

Keywords: dendritic, immunotherapy, matrix, proteoglycans, versican
Abstract
Cancer immunoediting progresses through elimination, equilibrium, and escape. Each of these phases is characterized by breaching, remodeling, and rebuilding tissue planes and structural barriers that engage extracellular matrix (ECM) components, in particular matrix proteoglycans. Some of the signals emanating from matrix proteoglycan remodeling are readily co-opted by the growing tumor to sustain an environment of tumor-promoting and immune-suppressive inflammation. Yet other matrix-derived cues can be viewed as part of a homeostatic response by the host, aiming to eliminate the tumor and restore tissue integrity. These latter signals may be harnessed for therapeutic purposes to tip the polarity of the tumor immune milieu toward anticancer immunity. In this review, we attempt to showcase the importance and complexity of matrix proteoglycan signaling in both cancer-restraining and cancer-promoting inflammation. We propose that the era of matrix diagnostics and therapeutics for cancer is fast approaching the clinic.
INTRODUCTION
Extracellular matrix (ECM) remodeling is a crucial regulator of tumor progression (1, 2). The ECM is a diverse, noncellular, three-dimensional network composed of proteoglycans (PGs) and glycosaminoglycans (GAGs) as well as fibrous proteins, including collagens, fibronectins, elastins, and laminins (3–5). Its composition is dynamic and changes substantially from organ to organ. The ECM is an integral structural component of the tumor microenvironment (TME), the latter also incorporating an intricate milieu of endothelial, mesenchymal, and immune cells that collectively host and support the tumor. The importance of ECM remodeling to local invasion, angiogenesis, and metastasis is well established (6, 7). However, the contribution of matrix composition in regulating the cancer-immunity cycle and shaping the tumor immune microenvironment is only beginning to be systematically explored (8).
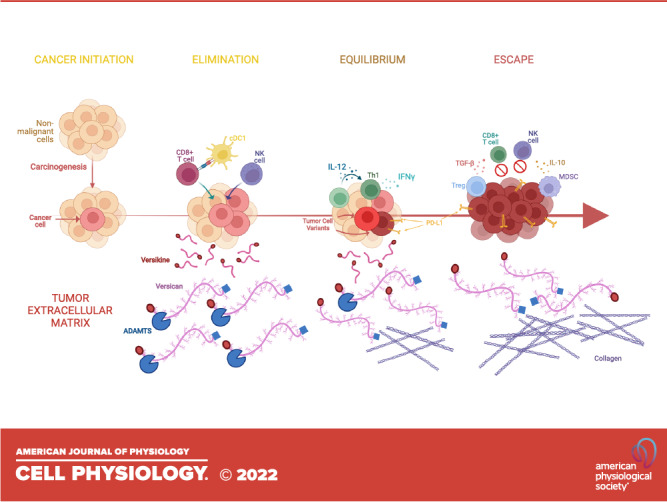
The dynamic interfacing between the developing tumor and the host immune response has been termed “immunoediting” and consists of three distinct stages: elimination, equilibrium, and escape (9) (Fig. 1). During the first stage, elimination, the host immune system is successfully able to detect and destroy cancer cells before the tumor reaches a detectable size. During the equilibrium stage, however, surviving tumor cells undergo genetic and/or epigenetic changes (“edits”) that compromise the host immune system’s ability to eliminate the tumor cells, leading to a “dormant” tumor with stagnant growth and a dynamic, albeit precarious, equilibrium between the tumor and host immune response. The final stage, escape, is characterized by tumor outgrowth, made possible by diminished or lost tumor antigen repertoire (which may also happen during the equilibrium stage), secretion of cytokines that promote cell growth, division, angiogenesis, and immune suppression as well as the recruitment of regulatory, protumor, immune cells (17). Each immunoediting stage is accompanied by extensive ECM breaching, deposition, and reshaping, resulting in the release or alteration of immunomodulatory ECM components, such as small leucine-rich proteoglycans (SLRPGs), large matrix proteoglycans (e.g., versican), hyaluronan (HA), and heparan sulfate fragments. True to their derivation from stroma and tissue plane restructuring, these mediators act as “danger signals” [termed damage-associated molecular patterns (DAMPs) or “alarmins”], to activate immune system sensor mechanisms (17–19). In this review, we shall specifically focus on the role of matrix proteoglycans and their products in shaping tumor inflammation and immunity.
Figure 1.
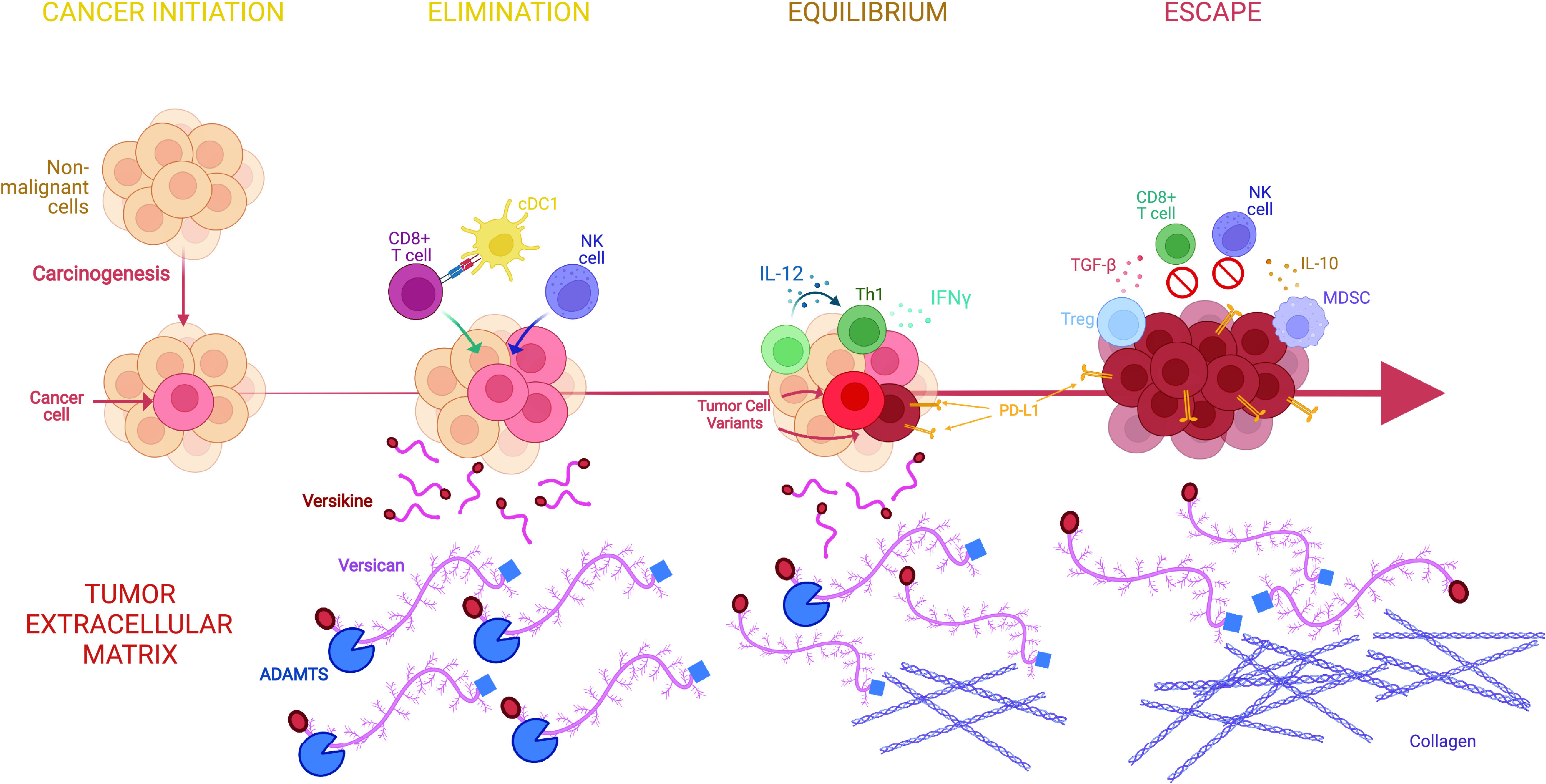
Matrix proteoglycans in cancer evolution and immunoediting. Cancers are “wounds that do not heal.” Immunoediting refers to the distinct stages of interfacing between the host immune system and the developing cancer. There are three stages: elimination, equilibrium, and escape (see text for details). Extracellular matrix remodeling regulates key signaling pathways at each step. The case is illustrated by versican (VCAN), a versatile large matrix proteoglycan that possesses diverse context-specific immunoregulatory or immunostimulatory roles, depending on its abundance, isoform structure, and proteolysis status (10). We propose the following model based on work by our group and others (11–13): during elimination, robust VCAN proteolysis releases the immunostimulatory fragment (matrikine), versikine, that regulates stimulatory dendritic cells (conventional type 1 dendritic cells, cDC1) promoting immune destruction of the tumor through enhanced neoantigen recognition. In equilibrium, a precarious balance is established between immunostimulatory proteolyzed VCAN products and parental, nonproteolyzed immunoregulatory VCAN that allows the propagation of tumors with active, but progressively ineffectual, immune surveillance. In escape, progressive fibrosis and immunosuppression [e.g., through TGF-β (14)] attenuates VCAN proteolysis and results in the unbalanced immunoregulatory activity of nonproteolyzed VCAN. In these latter stages, immunosuppressive signals from receptors recognizing collagen may further promote immune exclusion (15, 16). Created with BioRender.com.
PROTEOGLYCANS IN TUMOR IMMUNITY: AN OVERVIEW
PGs comprise a heterogeneous group of complex macromolecules that form an important structural and functional component of the ECM (20). Structurally, PGs consist of a core protein onto which one or more glycosaminoglycan (GAG) chains are covalently attached. Known PGs can be classified into four families based on criteria that include cellular and subcellular localization, overall gene/protein homology, and the utilization of specific protein modules: intracellular, cell surface, pericellular, and extracellular (21) (Table 1). GAGs, also known as mucopolysaccharides, are negatively charged polysaccharides composed of repeating disaccharide units. GAGs, based on the type of monosaccharides and presence or absence of modification by sulfation, are categorized into the following: chondroitin sulfate (CS)/dermatan sulfate (DS), keratan sulfate (KS), heparan sulfate (HS), and hyaluronan (HA). The sulfate-enriched negatively charged GAG chains in PGs bind water and positively charged soluble ligands such as cytokines, chemokines, growth factors, matrix metalloproteinases (MMPs), and cell surface receptors, facilitating the function of PGs as important mediators of cell proliferation, differentiation, adhesion, and dissemination within the ECM, as well as inflammation and angiogenesis (154). The bioactivity of PGs is further modulated by enzymes present within the tumor microenvironment, such as sheddases, proteases, and heparanases, that cleave these biomolecules and modify the structure and function of associated PG and their bound ligands.
Table 1.
