

Keywords: Aurora Kinase A, oncogene, transcription, mRNA processing, translation, cell cycle
Abstract
Aurora Kinase A (AURKA) is a positive regulator of mitosis with a strict cell cycle-dependent expression pattern. Recently, novel oncogenic roles of AURKA have been uncovered that are independent of the kinase activity and act within multiple signalling pathways, including cell proliferation, survival and cancer stem cell phenotypes. For this, cellular abundance of AURKA protein is per se crucial and must be tightly fine-tuned. Indeed, AURKA is found overexpressed in different cancers, typically as a result of gene amplification or enhanced transcription. It has however become clear that impaired processing, decay and translation of AURKA mRNA can also offer the basis for altered AURKA levels. Accordingly, the involvement of gene expression mechanisms controlling AURKA expression in human diseases is increasingly recognized and calls for much more research. Here, we explore and create an integrated view of the molecular processes regulating AURKA expression at the level of transcription, post-transcription and translation, intercalating discussion on how impaired regulation underlies disease. Given that targeting AURKA levels might affect more functions compared to inhibiting the kinase activity, deeper understanding of its gene expression may aid the design of alternative and therapeutically more successful ways of suppressing the AURKA oncogene.
1. Introduction
The AURKA gene (also known as STK6, STK15, IAK1, AIK) encodes a member of the human Aurora family of kinases that are critical regulators of cell division. This family comprises two other members, namely AURKB and AURKC, and is characterized by a highly conserved Serine/Threonine kinase domain. First discovered using genetic screens in Drosophila [1], Aurora Kinase A (AURKA) phosphorylates target substrates to modulate maturation of centrosomes as well as formation of the mitotic spindle, processes that are crucial for the correct segregation of chromosomes during mitosis (M phase) [2]. Persistent association of high expression of AURKA with cancer progression, poor prognosis and drug resistance has been reported to such an extent that AURKA represents a distinguished target in the development of anti-cancer drugs [3]. In recent years, growing evidence uncovered novel cancer-promoting roles of AURKA that are kinase-independent and occur in the nucleus [4–7]. These observations fortify the concept that deregulation of expression might alone be sufficient to drive AURKA oncogenic functions, since some of these can be exerted without the need for activation of the kinase or concomitant deregulation of kinase activators. For this, suppressing its expression might represent a more efficient way to target oncogenic AURKA than using kinase inhibitors [8].
AURKA overexpression in human malignancies is mostly reported to be caused by gene amplification, enhanced transcription, or loss of miRNA-mediated silencing. However, increased AURKA protein is not accompanied by changes in mRNA abundance in some cancers [9–11]. This implies that modulation of protein stability and translation also underlie AURKA altered expression in disease, although fewer examples are available in the literature. For instance, AURKA overexpression through reduced proteolysis has been observed in head and neck [12] and breast [13] cancer cells, and a new study reports that undegraded AURKA at mitotic exit enhances fragmentation of the mitochondrial network in the following interphase [14], with fragmented mitochondrial networks being a characteristic of some cancer cells, including human invasive breast cancer [15]. Similarly, deregulation of translation also contributed to the overexpression of AURKA in some cases [16,17]. Therefore, virtually every step regulating AURKA levels has the potential to be involved in its oncogenic activation, and the control of AURKA expression is more complex than anticipated in both physiological and pathological conditions. Regardless, it is surprising how the past decade has witnessed an explosion of studies on the functions and regulation of AURKA protein, rather neglecting fundamental questions about the modulation of its gene expression. At present, if control of AURKA protein stability and degradation have been characterized to a great extent, and multiple molecular mechanisms responsible for timely transcription of AURKA gene are known, fewer mechanisms of AURKA mRNA post-transcriptional and translational regulation have been described. Nor has the current state of knowledge of the regulation of AURKA gene expression been comprehensively reviewed. For this reason, in this review we first elucidate the molecular mechanisms of AURKA expression at the level of DNA and mRNA, highlighting the tight link between such mechanisms and disease. Finally, we integrate the current knowledge to offer an all-inclusive view of the temporal expression of AURKA during the cell cycle.
2. AURKA gene structure
The AURKA gene was mapped by a fluorescence in situ hybridization (FISH) experiment that yielded a signal in chromosome 20 (20q13.2) and one in chromosome 1 (1q41) [18]. Further analyses showed that the 20q13.2 band corresponded to the functional AURKA gene, as later corroborated by interspecific backcross mapping [19], and that the second band represented an AURKA processed pseudogene on chromosome 1. Three more pseudogenes were subsequently described for AURKA, located on different chromosomes [20], with the one located on chromosome 1 transcribing the long non-coding RNA AURKAPS1 [21]. The AURKA gene is located in the reverse strand orientation and consists of a total of 11 exons and 10 introns within a region 22 948 base pairs (bp) long, spanning from location 56 369 389 bp to 56 392 337 bp (GRCh38.p13). The open reading frame (ORF) of the full-length AURKA cDNA is 1212 bp and encodes a 403-amino acid protein of approximately 46 kDa. Exons IV to VI encode the unstructured N-terminal domain, whereas exons VII to XI code for the conserved central kinase domain and the C-terminal domain. The upstream regulatory region for AURKA gene, which includes the promoter, extends for 4.2 kb and is shared with the CSTF1 gene. It is worth noting that AURKA gene maps onto an intrinsically unstable chromosome region with frequent defects [22].
3. Transcriptional regulation
AURKA is expressed in almost all somatic cells, predominantly in dividing tissues such as haematopoietic cells, mammary gland, colon and testis. Conversely, AURKA expression is low in adult tissues with low or no rate of proliferation [22–24]. Low abundance of AURKA protein however does not correlate with lack of function. At least two lines of evidence support this notion, both based on discoveries of alternative non-mitotic and cell-specific functions of AURKA. Firstly, AURKA exerts important physiological functions during the interphase of cycling cells (reviewed in [25]), when its protein levels are much lower than in mitosis. Secondly, specific non-mitotic functions of AURKA have also been reported in non-cycling cells, such as neurons [26], in which the protein has intrinsically low expression levels (proteinatlas.org) [27].
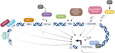
The existence of a variety of factors and signalling effectors that have been reported to modulate transcription of the AURKA gene (figure 1), both in normal and disease contexts, is very much a reflection of the plethora of cellular sources and experimental conditions adopted among the numerous studies. Several aspects of AURKA transcriptional regulation have been examined, and most investigations turned to classical luciferase reporters, Chromatin-immunoprecipitation (ChIP) and electrophoresis-based DNA-binding assays to uncover the minimal requirements for AURKA transcription, what dictates its cell cycle-dependency, and some of the transcriptional mechanisms responsible for pathological expression. However, many questions remain to be addressed (table 1). Nonetheless, one should be careful in assuming that the transcriptional mechanisms that we know today all co-exist, since many were uncovered using cancer cell lines or tissues and it is often not made clear whether the mechanisms described can be generalized to the normal context.
Figure 1.

Key regulators of AURKA transcription. ATR, AT-rich region. CDE, cell cycle-dependent element. CHR, cell cycle gene homology region. E-box, enhancer box. HRE, hypoxia response element. PRE, positive regulatory element. G0, G1, S, G2, M, cell cycle phases. *, factors that also interact with AURKA protein in potential regulatory feedback loops. Thick arrow indicates start site and direction of transcription. Figure created using BioRender.com.
Table 1.
List of questions that remain open on the regulation of AURKA gene expression in different pathological contexts, grouped by level of regulation.
| level of regulation | outstanding questions | pathological context | ref. |
|---|---|---|---|
| transcription | tissue-specific effects on AURKA transcription in response to hypoxic conditions | hypoxia, cystic renal disease | [28–31] |
| EGFR-mediated AURKA transcriptional regulation for cellular adaptation to EGF signalling | cancer | [32] | |
| mechanism of the viral early oncoprotein E6 in regulating AURKA transcription | HPV16-induced carcinogenesis | [33] | |
| EWS-Fli1-dependent transcriptional enhancement of AURKA | Ewing's sarcoma | [34] | |
| post-transcription | role of exon II in regulating AURKA translation and association with tumorigenesis | breast and colorectal cancer | [11,35] |
| inclusion of exon III as protective mechanism against tumorigenesis | breast cancer | [24] | |
| role of ERβ in controlling AS of AURKA exon II/III | breast cancer | [36] | |
| targeting AURKA AS via Spliceostatin A and Madrasin as a therapeutic intervention to reduce expression levels | cancer | [37–40] | |
| role of APA of AURKA mRNA in mediating AURKA overexpression and oncogenic activity | breast and lung cancer | [41–43] | |
| MCPIP1-mediated AURKA mRNA destabilization | neuroblastoma | [44] | |
| IGF2BP1-mediated AURKA mRNA stabilization | cancer | [45] | |
| miRNA-mediated targeting of AURKA mRNA | breast, liver, lung cancer | [46–49] | |
| translation | mechanism of IRES-dependent AURKA translation | breast cancer | [16] |
| combined roles of hnRNP Q1 and EGF/EGFR signalling in controlling AURKA translation | breast cancer | [17] |
3.1. Molecular mechanisms of AURKA transcription
3.1.1. Requirements for AURKA transcriptional activation
The fact that AURKA promoter lacks a conserved TATA-box prompted Tanaka et al. [20] to search for sequence elements necessary for the transcriptional activation of the human AURKA gene. By transiently transfecting HeLa and NIH3T3 cells with AURKA promoter-luciferase constructs containing a series of truncations or mutations, a positive regulatory element (PRE) (CTTCCGG, −85 to −79) was identified in the 5′ region flanking the +1 transcription start site (TSS) that is crucial for the transcriptional activity of AURKA gene. E4 Transcription Factor 1 (E4TF1), a member of the E26 transformation-specific (Ets) family of transcription factors, was found to bind to the PRE and to be predominantly responsible for AURKA transcriptional activation. In HeLa cells, deletion of the −124 to −90 sequence further decreased AURKA promoter activity, suggesting the presence of a cis-element for a tissue-specific factor that could modulate AURKA transcription by a yet unknown mechanism.
Another member of the Ets family of transcription factors that is thought to mediate activation of AURKA transcription is GA-Binding Protein (GABP), highly homologous to E4TF1 [50], in conjunction with TR-associated Protein/Mediator complex subunit 1 (TRAP220/MED1) [51]. TRAP220/MED1 is necessary for basal transcription of AURKA, and, again using luciferase reporter assays, it was shown that the TRAP220/MED1-mediated activation of AURKA transcription requires the PRE element in HeLa cells. Both TRAP220/MED1 and GABP were alone able to bind the PRE-containing region of AURKA promoter, and GABP binding to PRE was unaffected by TRAP220/MED1 silencing. Moreover, TRAP220/MED1 directly interacted with GABP in vivo and in vitro. From here, it was postulated that TRAP220/MED1 is recruited to AURKA promoter by PRE-bound GABP. These observations could be further substantiated by silencing GABP to establish that it is necessary for TRAP220/MED1 binding to AURKA promoter to activate transcription.
3.1.2. Cell cycle periodicity of AURKA transcription
Although the PRE is required for AURKA gene transcriptional activity, it is not responsible for its cell cycle-dependent regulation. For example, in electrophoretic mobility shift assays (EMSAs), E4TF1 remained bound to AURKA PRE throughout the cell cycle [20]. On the other hand, GABP has been reported to regulate genes in a cell cycle-dependent manner [52], but whether this is also the case for AURKA, and whether it also relies on the PRE, is not clear. Most genes whose expression peaks in G2 (late genes), such as Cyclin A, cdc2, cdc25C and Plk, are transcriptionally repressed in G0 and early G1, with their promoters being relieved from repression in late G1/early S phase [53,54]. In addition to de-repression, promoters of ‘late genes’ also undergo activation late in S phase, which is sustained until M phase [55]. A dual sequence module that consists of a Cell Cycle-Dependent Element (CDE) and a Cell Cycle Gene Homology Region (CHR) is crucial for this regulation: the CDE/CHR module is responsible both for transcriptional repression in G0 and early G1 and for transcriptional activation later in S phase, depending on the protein composition of CDE/CHR-binding Multi-vulval class B (MuvB)-based complex (figure 1). Additionally, transcriptional activation of G2/M genes is also mediated by CCAAT-boxes, recognized by the CCAAT-Binding Factor/Nuclear transcription Factor Y (CBF)/(NF-Y) complex.
