

Keywords: blindness, diabetic retinopathy, neurophysiology, potassium channels, retinal physiology
Abstract
Channel proteins are vital for conducting ions throughout the body and are especially relevant to retina physiology. Inward rectifier potassium (Kir) channels are a class of K+ channels responsible for maintaining membrane potential and extracellular K+ concentrations. Studies of the KCNJ gene (that encodes Kir protein) expression identified the presence of all of the subclasses (Kir 1–7) of Kir channels in the retina or retinal-pigmented epithelium (RPE). However, functional studies have established the involvement of the Kir4.1 homotetramer and Kir4.1/5.1 heterotetramer in Müller glial cells, Kir2.1 in bipolar cells, and Kir7.1 in the RPE cell physiology. Here, we propose the potential roles of Kir channels in the retina based on the physiological contributions to the brain, pancreatic, and cardiac tissue functions. There are several open questions regarding the expressed KCNJ genes in the retina and RPE. For example, why does not the Kir channel subtype gene expression correspond with protein expression? Catching up with multiomics or functional “omics” approaches might shed light on posttranscriptional changes that might influence Kir subunit mRNA translation within the retina that guides our vision.
INTRODUCTION
The retina is part of the central nervous system and is essential for the visual cycle (1). The neural retina lies in the back of the eye, and the retinal-pigmented epithelium (RPE) forms the blood-retina barrier (2, 3). Ion channel function within the retina’s complex neural circuitry is crucial to maintaining the visual cycle in the central nervous system’s light and dark and neurologic vision components (4). Inwardly rectifying potassium (Kir) channels are a subfamily of K+ channels expressed within the developing and mature retina and RPE (5). Seven distinct Kir channels (Kir 1–7) comprise the Kir family members in the mammalian system. Kir channels are regulated in multiple ways, such as membrane potential, ligand gating, and adenosine triphosphate (ATP) dependence (6). Within the retina, Kir channels mediate neural signaling, K+ homeostasis, membrane potentials, and the tight blood-retina barrier in the RPE (7, 8). Kir channel mutations within the retina reportedly cause early-onset blindness, including mutations in KCNJ13, which encode for Kir7.1, leading to Leber's congenital amaurosis (LCA16) (8). Kir channels also play a role in the neuronal response in Müller glial cells (Kir4.1) (9). The review provides detailed pathophysiology of Kir2.x, Kir4.x, and Kir7.1 channels in retinal disease.
MOLECULAR EXPRESSION OF Kir CHANNELS IN THE NEURAL RETINA AND RPE
The neural retina expresses most of the Kir channel subunits, with a higher level seen for Kir2.1, Kir3.1, and Kir4.1 than other subunits (5). A cell-type-specific subunit expression exists for these transcripts, yet Kir channels are not used as biomarkers. Transcripts for all subunits, except Kir2.3, Kir2.4, Kir3.2, Kir3.3, Kir4.1, Kir5.1, and Kir6.2, have been identified in the RPE. Kir channel proteins are present in retinal ganglion cells, amacrine cells, Müller cells, bipolar cells, and possibly photoreceptors within the neural retina (5). In the same paper, Yang et al. used immunoblot analysis to demonstrate the relatively low Kir1.1 and Kir3.1 expression in the RPE cells compared with other channel subunits (5). Immunostaining indicated that Kir2.1 protein is expressed in mature bipolar cells and horizontal cells in rodents (9, 10). The labeling for Kir2.1 was below the photoreceptor terminal and, more specifically, within the rod spherule proximity (9). Moreover, Kir2.1 does not colocalize with the Müller cell marker but is present in the rod bipolar cell termini (9). Kir2.1 staining was also noticed in ganglion cells, but it was mainly localized to the nuclei without an established significance (11). Kir3.2 protein is localized to the cytoplasm of rat retinal ganglion cell somata, proximal dendrites, and axons. When present in the membrane, Kir3.2 is associated with the G protein-coupled receptor (GPCR), the γ-aminobutyric acid B (GABAB) (11). Kir3.2 protein has also been identified within the inner plexiform layer of the chick retina during development based on neuronal activity (12).
Immunohistochemistry identified Kir4.1 localization in the Müller glial cells of the rat retina and interfacing with the apical RPE processes (13). Kir4.1 is colocalized with the glial fibrillary acidic protein in the Müller glial cells within the rat retina (9). Further confirmation of polarized Kir4.1 expression was due to its interactions with dystrophin-glycoprotein complex (DP71) and aquaporin 4 (AQP4) in both mouse and rat retina. The specific Kir4.1 localization was confined to the endfeet by the vitreal border and perivascular processes in the outer retinal layers (14–17). The Kir4.1/Kir5.1 heterotetramer is also present in the perisynaptic processes in the inner plexiform layer in addition to the Kir4.1 homotetramer (18). Kir6.1 protein is expressed in the RPE and may alter blood glucose homeostasis and retinal vasculature. Moreover, it maintains insulin sensitivity and reduces inflammation (19). Kir7.1 is present in the specialized apical RPE processes that establish communications with the adjacent photoreceptor cells (5, 7, 20, 21).
WHAT ROLES DO Kir CHANNELS PLAY IN THE RETINAL DISEASE STATES?
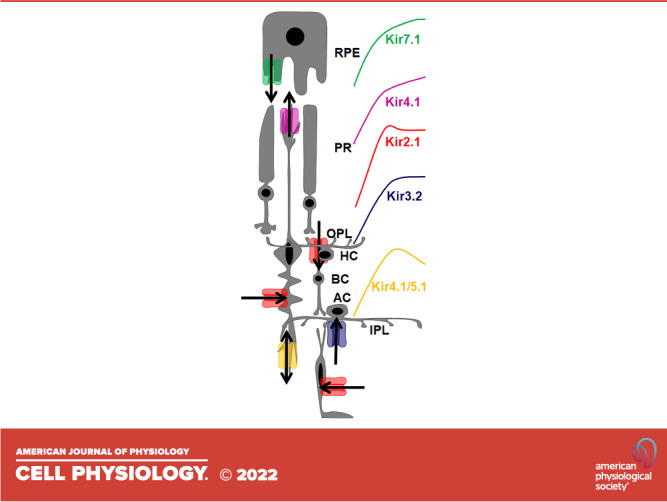
The existing model for Kir channels in the retina and RPE is based on the expression studies above and consists of a strong inward rectifier Kir2.1 expressed in bipolar cells (9), a weak inward rectifier Kir4.1 in Müller glial cells as a homotetramer, and a heterotetramer with Kir5.1, and a weak inward rectifier Kir7.1 in the apical RPE processes (Fig. 1). In contrast, Fig. 2 shows the expression of Kir channels in the diseased retina.
Figure 1.

The role of Kir channels in K+ spatial buffering within the retina and RPE. Kir7.1 releases a small outward current from the apical processes of the RPE into the subretinal space. Kir4.1 is expressed as a homotetramer in the Müller glial cell endfeet and releases K+ into the subretinal space. Kir4.1 is also expressed as a heterotetramer in the inner plexiform layer and exhibits a bidirectional K+ current. Müller glial cells, bipolar cells, and retinal glial cells all exhibit a strong inward K+ current from Kir2.1. Amacrine cells also express Kir3.2. Arrows indicate the direction of K+ flux through the respective channels. Rectification properties of individual retinal Kir channel subtypes is also color coded. AC, amacrine cells; BC, bipolar cells; GC, ganglion cells; HC, horizontal cells; IPL, inner plexiform layer; MGC, Müller glial cells; OPL, outer plexiform layer; PR, photoreceptors.
Figure 2.

Kir channels in the diseased retina. Kir2.1 is present in the apical processes of the RPE, in addition to the cone photoreceptor, and is believed to regulate VEGF secretion from the RPE and control retinal vasculature. Pathophysiology associated with Kir4.1 and Kir7.1 contributes to blindness. Shown are also blood vessels in the retina that benefit from expressing strong inward rectifier channels in the pericytes. RPE, retinal pigmented epithelium.
Kir2.1 Physiology and Pathophysiology
cKir2.1 is activated by hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) when the retina is illuminated (22). Previous evidence indicated Kir2.1 expression in Drosophila (22) and mouse (10) photoreceptors. These studies may have been undertaken at various retinal development stages, or the antibody was nonspecific for the Kir2.1 channel. In addition, researchers previously identified Kir2.1 in mouse photoreceptors colocalized with channel-associated protein synapse-110 and indicated its involvement in synaptic cell signaling (10). However, functional studies suggest that Kir2.1 exists only in bipolar cells (9, 23).
Research has recently identified Kir2.1 in the apical processes of rat RPE and the bipolar cells within the inner plexiform layer and Müller glial cells (Fig. 2) (22). However, one limitation of the study was that the cultured RPE cells were not mature monolayers, and the authors did not evaluate markers, such as RPE65, Best1, and TRP1, to determine monolayer maturity (23). The tight monolayers and polarity of the cultured RPE cells were also not evaluated by transepithelial electrical resistance (24). The only evidence provided that the Kir2.1 channel was trafficked to the apical process was the polarized distribution with more significant staining in the apical region of the cell compared with the basolateral region (22). However, this finding does not necessarily mean that the channel was trafficked to the cell membrane. Thus, a previously characterized polarized RPE model, such as human induced pluripotent stem cells (iPSC)-RPE monolayers, could address these limitations. It is also possible that Kir2.1 remain in the endosomal pathway and does not reach the cell membrane. This model aligns with what is observed in yeast, where the required endosomal sorting complex for transport pathways regulates the number of Kir2.1 channels that reach the cell membrane depending on the extracellular K+ concentration (25). We predict that Kir2.1 would only be trafficked to the membrane in disease conditions, such as diabetic retinopathy.
