

Abstract
Colorectal cancer (CRC) is among the most frequent forms of cancer, and new strategies for its prevention and therapy are urgently needed1. Here we identify a metabolite signalling pathway that provides actionable insights towards this goal. We perform a dietary screen in autochthonous animal models of CRC and find that ketogenic diets exhibit a strong tumour-inhibitory effect. These properties of ketogenic diets are recapitulated by the ketone body β-hydroxybutyrate (BHB), which reduces the proliferation of colonic crypt cells and potently suppresses intestinal tumour growth. We find that BHB acts through the surface receptor Hcar2 and induces the transcriptional regulator Hopx, thereby altering gene expression and inhibiting cell proliferation. Cancer organoid assays and single-cell RNA sequencing of biopsies from patients with CRC provide evidence that elevated BHB levels and active HOPX are associated with reduced intestinal epithelial proliferation in humans. This study thus identifies a BHB-triggered pathway regulating intestinal tumorigenesis and indicates that oral or systemic interventions with a single metabolite may complement current prevention and treatment strategies for CRC.
Colorectal cancer (CRC) incidence is associated with specific dietary patterns2. For example, a Western diet enhances the tumorigenicity of intestinal progenitor cells and suppresses anti-tumour immunity3,4. Similarly, high-sugar diets and hyperglycaemia predispose to CRC5. Furthermore, excessive intake of animal protein, particularly red meat, is associated with increased CRC risk6. By contrast, little is known about diets that promote the prevention or treatment of intestinal tumorigenesis. Although fasting and caloric restriction have shown powerful effects in animal models7,8, such measures may be challenging to widely implement in humans, particularly in patients with cachectic CRC. It is therefore essential to uncover new diet-induced endogenous mechanisms that are able to suppress tumour growth.
Ketogenic diet protects from CRC
To identify dietary interventions affecting intestinal tumour growth, we designed mouse diets with defined macronutrient sources, constant protein content and varying fat-to-carbohydrate ratios (Fig. 1a, b and Supplementary Table 1). We included two ketogenic diets (KDs) with 90% fat content from plant or animal sources. In mice fed these six different diets, we induced CRC by injecting azoxymethane (AOM) followed by three cycles of dextran sodium sulfate (DSS) (Fig. 1a). We noted that the tumour numbers and sizes were suppressed with increasing fat-to-carbohydrate ratios (Fig. 1c and Extended Data Fig. 1a–f) and that the KD improved long-term survival (Extended Data Fig. 1g). Furthermore, the KD inhibited tumour development in a genetic model of CRC (Cdx2creERTApcfl/fl; Fig. 1d, e and Extended Data Fig. 1h–j). This effect was observed across different animal housing conditions (Extended Data Fig. 1k–p).
Fig. 1 |. Inhibition of intestinal tumour growth by KDs.

a, Schematic of dietary exposure in AOM/DSS-treated mice. b, Macronutrient composition of diets. c, Tumour scores of AOM/DSS-treated mice fed six different diets. d, Schematic of dietary exposure of Cdx2CreERTApcfl/fl mice. e, Tumour score in Cdx2CreERTApcfl/fl mice fed a KD or control diet. f–k, Schematic of dietary exposure (f, i) and colonoscopy-based tumour quantification (g, h, j, k) in AOM/DSS-treated KD-fed mice in a treatment model (f–h) and a cessation model (i–k). Data are ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Exact n and P values are presented in Supplementary Table 3.
We also assessed whether KD could be used for the treatment of existing colorectal tumours. To this end, we subjected mice to a delayed exposure paradigm in which KD feeding was started after the second cycle of DSS (Fig. 1f). Notably, this treatment effectively suppressed the progression of tumour growth (Fig. 1g, h and Extended Data Fig. 1q, r). Conversely, discontinuation of KD feeding in a cessation model (Fig. 1i) led to tumour regrowth, even after the tumour was initially reduced efficiently (Fig. 1j, k and Extended Data Fig. 1s, t). Together, these results indicate that a KD potently suppressed colorectal tumour growth in both prevention and treatment models.
β-Hydroxybutyrate inhibits intestinal tumour growth
We next investigated the mechanisms underlying the effect of a KD on colonic tumour development. We first ruled out differences in caloric intake (Extended Data Fig. 2a, b). In the AOM/DSS model, cancer progression is driven by immune cells, including IL-17-producing cells9. A recent study demonstrated that KD feeding reduces the number of IL-17-producing T cells10. Indeed, transcript levels of Il17a in tumours were reduced in mice on a KD (Extended Data Fig. 2c); however, adaptive immune cells were not functionally required for the protective effect of the diet (Extended Data Fig. 2d–g). A KD has also been reported to inhibit the NLRP3 inflammasome11. We therefore generated bone marrow chimeric mice by reconstituting irradiated Cdx2CreERTApcfl/fl mice with bone marrow from Nlrp3-deficient or Nlrp3-sufficient donors. However, the tumour-inhibitory effect of the KD was observed regardless of the host genotype (Extended Data Fig. 2h–j).
Epithelial cell proliferation was markedly decreased in the KD-fed mice under both homeostatic conditions and in CRC (Extended Data Fig. 2k–n). Similarly, the rate of apoptosis was decreased, as indicated by an increase in anti-apoptotic protein Mcl1 (Extended Data Fig. 2o, p). This indicates an overall decreased turnover of epithelial cells. The turnover of the intestinal epithelium is regulated by Lgr5+ stem cells that reside at the bottom of intestinal crypts12. To investigate the effect of a KD on intestinal stem cells (ISCs), we used an organoid system that recapitulates the stem-cell-derived growth of the intestinal epithelium13 (Extended Data Fig. 3a). Crypts derived from mice on a KD gave rise to smaller organoids than crypts from control mice (Extended Data Fig. 3b, c), indicating that the diet inhibited the proliferation of crypt cells. To differentiate the effect of a KD on Lgr5+ stem cells from that on crypt-residing Paneth cells, we performed combinatorial culturing of stem cells and Paneth cells from mice on a KD or control diet. Stem cells from KD-fed mice showed reduced organoid growth regardless of the origin of the Paneth cells (Extended Data Fig. 3d, e), suggesting that a KD directly affects the function of Lgr5+ cells. Together, these results demonstrate that a KD decelerates intestinal crypt proliferation.
Carbohydrate-restricted diets containing a very high percentage of fats are referred to as ketogenic due to their ability to stimulate hepatic production of the ketone bodies acetoacetate (AcAc) and BHB, the body’s physiological response to starvation14. To determine whether the inhibitory effect of a KD on epithelial growth is mediated by ketone bodies, we monitored intestinal organoids cultured in the presence of AcAc or BHB (Fig. 2a). Although organoid growth was unchanged in the presence of AcAc (Fig. 2b, c), BHB reduced organoid size in a concentration-dependent manner (Fig. 2d, e). The effective concentration of BHB was similar to the tissue levels reached in the gut by KD feeding (Extended Data Fig. 3f) and did not affect organoid viability (Extended Data Fig. 3g, h). This effect of BHB was also observed in tumour organoids (Fig. 2f, g and Extended Data Fig. 3i, j) and is associated with lower proliferation rates (Extended Data Fig. 3k). In addition to inducing hepatic ketogenesis, KDs confer health benefits by lowering systemic glucose levels and enhancing insulin sensitivity15. To determine the relative contribution of BHB supplementation and glucose restriction, we treated organoids with BHB using varying glucose concentrations. The addition of BHB reduced organoid growth at each concentration of glucose (Extended Data Fig. 3l), indicating that glucose restriction and BHB supplementation may act through distinct mechanisms.
Fig. 2 |. Inhibition of intestinal tumour growth by BHB.
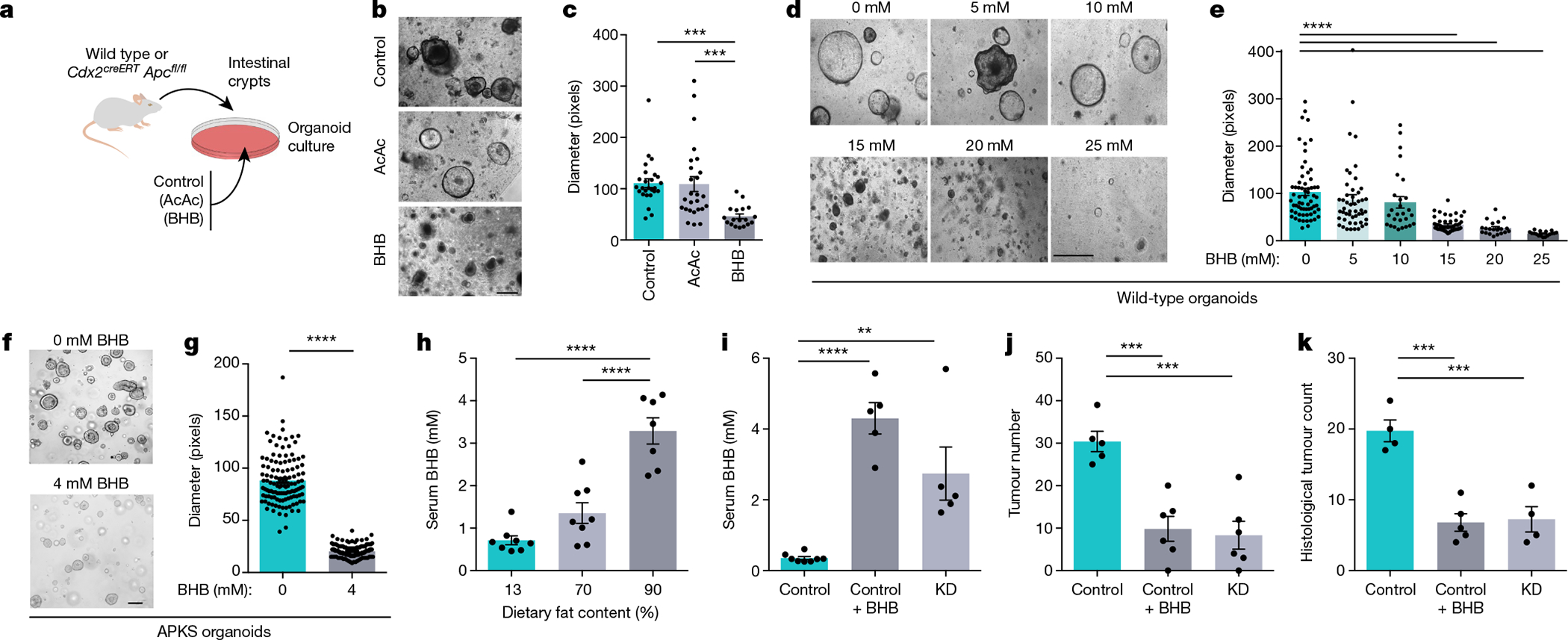
a, Schematic of organoid growth cultured with AcAc or BHB. b, c, Representative images (b) and quantification (c) of organoids exposed to AcAc or BHB. d–g, Representative images (d, f) and quantification (e, g) of wild-type (d, e) and APKS organoids (f, g) cultured with BHB. h, Serum concentrations of BHB in AOM/DSS-treated mice consuming diets with the indicated fat content. i–k, Serum BHB (i), colonoscopy-based tumour quantification (j) and histological tumour counts (k) of KD-fed or BHB-treated Cdx2CreERTApcfl/fl mice. Data are ± s.e.m. Scale bars, 68 μm (b, d) and 160 μm (f). **P < 0.01, ***P < 0.001, ****P < 0.0001. Exact n and P values are presented in Supplementary Table 3.
On the basis of these results, we hypothesized that BHB might be an effector metabolite by which a KD reduces intestinal crypt proliferation and tumour growth. Among the different diets we screened for their anti-cancer effects (Fig. 1a, b), those with fat contents of 70% and 90% slightly and strongly elevated serum BHB levels, respectively (Fig. 2h). To determine whether BHB was sufficient for reducing tumour growth in vivo, we administered BHB esters by oral gavage to Cdx2CreERTApcfl/fl mice on a control diet, achieving serum levels comparable to those induced by KD feeding (Fig. 2i). Strikingly, the BHB treatment alone led to reduced tumour burden in the colon (Fig. 2j, k and Extended Data Fig. 4a, b). By contrast, butyrate supplementation did not reduce tumour growth (Extended Data Fig. 4c). Furthermore, we supplemented AOM/DSS-treated mice with BHB (Extended Data Fig. 4d), which also reduced epithelial proliferation (Extended Data Fig. 4e, f) and led to decreased tumour growth (Extended Data Fig. 4g–i). BHB supplementation was also effective in a third model of CRC, in which the tumour suppressor Apc was deleted from Lgr5+ cells (Extended Data Fig. 4j, k). The glycaemic levels in the BHB-supplemented mice were identical to those in the control group (Extended Data Fig. 4l), which enabled us to differentiate the effect of this metabolite from the macronutrient content of the diet with 90% fat. Although oral administration of BHB esters is a suitable therapeutic modality, we also sought to mimic the endogenous release of BHB that is triggered by KD consumption. We therefore administered BHB salts using osmotic mini-pumps to Cdx2CreERTApcfl/fl mice, which was similarly effective in reducing tumour growth (Extended Data Fig. 4m–q). Together, these results indicate a potent tumour-suppressive effect of BHB.