Key proteoglycans organized per class, with cellular source(s), and selected functional roles indicated
| Proteoglycan | Expression | Source(s) | Functional Role(s) | References |
|---|---|---|---|---|
| Serglycin | Intracellular | Immune cells, endothelial cells, fibroblasts, chondrocytes, smooth muscle cells, fetal liver, nasopharyngeal, and hematopoietic cancer cells | Complexes with and modulates the activities of mast cell proteases, e.g., chymases, tryptases, and carboxypeptidase A; acts as a vehicle for delivery of granzyme B in cytotoxic cells; triggers CD44 signaling in tumor cells; regulates IL-8 secretion in cancers; via tumor-derived exosomes, it protects human multiple myeloma from complement attack by binding C1q and MBL in the TME | 21, 22–24, 25–42 |
| Syndecan-1 | Cell surface | Normal and malignant plasma cells; normal and malignant epithelial cells | Sequesters T-cell specific CC-chemokines, and in soluble form, with VEGFR2 and VLA-4; On the cell surface, engages CXCR4, VLA-4, and VEGFR2; upregulates IL-23 and Notch ligand DLL4, which polarizes CD4+ T cells toward the Treg, Th17, and Th1 phenotypes | 21, 43–56, 57–64 |
| Syndecan-2 | Cell surface | Mesenchymal cells, tumor-associated stromal cells | Modulates TFG-β signaling to control tumorigenesis via TFG-β-induced genes PD-L1 and CXCR4 in TASC | 21, 43–56, 65 |
| Syndecan-3 | Cell surface | Neuronal tissue; tumor-associated macrophages; tumor endothelial cells; bladder, prostate, breast, and pancreatic cancer cell lines | Accentuates the IFN-γ-regulome, including upregulation of CD274 and CD8; regulates hypoxia-resistance in a HIF1α-dependent manner | 21, 43–56, 66,67 |
| Syndecan-4 | Cell surface | Ubiquitous expression | Ligand for dendritic cell-associated heparan sulfate proteoglycan-integrin ligand (DC-HIL) to attenuate T-cell activation; sequesters TFG-β on the cell surface | 21, 43–56, 68–72 |
| Chondroitin sulfate proteoglycan 4 | Cell surface | Monocytes/macrophages | Activates monocytes to secrete IL-1β and induce monocyte-dependent B cell proliferation in vitro | 21, 73–75 |
| Betaglycan | Cell surface | Ubiquitous expression | Accentuates TGF-β signaling or interferes with TGF-β receptor type I and TGF- β receptor type II complexing; in soluble form, can sequester TGF-β; upregulates Cdc42 via β-arrestin-2 in vivo | 76–81 |
| Glypican-1 | Cell surface | Ubiquitous expression | FGF signaling; PTEN attenuation; Akt and β-catenin signaling; recognition by NK cells via NKp46 and NKp30; interacts with VEGF via HS chains | 21, 82, 83, 84 |
| Glypican-2 | Cell surface | Embryonic central nervous system tissue; neuroblastoma cells | Upregulates expression of N-Myc, which in turn promotes transcription of the glypican-2 in a positive feedback loop | 21, 82, 83 |
| Glypican-3 | Cell surface | Ovary, breast, lung, and kidney epithelial cells | Sequesters VEGF in TME; inhibits Zeb1 transcription factor; influences PI3K/Akt, p38, ERK1/2, and Wnt signaling pathways (context dependent); induces cytotoxic T-cell and macrophage recruitment in HCC | 21, 82, 83, 85–87 |
| Glypican-4 | Cell surface | Ubiquitous expression | Accentuates Wnt signaling and pluripotency; reduces sensitivity of pancreatic cancer cells to 5-fluorouracil | 21, 82, 83 |
| Glypican-5 | Cell surface | Central nervous system | Acts through FGF, HGF, and Wnt1α to promote cell division; tumor suppressor in certain contexts, e.g., glioma | 21, 82, 83 |
| Glypican-6 | Cell surface | Gallbladder, urinary bladder, liver, appendix, and kidney epithelial cells | CD8A mRNA transcription; Wnt signaling modulation; tumor infiltration of cytotoxic T-cells | 21, 82, 83, 88 |
| Perlecan | Pericellular | Stromal cells | Sequesters bioactive molecules (via GAG chains) | 21, 89–93 |
| Agrin | Pericellular | Stromal cells | Activates VEGF-VEGFR2 pathway; stimulates angiogenesis | 21, 94 |
| Biglycan | Extracellular | Stromal cells; macrophages; immune cells | Binds TLR2/4; forms complexes with CD14-TLR4 and P2X4/P2X7 respectively; promotes tumor progression | 95, 96–104, 105 |
| Decorin | Extracellular | Stromal cells | Binds TLR2/4; sequesters TGF-β; upregulates PDCD4; inhibits tumor growth | 95, 105–107 |
| Lumican | Extracellular | Stromal cells | Enhances LPS-dependent TLR4 signaling; tumor-dependent impact on EMT, immune infiltration and inflammation | 21, 97, 108–110 |
| Versican | Extracellular | Stromal cells; myeloid cells | Binds hyaluronan, P and L selectins, CD44, and TLR2; promotes accumulation of tumor-associated macrophages and decrease intraepithelial tumor infiltrating lymphocytes; positively correlated with Th2 and Treg transcriptional signatures, negatively correlated with cytotoxic T-cell signatures | 22, 10–13, 111–148 |
| Aggrecan | Extracellular | Cartilage and perineural net stromal cells | Forms aggregates for load-bearing structural support and mechanical properties to joints; protects neurons via cation sequestration and mechanically stabilization of synaptic connections | 21 |
| Brevican | Extracellular | Neural tissue stromal cells | Interacts with tenascin-R and fibulin-2; neural tissue injury and repair, Alzheimer’s disease, glioma tumorigenesis | 21 |
| Neurocan | Extracellular | Neural tissue stromal cells | Alleviates mechanical and ischemic damage in central nervous system; inhibits neurite outgrowth in vivo; assists in neural guidance during development | 21 |
| Hyaluronan and proteoglycan link protein 1 | Extracellular | Stromal cells | Binds to aggrecan complexes in cartilage to offer structural support; NF-κB pathway activation; myeloma drug resistance | 149–153 |
Tumor evolution results in significant alterations in PG expression and composition (21, 95, 155). Altered expression of GAG-synthesizing enzymes during malignant transformation affects the type and fine structure of GAG chains attached to PGs. Whereas these modifications have been classically thought to facilitate local invasion, angiogenesis, and metastasis, there is increasing evidence that PG remodeling—such as proteolysis of the protein core and alterations to the GAG sidechains—has further implications in shaping tumor-immune cross talk (22–24). As will be discussed in greater detail, these implications include the diversification of cell-surface PG functionalities through regulated shedding, the synthesis and degradation of cytokine- and chemokine-sequestering GAG chains, and the proteolytic release of immunomodulatory proteolytic fragments (matrikines). Our understanding of role that matrix PG remodeling plays in shaping tumor-immune cross talk will continue to increase as this growing field matures.
In recent years, the application of high throughput multiomics technologies has allowed the comprehensive cataloging and characterization of PG and other matrix components in diverse cancer types (5, 156, 157). For example, Naba et al. (156) developed a bioinformatic approach to predict the in silico “matrisome” consisting of ECM proteins and associated factors. They reported a detailed composition of human melanoma extracellular matrices (156). Both tumor and stromal cells were found to contribute, although differentially, to the secretion of the proteins making up the tumor ECM (156). Similar strategies could reveal novel molecular players, whose functional contributions can then be further studied, in addition to holding significant promise for biomarker identification in various cancer settings.
INTRACELLULAR PG—SERGLYCIN
Serglycin (Fig. 2) forms a class on its own, as it is the unique intracellular PG (21). Initially, it was identified as the dominant PG expressed in normal immune cells such as monocytes, macrophages as well as their malignant counterparts in hematopoietic cancers. However, serglycin is also expressed in nonhematopoietic normal and malignant cell types, such as endothelial cells, fibroblasts, chondrocytes, smooth muscle cells, fetal liver, murine embryonic stem cells, and nasopharyngeal cancer cells (22–25). The type and sulfation pattern of GAG chains attached to the serglycin core protein vary between cell types and different species (26). Serglycin-knockout mice have helped unravel its functional roles in both normal and pathological immunity (26, 27). Serglycin is localized in secretory granules and vesicles for proper intracellular storage and secretion of bioactive molecules. For example, serglycin forms complexes with, and modulates, the activities of mast cell proteases such as chymases, tryptases, and carboxypeptidase A among others (28, 29). Serglycin-deficient mast cells showed reduced sensitivity to apoptosis as a result of diminished storage and release of mast cell proteases into the cytosol and impaired caspase-3 activation (30). In cytotoxic T lymphocytes (CTLs), serglycin binds to granzyme B and acts as a vehicle for its delivery into the target cells during cytotoxic cell granule-mediated cell death (31). Lack of serglycin expression may affect CTL and NK cells’ ability to kill target cells (32, 33).
Figure 2.

Immunomodulatory roles of intracellular PG. Serglycin (SRGN) can modulate the activation status of the complement system. SRGN-CD44 interaction activates intracellular signaling pathways, such as NF-κB, promoting tumor aggressiveness. SRGN regulates IL-8 and TGF-β secretion, thus controlling crucial inflammatory networks in the TME. PG, proteoglycan. Created with BioRender.com.
Serglycin expression has been demonstrated in multiple cancer types including breast cancer, lung cancer, myeloma, lymphoma, mastocytoma, and thymoma. Secreted serglycin triggers CD44 signaling on tumor cells (34, 35). CD44 regulates various cellular processes such as migration, proliferation, and apoptosis (36). CD44 is also a primary receptor for HA, which will be discussed in further detail later. Guo et al. (35) showed that serglycin secreted by nonsmall cell lung cancers (NSCLCs) and/or cancer-associated fibroblasts (CAFs) in the TME can bind CD44 on tumor cells to promote malignant phenotypes by inducing stemness through the transcription factor Nanog, accompanied with chemoresistance and anoikis resistance. Serglycin, via the CD44/NF-κB/claudin-1 (CLDN1) axis, was shown to upregulate vimentin expression and hence promote epithelial-to-mesenchymal transition (EMT). Chu et al. (37) reported that in nasopharyngeal carcinoma (NPC), serglycin-CD44 interaction activates the MAPK/β-catenin axis to induce CD44 receptor expression in a positive feedback loop, resulting in NPC tumor cell stemness and chemoresistance. Serglycin is highly expressed in triple-negative breast cancer (TNBC) and it induces the expression and secretion of TGF-β2, which subsequently promotes EMT (38). Binding of serglycin to CD44 was shown to induce CREB phosphorylation, which in turn activated TGF-β2 transcription. Conversely, TGF-β2/TGFR1 signaling was shown to positively regulate serglycin expression by activating downstream SMAD2/3 signaling. Bouris et al. (39) showed that serglycin regulates IL-8 secretion in breast carcinoma cells and induces IL-8/CXCR2 signaling axis to activate PI3K, Src, and Rac GTPase activation. Furthermore, serglycin overexpression was shown to promote an aggressive phenotype, EMT, and drug resistance in estrogen receptor-α (ER-α) positive MCF-7 breast cancer cells. Skliris et al. (40) showed that serglycin secreted by human multiple myeloma cells may protect cancer cells from complement system attack by specifically inhibiting the classical and the lectin pathways through direct binding to C1q and MBL, without hampering alternative pathway activity. The data demonstrate a role for serglycin as a modulator of tumor immunity in the TME, where it can defend cancer cells from immunotherapy-induced complement attack. Recently, a novel function of serglycin was reported as a critical component of tumor-derived exosomes. Tumor-derived exosomes act as potent regulators of cell-to-cell interactions in the tumor microenvironment due to their ability to carry nucleic acids, proteins, and lipids and to transfer their cargo into the target cells (27, 41). They promote tumor growth through alteration of phenotypic and functional attributes of the recipient cells in the TME, resulting in drug resistance, inhibition of host antitumor responses, and attenuation of immunotherapy efficacy (41). Serglycin was detected in exosomes derived from the cell culture supernatants of human myeloma cell lines and from the serum of patients with myeloma (42). Exosomes derived from serglycin-knockdown cells, but not from control cells, lacked several proteins that are required for mediating crucial biological processes, such as translational initiation, cell adhesion, response to drug, and intracellular protein transport. Moreover, exosomes from serglycin-knockdown cells failed to induce an invasive phenotype in myeloma cells or promote migration of macrophages (42). Altogether these findings suggest that serglycin plays a key role in the encapsulation of proteins in exosomes as well as their transfer into target cells residing in the TME.
CELL SURFACE PGS—SYNDECANS, CSPG-4/NG2, BETAGLYCAN, AND GLYPICANS
Syndecans (Fig. 3) are a family of single-pass transmembrane PGs consisting of four members: syndecan-1 (Sdc-1, CD138), syndecan-2 (Sdc-2, fibroglycan), syndecan-3 (Sdc-3, N-syndecan), and syndecan-4 (Sdc-4, amphiglycan) (21). They are expressed in a developmentally and cell type-specific manner. Sdc-1 is expressed mainly by plasma cells and epithelial cells, whereas Sdc-2 and Sdc-3 are expressed by mesenchymal cells and neurons, respectively. Sdc-4 shows a ubiquitous expression pattern in vertebrates. Structurally, they consist of an ectodomain, a transmembrane domain, and an intracellular domain. The predominant GAG covalently attached to the ectodomain protein core is HS and occasionally CS, which makes syndecans “hybrid PGs.” Syndecans are involved in a wide variety of biological functions, including cell-cell and cell-ECM interactions, cell survival, adhesion, migration, and cell signaling through binding to numerous soluble molecules via their HS chains (43, 44). Many syndecans, if not all, can also function as soluble heparan sulfate PGs (HSPGs) via proteolysis-mediated release of their ectodomain. This shedding is an important posttranslational modification that regulates the amount of HSPG present on the cell surface versus the pericellular microenvironment. The role of syndecans in tumor initiation and progression has been extensively studied (45–47). Both cell-surface and soluble syndecans regulate multiple aspects of tumor cell adhesion and cell migration (44, 47). Cell-surface syndecans in co-operation with integrins control cell adhesion to extracellular fibronectin (48) and promote cell spreading through actin and fascin bundling (49). Cell-surface syndecans may enhance tumor endothelial cell proliferation and motility by binding growth factors (such as FGF-2 and VEGF), presenting them to their high-affinity receptors, and protecting them from inactivation (50, 51). Syndecans can also bind to and induce conformational changes to growth factor receptors, activating downstream signaling pathways that control cell proliferation and motility (46, 52). The ectodomain of soluble syndecans may also bind to certain chemoattractants, such as IL-8, effectively creating chemotactic gradients (53). Finally, ectodomains of soluble syndecans may increase endothelial cell membrane protrusion, migration, capillary tube formation, and cell-cell interactions by competitively inhibiting cell-surface syndecans (54).
Figure 3.

Immunomodulatory roles of transmembrane PG. Syndecan-1 (Sdc-1) regulates NF-κB and STAT-dependent inflammatory networks in tumor cells as well as TME immune cell polarization/activity through Notch. CSC, cancer stem cell; IBC, inflammatory breast cancer; PG, proteoglycan. Created with BioRender.com.
All four syndecans have been reported to be expressed in a variety of cancer types (46). They have been detected in varied components of the TME such as inflammatory cells, fibroblasts, blood vessels, and the ECM. Syndecan accumulation in the tumor stroma could be a consequence of either induction or shedding (55, 56). Many epithelial tumors exhibit stromal expression of syndecans; for example, Sdc-1 has been detected in the stromal compartment of breast cancer, colorectal cancer, endometrium cancer, and gastric cancer (57). In nonepithelial cancers such as multiple myeloma, the stroma stains intensely for Sdc-1, likely derived from tumor shedding (58). Notably, in myeloma, malignant plasma cells express high levels of Sdc-1 (CD138), which is often used as a lineage marker for this cancer.