3.1.2.1. AURKA transcriptional repression in G0 and early G1
Because of its transcriptional time window in dividing cells, AURKA is historically classified as a ‘G2/M’ gene [56,57] and, just downstream of the AURKA PRE, a CDE (−44 to −40) and a CHR (−39 to −35) are located. However, dissimilar to canonical CDE/CHR sites, AURKA CHR sequence (5′CTTAA3′) diverges from the consensus (5′TTTGAA3′) and CDE and CHR are located next to each other without the typical 4 nucleotides (nt) spacer. Mutations in the CDE or CHR on AURKA promoter-containing reporter constructs increased G1-specific transcription, suggesting that the CDE/CHR module functions as repressor of AURKA transcription in G1 [20]. As mentioned above, the transcriptional outcome dictated by CDE/CHR site depends on the contextual partners of the CDE/CHR-binding MuvB sub-complex. In G0 and early G1, AURKA CDE/CHR is expected to be bound by a complex comprising the core MuvB sub-complex (LIN9, LIN37, LIN52, LIN54, RBBP4) plus p130 or p107, DP, E2F4 and E2F5 factors (altogether the DREAM complex) [58–60]. In particular, the DREAM complex binds to the CDE sequence through E2F4 and E2F5, and to the CHR sequence through MuvB, resulting in transcriptional repression [61,62], although the precise mechanism through which this occurs is still enigmatic.
3.1.2.2. AURKA transcriptional activation in S to M
Once cells are in S phase, AURKA transcriptional program switches from repression to activation, and positive transcriptional activity lasts until M phase. It has been estimated that in NIH3T3 cells AURKA is transcribed at a rate of 5.39 mRNA molecules/(cell × hour) [63]. Contributing to AURKA activation are three classes of sequence motifs: CDE/CHR module, E2F sites and CCAAT-boxes. As we will explore, AURKA activation through CDE/CHR and CCAAT motifs has been delineated, whereas the role of E2F sites, as well as the functional interaction between the different classes of motifs, is still debated.
The suppressor DREAM complex is thought to disassemble from the CDE/CHR in G1 [61]. However, the activator complex initiates assembly onto the CDE/CHR site only in S phase [59,64,65], leaving a time window between release of DREAM complex and activation of transcription. This de-repression that precedes activation might be responsible for initial low-level transcription. Accordingly, AURKA mRNA molecules can be minimally detected already in G1 phase [24]. The AURKA CDE/CHR-binding activator complex comprises the MuvB sub-complex this time associated with other partners: B-MYB and Forkhead Box M1 (FOXM1) [62]. It is a matter of debate whether B-MYB and FOXM1 directly bind DNA when they are in complex with MuvB, and to which sites [61]. In S, MuvB first recruits B-MYB and this allows subsequent recruitment of FOXM1 in early G2 (MuvB-BMYB-FOXM1 complex). During the increase of AURKA transcription at the S/G2 transition, B-MYB undergoes phosphorylation and consequent proteasomal degradation, enabling the hyper-phosphorylation and full activation of FOXM1 in G2. In this way, transcription is only maximally activated by MuvB-phosphoFOXM1 complex after B-MYB is degraded, and the former is thought to sustain AURKA transcription until M phase [61,64,65]. However, analysis of global nuclear run-on followed by RNA sequencing (GRO-seq) on RNA extracted from thymidine- and nocodazole-blocked cells (synchronized in S and M phases, respectively) suggests that transcription of G2/M genes may be maximal already in S phase, and it is the steady-state mRNA levels that peak in M [66] (see §6).
E2F transcription factors include both activators (E2F1, E2F2 and E2F3) and repressors (E2F4 and E2F5) that recognize specific E2F sites (consensus 5′TTTCCCGC3′), although E2F4 and E2F5 can also bind CDEs (see above). E2Fs 1-3 typically concur to activate transcription of G1/S genes but have also been found implicated in the activation of G2/M genes in late S or G2 [67–69]. Upstream of PRE, two sites on AURKA promoter (−307 to −302 and −260 to −254) resemble the conserved binding site for E2Fs. This fostered the hypothesis that AURKA could be directly induced by E2Fs. However, AURKA gene was neither found among E2F1- or E2F2-induced genes nor among E2F3-induced genes in two studies that used similar DNA microarray methods to profile transcriptome changes following E2F1, E2F2 or E2F3 overexpression [67,68]. Nevertheless, this could be due to the stringency of criteria set for identifying E2F targets, such as expression fold change, if only minor transcriptional activation of AURKA was induced. In accordance with this supposition, AURKA promoter was later found to be bound weakly by E2F1, E2F2 and E2F3 in ChIP assays, albeit this result could be biased by lack of cell synchronization [70]. Suitably, E2F3 showed no binding to the same AURKA promoter region when this was assessed performing a ChIP experiment from cells synchronized in S [71]. One explanation for these disparate results could be that E2F3 binds to AURKA to activate transcription only in G2 but only to a limited extent, as observed for other G2/M genes [69]. Nevertheless, in other reports AURKA transcription has been shown to be induced by E2F1 following low concentration of arsenic treatment in immortalized keratinocytes and bladder cells [72,73]. In sum, these observations point to the conclusion that AURKA is likely to be positively but modestly regulated by E2F1-3, possibly through the two putative E2F sites, and this regulation probably adds to the MuvB-BMYM-FOXM1-mediated transcriptional activation.
The third known mechanism of AURKA cell cycle-dependent transcriptional activation relies on the two CCAAT-boxes on its promoter (−4 to +1 and +29 to +33), separated by a conserved spacer of approximately 30 bp. Using a dominant-negative approach, in conjunction with DNA-binding and luciferase assays, Hu et al. [71] demonstrated that the CBF/NF-Y transcription factor complex is needed for G2/M progression. It induces expression of late genes, including AURKA, and both CCAAT-boxes on AURKA promoter are needed for this induction. Even so, CBF/NF-Y was bound to these sequence elements throughout the cell cycle, and the authors could not explain the molecular mechanism that conferred temporal specificity to the CBF/NF-Y-mediated transcriptional activation of AURKA. It has been reported that CBF/NF-Y binding to rat CDK1 CCAAT-box is necessary for recruitment of activator E2Fs to their respective sites on CDK1 promoter in S phase to induce transcription [69]. A similar mechanism could apply to AURKA, although it is yet to be investigated. It is likely that CBF/NF-Y mediates AURKA activation by directly recruiting the RNA-Polymerase II [74] and/or by recruiting enzymes for positive chromatin modifications (see below). However, such processes need further study to better frame CBF/NF-Y-mediated AURKA activation.
Once AURKA mRNA levels reach a peak in M, transcription is switched off. To this end, FOXM1 is ubiquitinated through FZR1-activated Anaphase-Promoting Complex/Cyclosome (APC/CFZR1), which leads to its proteasomal degradation [75]. This contributes to decreasing AURKA mRNA levels at mitotic exit [61]. However, only in G1 does repression of transcription actively take over through the mechanisms discussed.
3.2. Signalling pathways modulating AURKA transcription
In addition to intrinsic cell cycle-dependent regulation, AURKA transcription is also modulated in response to internal and external stimuli, such as DNA damage, growth factors and environmental cues, both in normal and pathological conditions, ultimately offering a means of cell cycle control.
An important regulator of AURKA transcription is the p53 tumour suppressor, which represses AURKA expression following DNA damage through multiple mechanisms. Firstly, p53 is able to activate the DREAM complex to block transcription of G2/M genes outside G1 in the event of DNA damage [62]. Secondly, via the p53-Rb-E2F3 axis, p53 activation upon DNA damage increases the level of p21, reducing the activity of Cyclin-Dependent Kinase 2 (CDK2) and therefore blocking Retinoblastoma protein (Rb) hyper-phosphorylation, in turn promoting the sequestering of transcription factor E2F3; E2F3 becomes unable to bind to AURKA promoter at the CDE/CHR site, and this prevents activation of AURKA gene transcription [70]. Thirdly, p53 has been shown to constitutively interact with CBF/NF-Y at CCAAT-boxes of G2/M genes, and, upon DNA damage, it is rapidly acetylated resulting in release of Histone Acetyl-Transferases (HATs) and recruitment of histone deacetylases (HDACs) on the promoters, which induces transcriptional repression [76].
The critical transcription factor Myc has also been found implicated in positively regulating AURKA transcription. This is directly mediated by Myc binding to Enhancer-boxes (E-boxes) on AURKA promoter in mouse [77] and human [78] cells. Interestingly, Myc and its binding partner Max are associated with the AURKA promoter during G2. It seems that such association is prevented by topoisomerase I inhibition and results in downregulation of AURKA expression. With AURKA being implicated in centrosome dynamics, the study reported that topoisomerase I inhibition prevented separation of centrosomes, leading to G2 arrest and cellular senescence. Therefore, a model was proposed in which Myc bridges AURKA transcription to mechanisms of sensing DNA status [78].
A link between the hypoxia response and AURKA expression was first uncovered by a group that observed increased AURKA mRNA levels upon hypoxia in HepG2 hepatoma cells, and this occurred via Hypoxia-Inducible Factor 1 (HIF-1α) [28]. The AURKA promoter contains three hypoxia response elements (HREs) at positions −336 to −332 (HRE-1), −323 to −319 (HRE-2) and −240 to −236 (HRE-3), but HIF-1α-dependent induction of a luciferase reporter was most sensitive to mutation of HRE-2, suggesting that HRE-2 functionally represents the major HIF-1α binding site. In addition, AURKA silencing inhibited hypoxia-induced proliferation of HepG2 cells, suggesting that AURKA transcriptional up-regulation in hypoxic conditions is involved in controlling HepG2 cell proliferation. These results were later validated by Cui et al. [29], who also showed that HIF-1α induced AURKA expression by recruiting HATs to its promoter. In addition, they observed that expression of HIF-1α positively correlated with AURKA expression in hepato-cellular carcinoma (HCC) tissues. However, in other studies using cancer cells, hypoxia contrarily induced a down-regulation of AURKA expression [30], suggesting that the outcome on AURKA transcription in response to hypoxia might be tissue-specific. AURKA transcription is also up-regulated in cystic renal diseases where function of the HIF-1α destabilizer factor von Hippel-Lindau (VHL) is lost, and where formation of primary cilia and motility of renal cells is altered [31]. The regulation of AURKA expression by HIF-1α might occur not only in conditions of hypoxia, but also under other conditions that activate HIF-1α regardless of oxygen levels in the tissue environment, for example, under the influence of hormones and growth factors, cytokines, or other stresses. In summary, HIF-1α is a relevant factor linking AURKA expression to environmental cues.
A connection between AURKA transcription and growth factor signalling has also been reported [32]. In transformed cells with overexpression of epidermal growth factor (EGF), transcription of AURKA was enhanced following translocation of EGF receptor (EGFR) into the nucleus, where it is activated by phosphorylation. Phospho-EGFR then binds to AURKA promoter and facilitates its transcription. Since EGFR lacks a DNA-binding domain, its binding to AURKA promoter occurs via Signal Transducer and Activator of Transcription 5A (STAT5A), which is recruited to the AT-rich (ATR) region in the upstream sequence of AURKA. However, although some of these findings were replicated in multiple immortalized and cancer cell lines, it is not clear how cells use this EGFR-mediated AURKA transcriptional regulation to adapt their proliferation rates to EGF signalling.
AURKA is also a target for the oncogenic Human Papillomavirus 16 (HPV16) in cell carcinogenesis, due to the involvement of the viral early oncoprotein E6 in elevating AURKA transcription [33]. Furthermore, AURKA was found transcriptionally enhanced by Ewing sarcoma breakpoint region 1-Friend Leukaemia Integration 1 (EWS-Fli1) fusion protein, which results from a chromosomal translocation, in Ewing sarcoma cells following EWS-Fli1 binding to a Ets-binding site at −84 to −71 [34]. It would be interesting to test whether abnormal cellular phenotypes caused by EWS-Fli1 could be rescued by AURKA silencing.
3.3. Epigenetic regulation
In addition to the different transcription factors that regulate AURKA gene expression, greater fine-tuning at the transcriptional level is brought about by post-translational modifications of histones residing in proximity of the AURKA promoter. It is now well known that chromatin modifications affect expression of virtually all eukaryotic genes [79]. The observation that HDAC inhibitors diminished AURKA expression in lung cancer cells supports the idea that AURKA transcription is regulated by epigenetic mechanisms [80]. Indeed, it is known that activation of G2/M genes is linked to acetylation of promoter histones and nucleosome positioning, mediated by enzymes recruited by the diverse transcription factor complexes that bind promoter elements, including CBF/NF-Y and MuvB-BMYM-FOXM1 complexes [61]. It has been suggested that CBF/NF-Y binding to CCAAT-boxes allows recruitment of the p300 HAT and formation of an open chromatin state on target promoters [81,82]. However, it seems that, in this process, a distance between CCAAT-boxes of 33 bp is required to enable the correct orientation of the respective binding factors, whereas that between CCAAT-boxes on AURKA promoter is shorter. Other evidence links the MuvB-BMYM-FOXM1 complex to chromatin modifications, for example, deletion of BMYB resulted in reduced histone acetylation on AURKA promoter in cells entering the cell cycle [64].