BHB inhibits tumour growth through Hopx
We next investigated the mechanisms underlying BHB-mediated tumour inhibition. To this end, we performed RNA sequencing (RNA-seq) of BHB-treated organoids. BHB induced marked changes in global gene expression (Fig. 3a and Extended Data Fig. 5a). Among the genes that were upregulated by BHB was that encoding the homeodomain-only protein Hopx (Fig. 3a, b), a putative negative regulator of tumorigenicity16. We therefore hypothesized that the induction of Hopx may be important for BHB-mediated inhibition of tumour growth. To test this, we exposed organoids from Hopx-deficient mice and wild-type littermates to BHB. Whereas BHB reduced the growth of wild-type organoids (Fig. 3c, d), Hopx-deficient organoids were resistant to BHB treatment (Fig. 3e, f). Accordingly, epithelial proliferation was decreased by BHB in wild-type organoids but was not affected in the absence of Hopx (Extended Data Fig. 5b). Overexpression of Hopx in organoids was sufficient to reduce growth in both the wild-type and tumour organoids (Fig. 3g–j). Hopx expression was not induced by inhibition of glucose metabolism despite the effective inhibition of cancer organoid growth (Extended Data Fig. 5c, d). Accordingly, although Hopx-deficient organoids were resistant to the growth-inhibitory effect of BHB, glucose restriction still decelerated growth in the absence of Hopx (Extended Data Fig. 5e).
Fig. 3 |. BHB-mediated inhibition of tumour growth requires Hopx.
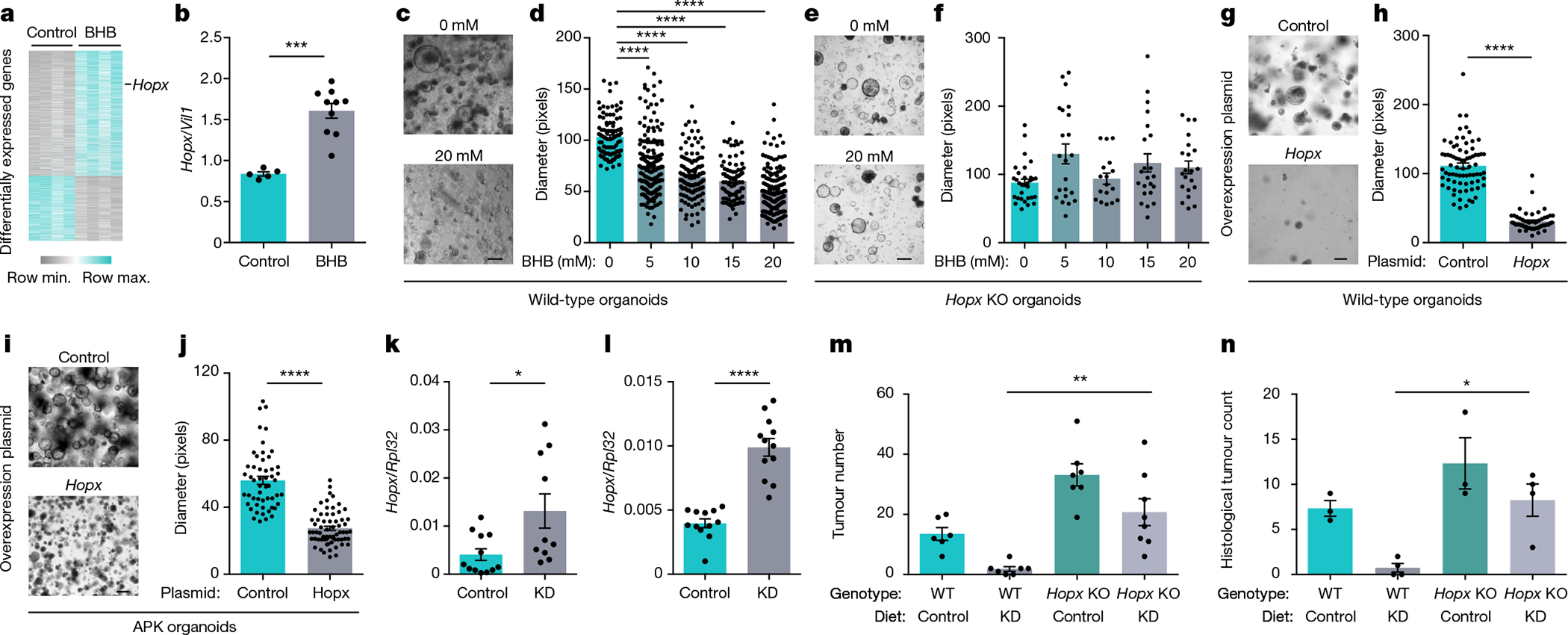
a, Heat map of differentially expressed genes in BHB-treated and control organoids. b, Hopx expression in BHB-treated organoids. c–f, Representative images (c, e) and quantification (d, f) of wild-type (c, d) and Hopx-deficient (e, f) organoids treated with BHB. g–j, Representative images (g, i) and quantification (h, j) of wild-type (g, h) and APKS (i, j) organoids overexpressing Hopx. k, l, Colonic transcript levels of Hopx in KD-fed mice under homeostatic conditions (k) and after tumour induction (l). m, n, Colonoscopy-based tumour quantification (m) and histological tumour counts (n) of KD-fed Hopx-deficient mice and controls. Data are ± s.e.m. Scale bars, 274 μm (c), 205 μm (e), 160 μm (g) and 320 μm (i). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Exact n and P values are presented in Supplementary Table 3.
We next sought to determine whether Hopx is also induced by KD feeding. Mice receiving a KD showed elevated levels of Hopx in colonic tissue (Fig. 3k), which was further accentuated after the induction of colonic tumours (Fig. 3l). This effect was unique to the colon and was not observed in other tissues (Extended Data Fig. 5f). Hopx has been identified as a marker for slowly dividing ‘reserve’ stem cells located adjacent to Lgr5+ cells. These cells may function as a quiescent reservoir for Lgr5+ cells and have been linked to regeneration after colonic injury17,18. Indeed, using HopxGFP reporter mice, we found its expression to be concentrated at the base of the colonic crypt (Extended Data Fig. 5g). In KD-fed mice, however, Hopx levels were increased, and the GFP signal extended to the transit amplifying compartment (Extended Data Fig. 5g, h).
To determine whether the tumour-suppressive effects of a KD and BHB required Hopx expression, we induced CRC in Hopx-deficient mice and wild-type littermates on a KD. Remarkably, although the wild-type group showed strongly decreased tumour growth after KD feeding, the diet was not effective in mice lacking Hopx (Fig. 3m, n and Extended Data Fig. 5i–l). We also tested whether the in vivo effects of BHB and glucose restriction were dependent on Hopx. A 30% caloric restriction reduced the glycaemic levels and slightly increased the levels of circulating BHB in the Cdx2CreERTApcfl/fl mice. By contrast, BHB supplementation strongly increased the systemic BHB levels under normoglycaemic conditions (Extended Data Fig. 5m, n). Notably, both caloric restriction and BHB supplementation reduced the tumour load. Even though the effect of BHB was entirely dependent on Hopx, caloric restriction was still effective in the absence of Hopx. This emphasizes that BHB supplementation and glucose restriction engage different downstream mechanisms to block intestinal tumour growth (Extended Data Fig. 5o, p).
To gain insight into the molecular mechanisms by which Hopx mediates the growth-inhibitory effect of a KD, we performed RNA-seq of colonic tissue from AOM/DSS-treated wild-type and Hopx-deficient mice. The functional annotation of differentially expressed genes showed a strong upregulation of proliferative pathways in mice lacking Hopx (Extended Data Fig. 6a, b), including several genes involved in cell cycle regulation (Extended Data Fig. 6c–f). Together, these results demonstrate that a KD and BHB-induced increased expression of Hopx reduce epithelial growth and colonic tumorigenesis.
Induction of Hopx by BHB requires Hcar2
We next sought to determine the mechanisms by which BHB induces Hopx expression. Because BHB inhibits histone deacetylases (HDACs)19, we explored whether HDAC inhibition mediates the effect of BHB on Hopx expression (Extended Data Fig. 7a). First, we verified that BHB treatment and KD feeding indeed reduced the HDAC activity (Extended Data Fig. 7b–k). The pharmacological HDAC blocker vorinostat and the HDAC-inhibiting metabolite butyrate20 reduced organoid growth in a manner similar to that of BHB (Extended Data Fig. 7l–n). However, RNA-seq indicated that the transcriptional profiles of organoids treated with vorinostat were largely distinct from those treated with BHB (Extended Data Fig. 7o, p). Hopx was among the genes that were upregulated only by BHB and not by vorinostat (Extended Data Fig. 7p, q). Because pharmacological HDAC inhibition did not recapitulate the effect of BHB on Hopx transcription, we reasoned that the ability of BHB to induce Hopx probably occurred independently of its HDAC-inhibitory effect. Indeed, acetylated histone H3 lysine 9 (H3K9ac) chromatin immunoprecipitation followed by sequencing (ChIP–sequencing) of intestinal epithelial cells from BHB-treated or KD-fed mice did not show changes in the acetylation pattern of the Hopx locus (Extended Data Fig. 7r). Furthermore, Hopx-deficient organoids were resistant to the effect of BHB but were susceptible to growth reduction mediated by vorinostat or butyrate (Extended Data Fig. 7s, t) despite the fact that BHB still reduced the HDAC activity in the absence of Hopx (Extended Data Fig. 7u). If the HDAC-inhibitory activity of BHB were dispensable for Hopx induction and growth suppression, then co-treatment with BHB and vorinostat should have shown additive effects on the intestinal epithelial cells. Indeed, organoids treated with BHB in addition to vorinostat showed a stronger reduction in growth compared with those treated with vorinostat alone (Extended Data Fig. 7l). Moreover, AOM/DSS-treated mice that received both a KD and vorinostat showed a further reduction in tumour growth compared with mice that received only the diet or vorinostat (Extended Data Fig. 7v, w). Together, these data indicate that although BHB inhibits HDACs in intestinal epithelial cells, this HDAC-inhibitory activity does not drive Hopx expression and Hopx-mediated growth arrest.
We next considered alternative explanations for BHB-mediated Hopx induction. The Hopx locus is frequently hypermethylated in cancer16. We thus reasoned that BHB induces changes in the DNA methylation status of Hopx(Extended Data Fig. 8a), and we performed bisulfite sequencing of intestinal epithelial cells from BHB-treated or KD-fed mice. Both treatments induced consistent global changes in DNA methylation (Extended Data Fig. 8b–d). However, the Hopx locus was unaffected and showed no difference in methylation in either group (Extended Data Fig. 8e), indicating that it is unlikely that methylation changes account for the effects of BHB and KD. Furthermore, the DNA methyltransferase inhibitor 5-azacitidine, a chemotherapeutical agent used for the treatment of myelodysplastic syndrome21, did not change the expression of Hopx in the organoids nor did it reverse the upregulation of Hopx when administered together with BHB (Extended Data Fig. 8f). Accordingly, BHB and 5-azacitidine showed an additive growth-inhibition effect on the organoids, and Hopx-deficient organoids responded to 5-azacitidine treatment but not to BHB (Extended Data Fig. 8g–j). Thus, these findings do not support that changes in DNA methylation mediate the effect of BHB on Hopx expression.
We thus considered an alternative approach to identify the mechanism by which BHB influences Hopx transcription (Extended Data Fig. 9a). We performed an organoid-based CRISPR screen of candidate genes that may act downstream of BHB (Supplementary Table 2). These genes include the BHB receptors Hcar2 and Ffar3 (also known as Gpr109a and Gpr41, respectively) as well as several genes reported to be influenced by BHB14. Organoids derived from Cas9-expressing mice were targeted with individual guide RNAs (gRNAs) using an average of four distinct gRNAs per candidate gene. Efficient gene targeting was confirmed by quantitative polymerase chain reaction (Extended Data Fig. 9b–g). We then exposed the organoids to BHB and measured their growth. Although the BHB effectively reduced growth in most of the gene-deficient organoids, all four gRNAs targeting Hcar2 conferred resistance to BHB-mediated size reduction (Fig. 4a and Extended Data Fig. 9h–m), thus suggesting Hcar2 as a possible mediator of the effect of BHB. To verify this finding, we exposed organoids from Hcar2-deficient mice to BHB, butyrate or vorinostat (Extended Data Fig. 9n). Although BHB strongly reduced the size of organoids in wild-type controls, organoids from Hcar2-deficient mice continued to grow in the presence of BHB (Fig. 4b and Extended Data Fig. 9o). By contrast, butyrate and vorinostat still suppressed organoid growth in the absence of Hcar2. Hcar2 deficiency also prevented the BHB-induced upregulation of Hopx (Fig. 4c), whereas butyrate, which also binds to Hcar2 in the intestinal epithelium22, did not induce Hopx (Fig. 4c). Other gene deficiencies did not affect Hopx induction in response to BHB (Extended Data Fig. 9p–v). Collectively, these results indicate that BHB acts through the surface receptor Hcar2 to induce Hopx expression and suppress epithelial growth.