Syndecans display diverse roles in cancer, acting as either tumor promoters or suppressors, depending on the type and stage of the tumor. Using a murine model of colitis-related colon carcinoma, Gallimidi et al. (59) demonstrated that Sdc-1-deficient mice showed higher susceptibility to malignant transformation due to increased local production of IL-6, activation of STAT3 and downstream effector genes with key roles in colonic tumorigenesis. In contrast, Sdc-1 silencing in inflammatory breast cancer (IBC) SUM-149 cells caused reduced colony formation and 3-D spheroid formation. Sdc-1 knockdown was shown to downregulate NF-κB and STAT3 signaling and reduce IL-6, IL-8, CCL20, gp130, and EGFR mRNA in both SUM-149 and non-IBC breast cancer SKBR3 cells (60, 61).
Sdc-1 shapes the cellular composition of the inflammatory environment by modulating soluble mediators. For example, Sdc-1 can sequester T-cell-specific CC-chemokines through its HS-chains and thereby inhibit T-cell-driven inflammation (62). Saleh et al. (63) focused on the immunomodulatory role of Sdc-1 in the polarization of CD4+ T cells within breast cancer TME. Conditioned media from Sdc-1-knockout SUM-149 breast cancer cells significantly enhanced ex vivo polarization of CD4+ T cells toward Treg (Foxp3 + CD4+), Th17 (IL-17 + CD4+), and Th1 (IFN-γ + CD4+) via upregulation of IL-23 and Notch ligand DLL4. The Rapraeger group (64) reported a novel mechanism by which Sdc-1 engages CXCR4, VLA-4 (very late antigen-4), and VEGFR2 (vascular endothelial growth factor receptor-2) to modulate cell migration across a variety of cell types. They showed that VEGFR2 and VLA-4 docks in the extracellular domain of sSdc-1 (shed Sdc-1) and VEGFR2 causes PKA-mediated phosphorylation of VLA-4 on S988, which promotes metastasis and immunosuppression.
Loftus et al. (65) showed that Sdc-2 expressed on the cell surface of tumor-associated stromal cells (TASCs), controls tumorigenesis via TGF-β signaling. Decreased Sdc-2 activity in TASCs inhibited TGF-β-induced immunosuppressive genes such as PD-L1 and CXCR4. This effect correlated with reduced ability of TASCs to suppress T-cell proliferation in vitro. Consistently, they observed T-cell activation and tumor inhibition in an immune-competent syngeneic breast cancer model.
Although Sdc-3 is known mainly to be expressed in neuronal tissues, some studies demonstrate it to be expressed in the TME as well as in cancer cell lines such as bladder cancer, prostate cancer, breast cancer, and pancreatic cancer (66). In the TME, Sdc-3 is expressed on tumor cells, TAMs, and endothelial cells. Sdc-3 expression was shown to be induced under hypoxic conditions in CT26 colon carcinoma cells in a HIF-1α-dependent manner (66). In addition, Sdc-3 expression was strongly upregulated in murine macrophage cell line RAW-264.7 treated with proinflammatory cytokine IFN-γ but not anti-inflammatory IL-4. This finding is in line with an earlier study by Takeda et al., which showed that IFN-γ stabilizes HIF1α in macrophages (67). Furthermore, Sdc-3 expression was shown to positively correlate with an IFN-γ-regulome, including CD274 and CD8, across the majority of TCGA tumor types and a better overall survival in melanoma tumors expressing a hypoxia transcriptional signature (66).
Sdc-4 is the major HSPG expressed on the dendritic cell (DC) surface. In the TME, DCs play an important role in T-cell priming and tumor rejection; however, tumor-associated DCs are often polarized into a tolerogenic state that allows cancer cells to escape immune surveillance. In Sdc-4-deficient mice, Lewis lung carcinoma (LLC) tumors were growth-impaired and were characterized by increased infiltration of mature DCs, NK cells, and NKT cells (68). Sdc-4 is a ligand for dendritic cell-associated heparan sulfate proteoglycan-integrin ligand (DC-HIL). DC-HIL is a highly glycosylated type I transmembrane protein of 125 and 95 kDa containing an extracellular Ig-like domain, constitutively expressed by APCs and acts as a negative regulator of T-cell activation (69). DC-HIL has been shown to bind HS chains on Sdc-4 expressed on activated T cells, and this binding attenuates T-cell activation (69–71). Chung et al. (72) showed that malignant T cells from patients with Sézary syndrome (SS) and cutaneous T-cell lymphoma (CTCL) cell lines constitutively expressed Sdc-4 featuring distinct HS moieties at high levels compared with those expressed by normal T cells activated in vitro. Using these HS moieties, Sdc-4 was shown to inhibit activation of T cells through two independent mechanisms: first, by binding to DC-HIL and second, by sequestering immunosuppressive cytokine TGF-β on the cell surface. These findings highlight the importance of HSPGs, like Sdc-4, in regulating tumor immunity.
Chondroitin sulfate proteoglycan 4 (CSPG-4) is a highly glycosylated, single-pass transmembrane PG that carries one CS chain and a large ectodomain (21). It is also referred to as melanoma-associated chondroitin sulfate proteoglycan (MCSP), high-molecular-weight melanoma-associated antigen (HMW-MAA), or neuron-glial antigen 2 (NG2). It was first reported in malignant melanoma as a highly immunogenic tumor antigen and subsequently implicated in the pathobiology of multiple solid tumors as well as hematological cancers. Due to its restricted or low expression in normal tissues, overexpression in certain tumor types, as well as functionally important role in supporting tumor growth and metastasis, it has been investigated as a potential immunotherapy target (73). Erfurt et al. (74) reported the presence of natural CSPG-4 specific CD4+ T cells that may confer immunosurveillance against melanoma. In some patients, attenuation of this response is associated with tumor progression. Activated human monocytes/macrophages also produce CS-containing PGs (75) that may subsequently act in a paracrine manner. CSPG-4 activates monocytes to secrete IL-1β and induce monocyte-dependent B cell proliferation in vitro, which can be inhibited by TGF-β and CD44 monoclonal antibodies (75).
Free GAGs can also have diverse immunomodulatory actions. Aoyama et al. (158) showed that chondroitin sulfate B (an older term for dermatan sulfate) could stimulate the proliferation of murine B cells in vitro via a mechanism involving translocation of protein kinase C (PKC) β from cytosol to membrane and increased AKT phosphorylation, but not extracellular signal-regulated kinase (ERK) activation. Yang et al. (159) showed that hyaluronic acid (HA) or chondroitin sulfate A (CSA) could promote the differentiation of immature human monocyte-derived dendritic cells in vitro, suggesting that HA or CSA have potential to modulate immune responses. A significant body of literature has established a clear role for distinct molecular weight HA polymers in regulating immune responses, acting through receptors CD44 and RHAMM (reviewed in Ref. 160).
Betaglycan, also known as TGF-β type III receptor (TGFBR3), is a single-pass transmembrane PG belonging to the TGF-β coreceptor superfamily. TGF-β is an immunosuppressive cytokine that represses dendritic cell antigen-presentation and inhibits T-cell function (76). Altered TGFBR3 expression has been reported in multiple tumor types. Reduced TGFBR3 has been shown in advanced neuroblastoma, ovarian carcinoma, endometrial carcinoma, nonsmall cell lung cancer, prostate cancer, breast cancer and pancreatic cancer (77). However, in high-grade non-Hodgkin’s lymphoma and B-CLL, betaglycan was found to be upregulated, suggesting a paradoxical tumor-promoting role. Eickelberg et al. (78) showed that betaglycan can prevent the complexing of TGF-β type I and type II receptors, whose sequential activation is necessary for TGF-β signaling, in the renal epithelial cell line LLC-PK1. They determined that the GAG chains of exogenously expressed betaglycan in LLC-PK1 cells had a higher molecular weight, relative to those of the L6 cell line, whose betaglycan promotes TGF-β signaling (78). Shedding of TGFBR3 from the tumor cell surface generates soluble sTGFBR3, which sequesters TGF-β, thus antagonizing downstream TGF-β signaling. Therefore, betaglycan may exert its function as a dual modulator of TGF-β signaling: in its membrane-bound form, it enhances TGF-β signaling, whereas in its soluble form, it acts as a decoy inhibitor (79). Hanks et al. (80) showed that loss of tumor-expressed TGFBR3/sTGFBR3 activated TGF-β-dependent expression and activity of immunomodulatory enzyme indoleamine 2, 3-dioxygenase (IDO) in plasmacytoid DCs (pDCs) and CCL22 chemokine expression in myeloid DCs (mDCs). This alteration within the locoregional DC population induced infiltration of immunosuppressive FOXP3+ Treg cells, correlated with a reduction in CD8+ T cells in the TME and suppressed tumor-associated antigen-specific T-cell responses, such as T-cell proliferation and IFNγ secretion. Finally, betaglycan may also have important roles in cell migration, irrespective of TGF-β signaling. Mythreye et al. (81) found that, in the absence of TGF-β, betaglycan was able to act through the scaffold protein β-arrestin-2 to upregulate Cdc42 and impair cell migration in the ovarian and breast cancer cell lines Ovca429 and MDA-MB231, as well as in the healthy ovarian epithelial cell line NOSE007. This effect was not related to sTGFBR3, and both the intracellular domain and GAG chains bound to extracellular moieties of betaglycan were important for this activity to occur (81).
Glypicans are HSPGs bound to the plasma membrane via a glycosylphosphatidylinositol (GPI) anchor (21). Glypicans are evolutionarily conserved and the human genome includes six distinct glypican genes (GPC-1 to GPC-6) that encode the six glypican proteins. Based on their amino acid sequence, GPC proteins can be further categorized into two families consisting of GPC-1, GPC-4 and GPC-6, and GPC-3 and GPC-5, respectively. Glypicans can regulate and interact with a wide gamut of soluble and insoluble factors such as cytokines, chemokines, growth factors, morphogens, enzymes, and receptors (82, 83). They are versatile PGs that play a dual role in either activating or inhibiting cellular signals, which may be attributed to their property to interact through their HS-GAG chains as well as their protein cores. They may act as either tumor suppressors or tumor promoters depending on the type and stage of tumor (83).
Glypican-3 is one of the most studied glypicans in human cancer and it has been proposed as a promising immunotherapy target. Glypican-3 antigenic peptide (amino acids 114–152) generates specific peptide-reactive cytotoxic T cells causing tumor regression without inducing autoimmunity (85). Wang et al. (86) showed that human dendritic cells genetically engineered to express GPC-3 were able to induce a cytotoxic T-cell-mediated antitumor response in vivo against GPC-3-expressing hepatocellular carcinoma cells (HCC). Furthermore, GPC-3 expression induces the recruitment of macrophages into human HCC tumors (87). Expression of GPC-6 positively correlated with mRNA levels of CD8A, a marker of T-cell infiltration, and improved overall survival in early stage ovarian cancer (88). Suppression of GPC-1 expression in pancreatic cancer cells rendered them resistant to recognition and lysis by NK cells through cytotoxicity receptors NKp46 and NKp30 (84).
PERICELLULAR AND BASEMENT MEMBRANE ZONE PGS—PERLECAN AND AGRIN
Perlecan or heparan sulfate proteoglycan 2 (HSPG2) is a complex modular HSPG consisting of ∼500 KDa protein core divided into five domains with homology to SEA (sea urchin sperm protein), N-CAM (neural cell adhesion molecule), IgG, LDL receptor, and laminin (21). It is a fully secreted PG that resides in the pericellular region and is a major component of the basement membrane. The majority of perlecan is produced by the cells of the reactive stroma in the TME, although it can also be produced by cancer cells (89, 90). It has been shown to be present in the desmoplastic stromal region surrounding lung, renal, prostate, and breast cancer lesions (91). Cytokines such as transforming growth factor-β (TGF-β) and tumor necrosis factor-α (TNF-α) regulate transcription levels of perlecan in normal stromal cells, tumor cells, and a subset of bone marrow stromal cells (91). In breast cancer, TGF-β was shown to positively regulate perlecan synthesis, whereas IFN-γ had a negative effect (92). Perlecan not only acts as a physical barrier but its HS-containing GAG chains sequester numerous bioactive molecules such as chemokines, cytokines, and enzymes. For example, perlecan forms a complex with FGF2 (fibroblast growth factor-2) and sequesters it in the basement membrane and stroma (93). Matrix metalloproteinases (MMPs), sulfatases, and heparanases present in the TME have been identified as the enzymes that modulate the molecular state of perlecan. For example, MMPs act on the perlecan core to release bioactive fragments, which influence cell adhesion, invasion, and angiogenesis. Sulfatases and heparanases act on the HS chains, thus releasing bound growth factors and cytokines to act on their target cells. Perlecan modifier enzymes promote degradation of the perlecan-rich stroma that may transform the TME from a “hostile” state to a “permissive” one that allows tumor growth, angiogenesis, and metastasis (reviewed in Ref. 89).
Agrin is another pericellular or basement membrane HSPG (21). It has a multimodal structure that is homologous to that of perlecan, with several splice variants. Agrin has been shown to stimulate angiogenesis in the TME by activating the VEGF-VEGFR2 pathway (94).
SMALL LEUCINE-RICH PGS—BIGLYCAN, DECORIN, AND LUMICAN
Small leucine-rich PGs (Fig. 4), encompassing 18 distinct gene products and multiple splice variants, form one of the largest families of extracellular PGs (21). They are characterized by a small protein core (36–42 kDa) and a central region composed of leucine-rich repeats (LRRs) (21, 95, 161). SLRPGs are grouped into five classes (I–V) based on several criteria such as evolutionary conservation, protein and gene homology, and chromosomal organization (21). SLRPGs bind to collagens, receptor tyrosine kinases (RTKs), TGF-β, TNF-α, and bone morphogenetic protein (BMP), thus modulating innate immune receptors and signaling pathways (161, 162). In this section, we will discuss well-studied major SLRPGs involved in regulating immune responses in the TME, such as biglycan, decorin, and lumican.
Figure 4.
