoenolpyruvate by the rate-limiting enzyme phosphoenolpyruvate carboxykinase (PEPCK) is what commits this substrate for use in gluconeogenesis (88). Transcriptomic analysis of rat kidneys suggests that these crucial gluconeogenic enzymes exhibit greater mRNA expression in all three PT segments than in any other region of the nephron, suggesting that the PT may be uniquely situated to perform gluconeogenesis in the kidney (90). Other studies have identified the greatest PEPCK mRNA expression specifically in the most distal segment of the PT that approaches the medulla (S3) yet found that mRNA expression could increase in the other segments under stress conditions like metabolic acidosis (88), further documenting the PT’s capacity for gluconeogenesis.
Figure 2.
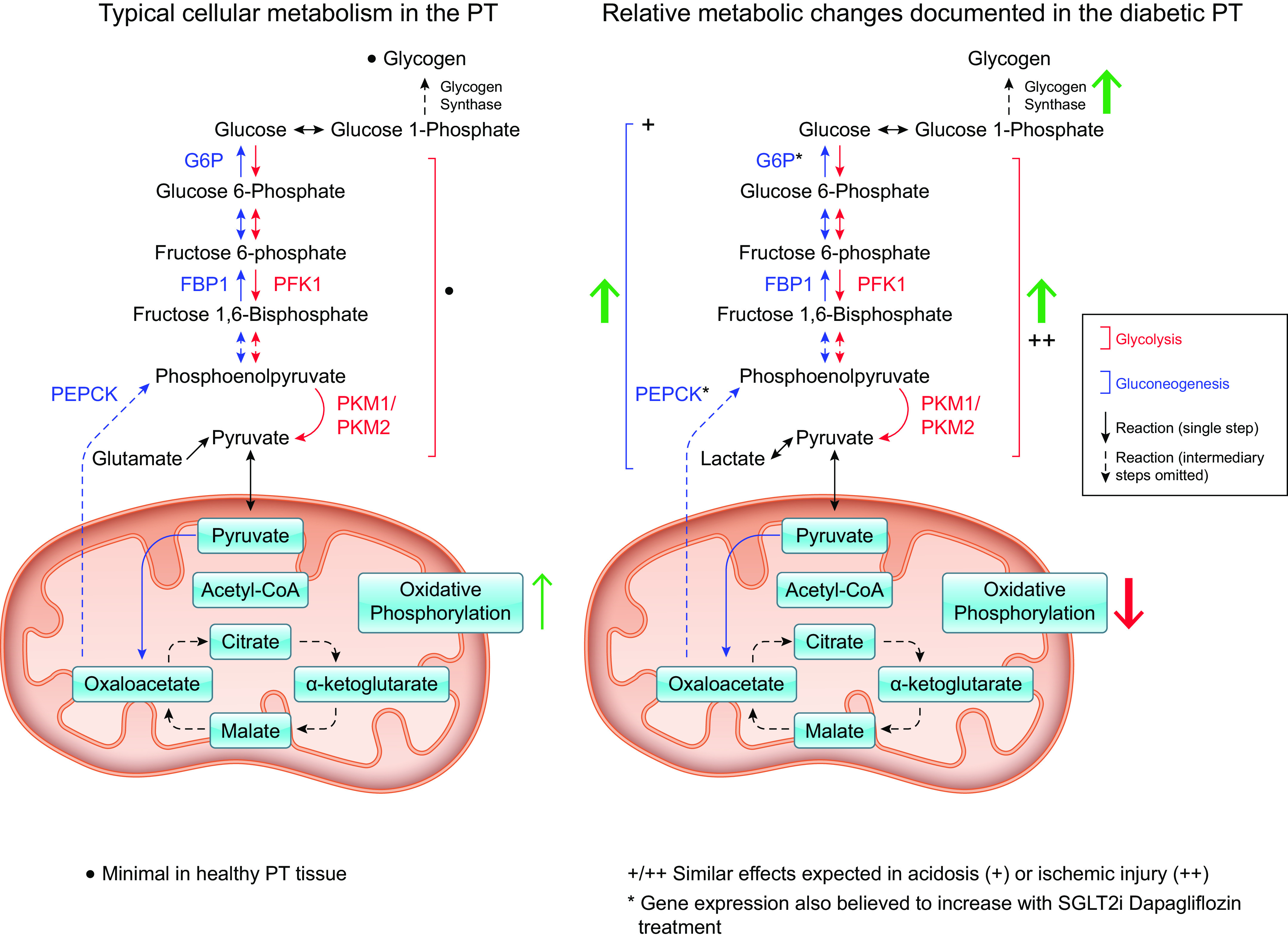
Major metabolic pathways within the proximal tubule (PT). Under healthy conditions (left), gluconeogenesis (blue arrows/enzyme names) is the prevalent metabolic pathway performed in the renal proximal tubule. Glycolysis (red arrows/enzyme names) is minimal. Under conditions of diabetes (right), the metabolic processes within the proximal tubule are altered. Green arrows indicate pathways that are increased while red arrows indicate downregulation. G6P, glucose 6-phosphatase; PEPCK, phosphoenolpyruvate carboxykinase; PFK1, phosphofructokinase 1; PKM1/2, pyruvate kinase muscle isoform 1/2.
Insulin signaling contributes to the modulation of gluconeogenic flux. In the fed state, when blood glucose availability is high and serum insulin is increased, expression of rate-limiting gluconeogenic enzymes like PEPCK and G6P are found to be downregulated in the renal cortex. In HK-2 cells (Table 1), insulin signaling resulted in phosphorylation of Akt and the forkhead box transcription factor 1 (FoxO1), which is associated with diminished gluconeogenic gene expression (89). Treatment with Akt inhibitors or Akt2 (but not Akt1) knockdown abrogated the ability of insulin to inhibit gluconeogenesis in the PT, demonstrating the vitality of Akt2-mediated signaling in the capacity of insulin to downregulate glucose synthesis. Furthermore, inhibition of mTORC1 (mammalian target of rapamycin) or mTORC2 via knockdown or rapamycin treatment was also demonstrated to abrogate the inhibition of cortical gluconeogenesis in the rat kidney in vivo; this effect was not observed in the liver, suggesting that the reliance of insulin signaling on the mTORC1/2 pathway is tissue specific (89). Interestingly, glucose itself appears to inhibit gluconeogenic gene expression by deacetylating peroxisome proliferator-activated receptor γ coactivator 1-α (PGC1 α), which contributes to FoxO1 activation; this glucose-mediated inhibition was reversed by phlorizin treatment to dampen renal glucose reabsorption (89). Conversely, acidosis is believed to activate renal gluconeogenesis, and inhibition of vacuolar H+-adenosine triphosphatase (H+-ATPase) activity via bafilomycin treatment successfully suppressed expression of PEPCK and G6P in diabetic rats. Baseline H+-ATPase activity is elevated in diabetic rats, yet inhibiting this ATPase resulted in hypoglycemic effects (seemingly independently of insulin signaling), presenting inhibition of H+-ATP as an interesting therapeutic direction in the treatment of hyperglycemia in diabetes (91).