Fig. 4 |. BHB induces Hopx expression and suppresses intestinal epithelial growth through Hcar2.

a, Size of organoids treated with 10 mM BHB after CRISPR-mediated gene deletion. Insets show representative organoid images from Hcar2-targeted organoids and controls. b, Size of organoids from wild-type and Hcar2-deficient mice treated with BHB, butyrate or vorinostat. c, Transcript levels of Hopx in organoids from wild-type or Hopx-deficient mice treated with 10 mM BHB or butyrate. Data are ± s.e.m. Scale bar, 68 μm. *P < 0.05, ***P < 0.001, ****P < 0.0001. Exact n and P values are presented in Supplementary Table 3.
BHB effects on human tumour cells
Next, we explored whether the BHB–HOPX pathway is similarly effective in suppressing human intestinal epithelial proliferation (Fig. 5a). Indeed, BHB reduced the growth of organoids in both healthy donors (Fig. 5b, c and Extended Data Fig. 10a, b) and patients with CRC (Extended Data Fig. 10c, d), indicating that the tumour organoids were not resistant to BHB-mediated inhibition. Similar to our findings in mice, BHB treatment caused elevated HOPX expression in human organoids (Fig. 5d).
Fig. 5 |. Effect of BHB and HOPX on human intestinal epithelial growth.
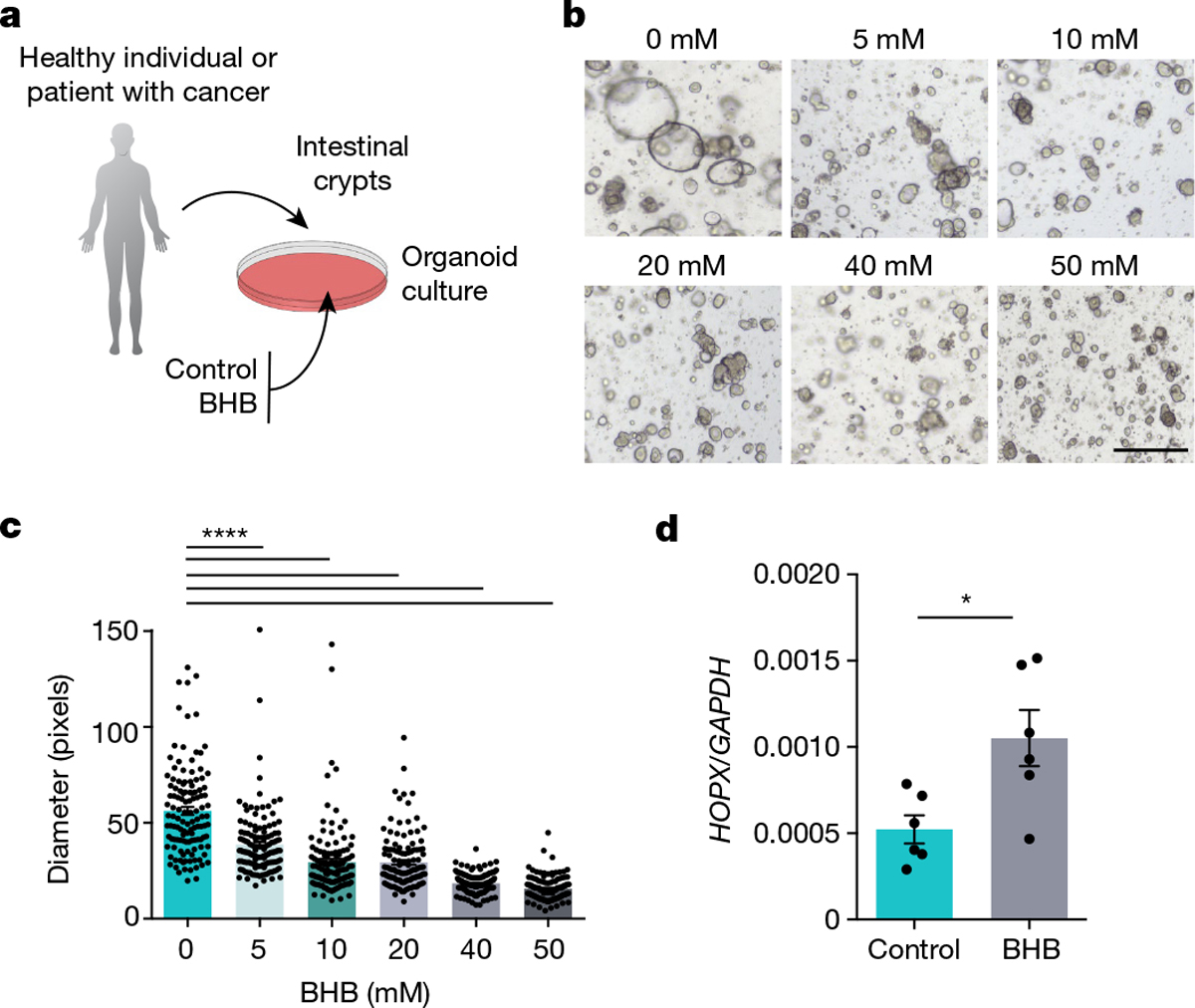
a, Schematic of intestinal organoid growth of crypts obtained from healthy donors or patients with CRC cultured with BHB or control medium. b, c, Representative images (b) and quantification (c) of organoids from a healthy adult donor after 10 days of culturing with the indicated concentrations of BHB. d, HOPX expression in BHB-treated and control organoids from patients with CRC. Data are ± s.e.m. Scale bar, 68 μm. *P < 0.05, ****P < 0.0001. Exact n and P values are presented in Supplementary Table 3.
The anti-proliferative effect of BHB was also observed in the human CRC cell line HT-29 (Extended Data Fig. 10e), along with a reduced fraction of cells in the S phase (Extended Data Fig. 10f), at different glucose concentrations (Extended Data Fig. 10g). By contrast, other CRC cells lines such as HCT116, Caco-2 and RKO, did not respond to BHB (Extended Data Fig. 10h–j). We hypothesized that the HCAR2–HOPX pathway may be involved in the differential responsiveness of CRC cell lines. Only HT-29 cells expressed both HCAR2 and HOPX (Extended Data Fig. 10k, l), indicating that these genes may determine the degree of responsiveness to BHB.
Finally, we sought to understand the association of serum BHB with colonic HOPX levels in the large intestine of patients with CRC. To this end, we collected sera from 41 patients with CRC, and we measured their BHB levels (Extended Data Fig. 11a, b). We then performed single-cell RNA-seq of tumour and normal tissue from nine patients with varying levels of BHB. We profiled and clustered a total of 11,208 cells from these patients and used marker genes to identify different cell populations (Extended Data Fig. 11c, d). Notably, in the epithelial subset, the BHB levels correlated positively with HOPX expression (Extended Data Fig. 11e) and negatively with cell cycle progression (Extended Data Fig. 11f–h). These data collectively indicate that BHB may elevate HOPX levels and antagonize proliferation in human intestinal epithelial cells.
Discussion
Our discovery of a BHB–Hcar2–Hopx signalling axis that decelerates intestinal tumour growth has several important implications. First, it adds an unexpected facet to the biology of ketone bodies. The ability of BHB to suppress intestinal epithelial growth might be part of its function to reallocate resources away from the energy-intensive renewal of peripheral tissues in times of starvation. Therefore, the very same metabolite may serve not only as an energy source for essential organs such as the brain but also as a signalling molecule that delays growth in the periphery during nutrient scarcity.
Second, our study extends the repertoire of dietary patterns associated with the prevention of CRC by demonstrating that BHB exerts its influence on the intestinal epithelium through a different pathway than glucose and butyrate (Extended Data Fig. 11i). A KD that restricts carbohydrates but contains sufficient quantities of dietary fibre may thus provide optimal protection from intestinal tumorigenesis. Notably, prospective randomized controlled studies are needed before specific recommendations for prevention and treatment can be made.
Third, together with studies investigating the differential effects of KD composition on various cancers23, our findings suggest a model whereby tumour-specific gene expression patterns determine the degree of responsiveness to particular dietary interventions. This model raises the possibility that optimal outcomes in cancer prevention and therapy may be achieved by adjusting the dietary macronutrient composition to the tumour type according to the expression of defined enzymes.
Finally, even though the nutritional interventions are attractive, chronic consumption of a specific diet is difficult to maintain and may cause systemic side effects. Our study demonstrates that the tumour-inhibitory effect of a diet can be recapitulated by metabolite supplementation. The BHB-mediated pathway may act synergistically with surgery, chemotherapy or immunotherapy and might emerge as an example of ‘metabotherapy’, a new pillar of cancer care.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-022-04649-6.
Methods
No statistical methods were used to predetermine sample size. Investigators were not blinded to allocation during experiments and outcome assessment with the exception of colonoscopy and histological analyses, which were performed by two independent researchers in a blinded manner.
Mice
C57BL/6J (000664) mice were purchased from The Jackson Laboratory and were allowed to acclimatize to the animal facility environment for 2 weeks before being used for experimentation. At the beginning of each experiment, mice were randomly allocated into experimental groups.
Hopx−/−, Hopx3xFlag-GFP and Hcar2−/− mice have been previously described24–26. Lgr5eGFP-IRES-creERT2 (008875), Apcfl/fl (029275) and Cdx2CreERT (022390) mice were purchased from The Jackson Laboratory. For tamoxifen-inducible tumorigenesis, Cdx2CreERTAPCfl/fl mice were injected once intraperitoneally with 1.5 mg of tamoxifen in 150 μl vehicle (Sigma; dissolved in 5% ethanol, 95% corn oil). Mice were killed 4–6 weeks after tamoxifen treatment for histological and tumour analyses.
In all experiments, age- and sex-matched littermates were used. Mice were 8–9 weeks of age at the beginning of experiments. Both male and female mice were used for experiments, but within each experiment, they were sex-matched. Mice were housed at 22.2 °C and 52.1% humidity. Mice were given access to food and water ad libitum and were maintained under a 12-h light–dark cycle. All mice were maintained in filter-topped cages and given autoclaved food and water at the University of Pennsylvania University Laboratory Animal Resources (PennULAR) facility. The alternative vivarium experiment was performed at Fox Chase Cancer Center (FCCC). All experiments were performed in accordance with the guidelines of the respective facilities and were approved by the regulations of the local institutional animal care and use committee (IACUC). All experiments used co-housed littermates to ensure consistency of common microbiota and genetic background. No methods were used to predetermine samples size; rather, sample sizes were determined by pilot experiments to assess effect sizes and variability. The precise number of animals used in each experiment is provided in Supplementary Table 3.
Diets
Protein-content-matched diets shown in Supplementary Table 1 were ordered from Envigo. Diets were matched for their source of macro- and micronutrient content and differed only in their fat-to-carbohydrate ratio. All experiments describing the KD with 90% fat used diets with plant fat source except where indicated in Fig. 1.
For caloric restriction experiments, food intake during ad libitum conditions was measured using a BioDAQ food-intake monitoring system (Research Diets) and was averaged for 24 h. Then 70% of the measured amount was provided daily for the duration of the caloric restriction period. To avoid competition between cage mates, only female mice were used in caloric restriction experiments.
Colitis-associated colon cancer (AOM/DSS)
To induce colitis-associated colon cancer, mice were intraperitoneally injected once with 10 mg kg−1 body weight of the mutagen AOM (A5486, Sigma-Aldrich), followed by three cycles of 2.5% DSS in the drinking water for 5 days (160110, MP Biomedicals). Each DSS cycle was followed by regular drinking water for 14 days. Mice were monitored for weight loss daily to assess disease progression, and 20% weight loss and body condition scoring were used as a humane end point (score of 2 + weight loss, or score of 1). The IACUC-approved limits were not exceeded in any experiment. Colonoscopy assessments were performed in a blinded manner after administration of each round of DSS to evaluate tumour progression.
BHB treatment and measurement
Mice were administered with BHB esters (HVMN), 200 μl by oral gavage daily, 80 mg per mouse.