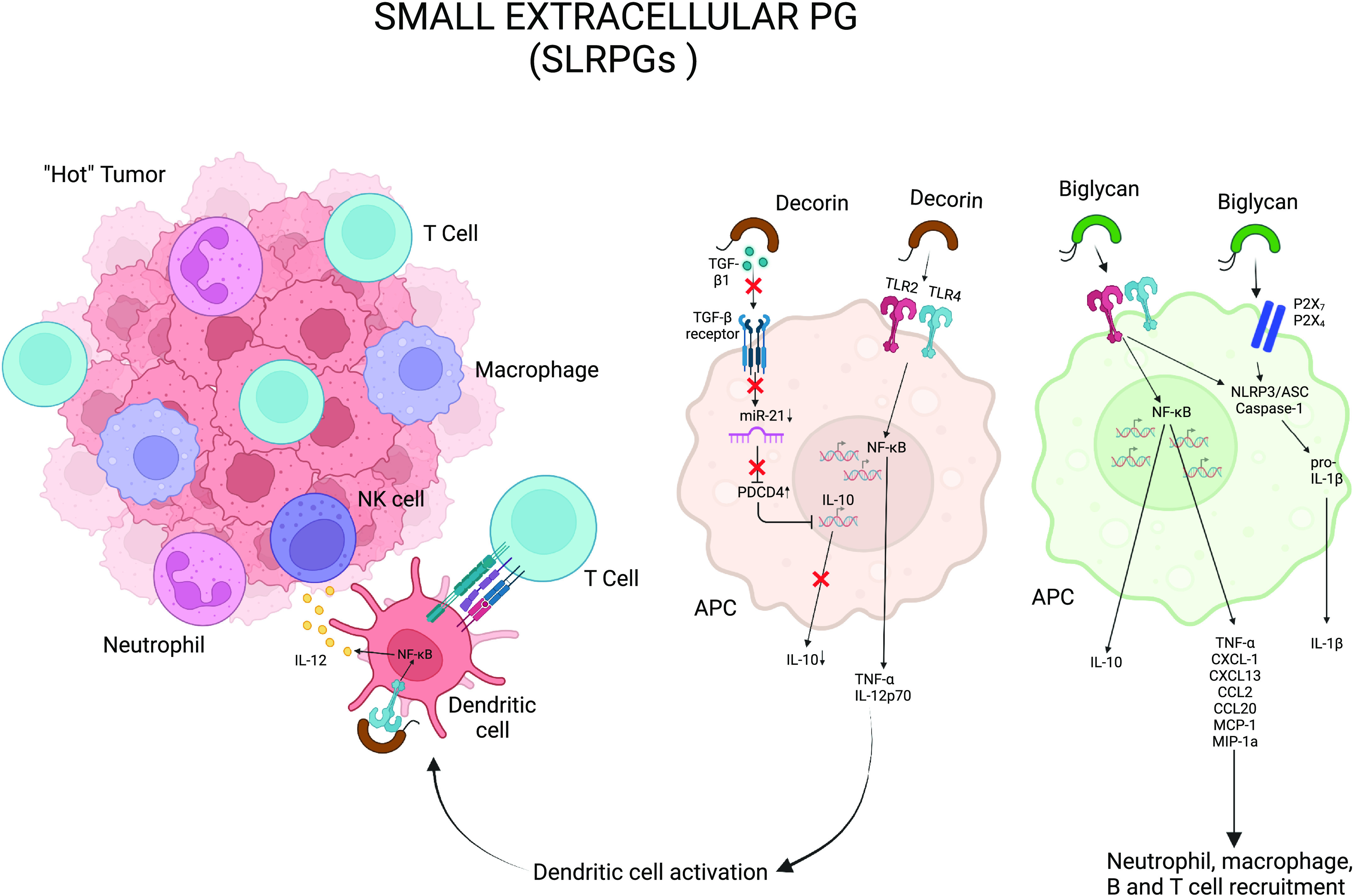
Immunomodulatory roles of small leucine-rich PG (SLRPGs). Soluble biglycan and decorin bind TLR-2 and -4, resulting in production of various cytokines and chemokines, such as proinflammatory TNF-α and IL-1β, as well as anti-inflammatory IL-10. Decorin sequesters TGF-β and reduces the availability of miR-21, which results in decrease in IL-10 secretion. Biglycan can cluster TLR2/4 and purinergic receptors, P2X4/P2X7. APC, antigen-presenting cell; PG, proteoglycan. Created with BioRender.com.
Biglycan, a class I SLRPG bearing chondroitin sulfate GAG chains, is well known for its structural and proinflammatory functions in the ECM (96). It is overexpressed in multiple cancer types such as gastric cancer, ovarian cancer, pancreatic cancer, colon cancer, and melanoma (97, 98). Biglycan can either be secreted de novo by activated macrophages and other resident immune cells or be proteolytially freed from the ECM on tissue injury or stress. Soluble biglycan can act as a danger signal through binding Toll-like receptors (TLR)-2 and -4, resulting in production of TNF-α, IL-1β, CCL2, CXCL1, CXCL-2, CXCL-13, and CCL5 (95). Using a transient transgenic mouse model where biglycan was overexpressed by hepatocytes and released into the bloodstream, Zeng-Brouwers et al. (99) demonstrated biglycan accumulation in the kidneys caused leukocyte infiltration in the renal parenchyma concurrent with abnormal renal levels of chemoattractants CXCL1, CXCL2, CCL2, and CCL5. Furthermore, using mice deficient in either TLR adaptor proteins MyD88 or TRIF, they discovered that biglycan utilizes TLR-2/4 signaling that engages the adaptor molecule MyD88 (myeloid differentiation primary response 88), whereas it modulates T-cell infiltration exclusively using TLR-4 and the adaptor molecule TRIF (Toll/interleukin 1 R domain-containing adapter inducing interferon-β) (99, 100). In macrophages, soluble biglycan has been reported to cluster TLR2/4 and purinergic receptors, P2X4/P2X7, which in turn triggers the activation of caspase-1 through the NLRP3 (NOD-like receptor protein 3) inflammasome, followed by the release of IL-1β and IL-18 (101). Another study showed that binding of biglycan to CD14 and TLR2 in macrophages induces TNF-α and HSP70 release, whereas biglycan-CD14-TLR4 complex causes CCL5 (RANTES) secretion (102). During tumorigenesis, interaction of biglycan and TLR receptors stabilizes NADPH oxidases (NOX-1, -2, and 4) and modulates reactive oxygen species (ROS) production, which in turn regulates IL-1β secretion (103). Biglycan-TLR2/4-mediated ROS generation activates macrophages and dendritic cells to release CXCL13, a major chemoattractant for B cells and B1 cells (104). Thus, biglycan acts at different stages of the cancer immune cycle where it can bridge innate and adaptive immune responses.
Decorin is another class-I SLRPG with dermatan sulfate side chains that possesses well-established antitumorigenic functions (95, 106). Like biglycan, it can bind TLR2/4 in macrophages to initiate a downstream signaling and secretion of TNF-α, IL-12p70, and CCL2 (95, 107). Decorin modulates the cytokine milieu toward proinflammation by regulating TGF-β/miR-21/PDCD4 (programmed cell death protein 4) axis, to control anti-inflammatory and immunosuppressive IL-10 secretion by macrophages. Decorin reduces the availability of TGF-β-regulated oncogenic microRNA miR-21 by sequestering TGF-β. The tumor suppressor PDCD4 is translationally inhibited by miR-21 and is itself a translational repressor of IL-10. Reduced oncogenic miR-21 causes an increased PDCD4 production, which results in decreased IL-10 secretion. This pathway, through modulation of the inflammatory cytokine milieu in the TME, inhibits tumor growth.
The actions of biglycan and decorin can often be characterized as antagonistic. For example, although TGF-β induces biglycan production by stromal fibroblasts, it inhibits decorin transcription. In addition, biglycan increases the affinity of TGF-β to its receptor and this biglycan-driven TGF-β signaling creates a permissive protumor TME, unlike decorin, which promotes an antitumor TME (105).
Lumican is a member of class-II SLRPGs bearing keratan sulfate GAGs. It is characterized by the presence of 10 leucine-rich repeats in its central region (21). Although several studies ascribe an antitumor role to lumican, its actual role in a particular tumor context is determined by the specific interplay among its abundance, distribution, and tumor stage/type (108). High lumican expression correlates with poor prognosis in colorectal cancer, pancreatic cancer, and breast cancer. Lumican has been shown to participate in inflammatory signaling and EMT (97). Interestingly, lumican affects peripheral monocyte extravasation and Fas-FasL signaling, thereby modulating tumor-associated inflammation (109). It also enhances LPS-dependent TLR4 signaling (108). Zhang et al. (110) studied the role of lumican in the colon cancer microenvironment: using in silico approaches, they showed that lumican expression is positively correlated with infiltration of immune cells such as B cells, monocytes, macrophages, TAMs, DCs, CD8+ T cells, CD4+ T cells, and Treg cells.
LARGE EXTRACELLULAR PGS—VERSICAN AND OTHER HYALECTANS
Versican (VCAN; Fig. 5) is a large matrix proteoglycan, whose actions have been extensively documented in embryonic morphogenesis, inflammation, as well as directing biochemical signaling and cell fate decisions (111). Unlike other Hyalectan (HA-binding) family members, such as aggrecan, brevican, and neurocan (21), VCAN is expressed in a wide variety of tissues throughout the body and has crucial nonredundant roles in organ development and disease (112, 113). The VCAN core protein consists of an N-terminal G1 domain, a C-terminal G3 domain, and CS chain-binding regions (113, 114). The G1 domain is composed of an immunoglobulin (Ig)-like module, followed by two hyaluronan (HA)-binding domains (link modules). VCAN’s G3 domain consists of two epidermal growth factor (EGF)-like repeats, followed by carbohydrate recognition domain (lectin-like, CRD) and complement binding protein (CBP)-like subunits (113). Depending on the number and species of GAG regions present, full-length (V0) and 3 common splice variants of VCAN have been described: V1, V2, and V3. An additional variant, V4, has been reported in breast cancer (115).
Figure 5.

Immunomodulatory roles of large extracellular PG. Tumor- or stromal-derived versican (VCAN) leads to DC dysfunction through TLR-2 activation. Stromal nonproteolyzed VCAN also promotes exclusion of CD8+ T cells from TME. Versikine, an N-terminal bioactive fragment (matrikine), promotes CD8+ influx in the TME through regulation of cDC1 abundance and activation. The balance between nonproteolyzed VCAN and versikine appears to critically influence tumor immune infiltration density. DC, dendritic cell; PG, proteoglycan. Created with BioRender.com.
Versican is of central relevance to several hallmarks of cancer (116) and plays important roles in both malignant transformation and tumor progression (117). There are various sources of VCAN production in the tumor microenvironment: tumor cells, stromal cells, tumor-associated myeloid cells, and possibly lymphoid cells in some contexts (10). Increased versican expression has been observed in a wide range of malignant tumors and has been associated with both cancer relapse and poor patient outcomes (10). Versican is a central partner in extracellular matrix (ECM) assembly and tissue homeostasis through key protein-protein or protein-carbohydrate interactions (118). VCAN engages binding partners both in the extracellular matrix, such as hyaluronan and link protein (HAPLN1, see below) through the N-terminal G1 domain (119) and on the surface of cells in the tumor microenvironment including P and L selectins (120), lymphoid tissue chemokines (121, 122), CD44 (115) and TLR2 (123, 124). VCAN, thus orchestrates a network of crucial intrinsic signals that dictate immune and inflammatory cell phenotype in the tumor microenvironment (125).
VCAN is not only produced by myeloid cells but also regulates the myeloid cell composition and regulatory networks in the tumor (98, 124). Tumor-derived VCAN leads to DC dysfunction through TLR2 activation. TLR2 ligation promotes secretion of autocrine IL-10 and IL-6 as well as sustained upregulation of the cell-surface receptors for these cytokines, which decreases the threshold for STAT3 activation. This positive feedback loop renders DCs dysfunctional and impedes downstream Th1 and cytotoxic lymphocyte (CTL) differentiation (11, 12).
VCAN plays a central role in the regulation of tumor-associated macrophages (TAMs). VCAN may function as a danger-associated molecular pattern (DAMP) molecule that interacts with Toll-like receptors (TLRs), such as TLR2 on macrophages and monocytes, to promote the production of inflammatory cytokines, including tumor necrosis factor-α (TNFα) and other proinflammatory cytokines in various tumor environments (123, 124, 126–129). In the setting of mesothelioma, tumor-derived versican promotes tumor progression by shaping an immunosuppressive milieu, mainly by impairing macrophage antitumor activities. Mice harboring versican-deficient tumors presented fewer tumor/pleural macrophages and neutrophils, and fewer pleural T-regulatory cells, compared with the control animals (130). In the 4T1 breast carcinoma model, versican expression, identified in late stages of progression, was associated with a high number of peritumoral infiltrating TAMs (131). The impact of VCAN in modulating the functional phenotypes of intratumoral macrophages was described in hematological malignancies as well. VCAN was shown to promote myeloma-associated monocytes/macrophages through TLR2/6 signaling in multiple myeloma (MM), thus triggering trophic IL-1β and IL-6 upregulation (132). In addition, the role of VCAN in loss of immune surveillance in human MM was further supported by the demonstration that myeloid-derived versican transcription was strongly associated with altered myeloid polarization toward an immunosuppressive phenotype, contraction of protective T-cell stem-like (Tcf1+) memory, expansion of dysfunctional/exhausted T effectors and consequently, MM progression (133). Moreover, macrophages with a unique immunosuppressive signature (expressing versican, ENTPD1, and STAB1) were associated with persistence of minimal residual disease postautologous stem cell transplant for myeloma and increased risk for relapse (134, 135).