Glycogen Regulation in the Proximal Tubule
The high mRNA expression of the aforementioned gluconeogenic enzymes throughout the three PT segments coupled with the absence of detectable glycogen synthase (GS) mRNA in the rat PT suggests that the glucose produced in the kidney is almost entirely maintained as free glucose (i.e., for utilization in the cells or release into the bloodstream) rather than synthesized into glycogen polymers (90). However, although the liver and muscle are primarily responsible for large-scale glycogen storage, the kidney maintains much smaller stores of glycogen under certain conditions. Healthy kidney tissue demonstrates negligible glycogen accumulation even in the fed condition, when hepatic GS is typically heightened under insulin signaling. Insulin activates enzymes like GS to promote polymerization of glucose monomers for storage while inhibiting enzymes like G6P whose activity consumes glycogenic substrates (92). When glycogen synthesis is active, this process enables metabolites like glucose 6-phosphate to be shunted toward a storage state until the resulting glycogen polymers are later broken down to mobilize tissue resources and release free glucose. However, although this process is minimal in healthy PT tissue, severe diabetes is sometimes associated with notable renal glycogen stores (93, 94) (Fig. 2). Although this accumulation first occurs in non-PT regions, it is observed in the PT later in the progression of diabetic nephropathies (in diabetic rats, approximately half of the PTs exhibited glycogen granules around 6 mo after challenge with alloxan, a known toxin to the pancreatic β cells) (93, 94). In insulin receptor substrate 2 (IRS2) knockout (KO) studies, which model diabetic phenotypes like insulin resistance and hyperglycemia, treatment with the antidiabetic agent sodium tungstate (NaW; an inorganic salt shown to mimic insulin function) resulted in surprising increases in glycogen accumulation in the renal tubules (94). This suggests that IRS2 may be involved in suppressing glycogen accumulation in the PT, potentially via ERK1/2 regulation. Indeed, the ERK1/2 pathway was shown to be activated in the PT of IRS2-KO mice (94). Furthermore, although gluconeogenesis is upregulated in diabetes (as previously discussed), NaW inhibits G6P, which catalyzes the final gluconeogenic reaction that converts glucose 6-phosphate into free glucose (94). When this final gluconeogenic step is inhibited, accumulating glucose 6-phosphate may be shunted toward glycogen storage instead (92). This metabolite is a positive regulator of glycogen synthase, which catalyzes the polymerization of glycogen (94). However, although its upregulation in diabetes may contribute to renal dysfunction, glycogen stores seem to exert protective effects on the kidneys in the context of ischemia. The transcription factor hypoxia-inducible factor (HIF), HIF-1α, is regulated by oxygen-dependent posttranslational hydroxylation by prolyl-hydroxylase domain (PHD)-containing enzymes and becomes stabilized as oxygen concentrations decline (95). This process can stimulate glycogen storage via upregulation of associated genes (96). When prolyl hydroxylase is inhibited (e.g., via enarodustat), resulting in greater HIF activation, renal PT epithelial cells (in culture) demonstrated increased glycogen storage; these cells exhibited improved survival following exposure to oxygen-glucose deprivation and lower levels of reactive oxygen species. These protective effects were abolished when glycogenesis was inhibited (96) suggesting that glycogen stores may be helpful in the PTs under conditions of fuel deprivation in which the kidney cannot sustain ATP needs via typical oxidative metabolism but has also depleted the free glucose stores necessary for glycolysis (97). Furthermore, inhibition of glycogen synthase kinase-3β (GSK3β)—which phosphorylates and inactivates glycogen synthase and therefore glycogenesis—is linked to improved tubular regeneration and protection against nephrotoxic injury and renal inflammation (98–101). GSK3β has myriad impacts on cellular metabolism and kinase signaling, so the impacts of its inhibition may not stem exclusively from increased glycogen synthesis, but the implications of glycogen storage in renal PT tissue present an interesting avenue for further research into the glucometabolic profile of the nephron in health and disease.
ATP Production in the Proximal Tubule
Meanwhile, in contrast to its role in glucose production, the PTs have limited capacity for glucose catabolism and rely mostly on mitochondrial oxidative metabolism to meet the high energy demands necessary to fuel their transport functions; the PTs (particularly the cortical segments S1 and S2) exhibit lower glycolytic flux in comparison to other sections of the nephron and only perform minimal glycolysis within fully differentiated cells (97). Although other kidney cell types are more reliant on glucose for cellular energy production (especially because medullary renal tissue receives less oxygen, limiting aerobic metabolism), the high vascularization and abundance of mitochondria within the PT enable greater utilization of more efficient energy producing pathways like oxidative phosphorylation. Models of oxygen consumption (102) demonstrate that the earlier PT segments S1 and S2 experience greater oxygen tension, supporting greater reliance on aerobic metabolic pathways (e.g., oxidative phosphorylation), whereas the later S3 segment (found within the outer renal medulla) exhibits lower oxygen consumption. This aligns with rat kidney transcriptome data demonstrating low gene expression of several rate-limiting glycolytic enzymes throughout the entire PT with a slight uptick in the enzyme pyruvate kinase (Pkm) within the S3 (90). In addition, although PT lines exhibit expression of glycolytic enzymes (Table 1), they shift away from this metabolic pathway (e.g., lower enzyme expression and increased flux through aerobic pathways) under conditions of mechanical stimulation via orbital shaking, which mimic the increased shear stress and oxygenation that might be experienced under conditions of greater renal blood flow in the body (35). This low glycolytic flux within the PT may also be beneficial based on their crucial resorptive function. Fluctuations in blood glucose (such as after a meal) translate into fluctuations in filtered glucose levels in the PT lumen and therefore in the PT cells that conduct its reabsorption. Thus, it is theorized that utilization of alternative metabolic pathways may provide more consistency in meeting metabolic needs in the PT than would glucose alone (35). Under healthy circumstances, then, the PTs rely mostly upon oxidative metabolism of various nutrients like pyruvate and fatty acids for the bulk of their ATP production to sufficiently fuel their cell transport functionality (35, 97). Molecules that can be moved across the mitochondrial membranes can be metabolized via various reactions such as the tricarboxylic acid cycle, electron transport chain, or β-oxidation to produce much greater ATP yields; for example, under aerobic conditions, a single molecule of the fatty acid palmitate (one lipid energy source present in the PT) can be catabolized to produce over 100 ATP molecules (97).
Thus, glycolysis is likely most crucial in the context of kidney injury in which ischemic and anoxic conditions may substantially impair typical oxidative metabolic processes by damaging PT cells’ mitochondria or restricting their access to oxygen. Cortical tubules are better oxygenated than medullary regions of the kidney; as such, in acute kidney injury, cortical mitochondria appear better able to adapt to conditions of insufficient renal blood flow and oxygenation. This is potentially partially mediated by HIF-1α (95). HIF-1α regulates expression of several glycolytic enzymes and pyruvate dehydrogenase kinase (PDK). The stabilization of HIF-1α further inhibits PPARα and acyl-CoA dehydrogenases further leading to a reduction in fatty acid oxidation (FAO). Upregulation of renal HIF subunits HIF-1α and HIF-2α has been documented in models for both acute and chronic kidney injury (103, 104). HIF activation is demonstrated to attenuate renal injury and protect adaptive regulation of glomerular filtration rate, and stimulates a variety of target genes including erythropoietin and glucose transporter-1 (GLUT-1) (104); this contributes to a metabolic shift toward promoting glycolytic ATP production while compensating for the diminished oxidative metabolism observed in renal injury (104, 105). Elevated expression of glycolytic enzymes such as phosphofructokinase 1 (PFK1) and pyruvate kinase muscle isoform 2 (PKM2) has been replicated in multiple mouse models for acute renal injury, and increased lactate and pyruvate in the renal cortex provide evidence that these altered expression patterns translate into heightened glycolytic activity in the tubules (106). Interestingly, following ischemic-reperfusion injury in the murine kidney, normally regenerating PT tissue demonstrated a return to typical reliance on oxidative metabolism within 2 wk of recovery (106). One contributor to this restoration may be activation of Akt (which has been demonstrated to stimulate mitochondrial activity in cultured PT cells); increasing the activity of this mitochondrial protein kinase was associated with restoration of mitochondrial function and oxidative metabolism in injured cultured PT cells (mediated by the application of the nephrotoxicant dichlorovinyl cysteine (107). This restored activity of several mitochondrial complexes (e.g., complexes I and III) that were attenuated by PT injury, promoting improved electron flow to strengthen mitochondrial oxidative metabolism (107).
However, tissue exhibiting unsuccessful regeneration of PT cells presented with increasingly severe glycolytic shift (106). Persistent increases in glycolytic flux in chronic kidney disease is associated with exacerbated fibrosis in chronic kidney disease, and inhibition of glycolysis via PKM inhibition resulted in reduced tubular damage in the fibrotic murine kidney following unilateral ureter obstruction surgery (108); thus, while glycolysis may represent a protective compensatory mechanism when the injured PT are unable to achieve energetic demand via oxidative metabolism, its chronic activation may be maladaptive to tubular tissue. Increased glycolytic intermediates coupled with dampened mitochondrial function results in the conversion of glucose into fructose, which can then be phosphorylated by fructokinase to produce uric acid; fructokinase knockout mice demonstrated reduced oxidative stress, ATP depletion, and renal inflammation following ischemic acute kidney injury, suggesting that this accumulation of glycolytic metabolites may contribute to the renal stress observed following glycolytic shift (109).
GLUCOSE HANDLING IN DISEASE AND THERAPEUTICS
Modulation of Glucose Handling
Advancements in therapeutics have allowed for novel strategies to target disorders that affect PT glucose handling. As mentioned previously, diabetes, and more specifically, type 2 diabetes (T2D) is one of the most common disorders involving glucose handling. As of recently, SGLT2 inhibitors (SGLT2i) have been one of the standards of treatment for glycemic control in type 2 diabetics. Specifically, SGLT2i inhibit renal glucose reabsorption with a binding preference to SGLT2 that is 200–2,500 times greater than SGLT1 (95, 110, 111). SGLT2i limit glucose reabsorption and subsequently result in excretion of glucose, lowering blood glucose levels (Fig. 1). SGLT2i may also lower albuminuria by restoring the charge/size selectivity of the glomerular basement membrane.
