

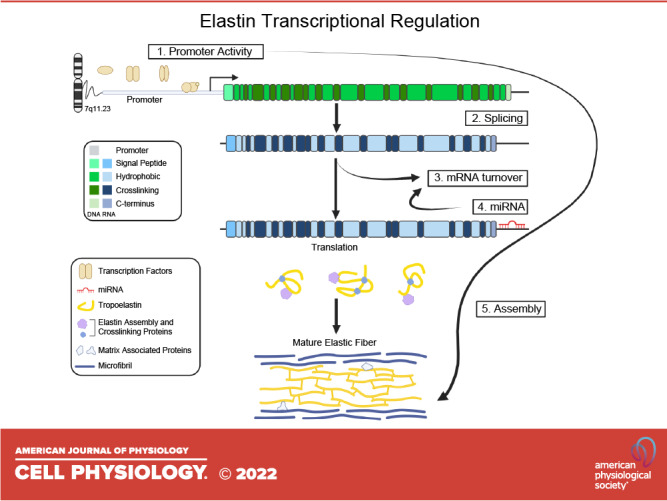
Keywords: elastin, promoter, supravalvar aortic stenosis, transcription, Williams syndrome
Abstract
Elastin provides recoil to tissues that stretch such as the lung, blood vessels, and skin. It is deposited in a brief window starting in the prenatal period and extending to adolescence in vertebrates, and then slowly turns over. Elastin insufficiency is seen in conditions such as Williams–Beuren syndrome and elastin-related supravalvar aortic stenosis, which are associated with a range of vascular and connective tissue manifestations. Regulation of the elastin (ELN) gene occurs at multiple levels including promoter activation/inhibition, mRNA stability, interaction with microRNAs, and alternative splicing. However, these mechanisms are incompletely understood. Better understanding of the processes controlling ELN gene expression may improve medicine’s ability to intervene in these rare conditions, as well as to replace age-associated losses by re-initiating elastin production. This review describes what is known about the ELN gene promoter structure, transcriptional regulation by cytokines and transcription factors, and posttranscriptional regulation via mRNA stability and micro-RNA and highlights new approaches that may influence regenerative medicine.
INTRODUCTION
Elastic fibers are macromolecular components of the extracellular matrix (ECM) that allow tissues such as blood vessels, lungs, and skin to stretch without being damaged and reduce mechanical work following stretch by returning to an entropically favored state during relaxation (1). Elastin, encoded by the ELN gene, makes up the amorphous core of these fibers, providing them with extensibility and recoil capability. Abnormalities of elastin quality or quantity occur in a range of health conditions, including rare diseases like elastin-related supravalvar aortic stenosis (SVAS), Williams–Beuren syndrome (Williams syndrome; WS), and elastin-related autosomal dominant cutis laxa (ADCL), named according to the dyadic principle (2). Understanding of the mechanisms of ELN transcript regulation, therefore, may suggest targets to reactivate elastin production and mitigate disease.
OVERVIEW OF ELASTIN
The ELN gene is located on chromosome 7 in humans. It consists of 34 in frame exons (Fig. 1) (for review, see Ref. 7) that permit alternative splicing of the ELN pre-mRNA without alteration of the open reading frame. This combination results in the generation of multiple mature mRNA isoforms (Fig. 1) (5). The canonical transcript lacks exon 22 and encodes an ∼70 kDa protein of repeating cross linking and hydrophobic domains.
Figure 1.
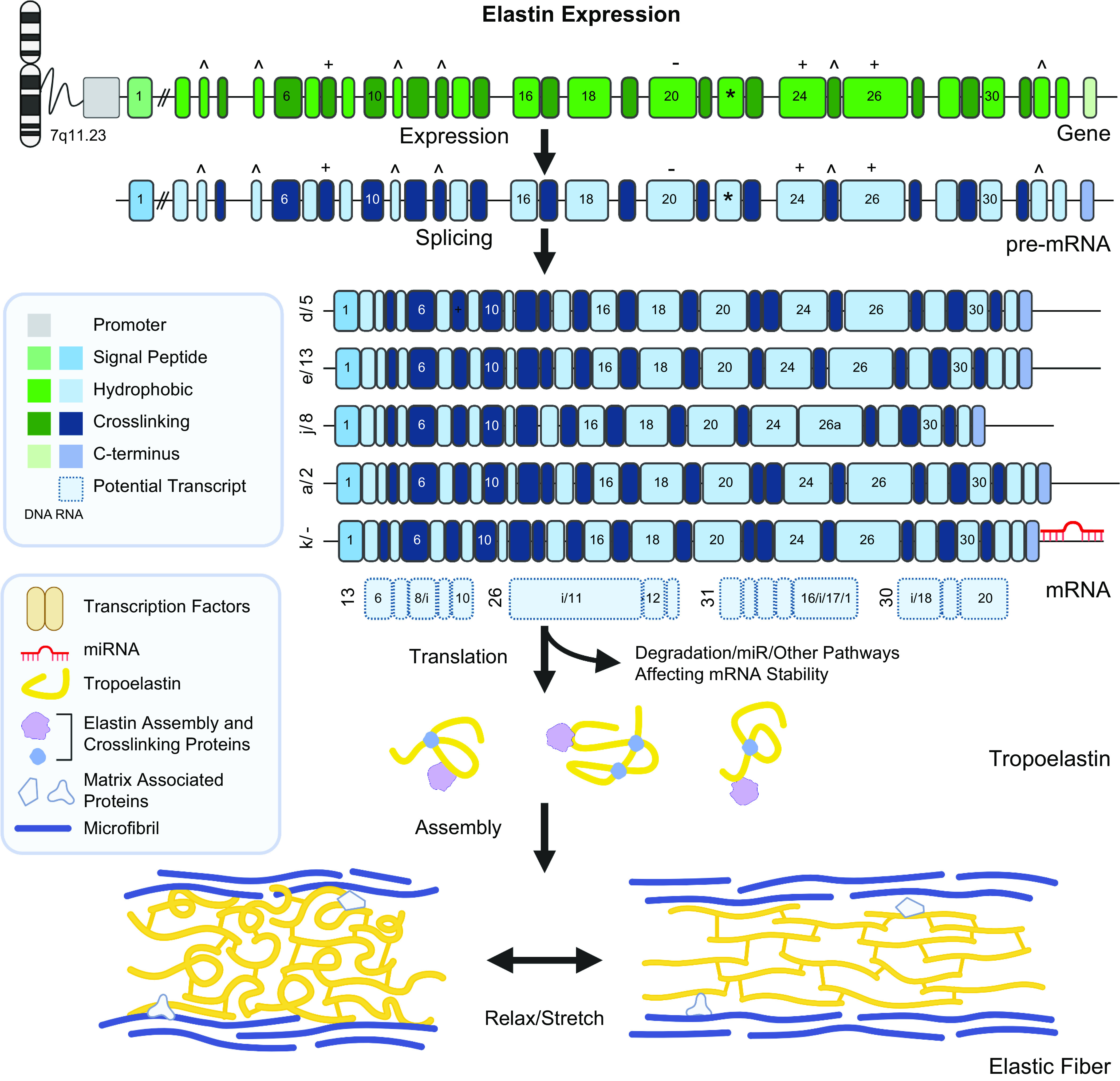
Elastin expression. The human elastin (ELN) gene is located on chromosome 7 at 7q11.23. It consists of 34 exons, by convention numbered 1–33 and 36 as most species contain an additional pair of exons, exons 34 and 35, that were lost in higher primates (3). The gene is transcribed as a large (47.7 kDa) pre-mRNA (NM_001278939, ENST00000692049.1) with a high intron/exon ratio (4). The exons are structured as alternating hydrophobic and crosslinking domains and are symmetric (in-phase) facilitating alternate splicing of exons. In microRNA (mRNA) studies, exon 22 (marked with an *) is almost uniformly removed, likely due to simple read-through (5). Other commonly spliced exons are marked with ^. There are also alternate splice donor and acceptor that can extend or shorten exons 8, 20, 24, and 26 (marked with + or −). Splice isoforms are present at varying relative ratios in different tissues (5) although mRNA and proteomic studies are not completely concordant (6). There are more than 30 splice isoforms identified to date. The five most common protein coding splice isoforms in human aorta are presented. Short isoforms using alternative transcription start sites and including intronic sequence (marked with an “i”) have also been identified through RNA studies (light blue with dashed border), but experimental confirmation of their presence and abundance is necessary. After splicing, mature tropoelastin coding isoforms are either translated into tropoelastin or degraded. mRNA degradation is an important pathway for regulation of tropoelastin expression. Tropoelastin monomers are able to undergo some self-assembly but the precise mechanisms underlying tropoelastin secretion and assembly into mature elastin fibers is unresolved, although crosslinking and assembly proteins such as lysyl oxidase (LOX) and FBLN4 and/or FBLN5 are involved. The mature elastic fiber consists of tropoelastin at the core within a microfibril (mainly consisting of FBN1 and FBN2) scaffold. While the ratio of elastin to microfibril changes through development; the mature elastin fiber contains approximately 90/10 elastin:microfibril content. This conformation allows for elastic fibers to stretch and relax without mechanical deformation. Created with BioRender.com.
Translation of the mature mRNA isoforms results in the production of tropoelastin (TE) monomers. TE is secreted into the ECM through a specialized acidic pathway (8). TE then multimerizes and is deposited into the microfibril-rich ECM as mature elastin (9, 10). As part of this process, the lysines in the cross-linking domains are cross linked with the help of the enzyme lysyl oxidase (LOX) resulting in a highly hydrophobic and insoluble protein.
The hydrophobic domains contribute to recoil capability as stretch exposes the hydrophobic domains to the hydrophilic environment and entropy drives them back (1, 11–13). The insoluble elastic fibers are highly stable, with a half-life of 74 years (14). Over time they can undergo glycosylation with advanced glycosylation end products (AGEs) that alter the recoil capability of the molecule making it brittle and increasing the rate of turnover (15–17).
ELASTIN IN DISEASE
Elastin is required for normal function of multiple tissues, with the most obvious effects seen in tissues that undergo recurrent deformations, such as the blood vessels, skin, and lungs. However, other tissues such as ligaments and elastic cartilages can also be impacted. Elastin-mediated disease can come about due to changes in either the quality or quantity of elastic fibers. Rare variation within ELN itself can cause ELN-related SVAS and ELN-related ADCL. SVAS is generally thought to result from reduced quantity of elastin while ADCL is thought to reflect the incorporation of abnormal elastic fibers into the ECM; however, this may be an over-simplification (18). In addition, common variation in ELN has been identified through multiple genome wide association studies (GWAS) to contribute to abnormalities of blood vessels (19–21) and other connective tissue functions (22–24) on a population basis. Given the long half-life of elastic fibers, environmental exposures such as UV radiation, cigarette smoke, and high glucose environments related to diabetes impact turnover rate in dramatic ways (25, 26).