BHB was measured in mouse serum. Blood was obtained by orbital vein bleed and was collected into a heparin-coated tube. Serum was separated by centrifugation at 1,000g for 10 min at 4 °C. Serum samples were frozen at −80 °C until thawed for BHB measurements using a colorimetric assay (700190, Cayman).
BHB mini-pump implantation
ALZET miniature osmotic infusion pumps were used to deliver sodium BHB subcutaneously. Because this model releases 0.25 μl h−1, we filled the capsule with 20 M sodium BHB dissolved in a sterile PBS solution. Control groups received osmotic pumps filled with PBS. Ten-week-old Cdx2ERTApcfl/fl mice were implanted with either osmotic pumps containing BHB or PBS under sterile surgical conditions. One day after the surgery, mice were injected with 1.5 mg of tamoxifen and were observed daily for 4 weeks. After 4 weeks, mice were subjected to colonoscopy and were then euthanized for tumour count and evaluation.
Bone-marrow transplantation
Six- to eight-week-old recipient mice were irradiated twice to achieve a lethal dose (600 rad) and were intravenously injected with a single-cell suspension of donor bone-marrow cells. Recipients were co-housed littermates, which were transplanted with either gene-deficient or wild-type bone marrow. After transplantation, the recipients were given neomycin and polymyxin B in the drinking water for 2 weeks, after which they were transferred to regular housing cages to reconstitute potentially depleted microbiota. Bone marrow was allowed to reconstitute for 7 weeks in Cdx2ERT-CreApcfl/fl recipient mice after which the animals were injected with tamoxifen. Subsequently, colon tumours were allowed to develop for 4–6 weeks.
Colonoscopy
Colonoscopy was performed using a high-resolution mouse video endoscopic system (Karl Storz). Tumour size was graded from 1 to 5 as follows: grade 1, very small but detectable tumour; grade 2, tumour covering up to one-eighth of the colonic circumference; grade 3, tumour covering up to one-fourth of the colonic circumference; grade 4, tumour covering up to half of the colonic circumference; and grade 5, tumour covering more than half of the colonic circumference. Tumours observed during endoscopy were counted to obtain the total number of tumours per animal. The size of each tumour in every mouse was recorded on the 1–5 scoring scale, and the sum of all tumour sizes per mouse was calculated to be the tumour score27.
HDAC inhibitor treatment
Mice were gavaged with 200 μl vorinostat (5 mg ml−1) or control solution (2% DMSO + 30% PEG300 + 10% Tween-80 + ddH2O) every day for 28 days.
Glucose measurements in mice
Glucose levels were measured in live mice with Nova Max Plus (53493) glucose test strips.
Caloric intake measurement
Caloric intake was measured using the BioDAQ food-intake monitoring system (Research Diets). Age- and weight-matched mice were acclimatized in the BioDAQ cages for 1 week. After acclimatization, food intake was measured continuously for each mouse. Caloric intake was calculated on the basis of food intake and the caloric content per gram of food for different diets.
Intestinal epithelial cell isolation for flow cytometry and protein analyses
Colonic tumours and normal tissues were cut into small pieces and subjected to mechanical shaking (200 rpm) with 10 mM EDTA in Ca2+/Mg2+-free 1× HBSS at 4 °C for 15 min for three rounds in total. After each round of shaking, the cells were vortexed for 20 s and filtered through a 70-μm metal strainer. The cell suspension was then collected and centrifuged at 1,600 rpm for 10 min at 4 °C. Pellets were resuspended in ice-cold PBS containing inhibitors of proteases and phosphatases and were then prepared for protein or nuclear extraction or were resuspended directly in FACS staining buffer for further cytometry analysis.
Intestinal epithelial stem cell isolation for organoid culture
Tissues from the colon or small intestines were washed with ice-cold PBS, opened longitudinally and cut into 0.5-cm pieces. Then the tissues were incubated on ice with HBSS–EDTA (10 mM) for 30 min with intermittent shaking, and the supernatant was removed at least three times. Crypts were then mechanically separated by shaking in HBSS–EDTA (10 mM) for 1 h at 4 °C and were then filtered through a 70-μm strainer into a 50-ml conical tube. Isolated crypts were embedded in Matrigel (BD356231, GFR) and cultured in a modified form of medium as described previously28,29.
Intestinal epithelial stem cell isolation for sorting of Lgr5+ intestinal stem cells and Paneth cells
For intestinal stem cell (ISC) isolation, crypt suspensions were dissociated into individual cells with TrypLE Express (Invitrogen). Cells were stained with an antibody cocktail consisting of CD45-PE (30-F11, eBioscience,), CD31-PE (Mec13.3, Biolegend), CD24-Pacific Blue (M1/69, Biolegend), EpCAM-APC (G8.8, eBioscience) and Kit (Biolegend). ISCs were isolated as Lgr5-eGFPhiEpCAM+CD31−CD45− Live/Dead-yellow−. Paneth cells from the small intestine were isolated as CD24hi SSChi c-Kithi Lgr5-eGFP−EpCAM+CD31−CD45− Live/Dead-yellow−. Cells were sorted with a BD FACS Aria Fusion cell sorter into complete crypt medium for culture. Dead cells were excluded from the analysis with the viability dye Live/Dead yellow (Life Technologies). ISCs and Paneth cells were mixed after sorting in a 1:1 ratio, centrifuged and then seeded onto Matrigel (Corning 356231 growth factor reduced) in a flat-bottom 24-well plate. The crypt medium was changed every third day. Organoids were quantified on days 3, 7 and 10 of culture unless otherwise specified.
Organoid treatment
Intestinal organoids from mice or humans were treated with freshly prepared solutions of BHB (Sigma, 54965) at concentrations of 5–50 mM for 4–14 days. The organoid culture medium was changed every third day, and the organoid size was quantified using ImageJ v.1.49 software.
In all experiments in which the concentration of BHB is not indicated otherwise, 10 mM was used. The concentration of BHB was aimed to match the amount of BHB molecules in vivo such that the colonic levels of BHB were approximately 0.03 μmol per gram of tissue, or roughly 14–18 nmol per colon. In mice fed a KD, these levels were about three times as high. Approximately 250 organoids were cultured per well in 500 μl of medium. To achieve 18 nmol BHB per organoid, and assuming free diffusion of BHB between the culture medium and the Matrigel sphere, a concentration of 9 mM was thus required.
2-Deoxyglucose was added to organoid cultures at a concentration of 100 μM.
In experiments in which the glucose concentrations in the media varied, the concentrations ranged from 5 mM to 20 mM. Butyrate was added to organoid cultures at concentrations of 1 mM, 5 mM or 10 mM.
Methylation inhibitor treatment
Colon organoids were treated with a freshly prepared solution of 10 μM 5-azacitidine for 24–72 h. Organoid size was monitored daily.
HDAC inhibitor treatment
Organoids were treated with 20 nM of the HDAC inhibitor vorinostat (SAHA, MK0683, Selleckchem, S1047). The inhibitor was added to the organoid culture on day 1 with a concentration range of 0.01× to 0.5× of the half maximal inhibitory concentration (IC50) determined individually for each inhibitor as indicated on the Selleckchem website (https://www.selleckchem.com). The organoid culture medium was changed every third day. Organoid size was monitored daily.
CellTiter-Glo 3D Cell Viability Assay
The CellTiter-Glo 3D Cell Viability Assay is a method for determining the number of viable cells in a 3D cell culture based on the quantification of the ATP present, which is a marker for the presence of metabolically active cells. Small intestine and colonic organoids were grown for 4–7 days in conditional media treated or untreated with BHB at concentrations of 20–40 mM. On the day of the experiment, the CellTiter-Glo reagent was added to the organoid culture, and luminescence was measured according to the manufacturer’s protocol.
Flow cytometric assessment of organoid viability
To determine the viability of organoids treated with BHB, single-cell suspensions of organoids were stained with Live/Dead-yellow for 30 min before FACS analysis at 1:300 dilution. Only cells gated for negative Live/Dead-yellow staining were used for further analysis.
Generation of lentiviral vectors and particles
To generate gRNA lentiviral vectors, Oligo-1 and Oligo-2 (Supplementary Table 2) were annealed and ligated into the BsmBI digested plasmid lentiCRISPRv2. For generation of lentivirus, HEK293FT cells were plated on six-well plates in high-glucose DMEM with addition of 3–4 μg plasmid DNA (cloned guides), with 2 μg psPAX2 and 1 μg pMD2.G plasmids mixed into 400 μl PBS to generate lentiviral particles. The packaging vectors were co-transfected using 7 μl polyethylenimine per well. After 5 h, the culture medium was replaced with DMEM. After 24 h and 48 h, the viral supernatants were collected and concentrated to 50–100 μl using a Lenti-X concentrator (Takara).
Transduction of lentivirus to mouse Cas9 organoids
Cas9-producing organoids were dissociated into single cells using TrypLE (Invitrogen). A total of 1 × 105 cells were mixed with polybrene (Millipore) and virus particles (1:50) in a 24-well plate, centrifuged at 600g for 1 h at 32 °C and incubated for 30 min at 37 °C in a CO2 incubator. Organoids were then embedded in 50 μl of Matrigel (BD Sciences) and cultured in 500 μl medium. At 24 h after transduction of gRNAs, blast antibiotics (1 μl ml−1) were added for selection of positive cells.
Organoid overexpression of Hopx
Organoids were dissociated into single cells using TrypLE. Organoid cell suspensions were resuspended with transfection mix (250 μl Opti-MEM + 10 μl lipofectamine 2000 + 10 μg plasmid). The mix was centrifuged at 600g at 32 °C for 1 h, transferred to a new 1.7-ml tube and spun down at 10,000g for 10 s. Then the pellet was mixed with complete DMEM F12 and Matrigel (1:1), seeded in a 24-well plate and overlain by 500 μl of medium. Organoid growth was analysed over the next 3–5 days, and Hopx overexpression was measured by quantitative polymerase chain reaction (qPCR).
Cell sorting
Colonic tumours and normal tissues were cut into small pieces and briefly washed in 1 mM DTT–HBSS followed by mechanical shaking with 10 mM EDTA–HBSS in a shaker (200 rpm) at 37 °C for 20 min for two rounds total. Then the intraepithelial lymphocyte/intestinal epithelial cell fraction was passed through a metal mesh, and the cells were collected. The single-cell suspension was then stained and sorted on a BD FACSAria Fusion sorter (BD Biosciences).
Flow cytometric assessment of histone acetylation
Intestinal epithelial cells from mice fed a KD or treated with BHB esters (for 4 weeks), or intestinal organoids treated with BHB (for 4 days) were collected. Single-cell suspension was resuspended, washed in FACS buffer (1× PBS, 1 mM EDTA, 2% FBS) and stained with the primary antibodies CD45, EpCAM, Live/Dead-yellow− followed by 45 min fixation at room temperature with fixation/permeabilization buffer (Foxp3 Transcription Factor Staining Kit, eBioscience). Concentrate and diluent (00-5521, eBioscience) were prepared according to the manufacturer’s protocol, and staining was performed with H3K9ac (9649T, Cell Signaling) or histone H3 (14269S, Cell Signaling) and Ki-67 antibody mix in permeabilization buffer (00-8333-56, eBioscience) overnight at 4 °C. On the next day, cells were washed twice and incubated for 2 h with a secondary PacBlue goat anti-rabbit antibody (P10994, Invitrogen), washed with FACS buffer and analysed by flow cytometry. The mean fluorescence intensity (MFI) level of H3K9ac staining was normalized to the MFI of total H3 using FlowJo v.9.7.6. The gating strategy is provided in Supplementary Fig. 1.
Nuclear extraction from intestinal epithelial stem cells
Intestinal epithelial cells were resuspended in lysis buffer (10 mM Tris-HCl at pH 7.5, 10 mM NaCl, 15 mM MgCl2, 250 mM sucrose, 0.1 mM EGTA, 0.5% NP-40), vortexed for 10 s, incubated on ice for 15 min and spun through 4 ml of a sucrose cushion at 1,300g for 10 min at 4 °C. The nucleus pellet was then washed with cold 10 mM Tris-HCl at pH 7.5, 10 mM NaCl, resuspended in 50 μl of extraction buffer (50 mM HEPES KOH at pH 7.5, 420 mM NaCl, 0.5 mM EDTA, 0.1 mM EGTA, 10% glycerol), sonicated for 30 s, left on ice for 30 min and centrifuged at maximum speed (13,000 rpm) for 10 min.
Histone deacetylase activity measurement
Intestinal organoids treated with BHB (for 5 days) or intestinal epithelial cells isolated from wild-type mice treated with BHB (for 3 weeks) were prepared for the isolation of nuclear extracts. Histone deacetylase was measured with the HDAC Activity Assay Kit (ab 156064, Abcam) according to the manufacturer’s instructions.