There is a well-established inverse correlation between stromal VCAN accumulation and cytotoxic lymphocyte infiltration and/or activity in multiple human cancers. Tumor-infiltrating lymphocyte (TIL) density has been shown to predict outcomes and correlate with antitumor therapeutic responses, particularly to checkpoint inhibition and other novel immunotherapies (136, 137). VCAN accumulation was inversely correlated with intraepithelial TIL abundance in a large cohort of samples obtained from patients with colorectal cancer (138). Similar conclusions were drawn from analysis of large cohorts of breast, pancreatic, esophageal, and neuroendocrine tumors (139). Earlier work in cervical cancer was also concordant with these observations (140). A meta-analysis of RNA-Seq data by Pearce et al. (141) demonstrated that VCAN, extrapolated from a global matrisome signature, positively correlated with Th2 and T regulatory responses and negatively correlated with cytotoxic T-cell markers (142). VCAN+ tumor-associated macrophage (TAM) accumulation conferred a survival disadvantage in patients with cutaneous skin melanoma; these TAMs were less abundant in ICB therapy responders (143). Preliminary data from a Phase II trial showed significantly lower recurrence rates of metastatic colorectal cancers expressing low to moderate levels of VCAN or high levels of VCAN proteolysis, following neoadjuvant stereotactic body radiation therapy (SBRT) and anti-PD-1 therapy (144). Altogether, these findings suggest that VCAN accumulation impedes antitumor CD8+ infiltration and/or antitumor responses.
Contrary to stromal nonproteolyzed VCAN, which promotes T-cell exclusion, VCAN proteolytic fragments (matrikines) appear to possess opposing, immunostimulatory properties (13, 145). Versikine, an N-terminal VCAN-matrikine, is generated through cleavage of a Glu441-Ala442 bond (V1 enumeration) in the GAG-β domain of V1 isoform, generating a neoepitope, DPEAAE (146). This bioactive fragment promotes conventional dendritic cell type 1 (cDC1) recruitment, survival, and activation across tumor types and genetic backgrounds (13). Therefore, the balance between nonproteolyzed VCAN and versikine appears to critically influence the levels of immune infiltration. VCAN proteolysis at Glu441-Ala442 is associated with robust CD8+ infiltration in multiple myeloma bone marrow (147, 148) as well as solid tumors (138).
Hyaluronan and proteoglycan link protein 1 (HAPLN1) is an intriguing VCAN binding partner and structural homolog (149, 150). It has a molecular weight of 45–52 kDa and consists of a signal peptide (SP), one immunoglobulin-like (Ig), and two HA-binding link modules (proteoglycan tandem repeats, PTR: PTR1, and PTR2) (151, 152). HAPLN1, produced by bone marrow stromal cells from patients with MM, activates an atypical NF-κB pathway in MM cells that may contribute to proteasome-inhibitor drug resistance (153).
CONCLUDING REMARKS
Protective immune responses by the organism against the existential threat posed by a nascent tumor can be understood in the discrete steps of the “cancer-immunity cycle.” This cycle has been classically studied from a cell- and soluble mediator-centric vantage point but the contribution of tumor matrix signaling appears essential. Disruption of the host tissue by the developing tumor is sensed as “danger” by a sentinel network featuring specialized antigen-presenting cells that subsequently migrate from the site of damage to the regional lymph nodes to prime effector lymphocytes. Seminar work by Schaefer and others demonstrated SLRPG as essential “danger” signals activating antigen-presenting cells. The next hurdle to be overcome concerns the trafficking of effectors back to the tumor site to fight the tumor and restore homeostasis. Later work highlighted large PG-derived matrikines, such as versikine, as regulators of chemokine networks controlling immune effector cell influx. Thus, tissue barriers are breached at both the early (“danger”) and latter (“effector”) stages of the cancer-immunity cycle, and cues emanating from extracellular matrix (ECM) remodeling regulate the activity of the cellular actors at each step (163). Successful tumors that overcome these defenses potently co-opt inflammatory networks to promote their own survival and propagation. In this review, we highlighted some of the essential roles of matrix proteoglycans in regulating cancer-immune cross talk as well as opportunities for therapeutic intervention.
GRANTS
The work in the authors’ laboratory is supported by the National Institutes of Health (NIH)/National Cancer Institute (R01CA252937), the American Cancer Society (127508-RSG-15-045-01-LIB), the Leukemia and Lymphoma Society (6551-18), the UW Trillium Myeloma Fund, and the Robert J. Shillman Foundation.
DISCLOSURES
F.A. is listed as inventor on US patent US20170258898A1: “Versikine for inducing or potentiating an immune response.” None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
G.D., A.C., A.P., and F.A. conceived and designed research; G.D., A.C., A.P., and F.A. performed experiments; G.D., A.C., A.P., and F.A. analyzed data; G.D., A.C., A.P., and F.A. interpreted results of experiments; G.D., A.C., A.P., and F.A. prepared figures; G.D., A.C., A.P., and F.A. drafted manuscript; G.D., A.C., A.P., and F.A. edited and revised manuscript; G.D., A.C., A.P., and F.A. approved final version of manuscript.
ACKNOWLEDGMENTS
This article is part of the special collection “Deciphering the Role of Proteoglycans and Glycosaminoglycans in Health and Disease.” Liliana Schaefer, MD, served as Guest Editor of this collection.
REFERENCES
- 1. Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol 196: 395–406, 2012. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep 15: 1243–1253, 2014. doi: 10.15252/embr.201439246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK. Extracellular matrix structure. Adv Drug Deliv Rev 97: 4–27, 2016. doi: 10.1016/j.addr.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 4. Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci 123: 4195–4200, 2010. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hynes RO, Naba A. Overview of the matrisome—an inventory of extracellular matrix constituents and functions. Cold Spring Harb Perspect Biol 4: a004903, 2012. doi: 10.1101/cshperspect.a004903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Labani-Motlagh A, Ashja-Mahdavi M, Loskog A. The tumor microenvironment: a milieu hindering and obstructing antitumor immune responses. Front Immunol 11: 940, 2020. doi: 10.3389/fimmu.2020.00940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hanahan D, Coussens Lisa M. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21: 309–322, 2012. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 8. Mushtaq MU, Papadas A, Pagenkopf A, Flietner E, Morrow Z, Chaudhary SG, Asimakopoulos F. Tumor matrix remodeling and novel immunotherapies: the promise of matrix-derived immune biomarkers. J Immunother Cancer 6: 65, 2018. doi: 10.1186/s40425-018-0376-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol 27: 16–25, 2014. doi: 10.1016/j.coi.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Papadas A, Asimakopoulos F. Versican in the tumor microenvironment. Adv Exp Med Biol 1272: 55–72, 2020. doi: 10.1007/978-3-030-48457-6_4. [DOI] [PubMed] [Google Scholar]
- 11. Tang M, Diao J, Gu H, Khatri I, Zhao J, Cattral MS. Toll-like receptor 2 activation promotes tumor dendritic cell dysfunction by regulating IL-6 and IL-10 receptor signaling. Cell Rep 13: 2851–2864, 2015. doi: 10.1016/j.celrep.2015.11.053. [DOI] [PubMed] [Google Scholar]
- 12. Tang M, Diao J, Cattral MS. Molecular mechanisms involved in dendritic cell dysfunction in cancer. Cell Mol Life Sci 74: 761–776, 2017. doi: 10.1007/s00018-016-2317-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Papadas A, Deb G, Officer A, Cicala A, Hope C, Emmerich P, Wiesner J, Pagenkopf A, Arauz G, Bansal V, Matkowskyj KA, Deming D, Politi K, Abrams SI, Harismendy O, Asimakopoulos F. Stromal remodeling regulates dendritic cell abundance and activity in the tumor microenvironment (Preprint). bioRxiv, 2021. doi: 10.1101/2021.11.10.467836. [DOI] [PMC free article] [PubMed]
- 14. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Yet al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554: 544–548, 2018. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peng DH, Rodriguez BL, Diao L, Chen L, Wang J, Byers LA, Wei Y, Chapman HA, Yamauchi M, Behrens C, Raso G, Soto LMS, Cuentes ERP, Wistuba II, Kurie JM, Gibbons DL. Collagen promotes anti-PD-1/PD-L1 resistance in cancer through LAIR1-dependent CD8(+) T cell exhaustion. Nat Commun 11: 4520, 2020. doi: 10.1038/s41467-020-18298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sun X, Wu B, Chiang HC, Deng H, Zhang X, Xiong W, Liu J, Rozeboom AM, Harris BT, Blommaert E, Gomez A, Garcia RE, Zhou Y, Mitra P, Prevost M, Zhang D, Banik D, Isaacs C, Berry D, Lai C, Chaldekas K, Latham PS, Brantner CA, Popratiloff A, Jin VX, Zhang N, Hu Y, Pujana MA, Curiel TJ, An Z, Li R. Tumour DDR1 promotes collagen fibre alignment to instigate immune exclusion. Nature 599: 673–678, 2021. doi: 10.1038/s41586-021-04057-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 10: 826–837, 2010. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm 2010: 1–21, 2010. doi: 10.1155/2010/672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun 11: 5120, 2020. doi: 10.1038/s41467-020-18794-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iozzo RV. Matrix proteoglycans: from molecular design to cellular function. Annu Rev Biochem 67: 609–652, 1998. doi: 10.1146/annurev.biochem.67.1.609. [DOI] [PubMed] [Google Scholar]
- 21. Iozzo RV, Schaefer L. Proteoglycan form and function: a comprehensive nomenclature of proteoglycans. Matrix Biol 42: 11–55, 2015. doi: 10.1016/j.matbio.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kolset SO, Gallagher JT. Proteoglycans in haemopoietic cells. Biochim Biophys Acta 1032: 191–211, 1990. doi: 10.1016/0304-419x(90)90004-k. [DOI] [PubMed] [Google Scholar]
- 23. Kolset SO, Pejler G. Serglycin: a structural and functional chameleon with wide impact on immune cells. J Immunol 187: 4927–4933, 2011. doi: 10.4049/jimmunol.1100806. [DOI] [PubMed] [Google Scholar]
- 24. Meen AJ, Øynebråten I, Reine TM, Duelli A, Svennevig K, Pejler G, Jenssen T, Kolset SO. Serglycin is a major proteoglycan in polarized human endothelial cells and is implicated in the secretion of the chemokine GROα/CXCL1. J Biol Chem 286: 2636–2647, 2011. doi: 10.1074/jbc.M110.151944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zernichow L, Abrink M, Hallgren J, Grujic M, Pejler G, Kolset SO. Serglycin is the major secreted proteoglycan in macrophages and has a role in the regulation of macrophage tumor necrosis factor-alpha secretion in response to lipopolysaccharide. J Biol Chem 281: 26792–26801, 2006. doi: 10.1074/jbc.M512889200. [DOI] [PubMed] [Google Scholar]
- 26. Korpetinou A, Skandalis SS, Labropoulou VT, Smirlaki G, Noulas A, Karamanos NK, Theocharis AD. Serglycin: at the crossroad of inflammation and malignancy. Front Oncol 3: 327–327, 2014. doi: 10.3389/fonc.2013.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Manou D, Karamanos NK, Theocharis AD. Tumorigenic functions of serglycin: regulatory roles in epithelial to mesenchymal transition and oncogenic signaling. Semin Cancer Biol 62: 108–115, 2020. doi: 10.1016/j.semcancer.2019.07.004. [DOI] [PubMed] [Google Scholar]
- 28. Abrink M, Grujic M, Pejler G. Serglycin is essential for maturation of mast cell secretory granule. J Biol Chem 279: 40897–40905, 2004. doi: 10.1074/jbc.M405856200. [DOI] [PubMed] [Google Scholar]
- 29. Braga T, Grujic M, Lukinius A, Hellman L, Abrink M, Pejler G. Serglycin proteoglycan is required for secretory granule integrity in mucosal mast cells. Biochem J 403: 49–57, 2007. doi: 10.1042/BJ20061257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Melo FR, Waern I, Rönnberg E, Åbrink M, Lee DM, Schlenner SM, Feyerabend TB, Rodewald HR, Turk B, Wernersson S, Pejler G. A role for serglycin proteoglycan in mast cell apoptosis induced by a secretory granule-mediated pathway. J Biol Chem 286: 5423–5433, 2011. doi: 10.1074/jbc.M110.176461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Metkar SS, Wang B, Aguilar-Santelises M, Raja SM, Uhlin-Hansen L, Podack E, Trapani JA, Froelich CJ. Cytotoxic cell granule-mediated apoptosis: perforin delivers granzyme B-serglycin complexes into target cells without plasma membrane pore formation. Immunity 16: 417–428, 2002. doi: 10.1016/s1074-7613(02)00286-8. [DOI] [PubMed] [Google Scholar]