RPE cells can be inferred to regulate the subretinal space osmolarity. In vitro, RPE cells regulate cell culture media osmolarity and alter the angiogenic vascular endothelial growth factor (VEGF) secretion (26). The angiogenic effects of extracellular VEGF in the human fetal retina are required for retinal vasculature development (27). Based on scientific facts, the current model suggests that hypoxic astrocytes secrete VEGF in the developing human retina, thereby facilitating angiogenesis from the optic disk (28). Excess VEGF is the primary cause of the blood-retina barrier breakdown (29). An increased extracellular VEGF is associated with Kir2.1 expression in the developing fetal retina (Fig. 2). Prenatal hypoxia by intrauterine growth restriction induces retinal dysfunction in the RPE caused by the excess release of VEGF (30). In addition, the increased Kir2.1 mRNA expression is correlated with increased VEGF mRNA (22). Previous studies have shown that Kir2.1 expression in the RPE, and potentially in the apical processes, primarily responds to hypoxia followed by sudden hyperoxia and RPE monolayer disruption.
Kir2.1 dysfunction causes cardiovascular (31), metabolic (32), and retinal diseases (22). Kir2.1 is trafficked to the cell membrane through the endosomal pathways by dynamin and Rac1, which regulates ion flux (33). The Andersen–Tawil syndrome due to nonfunctional Kir2.1 has also been linked to neurologic symptoms and paralysis (34). A single case report showed that a KCNJ5 mutation, which encodes Kir3.4, led to Andersen–Tawil syndrome through the indirect Kir2.1 channel function inhibition (35). Table 1 lists the known genetic mutations in KCNJ2 that lead to cardiovascular and neurologic diseases.
Table 1.
Disease-causing mutations in Kir2.1
| Channel | Disease | Mutation | Reference |
|---|---|---|---|
| Kir2.1 | Andersen-Tawil syndrome | R40X | (36) |
| G52V | (37) | ||
| R67Q | (38) | ||
| R67W | (39) | ||
| Y68D | (34) | ||
| D71N | (40) | ||
| D71V | (41) | ||
| D71Y | (42) | ||
| T74A | (43) | ||
| T75R | (40) | ||
| T75M | (34) | ||
| D78G | (34) | ||
| D78Y | (44) | ||
| R82Q | (34) | ||
| V123G | (34) | ||
| S136F | (41) | ||
| G138K | (37) | ||
| G144S | (41) | ||
| G144D | (45) | ||
| G144A | (43) | ||
| Y145C | (46) | ||
| G146S | (38) | ||
| G146R | (37) | ||
| G146D | (40) | ||
| G146A | (47) | ||
| C154F | (48) | ||
| P186T | (49) | ||
| P186L | (50) | ||
| K187R | (51) | ||
| R189S | (49) | ||
| R189I | (40) | ||
| T192I | (52) | ||
| T192A | (53) | ||
| G215D | (54) | ||
| N216H | (50) | ||
| L217P | (34) | ||
| R218Q | (41) | ||
| R218W | (41) | ||
| L222S | (55) | ||
| R260P | (56) | ||
| L298R | (51) | ||
| G300D | (40) | ||
| G300A | (57) | ||
| G300V | (41) | ||
| V302M | (50) | ||
| E303K | (41) | ||
| T305P | (51) | ||
| M307I | (58) | ||
| M307V | (59) | ||
| T309I | (48) | ||
| R312H | (51) | ||
| R312C | (40) | ||
| N318S | (60) | ||
| W322C | (60) | ||
| S369X | (61) | ||
| Long QT syndrome | T75A | (62) | |
| G206S | (63) | ||
| P351S | (38) | ||
| T400M | (64) | ||
| N410S | (65) | ||
| Short QT syndrome | D172N | (66) | |
| E299V | (67) | ||
| M301K | (68) | ||
| K346T | (69) | ||
| Ventricular tachycardia | R82W | (70) | |
| C101R | (71) | ||
| V227F | (70) | ||
| Arrythmia | T305A | (72) | |
| Atrial fibrillation | V93I | (73) |
Two primary retinal vasculature diseases are caused by Kir2.1 channelopathy, including retinopathy of prematurity (ROP) and diabetic retinopathy. Diabetic retinopathy results in an increased inward current in the retina pericytes (74). The responsible Kir subunits for this current are not yet determined. From 1997 to 2005, the incidence of ROP was 0.17% of the overall live births in the United States, which increased to 15.58% in premature births (75). In addition, 28.5% of diabetic adults in the United States develop diabetic retinopathy with significant vision loss (76). These retinal diseases are mediated by VEGF (77, 78). Anti-VEGF therapies have been routinely used to treat these conditions (79). These anti-VEGF therapies may indirectly reduce the Kir2.1 expression in the RPE. Moreover, increased Kir2.1 expression could be a biomarker for abnormal retinal vasculature.
The Surfactant Positive Airway Pressure and Pulse Oximetry Randomized Trial (SUPPORT) (80) in premature infants administered supplemental oxygen shortly after birth, in whom oxygen saturation was 91%–95% and revealed a significantly increased severe ROP frequency compared with infants whose oxygen saturations remained between 85% and 89%. This finding indicates that oxygen therapy affects pathogenic retinal neovascularization development. This finding is corroborated by a mouse model of oxygen-induced ROP, where mice from postnatal day 0 to eye-opening postnatal day 15 were exposed to 5 days of hyperoxia. Hyperoxia-induced neovascularization was identified as the increased presence of neovascular nuclei in the retinal vasculature (81). This altered neovascularization is an ROP characteristic. Considering VEGF as a critical angiogenic factor within the developing retina (82), we infer that hyperoxia induces VEGF expression and thus Kir2.1 expression in the RPE, resulting in retinal disease. However, we did not find any literature on the Kir2.1 expression in ROP.
Some evidence has suggested that normal Kir2.1 function is required for healthy retinal vascularization. One example is fetal alcohol syndrome, in which gestational alcohol exposure has multiple negative neurologic and cardiovascular effects on infant development. These include retinal dysplasia, abnormal vessel tortuosity, and altered ERG in nonhuman primates and humans (83, 84). Specifically, 49% of fundus images indicated a retinal vascular anomaly (85). In addition, Kir2.1 is inhibited by alcohol, and loss of function mutations mirror all the characteristic features of fetal alcohol syndrome (86). The effects of alcohol on Kir7.1 have not been evaluated; thus, whether inadequate K+ inward current in RPE is due to the Kir2.1 or Kir7.1 channels remains unclear. Inhibiting Kir2.1 would prevent the channel from counteracting the VEGF effects.
Missense mutations in Kir2.1 cause long-QT cardiac arrhythmias in cardiomyocytes, as shown in Table 1 (72). As demonstrated using stem cells, long-QT syndromes have also been associated with visual defects, including retinal vascular anomalies (87).
Kir4.1 PHYSIOLOGY AND PATHOPHYSIOLOGY
Glaucoma is associated with the Kir channel expression changes in the inner retina. Specifically, Kir4.1 expression (88) decreases under increasing pressures, whereas Kir2.1 increases (88). Elevated pressure induces retinal ganglion cell death in vitro and in vivo (89, 90). Kir4.1 is a weakly rectifying channel in the retinal Müller glial cells perisynaptic processes, which forms a heterotetramer with Kir5.1. Kir4.1 is also present as a homotetramer at the endfeet facing the subretinal space (6). In addition, the Kir4.1 homotetramer is only 85%–90% blocked by spermines compared with other Kir channels (91). VU0134992 is a Kir4.1-specific inhibitor that interacts with amino acid side chains E158 and I159 in the inner pore of the channel (115).
Previous researchers have observed that the Kir4.1/Kir5.1 heterotetramer increases inward and outward conductance compared with the Kir4.1 homotetramer, yielding a more bidirectional current flux (92). The gating mechanism for Kir4.1/Kir5.1 was recently described, and inward conductance is inversely dependent on exogenous PIP2 concentration and is sensitive to pH (93). The Kir4.1/Kir5.1 heterotetramer in primary renal cells modulates ENaC and regulates electrolyte homeostasis (94). This finding suggests that the Kir4.1/Kir5.1 heterotetramer may play a similar role in electrolyte homeostasis within the retina.
The role of Kir4.1 in maintaining K+ homeostasis changes throughout retinal development in mice between postnatal day 1 and eye-opening at postnatal day 15. The Kir4.1 and AQP4 channels regulate the osmolarity of the horizontal cells in the developing neural retina (95). Interestingly, immature horizontal cells are responsible for maintaining K+ buffering during retinogenesis before the postnatal differentiation of the Müller glial cells. The Kir4.1 channel and AQP4 had traveled from the horizontal cells and reached the endfeet of the Müller glial cells by day 15 (96). The Kir4.1 channel and AQP4 trafficking to the endfeet in Müller glial cells in mice is mediated by β-1 syntrophin (97) and DP71. Kir4.1 and AQP4 lose their polarization and localize throughout the Müller cell membrane in DP71-knockout mice (98), resulting in retinal edema. The absence of DP71 leads to retinal ischemia, ocular inflammation, retinal detachment, and blood-retina barrier degeneration. Given these, it is hypothesized that DP7.1 mutations would also affect Kir4.1 and AQP4 trafficking. Adeno-associated virus-mediated DP71 gene therapy restores the normal Kir4.1 and AQP4 distribution, the blood-retina barrier integrity, and retinal osmolarity (98).