RNA extraction and RT–qPCR analysis
Total RNA was extracted using TRIzol and reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (4368814, Applied Biosystems), and RT–qPCR was performed with the QuantiFast SYBR Green PCR Kit 2000 (204056, Qiagen) on an Applied Biosystems CFX96 machine. Expression data were normalized to Rpl32 mRNA levels. The data are presented in arbitrary units and were calculated as 2Ct(Rpl32−gene of interest). Primer sequences were obtained from the NIH Mouse qPrimerDepot.
Histology and immunofluorescence
For histology, colon rolls were fixed with 10% buffered formalin immediately after euthanasia and were kept overnight at room temperature. After fixation, the samples were transferred into 70% ethanol before paraffin embedding. Sections with a thickness of 5 μm were cut in the sagittal plane for staining with haematoxylin and eosin.
Sections were deparaffinized and re-hydrated using serial 5 min incubations in xylene and ethanol gradient (100% to 70%). Antigen retrieval was performed in citrate buffer (10 mM citrate, 0.05% Tween 20, pH 6.0) at 95 °C for 1 h. The slides were then rinsed in dH2O, dehydrated in 100% ethanol, blocked with 3% H2O2 in PBS, rinsed with water and blocked in 5% BSA and 5% normal goat serum in PBS for 20 min. Tissue sections were then incubated with anti-Ki-67 primary antibody (1:250) or MCL-1 (1:200) overnight at 4 °C and then washed three times in PBS with 1% BSA. Secondary biotinylated anti-rabbit antibodies were applied at a 1:200 dilution, incubated at room temperature for 1 h and then rinsed in PBS. Next, streptavidin–HRP was applied at 1:250 dilution in PBS with 1% BSA for 30 min at room temperature. The slides were then washed three times in PBS and developed with chromogen DAB solution for 7 min at room temperature. The slides were then rinsed and counterstained with haematoxylin, rinsed with water, immersed into 0.25% ammonium hydroxide and dehydrated with four changes of ethanol at 70% to 100% followed by four changes of xylene for 5 min each. Then the slides were mounted.
Immunofluorescence staining was performed in a manner similar to the immunohistochemistry protocol with a few modifications: the blocking buffer contained 5% goat serum, 10% BSA and 0.1% Triton X-100 in 1× PBS. Goat anti-rabbit AF488 was used as a secondary antibody. The slides were stained with DAPI for 10 min at room temperature, mounted with VectorShield and covered with a cover slip. After the slides had dried for 1–2 h at room temperature, images were acquired using a Nikon fluorescence microscope or a Zeiss confocal microscope.
To detect Hopx+ cells in the intestine, we used Hopx3XFlag-GFP mice. After 4 weeks of ketogenic (90% fat) or control (13% fat) diets, mice were killed, and frozen sections of the colon or organoids were generated. Colon Swiss rolls were made and briefly fixed in 2% PFA on ice for 1.5 h, immediately submerged into cryopreservation reagent and snap frozen on dry ice. Then 10–12 μm sections were cut using a cryostat, stained with DAPI and immediately analysed on a Zeiss confocal microscope.
ChIP–seq
For ChIP–seq, approximately half the colon of each mouse was minced on ice, cross-linked with 1% formaldehyde for 10 min and quenched with 125 mM glycine for 5 min. Nuclei were prepared by Dounce homogenization of cross-linked tissue in nucleus isolation buffer (50 mM Tris-HCl at pH 7.5, 25 mM KCl, 5 mM MgCl2, 0.25 M sucrose) with freshly added protease inhibitors and sodium butyrate. Nuclei were then lysed in nucleus lysis buffer (10 mM Tris-HCl at pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, 0.5% N-lauroylsarcosine) with freshly added protease inhibitors and sodium butyrate. Next, chromatin was sheared to approximately 250 bp in size using a Covaris S220 sonicator. Equal aliquots of sonicated chromatin were used per immunoprecipitation reaction with 5 μl H3K9ac antibody (active motif; 39137; 09811002) pre-conjugated to Protein G Dynabeads (Life Technologies). Ten per cent of the chromatin was saved as input DNA. ChIP reactions were incubated overnight at 4 °C with rotation and washed three times in wash buffer. Immunoprecipitated DNA was eluted from the beads, reversed cross-linked and purified together with the input DNA. Exactly 2 ng DNA (either ChIP or input) was used to construct sequencing libraries using the NEBNext Ultra II DNA library preparation kit for Illumina (New England Biolabs). Libraries were multiplexed using NEBNext Multiplex Oligos for Illumina (dual index primers) and single-end sequenced (75 bp) on the NextSeq 500 platform (Illumina) in accordance with the manufacturer’s protocol.
ChIP–seq analysis
ChIP–seq reads were aligned to mm9 reference genome assembly using Bowtie2 v.2.2.9; only reads with alignment scores greater than 10 were retained. Duplicated reads were removed using SAMtools (v.0.1.19, ‘rmdup -s’). Reads mapped to mitochondria and ENCODE blacklist regions (https://sites.google.com/site/anshulkundaje/projects/blacklists) were removed using BEDtools (v.2.25.0, ‘bedtools intersect -v’). Peaks were called using MACS2 (v.2.1.0) by comparing each immunoprecipitation library to its corresponding input control with a false discovery rate cutoff of 0.01 and broad peak configuration (‘--qvalue 0.01 --broad’), and bedGraph files normalized to sequencing depth were generated at the same time for visualization purposes (‘--bdg --SPMR’). The input control bedGraph files were subtracted from the IP library bedGraph file using MACS2 (‘bdgcmp’) and converted to bigWig files using the UCSC toolkit (‘bedGraphToBigWig’). The union of peaks called from all libraries were acquired using BEDtools (‘merge’) and were annotated to the nearest RefSeq gene using HOMER (‘annotatePeaks.pl’). For each library, the ChIP signal over each union peak region was quantified using bwtool (v.1.0, ‘bwtool summary -fill=0’), which was then used for generating PCA plots (in R) and passed on to the DESeq2 R package for statistical analysis.
Whole-genome bisulfite sequencing library preparation and analysis
First, 150 ng of purified genomic DNA was first diluted in 130 μl of low TE buffer (10 mM Tris pH 8.0, 0.1 mM EDTA) in a snap-cap microtube (520045, Covaris). Then the genomic DNA sample was fragmented to an average size of 200 bp using a Covaris M220 sonicator. Whole-genome bisulfite sequencing libraries were generated using an Ovation Ultralow Methyl-Seq kit (0541, NuGEN) following the manufacturer’s protocol. In brief, the ends were repaired and the methylated adaptors were ligated to the fragmented genomic DNA before bisulfite conversion. After bisulfite conversion, the converted libraries were amplified with ten PCR cycles to get the high sequence diversity and low duplicate rates. PhiX spike-in was added to the libraries before sequencing as unmethylated DNA to allow for the estimation of bisulfite conversion rates. Each condition for sequencing was repeated in technical triplicate (only one triplicate was sequenced). For analysis, the MethylPy pipeline was used to compute differential methylation, which reported 22,565 individual CpG sites as differential among the 3 experiment groups and 204 differentially methylated regions.
Bulk transcriptional profiling by RNA-seq
Libraries were prepared using the Illumina TruSeq stranded mRNA kit with Illumina TruSeq unique dual indices according to the manufacturer’s instructions. We performed quality and quantity control of RNA and libraries using an Agilent 4200 TapeStation and Qubit 4, respectively. Libraries were sequenced on an Illumina NextSeq instrument to produce 75-bp single-end reads with an average sequencing depth of 14 million reads per sample. Raw reads were mapped to the mouse reference transcriptome (GRCm38) using Kallisto v.0.46.030. Subsequent analysis was conducted using the statistical computing environment R v.3.6.131 in RStudio v.1.2.5019 and Bioconductor v.3.832 in an approach similar to that in a previous study33. Briefly, transcript quantification data were summarized to genes using the tximport package34 and normalized using the trimmed mean of M values method in edgeR35. Genes with <1 CPM in n + 1 of the samples, where n is the size of the smallest group of replicates, were filtered.
Single-cell RNA-seq
Paired human colon and tumour biopsies were obtained and made into single-cell suspensions. Cells were encapsulated into droplets and libraries prepared using the 10x Genomics platform, and libraries were sequenced on an Illumina NextSeq. BCL files were demultiplexed, aligned to mouse mm10 genome, filtered and unique molecular identifier-counted using CellRanger software (10x Genomics). Then downstream analysis was performed with Seurat v.3. Data were filtered to remove cells with high mitochondrial reads (>15%), low gene detection (<200) and high gene detection (>5,000). Normalization was performed with SCTransform and integrated. Subsequently, cells were clustered using Louvain clustering (0.5 resolution), and UMAP was used for visualization (assay, ‘SCT’; dims, 1:30). After normalization and clustering, we used adaptively thresholded low rank approximation36 to impute the RNA count matrix and fill in technical dropouts. Cells were then partitioned into epithelial, stromal and immune compartments based on expression of previously described marker genes in human colon single-cell RNA-seq data. The epithelial population for analysis of HOPX expression was positively selected for epithelial markers and negatively selected for markers of other populations. Cycling epithelial cells were identified by cell cycle scoring with canonical markers37–40.
Cell lines
HT-29, Caco-2, RKO and HCT116 (ATCC) cell lines were cultured in DMEM (11965-084, Life Technologies) supplemented with 100 μM sodium pyruvate (11-360-070, Thermo Fisher Scientific), non-essential amino acids (11140-050, Thermo Fisher Scientific), 55 μM 2-mercaptoethanol (21985023, Gibco) and 100 U ml−1 of penicillin–streptomycin (15140122, Invitrogen) or in DMEM, no glucose (11966-025, Life Technologies) supplemented with 100 U ml−1 of penicillin–streptomycin (15140122, Invitrogen) and 5 mM or 17.5 mM D-glucose (0188, Amresco). Cell cycle phase assays were performed with the Click-iT Plus EdU Alexa Fluor 647 Flow Cytometry Assay Kit (C10635, Life Technologies) according to the manufacturer’s instructions. All cell lines were confirmed as mycoplasma-negative, and identities were verified using short tandem repeat profiling.
To quantify proliferation, cells were plated at a seeding density of 100,000 cells per well for 12-well plates or 50,000 cells per 24-well plates on day 0. The medium was changed on the following day, and cells were treated with varying concentrations of BHB and a glucose gradient (0.5, 10, 17.5, 20, 40 mM) for 3 days or 6 days. On day 3 or day 6, proliferation was measured with a Click-iT EdU Cell Proliferation Kit (C10337, Thermo Fisher Scientific) by fluorescence microscopy imaging according to the manufacturer’s protocol. In brief, cells were fixed and permeabilized, and EdU detection reagent was used to stain DNA. Cells were imaged on the same day by fluorescence microscopy using the fluorescein isothiocyanate channel. Images were subsequently quantified with Fiji software using MFI.
Penn CRC cohort
Tissue and plasma samples were obtained from 41 patients with CRC seen at the Hospital of the University of Pennsylvania after informed consent was obtained. This biosample collection study was approved by the University of Pennsylvania Institutional Review Board, protocol number 827759. Briefly, a blood sample was obtained before colectomy from each patient from which serum was obtained. Fresh tissue from each patient, including benign colon and CRC, was obtained from the surgical specimen.
Human organoids
Adult human organoids were prepared from tissue biopsies obtained from the Penn CRC cohort (IRB 827759) and the Northwell Health cohort (IRB TAP1810).
All patients undergoing primary resection of a CRC tumour at the Hospital of the University of Pennsylvania were approached and asked if they would be willing to participate in a CRC biobanking study in which a blood sample and extra tumour tissue would be collected. This study did not provide compensation to participants, and the only biases in the patients enrolled would result from the fact that recruitment was performed at a tertiary referral centre.
Paediatric organoids were generated from mucosal biopsies from patients under a Children’s Hospital of Philadelphia Institutional Review Board-approved protocol (2002-07-2805), and parents of all patients provided written informed consent. Colonoscopy was performed for the following reasons: rectal bleeding, diarrhoea, abdominal pain or poor growth; however, healthy control designation was determined after confirmation of normal histology.
Statistical analysis and reproducibility
Data are presented as mean ± s.e.m. Replicates represent biologically independent samples. The significance of the differences between groups was evaluated using analysis of variance or Mann–Whitney U-test unless indicated otherwise, and P < 0.05 was considered significant. Two-sided testing was used. In the figures, asterisks denote statistical significance (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). The exact n and P values and the statistical test used for each panel are reported in Supplementary Table 3. Each experiment is representative of two or more independent repetitions. Statistical analysis was performed in GraphPad PRISM 6. Graphics were generated in BioRender or iStockphoto.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Raw sequencing data for this study are available at NCBI GEO under accession numbers PRJNA809364 (bulk RNA-seq), GSE196964 (single-cell RNA-seq), PRJNA808110 (ChIP–seq) and PRJNA809037 (bisulfite sequencing). Source data are provided with this paper.