- 32. Grujic M, Braga T, Lukinius A, Eloranta ML, Knight SD, Pejler G, Abrink M. Serglycin-deficient cytotoxic T lymphocytes display defective secretory granule maturation and granzyme B storage. J Biol Chem 280: 33411–33418, 2005. doi: 10.1074/jbc.M501708200. [DOI] [PubMed] [Google Scholar]
- 33. Grujic M, Christensen JP, Sørensen MR, Abrink M, Pejler G, Thomsen AR. Delayed contraction of the CD8+ T cell response toward lymphocytic choriomeningitis virus infection in mice lacking serglycin. J Immunol 181: 1043–1051, 2008. doi: 10.4049/jimmunol.181.2.1043. [DOI] [PubMed] [Google Scholar]
- 34. Toyama-Sorimachi N, Sorimachi H, Tobita Y, Kitamura F, Yagita H, Suzuki K, Miyasaka M. A novel ligand for CD44 is serglycin, a hematopoietic cell lineage-specific proteoglycan: possible involvement in lymphoid cell adherence and activation. J Biol Chem 270: 7437–7444, 1995. doi: 10.1074/jbc.270.13.7437. [DOI] [PubMed] [Google Scholar]
- 35. Guo JY, Hsu HS, Tyan SW, Li FY, Shew JY, Lee WH, Chen JY. Serglycin in tumor microenvironment promotes non-small cell lung cancer aggressiveness in a CD44-dependent manner. Oncogene 36: 2457–2471, 2017. doi: 10.1038/onc.2016.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zöller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer 11: 254–267, 2011. doi: 10.1038/nrc3023. [DOI] [PubMed] [Google Scholar]
- 37. Chu Q, Huang H, Huang T, Cao L, Peng L, Shi S, Zheng L, Xu L, Zhang S, Huang J, Li X, Qian C, Huang B. Extracellular serglycin upregulates the CD44 receptor in an autocrine manner to maintain self-renewal in nasopharyngeal carcinoma cells by reciprocally activating the MAPK/β-catenin axis. Cell Death Dis 7: e2456, 2016. doi: 10.1038/cddis.2016.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang Z, Deng Y, Zheng G, Jia X, Xiong Y, Luo K, Qiu Q, Qiu N, Yin J, Lu M, Liu H, Gu Y, He Z. SRGN-TGFβ2 regulatory loop confers invasion and metastasis in triple-negative breast cancer. Oncogenesis 6: e360, 2017. doi: 10.1038/oncsis.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bouris P, Manou D, Sopaki-Valalaki A, Kolokotroni A, Moustakas A, Kapoor A, Iozzo RV, Karamanos NK, Theocharis AD. Serglycin promotes breast cancer cell aggressiveness: induction of epithelial to mesenchymal transition, proteolytic activity and IL-8 signaling. Matrix Biol 74: 35–51, 2018. doi: 10.1016/j.matbio.2018.05.011. [DOI] [PubMed] [Google Scholar]
- 40. Skliris A, Happonen KE, Terpos E, Labropoulou V, Børset M, Heinegård D, Blom AM, Theocharis AD. Serglycin inhibits the classical and lectin pathways of complement via its glycosaminoglycan chains: implications for multiple myeloma. Eur J Immunol 41: 437–449, 2011. doi: 10.1002/eji.201040429. [DOI] [PubMed] [Google Scholar]
- 41. Whiteside TL. Tumor-derived exosomes and their role in cancer progression. Adv Clin Chem 74: 103–141, 2016. doi: 10.1016/bs.acc.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Purushothaman A, Bandari SK, Chandrashekar DS, Jones RJ, Lee HC, Weber DM, Orlowski RZ. Chondroitin sulfate proteoglycan serglycin influences protein cargo loading and functions of tumor-derived exosomes. Oncotarget 8: 73723–73732, 2017. doi: 10.18632/oncotarget.20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Piperigkou Z, Mohr B, Karamanos N, Götte M. Shed proteoglycans in tumor stroma. Cell Tissue Res 365: 643–655, 2016. doi: 10.1007/s00441-016-2452-4. [DOI] [PubMed] [Google Scholar]
- 44. Beauvais DM, Rapraeger AC. Syndecans in tumor cell adhesion and signaling. Reprod Biol Endocrinol 2: 3, 2004. doi: 10.1186/1477-7827-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Czarnowski D. Syndecans in cancer: a review of function, expression, prognostic value, and therapeutic significance. Cancer Treat Res Commun 27: 100312, 2021. doi: 10.1016/j.ctarc.2021.100312. [DOI] [PubMed] [Google Scholar]
- 46. Barbouri D, Afratis N, Gialeli C, Vynios D, Theocharis A, Karamanos N. Syndecans as modulators and potential pharmacological targets in cancer progression. Front Oncol 4: 4, 2014. doi: 10.3389/fonc.2014.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Theocharis AD, Skandalis SS, Tzanakakis GN, Karamanos NK. Proteoglycans in health and disease: novel roles for proteoglycans in malignancy and their pharmacological targeting. FEBS J 277: 3904–3923, 2010. doi: 10.1111/j.1742-4658.2010.07800.x. [DOI] [PubMed] [Google Scholar]
- 48. Munesue S, Kusano Y, Oguri K, Itano N, Yoshitomi Y, Nakanishi H, Yamashina I, Okayama M. The role of syndecan-2 in regulation of actin-cytoskeletal organization of Lewis lung carcinoma-derived metastatic clones. Biochem J 363: 201–209, 2002. doi: 10.1042/0264-6021:3630201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Beauvais DM, Rapraeger AC. Syndecan-1-mediated cell spreading requires signaling by αvβ3 integrins in human breast carcinoma cells. Exp Cell Res 286: 219–232, 2003. doi: 10.1016/s0014-4827(03)00126-5. [DOI] [PubMed] [Google Scholar]
- 50. Chang Z, Meyer K, Rapraeger AC, Friedl A. Differential ability of heparan sulfate proteoglycans to assemble the fibroblast growth factor receptor complex in situ. FASEB J 14: 137–144, 2000. doi: 10.1096/fasebj.14.1.137. [DOI] [PubMed] [Google Scholar]
- 51. Gospodarowicz D, Cheng J. Heparin protects basic and acidic FGF from inactivation. J Cell Physiol 128: 475–484, 1986. doi: 10.1002/jcp.1041280317. [DOI] [PubMed] [Google Scholar]
- 52. Fears CY, Woods A. The role of syndecans in disease and wound healing. Matrix Biol 25: 443–456, 2006. doi: 10.1016/j.matbio.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 53. Marshall LJ, Ramdin LSP, Brooks T, DPhil PC, Shute JK. Plasminogen activator inhibitor-1 supports IL-8-mediated neutrophil transendothelial migration by inhibition of the constitutive shedding of endothelial IL-8/heparan sulfate/syndecan-1 complexes. J Immunol 171: 2057–2065, 2003. doi: 10.4049/jimmunol.171.4.2057. [DOI] [PubMed] [Google Scholar]
- 54. Fears CY, Gladson CL, Woods A. Syndecan-2 is expressed in the microvasculature of gliomas and regulates angiogenic processes in microvascular endothelial cells. J Biol Chem 281: 14533–14536, 2006. doi: 10.1074/jbc.C600075200. [DOI] [PubMed] [Google Scholar]
- 55. Maeda T, Alexander CM, Friedl A. Induction of syndecan-1 expression in stromal fibroblasts promotes proliferation of human breast cancer cells. Cancer Res 64: 612–621, 2004. doi: 10.1158/0008-5472.can-03-2439. [DOI] [PubMed] [Google Scholar]
- 56. Su G, Blaine SA, Qiao D, Friedl A. Shedding of syndecan-1 by stromal fibroblasts stimulates human breast cancer cell proliferation via FGF2 activation. J Biol Chem 282: 14906–14915, 2007. doi: 10.1074/jbc.M611739200. [DOI] [PubMed] [Google Scholar]
- 57. Handra-Luca A. Syndecan-1 in the tumor microenvironment. Adv Exp Med Biol 1272: 39–53, 2020. doi: 10.1007/978-3-030-48457-6_3. [DOI] [PubMed] [Google Scholar]
- 58. Bayer-Garner IB, Sanderson RD, Dhodapkar MV, Owens RB, Wilson CS. Syndecan-1 (CD138) immunoreactivity in bone marrow biopsies of multiple myeloma: shed syndecan-1 accumulates in fibrotic regions. Mod Pathol 14: 1052–1058, 2001. doi: 10.1038/modpathol.3880435. [DOI] [PubMed] [Google Scholar]
- 59. Binder Gallimidi A, Nussbaum G, Hermano E, Weizman B, Meirovitz A, Vlodavsky I, Götte M, Elkin M. Syndecan-1 deficiency promotes tumor growth in a murine model of colitis-induced colon carcinoma. PLoS One 12: e0174343, 2017. doi: 10.1371/journal.pone.0174343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ibrahim SA, Gadalla R, El-Ghonaimy EA, Samir O, Mohamed HT, Hassan H, Greve B, El-Shinawi M, Mohamed MM, Götte M. Syndecan-1 is a novel molecular marker for triple negative inflammatory breast cancer and modulates the cancer stem cell phenotype via the IL-6/STAT3, Notch and EGFR signaling pathways. Mol Cancer 16: 57, 2017. doi: 10.1186/s12943-017-0621-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, Karin M. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 15: 103–113, 2009. [Erratum in Cancer Cell 15: 241, 2009]. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kharabi Masouleh B, Ten Dam GB, Wild MK, Seelige R, van der Vlag J, Rops AL, Echtermeyer FG, Vestweber D, van Kuppevelt TH, Kiesel L, Götte M. Role of the heparan sulfate proteoglycan syndecan-1 (CD138) in delayed-type hypersensitivity. J Immunol 182: 4985–4993, 2009. doi: 10.4049/jimmunol.0800574. [DOI] [PubMed] [Google Scholar]
- 63. Saleh ME, Gadalla R, Hassan H, Afifi A, Götte M, El-Shinawi M, Mohamed MM, Ibrahim SA. The immunomodulatory role of tumor syndecan-1 (CD138) on ex vivo tumor microenvironmental CD4+ T cell polarization in inflammatory and non-inflammatory breast cancer patients. PLoS One 14: e0217550, 2019. doi: 10.1371/journal.pone.0217550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jung O, Beauvais DM, Adams KM, Rapraeger AC. VLA-4 phosphorylation during tumor and immune cell migration relies on its coupling to VEGFR2 and CXCR4 by syndecan-1. J Cell Sci 132: jcs232645, 2019. doi: 10.1242/jcs.232645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Loftus PG, Watson L, Deedigan LM, Camarillo-Retamosa E, Dwyer RM, O’Flynn L, Alagesan S, Griffin M, O’Brien T, Kerin MJ, Elliman SJ, Barkley LR. Targeting stromal cell Syndecan-2 reduces breast tumour growth, metastasis and limits immune evasion. Int J Cancer 148: 1245–1259, 2021. doi: 10.1002/ijc.33383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Prieto-Fernández E, Egia-Mendikute L, Bosch A, García Del Río A, Jimenez-Lasheras B, Antoñana-Vildosola A, Lee SY, Palazon A. Hypoxia promotes syndecan-3 expression in the tumor microenvironment. Front Immunol 11: 586977–586977, 2020. doi: 10.3389/fimmu.2020.586977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Takeda N, O’Dea EL, Doedens A, Kim JW, Weidemann A, Stockmann C, Asagiri M, Simon MC, Hoffmann A, Johnson RS. Differential activation and antagonistic function of HIF-α isoforms in macrophages are essential for NO homeostasis. Genes Dev 24: 491–501, 2010. doi: 10.1101/gad.1881410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. El Ghazal R, Yin X, Johns SC, Swanson L, Macal M, Ghosh P, Zuniga EI, Fuster MM. Glycan sulfation modulates dendritic cell biology and tumor growth. Neoplasia 18: 294–306, 2016. doi: 10.1016/j.neo.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chung J-S, Sato K, Dougherty II, Cruz PD Jr, Ariizumi K. DC-HIL is a negative regulator of T lymphocyte activation. Blood 109: 4320–4327, 2007. doi: 10.1182/blood-2006-11-053769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chung JS, Dougherty I, Cruz PD Jr, Ariizumi K. Syndecan-4 mediates the coinhibitory function of DC-HIL on T cell activation. J Immunol 179: 5778–5784, 2007. doi: 10.4049/jimmunol.179.9.5778. [DOI] [PubMed] [Google Scholar]
- 71. Chung JS, Bonkobara M, Tomihari M, Cruz PD Jr, Ariizumi K. The DC-HIL/syndecan-4 pathway inhibits human allogeneic T-cell responses. Eur J Immunol 39: 965–974, 2009. doi: 10.1002/eji.200838990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chung J-S, Shiue LH, Duvic M, Pandya A, Cruz PD Jr, Ariizumi K. Sézary syndrome cells overexpress syndecan-4 bearing distinct heparan sulfate moieties that suppress T-cell activation by binding DC-HIL and trapping TGF-β on the cell surface. Blood 117: 3382–3390, 2011. doi: 10.1182/blood-2010-08-302034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ilieva KM, Cheung A, Mele S, Chiaruttini G, Crescioli S, Griffin M, Nakamura M, Spicer JF, Tsoka S, Lacy KE, Tutt ANJ, Karagiannis SN. Chondroitin sulfate proteoglycan 4 and its potential as an antibody immunotherapy target across different tumor types. Front Immunol 8: 1911, 2017. doi: 10.3389/fimmu.2017.01911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Erfurt C, Sun Z, Haendle I, Schuler-Thurner B, Heirman C, Thielemans K, van der Bruggen P, Schuler G, Schultz ES. Tumor-reactive CD4+ T cell responses to the melanoma-associated chondroitin sulphate proteoglycan in melanoma patients and healthy individuals in the absence of autoimmunity. J Immunol 178: 7703–7709, 2007. doi: 10.4049/jimmunol.178.12.7703. [DOI] [PubMed] [Google Scholar]