Elastin Haploinsufficiency (ELN-Related SVAS and Williams–Beuren Syndrome)
ELN-related SVAS typically comes about due to nonsense or frameshift variants, generally within the front half of the molecule, that are expected to produce haploinsufficiency for elastin (27–31). Splice variants have also been described with less consistent genotype-phenotype implications (32–34). More prevalent than ELN-related SVAS, though, is Williams syndrome (OMIM No. 194050), which is caused by the contiguous deletion of 25 to 27 coding genes on chromosome 7, including ELN. In one epidemiological study, it was reported in 1:7,500 individuals and causes haploinsufficiency for elastin as well as for the other deleted genes (35).
People with either ELN-related SVAS or WS exhibit a characteristic vascular disease called supravalvar aortic stenosis (36, 37) (for review, see Ref. 38). SVAS manifests as an area of focal narrowing in the aorta, just above the aortic valve. It is reportedly more severe in affected males (39, 40), although the reasons for this are unknown. Stenoses are also possible in other large elastic vessels such as the descending aorta, pulmonary arteries, and renal arteries (41). Long segment narrowing, where the entire vascular segment exhibits diminutive caliber rather than being focally stenotic, is also observed in a sizeable subset (42). Sudden death is present at a rate of 25 to 100 times higher than the general population (43–45) with increased rates of complications and death associated with anesthesia—features that have been attributed secondarily to the vascular disease. In addition to stenosis, individuals with these conditions often have attendant arterial stiffness and hypertension (46–49).
More recent data have suggested that individuals with these conditions may also exhibit abnormalities of other connective tissues. Ligamentous laxity (50) is seen as well as early wrinkling and facial drooping of the skin (51). More recently, lung phenotypes have been described in these individuals and in mouse models that suggest air trapping and lung obstruction that increases with age (52–57). Because WS impacts 24 to 26 genes beyond ELN, individuals with this condition manifest additional features attributable to those genes such as developmental delays, a characteristic hyper-social personality, and hypercalcemia (for review, see Ref. 58).
Mouse models of elastin insufficiency contain a deletion of exon 1 of Eln and 4 kb of the Eln promoter (59, 60). Those mice exhibit overlapping vascular features with humans with SVAS/WS, including narrow caliber vessels, arterial stiffness, and hypertension. Hemizygotes do not, however, exhibit the focal SVAS commonly seen in humans. A newer conditional deletion model, the Tagln-Cre; Elnf/f, in which Eln is deleted from both alleles in smooth muscle cells and displays a phenotype that is more similar to human SVAS, although the exact position of the stenotic lesion in the ascending aorta differs (61, 62).
Autosomal Dominant Cutis Laxa
Cutis laxa describes a heterogeneous class of disorders characterized by skin that is loose, redundant, and inelastic. It can be broadly divided into autosomal recessive or autosomal dominant forms although others, including acquired and X-linked CL, have also been characterized. Most of the genes whose variation imparts the cutis laxa phenotype impact components of the elastic fiber (ELN, EFEMP2, FBLN5, LTBP1, and LTBP4) or decrease the production of mechanically competent elastic fibers by modifying relevant amino acid concentrations, glycosylated side chain addition, or efficient transit through the specialized acidic secretory pathway (ATP6V1A, ALDH18A1, ATP6V0A2, ATP6V1E1, and PYCR1) (63).
The multiple forms of cutis laxa cannot be easily distinguished from one another based on skin presentation, though each carries a varying set of systemic manifestations (63). At the microscopic level, all forms are characterized by a decrease in the number of elastic fibers and fragmentation of those existing fibers (64). On an ultrastructural level, biopsies from patients with ADCL show bare microfibrils with irregular clumps of elastin and can display vacuolization, possibly due to increased fragmentation and/or turnover of the elastic fiber (64).
Unlike other ELN-related disorders, ADCL is thought to be caused by production of poor-quality elastin. The elastic fibers of patients with ADCL have reduced elastin content and are abnormally branched and fragmented (64, 65). As opposed to SVAS and WS that are caused by variants that cause ELN haploinsufficiency, most cases of ADCL result from frameshift variants in the last four exons (human) of elastin. Stable mRNA expression has been detected in human skin fibroblasts for multiple variants (64) and disrupted protein has been detected in the matrix. These frame shift variants are largely expected to extend the C-terminus of TE with disruption of the C-terminal structure and function. Callewaert et al. (65) demonstrate that the elastin in ADCL has impaired deposition into microfibrils and some variants can lead to increased TGFβ signaling and endoplasmic reticulum (ER) stress. Variation impacting exon 32 has been reported to yield less severe outcomes, potentially due to alternative splicing of this exon (64)
In addition to the characteristic skin features of ADCL, affected individuals are also at increased risk of pulmonary and vascular manifestations. Risk for aortic dilation/dissection, for example is 35%, and may or may not be accompanied by bicuspid aortic valve (65). Of particular importance is emphysema, which occurs in 55% of affected individuals and can be mild or severe and contributes significant morbidity and mortality to the condition (65, 66).
Diseases of Elastin Protein Dysfunction
In addition to elastin insufficiency (SVAS) and abnormal elastin deposition (ADCL), elastin turnover and protein dysfunction are involved in a number of common disease processes. Changes in elastin are seen with chronic obstructive pulmonary disease (COPD) (67) and rare ELN variants have been implicated in severe, early onset COPD (68). Exposure to environmental agents such as UV radiation in normal aging (69) and exposure to environmental toxins such as cigarette smoke and air pollution (70) can also disrupt elastin function. Elastin is also susceptible to glycation and disruptions in calcium homeostasis—processes important in the development of cardiovascular disease associated with diabetes (17). Diabetes is also associated with accelerated vascular aging and vascular ECM remodeling, leading to increased levels of elastin degradation peptides (EDP) that may lead to feedback inhibition of elastin synthesis (25, 71). Although conditions such as these are primarily caused by dysfunctional protein and increased rates of turnover, it is possible that the development of therapeutics that increase or re-initiate transcription outside of the usual developmental period could also correct tissues with protein destruction/dysfunction as well.
MECHANISMS OF ELASTIN TRANSCRIPTIONAL REGULATION
Overview
Elastin production is tightly temporally and spatially regulated, with elastin protein deposition largely restricted to fetal and early postnatal life. Although transcriptional controls direct where and when elastin assembly is initiated, fine tuning of elastic fiber quantity and quality is most heavily influenced by posttranscriptional mechanisms. Consequently, control of elastic tissue design is complex, and the balance of these pathways must be considered when estimating the elastogenic potential of a tissue.
ELN Promoter
The ELN promoter (Fig. 2) is GC rich, lacks a conventional TATA box, and utilizes multiple transcription start sites (72). The high GC content and lack of strong TATA box are features of conventional “housekeeping” genes that are ubiquitously and continuously transcribed. Compared with other structural matrix proteins, the coding region of ELN has relatively low sequence homology among mammalian species (73). However, there is a high degree of sequence homology in the 5′ flanking sequence, with 94% homology in the “proximal” promoter from −1 to −192 and 86% homology in the “distal” promoter from −193 to −588, suggesting that this region has an important functional role (74). However, no large-scale genomic studies have been completed on this region to investigate its impact.
Figure 2.
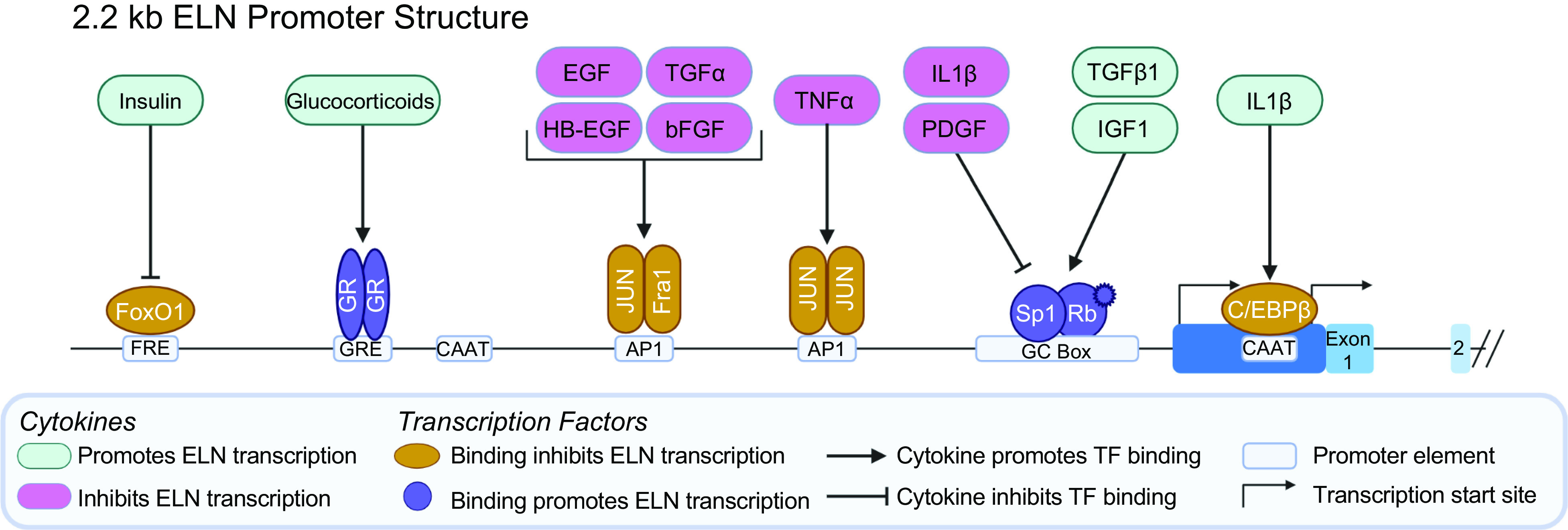
Elastin (ELN, 2.2 kb) gene promoter structure. The human ELN gene lacks a conventional TATA box but contains at least two CAAT boxes and multiple transcription start sites. Several cytokines are known to affect ELN promoter activity. Cytokines that are generally ELN transcription are in green whereas cytokines that generally lead to decreased ELN transcription are in magenta. Arrows represent interactions that promote transcription factor binding to the promoter whereas inhibitory interactions are denoted by arrows with flat heads. Transcription factors that promote ELN transcription are purple, those that inhibit transcription are orange. There are multiple GC boxes that enable Sp1 binding clustered close to the initiation methionine. SP1 interacts with activated Rb to promote transcription in response to TGFb1 and insulin like growth factor-1 (IGF1). Sp1 binding to the promoter is disrupted by IL1b and PDGF. IL1b also promotes binding of C/EBPb to the proximal CAAT box, inhibiting transcription. There are also multiple AP1 binding sites. A proximal AP1 binding site inhibits ELN transcription through the binding of JUN homodimers in response to TNFa while a more distal AP1 site inhibits ELN expression in response to multiple cytokines. There are three glucocorticoid response elements (GRE) that promote transcription in the presence of glucocorticoids such as dexamethasone or hydrocortisone although this appears to be cell type and state specific. Evidence also suggests that insulin can promote ELN transcription through release of inhibitory FoxO1 binding. Created with BioRender.com.