Physiological Consequences of SGLT2 Inhibitors
At the pharmaceutical level, SGLT2i are known as the -gliflozin class with four currently approved by the Food and Drug Administration (FDA): canagliflozin, dapagliflozin, empagliflozin, and ertugliflozin (Table 2). Each of these -gliflozin class pharmaceuticals is orally available with circulating half-lives of once per day at therapeutic doses. Clinical manifestation of SGLT2i treatment after 12 mo showed reduction in glycated hemoglobin (HbA1c) of 0.5%–0.9% (5–9 mmol/mol). Bodyweight reduction was also seen along with a systolic blood pressure reduction of 2.5–5.0 mmHg (110, 112, 113). There are also increased renal benefits with the treatment of canagliflozin resulting in a 30% reduction in composite renal failure. Canagliflozin can now be used to treat diabetic kidney disease (DKD) with an estimated glomerular filtration rate (Egfr) of more than 30 mL/min/1.73 m2 (110). Overall, there are significant beneficial changes to the cardiovascular system and renal system after extended treatment with an SGLT2i reducing both intraglomerular pressure and systolic blood pressure (114). This can be seen in Table 2 as 50% of SGLT2i drugs are now approved to treat patients with diabetes with heart failure with reduced ejection fraction (HfrEF) alongside those with chronic kidney disease (CKD) and DKD (115, 116).
Table 2.
Food and Drug Administration-approved SGLT2 inhibitors
| SGLT2 Inhibitor | SGLT2/SGLT1 IC50 Selectivity | Half-Life, h | Nondiabetic Treatment Approvals |
|---|---|---|---|
| Empagliflozin | ∼2,500-fold | 12.4 | Approved to treat HFrEF and HFpEF with an eGFR > 20 |
| Dapagliflozin | ∼1,200-fold | 12.9 | Approved to treat HFrEF, and CKD with an eGFR > 30 |
| Canagliflozin | ∼250-fold | 13.1 | Approved to treat DKD with an eGFR > 30 |
| Ertugliflozin | ∼2,000-fold | 16.6 | Not Approved to treat patients with an eGFR of <60 |
CKD, chronic kidney disease; DKD, diabetic kidney disease; eGFR, estimated glomerular filtration rate; HfrEF, heart failure with reduced ejection fraction; IC50, half-maximal inhibitory concentration; SGLT, sodium-glucose cotransporter.
Blockage of SGLT2, which accounts for >90% of glucose reabsorption, shifts the glucose load to SGLT1 within the S3 segment of the PT. This reduces overall glucose reabsorption by 50% and may induce antinatriuric effects because of sodium-glucose transport ratios of 1:1 and 2:1, respectively (117). As seen in canagliflozin and empagliflozin studies, the decrease in circulating glucose results in a loss of calories and subsequent weight loss of patients (117, 118). Overall, the decrease in reabsorption of glucose protects the body from excessive circulating glucose and high HbA1c (111). However, even with the inhibition of SGLT2, Vallon et al. identified that empagliflozin attenuates GLUT1/2 overexpression in a type I diabetic (T1D) murine models. T1D has upregulated SGLT2 expression while also decreasing SGLT1 expression. Empagliflozin further upregulates SGLT2 expression in a wild-type (WT) murine model and downregulates SGLT1 expression in diabetic mice (113, 117). SGLT2i has also been shown to decrease oxidative stress (119–121). This finding has been predicted to be a consequence of reduced oxygen consumption within the PT, although recent model analysis suggests otherwise (102). Furthermore, an empagliflozin study shows that there are reversible changes to eGFR after long-term use of an SGLT2i (110, 112). Identifying that SGLT2i, specifically empagliflozin, may initially alter eGFR during long-term treatment, but after discontinuation of the drug, the changes are reversible.
Data suggest dapagliflozin and empagliflozin beneficially alter renal inflammation in the PT. With decreased inflammation, there is also seen to be a reduction in urinary albumin excretion, decreasing intraglomerular pressure and improves eGFR with treatment of empagliflozin and dapagliflozin (122). Dapagliflozin’s ability to decrease intraglomerular pressure and subsequent reduction in eGFR can explain the albuminuria changes (123). Furthermore, SGLT2i decreases mTOR1 activity, a hallmark of tubule fibrosis and decreases renal function (124, 125). It has also been determined that the anti-inflammatory effects of dapagliflozin are independent of the SGLT2 inhibition. Dapagliflozin has been shown to decrease the amount of phosphorylated NF-κB p65, a common phosphorylated inflammatory marker of atherosclerogenesis (126). Dapagliflozin and empagliflozin have been proven to inhibit TLR-4/NF-κB axis activation and overexpression (113). SGLT2i have shown to provide structural improvements over time in the kidney and cardiovascular system and even after discontinuation of drug treatment as seen in an empagliflozin and dapagliflozin study with patients with T2D (110, 123, 126).
Dapagliflozin has also been shown to significantly decrease urinary KIM-1 and IL-6 excretion in patients with T2D compared with placebo (123). KIM-1 is a biomarker for hypoxic injury in the PT. Patients with diabetes have increased expression of SGLT2 in the PT. Reabsorption of organic soluble molecules and electrolytes like glucose and sodium require a large amount of oxygen production. The increased glucose reabsorption coupled with oxygen production has been linked to the induction of hypoxic injury and a subsequent expression of KIM-1. Therefore, a decrease in KIM-1 with treatment of dapagliflozin identifies a reduction in PT injury. IL-6 is an inflammatory cytokine associated with the progression of diabetic kidney disease with increased excretion of this cytokine because of increased albumin leakage in the PT. Dapagliflozin and canagliflozin have been seen to alleviate PT inflammation, decreasing the albumin leakage and subsequent decrease of IL-6, tumor necrosis factor receptor 1 (TNFR1), matrix metalloproteinase-7 (MMP7), and fibronectin FN1 excretion and plasma concentration (123, 127). Empagliflozin has also been shown to normalize elevated levels in diabetic mice of cytokine expression including CCL2, CD14, IL-6, and tissue inhibitor of metalloproteinase-2 (TIMP2) while having no change of expression in WT mice treated with the same dose of empagliflozin (113).
SGLT2i and Metabolism
As systemic glucose production is increased by up to 300% in diabetes (particularly in T2D) (48)—with substantial contributions from both the kidney and liver—targeting excessive increases in renal gluconeogenesis may present one promising way to reduce hyperglycemia. Although potential alterations to renal gluconeogenesis under conditions of SGLT2i have not been well described to date, initial PT cell line and in vivo mouse studies have identified upregulated renal gluconeogenesis following treatment with dapagliflozin (128, 129). This was linked to increased expression of gluconeogenic enzymes like PEPCK and G6P as well as changes to associated regulators [such as increased PGC-1α (128)] and depleted phosphorylated FoxO1 (129), potentially partially opposing the antihyperglycemic impacts of dapagliflozin. T2D is also associated with renal inflammation, whereas inhibition of key regulatory proteins like NF-κB (which promotes transcription of inflammatory molecules like TNF-α and macrophage inflammatory protein 1 alpha MIP-1 α) also resulted in reduced PEPCK expression in the renal cortex (130). Ultimately, typical insulin-mediated suppression of gluconeogenesis in the PT is dysregulated in the context of diabetes, whereas alterations in glucose reabsorption and renal inflammation further modulated gluconeogenic activity in the renal cortex.
Studies have also shown that SGLT2i, specifically dapagliflozin, abrogates the increase in HIF-1α typically seen in patients with DKD. This prevents a metabolic shift toward glycolysis and instead leads to an improvement in FAO through this same mechanism (95). Taken together, these data suggest that attenuation of glycolysis could play a role in the efficacy of SGLT2is in protecting tubular structure and function in the context of diabetes.
Type 1 Diabetes
Although the majority of studies focus on SGLT2i in the context of T2D, data from the DEPICT and TANDEM clinical programs have shown evidence that SGLT2i can be used to treat type 1 diabetes. To date, SGLT2i are now used as a treatment for T1D in Europe, but not in the United States, as dapagliflozin and sotagliflozin (an SGLT1 and 2 inhibitor that has 20:1 binding preference to SGLT2) have both been approved to treat patients with poor control on insulin and a body mass index (BMI) greater than 27 kg/m2 (110). Use in patients with a BMI of <27 kg/m2 is limited because of the increased risk of diabetic ketoacidosis. There have been positive data through meta-analysis that SGLT2i in patients with type 1 diabetes improves glycemic control and weight loss without increasing hypoglycemia risk (131). However, caution should be taken given the added complications that T1D presents with (glucose loss through ketoacidosis and required insulin dosing).