Kir4.1 antibody inhibition in the rat retina leads to a marked decrease in a posthyperpolarizing negative response in the electroretinogram (ERG) potential, suggesting Kir4.1 responsibility in the Müller glial cells (9). Mice with Kir4.1 mutations lack a slow PIII response by the Müller glial cells from light-evoked ERG, indicating a role for Kir4.1 in the light response. The PIII response is the dominant slowly developing potential as a light stimuli response. It has a direct effect on the ERG c-wave amplitude, which indicates the physiological interdependence between the Müller glial cells and the RPE (99). The inadequate, slow PIII response in these mice suggests that Kir4.1 is the primary K+ channel in Müller glial cells (100).
Selective serotonin reuptake inhibitors (SSRIs) inhibit Kir4.1 function in astroglia cells in the brain (101). However, the effects of SSRIs on the retina and retinal vasculature have not been evaluated. We predict that inhibition of Kir4.1 by SSRIs in the retina will result in ROP and diabetic retinopathy phenotype through the alterations in the retinal vasculature. Kir4.1 is a therapeutic target for diabetic retinopathy (102), which is consistent with other studies that defined the role of Kir4.1 in maintaining retinal vasculature. Metformin is a clinical drug used in diabetes therapy that regulates insulin sensitivity (103). Metformin targets Müller glial cells in mice and has been shown to correct altered circadian rhythm, mitigate diabetic retinopathy-associated neovascularization, and restore the normal Kir4.1 function (104).
Mutations in KCNJ10, which encodes Kir4.1, cause epilepsy, ataxia, sensorineural deafness, and tubulopathy (EAST) syndrome (105). Table 2 lists these specific missense mutations. Seizure disorder, developmental delays, and altered renal biochemical homeostasis have been observed in young children with Kir4.1 mutations (105). This is unsurprising because Kir4.1 is highly expressed in the brain (115) and the renal tubule (91). Kir4.1 is believed to coordinate with NaK–ATPase to maintain K+ homeostasis in the renal tubule (116), the proposed mechanism for altered urine chemistries in patients with Kir4.1 mutations (105). Visual defects are not considered a hallmark of EAST syndrome, but they occur in some patients. These include nystagmus in a few patients (106) and abnormal ERG (117). Abnormal ERG has been associated with the Müller cell component; thus, mutations may lead to loss of function. These findings indicate that vision assessment is warranted in individuals with EAST syndrome.
Table 2.
Disease-causing mutations in Kir4.1
| Channel | Disease | Mutation | Reference |
|---|---|---|---|
| Kir4.1 | EAST syndrome | T57I | (106) |
| R65P | (107) | ||
| R65C | (108) | ||
| L68P | (109) | ||
| F75C | (110) | ||
| F75L | (108) | ||
| G77R | (107) | ||
| I129V | (109) | ||
| C140R | (111) | ||
| T164I | (111) | ||
| A167V | (111) | ||
| R175Q | (112) | ||
| R199X | (111) | ||
| R297C | (111) | ||
| Hearing loss | P194H | (113) | |
| R348C | (113) | ||
| Seizures | R204H | (114) |
EAST, epilepsy, ataxia, sensorineural deafness, and tubulopathy syndrome.
Kir7.1 PHYSIOLOGY AND PATHOPHYSIOLOGY
NaK–ATPase pumps K+ ions into the RPE cells on light exposure, and Kir7.1 releases a small outward K+ current back into the subretinal space from the apical processes (118). Kir7.1 has a relatively small inward unitary conductance compared with other Kir channels, with ∼50 fS (119). The small Kir7.1 conductance is consistent with the notion that it primarily mediates small outward currents. The permeability characteristics of Kir7.1 could result from methionine at amino acid position 125, which is unique to Kir7.1 (120). The extracellular selectivity loop and narrow inner pore regulate ion permeability (120). In addition, VU590, a Kir7.1-specific blocker, has been identified to bind in the inner pore region of the Kir7.1 channel (121, 122). Thus, Kir7.1 has unique inner pore properties compared with other Kir channels, which warrants further investigation.
RPE K+ released into the subretinal space is essential in buffering K+ concentration changes produced by light-evoked photoreceptor activity (123). The PIP2 hydrolysis inhibits Kir7.1, which might be regulated by dark-light transitions (124). Using a patient iPSC-RPE model, the loss of Kir7.1 function altered phagocytosis of photoreceptor outer segments (125). Inward K+ conductance does not appear in the absence of Kir7.1 in the apical processes, and ERG c-wave amplitude is not maintained in mice with suppressed Kcnj13 expression in RPE (7).
Kir7.1 is regulated by GPCRs both directly by N-linked glycosylation (126) and indirectly by the RPE oxytocin receptor (127, 128) and progesterone brain receptor (129). Nonsense and missense mutations within the N-terminal cytoplasmic domain, selectivity loop, TM2 domain, and C-terminal cytoplasmic domain lead to early-onset blindness development, including snowflake vitreoretinal degeneration (SVD) and LCA16 (130–132). These effects are due to RPE, photoreceptor degeneration, retinal detachment, and nystagmus (130).
Table 3 lists the known KCNJ13 mutations that lead to blindness. Our laboratory has studied an autosomal recessive nonsense mutation at tryptophan 53, which yields a premature stop codon in the N-terminal cytoplasmic domain. This truncated protein does not reach the cell membrane and lacks K+ inward current (132). In addition, the W53X mutation yields a reduced ERG c-wave in mouse models compared with wild-type mice (7) but could be restored with gene therapy (138). Another study of a human iPSC cell disease-in-a-dish model revealed that read-through drug NB84 and gene augmentation via lentivirus fixed channel trafficking to the cell membrane restored K+ inward currents and demonstrated Rb+ enhanced current (125).
Table 3.
Disease-causing mutations in Kir7.1
Moreover, researchers currently evaluate the therapeutic potential of gene editing, base editing, and engineered tRNA for KCNJ13 point mutations (139). The glutamine to arginine mutation in the selectivity loop has been identified at amino acid position 117 (Q117R) and yields a nonfunctional channel that develops LCA16 (135). A missense mutation alters threonine to isoleucine at amino acid position 153 (T153I) (134). The T153I mutation within the inner pore region yields a dysfunctional channel that reaches the cell membrane. The T153I mutation highlights the importance of the narrow inner pore in channel conductance (140). Another allelic mutation in which arginine is changed to tryptophan at amino acid position 162 (R162W) leads to SVD because the channel is nonfunctional and does not reach the cell membrane (131). Notably, the R162Q mutation is not located in the signal sequence, amino acids 323–360 (141); thus, its mistrafficking likely comes from another mechanism, such as the endosomal pathways. An additional C-terminal cytoplasmic domain mutation included an arginine converted to a premature stop codon (R166X), which yields a truncated protein and leads to LCA16 development. Evidence suggests that mice’s proteosome degrades the R166X mutated channel (142). Together, the harmful effects of these point mutations on vision, including the RPE degeneration and loss of photoreceptors, highlight the importance of the Kir7.1 channel in retinal physiology.
CONCLUSIONS AND FUTURE IMPACT
Kir channels play a critical role in maintaining extracellular K+ concentrations within the retina and RPE in both rodents and humans. Gene expression studies have identified all subfamilies of Kir channels in the RPE and retina. This review mainly focused on functional Kir channels, including Kir2.1, Kir4.1, and Kir7.1. Recently, a role for Kir2.1 has been suggested in both normal and diseased RPE in retinal vasculature development and is closely associated with VEGF release from the RPE and retinal vasculature. This review evaluated how this novel finding alters the Kir channel function model within the RPE and retina in health and disease. We can identify several future research directions that would lead to our understanding of the role of Kir channel biology in the retina. We expect that a combination of blockers and in vivo and ex vivo electrophysiological measurements will prove channel subtype-specific role that contributes to retina physiology with the Kir channel subtype-specific blocker advancement. Future therapeutics development will rely on Kir channel tetramers’ specific structure (143). Another critical area would be the interactions between Kir channels and molecules that make up complex plasma membranes, such as cholesterol or other lipids/proteins. Therefore, studies should determine if the Kir channel function in the retina is similar to their role in other organs.
Defining the Kir channel expression levels in the retina cells will be critical in using cell transplantation as a treatment for several degenerative blindness. Hence, augmented expression of specific Kir channels in the heterologous cells would have a functional advantage therapeutically. The study of Kir channel biology will also increase our understanding of the correlation between gene expression and protein levels. As we have understood from this review, going by the several Kir channel subunits expression in the retina, it will be an overinterpretation that protein expression is proportional. Future multiomics or functional “omics” approaches might uncover mechanisms such as microRNA inhibition of mRNA that does not yield functional protein under physiological conditions but could have a role in disease pathophysiology.
Kir biology in the retina is an emerging field that benefited from gene mutation discovery that leads to blindness. Future work should focus on identifying mutations at intronic sites and their effects on gene expression/function. We reviewed the disease-causing mutations in Kir2.1, Kir4.1, and Kir7.1, as well as any associated visual defects. Kir2.1, Kir4.1, and Kir7.1 mainly play a role in maintaining subretinal K+ concentration and the RPE monolayer integrity. Specifically, Kir4.1 mutations cause neurologic diseases with and without visual defects. Neurologic disorders are generally associated with retinal defects, and our review indicates that the vision of individuals with EAST syndrome should be evaluated. Some of these mutations have been directly linked to visual defects, but others have not yet been considered. This review is our vision that identifies areas for further research to understand the biology of Kir channels and the roles of these channels in the RPE and retina.