Initial inquiries to identify the basic ELN promoter elements utilized fragments of the first 2.2 kb of the 5′ flanking sequence cloned into a chloramphenicol acetyltransferase (CAT) reporter plasmid and measured CAT activity as a surrogate for promotor activity (4, 72, 75) (summarized in Ref. 76). Using this technique, the basic promoter necessary for ELN transcription was defined as the first 128 bp 5′ to the initiation methionine. Multiple up and down regulatory elements were broadly identified using this technique with subsequent analysis further defining the sequences and transcription factors that modulate ELN promoter activity. The area beyond 2.2 kb upstream of ELN has yet to be studied.
Sequence analysis of the ELN 5′ flanking sequence yields multiple postulated transcription factor binding sequences including multiple Sp1, AP1, AP2, C/EBP binding sites, and TGFβ response elements that may be responsible for the positive and negative promoter activity identified by CAT reporter assay (4, 72, 75). However, modern sequencing and genomic techniques such as chromatin immunoprecipitation (ChIP) sequencing data available through the ENCODE Project (77) and predicted transcription factor binding sites available through the JASPAR CORE collection database (78) indicate a multitude of other transcription factors including WT1, transcription factors that interact with retinoblastoma, the multifunctional CTCF transcription factor, RAS responsive transcription factors, and members of histone modifying complexes may bind to the ELN promoter region.
TGFβ1 signaling is involved in numerous disease processes and is important in the regulation of the ECM. With regard to ELN, TGFβ1 can affect TE production through several mechanisms including regulation of transcriptional initiation, posttranscriptional mRNA processing and stability, and even mature elastin turnover. On a transcriptional level, TGFβ1 increases ELN mRNA transcription (79–83), potentially through the phosphatidylinostitol 3-kinase (PI3K)/AKT/p38 signaling pathway (84). However, the effect of TGFβ1 on transcription is cell type and cell state specific. For example, in the skin, exogenous administration of TGFβ1 in a transgenic mouse model leads to increased expression of a reporter construct (85), but in cultured human skin fibroblasts, a direct impact on transcription is no longer appreciated (80). Similarly, supplementation with TGFβ1 yields increased the levels of Eln mRNA and soluble TE in rat neonatal lung fibroblasts, but no change was observed in either mRNA or soluble TE in adult rat lung fibroblasts or smooth muscle cells (neonatal or adult) (81).
Insulin-like growth factor-I (IGF1) is also a positive regulator of Eln transcription in chicken and in rats (86–88). This effect also appears to be cell type specific, with no change in Eln mRNA levels in pulmonary fibroblasts despite an increase in mRNA levels in neonatal aortic smooth muscle cells (86, 89). IGF1 regulation of ELN mRNA levels is mediated via binding of Sp1 binding to the proximal Eln promoter in rats (88, 90). This effect is mediated by promotion of retinoblastoma (Rb) threonine-821 phosphorylation via a cyclinE-cyclin-dependent kinase 2 complex (91, 92).
Inhibition of ELN transcription is achieved through binding of the AP1 complex, composed of Fra1 and Jun heterodimers or Jun homodimers. There are two proposed AP1 binding sites, a site located more proximally at −229 to −223 and a more distal AP1 binding site at −564 to −558 (93). Binding of the AP1 complex is mediated by several cytokines and the proximal and distal site appear to act independently. Basic fibroblast growth factor (bFGF) decreases Eln mRNA levels in rat aortic smooth muscle cells (94, 95) and in rat pulmonary fibroblasts (96) via binding of the AP1 complex to the distal AP1 binding site. TNF-α similarly inhibits ELN mRNA via AP1 complex but acts via the more proximal AP1 binding site (93). Interestingly, stretch has been shown to activate Jun, but in rat pulmonary arterial adventitial fibroblasts, stretch increases Eln mRNA levels, an effect that may be attributed to increased activation of PKC having a stimulatory effect on Eln (97).
Glucocorticoids such as dexamethasone have long been used in cell culture to promote TE protein production (98). Del Monaco et al. (99) demonstrated the presence of three potential glucocorticoid response elements in the distal ELN promoter (at positions −1,018 to −1,023, −1,310 to −1,315, and −1,432 to −1,437) and demonstrated glucocorticoid receptor binding. They further demonstrated that those elements, when cloned into a CAT reporter construct and transfected into skin fibroblasts, increased CAT activity, thereby suggesting they are capable of initiating transcription. However, the effect of glucocorticoid on TE protein production is influenced by cell state (100) and that glucocorticoids also affect mRNA stability (101) and may involve transcriptional repression by microRNA (102).
Additional cytokines and signaling pathways have also been described, including insulin releasing FoxO1 mediated promoter inhibition and IL1β-mediated C/EBPβ binding to the proximal promoter (for review, see Ref. 103); more are likely yet to be discovered. These studies highlight the complexity of the elastogenic response depending on cell type and experimental model. The mechanisms that confer specificity to those responses have yet to be elucidated. Furthermore, although these studies demonstrate the effect of cytokines and promoter elements on ELN transcription, the “master switch” responsible for inducing ELN transcription in late gestation has yet to be determined. Studies of the ELN promoter to date have been limited to a region within 2.2 kb of the initiation methionine. There is a large (>150 kb) intragenic region between ELN and its closest 5′ genes (TMEM270, METTL27, and CLDN4) that has yet to be explored but may yield clues to the tissue and temporal specific regulation of ELN expression (104).
ELN mRNA Stability
Although a transcriptional switch is likely necessary to initiate TE production in elastogenic tissues, ELN pre-mRNA continues to be transcribed long after TE protein production ceases (105–108). This effect is seen across species (109). The decrease in protein production is secondary to a rapid decline in the half-life of mature Eln mRNA in the postadolescent period (110) (Fig. 1). The mechanisms that regulate mature ELN mRNA stability and the purpose of continuous transcription in the absence of TE production remain unclear.
In addition to its role in transcriptional regulation, TGFβ1 is also one of the best-studied factors influencing ELN mRNA stability. Its proelastogenic effect has been shown in multiple elastic cell types, including fibroblasts from lung (111) as well as vascular smooth muscle cells (79) and skin fibroblasts (80, 83). Data from these studies showed mature ELN mRNA levels increased and TE protein was produced even after the addition of a transcriptional inhibitor. The effect of TGFβ1 on ELN mRNA stability requires the protein kinase C, p38, and active SMADs (112).
In addition to TGFβ1, other cytokines have also been demonstrated to affect ELN mRNA stability. Glucocorticoids, for example, decrease ELN mRNA stability in some experiments (101). This may contribute to skin atrophy and loss of elastic fibers with prolonged topical glucocorticoid therapy (113). Transforming growth factor-α may have similar dual role in regulating Eln promoter activity and mRNA stability in rats (114).
The mechanisms by which ELN mRNA stability and turnover is regulated are incompletely understood. Zhang et al. (110) identified a sequence in exon 30 of Eln that interacts with a 50-kDa cytosolic protein in rats. Binding of the protein is associated with increased destruction of the mature TE mRNA. TGFβ1 decreases binding of the 50-kDa protein to the TE mRNA, thereby lengthening its half-life. Likewise, Hew et al. (108) identified a sequence in the 3′ untranslated region (UTR) of chicken Eln that has also been demonstrated to bind both nuclear and cytosolic proteins and is associated with increased tropoelastin mRNA stability. However, neither the exon 30 nor the 3′ UTR binding proteins have been identified; subsequent studies transitioned away from protein-mediated (in)stability mechanisms and instead focused on the impact of miRNAs that bind close to the 3′ protein-binding sequence.
Role of microRNA
MicroRNAs (miRNAs) are short (average of 22 nucleotide), noncoding RNA molecules that have been implicated in the regulation of a vast number of biologic processes and diseases. Their most well-established function is regulation of gene expression through binding to the 3′ UTR of target mRNA, thereby leading to posttranscriptional repression and via the miRISC complex leading to cleavage of the target mRNA (115, 116). MicroRNAs have long been implicated in a number of cardiovascular diseases caused or impacted by changes in ECM/elastic fibers including arterial stiffness (117), abdominal aortic aneurysms (AAA) (118), and AAA dissection (119, 120). The 3′ UTR of the ELN gene contains multiple miRNA binding sites and available evidence suggests that miRNA play an active role in the modulation of ELN mRNA steady-state concentrations and TE protein production.
The effect of the miR-29 family of microRNA is most well established but other microRNAs have also been linked to elastin regulation (118, 119). The miR-29 family consists of three miRNAs expressed from two bicistronic clusters where miR-29a and miR-29b1 are co-expressed from one cluster and miR-29b2 and miR-29c are expressed from a second (121). miR-29 family members have a conserved seed sequence (or DNA target) and their predicted targets are enriched for ECM proteins, including elastin (122–124), COL1A1, COL3A1, and VEGF-A (117, 121).
Experimental studies have validated the role of miR-29 family members in the regulation of elastin mRNA stability and TE protein production. Transfection of dermal fibroblasts and or pulmonary artery smooth muscle cells (SMCs) with miR-29a, miR-29b, and miR-29c mimics leads to decreased mature elastin mRNA levels (125). Conversely, treatment with miR-29a antigomers increases elastin mRNA levels (miR-29b and miR29c antigomers were not studied). The authors found that dermal fibroblasts from two patients with WS treated with anti-miR29a had an increase in ELN mRNA and TE protein (125).
Although miRNA is an attractive therapeutic target, the utility of miRNA mimetics or anti-miRNA is limited by the inherent lack of specificity. miRNA target sequences are short, and a single miRNA species can target multiple genes. This may enable coordinated modulation of the cumulative elastogenic machinery during development but limits specificity when applied to elastin-specific regenerative medicine strategies. Moreover, while in vitro studies suggest that miRNAs may orchestrate large amplitude changes in the concentration of their target mRNAs, the effect of miRNA in vivo is thought to produce more incremental changes in gene expression and thus may not be expected to account for the near total loss of TE protein production in adult tissues. Likewise, inhibiting miRNA may not be adequate to restore ELN temporally specific expression in cases of elastin insufficiency.
ELN Splicing
Across species, the ELN gene has significant variation in nucleotide sequence but maintains a characteristic pattern where every exon contains a multiple of three nucleotides, with the exception of exon 1 which contains 3n + 1 nucleotides (126–128). This facilitates alternative splicing of exons whereby an entire exon can be removed from the ELN pre-mRNA without changing the open reading frame, leaving the amino acid sequence following the splice unchanged. In addition to simple exon excision or alternative splicing, ELN isoforms also utilize alternate donor or acceptor sites that lengthen or shorten exons (specifically exons 8, 20, 24, and 26) while also retaining the open reading frame.