Extended Data
Extended Data Fig. 1 |. The impact of ketogenic diets on intestinal tumour growth.
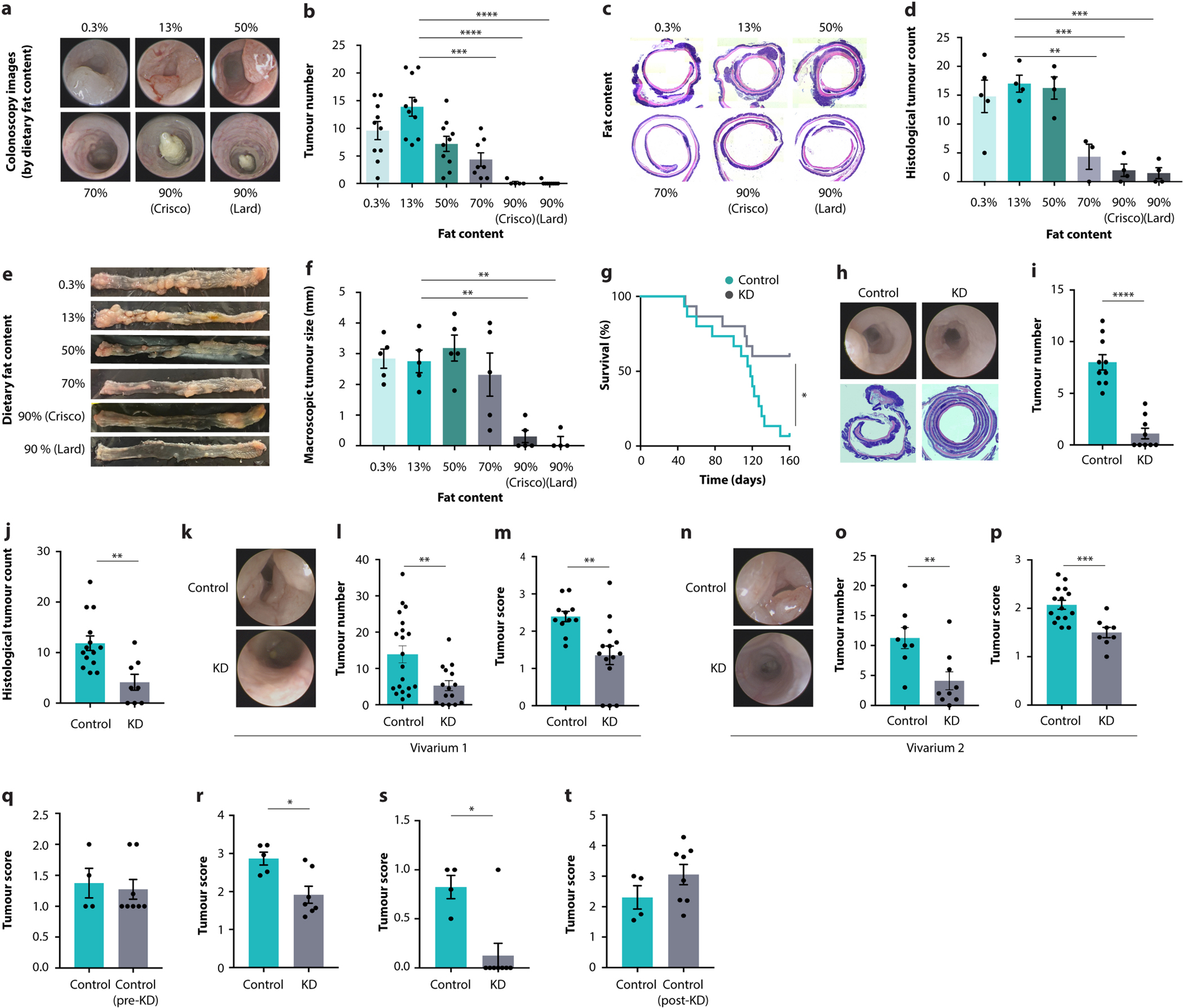
a–f, Representative colonoscopy images (a), colonoscopy-based tumour counts (b), representative histological images (c), histological tumour counts (d), representative macroscopic images (e), and tumour size quantification (f) of AOM/DSS-treated mice fed six different diets. g, Survival of AOM/DSS-treated mice fed a KD or control diet. h–j, Representative colonoscopy and histology images (h), colonoscopy-based tumour numbers (i) and histological tumour count (j) in CDX2CreERT-Apcfl/fl mice fed a KD or control diet. k–p, Representative colonoscopy images (k, n), colonoscopy-based tumour quantification (l, o), and tumour scores (m, p) from CDX2CreERT-Apcfl/fl mice fed a KD or control diet housed in different animal vivaria. q–t, Colonoscopy-based tumour scores of AOM/DSS-treated KD-fed mice in a treatment model initiated after tumorigenesis (q, r) and a cessation model with discontinued diet (s, t). Error bars indicate means ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Exact n and p-values are presented in Supplementary Table 3.
Extended Data Fig. 2 |. The immunological and epithelial impact of ketogenic diets.

a, b, Continuous recording (a) and quantification (b) of energy intake in mice fed six different diets with the indicated fat content. Grey areas indicate the dark phase. c, Transcript levels of Il17a in colonic tumours of AOM/DSS-treated mice on KD and control diet. d–g, Representative colonoscopy and histology images (d), colonoscopy-based tumour quantification (e), tumour score by colonoscopy (f), and histological tumour counts (g) in Rag1−/− mice fed a KD or control diet. h–j, Representative colonoscopy images (h), colonoscopy-based tumour quantification (i) and tumour score (j) in CDX2CreERT-Apcfl/fl mice reconstituted with Nlrp3-deficient or -sufficient bone marrow. k–n, Representative images (k, m) and quantification (l, n) of Ki67-stained colons in healthy (k, l) and AOM/DSS-treated (m, n) KD-fed mice and controls. o, p, Representative immunohistochemistry images (o) and quantification (p) of Mcl1 staining of tumours from AOM/DSS-treated KD-fed mice and controls. Error bars indicate means ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Exact n and p-values are presented in Supplementary Table 3.
Extended Data Fig. 3 |. Inhibition of intestinal epithelial growth by beta-hydroxybutyrate.
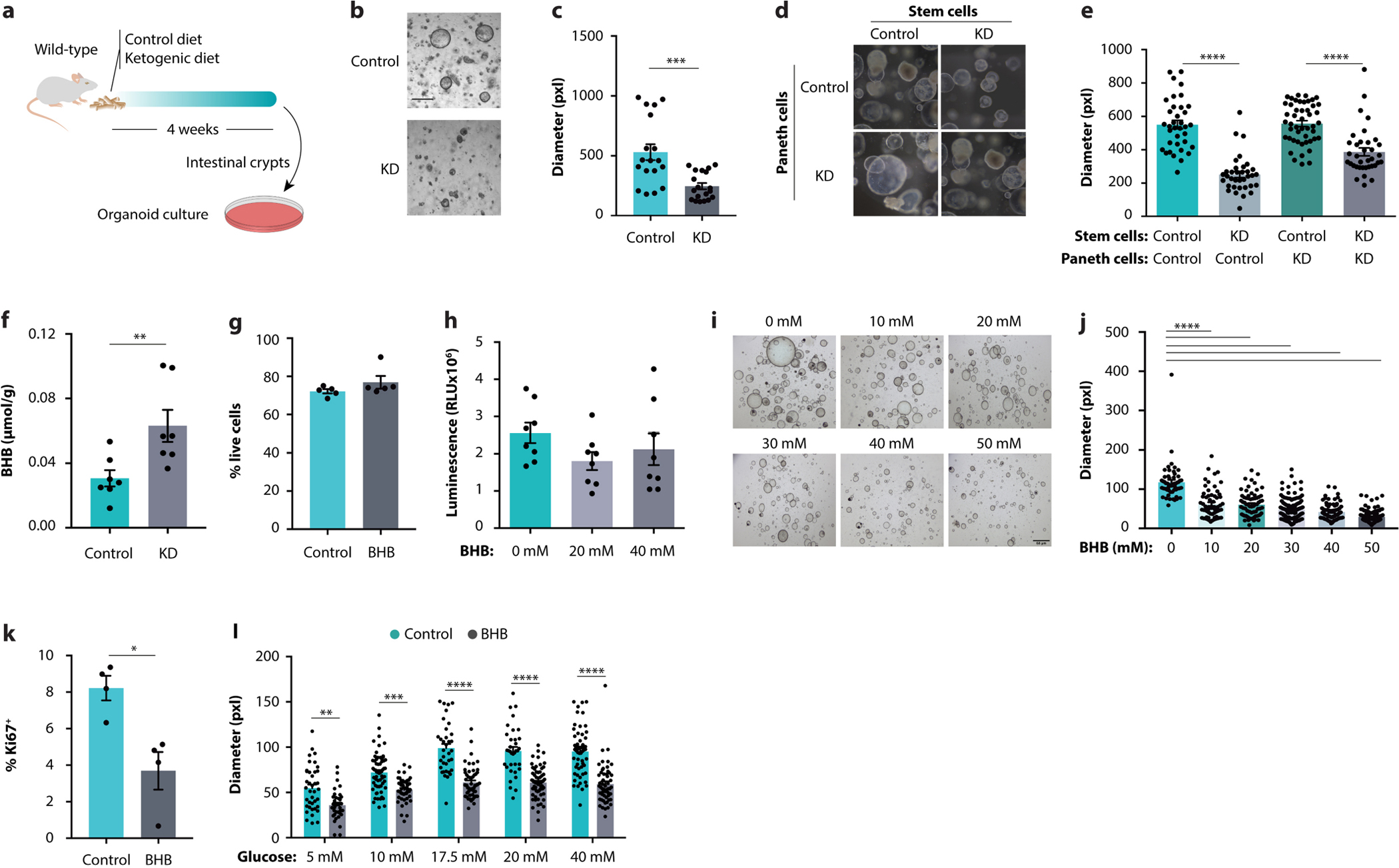
a–c, Schematic (a), representative images (b), and growth quantification (c) of intestinal organoids obtained from KD-fed mice and controls. d, e, Representative images (d) and growth quantification (e) of intestinal organoids after isolation of stem cells and Paneth cells from KD-fed mice and controls. f, Colonic concentrations of BHB in mice fed the indicated diets. g, h Viability assessments by flow cytometry (g) or luminescent ATP detection (h) of BHB-treated organoids and controls. i, j, Representative images (i) and size quantification (j) of organoids from CDX2CreERT-Apcfl/fl mice after culturing with the indicated concentrations of BHB. k, Ki67 quantification by flow cytometry in organoids treated with BHB. l, Size quantification of organoids grown in media containing different glucose concentrations, with or without BHB supplementation. Error bars indicate means ± SEM. Scale bars represent 68 μm. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Exact n and p-values are presented in Supplementary Table 3.
Extended Data Fig. 4 |. Inhibition of intestinal tumour growth by beta-hydroxybutyrate.