GLP-1 Receptor Agonists Compared with SGLT2i
Apart from SGLT2is, glucagon-like protein 1 (GLP-1) receptor agonists (GLP-1 RAs) have also emerged as a new therapeutic option for patients with T2D (132). GLP-1 is a gut-derived peptide that is released in response to glucose and fat. Acting as an incretin, GLP-1 promotes glucose-stimulated insulin secretion. Outside of the pancreas, GLP1 receptor is also found in other tissues including the kidney, and more specifically, within the PT (133). When it comes to diabetic treatment options, both SGLT2i and GLP-1 RAs have similar reductions in HbA1c (1.4%–1.6%) (134), although there are differing reports about reductions in body weight and their effectiveness in different patient populations (110, 134). With regard to renal function, GLP-1 RAs have been shown to increase natriuresis through the inhibition of sodium hydrogen exchanger 3 (NHE3) within the PT (133). Additional renoprotective mechanisms are linked to an increase in renal blood flow, a decrease in oxidative stress and blood pressure, and a modulation of atrial natriuretic peptide and renin-angiotensin-aldosterone signaling (133). Ongoing studies are evaluating the usefulness and safety of combination SGLT2i and GLP-1 (110) as yet another approach to combating T2D and renal complications.
CONCLUSIONS
Glucose is a small sugar that is widely used by all tissues in the human body. The maintenance of glucose homeostasis falls to the kidney, and specifically to the PT. The PT is particularly well suited to control this process as it does not itself catabolize the molecule. This allows for tight regulation of reabsorption without concern that its reabsorption will only be used to drive minimal ATP generation through glycolysis. Although the transporters involved in glucose reabsorption are well established, the appreciation of the kidney as a metabolic tissue is just being realized. How these processes influence, and are impacted by, pathophysiological changes are currently being explored. Finally, with the identification of both therapeutics and physiological model systems, the future research of glucose homeostasis is just beginning.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R03-DK123546 (to B. D. Shepard) and the Dekkers Endowed Chair in Human Science (to B. D. Shepard).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
B.D.S. and Z.S.H. prepared figures; Z.S.H., J.N.L.C., E.N.D.C., and B.D.S. drafted manuscript; Z.S.H., J.N.L.C., E.N.D.C., and B.D.S. edited and revised manuscript; Z.S.H., J.N.L.C., E.N.D.C., and B.D.S. approved final version of manuscript.
REFERENCES
- 1. Dalal R, Bruss ZS, Sehdev JS. Physiology, renal blood flow and filtration. In: StatPearls. Treasure Island, FL: StatPearls Publishing, 2022. [PubMed] [Google Scholar]
- 2. Nigam SK, Wu W, Bush KT, Hoenig MP, Blantz RC, Bhatnagar V. Handling of drugs, metabolites, and uremic toxins by kidney proximal tubule drug transporters. Clin J Am Soc Nephrol 10: 2039–2049, 2015. doi: 10.2215/CJN.02440314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jang K-J, Mehr AP, Hamilton GA, McPartlin LA, Chung S, Suh K-Y, Ingber DE. Human kidney proximal tubule-on-a-chip for drug transport and nephrotoxicity assessment. Integr Biol (Camb) 5: 1119–1129, 2013. doi: 10.1039/c3ib40049b. [DOI] [PubMed] [Google Scholar]
- 4. Liao J, Yu Z, Chen Y, Bao M, Zou C, Zhang H, Liu D, Li T, Zhang Q, Li J, Cheng J, Mo Z. Single-cell RNA sequencing of human kidney. Sci Data 7: 4, 2020. doi: 10.1038/s41597-019-0351-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Walmsley SJ, Broeckling C, Hess A, Prenni J, Curthoys NP. Proteomic analysis of brush-border membrane vesicles isolated from purified proximal convoluted tubules. Am J Physiol Renal Physiol 298: F1323–F1331, 2010. doi: 10.1152/ajprenal.00711.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu J, Sun Z, Yang S, Fu J, Fan Y, Wang N, Hu J, Ma L, Peng C, Wang Z, Lee K, He JC, Li Q. Kidney single-cell transcriptome profile reveals distinct response of proximal tubule cells to SGLT2i and ARB treatment in diabetic mice. Mol Ther 30: 1741–1753, 2022. doi: 10.1016/j.ymthe.2021.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mizuguchi K, Aoki H, Aoyama M, Kawaguchi Y, Waguri-Nagaya Y, Ohte N, Asai K. Three-dimensional spheroid culture induces apical-basal polarity and the original characteristics of immortalized human renal proximal tubule epithelial cells. Exp Cell Res 404: 112630, 2021. doi: 10.1016/j.yexcr.2021.112630. [DOI] [PubMed] [Google Scholar]
- 8. Kuppe C, Ibrahim MM, Kranz J, Zhang X, Ziegler S, Perales-Patón J, Jansen J, Reimer KC, Smith JR, Dobie R, Wilson-Kanamori JR, Halder M, Xu Y, Kabgani N, Kaesler N, Klaus M, Gernhold L, Puelles VG, Huber TB, Boor P, Menzel S, Hoogenboezem RM, Bindels EMJ, Steffens J, Floege J, Schneider RK, Saez-Rodriguez J, Henderson NC, Kramann R. Decoding myofibroblast origins in human kidney fibrosis. Nature 589: 281–286, 2021. doi: 10.1038/s41586-020-2941-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang L, Brooks CR, Xiao S, Sabbisetti V, Yeung MY, Hsiao L-L, Ichimura T, Kuchroo V, Bonventre JV. KIM-1-mediated phagocytosis reduces acute injury to the kidney. J Clin Invest 125: 1620–1636, 2015. doi: 10.1172/JCI75417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Al-Bataineh MM, Gong F, Marciszyn AL, Myerburg MM, Pastor-Soler NM. Regulation of proximal tubule vacuolar H+-ATPase by PKA and AMP-activated protein kinase. Am J Physiol Renal Physiol 306: F981–F995, 2014. doi: 10.1152/ajprenal.00362.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chichger H, Cleasby ME, Srai SK, Unwin RJ, Debnam ES, Marks J. Experimental type II diabetes and related models of impaired glucose metabolism differentially regulate glucose transporters at the proximal tubule brush border membrane. Exp Physiol 101: 731–742, 2016. doi: 10.1113/EP085670. [DOI] [PubMed] [Google Scholar]
- 12. Smith CP, Lee W-K, Haley M, Poulsen SB, Thévenod F, Fenton RA. Proximal tubule transferrin uptake is modulated by cellular iron and mediated by apical membrane megalin-cubilin complex and transferrin receptor 1. J Biol Chem 294: 7025–7036, 2019. doi: 10.1074/jbc.RA118.006390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ho KM, Morgan DJR. The proximal tubule as the pathogenic and therapeutic target in acute kidney injury. Nephron 1–9, 2022. doi: 10.1159/000522341. [DOI] [PubMed] [Google Scholar]
- 14. Conjard A, Martin M, Guitton J, Baverel G, Ferrier B. Gluconeogenesis from glutamine and lactate in the isolated human renal proximal tubule: longitudinal heterogeneity and lack of response to adrenaline. Biochem J 360: 371–377, 2001. doi: 10.1042/bj3600371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Corman B, Roinel N, de Rouffignac C. Dependence of water movement on sodium transport in kidney proximal tubule: a microperfusion study substituting lithium for sodium. J Membr Biol 62: 105–111, 1981. doi: 10.1007/BF01870204. [DOI] [PubMed] [Google Scholar]
- 16. Filipovic D, Sackin H. Stretch- and volume-activated channels in isolated proximal tubule cells. Am J Physiol Renal Physiol 262: F857–F870, 1992. doi: 10.1152/ajprenal.1992.262.5.F857. [DOI] [PubMed] [Google Scholar]
- 17. Hayden MR, Chowdhury NA, Cooper SA, Whaley-Connell A, Habibi J, Witte L, Wiedmeyer C, Manrique CM, Lastra G, Ferrario C, Stump C, Sowers JR. Proximal tubule microvilli remodeling and albuminuria in the Ren2 transgenic rat. Am J Physiol Renal Physiol 292: F861–F867, 2007. doi: 10.1152/ajprenal.00252.2006. [DOI] [PubMed] [Google Scholar]
- 18. Kahn AM, Weinman EJ. Urate transport in the proximal tubule: in vivo and vesicle studies. Am J Physiol Renal Physiol 249: F789–F798, 1985. doi: 10.1152/ajprenal.1985.249.6.F789. [DOI] [PubMed] [Google Scholar]