Direct analysis of the relationship between epigenetic modifications on AURKA promoter and transcriptional activity has been investigated in many cancer contexts. AT-Rich Interactive Domain 1A (ARID1A), a component of the Switch/Sucrose Non-Fermentable (SWI/SNF) chromatin-remodelling complex, which assembles nucleosomes to discourage access of transcription factors to chromatin, has been found to occupy the AURKA promoter and negatively regulate its transcription in colorectal cancer cells [83]. Others observed that p53 directly binds to an upstream region of the AURKA promoter in vivo and represses transcription through the recruitment of HDAC1 and of the mSin3 corepressor in non-small-cell lung cancer [84]. Furthermore, it was shown that INI1/SNF5, core component of the mammalian SWI/SNF complex, repressed AURKA transcription in rhabdoid but not in non-rhabdoid tumour cells, as it associated with AURKA promoter only in the former case [85]. These might only be few of the examples of how chromatin modifications and modifiers control AURKA expression in disease.
In conclusion, the number and variety of mechanisms discussed reflect how important it is for the cell to exercise a tight control of AURKA transcription. Despite this, characterization of AURKA transcription lags behind that of other cell cycle regulators such as Cyclins. AURKA is regulated by transcription factors belonging to the Ets family, such as EGFR, GABP, E4TF1 and many more unidentified factors. Most of these factors use AURKA promoter elements such as PRE, HRE, CDE and CHR, lying within a 400 bp region upstream of the coding sequence. However, only a fraction of the total 4.2 kb region immediately upstream AURKA TSS has been analysed, thus the presence of other regulatory elements further upstream is not to be excluded. In addition, the integrated function of all the different sequence elements is not known and constitutes a fundamental research quest of a complete framework of AURKA transcriptional regulation. It is also important to note the recurring phenomenon in which transcription of AURKA is regulated by some factors that AURKA engages with in protein-protein interactions, such as p53 [86], HIF1 [87], EGFR [88], Myc and FOXM1 [7] (figure 1). This suggests the presence of uncharted regulatory feedback loops, the exploration of which may reveal integrative circuits that control critical cellular functions, in addition to being exploited for therapeutic purposes [89,90].
4. Post-transcriptional regulation
The processing of AURKA gene transcript results in a precursor-mRNA (pre-mRNA) of length 2–2.4 kb. The events of AURKA pre-mRNA splicing and polyadenylation, which are, respectively, addressed in this section, are subject to regulation, leading to a heterogeneity of alternative mRNA isoforms that differ for the length and content of both untranslated regions (UTRs). Although genome-wide studies hint that splicing and polyadenylation are relevant steps of AURKA mRNA regulation, we still do not know how they affect AURKA expression and/or function in detail. Our final, short section on mechanisms controlling AURKA mRNA stability reflects how this aspect of AURKA mRNA regulation has been little explored. In future, the development of reporter assays that bypass transcriptional regulation to selectively focus on post-transcriptional events might be a key strategy to investigate AURKA mRNA modulation in vivo. Such assays could also offer a platform to screen for regulators or modulator drugs [91,92]. In addition, the large collection of RNA-sequencing data from different tumour types alongside clinical profiles of patients contained in The Cancer Genome Atlas (TCGA) could provide ample starting material to systematically study the biological significance of AURKA mRNA isoforms and their association with disease.
4.1. Pre-mRNA splicing
Early studies characterizing AURKA expression hinted that more than one transcript exists for AURKA [22,93], although, at the time, low-resolution Northern and RT-qPCR methods, mixed with relatively less standardized experimental conditions (i.e. RNA extraction methods, instruments, probe design, etc.), prohibited systematic studies of isoform expression among different cell types and tissues. Most of our knowledge on AURKA splicing derives from larger studies of high-throughput RNA sequencing or splicing-sensitive genome-wide microarrays. However, these are not conducive to a unifying hypothesis for the splicing regulation of AURKA, both physiological and disease-related, hence our sometime fragmented discussion on the topic.
Sixteen different high-scoring transcript isoforms resulting from alternative splicing (AS) have been annotated so far, 9 of which are listed in the NCBI database (N#1–9) and 7 in the Ensembl database (E#1–7) (figure 2a). All 16 isoforms are 5′UTR splicing variants, mainly via alternative splice sites and exon skipping, whereas the coding sequence and the 3′UTR follow canonical splicing. The fact that no Matched Annotation between NCBI and EBI (MANE) label was given to any of the 16 isoforms indicates that they are distinct transcripts independently annotated by Ensembl and NCBI. Other truncated and/or incomplete transcripts for AURKA gene are annotated, but since they are given a low annotation score and are not fully validated, they were not included in our discussion.
Figure 2.

Post-transcriptional regulation of AURKA mRNA. (a) Annotated 5′UTR splicing isoforms. (b) Regulators of AURKA mRNA translation (green) and decay (blue). I–IV, exons. An, polyA tail. ATG, start codon. CDS, coding sequence. m7G, 7-methyl-guanosine. miRISC, miRNA-induced silencing complex. PAS, polyadenylation site. UTR, untranslated region. Figure created using BioRender.com.
Given that the splicing process is tightly coupled to transcription, and given the cell cycle periodicity of AURKA transcription, it is not surprising that AURKA is among genes undergoing periodic AS during the cell cycle, presumably with retention of exon III occurring as early as G2 [94]. This inclusion event is likely to be important for engagement of ribosomes and translation [95], and is in accordance both with reports of a general coupling between AS and translation [96,97] and with some observations that AS is inhibited during M phase [98]. Nevertheless, AURKA AS was found to be regulated neither by CDC Like Kinase 1 (CLK1) nor by SON, a large Ser/Arg-rich protein, which are two known regulators of AS of genes with crucial roles in cell cycle control [94,99,100]. What is so peculiar about AURKA transcripts or transcription that sets the regulation of its AS apart from that of other cell cycle genes? Detailed mapping of splicing sites and splicing-associated sequence elements, together with addressing the mechanistic interaction of splicing factors with AURKA transcripts, could provide some clear answers.
Different studies seem to conclude that AURKA AS plays a role in cancer, although none go so far as reporting an AURKA AS-dependent mechanism of pathogenesis [101]. Shin et al. [35], who were the first to report a link between AURKA AS and disease, detected three 5′UTR splicing isoforms (N#1, N#2, N#7) (figure 2a) in a breast cancer cell line, whereas a normal cell line only expressed N#2. Because this is the only one of the three transcripts to lack exon II, the authors inferred that this exon might be implicated in tumorigenesis. However, all three AS isoforms supported equal AURKA protein translation, apparently excluding that exon II could account for AURKA protein overexpression in breast cancers. It is worth noting though that the in vitro translation assay used—in rabbit reticulocyte lysate—may not recapitulate the translational regulation of AURKA mRNA occurring in breast cancer cells. A separate study found that whereas overexpression of Serine(S)/Arginine(R)-rich Splicing Factor 1 (SRSF1), known to couple AS to translational regulation, correlated with inclusion of exon II on AURKA mRNA, AURKA was not amongst translational targets of SRSF1, adding evidence to the lack of correlation between exon II and translational regulation [97]. It can therefore be presumed that the two mechanisms of SRSF1-mediated AS and translational control are uncoupled in the case of AURKA mRNA. Contrary to these conclusions however, an exon II-dependent mechanism of AURKA translational activation was proposed for colorectal cancers in which both EGFR and AURKA are overexpressed [11]. In this work, two exon II-containing transcripts (N#3 and N#5) (figure 2a) were the major AURKA splicing isoforms expressed in human colorectal cancers. This study interestingly shows that exon II enables AURKA mRNA to become responsive to EGF stimulus, resulting in AURKA translational up-regulation. The result could perhaps explain why exon II-dependent AURKA overexpression could not be detected using in vitro translation assays.
Other AURKA exons have also been found to correlate with disease. Li et al. [24] used splicing-sensitive microarrays to analyse AS events of genes reported to play a role in cancer. They suggest that MDA-MB-231 breast cancer cells tend to splice AURKA pre-mRNA in a way to skip exon III, arguing that exon III might provide a protective function against tumorigenesis. MCF7 breast cancer cells also skipped exon III, although to a lesser extent. When both cell lines were grown in 3D culture, they now observed that exon III was more prevalently skipped in MCF7 cells compared to MDA-MB-231 cells, indicating that culture conditions and cellular environment could regulate AURKA splicing process. However, it would be interesting to assess whether the skipping of exon III in breast cancer cell lines is linked to the splicing dynamics of exon III occurring at G2/M [94] (see above) and thus linked to the cell cycle.
Dago et al. [36] report that stable expression of Estrogen Receptor β (Erβ) in MCF7 breast cancer cells induced skipping of exon II or III in different AURKA mRNA isoforms after oestradiol treatment. A truncated form of ERβ containing only the C-terminal domain could mediate skipping of exon III in transcript N#1 only, whereas a truncated form of ERβ containing only the N-terminal domain mediated the skipping of exon III in E#205, as well as skipping of exon II in isoforms N#3 and E#201 (figure 2a). This might suggest that ERβ C-terminal and N-terminal domains interact with the splicing machinery in different manners. Furthermore, the study reckoned that AURKA gene contains ERβ binding site(s), but surprisingly AURKA transcription was not itself regulated by ERβ. It is therefore unknown how ERβ mechanistically controls AURKA AS.
Other studies of AURKA AS shed light onto the possibility of targeting AURKA AS as a therapeutic intervention to control its expression levels. For example, it is known that skipping of exons VI to VIII leads to formation of a premature Stop codon that consequently triggers the Non-sense Mediated Decay (NMD) pathway of AURKA mRNA degradation: a mechanism that cells potentially put in place to prevent aberrant expression of AURKA proteins [37]. Insightfully, the same study also observed that skipping of exons VI–VIII can be induced by Spliceostatin A (SSA), through inhibition of the splice-site recognition Spliceosome Factor 3B 1 (SF3B1). This resulted in lower AURKA expression and constitutes an example of drugs that aim to reduce protein overexpression by means of modulating mRNA splicing to induce mRNA decay. Consistent with this finding in HeLa cells, other studies in K562 myelogenous leukaemia cells also observed skipping of exons VI to VIII on AURKA mRNA following SF3B1 silencing and mutation [38,39]. However, it remains to be seen whether SSA-mediated AURKA silencing can function in suppressing aberrant cell behaviour. Madrasin is a second drug shown to promote exon skipping (exon X) on AURKA mRNA in different cell lines [40]. It is possible that the induced defective splicing interferes with AURKA expression, but this remains to be investigated. It is however worth noting that madrasin induced cell cycle arrest at a lower concentration than that needed to induce exon X skipping. In other reports, AURKA was recovered among the top 50 transcripts encoding proteins regulating cell growth and survival that lacked exons as a result of exon skipping events, following treatment with CLK inhibitors [102]. Also in this case, it is conceivable that the frameshift caused by exon skipping may introduce premature Stop codons and thus enable faster degradation rate of the transcript.
To summarize, inclusions of exon II and of exon III seem to be linked to cancer, for example as they render AURKA mRNA responsive to growth factors signalling. It is however not to exclude that these two exons also play a role in normal AURKA expression, as may be the case for retention of exon III at G2/M. Furthermore, specific splicing events of AURKA mRNA, like skipping of exons VI to VIII, could be exploited therapeutically to promote mRNA decay and control AURKA expression levels. Curiously, recent new evidence has uncovered a role for AURKA in regulation of splicing. AURKA phosphorylates core proteins of the spliceosome in vitro and interacts with factors that regulate the spliceosome. In addition, AURKA promotes in vitro splicing of the β-globin pre-mRNA and inhibition of AURKA changed the AS of different genes [103]. It was not investigated if AURKA itself is among mRNAs whose splicing is altered by AURKA protein inhibition, which may represent the first evidence for an AURKA autoregulatory mechanism acting upon its mRNA.
4.2. Pre-mRNA cleavage and polyadenylation
The pre-mRNA processing step of cleavage and polyadenylation is mediated by Poly-Adenylation Signal (PAS) sites located within the 3′UTR. Two canonical PASs (5′AAUAAA3′) can be found in the AURKA 3′UTR (polyasite.unibas.ch [104]) (figure 2b). As a consequence of tandem 3′UTR Alternative cleavage and PolyAdenylation (APA), two mRNA isoforms that differ in 3′UTR length exist for AURKA mRNA. It has yet to be investigated which AURKA PAS site is preferentially used in which cellular context and to what extent, or whether a 3′UTR isoform switch is modulable. This information might be available from systematic searching for AURKA APA events within the many APA databases available in the literature, including deep learning predictive models [105,106] or libraries created from very diverse biological and pathological contexts such as cellular stress [107,108], immune cells [109,110], cellular senescence [111], cancer [112–114], embryonic development [115] or others [116–118]. More recently, AURKA was classified within the TNBC APA subtype with the highest median index of 3′UTR shortening events, and this correlated with increased AURKA gene expression [41,119]. This TNBC APA subtype also showed a high-intensity nuclear Ki-67 staining indicative of highly proliferative nature, and patients in this subtype had the worst disease-free survival [41]. Moreover, AURKA overexpression in TNBC identifies as a factor of early recurrence and poor prognosis [42] and AURKA showed 3′UTR shortening in poor-prognosis patients of both breast and lung cancer [43]. In their quest to define a periodically regulated AS program linked to the cell cycle, Dominguez et al. [94] preliminarily uncovered 94 genes involved in the cell cycle and/or proliferation that undergo periodic APA during the cell cycle. Although AURKA is not displayed in their gene list, potentially suggesting that AURKA APA is not temporally controlled, a more in-depth analysis using updated software tools and annotations may be required. Nonetheless, the existence of two PAS sites on AURKA mRNA suggests that APA could serve to regulate AURKA expression.