To date, more than 30 human isoforms have been identified through RNA studies [GTEx (https://www.gtexportal.org/home/), Ensembl https://useast.ensembl.org/Homo_sapiens/Info/Index, RefSeq (https://www.ncbi.nlm.nih.gov/refseq/)] (examples shown in Fig. 1). The relative quantity of each isoform varies across tissues, although exon 22 is almost uniformly removed (129). It has long been speculated that isoforms created by alternate splicing change the physical and mechanical properties of the elastic fiber (127, 129, 130). However, the functional consequences of the multiple isoforms, and whether each isoform is translated into protein at all, remains unclear.
In vitro studies have demonstrated alteration of the physical properties of elastin monomers and elastic fibers (131–134). Although not typically spliced in vivo, deletion of exon 30 alters elastin microassembly and can affect the stiffness and viscoelastic properties of elastic fibers (131, 132). Similarly, deletion of exon 36 of bovine Eln (analogous to exon 34 in humans) leads to elastic fibers with decreased desmosine crosslinks (132, 135). The importance of splicing is also illustrated by naturally occurring human splice site variants (mutations) associated with SVAS. Splice site variants, in particular those in splice donor sites, may lead to intron readthrough and creation of an alternate transcript with a premature stop codon, which would then be predicted to be destroyed by nonsense-mediated decay. For some variants, the impact is less clear. For example, there is evidence that the c.800-3C>G variant alters splicing, leading to production of a transcript that lacks exon 16 and 17 (exons that are not typically subjected to alternative splicing) and exhibits impaired incorporation into elastic fibers (34). Interestingly, work from another laboratory, using a different RT-PCR primer set suggests that this variant leads to the generation of a transcript that would undergo nonsense-mediated decay (32). There is also an ultrarare single nucleotide variant (ELN c.1358-3G>A) that alters the splice acceptor site in exon 22, which is almost ubiquitously removed (spliced out) that experimentally increases retention of exon 22. Inclusion of this exon increases the hydrodynamic radius of TE and results in decreased stress relaxation (134). Although numerous splice variants have been associated with SVAS, there is evidence of reduced penetrance with this variant type and overall experimental evidence is lacking. Furthermore, although they seem to be expressed, the correlation between differences in RNA species and actual differences in deposited TE is unclear (6).
Unfortunately, the existing studies largely fail to address the effect of splicing on those exons that most commonly undergo alternative splicing in vivo (exons 3, 5, 11, 13, 22, 25, and 32), whose impact on tissue mechanics and matrix associated protein interactions may be more subtle. Nor has any study investigated the role of short transcripts such as those identified in the GTEx project as having high expression in the adult human aorta (136). Given the continual transcription of the ELN gene once the gene is activated in a tissue, it is intriguing to consider a nonstructural role for these alternative transcripts. The effect of isoform expression and the effect on matrix associated proteins has not been studied, but must also be considered.
Emerging Mechanisms of ELN Regulation
ELN gene transcription occurs in tissue-specific and temporally regulated patterns, but the factors that give rise to that specificity remain elusive. Additional knowledge in this area is critical for the development of new therapies for both rare diseases of elastic fibers and more common conditions like emphysema, atherosclerosis, or even typical aging where elastic fibers are destroyed. To address these goals directly, we propose the need for additional investigation in the following areas.
Transcriptional switch.
Why the immediate ELN promoter is structured like an always on, “housekeeping” gene, but the mature ELN mRNA and TE protein production are tightly regulated remains uncertain. Targeted studies have identified cytokines and their downstream transcription factors that modulate ELN promoter activity, but the master regulator(s) that control the “on switch” timing and tissue specificity of elastin expression are unknown. There is emerging evidence that epigenetic modifications and noncoding variants play a role. To answer this question, it may be necessary to utilize new technologies and evaluate the genome beyond the established ELN promoter.
There is a large (roughly 160 kb) intergenic region upstream of ELN that has yet to be interrogated. ELN is embedded in a differentially methylated region (DMR) extending ∼61 kb upstream that has increased methylation in aged, atherosclerotic vessels (137). In addition, GWAS studies have identified single nucleotide polymorphisms (SNPs) from the 3′ UTR of ELN to 124 kb upstream of the ELN locus that are associated with vascular and connective tissue outcomes (19, 20, 22–24). Although the impact of the specific variants is unknown, linkage to SNPs at such a distance suggests that complex long-range modulation of ELN is likely. It is also possible that this intergenic large region contains important enhancer elements that control three-dimensional folding of chromatin that affects ELN expression. Focused genomic interrogation of the region surrounding elastin as well as ChIP assays and other modern modalities may shed light on the effect of noncoding variants on ELN expression and, importantly, may help us understand key drivers of ELN-mediated disease (138).
RNA stability.
Once the ELN gene is turned on, it is transcribed at a near constant rate despite a lack of TE protein production after adolescence due to rapid turnover of ELN mRNA. There is evidence that microRNAs play a role in targeting ELN mRNA for degradation, but it is unclear if microRNAs alone are capable of reducing the level of mature ELN mRNA to sufficiently inhibit production of TE protein. Sequences within the ELN gene have been identified that bind to cytosolic or nuclear proteins and confer stability or increase turnover, however those proteins have not been identified (108, 110). It is possible that one or more of these proteins is somehow related to the microRNA degradation machinery. However, this has not been investigated, nor have other mRNA stability/degradation pathways. RNA purification followed by mass-spectroscopy or other techniques such as in cell protein-RNA interaction (incPRINT) may help identify RNA binding proteins responsible for determining the fate of ELN mRNA transcripts (139, 140). Elucidation of these proteins and pathways may further identify possible targets to increase TE protein production in diseases of elastin insufficiency.
Splice variants.
The ELN gene structure allows for the production of a multitude of alternatively spliced isoforms. Indeed, over 30 splice isoforms have been described to date (5) including short splice isoforms and those with retained introns. It is also clear that alternative splicing changes with development and that deregulation of splicing occurs in numerous diseases (141, 142). However, ELN splicing across tissues and development has not been extensively studied. Comprehensive studies of ELN mRNA and TE protein splicing patterns using modern technologies such as long-read RNA sequencing are necessary to understand the tissue and temporal patterns of ELN splicing and the impact alternative splicing has on elastic tissue structure. Furthermore, additional studies are needed to evaluate the direct effect of various isoforms on tissue mechanics. These studies may be particularly informative for regenerative medicine projects aiming to create precisely mechanically tuned structures. Likewise, investigations of alternatively spliced isoforms may illustrate differential interactions with matrix-associated proteins.
Interestingly, there are multiple short splice isoforms transcribed, some at high levels, the function of which is entirely unknown. Emerging studies have highlighted the noncoding function of splice isoforms (141, 143, 144). Given the dichotomy of a TE protein that is only produced during a short temporal burst but ongoing gene transcription, it is possible that these short highly transcribed ELN isoforms are performing alternate functions within the cell. Modern techniques such as single molecule mRNA fluorescence in situ hybridization (FISH) may shed light on noncoding functions of the short splice isoforms observed in ELN.
Conclusions
Earlier studies provide a glimpse at the complex processes that underlie ELN regulation, but many questions remain and advances in genetic and molecular techniques are poised to further elucidate these mechanisms. Further understanding the systems that regulate ELN expression may help develop therapeutics to treat both rare ELN-mediated diseases such as WS, SVAS, and ADCL and may even help treat common conditions such as solar elastosis (aging skin), atherosclerosis, and wound healing.
GRANTS
This study was funded by National Heart, Lung, and Blood Institute Grant HL006212 (to B.A.K.) and Postdoctoral Fellowship T32 HL125241 (to S.S.P.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.S.P. and B.A.K. analyzed data; S.S.P. and B.A.K. interpreted results of experiments; S.S.P. prepared figures; S.S.P. and B.A.K. drafted manuscript; S.S.P. and B.A.K. edited and revised manuscript; S.S.P. and B.A.K. approved final version of manuscript.
ACKNOWLEDGMENTS
The data used for analysis of ELN splice isoform abundance described in this manuscript were obtained from the GTEx Portal on 04/22/22, dbGaP Accession Number phs000424.v8.p2.