GRANTS
This work was supported by Endocrinology and Reproductive Physiology NICHDT32 (to K.M.B.). The work was also supported by NIH R01 EY024995, NIH R24 EY032434, and Retina Research Foundation M.D. Matthews Research Professorship to B.R.P.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.M.B. and B.R.P. conceived and designed research; K.M.B. and B.R.P. performed experiments; K.M.B. and B.R.P. analyzed data; K.M.B. and B.R.P. interpreted results of experiments; K.M.B. and B.R.P. prepared figures; K.M.B. and B.R.P. drafted manuscript; K.M.B. and B.R.P. edited and revised manuscript; K.M.B. and B.R.P. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors recognize the contributions of past and present members of the Pattnaik lab to work presented in this review. We also acknowledge the many authors whose contributions to the Kir channel have not been included in this review. This article is part of the special collection “Inward Rectifying K+ Channels.” Drs. Jerod Denton and Eric Delpire served as Guest Editors of this collection.
REFERENCES
- 1. Ehinger B. Retina—one of the most known parts of the central nervous system. Lakartidningen 79: 2121–2124, 1982. [PubMed] [Google Scholar]
- 2. Masland RH. The neuronal organization of the retina. Neuron 76: 266–280, 2012. doi: 10.1016/j.neuron.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Campbell M, Humphries P. The blood-retina barrier: tight junctions and barrier modulation. Adv Exp Med Biol 763: 70–84, 2012. [PubMed] [Google Scholar]
- 4. Giblin JP, Comes N, Strauss O, Gasull X. Ion channels in the eye: involvement in ocular pathologies. Adv Protein Chem Struct Biol 104: 157–231, 2016. doi: 10.1016/bs.apcsb.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 5. Yang D, Zhang X, Hughes BA. Expression of inwardly rectifying potassium channel subunits in native human retinal pigment epithelium. Exp Eye Res 87: 176–183, 2008. doi: 10.1016/j.exer.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90: 291–366, 2010. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 7. Shahi PK, Liu X, Aul B, Moyer A, Pattnaik A, Denton J, Pillers DM, Pattnaik BR. Abnormal electroretinogram after Kir7.1 channel suppression suggests role in retinal electrophysiology. Sci Rep 7: 10651, 2017. doi: 10.1038/s41598-017-11034-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pattnaik BR, Asuma MP, Spott R, Pillers DA. Genetic defects in the hotspot of inwardly rectifying K(+) (Kir) channels and their metabolic consequences: a review. Mol Genet Metab 105: 64–72, 2012. doi: 10.1016/j.ymgme.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Raz-Prag D, Grimes WN, Fariss RN, Vijayasarathy C, Campos MM, Bush RA, Diamond JS, Sieving PA. Probing potassium channel function in vivo by intracellular delivery of antibodies in a rat model of retinal neurodegeneration. Proc Natl Acad Sci USA 107: 12710–12715, 2010. doi: 10.1073/pnas.0913472107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vila A, Shihabeddin E, Zhang Z, Santhanam A, Ribelayga CP, O'Brien J. Synaptic scaffolds, ion channels and polyamines in mouse photoreceptor synapses: anatomy of a signaling complex. Front Cell Neurosci 15: 667046, 2021. doi: 10.3389/fncel.2021.667046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen L, Yu YC, Zhao JW, Yang XL. Inwardly rectifying potassium channels in rat retinal ganglion cells. Eur J Neurosci 20: 956–964, 2004. doi: 10.1111/j.1460-9568.2004.03553.x. [DOI] [PubMed] [Google Scholar]
- 12. Drenhaus U, Morino P, Veh RW. On the development of the stratification of the inner plexiform layer in the chick retina. J Comp Neurol 460: 1–12, 2003. doi: 10.1002/cne.10602. [DOI] [PubMed] [Google Scholar]
- 13. Kusaka S, Inanobe A, Fujita A, Makino Y, Tanemoto M, Matsushita K, Tano Y, Kurachi Y. Functional Kir7.1 channels localized at the root of apical processes in rat retinal pigment epithelium. J Physiol 531: 27–36, 2001. doi: 10.1111/j.1469-7793.2001.0027j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Connors NC, Adams ME, Froehner SC, Kofuji P. The potassium channel Kir4.1 associates with the dystrophin-glycoprotein complex via alpha-syntrophin in glia. J Biol Chem 279: 28387–28392, 2004. doi: 10.1074/jbc.M402604200. [DOI] [PubMed] [Google Scholar]
- 15. Connors NC, Kofuji P. Dystrophin Dp71 is critical for the clustered localization of potassium channels in retinal glial cells. J Neurosci 22: 4321–4327, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Connors NC, Kofuji P. Potassium channel Kir4.1 macromolecular complex in retinal glial cells. Glia 53: 124–131, 2006. doi: 10.1002/glia.20271. [DOI] [PubMed] [Google Scholar]
- 17. Nagelhus EA, Horio Y, Inanobe A, Fujita A, Haug FM, Nielsen S, Kurachi Y, Ottersen OP. Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Muller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia 26: 47–54, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- 18. Ishii M, Fujita A, Iwai K, Kusaka S, Higashi K, Inanobe A, Hibino H, Kurachi Y. Differential expression and distribution of Kir5.1 and Kir4.1 inwardly rectifying K+ channels in retina. Am J Physiol Cell Physiol 285: C260–C267, 2003. doi: 10.1152/ajpcell.00560.2002. [DOI] [PubMed] [Google Scholar]
- 19. Du RH, Lu M, Wang C, Ding JH, Wu G, Hu G. The pore-forming subunit Kir6.1 of the K-ATP channel negatively regulates the NLRP3 inflammasome to control insulin resistance by interacting with NLRP3. Exp Mol Med 51: 1–13, 2019. doi: 10.1038/s12276-019-0291-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shimura M, Yuan Y, Chang JT, Zhang S, Campochiaro PA, Zack DJ, Hughes BA. Expression and permeation properties of the K(+) channel Kir7.1 in the retinal pigment epithelium. J Physiol 531: 329–346, 2001. doi: 10.1111/j.1469-7793.2001.0329i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang D, Swaminathan A, Zhang X, Hughes BA. Expression of Kir7.1 and a novel Kir7.1 splice variant in native human retinal pigment epithelium. Exp Eye Res 86: 81–91, 2008. doi: 10.1016/j.exer.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klose E, Kuhrt H, Kohen L, Wiedemann P, Bringmann A, Hollborn M. Hypoxic and osmotic expression of Kir2.1 potassium channels in retinal pigment epithelial cells: contribution to vascular endothelial growth factor expression. Exp Eye Res 211: 108741, 2021. doi: 10.1016/j.exer.2021.108741. [DOI] [PubMed] [Google Scholar]
- 23. Al-Ani A, Sunba S, Hafeez B, Toms D, Ungrin M. In vitro maturation of retinal pigment epithelium is essential for maintaining high expression of key functional genes. Int J Mol Sci 21: 6066, 2020. doi: 10.3390/ijms21176066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ghassemifar R, Lai CM, Rakoczy PE. VEGF differentially regulates transcription and translation of ZO-1alpha+ and ZO-1alpha- and mediates trans-epithelial resistance in cultured endothelial and epithelial cells. Cell Tissue Res 323: 117–125, 2006. doi: 10.1007/s00441-005-0046-7. [DOI] [PubMed] [Google Scholar]
- 25. Kolb AR, Needham PG, Rothenberg C, Guerriero CJ, Welling PA, Brodsky JL. ESCRT regulates surface expression of the Kir2.1 potassium channel. Mol Biol Cell 25: 276–289, 2014. doi: 10.1091/mbc.E13-07-0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hollborn M, Vogler S, Reichenbach A, Wiedemann P, Bringmann A, Kohen L. Regulation of the hyperosmotic induction of aquaporin 5 and VEGF in retinal pigment epithelial cells: involvement of NFAT5. Mol Vis 21: 360–377, 2015. [PMC free article] [PubMed] [Google Scholar]
- 27. Lutty GA, McLeod DS. Development of the hyaloid, choroidal and retinal vasculatures in the fetal human eye. Prog Retin Eye Res 62: 58–76, 2018. doi: 10.1016/j.preteyeres.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rattner A, Williams J, Nathans J. Roles of HIFs and VEGF in angiogenesis in the retina and brain. J Clin Invest 129: 3807–3820, 2019. doi: 10.1172/JCI126655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miller JW, Le Couter J, Strauss EC, Ferrara N. Vascular endothelial growth factor a in intraocular vascular disease. Ophthalmology 120: 106–114, 2013. doi: 10.1016/j.ophtha.2012.07.038. [DOI] [PubMed] [Google Scholar]