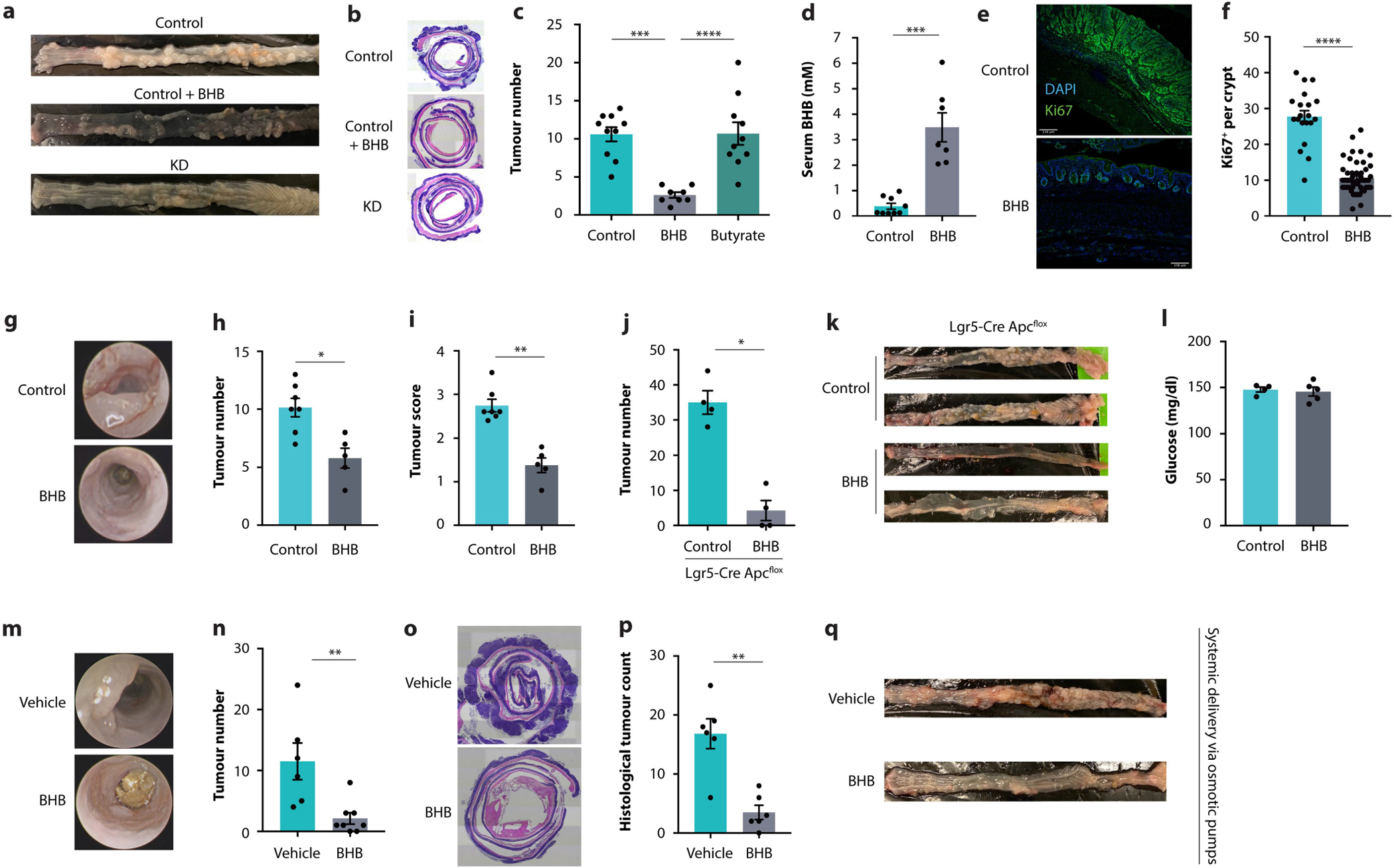
a, b, Representative macroscopic images (a) and representative histology images (b) of CDX2CreERT-Apcfl/fl mice fed either KD or control diet, with or without daily oral supplementation with BHB esters as indicated. c, Colonoscopy-based tumour quantification in AOM/DSS-treated mice receiving oral supplementation with either BHB or butyrate. d, BHB concentrations in the blood of AOM/DSS-treated mice on standard rodent diet receiving daily oral supplementation with BHB esters. e, f, Representative images (e) and quantification (f) of Ki67-stained tumours in AOM/DSS-treated mice supplemented with BHB. g–i, Representative colonoscopy images (g), colonoscopy-based tumour quantification (h), and tumour score by colonoscopy (i) in AOM/DSS-treated mice receiving daily oral supplementation with BHB esters. j, k, Macroscopic tumour counts (j) and representative macroscopic images (k) of Lgr5CreERT-Apcfl/fl mice receiving daily oral supplementation with BHB esters. l, Glucose concentrations in the blood of AOM/DSS-treated mice on standard chow receiving daily oral supplementation with BHB esters. m–q, Representative colonoscopy images (m), colonoscopy-based tumour quantification (n), representative histology images (o), histological tumour counts (p), and representative macroscopic images (q) of CDX2CreERT-Apcfl/fl mice supplemented with BHB salts by osmotic mini-pumps. Error bars indicate means ± SEM. Scale bars represent 135 μm. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Exact n and p-values are presented in Supplementary Table 3.
Extended Data Fig. 5 |. The role of Hopx in BHB-mediated tumour suppression.

a, PCA plot of global gene expression of BHB-treated and control organoids. b, Ki67 expression in organoids from wild-type and Hopx-deficient mice treated with BHB. c, Transcript levels of Hopx in organoids treated with 2-deoxyglucose (2-DG) or BHB. d, Size quantification of APKS organoids treated with 2-DG or BHB. e, Size quantification of organoids from wild-type or Hopx-deficient mice cultured at the indicated glucose concentrations, with or without BHB supplementation. f, Transcript levels of Hopx in the indicated tissues from KD-fed mice and controls. g, h, Representative images (g) and quantification (h) of GFP in the colons of Hopx-GFP reporter mice fed a KD or control diet. i–l, Representative colonoscopy images (i), macroscopic images (j), histological images (k), and colonoscopy-based tumour score (l) of AOM/DSS-treated Hopx-deficient and wild-type mice fed a KD or control diet. m–p, Blood glucose (m), circulating BHB (n), tumour number by colonoscopy (o), and colonoscopy-based tumour score (p) of Hopx-deficient mice and controls receiving BHB supplementation or 30% caloric restriction (CR). Error bars indicate means ± SEM. Scale bar represents 160 μm. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p<0.0001. Exact n and p-values are presented in Supplementary Table 3.
Extended Data Fig. 6 |. Regulation of epithelial proliferation by Hopx.

a, b, Heatmap (a) and KEGG pathway analysis (b) of differentially expressed genes determined by RNA-sequencing of AOM/DSS-treated Hopx-deficient mice and wild-type littermates on KD. c–f, Examples of genes upregulated in KD-fed AOM/DSS-treated Hopx-deficient mice compared to wild-type littermates. Error bars indicate means ± SEM. * p < 0.05, ** p < 0.01. Exact n and p-values are presented in Supplementary Table 3.
Extended Data Fig. 7 |. The impact of BHB on intestinal epithelial histone acetylation.

a, Model by which HOPX is induced through the HDAC-inhibitory activity of BHB. b, HDAC activity in organoids treated with BHB, with or without the pharmacological HDAC inhibitor vorinostat. c–e, Representative flow cytometry plot (c), relative proportion of H3K9ac (d), and normalized MFI of H3K9ac (e) in intestinal organoids treated with BHB. f, HDAC activity in intestinal epithelial cells from mice treated with BHB esters. g–i, Representative flow cytometry plot (g), relative proportion of H3K9ac (h), and normalized MFI of H3K9ac (i) in intestinal epithelial cells from KD-fed mice and controls. j, k, Representative immunofluorescence staining (j) and quantification (k) of pan-acetylation levels in colons from KD-fed mice and controls. l, Size of wild-type organoids treated with BHB and/or vorinostat. m, n, Size quantification of wild-type (m) and APKS organoids (n) exposed to BHB or butyrate. o, p, PCA plot (o) and k-means clustering (p) of global gene expression in organoids treated with BHB or the HDAC inhibitor (HDACi) vorinostat. q, Transcript abundance of Hopx determined by RNA-sequencing of organoids treated with BHB or vorinostat. r, Normalized profiles of H3K9ac in the Hopx locus from ChIP-sequencing of intestinal epithelial cells from mice treated with BHB or fed a 90% fat diet. s, Size of Hopx-deficient organoids after treatment with BHB and/or vorinostat. t, Size quantification of organoids from Hopx-deficient mice exposed to BHB or butyrate. u, Quantification of HDAC activity in wild-type and Hopx-deficient organoids treated with BHB and/or vorinostat. v, w, Tumour numbers (v) and representative colonoscopy images (w) in AOM/DSS-treated mice on KD and treated with the HDAC inhibitor vorinostat. Error bars indicate means ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Exact n and p-values are presented in Supplementary Table 3.
Extended Data Fig. 8 |. The impact of BHB on intestinal epithelial DNA methylation.

a, Hypothetical model by which HOPX is induced through BHB-mediated methylation changes. b–d, PCA plots (b, c) and scatter plot (d) of bisulphite sequencing of intestinal epithelial cells from mice treated with BHB or fed a KD compared to controls. e, Heatmap of CpG methylation status in the Hopx locus determined by bisulphite sequencing of intestinal epithelial cells from mice treated with BHB or fed a 90% fat diet. f, Transcript abundance of Hopx in organoids treated with BHB and/or 5-azacitidine (5-Aza). g, h, Size of wild-type (g) and Hopx-deficient organoids (h) after treatment with BHB and/or 5-Aza. i, j, Images of wild-type (i) and Hopx-deficient (j) organoids treated with BHB and 5-Aza. Error bars indicate means ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Exact n and p-values are presented in Supplementary Table 3.
Extended Data Fig. 9 |. A CRISPR-based approach to identify mediators of Hopx induction by BHB.
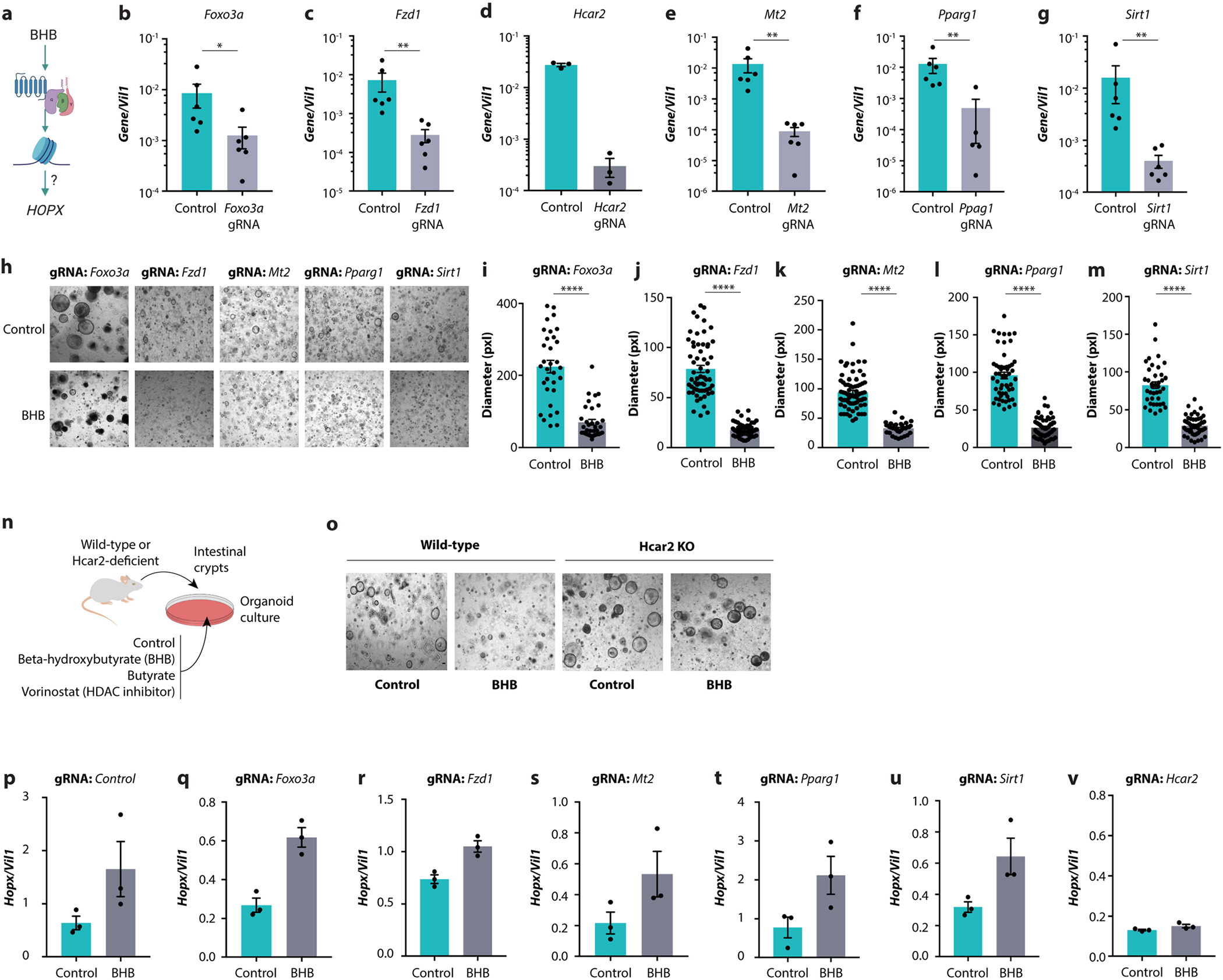
a, Hypothetical model by which HOPX is induced through BHB-mediated G protein-coupled receptor signalling. b–g, Expression of the indicated genes in organoids after CRISPR-mediated targeting thereof. h–m, Representative images (h) and size quantitation (i-m) of organoids treated with BHB after CRISPR-mediated targeting of the indicated genes. n, Schematic of organoid growth from Hcar2-deficient mice, cultured with BHB, butyrate, or vorinostat. o, Representative images of organoids from wild-type and Hcar2-deficient mice treated with BHB. p–v, Transcript levels of Hopx in organoids treated with BHB after CRISPR-mediated targeting of the indicated genes. Error bars indicate means ± SEM. **** p < 0.0001. Exact n and p-values are presented in Supplementary Table 3.
Extended Data Fig. 10 |. The impact of BHB on human organoids and colorectal cancer cell lines.