- 19. Kolman P, Pica A, Carvou N, Boyde A, Cockcroft S, Loesch A, Pizzey A, Simeoni M, Capasso G, Unwin RJ. Insulin uptake across the luminal membrane of the rat proximal tubule in vivo and in vitro. Am J Physiol Renal Physiol 296: F1227–F1237, 2009. doi: 10.1152/ajprenal.90351.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sackin H, Boulpaep EL. Isolated perfused salamander proximal tubule: methods, electrophysiology, and transport. Am J Physiol Renal Physiol 241: F39–F52, 1981. doi: 10.1152/ajprenal.1981.241.1.F39. [DOI] [PubMed] [Google Scholar]
- 21. Bgatova N, Taskaeva I, Makarova V. Influence of distant tumor growth and lithium treatment on ultrastructural organization of kidney proximal tubules. Ultrastruct Pathol 45: 212–223, 2021. doi: 10.1080/01913123.2021.1954735. [DOI] [PubMed] [Google Scholar]
- 22. Custer M, Lötscher M, Biber J, Murer H, Kaissling B. Expression of Na-P(i) cotransport in rat kidney: localization by RT-PCR and immunohistochemistry. Am J Physiol Renal Physiol 266: F767–F774, 1994. doi: 10.1152/ajprenal.1994.266.5.F767. [DOI] [PubMed] [Google Scholar]
- 23. Jouret F, Courtoy PJ, Devuyst O. Segmental and subcellular distribution of CFTR in the kidney. Methods Mol Biol 741: 285–299, 2011. doi: 10.1007/978-1-61779-117-8_19. [DOI] [PubMed] [Google Scholar]
- 24. Kamiyama M, Farragut KM, Garner MK, Navar LG, Kobori H. Divergent localization of angiotensinogen mRNA and protein in proximal tubule segments of normal rat kidney. J Hypertens 30: 2365–2372, 2012. doi: 10.1097/HJH.0b013e3283598eed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kobayashi M, Nishi K, Mizutani A, Okudaira H, Nakanishi T, Shikano N, Nishii R, Tamai I, Kawai K. Transport mechanism and affinity of [99mTc]Tc-mercaptoacetyltriglycine ([99mTc]MAG3) on the apical membrane of renal proximal tubule cells. Nucl Med Biol 84–85: 33–37, 2020. doi: 10.1016/j.nucmedbio.2020.01.002. [DOI] [PubMed] [Google Scholar]
- 26. Nagai J, Christensen EI, Morris SM, Willnow TE, Cooper JA, Nielsen R. Mutually dependent localization of megalin and Dab2 in the renal proximal tubule. Am J Physiol Renal Physiol 289: F569–F576, 2005. doi: 10.1152/ajprenal.00292.2004. [DOI] [PubMed] [Google Scholar]
- 27. Vormann MK, Gijzen L, Hutter S, Boot L, Nicolas A, van den Heuvel A, Vriend J, Ng CP, Nieskens TTG, van Duinen V, de Wagenaar B, Masereeuw R, Suter-Dick L, Trietsch SJ, Wilmer M, Joore J, Vulto P, Lanz HL. Nephrotoxicity and kidney transport assessment on 3D perfused proximal tubules. AAPS J 20: 90, 2018. doi: 10.1208/s12248-018-0248-z. [DOI] [PubMed] [Google Scholar]
- 28. Wilson BA, Cruz-Diaz N, Su Y, Rose JC, Gwathmey TM, Chappell MC. Angiotensinogen import in isolated proximal tubules: evidence for mitochondrial trafficking and uptake. Am J Physiol Renal Physiol 312: F879–F886, 2017. doi: 10.1152/ajprenal.00246.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang J, He J, Johnson JL, Rahman F, Gavathiotis E, Cuervo AM, Catz SD. Chaperone-mediated autophagy upregulation rescues megalin expression and localization in cystinotic proximal tubule cells. Front Endocrinol (Lausanne) 10: 21, 2019. doi: 10.3389/fendo.2019.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang Y, Li K, Li Y, Zhao W, Wang L, Chen Z, Ma X, Yao T, Wang J, Dong W, Li X, Tian X, Fu R. Profibrotic mechanisms of DPP8 and DPP9 highly expressed in the proximal renal tubule epithelial cells. Pharmacol Res 169: 105630, 2021. doi: 10.1016/j.phrs.2021.105630. [DOI] [PubMed] [Google Scholar]
- 31. Takasato M, Er PX, Chiu HS, Maier B, Baillie GJ, Ferguson C, Parton RG, Wolvetang EJ, Roost MS, Chuva de Sousa Lopes SM, Little MH. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 536: 238, 2016. doi: 10.1038/nature17982. [DOI] [PubMed] [Google Scholar]
- 32. Khundmiri SJ, Chen L, Lederer ED, Yang C-R, Knepper MA. Transcriptomes of major proximal tubule cell culture models. J Am Soc Nephrol 32: 86–97, 2021. doi: 10.1681/ASN.2020010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Birdsall HH, Hammond TG. Role of shear stress on renal proximal tubular cells for nephrotoxicity assays. J Toxicol 2021: 6643324, 2021. doi: 10.1155/2021/6643324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Duan Y, Weinstein AM, Weinbaum S, Wang T. Shear stress-induced changes of membrane transporter localization and expression in mouse proximal tubule cells. Proc Natl Acad Sci USA 107: 21860–21865, 2010. doi: 10.1073/pnas.1015751107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ren Q, Gliozzi ML, Rittenhouse NL, Edmunds LR, Rbaibi Y, Locker JD, Poholek AC, Jurczak MJ, Baty CJ, Weisz OA. Shear stress and oxygen availability drive differential changes in opossum kidney proximal tubule cell metabolism and endocytosis. Traffic 20: 448–459, 2019. doi: 10.1111/tra.12648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nieskens TTG, Persson M, Kelly EJ, Sjögren A-K. A multicompartment human kidney proximal tubule-on-a-chip replicates cell polarization-dependent cisplatin toxicity. Drug Metab Dispos 48: 1303–1311, 2020. doi: 10.1124/dmd.120.000098. [DOI] [PubMed] [Google Scholar]
- 37. Lin NYC, Homan KA, Robinson SS, Kolesky DB, Duarte N, Moisan A, Lewis JA. Renal reabsorption in 3D vascularized proximal tubule models. Proc Natl Acad Sci USA 116: 5399–5404, 2019. doi: 10.1073/pnas.1815208116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kirita Y, Wu H, Uchimura K, Wilson PC, Humphreys BD. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc Natl Acad Sci USA 117: 15874–15883, 2020. doi: 10.1073/pnas.2005477117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Digby JLM, Vanichapol T, Przepiorski A, Davidson AJ, Sander V. Evaluation of cisplatin-induced injury in human kidney organoids. Am J Physiol Renal Physiol 318: F971–F978, 2020. doi: 10.1152/ajprenal.00597.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Morizane R, Bonventre JV. Kidney organoids: a translational journey. Trends Mol Med 23: 246–263, 2017. doi: 10.1016/j.molmed.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sun G, Ding B, Wan M, Chen L, Jackson J, Atala A. Formation and optimization of three-dimensional organoids generated from urine-derived stem cells for renal function in vitro. Stem Cell Res Ther 11: 309, 2020. doi: 10.1186/s13287-020-01822-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Adedoyin O, Boddu R, Traylor A, Lever JM, Bolisetty S, George JF, Agarwal A. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am J Physiol Renal Physiol 314: F702–F714, 2018. doi: 10.1152/ajprenal.00044.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. García-Pérez E, Ryu D, Kim HY, Kim HD, Lee HJ. Human proximal tubule epithelial cells (HK-2) as a sensitive in vitro system for ochratoxin A induced oxidative stress. Toxins (Basel) 13: 787, 2021. doi: 10.3390/toxins13110787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Squires PE, Price GW, Mouritzen U, Potter JA, Williams BM, Hills CE. Danegaptide prevents TGFβ1-induced damage in human proximal tubule epithelial cells of the kidney. Int J Mol Sci 22: 2809, 2021. doi: 10.3390/ijms22062809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Daehn IS, Duffield JS. The glomerular filtration barrier: a structural target for novel kidney therapies. Nat Rev Drug Discov 20: 770–788, 2021. doi: 10.1038/s41573-021-00242-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Scott RP, Quaggin SE. Review series: the cell biology of renal filtration. J Cell Biol 209: 199–210, 2015. doi: 10.1083/jcb.201410017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Marsenic O. Glucose control by the kidney: an emerging target in diabetes. Am J Kidney Dis 53: 875–883, 2009. doi: 10.1053/j.ajkd.2008.12.031. [DOI] [PubMed] [Google Scholar]
- 48. Mather A, Pollock C. Glucose handling by the kidney. Kidney Int Suppl 79, Suppl 120: S1–S6, 2011. doi: 10.1038/ki.2010.509. [DOI] [PubMed] [Google Scholar]