- 30. Bourque SL, Kuny S, Reyes LM, Davidge ST, Sauve Y. Prenatal hypoxia is associated with long-term retinal dysfunction in rats. PLoS One 8: e61861, 2013. doi: 10.1371/journal.pone.0061861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reilly L, Eckhardt LL. Cardiac potassium inward rectifier Kir2: review of structure, regulation, pharmacology, and arrhythmogenesis. Heart Rhythm 18: 1423–1434, 2021. doi: 10.1016/j.hrthm.2021.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Riz M, Braun M, Wu X, Pedersen MG. Inwardly rectifying Kir2.1 currents in human beta-cells control electrical activity: characterisation and mathematical modelling. Biochem Biophys Res Commun 459: 284–287, 2015. doi: 10.1016/j.bbrc.2015.02.099. [DOI] [PubMed] [Google Scholar]
- 33. Hager NA, McAtee CK, Lesko MA, O'Donnell AF. Inwardly rectifying potassium channel Kir2.1 and its “Kir-ious” regulation by protein trafficking and roles in development and disease. Front Cell Dev Biol 9: 796136, 2021. doi: 10.3389/fcell.2021.796136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Davies NP, Imbrici P, Fialho D, Herd C, Bilsland LG, Weber A, Mueller R, Hilton-Jones D, Ealing J, Boothman BR, Giunti P, Parsons LM, Thomas M, Manzur AY, Jurkat-Rott K, Lehmann-Horn F, Chinnery PF, Rose M, Kullmann DM, Hanna MG. Andersen-Tawil syndrome: new potassium channel mutations and possible phenotypic variation. Neurology 65: 1083–1089, 2005. doi: 10.1212/01.wnl.0000178888.03767.74. [DOI] [PubMed] [Google Scholar]
- 35. Kokunai Y, Nakata T, Furuta M, Sakata S, Kimura H, Aiba T, Yoshinaga M, Osaki Y, Nakamori M, Itoh H, Sato T, Kubota T, Kadota K, Shindo K, Mochizuki H, Shimizu W, Horie M, Okamura Y, Ohno K, Takahashi MP. A Kir3.4 mutation causes Andersen-Tawil syndrome by an inhibitory effect on Kir2.1. Neurology 82: 1058–1064, 2014. doi: 10.1212/WNL.0000000000000239. [DOI] [PubMed] [Google Scholar]
- 36. Kimura H, Zhou J, Kawamura M, Itoh H, Mizusawa Y, Ding WG, Wu J, Ohno S, Makiyama T, Miyamoto A, Naiki N, Wang Q, Xie Y, Suzuki T, Tateno S, Nakamura Y, Zang WJ, Ito M, Matsuura H, Horie M. Phenotype variability in patients carrying KCNJ2 mutations. Circ Cardiovasc Genet 5: 344–353, 2012. doi: 10.1161/CIRCGENETICS.111.962316. [DOI] [PubMed] [Google Scholar]
- 37. Kostera-Pruszczyk A, Potulska-Chromik A, Pruszczyk P, Bieganowska K, Miszczak-Knecht M, Bienias P, Szczałuba K, Lee H-Y, Quinn E, Ploski R, Kaminska A, Ptáček LJ. Andersen-Tawil syndrome: report of 3 novel mutations and high risk of symptomatic cardiac involvement. Muscle Nerve 51: 192–196, 2015. doi: 10.1002/mus.24293. [DOI] [PubMed] [Google Scholar]
- 38. Haruna Y, Kobori A, Makiyama T, Yoshida H, Akao M, Doi T, Tsuji K, Ono S, Nishio Y, Shimizu W, Inoue T, Murakami T, Tsuboi N, Yamanouchi H, Ushinohama H, Nakamura Y, Yoshinaga M, Horigome H, Aizawa Y, Kita T, Horie M. Genotype-phenotype correlations of KCNJ2 mutations in Japanese patients with Andersen-Tawil syndrome. Hum Mutat 28: 208, 2007. doi: 10.1002/humu.9483. [DOI] [PubMed] [Google Scholar]
- 39. Andelfinger G, Tapper AR, Welch RC, Vanoye CG, George AL, Jr, Benson DW. KCNJ2 mutation results in Andersen syndrome with sex-specific cardiac and skeletal muscle phenotypes. Am J Hum Genet 71: 663–668, 2002. doi: 10.1086/342360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Donaldson MR, Jensen JL, Tristani-Firouzi M, Tawil R, Bendahhou S, Suarez WA, Cobo AM, Poza JJ, Behr E, Wagstaff J, Szepetowski P, Pereira S, Mozaffar T, Escolar DM, Fu YH, Ptacek LJ. PIP2 binding residues of Kir2.1 are common targets of mutations causing Andersen syndrome. Neurology 60: 1811–1816, 2003. doi: 10.1212/01.wnl.0000072261.14060.47. [DOI] [PubMed] [Google Scholar]
- 41. Plaster NM, Tawil R, Tristani-Firouzi M, Canun S, Bendahhou S, Tsunoda A, Donaldson MR, Iannaccone ST, Brunt E, Barohn R, Clark J, Deymeer F, George AL, Jr, Fish FA, Hahn A, Nitu A, Ozdemir C, Serdaroglu P, Subramony SH, Wolfe G, Fu YH, Ptacek LJ. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell 105: 511–519, 2001. doi: 10.1016/S0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 42. Marrus SB, Cuculich PS, Wang W, Nerbonne JM. Characterization of a novel, dominant negative KCNJ2 mutation associated with Andersen-Tawil syndrome. Channels (Austin) 5: 500–509, 2011. doi: 10.4161/chan.5.6.18524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ballester LY, Benson DW, Wong B, Law IH, Mathews KD, Vanoye CG, George AL, Jr.. Trafficking-competent and trafficking-defective KCNJ2 mutations in Andersen syndrome. Hum Mutat 27: 388, 2006. doi: 10.1002/humu.9418. [DOI] [PubMed] [Google Scholar]
- 44. Yoon G, Oberoi S, Tristani-Firouzi M, Etheridge SP, Quitania L, Kramer JH, Miller BL, Fu YH, Ptacek LJ. Andersen-Tawil syndrome: prospective cohort analysis and expansion of the phenotype. Am J Med Genet A 140: 312–321, 2006. doi: 10.1002/ajmg.a.31092. [DOI] [PubMed] [Google Scholar]
- 45. Lim BC, Kim GB, Bae EJ, Noh CI, Hwang H, Kim KJ, Hwang YS, Ko TS, Chae JH. Andersen cardiodysrhythmic periodic paralysis with KCNJ2 mutations: a novel mutation in the pore selectivity filter residue. J Child Neurol 25: 490–493, 2010. doi: 10.1177/0883073809357937. [DOI] [PubMed] [Google Scholar]
- 46. Tully I, Atherton J, Hunt L, Ingles J, Semsarian C, McGaughran J. Rarity and phenotypic heterogeneity provide challenges in the diagnosis of Andersen-Tawil syndrome: two cases presenting with ECGs mimicking catecholaminergic polymorphic ventricular tachycardia (CPVT). Int J Cardiol 201: 473–475, 2015. doi: 10.1016/j.ijcard.2015.07.069. [DOI] [PubMed] [Google Scholar]
- 47. Kim JB, Chung KW. Novel de novo mutation in the KCNJ2 gene in a patient with Andersen-Tawil syndrome. Pediatr Neurol 41: 464–466, 2009. doi: 10.1016/j.pediatrneurol.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 48. Bendahhou S, Fournier E, Sternberg D, Bassez G, Furby A, Sereni C, Donaldson MR, Larroque MM, Fontaine B, Barhanin J. In vivo and in vitro functional characterization of Andersen's syndrome mutations. J Physiol 565: 731–741, 2005. doi: 10.1113/jphysiol.2004.081620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Song J, Luo S, Cheng X, Yue D, Zhu W, Lin J, Huang J, Lu J, Zhao C, Qiao K. Clinical features and long exercise test in Chinese patients with Andersen-Tawil syndrome. Muscle Nerve 54: 1059–1063, 2016. doi: 10.1002/mus.25169. [DOI] [PubMed] [Google Scholar]
- 50. Tristani-Firouzi M, Jensen JL, Donaldson MR, Sansone V, Meola G, Hahn A, Bendahhou S, Kwiecinski H, Fidzianska A, Plaster N, Fu YH, Ptacek LJ, Tawil R. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J Clin Invest 110: 381–388, 2002. doi: 10.1172/JCI15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Delannoy E, Sacher F, Maury P, Mabo P, Mansourati J, Magnin I, Camous JP, Tournant G, Rendu E, Kyndt F, Haissaguerre M, Bezieau S, Guyomarch B, Le Marec H, Fressart V, Denjoy I, Probst V. Cardiac characteristics and long-term outcome in Andersen-Tawil syndrome patients related to KCNJ2 mutation. Europace 15: 1805–1811, 2013. doi: 10.1093/europace/eut160. [DOI] [PubMed] [Google Scholar]
- 52. Chan HF, Chen ML, Su JJ, Ko LC, Lin CH, Wu RM. A novel neuropsychiatric phenotype of KCNJ2 mutation in one Taiwanese family with Andersen-Tawil syndrome. J Hum Genet 55: 186–188, 2010. doi: 10.1038/jhg.2010.2. [DOI] [PubMed] [Google Scholar]
- 53. Ai T, Fujiwara Y, Tsuji K, Otani H, Nakano S, Kubo Y, Horie M. Novel KCNJ2 mutation in familial periodic paralysis with ventricular dysrhythmia. Circulation 105: 2592–2594, 2002. doi: 10.1161/01.cir.0000019906.35135.a3. [DOI] [PubMed] [Google Scholar]
- 54. Hosaka Y, Hanawa H, Washizuka T, Chinushi M, Yamashita F, Yoshida T, Komura S, Watanabe H, Aizawa Y. Function, subcellular localization and assembly of a novel mutation of KCNJ2 in Andersen's syndrome. J Mol Cell Cardiol 35: 409–415, 2003. doi: 10.1016/S0022-2828(03)00046-4. [DOI] [PubMed] [Google Scholar]
- 55. Rezazadeh S, Guo J, Duff HJ, Ferrier RA, Gerull B. Reversible dilated cardiomyopathy caused by a high burden of ventricular arrhythmias in Andersen-Tawil syndrome. Can J Cardiol 32: 1576.e15–1576.e18, 2016. doi: 10.1016/j.cjca.2016.07.587. [DOI] [PubMed] [Google Scholar]