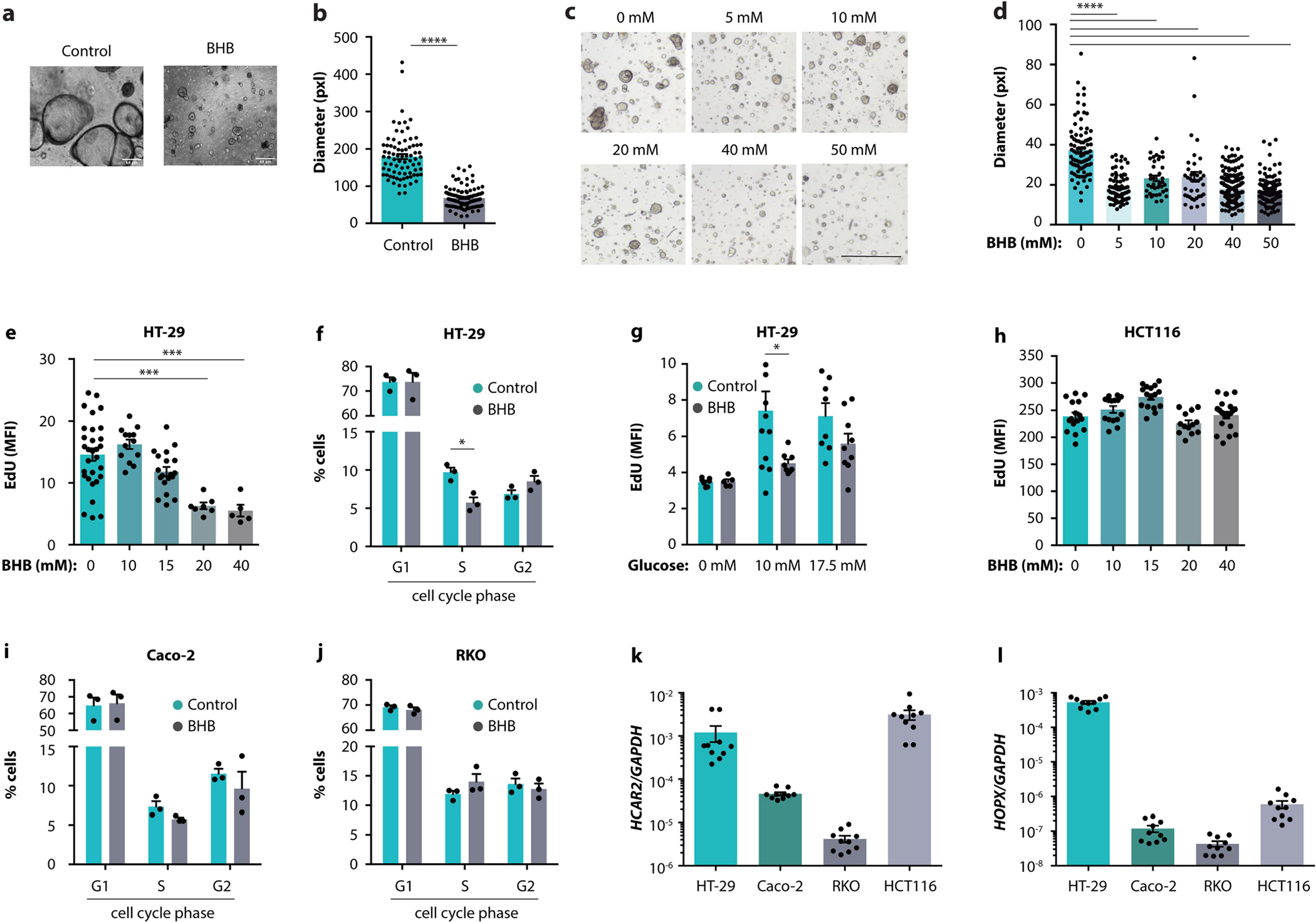
a, b, Representative images (a) and quantification (b) of organoids from a healthy paediatric donor after culturing with BHB. c, d, Representative images (c) and quantification (d) of organoids from a CRC patient after culturing with the indicated concentrations of BHB. e, EdU staining of HT-29 cells treated with the indicated concentrations of BHB. f, Cell cycle analysis of HT-29 cells treated with BHB. g, EdU staining of HT-29 cells cultured at the indicated concentrations of glucose, with or without addition of BHB. h, EdU staining of HCT116 cells treated with the indicated concentrations of BHB. i, j, Cell cycle analysis of Caco-2 cells (i) and RKO cells (j) treated with BHB. k, l, Expression of HCAR2 (g), and HOPX (h) by the indicated cell lines. Error bars indicate means ± SEM. * p < 0.05, *** p < 0.001, **** p < 0.0001. Exact n and p-values are presented in Supplementary Table 3.
Extended Data Fig. 11 |. The impact of BHB and HOPX on colorectal cancer biopsies.

a, Workflow of analysis of CRC patient cohort. b, Serum BHB levels in 41 colon cancer patients. c, d, UMAP clustering (c) and marker gene expression (d) in single-cell RNA-sequencing data of colonic biopsies from nine CRC patients. e–h, Epithelial HOPX expression binned by serum BHB levels (e), correlation of average epithelial HOPX expression with serum BHB (f), correlation of epithelial cell cycle progression with serum BHB (g), and correlation of the proportion of cells in S phase with the proportion of HOPX-expressing epithelial cells (h) as determined by single-cell RNA-sequencing of colonic biopsies of tumour and normal tissue from nine CRC patients. Error bars indicate means ± SEM. The p- and R-values were determined by linear regression. i, Cartoon illustrating that ketogenic diet induces BHB, which in turn signals via HCAR2 to induce HOPX, decelerate proliferation of the colonic crypt, and suppress cancer development. This pathway is contrasted with the mechanisms engaged by other tumour-modulatory diets. Exact n and p-values are presented in Supplementary Table 3.
Supplementary Material
Acknowledgements
We thank the members of the Levy and Thaiss laboratories for discussions and input. We acknowledge the University of Pennsylvania Molecular Pathology and Imaging Core, Department of Medicine, Gastroenterology Division, and the CDB Microscopy Core Facility for processing and imaging of tissue samples; S. Offermans, M. V. Kumar and N. Singh for providing mice; the Shalem laboratory (CHOP) for help with cell sorting; E. Krespan and D. Beiting (UPenn) for support with RNA-seq; P. W. Czyzewicz (Fox Chase Cancer Center) for assistance with animal experiments; X. Wang and A. Blanco (UPenn) for help with CRISPR experiments; J. Schug and the PSOM Next-Generation Sequencing Core for assistance with bisulfite sequencing analysis; E. Friedman and the Microbial Culture and Metabolomics Core of the PennCHOP Microbiome Program; and the Host Microbial Analytic and Repository Core of the Center for Molecular Studies in Digestive and Liver Diseases (P30DK050306). Human organoid support was from the Children’s Hospital of Philadelphia Gastrointestinal Epithelium Modelling Program. We thank J. H.-M. and S. Cherry for advice. J.A.E. is supported by NIH grant R35-HL-140018. R.J. is supported by NIH grant R01-HL-139783 and the Burroughs Wellcome Fund. A.C.W. is supported by training grant T32-AI-055400–19. P.L. is supported by training grant T32-AI-141393. S.I.G. is supported by NIH grants R01-CA-218133, R01-CA-227629 and P30-CA-006927 and by the BSF US–Israel Foundation. J.K. is supported by a Boehringer Ingelheim Fonds MD fellowship. K.M.S. is supported by DFG Fellowship DFG SCHN 1626/1–1. C.A.T. is a Pew biomedical scholar and a Kathryn W. Davis Aging Brain scholar and is supported by the NIH Director’s New Innovator Award (DP2-AG-067492), the Agilent Early Career Professor Award, and grants from the Edward Mallinckrodt Jr Foundation, the IDSA Foundation, the Thyssen Foundation, PennCHOP Microbiome Program, Penn Institute for Immunology, Penn Center for Molecular Studies in Digestive and Liver Diseases (P30-DK-050306), Penn Skin Biology and Diseases Resource-based Center (P30-AR-069589), Penn Diabetes Research Center (P30-DK-019525) and Penn Institute on Aging. M.L. is supported by the NIH Director’s New Innovator Award (DP2-AG-067511), an American Cancer Society Scholar Award, the Searle Scholar program, the Edward Mallinckrodt Jr Foundation and grants from the Abramson Cancer Center, Penn Institute for Immunology, Penn Center for Molecular Studies in Digestive and Liver Diseases, Penn Center for Precision Medicine, Penn Institute on Aging, Penn Center of Excellence in Environmental Toxicology (P30-ES-013508) and the Borrelli Family Pilot Grant in Lynch Syndrome.
Footnotes
Competing interests The authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-022-04649-6.
Peer review information Nature thanks Charles Mackay and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Reprints and permissions information is available at http://www.nature.com/reprints.
References
- 1.Dekker E, Tanis PJ, Vleugels JLA, Kasi PM & Wallace MB Colorectal cancer. Lancet 394, 1467–1480 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Brenner H, Kloor M & Pox CP Colorectal cancer. Lancet 383, 1490–1502 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Beyaz S et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 531, 53–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ringel AE et al. Obesity shapes metabolism in the tumor microenvironment to suppress anti-tumor immunity. Cell 183, 1848–1866 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu J et al. Association between markers of glucose metabolism and risk of colorectal cancer. BMJ Open 6, e011430 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aggarwal BB & Shishodia S Molecular targets of dietary agents for prevention and therapy of cancer. Biochem. Pharmacol. 71, 1397–1421 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Di Tano M et al. Synergistic effect of fasting-mimicking diet and vitamin C against KRAS mutated cancers. Nat. Commun. 11, 2332 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weng ML et al. Fasting inhibits aerobic glycolysis and proliferation in colorectal cancer via the Fdft1-mediated AKT/mTOR/HIF1α pathway suppression. Nat. Commun. 11, 1869 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Grivennikov SI et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 491, 254–258 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ang QY et al. Ketogenic diets alter the gut microbiome resulting in decreased intestinal Th17 cells. Cell 181, 1263–1275 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Youm YH et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 21, 263–269 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barker N et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Sato T et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Newman JC & Verdin E β-Hydroxybutyrate: a signaling metabolite. Annu. Rev. Nutr. 37, 51–76 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hall KD et al. Effect of a plant-based, low-fat diet versus an animal-based, ketogenic diet on ad libitum energy intake. Nat. Med. 27, 344–353 (2021). [DOI] [PubMed] [Google Scholar]
- 16.Yamashita K, Katoh H & Watanabe M The homeobox only protein homeobox (HOPX) and colorectal cancer. Int. J. Mol. Sci. 14, 23231–23243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takeda N et al. Interconversion between intestinal stem cell populations in distinct niches. Science 334, 1420–1424 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y et al. Long-term culture captures injury–repair cycles of colonic stem cells. Cell 179, 1144–1159 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimazu T et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339, 211–214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaiko GE et al. The colonic crypt protects stem cells from microbiota-derived metabolites. Cell 165, 1708–1720 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fenaux P et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 10, 223–232 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thangaraju M et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Canc. Res. 69, 2826–2832 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lien EC et al. Low glycaemic diets alter lipid metabolism to influence tumour growth. Nature 599, 302–307 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen F et al. Hop is an unusual homeobox gene that modulates cardiac development. Cell 110, 713–723 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Tunaru S et al. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat. Med. 9, 352–355 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Takeda N et al. Hopx expression defines a subset of multipotent hair follicle stem cells and a progenitor population primed to give rise to K6+ niche cells. Development 140, 1655–1664 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Becker C, Fantini MC & Neurath MF High resolution colonoscopy in live mice. Nat. Protoc. 1, 2900–2904 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Miyoshi H & Stappenbeck TS In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc. 8, 2471–2482 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato T et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Bray NL, Pimentel H, Melsted P & Pachter L Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Dessau RB & Pipper CB “R”–project for statistical computing. Ugeskr. Laeger 170, 328–330 (2008). [PubMed] [Google Scholar]
- 32.Huber W et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amorim CF et al. Variable gene expression and parasite load predict treatment outcome in cutaneous leishmaniasis. Sci. Transl. Med. 11, eaax4204 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soneson C, Love MI & Robinson MD Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 4, 1521 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robinson MD, McCarthy DJ & Smyth GK EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linderman GC et al. Zero-preserving imputation of scRNA-seq data. Nat Commun. 13, 192 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stuart T et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hafemeister C & Satija R Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smillie CS et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell 178, 714–730 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing data for this study are available at NCBI GEO under accession numbers PRJNA809364 (bulk RNA-seq), GSE196964 (single-cell RNA-seq), PRJNA808110 (ChIP–seq) and PRJNA809037 (bisulfite sequencing). Source data are provided with this paper.