Following cleavage at PAS sites, the AURKA mRNA is polyadenylated at the 3′end. The poly(A) tail enables stability and translation of mRNAs [120]. Park et al. propose that reduction in poly(A) tail length is coupled to translational suppression at mitotic entry only for poly(A) tails under approximately 20 nt [121]. From TAIL-seq analysis of the somatic cell cycle, they also revealed that AURKA mRNA has a median poly(A) length of 65 nt in S phase and of 59 nt in M phase, well above the approximately 20 nt median length threshold for the above correlation to occur. Such poly(A) tail length may however still be responsible for basal stability and translation of AURKA mRNA [122]. In sum, it is currently not known whether the poly(A) tail dynamics of AURKA mRNA is functional in regulating AURKA expression.
4.3. Regulation of mRNA stability and decay
Changes in abundance of AURKA mRNA are not exclusively due to activation or suppression of transcription. Events of mRNA stability control and decay also need to be accounted for when measuring overall mRNA levels (figure 2b). Like most mRNAs, that of AURKA might contain several sequence elements and structural motifs in the UTRs that dictate mRNA stability. However, to our knowledge no functional analysis providing a comprehensive view of AURKA UTR regulatory elements has been carried out to date.
AURKA mRNA average copy number has been estimated at 24 molecules/cell, and the transcript half-life around 5 h, in a population of non-synchronized cells [44,63]. Some evidence shows that depletion of the transcription factor ERG reduced decay of AURKA and AURKB mRNAs in S phase and caused accumulation of both transcripts in G2 and at mitotic entry, resulting in premature and higher induction and activation of AURKA and AURKB proteins, and consequential mitotic defects [123]. Collectively, the study clearly establishes that ERG-mediated degradation of AURKA and AURKB mRNAs is important to ensure proper cell cycle progression, although it does not discriminate between the individual involvement of AURKA and AURKB mRNA degradation. A recent study suggested that overexpression of the ribonuclease Monocyte Chemotactic Protein-1-Induced Protein-1 (MCPIP1) leads to the destabilization of AURKA mRNA in neuroblastoma cells, as it binds to and cleaves AURKA 3′UTR, although the precise mRNA sequence responsible for the observed interaction is still undetermined [44]. Moreover, enhanced CLIP (eCLIP) was used to validate that AURKA mRNA is a direct target of IGF2 mRNA-Binding Protein 1 (IGF2BP1), an important tumour and stem cell fate regulator, and is stabilized by IGF2BP1 binding [45]. IGF2BP1 could therefore promote AURKA oncogenic gene expression in a 3′UTR-dependent manner.
Evidence of microRNA (miRNA)-mediated regulation of the cell cycle continues to grow [124]. At least 50 different miRNAs are predicted to target AURKA 3′UTR, according to the microRNA target prediction database miRDB (mirdb.org), although only few have been validated as direct regulators of AURKA mRNA. Interestingly, many cases of miRNA targeting seem to be relevant particularly in those cancers for which AURKA overexpression is considered a promoting factor and a marker of poor prognosis, such as breast cancer, HCC and lung cancer. For example, Fadaka et al. [46] use molecular docking to suggest that AURKA might be regulated by miR-32-3p in breast cancer, and analyse binding energy and specific miRNA–mRNA interactions, although they did not directly probe the targeting. In HCC, miR-490-3p and miR-26a-5p could silence the expression of AURKA, allowing suppression of proliferation and migration properties of HCC cells [47], as well as reduction of chemoresistance [48]. Furthermore, it was demonstrated that up-regulating the expression of miR-32 via administration of tanshinone, could suppress AURKA expression leading to inhibition of Non-Small Cell Lung Cancer (NSCLC) [49]. These are only a few examples of direct miRNA targeting of AURKA mRNA, but many more are clearly coming, since the validation of miRNA targets can be achieved using simple and reliable methods such as in vivo 3′UTR reporter assays. Some interesting questions, such as the link between AURKA APA and miRNA targeting, remain—for now—totally unexplored. Since APA allows for sequences on the 3′UTR to be contextually displayed or removed, it potentially influences the targeting ability of miRNAs, offering a further layer of AURKA gene expression regulation [125].
RNA modifications also seem to have a role in influencing mRNA stability. For example, N6-methyladenosine (m6A) is selectively recognized by the human YTH Domain Family 2 (YTHDF2) ‘reader’ to positively regulate mRNA degradation [126]. AURKA mRNA is subject to m6A within the 3′UTR close to the STOP codon and is a high confident YTHDF2 target [127,128], although no significant m6A enrichment was detected in AURKA mRNA in any of the cell cycle phases. Nor did the mRNA display significantly higher accumulation in the absence of YTHDF2, suggesting that AURKA mRNA might be recognized by YTHDF2, but this does not mediate its degradation at any time during the cell cycle [128]. Therefore, the role of m6A in AURKA mRNA regulation is still unknown.
Our discussion on AURKA mRNA regulation makes evident that the study of this area has the potential to uncover novel exciting mechanisms of AURKA expression-dependent pathogenesis (table 1). It is easy to imagine that AS and APA processes are combined, and this would give rise not only to an even higher total number of isoforms for AURKA mRNA, but also to additional regulatory mechanisms of mRNA stability and translation. However, much more research is needed to fully characterize the repertoires of AURKA transcript isoforms in both physiological and pathological contexts. It has also been shown that proteins can acquire different localizations and functions depending on which 3′UTR isoform they are translated from [129–132]. For this reason, it is worth investigating whether changes in 3′UTR resulting from APA also affect AURKA protein stability, localization or function. Such a quest would show for the first time if and how 3′UTR APA controls AURKA properties.
5. Translational regulation
Regulation of AURKA gene expression at the level of translation has been less widely reported and little is known compared with its transcriptional and post-transcriptional mRNA processing. Nonetheless, it is now established that dysregulation of translation can also be linked to disease and the contributions of aberrant translation to cancer phenotypes are increasingly recognized [133–135]. The number of cell cycle regulators reported to be abnormally upregulated at the translational level in disease also includes AURKA, as we will argue below. Most of our knowledge on the topic derives from studies that make use of various genome-wide methods, which in recent years have generally shed light on translational control during the cell cycle [136–138]. On the other hand, only few are the studies conducting gene-specific investigations on AURKA translational regulation. In this section, we will focus our discussion initially on the temporal translation of AURKA in relation to the cell cycle, to then consider mechanisms known to modulate AURKA translation that are also linked to disease (table 1).
5.1. Cell cycle periodicity of AURKA translation
The average translation rate for AURKA mRNA was calculated to be 14 proteins per mRNA per hour [63]. However, such analysis, consisting of simultaneous measurements of absolute mRNA and protein copy numbers, as well as turnover rate of both, was carried out in exponentially growing mouse fibroblasts, averaging out any changes of AURKA translation rate through the cell cycle. Because AURKA protein levels peak at late G2 until M, when AURKA then starts to disappear, this would reasonably represent the period of highest translational activity of the AURKA mRNA before it shuts down in M phase. Accordingly, it has been known for many years that the global rate of protein synthesis is markedly reduced (by approx. 75%) in M phase compared to that in interphase [139–143]. However, this notion has become an issue of debate in recent years, following other studies that report smaller or minor variations in global translation rates between M phase and the rest of the cell cycle, possibly due to different cell synchronization methods [144–149]. Nonetheless, translation of hundreds of mRNAs is found to be specifically up- or down-regulated in M versus interphase in a significant manner [142,147,148,150]. Because no one to date has monitored AURKA translation tightly over the cell cycle, we can only infer from studies that have used genome-wide approaches to compare the translatome of different cell cycle phases.
Qin & Sarnow [142] show that, while most mRNAs are found in polysome fractions of lower molecular weight in mitotic extracts compared with extracts from unsynchronized cells, 49 mRNAs (3%) remained associated with either more or a similar number of ribosomes, suggesting that translation of these is up-regulated or constant in M, respectively. The fact that AURKA cannot be found in this group of mRNAs might indicate that its translation decreases in M as for most cellular mRNAs. In support of this, Aviner et al. [145] found AURKA within the subset of 339 proteins (7%) that show statistically significant changes in translation rate between cell cycle stages, with a peak at G2/M. Integration of cell cycle transcriptome, translatome and total proteome data from HeLa cells indicated that, concordantly with the trend of AURKA mRNA abundance, AURKA translation rate also increases through S to peak at M and is minimal in G1 [146]. Conversely however, the absence of AURKA within a list of 1255 mRNAs (12%) that exhibit significantly different levels of ribosome occupancy in any cell cycle phase compared to other phases [148] would suggest that its translation runs at a pace that is similar throughout the cell cycle. A similar result revealed no alteration of AURKA translation efficiency in M compared to S phase [121]. It is worth noting that the last two reports are based on quantifications of ribosomal footprints on mRNAs (ribosome profiling): there is evidence that ribosome occupancy is not necessarily correlated with the rate of translation, as mechanisms might interfere with elongation speed without altering the abundance of residing polysomes [144,151–154]. Moreover, since ribosome profiling typically requires that the amount of ribosome-protected mRNA fragments be normalized to the abundance of the mRNA itself [155], one possible interpretation of the two studies is that the results only reflect changes in the rate of AURKA transcription. By contrast, the studies hinting that AURKA translation rate might change over the cell cycle are primarily based on quantification of the nascent protein, which could perhaps report on translational status in a more accurate manner in some contexts. Overall, while we do not know precisely when AURKA translation is activated in interphase, we believe it is not up-regulated but either constant or down-regulated in M phase compared with S and/or G2.
The general decrease in translation observed in M phase typically applies to cap-dependent translation, which is thought to be the preferred mechanism of protein synthesis, while Internal Ribosome Entry Site (IRES)-dependent translation is proposed to take over in this phase [156]. Assays with bicistronic RNA constructs revealed that AURKA 5′UTR contains an IRES element, whose activity is regulated in a cell cycle-dependent manner and peaks at G2/M phase in immortalized and cancer cells only, where AURKA cap-dependent translation remained unchanged [16]. Because there is no evidence so far that AURKA translation is enhanced in M phase, when IRES-mediated translation is thought to be most active, IRES-dependent translational activation of AURKA could be decoupled from generic IRES-dependent mitotic translation and could exclusively be relevant as a mechanism for AURKA overexpression in cancer [134]. What sequence or structural element precisely constitutes the AURKA IRES, and whether it has a physiological role in regulating AURKA expression, are still open questions.
5.2. Molecular mechanisms of AURKA translation
Molecular mechanisms controlling AURKA translation (figure 2b) have been uncovered in studies that only focus on pathological contexts, consequently giving insights into how translation contributes to expression of oncogenic AURKA.
In a search for mechanisms underlying enhanced AURKA protein expression in breast cancer, one report found that none of the processes of transcription, mRNA stability, cap-dependent translation and protein stability were responsible for overexpression in some immortalized and tumorigenic breast cell lines [16]. In these, activity of AURKA IRES element was found to be positively correlated with its protein levels, suggesting that a switch from cap- to IRES-dependent translation contributes to overexpression of AURKA at the level of translation, probably marking an early event during cancer progression. It is however not known what might cause the switch.
Another study reported that heterogeneous nuclear Ribo-Nucleoprotein Particle Q1 (hnRNP Q1), which is overexpressed in colorectal cancer and can promote cell proliferation, translationally up-regulates AURKA both in cap- and IRES-dependent manner via binding to AURKA 5′UTR [17]. hnRNP Q1 may also regulate AURKA protein expression in a cell cycle-dependent manner, since silencing of hnRNP Q1 decreased mitosis-dependent AURKA expression, and hnRNP Q1 overexpression increased AURKA abundance at G2/M phase. However, as this was only assessed by western blot, and since AURKA translation in M phase is likely not sustained, it is not excluded that hnRNP Q1 increases AURKA protein levels in M phase via a different mechanism than translational regulation. Additionally, the study confirmed that the activity of AURKA mRNA IRES was elevated in G2/M phase compared to G1/S phase in a cancer cell line (see above) and showed that the AURKA mRNA 5′UTR variants containing exon II bore stronger IRES activity than the variants containing exon I only. Because overexpression of hnRNP Q1 positively correlated with AURKA overexpression in human colorectal cancer tissues, the authors suggest that hnRNP Q1 may contribute to the tumorigenesis of colorectal cancer via AURKA translational up-regulation.