- 56. Barajas-Martinez H, Hu D, Ontiveros G, Caceres G, Desai M, Burashnikov E, Scaglione J, Antzelevitch C. Biophysical and molecular characterization of a novel de novo KCNJ2 mutation associated with Andersen-Tawil syndrome and catecholaminergic polymorphic ventricular tachycardia mimicry. Circ Cardiovasc Genet 4: 51–57, 2011. doi: 10.1161/CIRCGENETICS.110.957696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Diaz-Manera J, Querol L, Clarimon J, Yague S, Illa I. Unique post-exercise electrophysiological test results in a new Andersen-Tawil syndrome mutation. Clin Neurophysiol 122: 2537–2539, 2011. doi: 10.1016/j.clinph.2011.04.025. [DOI] [PubMed] [Google Scholar]
- 58. Choi BO, Kim J, Suh BC, Yu JS, Sunwoo IN, Kim SJ, Kim GH, Chung KW. Mutations of KCNJ2 gene associated with Andersen-Tawil syndrome in Korean families. J Hum Genet 52: 280–283, 2007. doi: 10.1007/s10038-006-0100-7. [DOI] [PubMed] [Google Scholar]
- 59. Liu XL, Huang XJ, Luan XH, Zhou HY, Wang T, Wang JY, Shen JY, Chen SD, Tang HD, Cao L. Case report: a Chinese child with Andersen-Tawil syndrome due to a de novo KCNJ2 mutation. J Neurol Sci 352: 105–106, 2015. doi: 10.1016/j.jns.2015.02.027. [DOI] [PubMed] [Google Scholar]
- 60. Limberg MM, Zumhagen S, Netter MF, Coffey AJ, Grace A, Rogers J, Bockelmann D, Rinne S, Stallmeyer B, Decher N, Schulze-Bahr E. Non dominant-negative KCNJ2 gene mutations leading to Andersen-Tawil syndrome with an isolated cardiac phenotype. Basic Res Cardiol 108: 353, 2013. doi: 10.1007/s00395-013-0353-1. [DOI] [PubMed] [Google Scholar]
- 61. Doi T, Makiyama T, Morimoto T, Haruna Y, Tsuji K, Ohno S, Akao M, Takahashi Y, Kimura T, Horie M. A novel KCNJ2 nonsense mutation, S369X, impedes trafficking and causes a limited form of Andersen-Tawil syndrome. Circ Cardiovasc Genet 4: 253–260, 2011. doi: 10.1161/CIRCGENETICS.110.958157. [DOI] [PubMed] [Google Scholar]
- 62. Fodstad H, Swan H, Auberson M, Gautschi I, Loffing J, Schild L, Kontula K. Loss-of-function mutations of the K(+) channel gene KCNJ2 constitute a rare cause of long QT syndrome. J Mol Cell Cardiol 37: 593–602, 2004. doi: 10.1016/j.yjmcc.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 63. Lieve KV, Williams L, Daly A, Richard G, Bale S, Macaya D, Chung WK. Results of genetic testing in 855 consecutive unrelated patients referred for long QT syndrome in a clinical laboratory. Genet Test Mol Biomarkers 17: 553–561, 2013. doi: 10.1089/gtmb.2012.0118. [DOI] [PubMed] [Google Scholar]
- 64. Obeyesekere MN, Klein GJ, Conacher S, Krahn AD. KCNJ2 variant of unknown significance reclassified as long QT syndrome causing ventricular fibrillation. Can J Cardiol 27: 870 e811–e873, 2011. doi: 10.1016/j.cjca.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 65. Sabater-Molina M, Guillen-Navarro E, Garcia-Molina E, Ballesta-Martinez MJ, Escudero F, Ruiz-Espejo F. Barth syndrome in adulthood: a clinical case. Rev Esp Cardiol (Engl Ed) 66: 68–70, 2013. doi: 10.1016/j.recesp.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 66. Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, Napolitano C, Anumonwo J, di Barletta MR, Gudapakkam S, Bosi G, Stramba-Badiale M, Jalife J. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res 96: 800–807, 2005. doi: 10.1161/01.RES.0000162101.76263.8c. [DOI] [PubMed] [Google Scholar]
- 67. Deo M, Ruan Y, Pandit SV, Shah K, Berenfeld O, Blaufox A, Cerrone M, Noujaim SF, Denegri M, Jalife J, Priori SG. KCNJ2 mutation in short QT syndrome 3 results in atrial fibrillation and ventricular proarrhythmia. Proc Natl Acad Sci USA 110: 4291–4296, 2013. doi: 10.1073/pnas.1218154110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hattori T, Makiyama T, Akao M, Ehara E, Ohno S, Iguchi M, Nishio Y, Sasaki K, Itoh H, Yokode M, Kita T, Horie M, Kimura T. A novel gain-of-function KCNJ2 mutation associated with short-QT syndrome impairs inward rectification of Kir2.1 currents. Cardiovasc Res 93: 666–673, 2012. doi: 10.1093/cvr/cvr329. [DOI] [PubMed] [Google Scholar]
- 69. Ambrosini E, Sicca F, Brignone MS, D'Adamo MC, Napolitano C, Servettini I, Moro F, Ruan Y, Guglielmi L, Pieroni S, Servillo G, Lanciotti A, Valvo G, Catacuzzeno L, Franciolini F, Molinari P, Marchese M, Grottesi A, Guerrini R, Santorelli FM, Priori S, Pessia M. Genetically induced dysfunctions of Kir2.1 channels: implications for short QT3 syndrome and autism-epilepsy phenotype. Hum Mol Genet 23: 4875–4886, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tester DJ, Arya P, Will M, Haglund CM, Farley AL, Makielski JC, Ackerman MJ. Genotypic heterogeneity and phenotypic mimicry among unrelated patients referred for catecholaminergic polymorphic ventricular tachycardia genetic testing. Heart Rhythm 3: 800–805, 2006. doi: 10.1016/j.hrthm.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 71. Chun TU, Epstein MR, Dick M 2nd, Andelfinger G, Ballester L, Vanoye CG, George AL Jr, Benson DW. Polymorphic ventricular tachycardia and KCNJ2 mutations. Heart Rhythm 1: 235–241, 2004. doi: 10.1016/j.hrthm.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 72. Eckhardt LL, Farley AL, Rodriguez E, Ruwaldt K, Hammill D, Tester DJ, Ackerman MJ, Makielski JC. KCNJ2 mutations in arrhythmia patients referred for LQT testing: a mutation T305A with novel effect on rectification properties. Heart Rhythm 4: 323–329, 2007. doi: 10.1016/j.hrthm.2006.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Xia M, Jin Q, Bendahhou S, He Y, Larroque MM, Chen Y, Zhou Q, Yang Y, Liu Y, Liu B, Zhu Q, Zhou Y, Lin J, Liang B, Li L, Dong X, Pan Z, Wang R, Wan H, Qiu W, Xu W, Eurlings P, Barhanin J, Chen Y. A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem Biophys Res Commun 332: 1012–1019, 2005. doi: 10.1016/j.bbrc.2005.05.054. [DOI] [PubMed] [Google Scholar]
- 74. Matsushita K, Puro DG. Topographical heterogeneity of K(IR) currents in pericyte-containing microvessels of the rat retina: effect of diabetes. J Physiol 573: 483–495, 2006. doi: 10.1113/jphysiol.2006.107102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lad EM, Hernandez-Boussard T, Morton JM, Moshfeghi DM. Incidence of retinopathy of prematurity in the United States: 1997 through 2005. Am J Ophthalmol 148: 451–458, 2009. doi: 10.1016/j.ajo.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 76. Zhang X, Saaddine JB, Chou CF, Cotch MF, Cheng YJ, Geiss LS, Gregg EW, Albright AL, Klein BE, Klein R. Prevalence of diabetic retinopathy in the United States, 2005-2008. JAMA 304: 649–656, 2010. doi: 10.1001/jama.2010.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Movsas TZ, Muthusamy A. Associations between VEGF isoforms and impending retinopathy of prematurity. Int J Dev Neurosci 80: 586–593, 2020. doi: 10.1002/jdn.10054. [DOI] [PubMed] [Google Scholar]
- 78. Yang Y, Liu Y, Li Y, Chen Z, Xiong Y, Zhou T, Tao W, Xu F, Yang H, Yla-Herttuala S, Chaurasia SS, Adam WC, Yang K. MicroRNA-15b targets VEGF and inhibits angiogenesis in proliferative diabetic retinopathy. J Clin Endocrinol Metab 105: 3404–3415, 2020. doi: 10.1210/clinem/dgaa538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kim EJ, Lin WV, Rodriguez SM, Chen A, Loya A, Weng CY. Treatment of diabetic macular edema. Curr Diab Rep 19: 68, 2019. doi: 10.1007/s11892-019-1188-4. [DOI] [PubMed] [Google Scholar]
- 80. Gantz OB, Rynecki ND, Para A, Levidy M, Beebe KS. Postoperative negative pressure wound therapy is associated with decreased surgical site infections in all lower extremity amputations. J Orthop 21: 507–511, 2020. doi: 10.1016/j.jor.2020.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Smith LE, Wesolowski E, McLellan A, Kostyk SK, D'Amato R, Sullivan R, D'Amore PA. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci 35: 101–111, 1994. [PubMed] [Google Scholar]
- 82. Dai C, Webster KA, Bhatt A, Tian H, Su G, Li W. Concurrent physiological and pathological angiogenesis in retinopathy of prematurity and emerging therapies. Int J Mol Sci 22: 4809, 2021. doi: 10.3390/ijms22094809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bouskila J, Palmour RM, Bouchard JF, Ptito M. Retinal structure and function in monkeys with fetal alcohol exposure. Exp Eye Res 177: 55–64, 2018. doi: 10.1016/j.exer.2018.07.027. [DOI] [PubMed] [Google Scholar]
- 84. Hug TE, Fitzgerald KM, Cibis GW. Clinical and electroretinographic findings in fetal alcohol syndrome. J AAPOS 4: 200–204, 2000. doi: 10.1067/mpa.2000.105278. [DOI] [PubMed] [Google Scholar]