- 49. Rieg T, Masuda T, Gerasimova M, Mayoux E, Platt K, Powell DR, Thomson SC, Koepsell H, Vallon V. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am J Physiol Renal Physiol 306: F188–F193, 2014. doi: 10.1152/ajprenal.00518.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vallon V, Platt KA, Cunard R, Schroth J, Whaley J, Thomson SC, Koepsell H, Rieg T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol 22: 104–112, 2011. doi: 10.1681/ASN.2010030246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Balen D, Ljubojević M, Breljak D, Brzica H, ŽLender V, Koepsell H, Sabolić I. Revised immunolocalization of the Na+-d-glucose cotransporter SGLT1 in rat organs with an improved antibody. Am J Physiol Cell Physiol 295: C475–C489, 2008. doi: 10.1152/ajpcell.00180.2008. [DOI] [PubMed] [Google Scholar]
- 52. Vrhovac I, Balen Eror D, Klessen D, Burger C, Breljak D, Kraus O, Radović N, Jadrijević S, Aleksic I, Walles T, Sauvant C, Sabolić I, Koepsell H. Localizations of Na(+)-d-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflugers Arch 467: 1881–1898, 2015. doi: 10.1007/s00424-014-1619-7. [DOI] [PubMed] [Google Scholar]
- 53. Keljo DJ, Kleinzeller A, Murer H, Kinne R. Is hexokinase present in the basal lateral membranes of rat kidney proximal tubular epithelial cells? Biochim Biophys Acta 508: 500–512, 1978. doi: 10.1016/0005-2736(78)90095-0. [DOI] [PubMed] [Google Scholar]
- 54. Vallon V. Glucose transporters in the kidney in health and disease. Pflugers Arch 472: 1345–1370, 2020. doi: 10.1007/s00424-020-02361-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen L, Chou CL, Knepper MA. A comprehensive map of mrnas and their isoforms across all 14 renal tubule segments of mouse. J Am Soc Nephrol 32: 897–912, 2021. doi: 10.1681/ASN.2020101406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Limbutara K, Chou CL, Knepper MA. Quantitative proteomics of all 14 renal tubule segments in rat. J Am Soc Nephrol 31: 1255–1266, 2020. doi: 10.1681/ASN.2020010071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hediger MA, Coady MJ, Ikeda TS, Wright EM. Expression cloning and cDNA sequencing of the Na+/glucose co-transporter. Nature 330: 379–381, 1987. doi: 10.1038/330379a0. [DOI] [PubMed] [Google Scholar]
- 58. Turk E, Gasymov OK, Lanza S, Horwitz J, Wright EM. A reinvestigation of the secondary structure of functionally active vSGLT, the vibrio sodium/galactose cotransporter. Biochemistry 45: 1470–1479, 2006. doi: 10.1021/bi052160z. [DOI] [PubMed] [Google Scholar]
- 59. Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev 91: 733–794, 2011. doi: 10.1152/physrev.00055.2009. [DOI] [PubMed] [Google Scholar]
- 60. Adelman JL, Ghezzi C, Bisignano P, Loo DDF, Choe S, Abramson J, Rosenberg JM, Wright EM, Grabe M. Stochastic steps in secondary active sugar transport. Proc Natl Acad Sci USA 113: E3960–E3966, 2016. doi: 10.1073/pnas.1525378113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Barreto YB, Alencar AM. Random-walk model of the sodium-glucose transporter SGLT2 with stochastic steps and inhibition. J Phys Condens Matter 34: 184004, 2022. doi: 10.1088/1361-648X/ac4fea. [DOI] [PubMed] [Google Scholar]
- 62. Westman EC, Yancy WS Jr, Humphreys M. Dietary treatment of diabetes mellitus in the pre-insulin era (1914–1922). Perspect Biol Med 49: 77–83, 2006. doi: 10.1353/pbm.2006.0017. [DOI] [PubMed] [Google Scholar]
- 63. Yaribeygi H, Sathyapalan T, Maleki M, Jamialahmadi T, Sahebkar A. Molecular mechanisms by which SGLT2 inhibitors can induce insulin sensitivity in diabetic milieu: a mechanistic review. Life Sci 240: 117090, 2020. doi: 10.1016/j.lfs.2019.117090. [DOI] [PubMed] [Google Scholar]
- 64. Vidotti DB, Arnoni CP, Maquigussa E, Boim MA. Effect of long-term type 1 diabetes on renal sodium and water transporters in rats. Am J Nephrol 28: 107–114, 2008. doi: 10.1159/000109967. [DOI] [PubMed] [Google Scholar]
- 65. Tabatabai NM, Sharma M, Blumenthal SS, Petering DH. Enhanced expressions of sodium-glucose cotransporters in the kidneys of diabetic Zucker rats. Diabetes Res Clin Pract 83: e27–e30, 2009. doi: 10.1016/j.diabres.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Srinivasan Sridhar V, Ambinathan JPN, Kretzler M, Pyle LL, Bjornstad P, Eddy S, Cherney DZ, Reich HN; European Renal cDNA Bank (ERCB), Nephrotic Syndrome Study Network (NEPTUNE). Renal SGLT mRNA expression in human health and disease: a study in two cohorts. Am J Physiol Renal Physiol 317: F1224–F1230, 2019. doi: 10.1152/ajprenal.00370.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shepard BD, Pluznick JL. Saving the sweetness: renal glucose handling in health and disease. Am J Physiol Renal Physiol 313: F55–F61, 2017. doi: 10.1152/ajprenal.00046.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vallon V, Thomson SC. Targeting renal glucose reabsorption to treat hyperglycaemia: the pleiotropic effects of SGLT2 inhibition. Diabetologia 60: 215–225, 2017. doi: 10.1007/s00125-016-4157-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Amsler K, Cook JS. Development of Na+-dependent hexose transport in a cultured line of porcine kidney cells. Am J Physiol Cell Physiol 242: C94–C101, 1982. doi: 10.1152/ajpcell.1982.242.1.C94. [DOI] [PubMed] [Google Scholar]
- 70. Wacker D, Stevens RC, Roth BL. How ligands illuminate GPCR molecular pharmacology. Cell 170: 414–427, 2017. doi: 10.1016/j.cell.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lundstrom K. An overview on GPCRs and drug discovery: structure-based drug design and structural biology on GPCRs. Methods Mol Biol 552: 51–66, 2009. doi: 10.1007/978-1-60327-317-6_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shepard BD, Koepsell H, Pluznick JL. Renal olfactory receptor 1393 contributes to the progression of type 2 diabetes in a diet-induced obesity model. Am J Physiol Renal Physiol 316: F372–F381, 2019. doi: 10.1152/ajprenal.00069.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shepard BD, Cheval L, Peterlin Z, Firestein S, Koepsell H, Doucet A, Pluznick JL. A renal olfactory receptor aids in kidney glucose handling. Sci Rep 6: 35215, 2016. doi: 10.1038/srep35215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schiazza AR, Considine EG, Betcher M, Shepard BD. Loss of renal olfactory receptor 1393 leads to improved glucose homeostasis in a type 1 diabetic mouse model. Physiol Rep 9: e15007, 2021. doi: 10.14814/phy2.15007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Subramanian S, Glitz P, Kipp H, Kinne RKH, Castaneda F. Protein kinase-A affects sorting and conformation of the sodium-dependent glucose co-transporter SGLT1. J Cell Biochem 106: 444–452, 2009. doi: 10.1002/jcb.22025. [DOI] [PubMed] [Google Scholar]
- 76. Amsler K. Cyclic adenosine monophosphate modulates cell morphology and behavior of a cultured renal epithelial. Pediatr Nephrol 4: 378–386, 1990. doi: 10.1007/BF00862523. [DOI] [PubMed] [Google Scholar]
- 77. Sunilkumar S, Ford SM. Elevated glucose concentration in culture media decreases membrane trafficking of SGLT2 in LLC-PK1 cells via a cAMP/PKA-dependent pathway. Am J Physiol Cell Physiol 316: C913–C924, 2019. doi: 10.1152/ajpcell.00433.2018. [DOI] [PubMed] [Google Scholar]
- 78. Hirsch JR, Loo DD, Wright EM. Regulation of Na+/glucose cotransporter expression by protein kinases in Xenopus laevis oocytes. J Biol Chem 271: 14740–14746, 1996. doi: 10.1074/jbc.271.25.14740. [DOI] [PubMed] [Google Scholar]