An earlier report from the same group had shown that translation of AURKA mRNA is up-regulated downstream of EGF signalling in EGFR-overexpressed colorectal cancer, as pulse-chase assays confirmed increased de novo AURKA protein synthesis and AURKA mRNA was found more associated with the ribosomal S6 protein upon EGF treatment [11]. The study also demonstrated that the PI3 K/Akt/mTOR and MEK/ERK pathways mediated the EGF-induced translational up-regulation of AURKA, and that 5′UTR splice variants containing exon II were critical for such up-regulation (see previous section). The pathway of translational upregulation downstream of EGF/EGFR signalling seems to exist in addition to the nuclear EGF/EGFR pathway that upregulates AURKA transcription [32]. Interestingly, a follow-up study found that hnRNP Q1 may be the factor that links EGF/EGFR signalling to AURKA translation [157], since treatment with EGF enhanced binding of hnRNP Q1 to AURKA mRNA, as well as the activity of hnRNP Q1 in.inducing AURKA translation. In addition, the mTOR and ERK pathways mediated hnRNP Q1-induced translation of AURKA mRNA upon EGF treatment.
Altogether, the regulation of AURKA translation may be much more complex than initially thought. As discussed so far, AURKA UTRs bear different elements that control expression at the level of translation, the role of many of such sequence and structural motifs, including miRNA binding sites, RNA-Binding Proteins (RBPs) recognition sites and RNA modifications in the regulation of AURKA translation are unexplored to this date. Fortunately, several in vivo methods are being developed recently, such as Translating RNA Imaging by Coat Protein Knock-off (TRICK) [158] or Nascent Chain Tracking (NCT) techniques [159], that track translation with high temporal and spatial precision and allow to probe the functional interaction between mRNA sequence elements and potential regulators.
6. Integrated temporal view of AURKA expression
The regulatory steps of transcription, post-transcription, translation and post-translation combine to confer AURKA gene its characteristic cell cycle-dependent pattern of expression (figure 3). In the late 1990 s, initial research addressed the trend of AURKA protein expression during the cell cycle in mammalian and human cells, although only qualitatively [19,23,93]. In parallel, first studies on AURKA protein degradation elucidated that, at the end of M phase, AURKA protein is maximally degraded by the APC/C linked to the Ubiquitin Proteasome System (UPS) [160]. The pattern of AURKA expression during the cell cycle was later confirmed quantitatively [161], and DNA microarray analyses consistently found AURKA in the G2/M cluster of transcriptionally co-regulated genes [56,67,162]. Most recent analysis of time-resolved profiling of the cell cycle transcriptome using the FUCCI system confirmed AURKA to be downregulated during M/G1 transition [57]. AURKA is also among the genes with the highest cell cycle periodicity [56,66,94]. However, how transcription combines with mRNA stability, translation and protein dynamics to control AURKA's pattern of expression is not fully understood, especially in terms of the extent and the timing of the individual contributory mechanisms.
Figure 3.

Different stages of gene expression integrate into AURKA temporal expression. Activation of AURKA transcription and protein degradation are likely the drivers of the respective increase and decrease in AURKA protein levels during the cell cycle. Control of mRNA stabilization also contributes to AURKA expression pattern, whereas the precise timing and extent of AURKA translational activation and translational inhibition are not yet clear. Figure created using BioRender.com.
There is consolidated evidence that AURKA protein and mRNA levels are extremely low in G1 phase and start accumulating in S phase, to then peak at G2/M (figure 3). Indeed, S is the phase where AURKA transcription is switched on first by de-repression and then by activation (see above). For this, transcription is a key contributor to increasing AURKA levels during S until M. Mechanisms of mRNA stabilization might intervene in this phase to also facilitate the increase in mRNA abundance. Accordingly, Battich et al. [163] categorized AURKA amongst genes whose mRNA abundance results from a cooperative strategy between the rate of synthesis and that of degradation of mRNA, that is, increase in synthesis rate accompanied by a decrease in degradation rate, and vice versa. As soon as AURKA mRNA is transcribed and fully processed, translation initiates. Because the de novo transcribed AURKA mRNA copies can already appear in G1 [161], this could be the earliest time in which translation can begin. Although it has not been entirely proven, at this time translation of AURKA mRNA is probably only basal, to be actively enhanced later in S phase [146]. Once translation initiates, the contribution of the rate of protein stabilization must be considered as well, although the regulation of AURKA proteolysis in interphase is less understood compared with its APC/CFZR1-mediated degradation in M phase [164]. However, because the very peak of AURKA expression is reached only in late G2 (i.e. not immediately after the activation of transcription), eventual mechanisms of mRNA stabilization, translational enhancement and protein stabilization may altogether contribute less than transcription to rising AURKA levels in S and G2.
During M phase the whole program of AURKA expression changes abruptly to a shutdown, as the aim for the dividing cell now is to irreversibly eliminate mitotic AURKA activity in a very short time. This in fact represents a tightly regulated step during exit from M phase. Ubiquitin-mediated proteolysis is important for the decline in AURKA protein levels during this time [164]. AURKA degradation in M phase is strictly dependent upon the FZR1-activated version of APC/C [165,166]. Furthermore, AURKA transcription is turned off during M phase. While transcriptional arrest eventually contributes to a drop in mRNA levels, turnover of the existing pool of AURKA mRNAs must be accelerated. It is reasonable to assume that AURKA mRNA undergoes canonical mRNA decay pathways, however very little is known about what controls AURKA mRNA stability. By investigating the hypothesis that mRNA decay might be important to reset cell cycle gene expression at mitotic exit, similarly to timed protein degradation, Krenning et al. [57] identified two temporal ‘waves’ of mRNA decline during M-G1: one of immediate decrease that initiates during anaphase and one of delayed decrease set off during early G1. AURKA mRNA was found in the delayed decrease group, as its levels start to decline 1–4 h after the start of G1 with a computed half-life of about 40 min in this phase. The fact that the half-life of AURKA mRNA during this cell cycle window is shorter than that measured in asynchronously growing cells [44,57,63] suggests programmed mRNA degradation in early G1.
In sum, just as activation of transcription is the driver for the increasing AURKA levels in early S, activation of protein degradation is key to disappearance of AURKA protein in M. However, S phase transcription is assisted by mechanisms favouring mRNA stabilization, translation and protein stabilization, which become more prominent as the cell progresses through G2, whereas mitosis-dependent proteolysis is accompanied by the shutdown of transcription and of translation and enhancement of mRNA decay. These latter events seem to remain in place in G1 even after AURKA proteolysis is concluded, so that the absence or low levels of AURKA are ensured in early interphase of the daughter cells. Except for transcription and proteolysis, the molecular mechanisms responsible for the activation/repression switch of translation and mRNA decay have to date never been explored.
7. Conclusion
This literature review highlights how AURKA expression is tightly regulated at multiple levels to adopt a pattern strictly correlated to the cell cycle. Needless to say, understanding the mechanisms regulating AURKA gene expression is an important quest in the study of the eukaryotic cell cycle itself. At the transcriptional level, AURKA is controlled by several molecular mechanisms and transcription factor complexes, which ensure its timely activation in S phase and repression in M phase. AURKA promoter also responds to external stimuli such as oxygen levels and presence of growth factors. Post-transcriptionally, splicing of the 5′UTR accounts for the existence of at least 16 different mRNA isoforms, and alternative cleavage and polyadenylation generates two different 3′UTR isoforms. Only now are we starting to gain an understanding of how these isoforms control both AURKA mRNA and protein dynamics, and how they are involved in disease. Translation of AURKA remains a less explored step of gene expression, but recent analyses of cell cycle translatomes raise our expectations of the existence of active translational regulatory mechanisms for AURKA mRNA. Not to mention the increasingly recognized role of translational dysregulation in disease, some examples of which have been already reported for AURKA. Even though we could qualitatively assess the contribution of DNA, mRNA, and protein dynamics to the cell cycle-dependent expression of AURKA, several questions remain to be addressed. On the one hand, the cell cycle pattern of AURKA expression follows that of many other cell cycle genes, especially those with temporally overlapping cell cycle functions. On the other hand, some mechanisms of post-transcriptional control, for example, AS and alternative polyadenylation, might be exclusive to AURKA mRNA. Post-transcriptional regulatory events may specifically occur in interphase or in non-cycling cells, where AURKA exerts functions not related to cell division. It is plausible to state that the definition of AURKA as a ‘G2/M’ gene only refers to its pattern of gene expression in dividing cells, not to its period of activity. It is now broadly accepted that AURKA plays other physiological cell- and tissue-specific roles that are independent of mitosis (e.g. occur in G1 or S), of protein abundance (e.g. occur when AURKA expression is low) and of cellular proliferation rate (e.g. occur in non-dividing cells). Moreover, it is possible that there exist some non-pathological dividing cell populations in which AURKA is not classifiable as ‘G2/M’. Addressing the open questions highlighted in this review will be crucial to discern which stage of gene expression would be more efficient to target when the aim is to correct AURKA abundance in cancers where it is altered. Quantitatively and qualitatively aberrant AURKA expression in patients with different cancers have been observed at all levels of gene expression. This can only motivate deeper investigations into AURKA expression.
Acknowledgements
We are grateful to Giulia Guarguaglini and Hesna Begum Akman for insightful comments on this manuscript, and to Lindon Lab members for enriching discussions. We also thank Daniel Dominguez for sharing unpublished data.
Contributor Information
Roberta Cacioppo, Email: rc781@cam.ac.uk.
Catherine Lindon, Email: acl34@cam.ac.uk.
Data accessibility
This article has no additional data.
Authors' contributions
R.C.: writing—original draft, writing—review and editing; C.L.: resources, writing—review and editing.
Both authors gave final approval for publication and agreed to be held accountable for the work performed therein.
Conflict of interest declaration
We declare we have no competing interests.
Funding
R.C. is funded by a David James Studentship from the Department of Pharmacology. Research on AURKA in C.L.'s lab has been supported by Cancer Research UK (grant no. A10239) and BBSRC (grant no. BB/R004137/1).