REFERENCES
- 1. Hoeve CA, Flory PJ. The elastic properties of elastin. Biopolymers 13: 677–686, 1974. doi: 10.1002/bip.1974.360130404. [DOI] [PubMed] [Google Scholar]
- 2. Biesecker LG, Adam MP, Alkuraya FS, Amemiya AR, Bamshad MJ, Beck AE, et al. A dyadic approach to the delineation of diagnostic entities in clinical genomics. Am J Hum Genet 108: 8–15, 2021. doi: 10.1016/j.ajhg.2020.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Szabó Z, Levi-Minzi SA, Christiano AM, Struminger C, Stoneking M, Batzer MA, Boyd CD. Sequential loss of two neighboring exons of the tropoelastin gene during primate evolution. J Mol Evol 49: 664–671, 1999. doi: 10.1007/pl00006587. [DOI] [PubMed] [Google Scholar]
- 4. Kähäri VM, Fazio MJ, Chen YQ, Bashir MM, Rosenbloom J, Uitto J. Deletion analyses of 5'-flanking region of the human elastin gene. Delineation of functional promoter and regulatory cis-elements. J Biol Chem 265: 9485–9490, 1990. [PubMed] [Google Scholar]
- 5. Reichheld SE, Muiznieks LD, Lu R, Sharpe S, Keeley FW. Sequence variants of human tropoelastin affecting assembly, structural characteristics and functional properties of polymeric elastin in health and disease. Matrix Biol 84: 68–80, 2019. doi: 10.1016/j.matbio.2019.06.010. [DOI] [PubMed] [Google Scholar]
- 6. Hedtke T, Schräder CU, Heinz A, Hoehenwarter W, Brinckmann J, Groth T, Schmelzer CEH. A comprehensive map of human elastin cross-linking during elastogenesis. FEBS J 286: 3594–3610, 2019. doi: 10.1111/febs.14929. [DOI] [PubMed] [Google Scholar]
- 7. Duque Lasio ML, Kozel BA. Elastin-driven genetic diseases. Matrix Biol 71–72: 144–160, 2018. doi: 10.1016/j.matbio.2018.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Davis EC, Mecham RP. Intracellular trafficking of tropoelastin. Matrix Biol 17: 245–254, 1998. doi: 10.1016/s0945-053x(98)90078-6. [DOI] [PubMed] [Google Scholar]
- 9. Kozel BA, Rongish BJ, Czirok A, Zach J, Little CD, Davis EC, Knutsen RH, Wagenseil JE, Levy MA, Mecham RP. Elastic fiber formation: a dynamic view of extracellular matrix assembly using timer reporters. J Cell Physiol 207: 87–96, 2006. doi: 10.1002/jcp.20546. [DOI] [PubMed] [Google Scholar]
- 10. Ozsvar J, Yang C, Cain SA, Baldock C, Tarakanova A, Weiss AS. Tropoelastin and elastin assembly. Front Bioeng Biotechnol 9: 643110, 2021. doi: 10.3389/fbioe.2021.643110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dyksterhuis LB, Carter EA, Mithieux SM, Weiss AS. Tropoelastin as a thermodynamically unfolded premolten globule protein: the effect of trimethylamine N-oxide on structure and coacervation. Arch Biochem Biophys 487: 79–84, 2009. doi: 10.1016/j.abb.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 12. Urry DW, Long MM, Gross E. Conformations of the repeat peptides of elastin in solution: an application of proton and carbon-13 magnetic resonance to the determination of polypeptide secondary structure. CRC Crit Rev Biochem 4: 1–45, 1976. doi: 10.3109/10409237609102557. [DOI] [PubMed] [Google Scholar]
- 13. Muiznieks LD, Weiss AS, Keeley FW. Structural disorder and dynamics of elastin. Biochem Cell Biol 88: 239–250, 2010. doi: 10.1139/o09-161. [DOI] [PubMed] [Google Scholar]
- 14. Shapiro SD, Endicott SK, Province MA, Pierce JA, Campbell EJ. Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of d-aspartate and nuclear weapons-related radiocarbon. J Clin Invest 87: 1828–1834, 1991. doi: 10.1172/JCI115204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fhayli W, Boëté Q, Harki O, Briancon-Marjollet A, Jacob MP, Faury G. Rise and fall of elastic fibers from development to aging. Consequences on arterial structure-function and therapeutical perspectives. Matrix Biol 84: 41–56, 2019. doi: 10.1016/j.matbio.2019.08.005. [DOI] [PubMed] [Google Scholar]
- 16. Mammoto A, Matus K, Mammoto T. Extracellular matrix in aging aorta. Front Cell Dev Biol 10: 822561, 2022. doi: 10.3389/fcell.2022.822561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang C, Weiss AS, Tarakanova A. Changes in elastin structure and extensibility induced by hypercalcemia and hyperglycemia. Acta Biomater 2022. doi: 10.1016/j.actbio.2022.03.041. [DOI] [PubMed] [Google Scholar]
- 18. Graul-Neumann LM, Hausser I, Essayie M, Rauch A, Kraus C. Highly variable cutis laxa resulting from a dominant splicing mutation of the elastin gene. Am J Med Genet A 146: 977–983, 2008. doi: 10.1002/ajmg.a.32242. [DOI] [PubMed] [Google Scholar]
- 19. Pirruccello JP, Chaffin MD, Chou EL, Fleming SJ, Lin H, Nekoui M, Khurshid S, Friedman SF, Bick AG, Arduini A, Weng LC, Choi SH, Akkad AD, Batra P, Tucker NR, Hall AW, Roselli C, Benjamin EJ, Vellarikkal SK, Gupta RM, Stegmann CM, Juric D, Stone JR, Vasan RS, Ho JE, Hoffmann U, Lubitz SA, Philippakis AA, Lindsay ME, Ellinor PT. Deep learning enables genetic analysis of the human thoracic aorta. Nat Genet 54: 40–51, 2022. doi: 10.1038/s41588-021-00962-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Benjamins JW, Yeung MW, van de Vegte YJ, Said MA, van der Linden T, Ties D, Juarez-Orozco LE, Verweij N, van der Harst P. Genomic insights in ascending aortic size and distensibility. EBioMedicine 75: 103783, 2022. doi: 10.1016/j.ebiom.2021.103783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Surendran P, Feofanova EV, Lahrouchi N, Ntalla I, Karthikeyan S, Cook J, et al. Discovery of rare variants associated with blood pressure regulation through meta-analysis of 1.3 million individuals. Nat Genet 52: 1314–1332, 2020. doi: 10.1038/s41588-020-00713-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schafmayer C, Harrison JW, Buch S, Lange C, Reichert MC, Hofer P, et al. Genome-wide association analysis of diverticular disease points towards neuromuscular, connective tissue and epithelial pathomechanisms. Gut 68: 854–865, 2019. doi: 10.1136/gutjnl-2018-317619. [DOI] [PubMed] [Google Scholar]
- 23. Kichaev G, Bhatia G, Loh PR, Gazal S, Burch K, Freund MK, Schoech A, Pasaniuc B, Price AL. Leveraging polygenic functional enrichment to improve GWAS power. Am J Hum Genet 104: 65–75, 2019. doi: 10.1016/j.ajhg.2018.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sakaue S, Kanai M, Tanigawa Y, Karjalainen J, Kurki M, Koshiba S, et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet 53: 1415–1424, 2021. doi: 10.1038/s41588-021-00931-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Duca L, Blaise S, Romier B, Laffargue M, Gayral S, El Btaouri H, Kawecki C, Guillot A, Martiny L, Debelle L, Maurice P. Matrix ageing and vascular impacts: focus on elastin fragmentation. Cardiovasc Res 110: 298–308, 2016. doi: 10.1093/cvr/cvw061. [DOI] [PubMed] [Google Scholar]
- 26. Schmelzer CEH, Duca L. Elastic fibers: formation, function, and fate during aging and disease. FEBS J 66: 101255, 2021. doi: 10.1016/j.arr.2021.101255. [DOI] [PubMed] [Google Scholar]
- 27. Metcalfe K, Rucka AK, Smoot L, Hofstadler G, Tuzler G, McKeown P, Siu V, Rauch A, Dean J, Dennis N, Ellis I, Reardon W, Cytrynbaum C, Osborne L, Yates JR, Read AP, Donnai D, Tassabehji M. Elastin: mutational spectrum in supravalvular aortic stenosis. Eur J Hum Genet 8: 955–963, 2000. doi: 10.1038/sj.ejhg.5200564. [DOI] [PubMed] [Google Scholar]
- 28. Park S, Seo EJ, Yoo HW, Kim Y. Novel mutations in the human elastin gene (ELN) causing isolated supravalvular aortic stenosis. Int J Mol Med 18: 329–332, 2006. doi: 10.3892/ijmm.18.2.329. [DOI] [PubMed] [Google Scholar]
- 29. Micale L, Turturo MG, Fusco C, Augello B, Jurado LA, Izzi C, Digilio MC, Milani D, Lapi E, Zelante L, Merla G. Identification and characterization of seven novel mutations of elastin gene in a cohort of patients affected by supravalvular aortic stenosis. Eur J Hum Genet 18: 317–323, 2010. doi: 10.1038/ejhg.2009.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li DY, Toland AE, Boak BB, Atkinson DL, Ensing GJ, Morris CA, Keating MT. Elastin point mutations cause an obstructive vascular disease, supravalvular aortic stenosis. Hum Mol Genet 6: 1021–1028, 1997. doi: 10.1093/hmg/6.7.1021. [DOI] [PubMed] [Google Scholar]
- 31. Tassabehji M, Metcalfe K, Donnai D, Hurst J, Reardon W, Burch M, Read AP. Elastin: genomic structure and point mutations in patients with supravalvular aortic stenosis. Hum Mol Genet 6: 1029–1036, 1997. doi: 10.1093/hmg/6.7.1029. [DOI] [PubMed] [Google Scholar]
- 32. Urbán Z, Michels VV, Thibodeau SN, Donis-Keller H, Csiszár K, Boyd CD. Supravalvular aortic stenosis: a splice site mutation within the elastin gene results in reduced expression of two aberrantly spliced transcripts. Hum Genet 104: 135–142, 1999. doi: 10.1007/s004390050926. [DOI] [PubMed] [Google Scholar]
- 33. Urbán Z, Zhang J, Davis EC, Maeda GK, Kumar A, Stalker H, Belmont JW, Boyd CD, Wallace MR. Supravalvular aortic stenosis: genetic and molecular dissection of a complex mutation in the elastin gene. Hum Genet 109: 512–520, 2001. doi: 10.1007/s00439-001-0608-z. [DOI] [PubMed] [Google Scholar]
- 34. Wachi H, Sato F, Nakazawa J, Nonaka R, Szabo Z, Urban Z, Yasunaga T, Maeda I, Okamoto K, Starcher BC, Li DY, Mecham RP, Seyama Y. Domains 16 and 17 of tropoelastin in elastic fibre formation. Biochem J 402: 63–70, 2007. doi: 10.1042/BJ20061145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Strømme P, Bjørnstad PG, Ramstad K. Prevalence estimation of Williams syndrome. J Child Neurol 17: 269–271, 2002. doi: 10.1177/088307380201700406. [DOI] [PubMed] [Google Scholar]
- 36. Beuren AJ, Apitz J, Harmjanz D. Supravalvular aortic stenosis in association with mental retardation and a certain facial appearance. Circulation 26: 1235–1240, 1962. doi: 10.1161/01.cir.26.6.1235. [DOI] [PubMed] [Google Scholar]
- 37. Williams JC, Barratt-Boyes BG, Lowe JB. Supravalvular aortic stenosis. Circulation 24: 1311–1318, 1961. doi: 10.1161/01.cir.24.6.1311. [DOI] [PubMed] [Google Scholar]
- 38. Collins RT. Cardiovascular disease in Williams syndrome. Curr Opin Pediatr 30: 609–615, 2018. doi: 10.1097/MOP.0000000000000664. [DOI] [PubMed] [Google Scholar]