- 79. Ghezzi C, Wright EM. Regulation of the human Na+-dependent glucose cotransporter hSGLT2. Am J Physiol Cell Physiol 303: C348–C354, 2012. doi: 10.1152/ajpcell.00115.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lee YJ, Kim MO, Ryu JM, Han HJ. Regulation of SGLT expression and localization through Epac/PKA-dependent caveolin-1 and F-actin activation in renal proximal tubule cells. Biochim Biophys Acta 1823: 971–982, 2012. doi: 10.1016/j.bbamcr.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 81. Krawczyk KM, Hansson J, Nilsson H, Krawczyk KK, Swärd K, Johansson ME. Injury induced expression of caveolar proteins in human kidney tubules—role of megakaryoblastic leukemia 1. BMC Nephrol 18: 320, 2017. doi: 10.1186/s12882-017-0738-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhuang Z, Marshansky V, Breton S, Brown D. Is caveolin involved in normal proximal tubule function? Presence in model PT systems but absence in situ. Am J Physiol Renal Physiol 300: F199–F206, 2011. doi: 10.1152/ajprenal.00513.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Danielsen EM, Hansen GH. Lipid rafts in epithelial brush borders: atypical membrane microdomains with specialized functions. Biochim Biophys Acta 1617: 1–9, 2003. doi: 10.1016/j.bbamem.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 84. Gu M, Tan M, Zhou L, Sun X, Lu Q, Wang M, Jiang H, Liang Y, Hou Q, Xue X, Xu Z, Dai C. Protein phosphatase 2Acα modulates fatty acid oxidation and glycolysis to determine tubular cell fate and kidney injury. Kidney Int 102: 321–336, 2022. doi: 10.1016/j.kint.2022.03.024. [DOI] [PubMed] [Google Scholar]
- 85. Lewis S, Chen L, Raghuram V, Khundmiri SJ, Chou C-L, Yang C-R, Knepper MA. SLC-“omics” of the kidney: solute transporters along the nephron. Am J Physiol Cell Physiol 321: C507–C518, 2021. doi: 10.1152/ajpcell.00197.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Diez-Sampedro A, Hirayama BA, Osswald C, Gorboulev V, Baumgarten K, Volk C, Wright EM, Koepsell H. A glucose sensor hiding in a family of transporters. Proc Natl Acad Sci USA 100: 11753–11758, 2003. doi: 10.1073/pnas.1733027100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kothinti RK, Blodgett AB, North PE, Roman RJ, Tabatabai NM. A novel SGLT is expressed in the human kidney. Eur J Pharmacol 690: 77–83, 2012. doi: 10.1016/j.ejphar.2012.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Drewnowsk KD, Craig MR, Digiovanni SR, McCarty JM, Moorman AFM, Lamers WH, Schoolwerth AC. PEPCK mRNA localization in proximal tubule and gene regulation during metabolic acidosis. J Physiol Pharmacol 53: 3–20, 2002. [PubMed] [Google Scholar]
- 89. Sasaki M, Sasako T, Kubota N, Sakurai Y, Takamoto I, Kubota T, Inagi R, Seki G, Goto M, Ueki K, Nangaku M, Jomori T, Kadowaki T. Dual regulation of gluconeogenesis by insulin and glucose in the proximal tubules of the kidney. Diabetes 66: 2339–2350, 2017. doi: 10.2337/db16-1602. [DOI] [PubMed] [Google Scholar]
- 90. Lee JW, Chou CL, Knepper MA. Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J Am Soc Nephrol 26: 2669–2677, 2015. doi: 10.1681/ASN.2014111067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tojo A, Hatakeyama S, Nangaku M, Ishimitsu T. H+-ATPase blockade reduced renal gluconeogenesis and plasma glucose in a diabetic rat model. Med Mol Morphol 51: 89–95, 2018. doi: 10.1007/s00795-017-0175-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Clar J, Gri B, Calderaro J, Birling M-C, Hérault Y, Smit GPA, Mithieux G, Rajas F. Targeted deletion of kidney glucose-6 phosphatase leads to nephropathy. Kidney Int 86: 747–756, 2014. doi: 10.1038/ki.2014.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kang J, Dai X-S, Yu T-B, Wen B, Yang Z-W. Glycogen accumulation in renal tubules, a key morphological change in the diabetic rat kidney. Acta Diabetol 42: 110–116, 2005. doi: 10.1007/s00592-005-0188-9. [DOI] [PubMed] [Google Scholar]
- 94. Bertinat R, Westermeier F, Silva P, Gatica R, Oliveira JM, Nualart F, Gomis R, Yáñez AJ. The antidiabetic agent sodium tungstate induces abnormal glycogen accumulation in renal proximal tubules from diabetic IRS2-knockout mice. J Diabetes Res 2018: 5697970, 2018. doi: 10.1155/2018/5697970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Cai T, Ke Q, Fang Y, Wen P, Chen H, Yuan Q, Luo J, Zhang Y, Sun Q, Lv Y, Zen K, Jiang L, Zhou Y, Yang J. Sodium-glucose cotransporter 2 inhibition suppresses HIF-1α-mediated metabolic switch from lipid oxidation to glycolysis in kidney tubule cells of diabetic mice. Cell Death Dis 11: 390, 2020. doi: 10.1038/s41419-020-2544-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ito M, Tanaka T, Ishii T, Wakashima T, Fukui K, Nangaku M. Prolyl hydroxylase inhibition protects the kidneys from ischemia via upregulation of glycogen storage. Kidney Int 97: 687–701, 2020. doi: 10.1016/j.kint.2019.10.020. [DOI] [PubMed] [Google Scholar]
- 97. Schaub JA, Venkatachalam MA, Weinberg JM. Proximal tubular oxidative metabolism in acute kidney injury and the transition to CKD. Kidney360 2: 355–364, 2021. doi: 10.34067/KID.0004772020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther 148: 114–131, 2015. doi: 10.1016/j.pharmthera.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Howard C, Tao S, Yang H-C, Fogo AB, Woodgett JR, Harris RC, Rao R. Specific deletion of glycogen synthase kinase-3β in the renal proximal tubule protects against acute nephrotoxic injury in mice. Kidney Int 82: 1000–1009, 2012. doi: 10.1038/ki.2012.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sinha S, Dwivedi N, Woodgett J, Tao S, Howard C, Fields TA, Jamadar A, Rao R. Glycogen synthase kinase-3β inhibits tubular regeneration in acute kidney injury by a FoxM1-dependent mechanism. FASEB J 34: 13597–13608, 2020. doi: 10.1096/fj.202000526RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Hsing C-H, Tsai C-C, Chen C-L, Lin Y-H, Tseng P-C, Satria RD, Lin C-F. Pharmacologically inhibiting glycogen synthase kinase-3β ameliorates renal inflammation and nephrotoxicity in an animal model of cisplatin-induced acute kidney injury. Biomedicines 9: 887, 2021. doi: 10.3390/biomedicines9080887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Layton AT, Vallon V, Edwards A. Modeling oxygen consumption in the proximal tubule: effects of NHE and SGLT2 inhibition. Am J Physiol Renal Physiol 308: F1343–F1357, 2015. doi: 10.1152/ajprenal.00007.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kapitsinou PP, Sano H, Michael M, Kobayashi H, Davidoff O, Bian A, Yao B, Zhang M-Z, Harris RC, Duffy KJ, Erickson-Miller CL, Sutton TA, Haase VH. Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J Clin Invest 124: 2396–2409, 2014. doi: 10.1172/JCI69073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Thomas JL, Pham H, Li Y, Hall E, Perkins GA, Ali SS, Patel HH, Singh P. Hypoxia-inducible factor-1α activation improves renal oxygenation and mitochondrial function in early chronic kidney disease. Am J Physiol Renal Physiol 313: F282–F290, 2017. doi: 10.1152/ajprenal.00579.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Li Y, Nourbakhsh N, Pham H, Tham R, Zuckerman JE, Singh P. Evolution of altered tubular metabolism and mitochondrial function in sepsis-associated acute kidney injury. Am J Physiol Renal Physiol 319: F229–F244, 2020. [Erratum in Am J Physiol Renal Physiol 320: F1019, 2021]. doi: 10.1152/ajprenal.00390.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Lan R, Geng H, Singha PK, Saikumar P, Bottinger EP, Weinberg JM, Venkatachalam MA. Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J Am Soc Nephrol 27: 3356–3367, 2016. doi: 10.1681/ASN.2015020177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Shaik ZP, Fifer EK, Nowak G. Akt activation improves oxidative phosphorylation in renal proximal tubular cells following nephrotoxicant injury. Am J Physiol Renal Physiol 294: F423–F432, 2008. doi: 10.1152/ajprenal.00463.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ding H, Jiang L, Xu J, Bai F, Zhou Y, Yuan Q, Luo J, Zen K, Yang J. Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am J Physiol Renal Physiol 313: F561–F575, 2017. doi: 10.1152/ajprenal.00036.2017. [DOI] [PubMed] [Google Scholar]