References
- 1.Glover DM, Leibowitz MH, McLean DA, Parry H. 1995. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell 81, 95-105. ( 10.1016/0092-8674(95)90374-7) [DOI] [PubMed] [Google Scholar]
- 2.Willems E, Dedobbeleer M, Digregorio M, Lombard A, Lumapat PN, Rogister B. 2018. The functional diversity of Aurora kinases: a comprehensive review. Cell Div. 13, 1-17. ( 10.1186/s13008-018-0040-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bavetsias V, Linardopoulos S. 2015. Aurora kinase inhibitors: current status and outlook. Front. Oncol. 5, 1-10. ( 10.3389/fonc.2015.00278) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang N, et al. 2017. FOXM1 recruits nuclear Aurora kinase A to participate in a positive feedback loop essential for the self-renewal of breast cancer stem cells. Oncogene 36, 3428-3440. ( 10.1038/onc.2016.490) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng F, et al. 2016. Nuclear AURKA acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nat. Commun. 7, 1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Büchel G, et al. 2017. Association with Aurora-A controls N-MYC-dependent promoter escape and pause release of RNA polymerase II during the cell cycle. Cell Rep. 21, 3483-3497. ( 10.1016/j.celrep.2017.11.090) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naso FD, Boi D, Ascanelli C, Pamfil G, Lindon C, Paiardini A, Guarguaglini G. 2021. Nuclear localisation of Aurora-A: its regulation and significance for Aurora-A functions in cancer. Oncogene 40, 3917-3928. ( 10.1038/s41388-021-01766-w) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanaka H, Nakashiro KI, Iwamoto K, Tokuzen N, Fujita Y, Shirakawa R, Oka R, Goda H, Hamakawa H. 2013. Targeting Aurora kinase A suppresses the growth of human oral squamous cell carcinoma cells in vitro and in vivo. Oral Oncol. 49, 551-559. ( 10.1016/j.oraloncology.2013.02.002) [DOI] [PubMed] [Google Scholar]
- 9.Gritsko TM, Coppola D, Paciga JE, Yang L, Sun M, Shelley SA, Fiorica JV, Nicosia SV, Cheng JQ. 2003. Activation and overexpression of centrosome kinase BTAK/Aurora-A in human ovarian cancer. Clin. Cancer Res. 9, 1420-1426. [PubMed] [Google Scholar]
- 10.Jeng YM, Peng SY, Lin CY, Hsu HC. 2004. Overexpression and amplification of Aurora-A in hepatocellular carcinoma. Clin. Cancer Res. 10, 2065-2071. ( 10.1158/1078-0432.CCR-1057-03) [DOI] [PubMed] [Google Scholar]
- 11.Lai CH, et al. 2010. Translational up-regulation of Aurora-A in EGFR-overexpressed cancer. J. Cell. Mol. Med. 14, 1520-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitajima S, Kudo Y, Ogawa I, Tatsuka M, Kawai H, Pagano M, Takata T. 2007. Constitutive phosphorylation of Aurora-A on Ser51 induces its stabilization and consequent overexpression in cancer. PLoS ONE 2, e944. ( 10.1371/journal.pone.0000944) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D'Assoro AB, et al. 2014. The mitotic kinase aurora-A promotes distant metastases by inducing epithelial-to-mesenchymal transition in ER + breast cancer cells. Oncogene 33, 599-610. ( 10.1038/onc.2012.628) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abdelbaki A, Akman HB, Poteau M, Grant R, Gavet O, Guarguaglini G, Lindon C. 2020. AURKA destruction is decoupled from its activity at mitotic exit but is essential to suppress interphase activity. J. Cell Sci. 133, jcs243071. ( 10.1242/jcs.243071) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, Abel PW, Tu Y. 2013. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 32, 4814-4824. ( 10.1038/onc.2012.494) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dobson T, Chen J, Krushel LA. 2013. Dysregulating IRES-dependent translation contributes to overexpression of oncogenic aurora a kinase. Mol. Cancer Res. 11, 887-900. ( 10.1158/1541-7786.MCR-12-0707) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lai CH, Solesio ME, Pavlov EV. 2017. Translational upregulation of Aurora-A by hnRNP Q1 contributes to cell proliferation and tumorigenesis in colorectal cancer. Cell Death Dis. 8, 1-12. ( 10.1038/s41419-017-0042-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kimura M, Matsuda Y, Okumura K, Okano Y. 1998. Assignment of STK6 to human chromosome. Cytogenet. Cell Genet. 3–4, 201-203. [DOI] [PubMed] [Google Scholar]
- 19.Shindo M, et al. 1998. cDNA cloning, expression, subcellular localization, and chromosomal assignment of mammalian aurora homologues, aurora-related kinase (ARK) 1 and 2. Biochem. Biophys. Res. Commun. 244, 285-292. ( 10.1006/bbrc.1998.8250) [DOI] [PubMed] [Google Scholar]
- 20.Tanaka M, Ueda A, Kanamori H, Ideguchi H, Yang J, Kitajima S, Ishigatsubo Y. 2002. Cell-cycle-dependent regulation of human aurora A transcription is mediated by periodic repression of E4TF1. J. Biol. Chem. 277, 10 719-10 726. ( 10.1074/jbc.M108252200) [DOI] [PubMed] [Google Scholar]
- 21.Li J, Guo W, Xue W, Xu P, Deng Z, Zhang D, Zheng S, Qiu X. 2019. Long noncoding RNA AURKAPS1 potentiates malignant hepatocellular carcinoma progression by regulating miR-142, miR-155 and miR-182. Sci. Rep. 9, 1-11. ( 10.1038/s41598-018-37186-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bischoff JR, et al. 1998. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 17, 3052-3065. ( 10.1093/emboj/17.11.3052) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kimura M, Kotani S, Hattori T, Sumi N, Yoshioka T, Todokoro K, Okano Y. 1997. Cell cycle-dependent expression and spindle pole localization of a novel human protein kinase, aik, related to aurora of Drosophila and yeast Ipl1. J. Biol. Chem. 272, 13 766-13 771. ( 10.1074/jbc.272.21.13766) [DOI] [PubMed] [Google Scholar]
- 24.Li C, Kato M, Shiue L, Shively JE, Ares M Jr, Lin RJ. 2006. Cell type and culture condition-dependent alternative splicing in human breast cancer cells revealed by splicing-sensitive microarrays. Cancer Res. 66, 1990-1999. ( 10.1158/0008-5472.CAN-05-2593) [DOI] [PubMed] [Google Scholar]
- 25.Bertolin G, Tramier M. 2020. Insights into the non-mitotic functions of Aurora kinase A: more than just cell division. Cell. Mol. Life Sci. 77, 1031-1047. ( 10.1007/s00018-019-03310-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mori D, Yamada M, Mimori-Kiyosue Y, Shirai Y, Suzuki A, Ohno S, Saya H, Wynshaw-Boris A, Hirotsune S. 2009. An essential role of the aPKC-Aurora A-NDEL1 pathway in neurite elongation by modulation of microtubule dynamics. Nat. Cell Biol. 11, 1057-1068. ( 10.1038/ncb1919) [DOI] [PubMed] [Google Scholar]
- 27.Sjöstedt E, et al. 2020. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 367, eaay5947. ( 10.1126/science.aay5947) [DOI] [PubMed] [Google Scholar]
- 28.Alexandra K, Flügel D, Kietzmann T. 2008. Transcriptional regulation of STK15 expression by hypoxia and HIF-1. Mol. Biol. Cell 19, 3667-3675. ( 10.1091/mbc.e08-01-0042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui SY, Huang JY, Chen YT, Song HZ, Huang GC, De W, Wang R, Chen LB. 2013. The role of Aurora A in hypoxia-inducible factor 1α-promoting malignant phenotypes of hepatocelluar carcinoma. Cell Cycle 12, 2849-2866. ( 10.4161/cc.25916) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fanale D, Bazan V, Corsini LR, Caruso S, Insalaco L, Castiglia M, Cicero G, Bronte G, Russo A. 2013. HIF-1 is involved in the negative regulation of AURKA expression in breast cancer cell lines under hypoxic conditions. Breast Cancer Res. Treat. 140, 505-517. ( 10.1007/s10549-013-2649-0) [DOI] [PubMed] [Google Scholar]
- 31.Xu J, et al. 2010. VHL inactivation induces HEF1 and Aurora kinase A. J. Am. Soc. Nephrol. 21, 2041-2046. ( 10.1681/ASN.2010040345) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hung LY, Tseng JT, Lee YC, Xia W, Wang YN, Wu ML, Chuang YH, Lai CH, Chang W. 2008. Nuclear epidermal growth factor receptor (EGFR) interacts with signal transducer and activator of transcription 5 (STAT5) in activating Aurora-A gene expression. Nucleic Acids Res. 36, 4337-4351. ( 10.1093/nar/gkn417) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo Y, Ma J, Zheng Y, Li L, Gui X, Wang Q, Meng X, Shang H. 2016. HPV16 E6 upregulates Aurora A expression. Oncol. Lett. 12, 1387-1393. ( 10.3892/ol.2016.4786) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wakahara K, et al. 2008. EWS-Fli1 up-regulates expression of the Aurora A and Aurora B kinases. Mol. Cancer Res. 6, 1937-1945. ( 10.1158/1541-7786.MCR-08-0054) [DOI] [PubMed] [Google Scholar]
- 35.Shin SO, Lee KH, Kim JH, Baek SH, Park JW, Gabrielson EW, Kwon TK. 2000. Alternative splicing in 5′-untranslational region of STK-15 gene, encoding centrosome associated kinase, in breast cancer cell lines. Exp. Mol. Med. 32, 193-196. ( 10.1038/emm.2000.31) [DOI] [PubMed] [Google Scholar]
- 36.Dago DN, et al. 2015. Estrogen receptor beta impacts hormone-induced alternative mRNA splicing in breast cancer cells. BMC Genomics 16, 1-13. ( 10.1186/1471-2164-16-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Corrionero A, Miñana B, Valcárcel J. 2011. Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev. 25, 445-459. ( 10.1101/gad.2014311) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dolatshad H, et al. 2015. Disruption of SF3B1 results in deregulated expression and splicing of key genes and pathways in myelodysplastic syndrome hematopoietic stem and progenitor cells. Leukemia 29, 1092-1103. ( 10.1038/leu.2014.331) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bergot T, Lippert E, Douet-Guilbert N, Commet S, Corcos L, Bernard DG. 2020. Human cancer-associated mutations of SF3B1 lead to a splicing modification of its own RNA. Cancers (Basel). 12, 1-16. ( 10.3390/cancers12030652) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pawellek A, McElroy S, Samatov T, Mitchell L, Woodland A, Ryder U, Gray D, Lührmann R, Lamond AI. 2014. Identification of small molecule inhibitors of pre-mRNA splicing. J. Biol. Chem. 289, 34683-34 698. ( 10.1074/jbc.M114.590976) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, et al. 2020. Dissecting the heterogeneity of the alternative polyadenylation profiles in triple-negative breast cancers. Theranostics 10, 10 531-10 547. ( 10.7150/thno.40944) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu J, et al. 2013. Aurora-A identifies early recurrence and poor prognosis and promises a potential therapeutic target in triple negative breast cancer. PLoS ONE 8, 1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lembo A, Di Cunto F, Provero P. 2012. Shortening of 3′UTRs correlates with poor prognosis in breast and lung cancer. PLoS ONE 7, e31129. ( 10.1371/journal.pone.0031129) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nowak I, Boratyn E, Student S, Bernhart SF, Fallmann J, Durbas M, Stadler PF, Rokita H. 2021. MCPIP1 ribonuclease can bind and cleave AURKA mRNA in MYCN-amplified neuroblastoma cells. RNA Biol. 18, 144-156. ( 10.1080/15476286.2020.1804698) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Glaß M, Misiak D, Bley N, Müller S, Hagemann S, Busch B, Rausch A, Hüttelmaier S. 2021. IGF2BP1, a conserved regulator of RNA turnover in cancer. Front. Mol. Biosci. 8, 1-16. ( 10.3389/fmolb.2021.632219) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fadaka AO, Sibuyi NRS, Madiehe AM, Meyer M. 2020. MicroRNA-based regulation of Aurora A kinase in breast cancer. Oncotarget 11, 4306-4324. ( 10.18632/oncotarget.27811) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang H, Bao J, Zhao S, Huo Z, Li B. 2020. MicroRNA-490-3p suppresses hepatocellular carcinoma cell proliferation and migration by targeting the aurora kinase A gene (AURKA). Arch. Med. Sci. 16, 395-406. ( 10.5114/aoms.2019.91351) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuan YL, Yu H, Mu SM, Dong YD, Li DY. 2019. MiR-26a-5p inhibits cell proliferation and enhances doxorubicin sensitivity in HCC cells via targeting AURKA. Technol. Cancer Res. Treat. 18, 1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma ZL, Zhang BJ, Wang DT, Li X, Wei JL, Zhao BT, Jin Y, Li YL, Jin YX. 2015. Tanshinones suppress AURKA through up-regulation of miR-32 expression in non-small cell lung cancer. Oncotarget 6, 20 111-20 120. ( 10.18632/oncotarget.3933) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watanabe H, Sawada JI, Yano KI, Yamaguchi K, Goto M, Handa H. 1993. cDNA cloning of transcription factor E4TF1 subunits with Ets and notch motifs. Mol. Cell. Biol. 13, 1385-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Udayakumar TS, Belakavadi M, Choi KH, Pandey PK, Fondell JD. 2006. Regulation of Aurora-A kinase gene expression via GABP recruitment of TRAP220/MED1. J. Biol. Chem. 281, 14 691-14 699. ( 10.1074/jbc.M600163200) [DOI] [PubMed] [Google Scholar]
- 52.Imaki H, Nakayama K, Delehouzee S, Handa H, Kitagawa M, Kamura T, Nakayama KI. 2003. Cell cycle-dependent regulation of the Skp2 promoter by GA-binding protein. Cancer Res. 63, 4607-4613. [PubMed] [Google Scholar]