- 39. Parrish PCR, Liu D, Knutsen RH, Billington CJ, Mecham RP, Fu YP, Kozel BA. Whole exome sequencing in patients with Williams-Beuren syndrome followed by disease modeling in mice points to four novel pathways that may modify stenosis risk. Hum Mol Genet 29: 2035–2050, 2020. doi: 10.1093/hmg/ddaa093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sadler LS, Pober BR, Grandinetti A, Scheiber D, Fekete G, Sharma AN, Urbán Z. Differences by sex in cardiovascular disease in Williams syndrome. J Pediatr 139: 849–853, 2001. doi: 10.1067/mpd.2001.118889. [DOI] [PubMed] [Google Scholar]
- 41. Cha SG, Song MK, Lee SY, Kim GB, Kwak JG, Kim WH, Bae EJ. Long-term cardiovascular outcome of Williams syndrome. Congenit Heart Dis 14: 684–690, 2019. doi: 10.1111/chd.12810. [DOI] [PubMed] [Google Scholar]
- 42. Radford DJ, Pohlner PG. The middle aortic syndrome: an important feature of Williams' syndrome. Cardiol Young 10: 597–602, 2000. doi: 10.1017/s1047951100008878. [DOI] [PubMed] [Google Scholar]
- 43. Wessel A, Gravenhorst V, Buchhorn R, Gosch A, Partsch CJ, Pankau R. Risk of sudden death in the Williams-Beuren syndrome. Am J Med Genet A 127A: 234–237, 2004. doi: 10.1002/ajmg.a.30012. [DOI] [PubMed] [Google Scholar]
- 44. Gupta P, Tobias JD, Goyal S, Miller MD, Melendez E, Noviski N, De Moor MM, Mehta V. Sudden cardiac death under anesthesia in pediatric patient with Williams syndrome: a case report and review of literature. Ann Card Anaesth 13: 44–48, 2010. doi: 10.4103/0971-9784.58834. [DOI] [PubMed] [Google Scholar]
- 45. Bird LM, Billman GF, Lacro RV, Spicer RL, Jariwala LK, Hoyme HE, Zamora-Salinas R, Morris C, Viskochil D, Frikke MJ, Jones MC. Sudden death in Williams syndrome: report of ten cases. J Pediatr 129: 926–931, 1996. doi: 10.1016/s0022-3476(96)70042-2. [DOI] [PubMed] [Google Scholar]
- 46. Bouchireb K, Boyer O, Bonnet D, Brunelle F, Decramer S, Landthaler G, Liutkus A, Niaudet P, Salomon R. Clinical features and management of arterial hypertension in children with Williams-Beuren syndrome. Nephrol Dial Transplant 25: 434–438, 2010. doi: 10.1093/ndt/gfp522. [DOI] [PubMed] [Google Scholar]
- 47. Kozel BA, Danback JR, Waxler JL, Knutsen RH, de Las Fuentes L, Reusz GS, Kis E, Bhatt AB, Pober BR. Williams syndrome predisposes to vascular stiffness modified by antihypertensive use and copy number changes in NCF1. Hypertension 63: 74–79, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rose C, Wessel A, Pankau R, Partsch CJ, Bürsch J. Anomalies of the abdominal aorta in Williams-Beuren syndrome—another cause of arterial hypertension. Eur J Pediatr 160: 655–658, 2001. doi: 10.1007/s004310100835. [DOI] [PubMed] [Google Scholar]
- 49. Furusawa EA, Esposito CSL, Honjo RS, Suzuki L, Leal GN, Kim CA, Schvartsman BGS. Diagnosis and management of systemic hypertension due to renovascular and aortic stenosis in patients with Williams-Beuren syndrome. Rev Assoc Med Bras (1992) 64: 723–728, 2018. doi: 10.1590/1806-9282.64.08.723. [DOI] [PubMed] [Google Scholar]
- 50. Morris CA, Demsey SA, Leonard CO, Dilts C, Blackburn BL. Natural history of Williams syndrome: physical characteristics. J Pediatr 113: 318–326, 1988. doi: 10.1016/s0022-3476(88)80272-5. [DOI] [PubMed] [Google Scholar]
- 51. Kozel BA, Bayliss SJ, Berk DR, Waxler JL, Knutsen RH, Danback JR, Pober BR. Skin findings in Williams syndrome. Am J Med Genet A 164: 2217–2225, 2014. doi: 10.1002/ajmg.a.36628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wojcik MH, Carmichael N, Bieber FR, Wiener DC, Madan R, Pober BR, Raby BA. A new diagnosis of Williams-Beuren syndrome in a 49-year-old man with severe bullous emphysema. Am J Med Genet A 173: 2235–2239, 2017. doi: 10.1002/ajmg.a.38289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kronquist EK, Kaur M, Gober LM, Knutsen RH, Fu YP, Yu ZX, Donahue DR, Chen MY, Osgood S, Raja N, Levin MD, Barochia A, Kozel BA. Airflow obstruction in adults with Williams syndrome and mice with elastin insufficiency. Diagnostics (Basel) 12: 1438, 2022. doi: 10.3390/diagnostics12061438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cho MH, Ciulla DM, Klanderman BJ, Hersh CP, Litonjua AA, Sparrow D, Raby BA, Silverman EK. Analysis of exonic elastin variants in severe, early-onset chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 40: 751–755, 2009. doi: 10.1165/rcmb.2008-0340OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kelleher CM, Silverman EK, Broekelmann T, Litonjua AA, Hernandez M, Sylvia JS, Stoler J, Reilly JJ, Chapman HA, Speizer FE, Weiss ST, Mecham RP, Raby BA. A functional mutation in the terminal exon of elastin in severe, early-onset chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 33: 355–362, 2005. doi: 10.1165/rcmb.2005-0206OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wan ES, Pober BR, Washko GR, Raby BA, Silverman EK. Pulmonary function and emphysema in Williams-Beuren syndrome. Am J Med Genet A 152: 653–656, 2010. doi: 10.1002/ajmg.a.33300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pangallo E, Cianci P, Favuzza F, Milani D, Vimercati C, Moretti A, Picchi R, De Paoli A, Agosti M, Selicorni A. Pulmonary function in Williams-Beuren syndrome: spirometric data of 22 Italian patients. Am J Med Genet A 185: 390–396, 2021. doi: 10.1002/ajmg.a.61966. [DOI] [PubMed] [Google Scholar]
- 58. Kozel BA, Barak B, Kim CA, Mervis CB, Osborne LR, Porter M, Pober BR. Williams syndrome. Nat Rev Dis Primers 7: 42, 2021. doi: 10.1038/s41572-021-00276-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li DY, Faury G, Taylor DG, Davis EC, Boyle WA, Mecham RP, Stenzel P, Boak B, Keating MT. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest 102: 1783–1787, 1998. doi: 10.1172/JCI4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature 393: 276–280, 1998. doi: 10.1038/30522. [DOI] [PubMed] [Google Scholar]
- 61. Lin CJ, Staiculescu MC, Hawes JZ, Cocciolone AJ, Hunkins BM, Roth RA, Lin CY, Mecham RP, Wagenseil JE. Heterogeneous cellular contributions to elastic laminae formation in arterial wall development. Circ Res 125: 1006–1018, 2019. doi: 10.1161/CIRCRESAHA.119.315348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lin CJ, Hunkins BM, Roth RA, Lin CY, Wagenseil JE, Mecham RP. Vascular smooth muscle cell subpopulations and neointimal formation in mouse models of elastin insufficiency. Arterioscler Thromb Vasc Biol 41: 2890–2905, 2021. doi: 10.1161/ATVBAHA.120.315681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Beyens A, Boel A, Symoens S, Callewaert B. Cutis laxa: a comprehensive overview of clinical characteristics and pathophysiology. Clin Genet 99: 53–66, 2021. doi: 10.1111/cge.13865. [DOI] [PubMed] [Google Scholar]
- 64. Tassabehji M, Metcalfe K, Hurst J, Ashcroft GS, Kielty C, Wilmot C, Donnai D, Read AP, Jones CJ. An elastin gene mutation producing abnormal tropoelastin and abnormal elastic fibres in a patient with autosomal dominant cutis laxa. Hum Mol Genet 7: 1021–1028, 1998. doi: 10.1093/hmg/7.6.1021. [DOI] [PubMed] [Google Scholar]
- 65. Callewaert B, Renard M, Hucthagowder V, Albrecht B, Hausser I, Blair E, Dias C, Albino A, Wachi H, Sato F, Mecham RP, Loeys B, Coucke PJ, De Paepe A, Urban Z. New insights into the pathogenesis of autosomal-dominant cutis laxa with report of five ELN mutations. Hum Mutat 32: 445–455, 2011. doi: 10.1002/humu.21462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Corbett E, Glaisyer H, Chan C, Madden B, Khaghani A, Yacoub M. Congenital cutis laxa with a dominant inheritance and early onset emphysema. Thorax 49: 836–837, 1994. doi: 10.1136/thx.49.8.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Merrilees MJ, Ching PS, Beaumont B, Hinek A, Wight TN, Black PN. Changes in elastin, elastin binding protein and versican in alveoli in chronic obstructive pulmonary disease. Respir Res 9: 41, 2008. doi: 10.1186/1465-9921-9-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Deslee G, Woods JC, Moore CM, Liu L, Conradi SH, Milne M, Gierada DS, Pierce J, Patterson A, Lewit RA, Battaile JT, Holtzman MJ, Hogg JC, Pierce RA. Elastin expression in very severe human COPD. Eur Respir J 34: 324–331, 2009. doi: 10.1183/09031936.00123008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bernstein EF, Chen YQ, Tamai K, Shepley KJ, Resnik KS, Zhang H, Tuan R, Mauviel A, Uitto J. Enhanced elastin and fibrillin gene expression in chronically photodamaged skin. J Invest Dermatol 103: 182–186, 1994. doi: 10.1111/1523-1747.ep12392693. [DOI] [PubMed] [Google Scholar]
- 70. Churg A, Dai J, Tai H, Xie C, Wright JL. Tumor necrosis factor-alpha is central to acute cigarette smoke-induced inflammation and connective tissue breakdown. Am J Respir Crit Care Med 166: 849–854, 2002. doi: 10.1164/rccm.200202-097OC. [DOI] [PubMed] [Google Scholar]
- 71. Wachi H, Seyama Y, Yamashita S, Suganami H, Uemura Y, Okamoto K, Yamada H, Tajima S. Stimulation of cell proliferation and autoregulation of elastin expression by elastin peptide VPGVG in cultured chick vascular smooth muscle cells. FEBS Lett 368: 215–219, 1995. doi: 10.1016/0014-5793(95)00641-l. [DOI] [PubMed] [Google Scholar]
- 72. Bashir MM, Indik Z, Yeh H, Ornstein-Goldstein N, Rosenbloom JC, Abrams W, Fazio M, Uitto J, Rosenbloom J. Characterization of the complete human elastin gene. Delineation of unusual features in the 5'-flanking region. J Biol Chem 264: 8887–8891, 1989. [PubMed] [Google Scholar]
- 73. Piontkivska H, Zhang Y, Green ED; NISC Comparative Sequencing Program, Elnitski L. Multi-species sequence comparison reveals dynamic evolution of the elastin gene that has involved purifying selection and lineage-specific insertions/deletions. BMC Genomics 5: 31, 2004. doi: 10.1186/1471-2164-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rosenbloom J, Abrams WR, Mecham R. Extracellular matrix 4: the elastic fiber. FASEB J 7: 1208–1218, 1993. [PubMed] [Google Scholar]