- 109. Andres-Hernando A, Li N, Cicerchi C, Inaba S, Chen W, Roncal-Jimenez C, Le MT, Wempe MF, Milagres T, Ishimoto T, Fini M, Nakagawa T, Johnson RJ, Lanaspa MA. Protective role of fructokinase blockade in the pathogenesis of acute kidney injury in mice. Nat Commun 8: 14181, 2017. doi: 10.1038/ncomms14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Brown E, Heerspink HJL, Cuthbertson DJ, Wilding JPH. SGLT2 inhibitors and GLP-1 receptor agonists: established and emerging indications. Lancet 398: 262–276, 2021. doi: 10.1016/S0140-6736(21)00536-5. [DOI] [PubMed] [Google Scholar]
- 111. Rieg T, Vallon V. Development of SGLT1 and SGLT2 inhibitors. Diabetologia 61: 2079–2086, 2018. doi: 10.1007/s00125-018-4654-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Barnett AH, Mithal A, Manassie J, Jones R, Rattunde H, Woerle HJ, Broedl UC. Efficacy and safety of empagliflozin added to existing antidiabetes treatment in patients with type 2 diabetes and chronic kidney disease: a randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol 2: 369–384, 2014. doi: 10.1016/S2213-8587(13)70208-0. [DOI] [PubMed] [Google Scholar]
- 113. Vallon V, Gerasimova M, Rose MA, Masuda T, Satriano J, Mayoux E, Koepsell H, Thomson SC, Rieg T. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am J Physiol Renal Physiol 306: F194–F204, 2014. doi: 10.1152/ajprenal.00520.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, Edwards R, Agarwal R, Bakris G, Bull S, Cannon CP, Capuano G, Chu P-L, de Zeeuw D, Greene T, Levin A, Pollock C, Wheeler DC, Yavin Y, Zhang H, Zinman B, Meininger G, Brenner BM, Mahaffey KW; CREDENCE Trial Investigators. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med 380: 2295–2306, 2019. doi: 10.1056/NEJMoa1811744. [DOI] [PubMed] [Google Scholar]
- 115. Borges-Junior FA, Silva Dos Santos D, Benetti A, Polidoro JZ, Wisnivesky ACT, Crajoinas RO, Antônio EL, Jensen L, Caramelli B, Malnic G, Tucci PJ, Girardi ACC. Empagliflozin inhibits proximal tubule NHE3 activity, preserves GFR, and restores euvolemia in nondiabetic rats with induced heart failure. J Am Soc Nephrol 32: 1616–1629, 2021. doi: 10.1681/ASN.2020071029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Jhalani NB. Clinical considerations for use of SGLT2 inhibitor therapy in patients with heart failure and reduced ejection fraction: a review. Adv Ther 39: 3472–3487, 2022. doi: 10.1007/s12325-022-02169-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Onishi A, Fu Y, Patel R, Darshi M, Crespo-Masip M, Huang W, Song P, Freeman B, Kim YC, Soleimani M, Sharma K, Thomson SC, Vallon V. A role for tubular Na+/H+ exchanger NHE3 in the natriuretic effect of the SGLT2 inhibitor empagliflozin. Am J Physiol Renal Physiol 319: F712–F728, 2020. doi: 10.1152/ajprenal.00264.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Barrios V, Escobar C. Canagliflozin: metabolic, cardiovascular and renal protection. Future Cardiol 17: 443–458, 2021. doi: 10.2217/fca-2020-0192. [DOI] [PubMed] [Google Scholar]
- 119. Andreadi A, Bellia A, Di Daniele N, Meloni M, Lauro R, Della-Morte D, Lauro D. The molecular link between oxidative stress, insulin resistance, and type 2 diabetes: a target for new therapies against cardiovascular diseases. Curr Opin Pharmacol 62: 85–96, 2022. doi: 10.1016/j.coph.2021.11.010. [DOI] [PubMed] [Google Scholar]
- 120. Steven S, Oelze M, Hanf A, Kröller-Schön S, Kashani F, Roohani S, Welschof P, Kopp M, Gödtel-Armbrust U, Xia N, Li H, Schulz E, Lackner KJ, Wojnowski L, Bottari SP, Wenzel P, Mayoux E, Münzel T, Daiber A. The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol 13: 370–385, 2017. doi: 10.1016/j.redox.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Uthman L, Li X, Baartscheer A, Schumacher CA, Baumgart P, Hermanides J, Preckel B, Hollmann MW, Coronel R, Zuurbier CJ, Weber NC. Empagliflozin reduces oxidative stress through inhibition of the novel inflammation/NHE/[Na(+)]c/ROS-pathway in human endothelial cells. Biomed Pharmacother 146: 112515, 2022. doi: 10.1016/j.biopha.2021.112515. [DOI] [PubMed] [Google Scholar]
- 122. Cherney D, Lund SS, Perkins BA, Groop P-H, Cooper ME, Kaspers S, Pfarr E, Woerle HJ, von Eynatten M. The effect of sodium glucose cotransporter 2 inhibition with empagliflozin on microalbuminuria and macroalbuminuria in patients with type 2 diabetes. Diabetologia 59: 1860–1870, 2016. doi: 10.1007/s00125-016-4008-2. [DOI] [PubMed] [Google Scholar]
- 123. Dekkers CCJ, Petrykiv S, Laverman GD, Cherney DZ, Gansevoort RT, Heerspink HJL. Effects of the SGLT-2 inhibitor dapagliflozin on glomerular and tubular injury markers. Diabetes Obes Metab 20: 1988–1993, 2018. doi: 10.1111/dom.13301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Fukushima K, Kitamura S, Tsuji K, Sang Y, Wada J. Sodium glucose co-transporter 2 inhibitor ameliorates autophagic flux impairment on renal proximal tubular cells in obesity mice. Int J Mol Sci 21: 4054, 2020. doi: 10.3390/ijms21114054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kogot-Levin A, Hinden L, Riahi Y, Israeli T, Tirosh B, Cerasi E, Mizrachi EB, Tam J, Mosenzon O, Leibowitz G. Proximal tubule mTORC1 is a central player in the pathophysiology of diabetic nephropathy and its correction by SGLT2 inhibitors. Cell Rep 32: 107954, 2020. doi: 10.1016/j.celrep.2020.107954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Abdollahi E, Keyhanfar F, Delbandi A-A, Falak R, Hajimiresmaiel SJ, Shafiei M. Dapagliflozin exerts anti-inflammatory effects via inhibition of LPS-induced TLR-4 overexpression and NF-κB activation in human endothelial cells and differentiated macrophages. Eur J Pharmacol 918: 174715, 2022. doi: 10.1016/j.ejphar.2021.174715. [DOI] [PubMed] [Google Scholar]
- 127. Heerspink HJL, Stefánsson BV, Correa-Rotter R, Chertow GM, Greene T, Hou F-F, Mann JFE, McMurray JJV, Lindberg M, Rossing P, Sjöström CD, Toto RD, Langkilde A-M, Wheeler DC. Dapagliflozin in patients with chronic kidney disease. N Engl J Med 383: 1436–1446, 2020. doi: 10.1056/NEJMoa2024816. [DOI] [PubMed] [Google Scholar]
- 128. Sun X, Cao Z, Ma Y, Shao Y, Zhang J, Yuan G, Guo X. Resveratrol attenuates dapagliflozin-induced renal gluconeogenesis via activating the PI3K/Akt pathway and suppressing the FoxO1 pathway in type 2 diabetes. Food Funct 12: 1207–1218, 2021. doi: 10.1039/d0fo02387f. [DOI] [PubMed] [Google Scholar]
- 129. Kim JH, Ko HY, Wang HJ, Lee H, Yun MJ, Kang ES. Effect of dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, on gluconeogenesis in proximal renal tubules. Diabetes Obes Metab 22: 373–382, 2020. doi: 10.1111/dom.13905. [DOI] [PubMed] [Google Scholar]
- 130. Liu Q, Zhang L, Zhang W, Hao Q, Qiu W, Wen Y, Wang H, Li X. Inhibition of NF-κB reduces renal inflammation and expression of PEPCK in type 2 diabetic mice. Inflammation 41: 2018–2029, 2018. doi: 10.1007/s10753-018-0845-0. [DOI] [PubMed] [Google Scholar]
- 131. Yamada T, Shojima N, Noma H, Yamauchi T, Kadowaki T. Sodium-glucose co-transporter-2 inhibitors as add-on therapy to insulin for type 1 diabetes mellitus: systematic review and meta-analysis of randomized controlled trials. Diabetes Obes Metab 20: 1755–1761, 2018. doi: 10.1111/dom.13260. [DOI] [PubMed] [Google Scholar]
- 132. Seino Y, Fukushima M, Yabe D. GIP and GLP-1, the two incretin hormones: similarities and differences. J Diabetes Investig 1: 8–23, 2010. doi: 10.1111/j.2040-1124.2010.00022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Skov J. Effects of GLP-1 in the kidney. Rev Endocr Metab Disord 15: 197–207, 2014. doi: 10.1007/s11154-014-9287-7. [DOI] [PubMed] [Google Scholar]
- 134. Shepard BD, Ecelbarger CM. Sodium glucose transporter, type 2 (SGLT2) inhibitors (SGLT2i) and glucagon-like peptide 1-receptor agonists: newer therapies in whole-body glucose stabilization. Semin Nephrol 41: 331–348, 2021. doi: 10.1016/j.semnephrol.2021.06.005. [DOI] [PubMed] [Google Scholar]