- 85. Stromland K. Visual impairment and ocular abnormalities in children with fetal alcohol syndrome. Addict Biol 9: 153–157, 2004. doi: 10.1080/13556210410001717024. [DOI] [PubMed] [Google Scholar]
- 86. Bates EA. A potential molecular target for morphological defects of fetal alcohol syndrome: Kir2.1. Curr Opin Genet Dev 23: 324–329, 2013. doi: 10.1016/j.gde.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 87. Matsa E, Dixon JE, Medway C, Georgiou O, Patel MJ, Morgan K, Kemp PJ, Staniforth A, Mellor I, Denning C. Allele-specific RNA interference rescues the long-QT syndrome phenotype in human-induced pluripotency stem cell cardiomyocytes. Eur Heart J 35: 1078–1087, 2014. doi: 10.1093/eurheartj/eht067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Fischer RA, Roux AL, Wareham LK, Sappington RM. Pressure-dependent modulation of inward-rectifying K(+) channels: implications for cation homeostasis and K(+) dynamics in glaucoma. Am J Physiol Cell Physiol 317: C375–C389, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sappington RM, Sidorova T, Long DJ, Calkins DJ. TRPV1: contribution to retinal ganglion cell apoptosis and increased intracellular Ca2+ with exposure to hydrostatic pressure. Invest Ophthalmol Vis Sci 50: 717–728, 2009. doi: 10.1167/iovs.08-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sappington RM, Chan M, Calkins DJ. Interleukin-6 protects retinal ganglion cells from pressure-induced death. Invest Ophthalmol Vis Sci 47: 2932–2942, 2006. doi: 10.1167/iovs.05-1407. [DOI] [PubMed] [Google Scholar]
- 91. Kharade SV, Kurata H, Bender AM, Blobaum AL, Figueroa EE, Duran A, Kramer M, Days E, Vinson P, Flores D, Satlin LM, Meiler J, Weaver CD, Lindsley CW, Hopkins CR, Denton JS. Discovery, characterization, and effects on renal fluid and electrolyte excretion of the Kir4.1 potassium channel pore blocker, VU0134992. Mol Pharmacol 94: 926–937, 2018. doi: 10.1124/mol.118.112359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pessia M, Tucker SJ, Lee K, Bond CT, Adelman JP. Subunit positional effects revealed by novel heteromeric inwardly rectifying K+ channels. EMBO J 15: 2980–2987, 1996. [PMC free article] [PubMed] [Google Scholar]
- 93. Marmolejo-Murillo LG, Arechiga-Figueroa IA, Moreno-Galindo EG, Ferrer T, Zamora-Cardenas R, Navarro-Polanco RA, Sanchez-Chapula JA, Rodriguez-Menchaca AA. Kir4.1/Kir5.1 channels possess strong intrinsic inward rectification determined by a voltage-dependent K+-flux gating mechanism. J Gen Physiol 153: e201912540, 2021. doi: 10.1085/jgp.201912540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Isaeva E, Bohovyk R, Fedoriuk M, Shalygin A, Klemens CA, Zietara A, Levchenko V, Denton JS, Staruschenko A, Palygin O. Crosstalk between epithelial sodium channels (ENaC) and basolateral potassium channels (Kir 4.1/Kir 5.1) in the cortical collecting duct. Br J Pharmacol 34: 1–1, 2020. doi: 10.1096/fasebj.2020.34.s1.02233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Reichenbach A, Wurm A, Pannicke T, Iandiev I, Wiedemann P, Bringmann A. Muller cells as players in retinal degeneration and edema. Graefes Arch Clin Exp Ophthalmol 245: 627–636, 2007. doi: 10.1007/s00417-006-0516-y. [DOI] [PubMed] [Google Scholar]
- 96. Bosco A, Cusato K, Nicchia GP, Frigeri A, Spray DC. A developmental switch in the expression of aquaporin-4 and Kir4.1 from horizontal to Muller cells in mouse retina. Invest Ophthalmol Vis Sci 46: 3869–3875, 2005. doi: 10.1167/iovs.05-0385. [DOI] [PubMed] [Google Scholar]
- 97. Rao SB, Katoozi S, Skauli N, Froehner SC, Ottersen OP, Adams ME, Amiry-Moghaddam M. Targeted deletion of beta1-syntrophin causes a loss of Kir 4.1 from Muller cell endfeet in mouse retina. Glia 67: 1138–1149, 2019. doi: 10.1002/glia.23600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Vacca O, Charles-Messance H, El Mathari B, Sene A, Barbe P, Fouquet S, Aragon J, Darche M, Giocanti-Auregan A, Paques M, Sahel JA, Tadayoni R, Montanez C, Dalkara D, Rendon A. AAV-mediated gene therapy in Dystrophin-Dp71 deficient mouse leads to blood-retinal barrier restoration and oedema reabsorption. Hum Mol Genet 25: 3070–3079, 2016. doi: 10.1093/hmg/ddw159. [DOI] [PubMed] [Google Scholar]
- 99. Dmitriev AV, Dmitriev AA, Linsenmeier RA. K(+)dependent Muller cell-generated components of the electroretinogram. Vis Neurosci 38: E010, 2021. doi: 10.1017/S0952523821000092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci 20: 5733–5740, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ohno Y, Hibino H, Lossin C, Inanobe A, Kurachi Y. Inhibition of astroglial Kir4.1 channels by selective serotonin reuptake inhibitors. Brain Res 1178: 44–51, 2007. doi: 10.1016/j.brainres.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 102. Li X, Lv J, Li J, Ren X. Kir4.1 may represent a novel therapeutic target for diabetic retinopathy (Review). Exp Ther Med 22: 1021, 2021. doi: 10.3892/etm.2021.10453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Cree-Green M, Bergman BC, Cengiz E, Fox LA, Hannon TS, Miller K, Nathan B, Pyle L, Kahn D, Tansey M, Tichy E, Tsalikian E, Libman I, Nadeau KJ. Metformin improves peripheral insulin sensitivity in youth with type 1 diabetes. J Clin Endocrinol Metab 104: 3265–3278, 2019. doi: 10.1210/jc.2019-00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Alex A, Luo Q, Mathew D, Di R, Bhatwadekar AD. Metformin corrects abnormal circadian rhythm and Kir4.1 channels in diabetes. Invest Ophthalmol Vis Sci 61: 46, 2020. doi: 10.1167/iovs.61.6.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Abdelhadi O, Iancu D, Stanescu H, Kleta R, Bockenhauer D. EAST syndrome: clinical, pathophysiological, and genetic aspects of mutations in KCNJ10. Rare Dis 4: e1195043, 2016. doi: 10.1080/21675511.2016.1195043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Scholl UI, Dave HB, Lu M, Farhi A, Nelson-Williams C, Listman JA, Lifton RP. SeSAME/EAST syndrome–phenotypic variability and delayed activity of the distal convoluted tubule. Pediatr Nephrol 27: 2081–2090, 2012. doi: 10.1007/s00467-012-2219-4. [DOI] [PubMed] [Google Scholar]
- 107. Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, Arora R, Sirimanna T, Thompson D, Cross JH, van't Hoff W, Al Masri O, Tullus K, Yeung S, Anikster Y, Klootwijk E, Hubank M, Dillon MJ, Heitzmann D, Arcos-Burgos M, Knepper MA, Dobbie A, Gahl WA, Warth R, Sheridan E, Kleta R. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med 360: 1960–1970, 2009. doi: 10.1056/NEJMoa0810276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Freudenthal B, Kulaveerasingam D, Lingappa L, Shah MA, Brueton L, Wassmer E, Ognjanovic M, Dorison N, Reichold M, Bockenhauer D, Kleta R, Zdebik AA. KCNJ10 mutations disrupt function in patients with EAST syndrome. Nephron Physiol 119: p40–p48, 2011. doi: 10.1159/000330250. [DOI] [PubMed] [Google Scholar]
- 109. Lemke JR, Riesch E, Scheurenbrand T, Schubach M, Wilhelm C, Steiner I, , et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia 53: 1387–1398, 2012. doi: 10.1111/j.1528-1167.2012.03516.x. [DOI] [PubMed] [Google Scholar]
- 110. Parrock S, Hussain S, Issler N, Differ AM, Lench N, Guarino S, Oosterveld MJ, Keijzer-Veen M, Brilstra E, van Wieringen H, Konijnenberg AY, Amin-Rasip S, Dumitriu S, Klootwijk E, Knoers N, Bockenhauer D, Kleta R, Zdebik AA. KCNJ10 mutations display differential sensitivity to heteromerisation with KCNJ16. Nephron Physiol 123: 7–14, 2013. doi: 10.1159/000356353. [DOI] [PubMed] [Google Scholar]
- 111. Scholl UI, Choi M, Liu T, Ramaekers VT, Hausler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci USA 106: 5842–5847, 2009. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Reichold M, Zdebik AA, Lieberer E, Rapedius M, Schmidt K, Bandulik S, Sterner C, Tegtmeier I, Penton D, Baukrowitz T, Hulton SA, Witzgall R, Ben-Zeev B, Howie AJ, Kleta R, Bockenhauer D, Warth R. KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc Natl Acad Sci USA 107: 14490–14495, 2010. doi: 10.1073/pnas.1003072107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Yang T, Gurrola JG 2nd, Wu H, Chiu SM, Wangemann P, Snyder PM, Smith RJ. Mutations of KCNJ10 together with mutations of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. Am J Hum Genet 84: 651–657, 2009. doi: 10.1016/j.ajhg.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Kara B, Ekici B, Ipekci B, Aslanger AK, Scholl U. KCNJ10 gene mutation in an 8-year-old boy with seizures. Acta Neurol Belg 113: 75–77, 2013. doi: 10.1007/s13760-012-0113-2. [DOI] [PubMed] [Google Scholar]