- 75. Rachmilewitz J, Tykocinski ML. Differential effects of chondroitin sulfates A and B on monocyte and B-cell activation: evidence for B-cell activation via a CD44-dependent pathway. Blood 92: 223–229, 1998. [PubMed] [Google Scholar]
- 76. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumour environment by TGFβ. Nat Rev Immunol 10: 554–567, 2010. doi: 10.1038/nri2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gatza CE, Oh SY, Blobe GC. Roles for the type III TGF-β receptor in human cancer. Cell Signal 22: 1163–1174, 2010. doi: 10.1016/j.cellsig.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Eickelberg O, Centrella M, Reiss M, Kashgarian M, Wells RG. Betaglycan inhibits TGF-β signaling by preventing type I-type II receptor complex formation. Glycosaminoglycan modifications alter betaglycan function. J Biol Chem 277: 823–829, 2002. doi: 10.1074/jbc.M105110200. [DOI] [PubMed] [Google Scholar]
- 79. Huang JJ, Corona AL, Dunn BP, Cai EM, Prakken JN, Blobe GC. Increased type III TGF-β receptor shedding decreases tumorigenesis through induction of epithelial-to-mesenchymal transition. Oncogene 38: 3402–3414, 2019. doi: 10.1038/s41388-018-0672-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hanks BA, Holtzhausen A, Evans KS, Jamieson R, Gimpel P, Campbell OM, Hector-Greene M, Sun L, Tewari A, George A, Starr M, Nixon AB, Augustine C, Beasley G, Tyler DS, Osada T, Morse MA, Ling L, Lyerly HK, Blobe GC. Type III TGF-β receptor downregulation generates an immunotolerant tumor microenvironment. J Clin Invest 123: 3925–3940, 2013. doi: 10.1172/JCI65745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Mythreye K, Blobe GC. The type III TGF-β receptor regulates epithelial and cancer cell migration through β-arrestin2-mediated activation of Cdc42. Proc Natl Acad Sci USA 106: 8221–8226, 2009. doi: 10.1073/pnas.0812879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Filmus J, Selleck SB. Glypicans: proteoglycans with a surprise. J Clin Invest 108: 497–501, 2001. doi: 10.1172/JCI13712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kaur SP, Cummings BS. Role of glypicans in regulation of the tumor microenvironment and cancer progression. Biochem Pharmacol 168: 108–118, 2019. doi: 10.1016/j.bcp.2019.06.020. [DOI] [PubMed] [Google Scholar]
- 84. Bloushtain N, Qimron U, Bar-Ilan A, Hershkovitz O, Gazit R, Fima E, Korc M, Vlodavsky I, Bovin NV, Porgador A. Membrane-associated heparan sulfate proteoglycans are involved in the recognition of cellular targets by NKp30 and NKp46. J Immunol 173: 2392–2401, 2004. doi: 10.4049/jimmunol.173.4.2392. [DOI] [PubMed] [Google Scholar]
- 85. Shimizu Y, Suzuki T, Yoshikawa T, Endo I, Nakatsura T. Next-generation cancer immunotherapy targeting glypican-3. Front Oncol 9: 248, 2019. doi: 10.3389/fonc.2019.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wang Y, Wang Y, Mu H, Liu T, Chen X, Shen Z. Enhanced specific antitumor immunity of dendritic cells transduced with the glypican 3 gene and co-cultured with cytokine-induced killer cells against hepatocellular carcinoma cells. Mol Med Rep 11: 3361–3367, 2015. doi: 10.3892/mmr.2015.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Takai H, Ashihara M, Ishiguro T, Terashima H, Watanabe T, Kato A, Suzuki M. Involvement of glypican-3 in the recruitment of M2-polarized tumor-associated macrophages in hepatocellular carcinoma. Cancer Biol Ther 8: 2329–2338, 2009. doi: 10.4161/cbt.8.24.9985. [DOI] [PubMed] [Google Scholar]
- 88. Karapetsas A, Giannakakis A, Dangaj D, Lanitis E, Kynigopoulos S, Lambropoulou M, Tanyi JL, Galanis A, Kakolyris S, Trypsianis G, Coukos G, Sandaltzopoulos R. Overexpression of GPC6 and TMEM132D in early stage ovarian cancer correlates with CD8+ T-lymphocyte infiltration and increased patient survival. Biomed Res Int 2015: 712438, 2015. doi: 10.1155/2015/712438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Cruz LA, Tellman TV, Farach-Carson MC. Flipping the molecular switch: influence of perlecan and its modifiers in the tumor microenvironment. Adv Exp Med Biol 1245: 133–146, 2020. doi: 10.1007/978-3-030-40146-7_6. [DOI] [PubMed] [Google Scholar]
- 90. Elgundi Z, Papanicolaou M, Major G, Cox TR, Melrose J, Whitelock JM, Farrugia BL. Cancer metastasis: the role of the extracellular matrix and the heparan sulfate proteoglycan perlecan. Front Oncol 9: 1482, 2020. doi: 10.3389/fonc.2019.01482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Warren CR, Grindel BJ, Francis L, Carson DD, Farach-Carson MC. Transcriptional activation by NFκB increases perlecan/HSPG2 expression in the desmoplastic prostate tumor microenvironment. J Cell Biochem 115: 1322–1333, 2014. [Erratum in J Cell Biochem 117: 2669, 2016]. doi: 10.1002/jcb.24788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Poluzzi C, Iozzo RV, Schaefer L. Endostatin and endorepellin: a common route of action for similar angiostatic cancer avengers. Adv Drug Deliv Rev 97: 156–173, 2016. doi: 10.1016/j.addr.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Aviezer D, Hecht D, Safran M, Eisinger M, David G, Yayon A. Perlecan, basal lamina proteoglycan, promotes basic fibroblast growth factor-receptor binding, mitogenesis, and angiogenesis. Cell 79: 1005–1013, 1994. doi: 10.1016/0092-8674(94)90031-0. [DOI] [PubMed] [Google Scholar]
- 94. Chakraborty S, Njah K, Hong W. Agrin mediates angiogenesis in the tumor microenvironment. Trends Cancer 6: 81–85, 2020. doi: 10.1016/j.trecan.2019.12.002. [DOI] [PubMed] [Google Scholar]
- 95. Schaefer L, Tredup C, Gubbiotti MA, Iozzo RV. Proteoglycan neofunctions: regulation of inflammation and autophagy in cancer biology. FEBS J 284: 10–26, 2017. doi: 10.1111/febs.13963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, Marsche G, Young MF, Mihalik D, Götte M, Malle E, Schaefer RM, Gröne HJ. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest 115: 2223–2233, 2005. doi: 10.1172/JCI23755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Appunni S, Anand V, Khandelwal M, Gupta N, Rubens M, Sharma A. Small leucine rich proteoglycans (decorin, biglycan and lumican) in cancer. Clin Chim Acta 491: 1–7, 2019. doi: 10.1016/j.cca.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 98. Tzanakakis G, Neagu M, Tsatsakis A, Nikitovic D. Proteoglycans and immunobiology of cancer-therapeutic implications. Front Immunol 10: 875, 2019. doi: 10.3389/fimmu.2019.00875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zeng-Brouwers J, Beckmann J, Nastase M-V, Iozzo RV, Schaefer L. De novo expression of circulating biglycan evokes an innate inflammatory tissue response via MyD88/TRIF pathways. Matrix Biol 35: 132–142, 2014. doi: 10.1016/j.matbio.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Frey H, Schroeder N, Manon-Jensen T, Iozzo RV, Schaefer L. Biological interplay between proteoglycans and their innate immune receptors in inflammation. Febs J 280: 2165–2179, 2013. doi: 10.1111/febs.12145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Babelova A, Moreth K, Tsalastra-Greul W, Zeng-Brouwers J, Eickelberg O, Young MF, Bruckner P, Pfeilschifter J, Schaefer RM, Gröne HJ, Schaefer L. Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J Biol Chem 284: 24035–24048, 2009. doi: 10.1074/jbc.M109.014266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Roedig H, Nastase MV, Frey H, Moreth K, Zeng-Brouwers J, Poluzzi C, Hsieh LT, Brandts C, Fulda S, Wygrecka M, Schaefer L. Biglycan is a new high-affinity ligand for CD14 in macrophages. Matrix Biol 77: 4–22, 2019. doi: 10.1016/j.matbio.2018.05.006. [DOI] [PubMed] [Google Scholar]
- 103. Hsieh LT-H, Frey H, Nastase M-V, Tredup C, Hoffmann A, Poluzzi C, Zeng-Brouwers J, Manon-Jensen T, Schröder K, Brandes RP, Iozzo RV, Schaefer L. Bimodal role of NADPH oxidases in the regulation of biglycan-triggered IL-1β synthesis. Matrix Biol 49: 61–81, 2016. doi: 10.1016/j.matbio.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Moreth K, Brodbeck R, Babelova A, Gretz N, Spieker T, Zeng-Brouwers J, Pfeilschifter J, Young MF, Schaefer RM, Schaefer L. The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis. J Clin Invest 120: 4251–4272, 2010. doi: 10.1172/JCI42213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Diehl V, Huber LS, Trebicka J, Wygrecka M, Iozzo RV, Schaefer L. The role of decorin and biglycan signaling in tumorigenesis. Front Oncol 11: 801801, 2021. doi: 10.3389/fonc.2021.801801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Gubbiotti MA, Vallet SD, Ricard-Blum S, Iozzo RV. Decorin interacting network: a comprehensive analysis of decorin-binding partners and their versatile functions. Matrix Biol 55: 7–21, 2016. doi: 10.1016/j.matbio.2016.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Merline R, Moreth K, Beckmann J, Nastase MV, Zeng-Brouwers J, Tralhão JG, Lemarchand P, Pfeilschifter J, Schaefer RM, Iozzo RV, Schaefer L. Signaling by the matrix proteoglycan decorin controls inflammation and cancer through PDCD4 and MicroRNA-21. Sci Signal 4: ra75, 2011. doi: 10.1126/scisignal.2001868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Nikitovic D, Papoutsidakis A, Karamanos NK, Tzanakakis GN. Lumican affects tumor cell functions, tumor-ECM interactions, angiogenesis and inflammatory response. Matrix Biol 35: 206–214, 2014. doi: 10.1016/j.matbio.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 109. Vij N, Roberts L, Joyce S, Chakravarti S. Lumican regulates corneal inflammatory responses by modulating Fas-Fas ligand signaling. Invest Ophthalmol Vis Sci 46: 88–95, 2005. doi: 10.1167/iovs.04-0833. [DOI] [PubMed] [Google Scholar]
- 110. Zang Y, Dong Q, Lu Y, Dong K, Wang R, Liang Z. Lumican inhibits immune escape and carcinogenic pathways in colorectal adenocarcinoma. Aging (Albany NY) 13: 4388–4408, 2021. doi: 10.18632/aging.202401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Papadas A, Arauz G, Cicala A, Wiesner J, Asimakopoulos F. Versican and versican-matrikines in cancer progression, inflammation, and immunity. J Histochem Cytochem 68: 871–885, 2020. doi: 10.1369/0022155420937098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wight TN. Versican: a versatile extracellular matrix proteoglycan in cell biology. Curr Opin Cell Biol 14: 617–623, 2002. doi: 10.1016/s0955-0674(02)00375-7. [DOI] [PubMed] [Google Scholar]
- 113. Wu YJ, La Pierre DP, Wu J, Yee AJ, Yang BB. The interaction of versican with its binding partners. Cell Res 15: 483–494, 2005. doi: 10.1038/sj.cr.7290318. [DOI] [PubMed] [Google Scholar]
- 114. Iozzo RV, Naso MF, Cannizzaro LA, Wasmuth JJ, McPherson JD. Mapping of the versican proteoglycan gene (CSPG2) to the long arm of human chromosome 5 (5q12-5q14). Genomics 14: 845–851, 1992. doi: 10.1016/S0888-7543(05)80103-X. [DOI] [PubMed] [Google Scholar]
- 115. Kischel P, Waltregny D, Dumont B, Turtoi A, Greffe Y, Kirsch S, De Pauw E, Castronovo V. Versican overexpression in human breast cancer lesions: known and new isoforms for stromal tumor targeting. Int J Cancer 126: 640–650, 2010. doi: 10.1002/ijc.24812. [DOI] [PubMed] [Google Scholar]
- 116. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 144: 646–674, 2011. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 117. Wight TN, Kang I, Evanko SP, Harten IA, Chang MY, Pearce OMT, Allen CE, Frevert CW. Versican-a critical extracellular matrix regulator of immunity and inflammation. Front Immunol 11: 512, 2020. doi: 10.3389/fimmu.2020.00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Wight TN. Provisional matrix: a role for versican and hyaluronan. Matrix Biol 60–61: 38–56, 2017. doi: 10.1016/j.matbio.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Matsumoto K, Shionyu M, Go M, Shimizu K, Shinomura T, Kimata K, Watanabe H. Distinct interaction of versican/PG-M with hyaluronan and link protein. J Biol Chem 278: 41205–41212, 2003. doi: 10.1074/jbc.M305060200. [DOI] [PubMed] [Google Scholar]
- 120. Kawashima H, Hirose M, Hirose J, Nagakubo D, Plaas AH, Miyasaka M. Binding of a large chondroitin sulfate/dermatan sulfate proteoglycan, versican, to L-selectin, P-selectin, and CD44. J Biol Chem 275: 35448–35456, 2000. doi: 10.1074/jbc.M003387200. [DOI] [PubMed] [Google Scholar]
- 121. Hirose J, Kawashima H, Yoshie O, Tashiro K, Miyasaka M. Versican interacts with chemokines and modulates cellular responses. J Biol Chem 276: 5228–5234, 2001. doi: 10.1074/jbc.M007542200. [DOI] [PubMed] [Google Scholar]