- 53.Zwicker J, Lucibello FC, Wolfraim LA, Gross C, Truss M, Engeland K, Müller R. 1995. Cell cycle regulation of the cyclin A, cdc25C and cdc2 genes is based on a common mechanism of transcriptional repression. EMBO J. 14, 4514-4522. ( 10.1002/j.1460-2075.1995.tb00130.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uchiumi T, Longo DL, Ferris DK. 1997. Cell cycle regulation of the human polo-like kinase (PLK) promoter. J. Biol. Chem. 272, 9166-9174. ( 10.1074/jbc.272.14.9166) [DOI] [PubMed] [Google Scholar]
- 55.Müller GA, Engeland K. 2010. The central role of CDE/CHR promoter elements in the regulation of cell cycle-dependent gene transcription: review article. FEBS J. 277, 877-893. ( 10.1111/j.1742-4658.2009.07508.x) [DOI] [PubMed] [Google Scholar]
- 56.Whitfield ML, et al. 2002. Human cell cycle and their expression in tumors. Mol. Biol. Cell 13, 1977-2000. ( 10.1091/mbc.02-02-0030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krenning L, Sonneveld S, Tanenbaum ME. 2021. Time-resolved single-cell sequencing identifies multiple waves of mRNA decay during mitotic exit. bioRxiv 2021.04.17.440266. [DOI] [PMC free article] [PubMed]
- 58.Osterloh L, von Eyss B, Schmit F, Rein L, Hübner D, Samans B, Hauser S, Gaubatz S. 2007. The human synMuv-like protein LIN-9 is required for transcription of G2/M genes and for entry into mitosis. EMBO J. 26, 144-157. ( 10.1038/sj.emboj.7601478) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Litovchick L, et al. 2007. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol. Cell 26, 539-551. ( 10.1016/j.molcel.2007.04.015) [DOI] [PubMed] [Google Scholar]
- 60.Reichert N, et al. 2010. Lin9, a subunit of the mammalian DREAM complex, is essential for embryonic development, for survival of adult mice, and for tumor suppression. Mol. Cell. Biol. 30, 2896-2908. ( 10.1128/MCB.00028-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fischer M, Müller GA. 2017. Cell cycle transcription control: DREAM/MuvB and RB-E2F complexes. Crit. Rev. Biochem. Mol. Biol. 52, 638-662. ( 10.1080/10409238.2017.1360836) [DOI] [PubMed] [Google Scholar]
- 62.Engeland K. 2018. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 25, 114-132. ( 10.1038/cdd.2017.172) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schwanhüusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. 2011. Global quantification of mammalian gene expression control. Nature 473, 337-342. ( 10.1038/nature10098) [DOI] [PubMed] [Google Scholar]
- 64.Down CF, Millour J, Lam EWF, Watson RJ. 2012. Binding of FoxM1 to G2/M gene promoters is dependent upon B-Myb. Biochim. Biophys. Acta - Gene Regul. Mech. 1819, 855-862. ( 10.1016/j.bbagrm.2012.03.008) [DOI] [PubMed] [Google Scholar]
- 65.Sadasivam S, Duan S, DeCaprio JA. 2012. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev. 26, 474-489. ( 10.1101/gad.181933.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu Y, et al. 2017. Transcriptional landscape of the human cell cycle. Proc. Natl Acad. Sci. USA 114, 3473-3478. ( 10.1073/pnas.1617636114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ishida S, Huang E, Zuzan H, Spang R, Leone G, West M, Nevins JR. 2001. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol. Cell. Biol. 21, 4684-4699. ( 10.1128/MCB.21.14.4684-4699.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Polager S, Kalma Y, Berkovich E, Ginsberg D. 2002. E2fs up-regulate expression of genes involved in DNA replication, DNA repair and mitosis. Oncogene 21, 437-446. ( 10.1038/sj.onc.1205102) [DOI] [PubMed] [Google Scholar]
- 69.Zhu W, Giangrande PH, Nevins JR. 2004. E2Fs link the control of G1/S and G2/M transcription. EMBO J. 23, 4615-4626. ( 10.1038/sj.emboj.7600459) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu CC, Yang TY, Yu CT, Phan L, Ivan C, Sood AK, Hsu SL, Lee MH. 2012. p53 negatively regulates Aurora A via both transcriptional and posttranslational regulation. Cell Cycle 11, 3433-3442. ( 10.4161/cc.21732) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hu Q, Lu JF, Luo R, Sen S, Maity SN. 2006. Inhibition of CBF/NF-Y mediated transcription activation arrests cells at G2/M phase and suppresses expression of genes activated at G2/M phase of the cell cycle. Nucleic Acids Res. 34, 6272-6285. ( 10.1093/nar/gkl801) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu CH, Tseng YS, Kao YT, Sheu HM, Liu HS. 2013. Low concentration of arsenic-induced aberrant mitosis in keratinocytes through E2F1 transcriptionally regulated aurora-A. Toxicol. Sci. 132, 43-52. ( 10.1093/toxsci/kfs322) [DOI] [PubMed] [Google Scholar]
- 73.Kao YT, Wu CH, Wu SY, Lan SH, Liu HS, Tseng YS. 2017. Arsenic treatment increase Aurora-A overexpression through E2F1 activation in bladder cells. BMC Cancer 17, 1-10. ( 10.1186/s12885-016-3022-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kabe Y, Yamada J, Uga H, Yamaguchi Y, Wada T, Handa H. 2005. NF-Y is essential for the recruitment of RNA polymerase II and inducible transcription of several CCAAT box-containing genes. Mol. Cell. Biol. 25, 512-522. ( 10.1128/MCB.25.1.512-522.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Laoukili J, Alvarez-Fernandez M, Stahl M, Medema RH. 2008. FoxM1 is degraded at mitotic exit in a Cdh1-dependent manner. Cell Cycle 7, 2720-2726. ( 10.4161/cc.7.17.6580) [DOI] [PubMed] [Google Scholar]
- 76.Imbriano C, et al. 2005. Direct p53 transcriptional repression: in vivo analysis of CCAAT-containing G 2 /M promoters. Mol. Cell. Biol. 25, 3737-3751. ( 10.1128/MCB.25.9.3737-3751.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Den Hollander J, et al. 2010. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood 116, 1498-1505. ( 10.1182/blood-2009-11-251074) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Courapied S, et al. 2010. Regulation of the Aurora-A gene following topoisomerase I inhibition: implication of the Myc transcription Factor. Mol. Cancer 9, 1-15. ( 10.1186/1476-4598-9-205) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Allis CD, Jenuwein T. 2016. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17, 487-500. ( 10.1038/nrg.2016.59) [DOI] [PubMed] [Google Scholar]
- 80.Zhang XH, et al. 2008. Aurora A, aurora B and survivin are novel targets of transcriptional regulation by histone deacetylase inhibitors in non-small cell lung cancer. Cancer Biol. Ther. 7, 1388-1397. ( 10.4161/cbt.7.9.6415) [DOI] [PubMed] [Google Scholar]
- 81.Gurtner A, Fuschi P, Magi F, Colussi C, Gaetano C, Dobbelstein M, Sacchi A, Piaggio G. 2008. NF-Y dependent epigenetic modifications discriminate between proliferating and postmitotic tissue. PLoS ONE 3, e2047. ( 10.1371/journal.pone.0002047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Salsi V, Caretti G, Wasner M, Reinhard W, Haugwitz U, Engeland K, Mantovani R. 2003. Interactions between p300 and multiple NF-Y trimers govern cyclin B2 promoter function. J. Biol. Chem. 278, 6642-6650. ( 10.1074/jbc.M210065200) [DOI] [PubMed] [Google Scholar]
- 83.Wu C, Lyu J, Yang EJ, Liu Y, Zhang B, Shim JS. 2018. Targeting AURKA-CDC25C axis to induce synthetic lethality in ARID1A-deficient colorectal cancer cells. Nat. Commun. 9, 1-4. ( 10.1038/s41467-017-02088-w) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang TY, Teng CL, Lin TC, Chen KC, Hsu SL, Wu CC. 2018. Transcriptional repression of Aurora-A gene by wild-type p53 through directly binding to its promoter with histone deacetylase 1 and mSin3a. Int. J. Cancer 142, 92-108. ( 10.1002/ijc.31035) [DOI] [PubMed] [Google Scholar]
- 85.Lee SJ, Cimica V, Ramachandra N, Zagzag D, Kalpana GV. 2011. Aurora A is a repressed effector target of the chromatin remodeling protein INI1/hSNF5 required for rhabdoid tumor cell survival. Cancer Res. 71, 3225-3235. [DOI] [PubMed] [Google Scholar]
- 86.Sasai K, Treekitkarnmongkol W, Kai K, Katayama H, Sen S. 2016. Functional significance of Aurora kinases-p53 protein family interactions in cancer. Front. Oncol. 6, 247. ( 10.3389/fonc.2016.00247) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Whately KM, et al. 2021. Nuclear Aurora-A kinase-induced hypoxia signaling drives early dissemination and metastasis in breast cancer: implications for detection of metastatic tumors. Oncogene 40, 5651-5664. ( 10.1038/s41388-021-01969-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang YX, Feige P, Brun CE, Hekmatnejad B, Dumont NA, Renaud JM, Faulkes S, Guindon DE, Rudnicki MA. 2019. EGFR-Aurka signaling rescues polarity and regeneration defects in dystrophin-deficient muscle stem cells by increasing asymmetric divisions. Cell Stem Cell 24, 419-432. ( 10.1016/j.stem.2019.01.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Boi D, et al. 2021. Pha-680626 is an effective inhibitor of the interaction between aurora-a and n-myc. Int. J. Mol. Sci. 22, 1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shah KN, et al. 2019. Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat. Med. 25, 111-118. ( 10.1038/s41591-018-0264-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Moore MJ, Wang Q, Kennedy CJ, Silver PA. 2010. An alternative splicing network links cell-cycle control to apoptosis. Cell 142, 625-636. ( 10.1016/j.cell.2010.07.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stoilov P, Lin CH, Damoiseaux R, Nikolic J, Black DL. 2008. A high-throughput screening strategy identifies cardiotonic steroids as alternative splicing modulators. Proc. Natl Acad. Sci. USA 105, 11 218-11 223. ( 10.1073/pnas.0801661105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gopalan G, Chan CSM, Donovan PJ. 1997. A novel mammalian, mitotic spindle-associated kinase is related to yeast and fly chromosome segregation regulators. J. Cell Biol. 138, 643-656. ( 10.1083/jcb.138.3.643) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dominguez D, Tsai YH, Weatheritt R, Wang Y, Blencowe BJ, Wang Z. 2016. An extensive program of periodic alternative splicing linked to cell cycle progression. Elife 5, 1-19. ( 10.7554/eLife.10288) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weatheritt RJ, Sterne-Weiler T, Blencowe BJ. 2016. The ribosome-engaged landscape of alternative splicing. Nat. Struct. Mol. Biol. 23, 1117-1123. ( 10.1038/nsmb.3317) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sterne-Weiler T, Martinez-Nunez RT, Howard JM, Cvitovik I, Katzman S, Tariq MA, Pourmand N, Sanford JR. 2013. Frac-seq reveals isoform-specific recruitment to polyribosomes. Genome Res. 23, 1615-1623. ( 10.1101/gr.148585.112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Maslon MM, Heras SR, Bellora N, Eyras E, Cáceres JF. 2014. The translational landscape of the splicing factor SRSF1 and its role in mitosis. Elife 3, e02028. ( 10.7554/eLife.02028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shin C, Manley JL. 2002. The SR protein SRp38 represses splicing in M phase cells. Cell 111, 407-417. ( 10.1016/S0092-8674(02)01038-3) [DOI] [PubMed] [Google Scholar]
- 99.Ahn EY, DeKelver RC, Lo MC, Nguyen TA, Matsuura S, Boyapati A, Pandit S, Fu XD, Zhang DE. 2011. SON controls cell-cycle progression by coordinated regulation of RNA splicing. Mol. Cell 42, 185-198. ( 10.1016/j.molcel.2011.03.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sharma A, Markey M, Torres-Munoz K, Varia S, Kadakia M, Bubulya A, Bubulya PA. 2011. Son maintains accurate splicing for a subset of human pre-mRNAs. J. Cell Sci. 124, 4286-4298. ( 10.1242/jcs.092239) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Druillennec S, Dorard C, Eychène A. 2012. Alternative splicing in oncogenic kinases: from physiological functions to cancer. J. Nucleic Acids 2012, 639062. ( 10.1155/2012/639062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Araki S, Dairiki R, Nakayama Y, Murai A, Miyashita R, Iwatani M, Nomura T, Nakanishi O. 2015. Inhibitors of CLK protein kinases suppress cell growth and induce apoptosis by modulating pre-mRNA splicing. PLoS ONE 10, 1-18. ( 10.1371/journal.pone.0116929) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Damodaran AP, et al. 2020. Aurora-A phosphorylates splicing factors and regulates alternative splicing. bioRxiv. ( 10.1101/2020.11.04.368498) [DOI]
- 104.Herrmann CJ, Schmidt R, Kanitz A, Artimo P, Gruber AJ, Zavolan M. 2020. PolyASite 2.0: a consolidated atlas of polyadenylation sites from 3′ end sequencing. Nucleic Acids Res. 48, D174-D179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Leung MKK, Delong A, Frey BJ. 2018. Inference of the human polyadenylation code. Bioinformatics 34, 2889-2898. ( 10.1093/bioinformatics/bty211) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li Z, et al. In press. DeeReCT-APA: prediction of alternative polyadenylation site usage through deep learning. Genomics, Proteomics & Bioinformatics. ( 10.1016/j.gpb.2020.05.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zheng D, Wang R, Ding Q, Wang T, Xie B, Wei L, Zhong Z, Tian B. 2018. Cellular stress alters 3′UTR landscape through alternative polyadenylation and isoform-specific degradation. Nat. Commun. 9, 1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hollerer I, Curk T, Haase B, Benes V, Hauer C, Neu-Yilik G, Bhuvanagiri M, Hentze MW, Kulozik AE. 2016. The differential expression of alternatively polyadenylated transcripts is a common stress-induced response mechanism that modulates mammalian mRNA expression in a quantitative and qualitative fashion. Rna 22, 1441-1453. ( 10.1261/rna.055657.115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Singh I, Lee SH, Sperling AS, Samur MK, Tai YT, Fulciniti M, Munshi NC, Mayr C, Leslie CS. 2018. Widespread intronic polyadenylation diversifies immune cell transcriptomes. Nat. Commun. 9, 1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gruber AR, et al. 2014. Global 3′ UTR shortening has a limited effect on protein abundance in proliferating T cells. Nat. Commun. 5, 1-10. ( 10.1038/ncomms6465) [DOI] [PubMed] [Google Scholar]