- 75. Fazio MJ, Kahari VM, Bashir MM, Saitta B, Rosenbloom J, Uitto J. Regulation of elastin gene expression: evidence for functional promoter activity in the 5'-flanking region of the human gene. J Invest Dermatol 94: 191–196, 1990. doi: 10.1111/1523-1747.ep12874495. [DOI] [PubMed] [Google Scholar]
- 76. Rosenbloom J, Bashir M, Yeh H, Rosenbloom J, Ornstein-Goldstein N, Fazio M, Kahari VM, Uitto J. Regulation of elastin gene expression. Ann N Y Acad Sci 624: 116–136, 1991. doi: 10.1111/j.1749-6632.1991.tb17012.x. [DOI] [PubMed] [Google Scholar]
- 77. Luo Y, Hitz BC, Gabdank I, Hilton JA, Kagda MS, Lam B, Myers Z, Sud P, Jou J, Lin K, Baymuradov UK, Graham K, Litton C, Miyasato SR, Strattan JS, Jolanki O, Lee JW, Tanaka FY, Adenekan P, O'Neill E, Cherry JM. New developments on the encyclopedia of DNA elements (ENCODE) data portal. Nucleic Acids Res 48: D882–D889, 2020. doi: 10.1093/nar/gkz1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, Lemma RB, Turchi L, Blanc-Mathieu R, Lucas J, Boddie P, Khan A, Manosalva Pérez N, Fornes O, Leung TY, Aguirre A, Hammal F, Schmelter D, Baranasic D, Ballester B, Sandelin A, Lenhard B, Vandepoele K, Wasserman WW, Parcy F, Mathelier A. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res 50: D165–D173, 2022. doi: 10.1093/nar/gkab1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Liu JM, Davidson JM. The elastogenic effect of recombinant transforming growth factor-beta on porcine aortic smooth muscle cells. Biochem Biophys Res Commun 154: 895–901, 1988. doi: 10.1016/0006-291x(88)90224-0. [DOI] [PubMed] [Google Scholar]
- 80. Kähäri VM, Olsen DR, Rhudy RW, Carrillo P, Chen YQ, Uitto J. Transforming growth factor-beta up-regulates elastin gene expression in human skin fibroblasts. Evidence for post-transcriptional modulation. Lab Invest 66: 580–588, 1992. [PubMed] [Google Scholar]
- 81. McGowan SE. Influences of endogenous and exogenous TGF-beta on elastin in rat lung fibroblasts and aortic smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 263: L257–L263, 1992. doi: 10.1152/ajplung.1992.263.2.L257. [DOI] [PubMed] [Google Scholar]
- 82. McGowan SE, Jackson SK, Olson PJ, Parekh T, Gold LI. Exogenous and endogenous transforming growth factors-beta influence elastin gene expression in cultured lung fibroblasts. Am J Respir Cell Mol Biol 17: 25–35, 1997. doi: 10.1165/ajrcmb.17.1.2686. [DOI] [PubMed] [Google Scholar]
- 83. Zhang MC, Giro M, Quaglino D Jr, Davidson JM. Transforming growth factor-beta reverses a posttranscriptional defect in elastin synthesis in a cutis laxa skin fibroblast strain. J Clin Invest 95: 986–994, 1995. doi: 10.1172/JCI117808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kuang PP, Zhang XH, Rich CB, Foster JA, Subramanian M, Goldstein RH. Activation of elastin transcription by transforming growth factor-beta in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 292: L944–L952, 2007. doi: 10.1152/ajplung.00184.2006. [DOI] [PubMed] [Google Scholar]
- 85. Katchman SD, Hsu-Wong S, Ledo I, Wu M, Uitto J. Transforming growth factor-beta up-regulates human elastin promoter activity in transgenic mice. Biochem Biophys Res Commun 203: 485–490, 1994. doi: 10.1006/bbrc.1994.2208. [DOI] [PubMed] [Google Scholar]
- 86. Foster JA, Miller ML, Benedict MR, Richmann RA, Rich CB. Evidence for insulin-like growth factor-I regulation of chick aortic elastogenesis. Matrix 9: 328–335, 1989. doi: 10.1016/s0934-8832(89)80009-5. [DOI] [PubMed] [Google Scholar]
- 87. Rich CB, Goud HD, Bashir M, Rosenbloom J, Foster JA. Developmental regulation of aortic elastin gene expression involves disruption of an IGF-I sensitive repressor complex. Biochem Biophys Res Commun 196: 1316–1322, 1993. doi: 10.1006/bbrc.1993.2396. [DOI] [PubMed] [Google Scholar]
- 88. Wolfe BL, Rich CB, Goud HD, Terpstra AJ, Bashir M, Rosenbloom J, Sonenshein GE, Foster JA. Insulin-like growth factor-I regulates transcription of the elastin gene. J Biol Chem 268: 12418–12426, 1993. doi: 10.1016/S0021-9258(18)31406-6. [DOI] [PubMed] [Google Scholar]
- 89. Rich CB, Ewton DZ, Martin BM, Florini JR, Bashir M, Rosenbloom J, Foster JA. IGF-I regulation of elastogenesis: comparison of aortic and lung cells. Am J Physiol Lung Cell Mol Physiol 263: L276–L282, 1992. doi: 10.1152/ajplung.1992.263.2.L276. [DOI] [PubMed] [Google Scholar]
- 90. Conn KJ, Rich CB, Jensen DE, Fontanilla MR, Bashir MM, Rosenbloom J, Foster JA. Insulin-like growth factor-I regulates transcription of the elastin gene through a putative retinoblastoma control element. A role for Sp3 acting as a repressor of elastin gene transcription. J Biol Chem 271: 28853–28860, 1996. doi: 10.1074/jbc.271.46.28853. [DOI] [PubMed] [Google Scholar]
- 91. Sen S, Bunda S, Shi J, Wang A, Mitts TF, Hinek A. Retinoblastoma protein modulates the inverse relationship between cellular proliferation and elastogenesis. J Biol Chem 286: 36580–36591, 2011. doi: 10.1074/jbc.M111.269944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jensen DE, Rich CB, Terpstra AJ, Farmer SR, Foster JA. Transcriptional regulation of the elastin gene by insulin-like growth factor-I involves disruption of Sp1 binding. Evidence for the role of Rb in mediating Sp1 binding in aortic smooth muscle cells. J Biol Chem 270: 6555–6563, 1995. doi: 10.1074/jbc.270.12.6555. [DOI] [PubMed] [Google Scholar]
- 93. Kähäri VM, Chen YQ, Bashir MM, Rosenbloom J, Uitto J. Tumor necrosis factor-alpha down-regulates human elastin gene expression. Evidence for the role of AP-1 in the suppression of promoter activity. J Biol Chem 267: 26134–26141, 1992. [PubMed] [Google Scholar]
- 94. Carreras I, Rich CB, Jaworski JA, Dicamillo SJ, Panchenko MP, Goldstein R, Foster JA. Functional components of basic fibroblast growth factor signaling that inhibit lung elastin gene expression. Am J Physiol Lung Cell Mol Physiol 281: L766–L775, 2001. doi: 10.1152/ajplung.2001.281.4.L766. [DOI] [PubMed] [Google Scholar]
- 95. Carreras I, Rich CB, Panchenko MP, Foster JA. Basic fibroblast growth factor decreases elastin gene transcription in aortic smooth muscle cells. J Cell Biochem 85: 592–600, 2002. doi: 10.1002/jcb.10163. [DOI] [PubMed] [Google Scholar]
- 96. Rich CB, Nugent MA, Stone P, Foster JA. Elastase release of basic fibroblast growth factor in pulmonary fibroblast cultures results in down-regulation of elastin gene transcription. A role for basic fibroblast growth factor in regulating lung repair. J Biol Chem 271: 23043–23048, 1996. doi: 10.1074/jbc.271.38.23043. [DOI] [PubMed] [Google Scholar]
- 97. Wang A, Cao S, Stowe JC, Valdez-Jasso D. Substrate stiffness and stretch regulate profibrotic mechanosignaling in pulmonary arterial adventitial fibroblasts. Cells 10: 1000, 2021. doi: 10.3390/cells10051000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mecham RP, Lange G, Madaras J, Starcher B. Elastin synthesis by ligamentum nuchae fibroblasts: effects of culture conditions and extracellular matrix on elastin production. J Cell Biol 90: 332–338, 1981. doi: 10.1083/jcb.90.2.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Del Monaco M, Covello SP, Kennedy SH, Gilinger G, Litwack G, Uitto J. Identification of novel glucocorticoid-response elements in human elastin promoter and demonstration of nucleotide sequence specificity of the receptor binding. J Invest Dermatol 108: 938–942, 1997. doi: 10.1111/1523-1747.ep12295241. [DOI] [PubMed] [Google Scholar]
- 100. Keeley FW, Johnson DJ. Age differences in the effect of hydrocortisone on the synthesis of insoluble elastin in aortic tissue of growing chicks. Connect Tissue Res 16: 259–268, 1987. doi: 10.3109/03008208709006980. [DOI] [PubMed] [Google Scholar]
- 101. Kähäri VM. Dexamethasone suppresses elastin gene expression in human skin fibroblasts in culture. Biochem Biophys Res Commun 201: 1189–1196, 1994. doi: 10.1006/bbrc.1994.1831. [DOI] [PubMed] [Google Scholar]
- 102. Chuang TD, Pearce WJ, Khorram O. miR-29c induction contributes to downregulation of vascular extracellular matrix proteins by glucocorticoids. Am J Physiol Cell Physiol 309: C117–C125, 2015. doi: 10.1152/ajpcell.00254.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sproul EP, Argraves WS. A cytokine axis regulates elastin formation and degradation. Matrix Biol 32: 86–94, 2013. doi: 10.1016/j.matbio.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Long HK, Prescott SL, Wysocka J. Ever-changing landscapes: transcriptional enhancers in development and evolution. Cell 167: 1170–1187, 2016. doi: 10.1016/j.cell.2016.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Swee MH, Parks WC, Pierce RA. Developmental regulation of elastin production. Expression of tropoelastin pre-mRNA persists after down-regulation of steady-state mRNA levels. J Biol Chem 270: 14899–14906, 1995. doi: 10.1074/jbc.270.25.14899. [DOI] [PubMed] [Google Scholar]
- 106. Parks WC, Kolodziej ME, Pierce RA. Phorbol ester-mediated downregulation of tropoelastin expression is controlled by a posttranscriptional mechanism. Biochemistry 31: 6639–6645, 1992. doi: 10.1021/bi00144a003. [DOI] [PubMed] [Google Scholar]
- 107. Pierce RA, Kolodziej ME, Parks WC. 1,25-Dihydroxyvitamin D3 represses tropoelastin expression by a posttranscriptional mechanism. J Biol Chem 267: 11593–11599, 1992. [PubMed] [Google Scholar]
- 108. Hew Y, Lau C, Grzelczak Z, Keeley FW. Identification of a GA-rich sequence as a protein-binding site in the 3'-untranslated region of chicken elastin mRNA with a potential role in the developmental regulation of elastin mRNA stability. J Biol Chem 275: 24857–24864, 2000. doi: 10.1074/jbc.M002776200. [DOI] [PubMed] [Google Scholar]
- 109. Johnson DJ, Robson P, Hew Y, Keeley FW. Decreased elastin synthesis in normal development and in long-term aortic organ and cell cultures is related to rapid and selective destabilization of mRNA for elastin. Circ Res 77: 1107–1113, 1995. doi: 10.1161/01.res.77.6.1107. [DOI] [PubMed] [Google Scholar]