- 115. Brasko C, Hawkins V, De La Rocha IC, Butt AM. Expression of Kir4.1 and Kir5.1 inwardly rectifying potassium channels in oligodendrocytes, the myelinating cells of the CNS. Brain Struct Funct 222: 41–59, 2017. doi: 10.1007/s00429-016-1199-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Feraille E, Dizin E. Coordinated control of ENaC and Na+,K+-ATPase in renal collecting duct. J Am Soc Nephrol 27: 2554–2563, 2016. doi: 10.1681/ASN.2016020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Thompson DA, Feather S, Stanescu HC, Freudenthal B, Zdebik AA, Warth R, Ognjanovic M, Hulton SA, Wassmer E, van't Hoff W, Russell-Eggitt I, Dobbie A, Sheridan E, Kleta R, Bockenhauer D. Altered electroretinograms in patients with KCNJ10 mutations and EAST syndrome. J Physiol 589: 1681–1689, 2011. doi: 10.1113/jphysiol.2010.198531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Yang D, Pan A, Swaminathan A, Kumar G, Hughes BA. Expression and localization of the inwardly rectifying potassium channel Kir7.1 in native bovine retinal pigment epithelium. Invest Ophthalmol Vis Sci 44: 3178–3185, 2003. doi: 10.1167/iovs.02-1189. [DOI] [PubMed] [Google Scholar]
- 119. Krapivinsky G, Medina I, Eng L, Krapivinsky L, Yang Y, Clapham DE. A novel inward rectifier K+ channel with unique pore properties. Neuron 20: 995–1005, 1998. doi: 10.1016/s0896-6273(00)80480-8. [DOI] [PubMed] [Google Scholar]
- 120. Doring F, Derst C, Wischmeyer E, Karschin C, Schneggenburger R, Daut J, Karschin A. The epithelial inward rectifier channel Kir7.1 displays unusual K+ permeation properties. J Neurosci 18: 8625–8636, 1998. doi: 10.1523/JNEUROSCI.18-21-08625.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Kharade SV, Sheehan JH, Figueroa EE, Meiler J, Denton JS. Pore polarity and charge determine differential block of Kir1.1 and Kir7.1 potassium channels by small-molecule inhibitor VU590. Mol Pharmacol 92: 338–346, 2017. doi: 10.1124/mol.117.108472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Denton JS, Kharade SV. Plight of the pore polar bar(rier). Channels (Austin) 11: 502–503, 2017. doi: 10.1080/19336950.2017.1367234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Miller SS, Edelman JL. Active ion transport pathways in the bovine retinal pigment epithelium. J Physiol 424: 283–300, 1990. doi: 10.1113/jphysiol.1990.sp018067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Pattnaik BR, Hughes BA. Regulation of Kir channels in bovine retinal pigment epithelial cells by phosphatidylinositol 4,5-bisphosphate. Am J Physiol Cell Physiol 297: C1001–C1011, 2009. doi: 10.1152/ajpcell.00250.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Shahi PK, Hermans D, Sinha D, Brar S, Moulton H, Stulo S, Borys KD, Capowski E, Pillers DM, Gamm DM, Pattnaik BR. Gene augmentation and readthrough rescue channelopathy in an iPSC-RPE model of congenital blindness. Am J Hum Genet 104: 310–318, 2019. doi: 10.1016/j.ajhg.2018.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Carrington S, Hernandez C, Swale D, Aluko OA, Denton JS, Cone R. G protein-coupled receptors differentially regulate glycosylation and activity of the inwardly rectifying potassium channel Kir7.1. J Biol Chem 293: 17739–17753, 2018. doi: 10.1074/jbc.RA118.003238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Halbach P, Pillers DA, York N, Asuma MP, Chiu MA, Luo W, Tokarz S, Bird IM, Pattnaik BR. Oxytocin expression and function in the posterior retina: a novel signaling pathway. Invest Ophthalmol Vis Sci 56: 751–760, 2015. doi: 10.1167/iovs.14-15646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. York N, Halbach P, Chiu MA, Bird IM, Pillers DM, Pattnaik BR. Oxytocin (OXT)-stimulated inhibition of Kir7.1 activity is through PIP2-dependent Ca(2+) response of the oxytocin receptor in the retinal pigment epithelium in vitro. Cell Signal 37: 93–102, 2017. doi: 10.1016/j.cellsig.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Bjorkgren I, Mendoza S, Chung DH, Haoui M, Petersen NT, Lishko PV. The epithelial potassium channel Kir7.1 is stimulated by progesterone. J Gen Physiol 153: e202112924, 2021. doi: 10.1085/jgp.202112924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Kumar M, Pattnaik BR. Focus on Kir7.1: physiology and channelopathy. Channels (Austin) 8: 488–495, 2014. doi: 10.4161/19336950.2014.959809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Pattnaik BR, Tokarz S, Asuma MP, Schroeder T, Sharma A, Mitchell JC, Edwards AO, Pillers DA. Snowflake vitreoretinal degeneration (SVD) mutation R162W provides new insights into Kir7.1 ion channel structure and function. PLoS One 8: e71744, 2013. doi: 10.1371/journal.pone.0071744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Pattnaik BR, Shahi PK, Marino MJ, Liu X, York N, Brar S, Chiang J, Pillers DA, Traboulsi EI. A novel KCNJ13 nonsense mutation and loss of Kir7.1 channel function causes Leber congenital amaurosis (LCA16). Hum Mutat 36: 720–727, 2015. doi: 10.1002/humu.22807. [DOI] [PubMed] [Google Scholar]
- 133. Perez-Roustit S, Marquette V, Bocquet B, Kaplan J, Perrault I, Meunier I, Hamel CP. Leber congenital amaurosis with large retinal pigment clumps caused by compound heterozygous mutations in Kcnj13. Retin Cases Brief Rep 11: 221–226, 2017. doi: 10.1097/ICB.0000000000000326. [DOI] [PubMed] [Google Scholar]
- 134. Toms M, Dubis AM, Lim WS, Webster AR, Gorin MB, Moosajee M. Missense variants in the conserved transmembrane M2 protein domain of KCNJ13 associated with retinovascular changes in humans and zebrafish. Exp Eye Res 189: 107852, 2019. doi: 10.1016/j.exer.2019.107852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Sergouniotis PI, Davidson AE, Mackay DS, Li Z, Yang X, Plagnol V, Moore AT, Webster AR. Recessive mutations in KCNJ13, encoding an inwardly rectifying potassium channel subunit, cause leber congenital amaurosis. Am J Hum Genet 89: 183–190, 2011. doi: 10.1016/j.ajhg.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Khan AO, Bergmann C, Neuhaus C, Bolz HJ. A distinct vitreo-retinal dystrophy with early-onset cataract from recessive KCNJ13 mutations. Ophthalmic Genet 36: 79–84, 2015. doi: 10.3109/13816810.2014.985846. [DOI] [PubMed] [Google Scholar]
- 137. Hejtmancik JF, Jiao X, Li A, Sergeev YV, Ding X, Sharma AK, Chan CC, Medina I, Edwards AO. Mutations in KCNJ13 cause autosomal-dominant snowflake vitreoretinal degeneration. Am J Hum Genet 82: 174–180, 2008. doi: 10.1016/j.ajhg.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Jiao X, Ma Z, Lei J, Liu P, Cai X, Shahi PK, Chan CC, Fariss R, Pattnaik BR, Dong L, Hejtmancik JF. Retinal development and pathophysiology in Kcnj13 knockout mice. Front Cell Dev Biol 9: 810020, 2021. doi: 10.3389/fcell.2021.810020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Kabra M, Pattnaik BR. Sensing through non-sensing ocular ion channels. Int J Mol Sci 21: 6925, 2020. doi: 10.3390/ijms21186925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Beverley K, Shahi P, Kabra M, Zhao Q, Heyrman J, Steffen J, Pattnaik B. Kir7.1 disease mutant T153I within the inner pore affects K+ conduction. Am J Physiol Cell Physiol 323: C56–C68, 2022. doi: 10.1152/ajpcell.00093.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Tateno T, Nakamura N, Hirata Y, Hirose S. Role of C-terminus of Kir7.1 potassium channel in cell-surface expression. Cell Biol Int 30: 270–277, 2006. doi: 10.1016/j.cellbi.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 142. Vera E, Cornejo I, Burgos J, Niemeyer MI, Sepulveda FV, Cid LP. A novel Kir7.1 splice variant expressed in various mouse tissues shares organisational and functional properties with human Leber amaurosis-causing mutations of this K(+) channel. Biochem Biophys Res Commun 514: 574–579, 2019. doi: 10.1016/j.bbrc.2019.04.169. [DOI] [PubMed] [Google Scholar]
- 143. Cui M, Cantwell L, Zorn A, Logothetis DE. Kir channel molecular physiology, pharmacology, and therapeutic implications. Handb Exp Pharmacol 267: 277–356, 2021. doi: 10.1007/164_2021_501. [DOI] [PubMed] [Google Scholar]