- 111.Chen M, et al. 2020. Erratum: 3′ UTR lengthening as a novel mechanism in regulating cellular senescence (Genome Research (2018) 28 (285–294) (doi:10.1101/gr.224451.117). Genome Res. 30, 1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xiang Y, et al. 2018. Comprehensive characterization of alternative polyadenylation in human cancer. J. Natl. Cancer Inst. 110, 379-389. ( 10.1093/jnci/djx223) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yang X, Wu J, Xu W, Tan S, Chen C, Wang X, Sun J, Kang Y. 2018. Genome-wide profiling reveals cancer-related genes with switched alternative polyadenylation sites in colorectal cancer. Onco. Targets. Ther. 11, 5349-5357. ( 10.2147/OTT.S164233) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lin Y, et al. 2012. An in-depth map of polyadenylation sites in cancer. Nucleic Acids Res. 40, 8460-8471. ( 10.1093/nar/gks637) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ji Z, Ju YL, Pan Z, Jiang B, Tian B. 2009. Progressive lengthening of 3′ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development (Proceedings of the National Academy of Sciences of the United States of America (2009) 106, 17, (7028–7033) (doi:10.1073/pnas.090). Proc. Natl Acad. Sci. USA 106, 9535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hoque M, Ji Z, Zheng D, Luo W, Li W, You B, Park JY, Yehia G, Tian B. 2013. Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat. Methods 10, 133-139. ( 10.1038/nmeth.2288) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tian B, Hu J, Zhang H, Lutz CS. 2005. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 33, 201-212. ( 10.1093/nar/gki158) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lianoglou S, Garg V, Yang JL, Leslie CS, Mayr C. 2013. Ubiquitously transcribed genes use alternative polyadenylation to achieve tissue-specific expression. Genes Dev. 27, 2380-2396. ( 10.1101/gad.229328.113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Akman HB, Oyken M, Tuncer T, Can T, Erson-Bensan AE. 2015. 3′UTR shortening and EGF signaling: implications for breast cancer. Hum. Mol. Genet. 24, 6910-6920. [DOI] [PubMed] [Google Scholar]
- 120.Nicholson AL, Pasquinelli AE. 2019. Tales of detailed poly(A) tails. Trends Cell Biol. 29, 191-200. ( 10.1016/j.tcb.2018.11.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Park JE, Yi H, Kim Y, Chang H, Kim VN. 2016. Regulation of poly(A) tail and translation during the somatic cell cycle. Mol. Cell 62, 462-471. ( 10.1016/j.molcel.2016.04.007) [DOI] [PubMed] [Google Scholar]
- 122.Lima SA, Chipman LB, Nicholson AL, Chen YH, Yee BA, Yeo GW, Coller J, Pasquinelli AE. 2017. Short poly(A) tails are a conserved feature of highly expressed genes. Nat. Struct. Mol. Biol. 24, 1057-1063. ( 10.1038/nsmb.3499) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rambout X, et al. 2016. The transcription factor ERG recruits CCR4-NOT to control mRNA decay and mitotic progression. Nat. Struct. Mol. Biol. 23, 663-672. ( 10.1038/nsmb.3243) [DOI] [PubMed] [Google Scholar]
- 124.Bueno MJ, Malumbres M. 2011. MicroRNAs and the cell cycle. Biochim. Biophys. Acta - Mol. Basis Dis. 1812, 592-601. ( 10.1016/j.bbadis.2011.02.002) [DOI] [PubMed] [Google Scholar]
- 125.Mayr C, Bartel DP. 2009. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138, 673-684. ( 10.1016/j.cell.2009.06.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang X, et al. 2014. N 6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117-120. ( 10.1038/nature12730) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dominissini D, et al. 2012. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201-206. ( 10.1038/nature11112) [DOI] [PubMed] [Google Scholar]
- 128.Fei Q, Zou Z, Roundtree IA, Sun HL, He C. 2020. YTHDF2 promotes mitotic entry and is regulated by cell cycle mediators. PLoS Biol. 18, 1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Berkovits BD, Mayr C. 2015. Alternative 3′ UTRs act as scaffolds to regulate membrane protein localization. Nature 522, 363-367. ( 10.1038/nature14321) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ni TK, Kuperwasser C. 2016. Premature polyadenylation of MAGI3 produces a dominantly-acting oncogene in human breast cancer. Elife 5, 1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gillen AE, Brechbuhl HM, Yamamoto TM, Kline E, Pillai MM, Hesselberth JR, Kabos P. 2017. Alternative polyadenylation of PRELID1 regulates mitochondrial ros signaling and cancer outcomes. Mol. Cancer Res. 15, 1741-1751. ( 10.1158/1541-7786.MCR-17-0010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lau AG, et al. 2010. Distinct 3′UTRs differentially regulate activity-dependent translation of brain-derived neurotrophic factor (BDNF). Proc. Natl Acad. Sci. USA 107, 15 945-15 950. ( 10.1073/pnas.1002929107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Xu Y, Ruggero D. 2020. The role of translation control in tumorigenesis and its therapeutic implications. Annu. Rev. Cancer Biol. 4, 437-457. ( 10.1146/annurev-cancerbio-030419-033420) [DOI] [Google Scholar]
- 134.Sriram A, Bohlen J, Teleman AA. 2018. Translation acrobatics: how cancer cells exploit alternate modes of translational initiation. EMBO Rep. 19, e45947. ( 10.15252/embr.201845947) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Vislovukh A. 2014. Role of 3′-untranslated region translational control in cancer development, diagnostics and treatment. World J. Biol. Chem. 5, 40. ( 10.4331/wjbc.v5.i1.40) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhao J, Qin B, Nikolay R, Spahn CMT, Zhang G. 2019. Translatomics: the global view of translation. Int. J. Mol. Sci. 20, 212. ( 10.3390/ijms20010212) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Anda S, Grallert B. 2019. Cell-cycle-dependent regulation of translation: new interpretations of old observations in light of new approaches. BioEssays 41, 1-7. ( 10.1002/bies.201900022) [DOI] [PubMed] [Google Scholar]
- 138.Kronja I, Orr-Weaver TL. 2011. Translational regulation of the cell cycle: when, where, how and why? Phil. Trans. R. Soc. B 366, 3638-3652. ( 10.1098/rstb.2011.0084) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fan H, Penman S. 1970. Regulation of protein synthesis in mammalian cells. J. Mol. Biol. 50, 655-670. ( 10.1016/0022-2836(70)90091-4) [DOI] [PubMed] [Google Scholar]
- 140.Tarnowka MA, Baglioni C. 1979. Regulation of protein synthesis in mitotic HeLa cells. J. Cell. Physiol. 99, 359-367. ( 10.1002/jcp.1040990311) [DOI] [PubMed] [Google Scholar]
- 141.Bonneau AM, Sonenberg N. 1987. Involvement of the 24-kDa cap-binding protein in regulation of protein synthesis in mitosis. J. Biol. Chem. 262, 11 134-11 139. ( 10.1016/S0021-9258(18)60935-4) [DOI] [PubMed] [Google Scholar]
- 142.Qin X, Sarnow P. 2004. Preferential translation of internal ribosome entry site-containing mRNAs during the mitotic cycle in mammalian cells. J. Biol. Chem. 279, 13 721-13 728. ( 10.1074/jbc.M312854200) [DOI] [PubMed] [Google Scholar]
- 143.Wilker EW, et al. 2007. 14-3-3Σ controls mitotic translation to facilitate cytokinesis. Nature 446, 329-332. ( 10.1038/nature05584) [DOI] [PubMed] [Google Scholar]
- 144.Sivan G, Kedersha N, Elroy-Stein O. 2007. Ribosomal slowdown mediates translational arrest during cellular division. Mol. Cell. Biol. 27, 6639-6646. ( 10.1128/MCB.00798-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Aviner R, Geiger T, Elroy-Stein O. 2013. Novel proteomic approach (PUNCH-P) reveals cell cycle-specific fluctuations in mRNA translation. Genes Dev. 27, 1834-1844. ( 10.1101/gad.219105.113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Aviner R, Shenoy A, Elroy-Stein O, Geiger T. 2015. Uncovering hidden layers of cell cycle regulation through integrative multi-omic analysis. PLoS Genet. 11, 1-23. ( 10.1371/journal.pgen.1005554) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tanenbaum ME, Stern-Ginossar N, Weissman JS, Vale RD. 2015. Regulation of mRNA translation during mitosis. Elife 4, 1-19. ( 10.7554/eLife.07957) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Stumpf CR, Moreno MV, Olshen AB, Taylor BS, Ruggero D. 2013. The translational landscape of the mammalian cell cycle. Mol. Cell 52, 574-582. ( 10.1016/j.molcel.2013.09.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Stonyte V, Boye E, Grallert B. 2018. Regulation of global translation during the cell cycle. J. Cell Sci. 131, 1-9. [DOI] [PubMed] [Google Scholar]
- 150.Imami, K., Milek M, Bogdanow B, Yasuda T, Kastelic N, Zauber H, Ishihama Y, Landthaler M, Selbach M. 2018. Phosphorylation of the ribosomal protein RPL12/uL11 affects translation during mitosis. Mol. Cell 72, 84-98. ( 10.1016/j.molcel.2018.08.019) [DOI] [PubMed] [Google Scholar]
- 151.Clark IE, Wyckoff D, Gavis ER. 2000. Synthesis of the posterior determinant Nanos is spatially restricted by a novel cotranslational regulatory mechanism. Curr. Biol. 10, 1311-1314. ( 10.1016/S0960-9822(00)00754-5) [DOI] [PubMed] [Google Scholar]
- 152.Nottrott S, Simard MJ, Richter JD. 2006. Human let-7a miRNA blocks protein production on actively translating polyribosomes. Nat. Struct. Mol. Biol. 13, 1108-1114. ( 10.1038/nsmb1173) [DOI] [PubMed] [Google Scholar]
- 153.Petersen CP, Bordeleau ME, Pelletier J, Sharp PA. 2006. Short RNAs repress translation after initiation in mammalian cells. Mol. Cell 21, 533-542. ( 10.1016/j.molcel.2006.01.031) [DOI] [PubMed] [Google Scholar]
- 154.Neelagandan N, Lamberti I, Carvalho HJF, Gobet C, Naef F. 2020. What determines eukaryotic translation elongation: recent molecular and quantitative analyses of protein synthesis: determinants of eukaryotic translation. Open Biol. 10, 200292. ( 10.1098/rsob.200292) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218-223. ( 10.1126/science.1168978) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Komar AA, Hatzoglou M. 2011. Cellular IRES-mediated translation: the war of ITAFs in pathophysiological states. Cell Cycle 10, 229-240. ( 10.4161/cc.10.2.14472) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wang YC, et al. 2018. The EGF/hnRNP Q1 axis is involved in tumorigenesis via the regulation of cell cycle-related genes. Exp. Mol. Med. 50, 1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Halstead JM, Lionnet T, Wilbertz JH, Wippich F, Ephrussi A, Singer RH, Chao JA. 2015. An RNA biosensor for imaging the first round of translation from single cells to living animals. Science 347, 1367-1370. ( 10.1126/science.aaa3380) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Biswas J, Liu Y, Singer RH, Wu B. 2019. Fluorescence imaging methods to investigate translation in single cells. Cold Spring Harb. Perspect. Biol. 11, a032722. ( 10.1101/cshperspect.a032722) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Honda K, Mihara H, Kato Y, Yamaguchi A, Tanaka H, Yasuda H, Furukawa K, Urano T. 2000. Degradation of human Aurora2 protein kinase by the anaphase-promoting complex-ubiquitin-proteasome pathway. Oncogene 19, 2812-2819. ( 10.1038/sj.onc.1203609) [DOI] [PubMed] [Google Scholar]
- 161.Lin Y, et al. 2006. Gene expression profiles of the aurora family kinases. Gene Expr. 13, 15-26. ( 10.3727/000000006783991962) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Crawford DF, Piwnica-Worms H. 2001. The G2 DNA damage checkpoint delays expression of genes encoding mitotic regulators. J. Biol. Chem. 276, 37 166-37 177. ( 10.1074/jbc.M103414200) [DOI] [PubMed] [Google Scholar]
- 163.Battich N, Beumer J, de Barbanson B, Krenning L, Baron CS, Tanenbaum ME, Clevers H, van Oudenaarden A. 2020. Sequencing metabolically labeled transcripts in single cells reveals mRNA turnover strategies. Science 367, 1151-1156. ( 10.1126/science.aax3072) [DOI] [PubMed] [Google Scholar]
- 164.Lindon C, Grant R, Min M. 2016. Ubiquitin-mediated degradation of Aurora kinases. Front. Oncol. 5, 1-13. ( 10.3389/fonc.2015.00307) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Floyd S, Pines J, Lindon C. 2008. APC/CCdh1 targets aurora kinase to control reorganization of the mitotic spindle at anaphase. Curr. Biol. 18, 1649-1658. ( 10.1016/j.cub.2008.09.058) [DOI] [PubMed] [Google Scholar]
- 166.Min M, Mevissen TET, De Luca M, Komander D, Lindon C. 2015. Effcient APC/C substrate degradation in cells undergoing mitotic exit depends on K11 ubiquitin linkages. Mol. Biol. Cell 26, 4325-4332. ( 10.1091/mbc.E15-02-0102) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.