- 110. Zhang M, Pierce RA, Wachi H, Mecham RP, Parks WC. An open reading frame element mediates posttranscriptional regulation of tropoelastin and responsiveness to transforming growth factor beta1. Mol Cell Biol 19: 7314–7326, 1999. doi: 10.1128/MCB.19.11.7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kucich U, Rosenbloom JC, Abrams WR, Bashir MM, Rosenbloom J. Stabilization of elastin mRNA by TGF-beta: initial characterization of signaling pathway. Am J Respir Cell Mol Biol 17: 10–16, 1997. doi: 10.1165/ajrcmb.17.1.2816. [DOI] [PubMed] [Google Scholar]
- 112. Kucich U, Rosenbloom JC, Abrams WR, Rosenbloom J. Transforming growth factor-beta stabilizes elastin mRNA by a pathway requiring active Smads, protein kinase C-delta, and p38. Am J Respir Cell Mol Biol 26: 183–188, 2002. doi: 10.1165/ajrcmb.26.2.4666. [DOI] [PubMed] [Google Scholar]
- 113. Niculet E, Bobeica C, Tatu AL. Glucocorticoid-induced skin atrophy: the old and the new. Clin Cosmet Investig Dermatol 13: 1041–1050, 2020. doi: 10.2147/CCID.S224211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. DiCamillo SJ, Yang S, Panchenko MV, Toselli PA, Naggar EF, Rich CB, Stone PJ, Nugent MA, Panchenko MP. Neutrophil elastase-initiated EGFR/MEK/ERK signaling counteracts stabilizing effect of autocrine TGF-β on tropoelastin mRNA in lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 291: L232–L243, 2006. doi: 10.1152/ajplung.00530.2005. [DOI] [PubMed] [Google Scholar]
- 115. Ipsaro JJ, Joshua-Tor L. From guide to target: molecular insights into eukaryotic RNA-interference machinery. Nat Struct Mol Biol 22: 20–28, 2015. doi: 10.1038/nsmb.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Sudo R, Sato F, Azechi T, Wachi H. MiR-29-mediated elastin down-regulation contributes to inorganic phosphorus-induced osteoblastic differentiation in vascular smooth muscle cells. Genes Cells 20: 1077–1087, 2015. doi: 10.1111/gtc.12311. [DOI] [PubMed] [Google Scholar]
- 117. Nanoudis S, Pikilidou M, Yavropoulou M, Zebekakis P. The role of microRNAs in arterial stiffness and arterial calcification. An update and review of the literature. Front Genet 8: 209, 2017. doi: 10.3389/fgene.2017.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Di Gregoli K, Mohamad Anuar NN, Bianco R, White SJ, Newby AC, George SJ, Johnson JL. MicroRNA-181b controls atherosclerosis and aneurysms through regulation of TIMP-3 and elastin. Circ Res 120: 49–65, 2017. doi: 10.1161/CIRCRESAHA.116.309321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Qi YF, Shu C, Xiao ZX, Luo MY, Fang K, Guo YY, Zhang WB, Yue J. Post-transcriptional control of tropoelastin in aortic smooth muscle cells affects aortic dissection onset. Mol Cells 41: 198–206, 2018. doi: 10.14348/molcells.2018.2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Yu Y, Shi E, Gu T, Tang R, Gao S, Wang Y, Liu H. Overexpression of microRNA-30a contributes to the development of aortic dissection by targeting lysyl oxidase. J Thorac Cardiovasc Surg 154: 1862–1869, 2017. doi: 10.1016/j.jtcvs.2017.06.019. [DOI] [PubMed] [Google Scholar]
- 121. van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA 105: 13027–13032, 2008. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Boon RA, Seeger T, Heydt S, Fischer A, Hergenreider E, Horrevoets AJ, Vinciguerra M, Rosenthal N, Sciacca S, Pilato M, van Heijningen P, Essers J, Brandes RP, Zeiher AM, Dimmeler S. MicroRNA-29 in aortic dilation: implications for aneurysm formation. Circ Res 109: 1115–1119, 2011. doi: 10.1161/CIRCRESAHA.111.255737. [DOI] [PubMed] [Google Scholar]
- 123. Ott CE, Grünhagen J, Jäger M, Horbelt D, Schwill S, Kallenbach K, Guo G, Manke T, Knaus P, Mundlos S, Robinson PN. MicroRNAs differentially expressed in postnatal aortic development downregulate elastin via 3' UTR and coding-sequence binding sites. PLoS One 6: e16250, 2011. doi: 10.1371/journal.pone.0016250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Liang K, Cui M, Fu X, Ma J, Zhang K, Zhang D, Zhai S. LncRNA Xist induces arterial smooth muscle cell apoptosis in thoracic aortic aneurysm through miR-29b-3p/Eln pathway. Biomed Pharmacother 137: 111163, 2021. doi: 10.1016/j.biopha.2020.111163. [DOI] [PubMed] [Google Scholar]
- 125. Zhang P, Huang A, Ferruzzi J, Mecham RP, Starcher BC, Tellides G, Humphrey JD, Giordano FJ, Niklason LE, Sessa WC. Inhibition of microRNA-29 enhances elastin levels in cells haploinsufficient for elastin and in bioengineered vessels—brief report. Arterioscler Thromb Vasc Biol 32: 756–759, 2012. doi: 10.1161/ATVBAHA.111.238113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Fazio MJ, Olsen DR, Kuivaniemi H, Chu ML, Davidson JM, Rosenbloom J, Uitto J. Isolation and characterization of human elastin cDNAs, and age-associated variation in elastin gene expression in cultured skin fibroblasts. Lab Invest 58: 270–277, 1988. [PubMed] [Google Scholar]
- 127. Indik Z, Yeh H, Ornstein-Goldstein N, Sheppard P, Anderson N, Rosenbloom JC, Peltonen L, Rosenbloom J. Alternative splicing of human elastin mRNA indicated by sequence analysis of cloned genomic and complementary DNA. Proc Natl Acad Sci USA 84: 5680–5684, 1987. doi: 10.1073/pnas.84.16.5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Yeh H, Ornstein-Goldstein N, Indik Z, Sheppard P, Anderson N, Rosenbloom JC, Cicila G, Yoon K, Rosenbloom J. Sequence variation of bovine elastin mRNA due to alternative splicing. Coll Relat Res 7: 235–247, 1987. doi: 10.1016/s0174-173x(87)80030-4. [DOI] [PubMed] [Google Scholar]
- 129. Fazio MJ, Olsen DR, Kauh EA, Baldwin CT, Indik Z, Ornstein-Goldstein N, Yeh H, Rosenbloom J, Uitto J. Cloning of full-length elastin cDNAs from a human skin fibroblast recombinant cDNA library: further elucidation of alternative splicing utilizing exon-specific oligonucleotides. J Invest Dermatol 91: 458–464, 1988. doi: 10.1111/1523-1747.ep12476591. [DOI] [PubMed] [Google Scholar]
- 130. Parks WC, Deak SB. Tropoelastin heterogeneity: implications for protein function and disease. Am J Respir Cell Mol Biol 2: 399–406, 1990. doi: 10.1165/ajrcmb/2.5.399. [DOI] [PubMed] [Google Scholar]
- 131. Muiznieks LD, Miao M, Sitarz EE, Keeley FW. Contribution of domain 30 of tropoelastin to elastic fiber formation and material elasticity. Biopolymers 105: 267–275, 2016. doi: 10.1002/bip.22804. [DOI] [PubMed] [Google Scholar]
- 132. Kozel BA, Wachi H, Davis EC, Mecham RP. Domains in tropoelastin that mediate elastin deposition in vitro and in vivo. J Biol Chem 278: 18491–18498, 2003. doi: 10.1074/jbc.M212715200. [DOI] [PubMed] [Google Scholar]
- 133. Yeo GC, Tarakanova A, Baldock C, Wise SG, Buehler MJ, Weiss AS. Subtle balance of tropoelastin molecular shape and flexibility regulates dynamics and hierarchical assembly. Sci Adv 2: e1501145, 2016. doi: 10.1126/sciadv.1501145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Miao M, Reichheld SE, Muiznieks LD, Sitarz EE, Sharpe S, Keeley FW. Single nucleotide polymorphisms and domain/splice variants modulate assembly and elastomeric properties of human elastin. Implications for tissue specificity and durability of elastic tissue. Biopolymers 107: e23007, 2017. doi: 10.1002/bip.23007. [DOI] [PubMed] [Google Scholar]
- 135. Hsiao H, Stone PJ, Toselli P, Rosenbloom J, Franzblau C, Schreiber BM. The role of the carboxy terminus of tropoelastin in its assembly into the elastic fiber. Connect Tissue Res 40: 83–95, 1999. doi: 10.3109/03008209909029104. [DOI] [PubMed] [Google Scholar]
- 136.The eGTEx Project. Enhancing GTEx by bridging the gaps between genotype, gene expression, and disease. Nat Genet 49: 1664–1670, 2017. doi: 10.1038/ng.3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Lacey M, Baribault C, Ehrlich KC, Ehrlich M. Atherosclerosis-associated differentially methylated regions can reflect the disease phenotype and are often at enhancers. Atherosclerosis 280: 183–191, 2019. doi: 10.1016/j.atherosclerosis.2018.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Wainschtein P, Jain D, Zheng Z; TOPMed Anthropometry Working Group, NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium, Cupples LA, Shadyab AH, et al. Assessing the contribution of rare variants to complex trait heritability from whole-genome sequence data. Nat Genet 54: 263–273, 2022. doi: 10.1038/s41588-021-00997-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Jazurek M, Ciesiolka A, Starega-Roslan J, Bilinska K, Krzyzosiak WJ. Identifying proteins that bind to specific RNAs—focus on simple repeat expansion diseases. Nucleic Acids Res 44: 9050–9070, 2016. doi: 10.1093/nar/gkw803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Graindorge A, Pinheiro I, Nawrocka A, Mallory AC, Tsvetkov P, Gil N, Carolis C, Buchholz F, Ulitsky I, Heard E, Taipale M, Shkumatava A. In-cell identification and measurement of RNA-protein interactions. Nat Commun 10: 5317, 2019. [Erratum in Nat Commun 11: 3498, 2020]. doi: 10.1038/s41467-019-13235-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Baralle FE, Giudice J. Alternative splicing as a regulator of development and tissue identity. Nat Rev Mol Cell Biol 18: 437–451, 2017. doi: 10.1038/nrm.2017.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Chabot B, Shkreta L. Defective control of pre-messenger RNA splicing in human disease. J Cell Biol 212: 13–27, 2016. doi: 10.1083/jcb.201510032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Damianov A, Black DL. Autoregulation of Fox protein expression to produce dominant negative splicing factors. RNA 16: 405–416, 2010. doi: 10.1261/rna.1838210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Baralle D, Baralle M. Splicing in action: assessing disease causing sequence changes. J Med Genet 42: 737–748, 2005. doi: 10.1136/jmg.2004.029538. [DOI] [PMC free article] [PubMed] [Google Scholar]