- 122. Zheng PS, Vais D, Lapierre D, Liang YY, Lee V, Yang BL, Yang BB. PG-M/versican binds to P-selectin glycoprotein ligand-1 and mediates leukocyte aggregation. J Cell Sci 117: 5887–5895, 2004. doi: 10.1242/jcs.01516. [DOI] [PubMed] [Google Scholar]
- 123. Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, Luo JL, Karin M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 457: 102–106, 2009. doi: 10.1038/nature07623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Wang W, Xu GL, Jia WD, Ma JL, Li JS, Ge YS, Ren WH, Yu JH, Liu WB. Ligation of TLR2 by versican: a link between inflammation and metastasis. Arch Med Res 40: 321–323, 2009. doi: 10.1016/j.arcmed.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 125. Wight TN, Kang I, Merrilees MJ. Versican and the control of inflammation. Matrix Biol 35: 152–161, 2014. doi: 10.1016/j.matbio.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Bögels M, Braster R, Nijland PG, Gül N, van de Luijtgaarden W, Fijneman RJ, Meijer GA, Jimenez CR, Beelen RH, van Egmond M. Carcinoma origin dictates differential skewing of monocyte function. Oncoimmunology 1: 798–809, 2012. doi: 10.4161/onci.20427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Li D, Wang X, Wu JL, Quan WQ, Ma L, Yang F, Wu KY, Wan HY. Tumor-produced versican V1 enhances hCAP18/LL-37 expression in macrophages through activation of TLR2 and vitamin D3 signaling to promote ovarian cancer progression in vitro. PLoS One 8: e56616, 2013. doi: 10.1371/journal.pone.0056616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Said N, Theodorescu D. RhoGDI2 suppresses bladder cancer metastasis via reduction of inflammation in the tumor microenvironment. Oncoimmunology 1: 1175–1177, 2012. doi: 10.4161/onci.20594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Said N, Sanchez-Carbayo M, Smith SC, Theodorescu D. RhoGDI2 suppresses lung metastasis in mice by reducing tumor versican expression and macrophage infiltration. J Clin Invest 122: 1503–1518, 2012. doi: 10.1172/JCI61392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Pappas AG, Magkouta S, Pateras IS, Skianis I, Moschos C, Vazakidou ME, Psarra K, Gorgoulis VG, Kalomenidis I. Versican modulates tumor-associated macrophage properties to stimulate mesothelioma growth. Oncoimmunology 8: e1537427, 2019. doi: 10.1080/2162402X.2018.1537427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Dos Reis DC, Damasceno KA, de Campos CB, Veloso ES, Pêgas GRA, Kraemer LR, Rodrigues MA, Mattos MS, Gomes DA, Campos PP, Ferreira E, Russo RC, Cassali GD. Versican and tumor-associated macrophages promotes tumor progression and metastasis in canine and murine models of breast carcinoma. Front Oncol 9: 577, 2019. doi: 10.3389/fonc.2019.00577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Hope C, Ollar SJ, Heninger E, Hebron E, Jensen JL, Kim J, Maroulakou I, Miyamoto S, Leith C, Yang DT, Callander N, Hematti P, Chesi M, Bergsagel PL, Asimakopoulos F. TPL2 kinase regulates the inflammatory milieu of the myeloma niche. Blood 123: 3305–3315, 2014. doi: 10.1182/blood-2014-02-554071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Bailur JK, McCachren SS, Doxie DB, Shrestha M, Pendleton K, Nooka AK, Neparidze N, Parker TL, Bar N, Kaufman JL, Hofmeister CC, Boise LH, Lonial S, Kemp ML, Dhodapkar KM, Dhodapkar MV. Early alterations in stem-like/resident T cells, innate and myeloid cells in the bone marrow in preneoplastic gammopathy. JCI Insight 4: e127807, 2019. doi: 10.1172/jci.insight.127807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Arana P, Zabaleta A, Lasa M, Maiso P, Alignani D, Jelinek T, Segura V, van den Bossche W, Teodosio C, Damasceno D, Rodriguez-Otero P, Prosper F, Mateos M-V, Lahuerta JJ, Bladé J, van Dongen JJM, San Miguel J, Orfao A, Paiva B. High-throughput characterization and new insight into the role of tumor associated macrophages (TAMs) in multiple myeloma (MM). Blood 128: 482, 2016. doi: 10.1182/blood.V128.22.482.482. [DOI] [Google Scholar]
- 135. Nakamura K, Kassem S, Cleynen A, Chrétien ML, Guillerey C, Putz EM, Bald T, Förster I, Vuckovic S, Hill GR, Masters SL, Chesi M, Bergsagel PL, Avet-Loiseau H, Martinet L, Smyth MJ. Dysregulated IL-18 is a key driver of immunosuppression and a possible therapeutic target in the multiple myeloma microenvironment. Cancer Cell 33: 634–648.e5, 2018. doi: 10.1016/j.ccell.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 136. Bai R, Lv Z, Xu D, Cui J. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomark Res 8: 34, 2020. doi: 10.1186/s40364-020-00209-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Gooden MJ, de Bock GH, Leffers N, Daemen T, Nijman HW. The prognostic influence of tumour-infiltrating lymphocytes in cancer: a systematic review with meta-analysis. Br J Cancer 105: 93–103, 2011. doi: 10.1038/bjc.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Hope C, Emmerich PB, Papadas A, Pagenkopf A, Matkowskyj KA, Van De Hey DR, Payne SN, Clipson L, Callander NS, Hematti P, Miyamoto S, Johnson MG, Deming DA, Asimakopoulos F. Versican-derived matrikines regulate Batf3-dendritic cell differentiation and promote T cell infiltration in colorectal cancer. J Immunol 199: 1933–1941, 2017. doi: 10.4049/jimmunol.1700529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Emmerich P, Matkowskyj KA, McGregor S, Kraus S, Bischel K, Qyli T, Buehler D, Pasch C, Babiarz C, Depke M, Clipson L, Wisinski KB, Burkard ME, Baschnagel A, Uboha NV, Asimakopoulos F, Deming DA. VCAN accumulation and proteolysis as predictors of T lymphocyte-excluded and permissive tumor microenvironments. J Cin Oncol 38: 3127, 2020. doi: 10.1200/JCO.2020.38.15_suppl.3127. [DOI] [Google Scholar]
- 140. Gorter A, Zijlmans HJ, van Gent H, Trimbos JB, Fleuren GJ, Jordanova ES. Versican expression is associated with tumor-infiltrating CD8-positive T cells and infiltration depth in cervical cancer. Mod Pathol 23: 1605–1615, 2010. doi: 10.1038/modpathol.2010.154. [DOI] [PubMed] [Google Scholar]
- 141. Pearce OMT, Delaine-Smith RM, Maniati E, Nichols S, Wang J, Böhm S, Rajeeve V, Ullah D, Chakravarty P, Jones RR, Montfort A, Dowe T, Gribben J, Jones JL, Kocher HM, Serody JS, Vincent BG, Connelly J, Brenton JD, Chelala C, Cutillas PR, Lockley M, Bessant C, Knight MM, Balkwill FR. Deconstruction of a metastatic tumor microenvironment reveals a common matrix response in human cancers. Cancer Discov 8: 304–319, 2018. doi: 10.1158/2159-8290.CD-17-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Hirani P, Gauthier V, Allen CE, Wight TN, Pearce OMT. Targeting versican as a potential immunotherapeutic strategy in the treatment of cancer. Front Oncol 11: 712807, 2021. doi: 10.3389/fonc.2021.712807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, Qin S, Zhang L, Ouyang H, Du P, Jiang L, Zhang B, Yang Y, Wang X, Ren X, Bei JX, Hu X, Bu Z, Ji J, Zhang Z. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184: 792–809.e23, 2021. doi: 10.1016/j.cell.2021.01.010. [DOI] [PubMed] [Google Scholar]
- 144. Deming DA, Emmerich P, Turk AA, Lubner SJ, Uboha NV, LoConte NK, Mulkerin D, Kim DH, Matkowskyj KA, Weber SM, Abbott D, Eickhoff JC, Bassetti MF. Pembrolizumab (Pem) in combination with stereotactic body radiotherapy (SBRT) for resectable liver oligometastatic MSS/MMR proficient colorectal cancer (CRC). J Clin Oncol 38: 4046, 2020. doi: 10.1200/JCO.2020.38.15_suppl.4046. [DOI] [Google Scholar]
- 145. Schmitt M. Versican vs versikine: tolerance vs attack. Blood 128: 612–613, 2016. doi: 10.1182/blood-2016-06-721092. [DOI] [PubMed] [Google Scholar]
- 146. Sandy JD, Westling J, Kenagy RD, Iruela-Arispe ML, Verscharen C, Rodriguez-Mazaneque JC, Zimmermann DR, Lemire JM, Fischer JW, Wight TN, Clowes AW. Versican V1 proteolysis in human aorta in vivo occurs at the Glu441-Ala442 bond, a site that is cleaved by recombinant ADAMTS-1 and ADAMTS-4. J Biol Chem 276: 13372–13378, 2001. doi: 10.1074/jbc.M009737200. [DOI] [PubMed] [Google Scholar]
- 147. Dhakal B, Pagenkopf A, Mushtaq MU, Cunningham AM, Flietner E, Morrow Z, Papadas A, Hope C, Leith C, Hematti P, Hari P, Callander NS, Asimakopoulos F. Versican proteolysis predicts immune effector infiltration and post-transplant survival in myeloma. Leuk Lymphoma 60: 2558–2562, 2019. doi: 10.1080/10428194.2019.1585836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Hope C, Foulcer S, Jagodinsky J, Chen SX, Jensen JL, Patel S, Leith C, Maroulakou I, Callander N, Miyamoto S, Hematti P, Apte SS, Asimakopoulos F. Immunoregulatory roles of versican proteolysis in the myeloma microenvironment. Blood 128: 680–685, 2016. doi: 10.1182/blood-2016-03-705780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Hardingham TE. The role of link-protein in the structure of cartilage proteoglycan aggregates. Biochem J 177: 237–247, 1979. doi: 10.1042/bj1770237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Binette F, Cravens J, Kahoussi B, Haudenschild DR, Goetinck PF. Link protein is ubiquitously expressed in non-cartilaginous tissues where it enhances and stabilizes the interaction of proteoglycans with hyaluronic acid. J Biol Chem 269: 19116–19122, 1994. [PubMed] [Google Scholar]
- 151. Périn JP, Bonnet F, Thurieau C, Jollès P. Link protein interactions with hyaluronate and proteoglycans. Characterization of two distinct domains in bovine cartilage link proteins. J Biol Chem 262: 13269–13272, 1987. [PubMed] [Google Scholar]
- 152. Perkins SJ, Nealis AS, Dudhia J, Hardingham TE. Immunoglobulin fold and tandem repeat structures in proteoglycan N-terminal domains and link protein. J Mol Biol 206: 737–753, 1989. doi: 10.1016/0022-2836(89)90580-9. [DOI] [PubMed] [Google Scholar]
- 153. Huynh M, Pak C, Markovina S, Callander NS, Chng KS, Wuerzberger-Davis SM, Bakshi DD, Kink JA, Hematti P, Hope C, Asimakopoulos F, Rui L, Miyamoto S. Hyaluronan and proteoglycan link protein 1 (HAPLN1) activates bortezomib-resistant NF-κB activity and increases drug resistance in multiple myeloma. J Biol Chem 293: 2452–2465, 2018. doi: 10.1074/jbc.RA117.000667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Theocharis AD, Karamanos NK. Proteoglycans remodeling in cancer: Underlying molecular mechanisms. Matrix Biol 75–76: 220–259, 2019. doi: 10.1016/j.matbio.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 155. Nikitovic D, Berdiaki A, Spyridaki I, Krasanakis T, Tsatsakis A, Tzanakakis GN. Proteoglycans-biomarkers and targets in cancer therapy. Front Endocrinol (Lausanne) 9: 69, 2018. doi: 10.3389/fendo.2018.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics 11: M111.014647, 2012. doi: 10.1074/mcp.M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Zielinski JM, Luke JJ, Guglietta S, Krieg C. High throughput multi-omics approaches for clinical trial evaluation and drug discovery. Front Immunol 12: 590742, 2021. doi: 10.3389/fimmu.2021.590742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Aoyama E, Yoshihara R, Tai A, Yamamoto I, Gohda E. PKC- and PI3K-dependent but ERK-independent proliferation of murine splenic B cells stimulated by chondroitin sulfate B. Immunol Lett 99: 80–84, 2005. doi: 10.1016/j.imlet.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 159. Yang R, Yan Z, Chen F, Hansson GK, Kiessling R. Hyaluronic acid and chondroitin sulphate A rapidly promote differentiation of immature DC with upregulation of costimulatory and antigen-presenting molecules, and enhancement of NF-κB and protein kinase activity. Scand J Immunol 55: 2–13, 2002. doi: 10.1046/j.0300-9475.2001.01033.x. [DOI] [PubMed] [Google Scholar]
- 160. Tavianatou AG, Caon I, Franchi M, Piperigkou Z, Galesso D, Karamanos NK. Hyaluronan: molecular size-dependent signaling and biological functions in inflammation and cancer. FEBS J 286: 2883–2908, 2019. doi: 10.1111/febs.14777. [DOI] [PubMed] [Google Scholar]
- 161. Schaefer L, Iozzo RV. Small leucine-rich proteoglycans, at the crossroad of cancer growth and inflammation. Curr Opin Genet Dev 22: 56–57, 2012. doi: 10.1016/j.gde.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 162. Merline R, Schaefer RM, Schaefer L. The matricellular functions of small leucine-rich proteoglycans (SLRPs). J Cell Commun Signal 3: 323–335, 2009. doi: 10.1007/s12079-009-0066-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Vyas M, Demehri S. The extracellular matrix and immunity: breaking the old barrier in cancer. Trends Immunol 43: 423–425, 2022. doi: 10.1016/j.it.2022.04.004. [DOI] [PubMed] [Google Scholar]